

Epstein-Barr virus (EBV) infects most people worldwide and is causatively associated with several types of cancer, including ∼10% of gastric carcinomas. EBV encodes ∼80 proteins, many of which are believed to manipulate cellular regulatory pathways but are poorly characterized. The DNA damage response (DDR) is one such pathway that is critical for maintaining genome integrity and preventing cancer-associated mutations. In this study, a screen for EBV proteins that inhibit the DDR identified BKRF4 as a DDR inhibitor that binds histones and blocks their ubiquitylation at the DNA damage sites. We also present evidence that BKRF4 is expressed in both latent and lytic forms of EBV infection, where it downregulates the DDR, as well as in EBV-positive gastric tumors. The results suggest that BKRF4 could contribute to the development of gastric carcinoma through its ability to inhibit the DDR.
KEYWORDS: BKRF4, double-stranded break signaling, RNF168, histone ubiquitylation, DNA damage response, Epstein-Barr virus, histones
ABSTRACT
To replicate and persist in human cells, linear double-stranded DNA (dsDNA) viruses, such as Epstein-Barr virus (EBV), must overcome the host DNA damage response (DDR) that is triggered by the viral genomes. Since this response is necessary to maintain cellular genome integrity, its inhibition by EBV is likely an important factor in the development of cancers associated with EBV infection, including gastric carcinoma. Here we present the first extensive screen of EBV proteins that inhibit dsDNA break signaling. We identify the BKRF4 tegument protein as a DDR inhibitor that interferes with histone ubiquitylation at dsDNA breaks and recruitment of the RNF168 histone ubiquitin ligase. We further show that BKRF4 binds directly to histones through an acidic domain that targets BKRF4 to cellular chromatin and is sufficient to inhibit dsDNA break signaling. BKRF4 transcripts were detected in EBV-positive gastric carcinoma cells (AGS-EBV), and these increased in lytic infection. Silencing of BKRF4 in both latent and lytic AGS-EBV cells (but not in EBV-negative AGS cells) resulted in increased dsDNA break signaling, confirming a role for BKRF4 in DDR inhibition in the context of EBV infection and suggesting that BKRF4 is expressed in latent cells. BKRF4 was also found to be consistently expressed in EBV-positive gastric tumors in the absence of a full lytic infection. The results suggest that BKRF4 plays a role in inhibiting the cellular DDR in latent and lytic EBV infection and that the resulting accumulation of DNA damage might contribute to development of gastric carcinoma.
IMPORTANCE Epstein-Barr virus (EBV) infects most people worldwide and is causatively associated with several types of cancer, including ∼10% of gastric carcinomas. EBV encodes ∼80 proteins, many of which are believed to manipulate cellular regulatory pathways but are poorly characterized. The DNA damage response (DDR) is one such pathway that is critical for maintaining genome integrity and preventing cancer-associated mutations. In this study, a screen for EBV proteins that inhibit the DDR identified BKRF4 as a DDR inhibitor that binds histones and blocks their ubiquitylation at the DNA damage sites. We also present evidence that BKRF4 is expressed in both latent and lytic forms of EBV infection, where it downregulates the DDR, as well as in EBV-positive gastric tumors. The results suggest that BKRF4 could contribute to the development of gastric carcinoma through its ability to inhibit the DDR.
INTRODUCTION
Epstein-Barr virus (EBV) is a gammaherpesvirus that has established lifelong infections in most people due to the ability to alternate between latent and lytic modes of infection. While usually asymptomatic, EBV infection induces a variety of B-cell lymphomas as well as nasopharyngeal carcinoma and gastric carcinoma (GC), accounting for ∼10% of GCs (1–3). Due to the expression of a small subset of viral proteins and many viral microRNAs, latent infection induces cell immortalization, which is an important step in oncogenesis. However, increasing evidence indicates that sporadic switches to lytic infection and/or expression of subsets of lytic proteins drives oncogenesis (4–6). The EBV genome encodes approximately 80 proteins, ∼70 of which are expressed only in lytic infection. Combined, these proteins manipulate many cellular processes, including innate immune responses, cell cycle progression, and DNA damage responses (DDRs), to promote both cell survival and viral production. However, the functions of most EBV lytic proteins are incompletely understood or completely unknown.
EBV and herpesviruses in general have a complicated relationship with the cellular DNA damage response (DDR), both activating and inactivating different steps in the response (7–10). The cell responds to DNA double-strand breaks (DSBs) by a cascade of events beginning with MRE11-RAD50-NBS1 (MRN) binding to the double-stranded DNA (dsDNA) break, which recruits and activates ATM, a phosphatidylinositol 3-kinase (PI3K) protein kinase. Phosphorylation of the histone variant H2AX by ATM generates a histone mark, γ-H2AX, that is then read by MDC1. Next, the phosphorylation of MDC1 by ATM induces the recruitment of RNF8, an E3 ligase that initiates a ubiquitylation cascade at the break by polyubiquitylating histone H1 (11–14). Following its recruitment to DSBs through its interaction with polyubiquitylating histone H1, the E3 ligase RNF168 monoubiquitylates histone H2A at lysines 13 and 15 (15), a histone mark that is subsequently read by 53BP1, a DNA repair factor that promotes nonhomologous end joining by preventing resection of the broken ends (16, 17). DSB signaling also induces a transient p53 induction and activation and cell cycle arrest until the DNA break is repaired. Failure to repair the break or aberrant DDR signaling results in sustained p53 activation and apoptosis.
As their DNA ends mimic dsDNA breaks, linear dsDNA viruses, such as EBV, would typically elicit a DDR upon entry into the nucleus if not countered by viral proteins. Indeed, it has been shown that incoming herpes simplex virus 1 (HSV-1) genomes are detected by the DNA repair machinery but that histone ubiquitylation and recruitment of repair proteins are inhibited by the action of the viral immediate early (IE) ubiquitin ligase ICP0, which targets RNF8 and RNF168 for degradation (18, 19). For EBV and Kaposi's sarcoma-associated herpesvirus (KSHV), it has been shown that ATM kinase becomes activated upon initial infection of primary B cells and endothelial cells, respectively (20–22). For EBV, this host response activates cell cycle arrest and limits cell transformation (20, 21). There is also reason to believe that ATM activation by EBV may be intentional, as ATM activation is required for EBV lytic infection. Indeed, ATM inhibition or silencing has been reported by several labs to inhibit reactivation of EBV to the lytic cycle in gastric carcinoma, nasopharyngeal carcinoma, and Burkitt's lymphoma cells (23–25). Moreover, ATM and several other proteins from the DDR pathway have been detected at the sites of EBV lytic DNA replication, suggesting that they play roles in EBV DNA replication (24, 26, 27). The EBV BZLF1 immediate early protein and the BGLF4 viral kinase have been implicated in EBV-induced ATM activation, as either protein expressed on its own induces ATM activation (25, 28).
Despite ATM activation by EBV infection, activation of p53 and apoptosis are avoided, indicating that EBV has mechanisms to interfere with sustained DSB signaling. Interestingly, BZLF1 has been shown to concomitantly activate ATM and interfere with the formation of 53BP1 foci in response to DNA damage (29). Inhibition of the DDR was reported to involve failure to recruit RNF8 to DNA damage sites, due to the ability of BZLF1 to interfere with the Mdc-RNF8 interaction (29). However, few EBV proteins have been studied for their effects on the DDR, and given the importance of DDR modulation for EBV infection, it seemed likely that multiple mechanisms would exist to commandeer this pathway.
The functions of many of the ∼70 EBV lytic proteins are poorly characterized, although it is assumed that they have roles in manipulating cellular processes. To identify cellular functions for EBV proteins, we generated a library for expression of individual EBV lytic proteins in human cells with C-terminal FLAG tags (30). The library was characterized for efficiency of expression of full-length proteins and subcellular localization (30). Viable constructs were then screened for the ability to disrupt or alter PML nuclear bodies and contribute to cell cycle arrest at the G1/S interface (as is typical of EBV lytic infection), leading to new functions for several EBV proteins (30, 31). In this study, we screened the library of 47 viable EBV proteins for the ability to interfere with cellular DSB signaling as detected by 53BP1 foci. This led to the identification of an EBV tegument protein that binds to histones through an acidic region and inhibits the ubiquitylation cascade that occurs at DSBs.
RESULTS
Screen of EBV proteins for inhibition of 53BP1 focus formation.
To identify EBV proteins that inhibit DSB signaling, 47 different epitope-tagged EBV lytic cycle proteins were expressed individually in U2OS cells, followed by a brief etoposide treatment to induce DNA damage. Cells were then stained with anti-FLAG to identify cells expressing and not expressing the FLAG-tagged viral protein (or with anti-myc in the case of myc-tagged BBLF2/3), as well as with 53BP1 antibody. All visible 53BP1 foci (regardless of intensity) were counted in 50 FLAG-positive and 50 FLAG-negative cells on the same slide for each viral protein, and average values were compared (Table 1). The ICP0 protein of HSV-1 was included as a positive control, as it is known to induce the degradation of RNF8 and RNF168 (18), which should inhibit formation of 53BP1 foci. As expected, the 53BP1 focus numbers in cells expressing ICP0 were 3- to 4-fold lower than those in neighboring cells not expressing ICP0 (ratio, 0.29). For most EBV proteins, the ratio of FLAG-positive over FLAG-negative cells was close to 1, indicating that the viral proteins did not affect formation of 53BP1 foci. For EBV proteins that appeared to increase or decrease 53BP1 foci in the initial screen, the experiment was repeated two additional times and overall fold changes, standard deviations, and P values for foci were calculated (Table 1). These analyses identified 9 EBV proteins that consistently downregulated 53BP1 foci by 30% or more (with P values of <0.05), including BZLF1, which was previously reported to inhibit recruitment of RNF8 (29). These are indicated by shading in Table 1.
TABLE 1.
Effects of EBV proteins on 53BP1 focus formation after etoposide treatmenta

Number of 53BP1 foci in cells expressing the indicated protein (FLAG positive) were counted for 50 cells and compared to 50 cells on the same slide that are not expressing the protein (FLAG negative). Ratios of FLAG-positive to FLAG-negatve focus numbers are shown. Standard deviations and P values were calculated from 3 to 4 independent experiments. EBV proteins that consistently inhibited 53BP1 focus formation (P value < 0.05) are shaded. NA, not applicable.
BKRF4 inhibits ubiquitylation at DSBs through its N-terminal domain.
One of the EBV proteins that consistently inhibited the formation of 53BP1 foci was a tegument protein (32). Tegument proteins are of interest because proteins in this compartment of the virion typically function to alter cellular processes immediately upon viral infection. Figure 1A (top row) shows the decrease in 53BP1 foci observed in cells expressing FLAG-BKRF4 compared to the neighboring cells. We then examined two earlier steps in the DDR. Ubiquitin conjugation to histones and other proteins at DNA breaks can be detected with the FK2 antibody. Comparison of the numbers of FK2 foci in cells expressing and not expressing BKRF4 after etoposide treatment showed that this step of the DDR was also severely impaired by BKRF4 (Fig. 1A, second row). However, the formation of γ-H2AX foci, which occurs earlier in the DDR, was unaffected by BKRF4 expression (Fig. 1A, bottom row).
FIG 1.

Effect of BKRF4 on DSB signaling. (A) U2OS cells were transiently transfected with FLAG-BKRF4 expression plasmid, and then cells were treated for 2 h with etoposide (10 μg/ml for 53BP1 and FK2 foci and 1 μg/ml for γ-H2AX foci) to induce DNA damage. Cells were stained with antibody against FLAG and antibody against either 53BP1, conjugated ubiquitin (FK2), or γ-H2AX to mark DDR foci and then counterstained with DAPI. DDR foci were counted in 50 FLAG-positive and 50 FLAG-negative cells on the same slides. The distribution of the cell focus numbers for one experiment is shown in each cluster diagram. The bar graphs show the average focus values relative to FLAG-negative control (ctl) cells, with SDs and P values (*, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001) from 3 independent experiments. (B) Same experiment as for panel A except that FLAG-BKRF4N and FLAG-BKRF4C expression plasmids were used. Scale bars = 10 μm.
The sequence of BKRF4 is shown in Fig. 2A. Although it contains no recognizable enzymatic or structural domains, the N-terminal half (BKRF4N) has many acidic residues, while the C-terminal half (BKRF4C) is very proline rich. Therefore, we examined the effects of these two halves of BKRF4 (Fig. 2B). As shown in Fig. 1B, FLAG-BKRF4N but not FLAG-BKRF4C inhibited the formation of 53BP1 and conjugated ubiquitin (FK2) foci in response to etoposide treatment.
FIG 2.
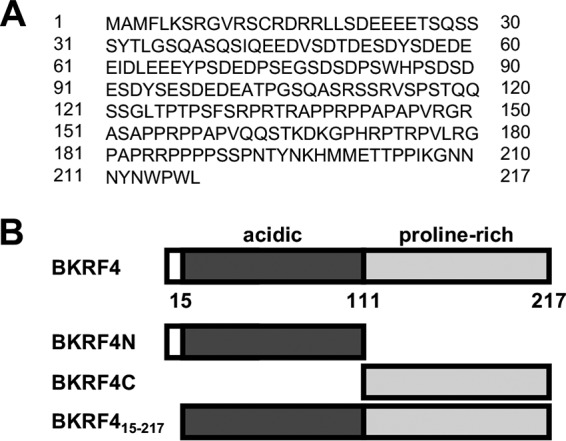
BKRF4 amino acid sequence. (A) The amino acid sequence of BKRF4 is shown. (B) Schematic of the arrangement of the BKRF4 acidic and proline-rich domains and the BKRF4 mutants generated.
One possible reason for the inhibition of FK2 foci by BKRF4 would be failure to recruit RNF168 at DSBs. Currently available RNF168 antibodies are not of sufficient specificity to reliably detect RNF168 in DDR foci in the above-described assay. We generated a doxycycline (Dox)-inducible green fluorescent protein (GFP)-RNF168 U2OS 2-6-5 cell line to visualize the recruitment of the protein to DSBs. In these experiments, cells transfected with plasmids expressing BKRF4 or BKRF4N or empty plasmid were Dox treated to induce GFP-RNF168 expression and then treated with etoposide, and the recruitment of GFP-RNF168 to DSBs was quantified by counting the γ-H2AX-positive GFP-RNF168 foci in 50 cells in three independent experiments (Fig. 3A and B). Cells expressing BKRF4 or BKRF4N were found to have decreased GFP-RNF168 foci at the DNA breaks, although the expression of GFP-RNF168 was not significantly affected (Fig. 3C). These results support a model where BKRF4 inhibits RNF168-induced ubiquitylation of H2A on the chromatin surrounding the break, at least in part by inhibiting the recruitment of RNF168.
FIG 3.
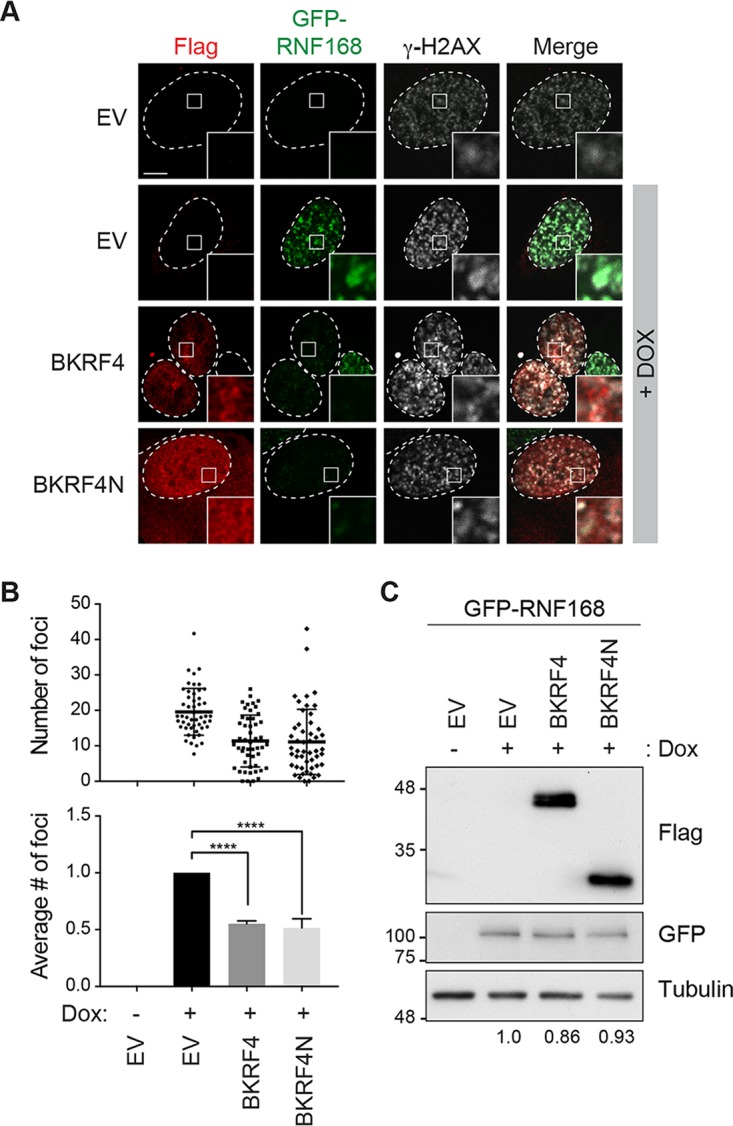
Effect of BKRF4 on RNF168 recruitment to dsDNA breaks. (A) U2OS 2-6-5 cells with inducible GFP-RNF168 were transiently transfected with plasmids expressing FLAG-tagged BKRF4 or BKRF4N or empty plasmid (EV), GFP-RNF168 expression was induced with doxycycline for 24 h, and cells were then treated with 10 μg/ml of etoposide for 2 h to induce DNA damage. Cells were processed for FLAG (red) and γ-H2AX (white) immunofluorescence, and GFP foci were also visualized by fluorescence microscopy. In all micrographs, dashed lines indicate the nucleus outline (as determined by DAPI staining; not shown), and insets represent ×10 magnifications of the indicated fields. Scale bar = 5 μm. (B) GFP-RNF168 foci were counted in at least 50 cells expressing the indicated FLAG-tagged protein (or EV control) for three independent experiments; distributions of the focus numbers are shown in the cluster diagram. Mean values ± SDs relative to control cells for three independent experiments are also shown in the bar graph. P values are indicated as in Fig. 1. (C) Western blots are shown for samples generated as for panel A using antibodies against GFP and FLAG (and tubulin as a loading control). Values under the lanes show expression levels of GFP-RNF168 (normalized to tubulin) relative to empty vector control.
BKRF4 is highly associated with histones.
To gain insight into how BKRF4 might inhibit RNF168 recruitment, we conducted affinity purification-mass spectrometry (AP-MS) experiments on BKRF4 to identify its cellular protein interactions. To this end, FLAG-tagged BKRF4 was expressed in 293T cells, then BKRF4 and associated cellular proteins were recovered on anti-FLAG resin, and eluted proteins were trypsinized and analyzed by liquid chromatography-tandem mass spectroscopy (LC-MS/MS). 293T cells are the cells of choice for AP-MS experiments because their extensive use by proteomics experts has led to the development of a database (CRAPome [33]) for assessing and eliminating nonspecific/background interactions (http://crapome.org/). We conducted three independent experiments with BKRF4 under stringent salt conditions (250 mM NaCl) designed to limit nonspecific interactions. Interactors that were recovered in all three BKRF4 experiments at levels significantly above that seen in the FLAG-LacZ negative control or average values in the CRAPome database of nonspecific interactions are shown in Table 2. Histones were overwhelmingly the most frequently recovered proteins, and most of the other recovered proteins were chromatin associated. This profile suggested that BKRF4 might bind directly to histones and recover other proteins indirectly through their association with chromatin.
TABLE 2.
Affinity purification-mass spectrometry of BKRF4 reveals histone interactionsa

Total spectral counts for three independent experiments with FLAG-BKRF4 or FLAG-LacZ (negative control) are shown. Histone interactors are shaded. Average spectral counts reported for each protein in the CRAPome database are also shown for comparison.
BKRF4 binds directly to histones through its N-terminal domain.
The AP-MS profile of BKRF4 suggested that it might bind directly to histones. To test this, we expressed 6×His-BKRF4-glutathione S-transferase (GST) in Escherichia coli and partially purified it on metal-chelating resin. In parallel, we generated histone octamers by assembling histones that were produced in E. coli (34). The BKRF4 protein was immobilized on glutathione resin and then incubated with the purified histone octamers. After washing, bound proteins were eluted from the resin with glutathione and compared to proteins that had flowed through the resin. Identical experiments were performed with 6×His-GST purified on metal chelating resin as a negative control. Figure 4A shows that histone octamers were efficiently bound by BKRF4 but not by the 6×His-GST negative control.
FIG 4.

In vitro histone binding assays. 6×His-BKRF4-GST (A, B, and C), 6×His-BKRF4N-GST (D, E, F, and G), or 6×His-GST (left sections of all panels) were generated in E. coli, purified on metal-chelating resin, and then loaded onto glutathione resin. The resin was then incubated with purified recombinant histone octamers (A and D), H3/H4 tetramers (B and E), or H2A/H2B dimers (C, F, and G). Unbound proteins were collected in the flowthrough (FT), and the resin was eluted with glutathione (Elution). Samples of these fractions along with input GST proteins and histones were analyzed by SDS-PAGE and Coomassie staining. In panel G, BKRF4N binding to H2A/H2B dimers with acidic patch mutations (BKRF4N + mutant) is compared to binding to WT H2A/H2B dimers (BKRF4N + H2A/H2B).
Histone octamers are comprised of H2A/H2B dimers and H3/H4 tetramers. To determine whether BKRF4 had specificity for either of these subcomplexes, H2A/H2B dimers and H3/H4 tetramers were assembled from recombinant histones, and the binding experiments with 6×His-BKRF4-GST and 6×His-GST were repeated (Fig. 4B and C). Both the H2A/H2B dimers and H3/H4 tetramers were efficiently bound by BKRF4 but not by 6×His-GST.
Since BKRF4N was sufficient to inhibit the DDR, we also asked whether it mediated the interaction with histones. To determine this, we generated and partially purified 6×His-BKRF4N-GST from E. coli and tested its ability to bind purified recombinant histones in GST pulldown assays. Like BKRF4, BKRF4N efficiently bound histone octamers, H3/H4 tetramers, and H2A/H2B dimers (Fig. 4D, E, and F), indicating that the N-terminal acid domain is responsible for histone interactions.
Two herpesvirus proteins have been reported to bind histones octamers, LANA from KSHV and IE1 from cytomegalovirus (35, 36). In both cases these interactions involve contact with the acid patch in H2A/H2B dimers. Our finding that BKRF4 could bind to both H2A/H2B dimers and H3/H4 tetramers suggested that the acid patch was not required for binding. However, it was also possible that the dimers and tetramers were bound through two different mechanisms. To test the importance of the acid patch for H2A/H2B binding, we generated H2A/H2B dimers from a mutant form of H2A in which 5 acidic residues that form the acid patch were replaced with alanines and compared the retention of these dimers by BKRF4N to wild-type (WT) H2A/H2B dimers (Fig. 4G). Both forms of H2A/H2B dimers were efficiently bound by BKRF4N, confirming that BKRF4 interacts with histones through a different mechanism than LANA and IE1.
BKRF4 localizes to cellular chromatin through its N-terminal domain.
The ability of BKRF4 to bind histones suggested that BKRF4 may be associated with cellular chromatin. We conducted two sets of experiments to test this possibility. First, we expressed FLAG-tagged BKRF4 in a nasopharyngeal carcinoma cell line (CNE2Z), stained the cells with 4′,6-diamidino-2-phenylindole (DAPI) and anti-FLAG antibody, and looked for colocalization of FLAG-BKRF4 with the condensed DNA in mitotic cells. As shown in Fig. 5A (top row), a significant proportion of BKRF4 clearly localized to metaphase chromatin, and this was observed in all 8 of the metaphase cells expressing FLAG-BKRF4 that we found.
FIG 5.

Chromatin localization of BKRF4 and BKRF4N. (A) CNE2Z cells were transiently transfected with plasmids expressing FLAG-BKRF4 or the indicated FLAG-tagged BKRF4 mutant. Log-phase cells were fixed and stained with anti-FLAG antibody and DAPI. Scale bar = 10 μm. (B and C) 293T cells were transiently transfected with plasmids expressing FLAG-BKRF4 (B) or the indicated FLAG-tagged BKRF4 mutant (C). Log-phase cells were lysed (total fraction in panel C) and separated into soluble and insoluble fractions. Chromatin-associated proteins in the insoluble fraction were then solubilized by DNase digestions to generate the chromatin-associated fraction (chromatin). Equal cell equivalents of each fraction were analyzed by Western blotting with anti-FLAG antibody. The same fractions were probed with antibodies against histone H4 and Nm23-H1 as markers of chromatin-associated and soluble proteins.
We also examined chromatin association of BKRF4 by biochemical fractionation. In this case, FLAG-BKRF4 was expressed in 293T cells, and then lysates from log-phase cells were separated into soluble and pellet fractions. The pellet fraction was then treated with DNase to resolubilize chromatin-associated proteins (chromatin-associated fraction). Equal cell equivalents of the soluble and chromatin-associated fractions were then compared by Western blotting with anti-FLAG antibody (Fig. 5B). To verify the clean separation of soluble and chromatin fractions, these fractions were also probed with antibodies against histone H4 and NM23-H1 (37), as markers for chromatin-associated and cytoplasmic proteins, respectively. The results show that BKRF4 was distributed approximately evenly between the soluble and chromatin-bound fractions. We also conducted the same experiment after etoposide treatment of the cells but found that this did not change the distribution of BKRF4 (Fig. 5B).
The chromatin localization of FLAG-BKRF4N and FLAG-BKRF4C was also examined. Immunofluorescence microscopy (in CNE2Z cells) showed that BKRFN was tightly associated with the condensed chromatin in metaphase cells, whereas BKRFC was excluded from the chromatin (Fig. 5A). The same localization was observed in all metaphase cells that we found expressing FLAG-BKRF4N or FLAG-BKRF4C. Similarly, biochemical fractionation of log-phase 293T cells showed that most of the BKRFN was chromatin associated, while BKRF4C was in the soluble fraction (Fig. 5C). In addition to the large acidic region, BKRF4N has a sequence at its N terminus (3-MFLKS-7) with similarity to the LANA peptide (6-MRLRS-10) that mediates interactions with the H2A/H2B acid patch (35). The finding that BKRF4 binding to histones does not require the acid patch suggests that this sequence is not important for histone binding. However, to determine if it made some contribution to chromatin targeting, we examined the chromatin association of FLAG-BKRF415–217, which lacks this sequence (Fig. 2B). Both immunofluorescence microscopy and biochemical fractionation approaches showed that FLAG-BKRF415–217 was tightly associated with cellular chromatin (Fig. 5A and C). Together, the results indicate that the BKRF4 acidic region mediates interactions with cellular chromatin.
BKRF4 expression and effects on the DDR in EBV infection.
We previously analyzed transcriptome sequencing (RNA-Seq) data on purified-poly(A)-containing RNA from eight EBV-positive gastric carcinomas and showed that in addition to the expected EBV latency genes, these tumors consistently express 18 lytic EBV transcripts, representing subsets of immediate early, early, and late genes (5). This pattern was not consistent with a lytic infection but rather suggested that specific EBV lytic proteins were being expressed in the absence of lytic infection. Interestingly, BKRF4 was one of these EBV proteins. The level of poly(A)-containing BKRF4 mRNA in each tumor sample is shown in Fig. 6A.
FIG 6.
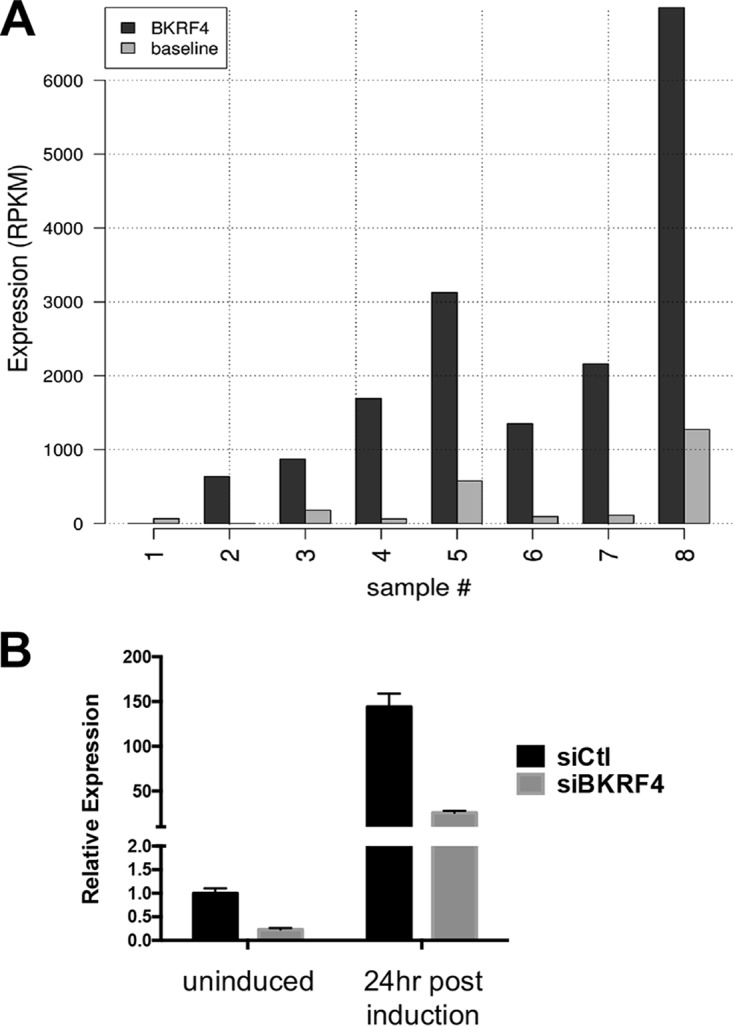
Expression of BKRF4 in AGS-EBV cells and gastric tumors. (A) Transcriptome (RNA-Seq) data from eight EBV-positive gastric carcinomas were analyzed for BKRF4 transcripts (dark bars) and shown relative to baseline values (light bars), calculated as the average expression of EBV-encoded EBNA 2, EBNA3A, EBNA3B, and EBNA3C transcripts, known not to be expressed in gastric carcinoma. (B) AGS-EBV cells were transfected with siRNA against BKRF4 (siBKRF4) or AllStars negative-control siRNA (siCtl). Twenty-four hours later, cells were either lysed (uninduced) or induced with butyrate-TPA treatment for 24 h prior to harvesting. BKRF4 transcripts in each sample were quantified by qRT-PCR and normalized to actin. Values shown (from three independent experiments) are relative to siCtl uninduced samples.
The BKRF4 expression in gastric tumors prompted us to examine the expression of BKRF4 in the EBV-positive gastric carcinoma cell line AGS-EBV. Transcripts corresponding to the BKRF4 coding sequence were quantified in AGS-EBV cells before and 24 h after reactivation to the lytic cycle by treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA) and sodium butyrate. As expected, BKRF4 transcripts increased dramatically after lytic reactivation (Fig. 6B). This signal may also include primary transcripts from BKRF3, since its 3′ untranslated region (UTR) overlaps with BKRF4. Interestingly, we also consistently detected a low level of BKRF4 transcripts in the uninduced cells. This signal in uninduced cells could be due either to BKRF4 expression in the 1 to 3% of AGS-EBV cells that spontaneously enter the lytic cycle or to a low level of BKRF4 transcripts in the latent cells. We also repeated these experiments with AGS-EBV cells after treatment with small interfering RNA (siRNA) targeted to BKRF4, confirming that these transcripts can be efficiently depleted by this approach in both latent and lytic AGS-EBV cells (Fig. 6B).
To determine whether BKRF4 impacts cellular DSB signaling in the context of EBV infection, we silenced BKRF4 in AGS-EBV cells with siRNA and then either reactivated EBV to the lytic cycle (with sodium butyrate-TPA treatment) or left the cells in the latent state. Efficient silencing of BKRF4 transcripts was confirmed by quantitative real-time PCR (qRT-PCR) as shown in Fig. 6B. Cells were then stained for 53BP1 to identify endogenous DSB signaling, as well as for BMRF1 as a marker of early lytic gene expression. Because butyrate-TPA treatment causes viral reactivation in only a subset of the cells, BMRF1 staining allowed us to distinguish the cells that entered the lytic cycle after this treatment from those cells on the same slide that remained latent. 53BP1 foci were counted separately in BMRF1-positive and BMRF1-negative cells that had received the butyrate-TPA treatment. In both cases, BKRF4 silencing caused a degree of increase in 53BP1 foci similar to that with negative-control siRNA (P values were 0.008 and 0.07 for latent and lytic cells, respectively), suggesting that BKRF4 was expressed in both cell populations (Fig. 7A). In contrast, any effect on BKRF3 expression would not have been expected to affect 53BP1 foci, since our screen showed that BKRF3 did not affect 53BP1 foci (Table 1). Latent AGS-EBV cells that had not received the butyrate-TPA treatment also showed a statistically significant increase in 53BP1 foci upon BKRF4 silencing (P = 0.0017 [Fig. 7A]). This effect of BKRF4 siRNA was not seen in parental EBV-negative AGS cells (Fig. 7A), indicating that the effect on 53BP1 foci was not due to an off-target effect of the siRNA on a cellular protein. Interestingly, the lytic cells had a reduced number of 53BP1 foci compared to the latent cells, consistent with inhibitory effects of lytic proteins on DSB signaling.
FIG 7.

BKRF4 silencing increases DSB signaling in AGS-EBV latent and lytic cells. (A) AGS-EBV cells or EBV-negative AGS cells (right side) were transfected with siRNA targeting BKRF4 (siBKRF4) or negative-control siRNA (siCtl). Twenty-four hours later, cells were harvested directly or treated with butyrate and TPA (Bu/TPA) for 24 h to reactivate EBV. Cells were then stained with antibodies against BMRF1 (to identify cells in lytic infection) and 53BP1 to mark DDR foci. DDR foci were counted in 50 cells in each category in three independent experiments. Average values, SDs, and P values (*, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001) are shown. In the Bu/TPA-treated samples, DDR focus counts are shown separately for BMRF1-positive (lytic) and BMRF1-negative (latent) cells. (B) Three independent experiments with AGS-EBV cells were performed as for panel A, except that cells were treated with 10 μg/ml of etoposide for 2 h to induce DNA damage. (C) Sample images of Bu/TPA-treated siCtl cells in B showing 53BP1 foci in lytic (BMRF1-positive) and latent (BMRF1-negative) cells. Scale bar = 10 μm.
We also repeated the BKRF4 silencing experiment (with and without reactivation of EBV to the lytic cycle) but included a mild etoposide treatment (as in the initial screen) to induce the DDR. Again, the latently infected cells that did not receive the butyrate-TPA treatment or that did not reactivate after this treatment showed a statistically significant increase in 53BP1 foci with BKRF4 silencing (P values of 0.009 and 0.003, respectively [Fig. 7B]). Note that the mild etoposide treatment did not lead to EBV reactivation, as cells did not stain for the BMRF1 lytic protein marker (data not shown). Again, the AGS-EBV cells that reactivated to the lytic cycle had fewer 53BP1 foci than the latent cells, indicating that DSB signaling was suppressed in the EBV lytic cycle. Sample images of this effect are shown in Fig. 7C. This is consistent with our finding that multiple EBV lytic proteins inhibit DSB signaling. However, silencing of BKRF4 still resulted in a detectable increase in 53BP1 foci (P = 0.08). Together the results suggest that BKRF4 plays a role in inhibiting DSB signaling in both latent and lytic EBV infection.
DISCUSSION
EBV manipulates multiple cellular processes in order to promote viral infection and the survival of infected cells. While some aspects of EBV lytic infection induce ATM kinase signaling, downstream steps of the DDR must be inhibited in order to avoid apoptosis. However, little is known about which EBV proteins function to inhibit the host DDR. Here we present an unbiased screen of EBV lytic cycle proteins that inhibit DSB signaling as detected by downregulation of 53BP1 foci after induction of DNA damage. We identify nine EBV lytic proteins with some capacity to inhibit this pathway, including one (BZLF1) that was previously reported to do so (29). While some of these proteins might affect DSB signaling indirectly, we have shown that BKRF4 binds directly to histones and interferes with ubiquitylation at sites of DNA damage.
Analysis of the steps of the DDR that are affected by BKRF4 showed that H2AX phosphorylation occurred normally but that subsequent ubiquitylation events at the DSB breaks was impaired. This ubiquitylation is carried out by two E3 ubiquitin ligases, RNF8 and RNF168. RNF8 initiates the ubiquitylation cascade by polyubiquitylating histone H1 (11–14), which serves to recruit RNF168. RNF168 then monoubiquitylates histone H2A (at lysines 13 and 15) (15). Assays measuring recruitment of RNF168 to sites of DNA damage showed that BKRF4 interfered with this recruitment, which would explain the effect of BKRF4 on the inhibition of ubiquitylation signaling and the recruitment of 53BP1 at the break. It is possible that recruitment of RNF8 might also be affected, although this remains to be tested.
In keeping with the ability of BKRF4 to interfere with RNF168 recruitment and histone ubiquitylation, we found that BKRF4 directly binds to histones. This supports a model in which BKRF4 binding to histones at DSBs physically blocks recruitment of RNF168, and possibly other factors, thereby inhibiting further DSB signaling and DNA repair. Two other viral proteins have been reported to inhibit DSB signaling through nucleosome interactions. Adenovirus core protein VII interacts with nucleosomes and downregulates DSB signaling by inhibiting phosphorylation of H2AX (38, 39). The KSHV LANA protein interacts directly with histones and inhibits H2A ubiquitylation by RNF168 (35, 40). This resembles the mechanism of DDR inhibition that we have discovered for BKRF4. However, LANA interacts specifically with H2A/H2B dimers through the acid patch on H2A, blocking the RNF168-acid patch interaction that is required for H2A ubiquitylation (40, 41). In contrast, BKRF4 can interact with H3/H4 tetramers or H2A/H2B dimers independent of the acid patch. The cytomegalovirus IE protein also interacts with the acid patch on H2A/H2B dimers (36), although its effect on DSB signaling has not been reported. Both LANA and IE1 use a short basic peptide to target the H2A/H2B acid patch. BKRF4 is unique among the known histone binding viral proteins in using an acidic region to target histones. This resembles the mechanism by which the cellular histone chaperone Nap1 targets histones. Like BKRF4, Nap1 uses an acidic domain to interact with histone and can bind both H2A/H2B dimers and H3/H4 tetramers in vitro (42–44).
BKRF4 has no homologues in alpha- or betaherpesviruses but is distantly related to ORF45 in KSHV. However, ORF45 is twice as big (407 amino acids) as BKRF4 and only 20% similar (14% identical). BKRF4 and ORF45 appear to have conserved structural functions in the virion, as they both interact with homologous virion components (EBV BGLF2 and KSHV ORF33, respectively) through a conserved C-terminal sequence, are packaged in the virion tegument, and affect the production of progeny virions (45, 46). ORF45 was also found to bind USP7 and use it to stabilize ORF33 (45). Our AP-MS data with BKRF4 also revealed an interaction with USP7, suggesting that this interaction might similarly be used to stabilize BGLF2. While BKRF4 and ORF45 may have similar structural roles in the virion, there is no evidence of conservation of their cellular effects. ORF45 functions in activating extracellular signal-related kinase (ERK) and p90 ribosomal S6 kinase to facilitate viral translation (47, 48). Unlike with BKRF4, AP-MS performed with ORF45 does not identify any significant interaction with histones (reference 49 and our unpublished data). Therefore, it is likely that these proteins have distinct cellular effects.
Our data are consistent with a low level of BKRF4 expression in latently infected AGS-EBV cells. We consistently detected BKRF4-containing transcripts in AGS-EBV. While this could be due to BKRF4 expression in the small percentage (1 to 3%) of cells spontaneously entering the lytic cycle, our finding that BKRF4 silencing increased the DSB break signaling in the latent population of AGS-EBV cells strongly suggests that some BKRF4 was being expressed in these cells. This effect was seen on both spontaneous 53BP1 foci and DNA damage-induced 53BP1 foci in latent AGS-EBV cells, whereas no effect of BKRF4 silencing was evident in EBV-negative AGS cells, arguing against an off-target effect on a cellular protein. Note that while an antibody against BKRF4 was recently developed, it appears to require high levels of BKRF4, such as those that accumulate 2 days after reactivation, for detection by immunofluorescence microscopy (reference 46 and our unpublished data). The expression of BKRF4 in AGS-EBV cells was also reported by Liang et al. (50), who further verified the presence of BKRF4 transcripts in two additional EBV-positive gastric carcinoma cell lines (SNU179 and YCCEL1). In addition, BKRF4 transcripts have been detected in the EBV-positive nasopharyngeal carcinoma cell line C666-1, which is tightly latent (51). Furthermore, our analysis of purified poly(A) mRNA from eight gastric carcinoma samples showed that BKRF4 was expressed in 7 out of 8 of these samples (reference 5 and Fig. 6A). Together, the data suggest that BKRF4 not only plays a role in lytic infection but also makes contributions to latent infection.
We have shown that BKRF4 downregulates DSB signaling in the context of both latent and lytic EBV infection. The finding that the magnitude of the effect is small is likely due to the expression of multiple EBV proteins in these cells, some of which would also impact DDRs. Indeed, we identified nine EBV proteins expressed in lytic infection that downregulate DSB signaling, suggesting some degree of redundancy in inhibiting this cellular pathway. We expect that these proteins function through a variety of mechanisms and that some may affect DSB signaling indirectly. The efficient inhibition of DSB signaling by EBV lytic proteins was demonstrated by the fact that 53BP1 foci that formed in response to DNA damage were significantly decreased in lytically infected cells compared to latent cells (Fig. 7B). This efficient inhibition is most likely due to the action of a combination of the EBV lytic proteins that we have identified as inhibiting DSB signaling. Since previous studies have shown that EBV lytic infection activates ATM (20, 23, 24), inhibition of the ATM signaling pathway downstream of ATM activation is likely important to ensure cell survival.
The finding that BKRF4 is consistently expressed in gastric carcinoma raises the possibility that BKRF4 contributes to oncogenesis. Its ability to inhibit the DDR would be expected to lead to an accumulation of DNA damage, which is an important factor in oncogenesis (52). While our screen identified BKRF4 as a DDR inhibitor, it seems likely that a protein that binds histones so efficiently may impact additional chromatin-associated functions. For example, BKRF4 might interfere with the recruitment of histone readers and writers that regulate the transcription of cellular and viral genes. Therefore, it will be interesting to investigate the full impact of BKRF4 on histones and chromatin-related processes and how these relate to EBV infection and oncogenesis.
MATERIALS AND METHODS
Expression constructs.
The library of EBV proteins was generated in pMZ3F for expression with C-terminal calmodulin binding peptide and triple FLAG tags, as previously described (30). The DDR screen also used pDH318 (53) expressing Myc-BBLF2/3 (a gift from Diane Hayward) and pCMV3FC expressing SM (two EBV proteins not in our initial library). pCMV3FC-SM was generated by PCR amplification of SM from pCDNA3 (a gift from Sankar Swaminathan) and insertion between SalI and XbaI sites in pCMV3FC. Full-length BKRF4 was subcloned into pCMV3FC from pMZS3F-BKRF4 using XbaI/XhoI sites. For the expression of BKRF4N (amino acids 1 to 111), BKRF4C (amino acids 112 to 217), and BKRF415–217 (amino acids 15 to 217) in human cells, relevant BKRF4 fragments were PCR amplified and inserted between the HindIII and BamHI sties in pCMV3FC using the following primer pairs: for BKRF4N, 5′-ACGAAGCTTATGGCCATGTTTCTGAAGTC-3′ and 5′-ACGGGATCCAGATCGTGAGGCCTGAG-3′; for BKRF4, 5′-ACGAAGCTTATGTCAAGAGTCTCGCCATCTAC-3′ and 5′-ACGGGATCCCAGCCATGGCCAATTG-3′; and for BKRF415–217, 5′-ACGAAGCTTATGGACCGGCGCCTCT-3′ and 5′-ACGGGATCCCAGCCATGGCCAATTG-3′.
To construct the BKRF4 clones for expression in bacteria, a modified version of pET15b (pET15b-GST) was made by inserting a multiple-cloning site followed by a GST tag. The primers 5′-ACGCATATGGGATCCCGGGTCGACTCGAGAATGTCCCCTATACTAGGTTAT-3′ and 5′-CGTAGATCTTCATTTTGGAGGATGGTCGC-3′ were used to PCR amplify a GST tag with a multiple-cloning site, and this fragment was inserted between the NdeI and BglII sites of pET15b. The coding sequences of full-length BKRF4 and BKRF4N were PCR amplified and inserted between the NcoI and SalI sites of pET15b-GST vector (to generate proteins with N-terminal 6×His and C-terminal GST tags) using the following primer pairs: for full-length BKRF4, 5′-ATACCATGGGCAGCAGCCATCATCATCATCATCACAGCAGCGGCCTGGTGCCGCGCGGCAGCATGGCAATGTTTCTGAAAAGC-3′ and 5′-CGTGTCGACCAGCCACGGCCAGTTATAATT-3′, and for BKRF4N, 5′ATACCATGGGCAGCAGCCATCATCATCATCATCACAGCAGCGGCCTGGTGCCGCGCGGCAGCATGGCAATGTTTCTGAAAAGC-3′ and 5′-CGTGTCGACGCTACGGCTGGCCTG-3′.
The PiggyBac (PB) transposon-based PB-TetO-GFP-RNF168 expression vector was generated in three steps. (i) The OSKM genes from PB-TAC-OSKM (kind gift from Knut Woltjen; Addgene plasmid 80481) was exchanged for a ccdB gene in a BP clonase reaction with pDONR-221 to generate PB-TAC-gateway plasmid. (ii) The IRES-mCherry reporter cassette was then removed from PB-TAC-gateway by Sfo I/ApaI restriction digestion, Klenow fill-in reaction, and ligation to generate PB-TetO-gateway. (iii) GFP-RNF168 was PCR amplified from pCDNA FRT-TO-GFP-RNF168 (54), gateway cloned into pDONR-221, and recombined into PB-TetO-gateway with LR clonase. The PB-CA-rtTA-IRES-NEO plasmid was generated by cloning an rtTA-IRES-neo fragment (obtained by NotI/SalI restriction enzyme digestion of pRetroX-Tet-On Adv [632104 from Clonetech]) into PB-CA (kind gift from Andras Nagy; Addgene plasmid 20960) by NotI/SalI restriction enzyme digestion and ligation.
Cell culture.
Human embryonic kidney 293T cells and U2OS osteosarcoma cells were cultured in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). CNE2Z EBV-negative nasopharyngeal carcinoma cells (55) were grown in α-MEM with 10% FBS, and AGS-EBV cells (EBV-positive gastric carcinoma [56]; a gift from Lindsay Hutt-Fletcher) were grown in RPMI medium with 10% FBS. U2OS 2-6-5 cells (57) (a gift from Roger Greenberg) were cultured in McCoy's medium supplemented with 10% FBS. U2OS 2-6-5 cells with Tet-inducible GFP-RNF168 were generated with a PB transposon system (58, 59). These cells were transfected with 125 ng of plasmid expressing the transposase (pCMV hypBase; Wellcome Sanger Institute), 125 ng of PB-CA-rTTA-IRES-NEO, and 1 μg of PB-TAC-GFP-RNF168. Forty-eight hours posttransfection, cells were treated with 2 μg/ml of G-418 (Sigma-Aldrich) for 5 days to select positive clones. Cells were then sorted for GFP expression with a BD SORP FACS Aria II cytometer.
DDR focus analysis in U2OS cells.
U2OS cells were grown in 6-well cluster plates on glass coverslips and transfected with 0.5 μg of expression plasmids using PolyJet in vitro DNA transfection reagent (FroggaBio) with a DNA-to-PolyJet ratio of 1:2. Twenty-four hours posttransfection, cells were treated for 2 h with 10 μg/ml (for 53BP1 and FK2 foci) or 1 μg/ml (for γ-H2AX foci) of etoposide. Cells were then fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS; 20 min), permeabilized with 0.5% Triton X-100 in PBS (5 min), and blocked with 4% bovine serum albumin (BSA) in PBS (20 min) prior to incubation with primary and secondary antibodies in 4% BSA in PBS. EBV proteins were detected by virtue of their FLAG tag using either mouse anti-FLAG M2 antibody at 1:1,000 (Sigma-Aldrich) or rabbit anti-DDDDK antibody at 1:1,000 (Bethyl Laboratories). EBV protein BBLF2/3 was detected through its myc tag with anti-c-myc monoclonal antibody 9E10 (Santa Cruz Biotechnology; 1:1,000 dilution), and ICP0 was detected with mouse anti-ICP0 antibody (Virusys; 1:2,000). γ-H2AX, conjugated ubiquitin, and 53BP1 foci were visualized using rabbit anti-γ-H2AX (Bethyl Laboratories; 1:1,000 dilution), mouse anti-ubiquitinylated protein antibody clone FK2 (EMD Millipore; 1:500 dilution), and rabbit anti-53BP1 (Bethyl Laboratories; 1:500 dilution), respectively. Primary antibodies were detected using either goat anti-mouse or goat anti-rabbit Alexa Fluor 488 or 555 secondary antibodies (Molecular Probes) at a 1:700 dilution. Coverslips were mounted onto slides using ProLong Gold antifade fluorescent mounting medium (Invitrogen) containing DAPI for visualization of nuclear DNA. Images were acquired using a 63× oil objective (NA 1.4) on a Leica DM IRE2 inverted fluorescence microscope. Images were processed using OpenLAB (version 4.0.2). For quantification, 50 FLAG-positive and 50 FLAG-negative cells were counted for each sample and graphs were plotted using Prism software.
RNF168 recruitment assay.
U2OS 2-6-5 cells with Tet-inducible GFP-RNF168 were grown in 24-well plates on glass coverslips. Cells were transfected with 250 ng of pCMV3FC-BKRF4 or pCMV3FC-BKRF4N using Lipofectamine 2000. After 6 h, expression of GFP-RNF168 was induced with 5 μg/ml of doxycycline (Sigma). Twenty-four hours posttransfection, cells were treated for 2 h with 10 μg/ml of etoposide (Cederlane). Cells were fixed with 2% (wt/vol) paraformaldehyde in PBS for 20 min at room temperature, permeabilized with 0.3% (vol/vol) Triton X-100 for 20 min at room temperature, and blocked with antibody dilution buffer (ADB; 10% normal goat serum, 0.5% NP-40, 0.5% saponin in PBS) for 30 min at room temperature. Cells were incubated with rabbit anti-FLAG at a 1:1,000 dilution (2368S; Cell Signaling) and mouse anti-γ-H2AX at 1:5,000 dilution (clone JBW301; Millipore) diluted in ADB for 2 h at room temperature, followed by a washing in PBS. Next, cells were incubated for 1 h at room temperature with secondary antibodies at a 1:1,000 dilution (Alexa Fluor 555 goat anti-rabbit or Alexa Fluor 647 goat anti-rabbit [Molecular Probes]) diluted in ADB and counterstained with DAPI (0.4 μg/ml). Cells were washed with PBS and the coverslips were mounted onto glass slides with Prolong Gold mounting agent (Invitrogen). Micrograph images were taken using a Zeiss LSM700 laser-scanning microscope equipped with a 63× oil lens.
Affinity purification-mass spectrometry.
Five 10-cm plates of 293T cells were transfected with 5 μg of pMZ3F-BKRF4 plasimd (or with pMZ3F-LacZ as a negative control) using polyethyleneimine (PEI) cellulose (Polysciences) as per the manufacturer's instructions. The cells were moved to 15-cm plates 24 h later and harvested 72 h posttransfection. Cells were lysed in 5 volumes of modified RIPA buffer (50 mM Tris-HCl [pH 8], 300 mM NaCl, 0.1% sodium deoxycholate, 0.5% NP-40, 2 mM EDTA) containing P8340 protease inhibitor cocktail (Invitrogen). BKRF4 and associated proteins were recovered on anti-FLAG resin, eluted, and trypsinized as described by Cao et al. (60). Each peptide mixture was individually loaded onto a reverse-phase microcapillary liquid trap precolumn and separated on an analytical column using an EASY-nLC high-performance liquid chromatography (HPLC) system (Proxeon) as previously described (61). Eluted peptides were directly sprayed into an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) with the collision-induced dissociation (CID) fragment method using a nanospray ion source (Proxeon) (61). For protein and peptide identification, RAW files were submitted for database searching using X! TANDEM (version 2007.07.01.3) and TPP (version 4.3) under standard workflow and a modified UniProt/Swiss-Prot protein database FASTA file. The modification consisted of adding BSA (Swiss-Prot accession number P02769). Search parameters were set to allow for two missed cleavage sites, variable modification by methionine oxidation, and one fixed modification by cysteine carbamidomethylation. A 10-ppm filter was used for peptide identification. The search results were uploaded to Prohits (62) and compared using at least 99% TPP probability.
Biochemical fractionation.
293T cells in 10-cm dishes were transfected with 5 μg of BKRF4 expression plasmid using PolyJet reagent. Cells were harvested 24 h posttransfection, washed once with PBS, lysed in 100 μl of lysis buffer (20 mM Tris-HCl [pH 7.5], 75 mM KCl, 30 mM MgCl2, 0.5% NP-40, 1 mM dithiothreitol [DTT], P8849 protease inhibitor cocktail [Sigma-Aldrich]) for 30 min on ice and fractionated as described by Kapoor et al. (63). Fifty microliters of the lysate was removed and kept on ice (whole-cell lysate sample). The remaining 50 μl of the lysate was subjected to centrifugation at 10,000 rpm for 10 min at 4°C. The supernatant was removed and kept on ice (soluble fraction). The pellet was resuspended in 50 μl of lysis buffer, incubated with 2 U of DNase I (New England BioLabs) at 37°C for 10 min, and then subjected to centrifugation at 10,000 rpm in a microcentrifuge for 10 min at 4°C. The supernatant was kept as the chromatin-associated fraction. Twenty microliters of the whole-cell lysate, soluble fraction, and chromatin-associated fraction were then boiled in SDS loading buffer and separated by SDS-PAGE.
Purification of BKRF4 for in vitro assays.
To generate recombinant 6×His-BKRF4-GST and 6×His-BKRF4N-GST proteins, E. coli BL21-pLysS containing pET15b-BKRF4-GST or pET15b-BKRF4N-GST at an optical density at 600 nm (OD600) of 0.6 was induced overnight at 18°C by addition of 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG). E. coli organisms harvested from 1 liter of culture were resuspended in 10 ml of extraction buffer (50 mM sodium phosphate [pH 7.0], 300 mM NaCl, P8849 protease inhibitor cocktail) and lysed by 3 rounds of sonication (20 s each). The lysate was clarified by centrifugation at 10,000 × g for 20 min at 4°C and then incubated with 500 μl of TALON metal affinity resin (Clontech) for 1 h at 4°C with mixing. The resin was washed 3 times with 5 ml of extraction buffer, then poured into a gravity flow column, and washed once more with 2.5 ml of extraction buffer. Protein was eluted from the column with 1 ml of elution buffer (50 mM sodium phosphate [pH 7.0], 300 mM NaCl, 150 mM imidazole, 1% glycerol, P8849 protease inhibitor cocktail) and collected in 100-μl fractions.
Purification of histones for in vitro assays.
Histones were expressed in E. coli BL21 from pET vectors as previously described (34), with a few minor modifications. Briefly, cultures at OD600s of 0.5 to 0.7 were induced at 37°C with either 0.2 or 0.4 mM IPTG for 2 or 3 h, depending on the histone. Cell pellets were resuspended in histone wash buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1 mM EDTA, 1 mM DTT) and lysed by freeze-thawing. Lysate were kept at 4°C, digested with lysozyme or homogenized (using a type B pestle), and briefly sonicated. Inclusion bodies were pelleted by centrifugation for 10 min at 25,000 × g and then washed by rehomogenizing in 0.1× culture volume in histone wash buffer containing 1% Triton X-100. The washing step was repeated at least two more times with Triton X-100 and twice without detergent. Inclusion bodies were finally dissolved in a third of the wash volume in protein unfolding buffer (7 M guanidinium, 20 mM Tris [pH 7.5], 5 mM β-mercaptoethanol) and left for gentle stirring for 1 h at room temperature. Insoluble material was removed by centrifugation at 25,000 × g for 30 min at 20°C. Following their purification on a HiTrap SP HP (GE Healthcare) cation exchange column, histones were refolded by mixing nucleosomal histones in equimolar amounts (H2A/H2B, H3/H4, or octamers), dialyzing at 4°C against at least 3 changes of 2 liters of refolding buffer (10 mM Tris-HCl [pH 7.5], 2 M NaCl, 10% glycerol, 2 mM EDTA, 5 mM β-mercaptoethanol), and slowly redialyzed against the same buffer containing 100 mM NaCl. Precipitated material was removed by centrifugation (25,000 × g for 30 min at 4°C), and H2A/H2B dimers and H3/H4 tetramers were further resolved by gel filtration. For H2A/H2B dimers with and without acid patch mutations in Fig. 4G, previously described plasmids (pET15b His-H2A and pET15b His-H2B [64]) were used to produce the histones in E. coli. Mutations in the acidic patch of H2A (E61A, E64A, D90A, E91A, and E92A) were introduced by site-directed mutagenesis using QuikChange (Stratagene), and all plasmids were sequence verified.
In vitro histone binding assay.
Thirty micrograms of purified 6×His-BKRF4-GST, 6×His-BKRF4N-GST, or 6×His-GST was combined with 10 μl of Pierce glutathione agarose (Thermo Fisher Scientific), preblocked with 2% BSA in a 50-μl final volume of binding buffer (50 mM Tris-HCl [pH 7.0], 25 mM sodium phosphate, 150 mM NaCl, 0.5% glycerol, and P8849 protease cocktail inhibitor), and mixed for 2 h at 4°C. Then 10 μg of purified histones was added in a 150-μl final volume of binding buffer and incubated with mixing for 30 min at 4°C. Proteins were eluted from the glutathione agarose by incubation with 20 μl of elution buffer (50 mM Tris-HCl [pH 8.0], 33 mM glutathione, 5% glycerol, and P8849 protease inhibitor cocktail) for 1 h at 37°C, with frequent mixing.
BKRF4 localization to mitotic chromatin.
CNE2Z cells were grown on coverslips, transfected with 0.5 μg of pCMV3FC-BKRF4 expression plasmid, and fixed 24 h posttransfection as described above for U2OS cells. Cells were stained with anti-FLAG M2 antibody at 1:1,000 (Sigma-Aldrich), followed by goat anti-mouse Alexa Fluor 488 secondary antibody, and then coverslips were mounted onto slides using ProLong Gold antifade fluorescent mounting medium (Invitrogen) containing DAPI. Images were acquired as described above for DDR foci, and mitotic cells were identified due to the condensed chromatin staining.
Detection of BKRF4 transcripts in gastric carcinoma.
EBV transcripts were analyzed from total transcriptome [RNA-Seq; poly(A)-containing RNA] data available for eight EBV-positive gastric carcinoma samples from PCAWG (65) (stomach adenocarcinoma, project code STAD-US [66]) as previously described (5). In summary, reads that did not map to the human reference sequence and that were not filtered out for low complexity and quality were aligned to the annotated EBV NCBI reference genome NC_007605 using the Bowtie2 (67) alignment algorithm in single-end mode with the -very-sensitive-local option. Levels of BKRF4 expression (in units of reads per kilobase of transcript per million mapped reads [RPKM]) in each sample were then calculated using the formula RPKM = (109 × C)/(N × L), where C is the number of reads mapping to the BKRF4 coding sequence (CDS) regions, N is the total number of mapped reads in the experiment, and L is the transcript length in base pairs. Since it is well established that EBV-encoded EBNA2, EBNA3A, EBNA3B, and EBNA3C are not expressed in gastric carcinoma, the average RPKM value for these transcripts (i.e., over their CDS regions) was used as the baseline expression to which the BKRF4 expression was compared.
Detection of BKRF4 transcripts in AGS-EBV.
AGS-EBV cells in 6-well plates were transfected with 100 pmol of BKRF4-targeted siRNA (GAAGACCCAUCUGAGGGCAGUGAUA) or AllStars negative-control siRNA (Qiagen) using Lipofectamine 2000 reagent (Thermo Fisher Scientific), and this transfection was repeated 24 h later. After an additional 24 h, cells were either harvested or induced into the lytic cycle by incubation with 3 mM sodium butyrate and 0.02 μg/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA). Twenty-four hours postinduction, cells were harvested and total RNA was extracted using TRIzol reagent (Sigma-Aldrich) according to the manufacturer's protocol. One microgram of extracted RNA from each sample was treated with 2 U of DNase I (New England BioLabs) for 10 min and then reverse transcribed in a 20-μl reaction using SuperScript IV reverse transcriptase (Invitrogen) with random hexamer primers according to the manufacturer's instructions. Quantitative real-time PCR was performed with 1/10 of the sample using SsoFastTM EvaGreen Supermix (Bio-Rad) with a total reaction volume of 10 μl in a Bio-Rad CFX384 real-time system. Primers used for mRNA quantification of viral genes and BKRF4 were as previously described (68). The relative mRNA expression level was derived from 2−ΔΔCT by use of the comparative threshold cycle (CT) method. The amount of mRNA in each sample was normalized to the amount of actin mRNA.
DDR focus analysis in AGS-EBV cells.
AGS-EBV cells were transfected with BKRF4-targeted siRNA or AllStars negative-control siRNA as described above. Cells were then either treated for 2 h with 10 μg/ml of etoposide directly or induced into the lytic cycle with sodium butyrate-TPA treatment (as described above) followed 24 h later by the etoposide treatment. Cells were then fixed and stained with 53BP1 antibody (as described above for U2OS cells) and with mouse anti-EBV EA-D-p52/50 (BMRF1; EMD Millipore; 1:1,000) to identify reactivated cells. 53BP1 foci were counted in 50 reactivated (BMRF1-positive) or latent (BMRF1-negative) cells in three independent experiments.
ACKNOWLEDGMENTS
We thank James Roh and Stephanie Gulstene for early work on the DDR screen, Daniel Durocher for helpful discussions, and Roger Greenberg for U2OS 2-6-5 cells.
This work was supported by Canadian Institutes of Health Research project grant 153014 (to L.F.), Natural Sciences and Engineering Research Council of Canada operating grant RGPIN-2016-05844, and a Scholarship for the Next Generation of Scientists from the Cancer Research Society to A.F.-T. L.F. is a tier 1 Canada Research Chair in Molecular Virology. A.F.-T. is a tier 2 Canada Research Chair in Molecular Virology and Genomic Instability and is supported by the Foundation J.-Louis Lévesque.
REFERENCES
- 1.Delecluse HJ, Feederle R, O'Sullivan B, Taniere P. 2007. Epstein Barr virus-associated tumours: an update for the attention of the working pathologist. J Clin Pathol 60:1358–1364. doi: 10.1136/jcp.2006.044586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takada K. 2000. Epstein-Barr virus and gastric carcinoma. Mol Pathol 53:255–261. doi: 10.1136/mp.53.5.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raab-Traub N. 2002. Epstein-Barr virus in the pathogenesis of NPC. Semin Cancer Biol 12:431–441. doi: 10.1016/S1044579X0200086X. [DOI] [PubMed] [Google Scholar]
- 4.Murata T. 2014. Regulation of Epstein-Barr virus reactivation from latency. Microbiol Immunol 58:307–317. doi: 10.1111/1348-0421.12155. [DOI] [PubMed] [Google Scholar]
- 5.Borozan I, Zapatka M, Frappier L, Ferretti V. 2018. Analysis of Epstein-Barr virus genomes and expression profiles in gastric adenocarcinoma. J Virol 92:e01239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE, Kenney SC. 2011. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol 85:165–177. doi: 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luftig MA. 2014. Viruses and the DNA damage response: activation and antagonism. Annu Rev Virol 1:605–625. doi: 10.1146/annurev-virology-031413-085548. [DOI] [PubMed] [Google Scholar]
- 8.Frappier L. 2016. Manipulation of PML nuclear bodies and DNA damage responses by DNA viruses, p 283–312. In Bazett-Jones DJ, Dellaire G (ed), The functional nucleus. Springer International Publishing, Basel, Switzerland. [Google Scholar]
- 9.Xiaofei E, Kowalik TF. 2014. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 6:2155–2185. doi: 10.3390/v6052155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith S, Weller SK. 2015. HSV-I and the cellular DNA damage response. Future Virol 10:383–397. doi: 10.2217/fvl.15.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D. 2007. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. 2007. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 13.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. 2007. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, Mailand N. 2015. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527:389–393. doi: 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- 15.Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. 2012. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 150:1182–1195. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 2013. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson MD, Benlekbir S, Fradet-Turcotte A, Sherker A, Julien JP, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, Durocher D. 2016. The structural basis of modified nucleosome recognition by 53BP1. Nature 536:100–103. doi: 10.1038/nature18951. [DOI] [PubMed] [Google Scholar]
- 18.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. 2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog 7:e1002084. doi: 10.1371/journal.ppat.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. 2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J 29:943–955. doi: 10.1038/emboj.2009.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, Allday MJ, Patel A, Dave SS, Kim W, Hu K, Guo J, Tainter D, Rusyn E, Luftig MA. 2010. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8:510–522. doi: 10.1016/j.chom.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikitin PA, Price AM, McFadden K, Yan CM, Luftig MA. 2014. Mitogen-induced B-cell proliferation activates Chk2-dependent G1/S cell cycle arrest. PLoS One 9:e87299. doi: 10.1371/journal.pone.0087299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh VV, Dutta D, Ansari MA, Dutta S, Chandran B. 2014. Kaposi's sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J Virol 88:2821–2834. doi: 10.1128/JVI.03126-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagemeier SR, Barlow EA, Meng Q, Kenney SC. 2012. The cellular ataxia telangiectasia-mutated kinase promotes Epstein-Barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J Virol 86:13360–13370. doi: 10.1128/JVI.01850-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hau PM, Deng W, Jia L, Yang J, Tsurumi T, Chiang AK, Huen MS, Tsao SW. 2015. Role of ATM in the formation of the replication compartment during lytic replication of Epstein-Barr virus in nasopharyngeal epithelial cells. J Virol 89:652–668. doi: 10.1128/JVI.01437-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang'ondu R, Teal S, Park R, Heston L, Delecluse H, Miller G. 2015. DNA damage signaling is induced in the absence of Epstein-Barr virus (EBV) lytic DNA replication and in response to expression of ZEBRA. PLoS One 10:e0126088. doi: 10.1371/journal.pone.0126088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem 280:8156–8163. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- 27.Kudoh A, Iwahori S, Sato Y, Nakayama S, Isomura H, Murata T, Tsurumi T. 2009. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J Virol 83:6641–6651. doi: 10.1128/JVI.00049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li R, Liao G, Nirujogi RS, Pinto SM, Shaw PG, Huang TC, Wan J, Qian J, Gowda H, Wu X, Lv DW, Zhang K, Manda SS, Pandey A, Hayward SD. 2015. Phosphoproteomic profiling reveals Epstein-Barr virus protein kinase integration of DNA damage response and mitotic signaling. PLoS Pathog 11:e1005346. doi: 10.1371/journal.ppat.1005346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Deng W, Hau PM, Liu J, Lau VM, Cheung AL, Huen MS, Tsao SW. 2015. Epstein-Barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab Invest 95:937–950. doi: 10.1038/labinvest.2015.69. [DOI] [PubMed] [Google Scholar]
- 30.Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L. 2008. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog 4:e1000100. doi: 10.1371/journal.ppat.1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paladino P, Marcon E, Greenblatt J, Frappier L. 2014. Identification of herpesvirus proteins that contribute to G1/S arrest. J Virol 88:4480–4492. doi: 10.1128/JVI.00059-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, Al-Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin ZY, Badillo BG, Pawson T, Durocher D, Coulombe B, Aebersold R, Superti-Furga G, Colinge J, Heck AJ, Choi H, Gstaiger M, Mohammed S, Cristea IM, Bennett KL, Washburn MP, Raught B, Ewing RM, Gingras AC, Nesvizhskii AI. 2013. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. 1997. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol 272:301–311. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 35.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 36.Mücke K, Paulus C, Bernhardt K, Gerrer K, Schon K, Fink A, Sauer EM, Asbach-Nitzsche A, Harwardt T, Kieninger B, Kremer W, Kalbitzer HR, Nevels M. 2014. Human cytomegalovirus major immediate early 1 protein targets host chromosomes by docking to the acidic pocket on the nucleosome surface. J Virol 88:1228–1248. doi: 10.1128/JVI.02606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murakami M, Lan K, Subramanian C, Robertson ES. 2005. Epstein-Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J Virol 79:1559–1568. doi: 10.1128/JVI.79.3.1559-1568.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avgousti DC, Della Fera AN, Otter CJ, Herrmann C, Pancholi NJ, Weitzman MD. 2017. Adenovirus core protein VII downregulates the DNA damage response on the host genome. J Virol 91:e01089-17. doi: 10.1128/JVI.01089-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avgousti DC, Herrmann C, Kulej K, Pancholi NJ, Sekulic N, Petrescu J, Molden RC, Blumenthal D, Paris AJ, Reyes ED, Ostapchuk P, Hearing P, Seeholzer SH, Worthen GS, Black BE, Garcia BA, Weitzman MD. 2016. A core viral protein binds host nucleosomes to sequester immune danger signals. Nature 535:173–177. doi: 10.1038/nature18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung JW, Agarwal P, Canny MD, Gong F, Robison AD, Finkelstein IJ, Durocher D, Miller KM. 2014. Nucleosome acidic patch promotes RNF168- and RING1B/BMI1-dependent H2AX and H2A ubiquitination and DNA damage signaling. PLoS Genet 10:e1004178. doi: 10.1371/journal.pgen.1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattiroli F, Uckelmann M, Sahtoe DD, van Dijk WJ, Sixma TK. 2014. The nucleosome acidic patch plays a critical role in RNF168-dependent ubiquitination of histone H2A. Nat Commun 5:3291. doi: 10.1038/ncomms4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andrews AJ, Downing G, Brown K, Park YJ, Luger K. 2008. A thermodynamic model for Nap1-histone interactions. J Biol Chem 283:32412–32418. doi: 10.1074/jbc.M805918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowman A, Ward R, Wiechens N, Singh V, El-Mkami H, Norman DG, Owen-Hughes T. 2011. The histone chaperones Nap1 and Vps75 bind histones H3 and H4 in a tetrameric conformation. Mol Cell 41:398–408. doi: 10.1016/j.molcel.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aguilar-Gurrieri C, Larabi A, Vinayachandran V, Patel NA, Yen K, Reja R, Ebong IO, Schoehn G, Robinson CV, Pugh BF, Panne D. 2016. Structural evidence for Nap1-dependent H2A-H2B deposition and nucleosome assembly. EMBO J 35:1465–1482. doi: 10.15252/embj.201694105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gillen J, Li W, Liang Q, Avey D, Wu J, Wu F, Myoung J, Zhu F. 2015. A survey of the interactome of Kaposi's sarcoma-associated herpesvirus ORF45 revealed its binding to viral ORF33 and cellular USP7, resulting in stabilization of ORF33 that is required for production of progeny viruses. J Virol 89:4918–4931. doi: 10.1128/JVI.02925-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masud H, Watanabe T, Yoshida M, Sato Y, Goshima F, Kimura H, Murata T. 2017. Epstein-Barr virus BKRF4 gene product is required for efficient progeny production. J Virol 91:e00975-17. doi: 10.1128/JVI.00975-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avey D, Tepper S, Li W, Turpin Z, Zhu F. 2015. Phosphoproteomic analysis of KSHV-infected cells reveals roles of ORF45-activated RSK during lytic replication. PLoS Pathog 11:e1004993. doi: 10.1371/journal.ppat.1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuang E, Tang Q, Maul GG, Zhu F. 2008. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J Virol 82:1838–1850. doi: 10.1128/JVI.02119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang Q, Yao X, Tang S, Zhang J, Yau TO, Li X, Tang CM, Kang W, Lung RW, Li JW, Chan TF, Xing R, Lu Y, Lo KW, Wong N, To KF, Yu C, Chan FK, Sung JJ, Yu J. 2014. Integrative identification of Epstein-Barr virus-associated mutations and epigenetic alterations in gastric cancer. Gastroenterology 147:1350–1362.e4. doi: 10.1053/j.gastro.2014.08.036. [DOI] [PubMed] [Google Scholar]
- 51.Hu L, Lin Z, Wu Y, Dong J, Zhao B, Cheng Y, Huang P, Xu L, Xia T, Xiong D, Wang H, Li M, Guo L, Kieff E, Zeng Y, Zhong Q, Zeng M. 2016. Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front Med 10:61–75. doi: 10.1007/s11684-016-0436-0. [DOI] [PubMed] [Google Scholar]
- 52.O'Connor MJ. 2015. Targeting the DNA damage response in cancer. Mol Cell 60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- 53.Gao Z, Krithivas A, Finan JE, Semmes OJ, Zhou S, Wang Y, Hayward SD. 1998. The Epstein-Barr virus lytic transactivator Zta interacts with the helicase-primase replication proteins. J Virol 72:8559–8567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Panier S, Ichijima Y, Fradet-Turcotte A, Leung CC, Kaustov L, Arrowsmith CH, Durocher D. 2012. Tandem protein interaction modules organize the ubiquitin-dependent response to DNA double-strand breaks. Mol Cell 47:383–395. doi: 10.1016/j.molcel.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 55.Sun Y, Hegamyer G, Cheng YJ, Hildesheim A, Chen JY, Chen IH, Cao Y, Yao KT, Colburn NH. 1992. An infrequent point mutation of the p53 gene in human nasopharyngeal carcinoma. Proc Natl Acad Sci U S A 89:6516–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- 57.Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. 2013. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20:317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yusa K, Zhou L, Li MA, Bradley A, Craig NL. 2011. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim SI, Oceguera-Yanez F, Hirohata R, Linker S, Okita K, Yamada Y, Yamamoto T, Yamanaka S, Woltjen K. 2015. KLF4 N-terminal variance modulates induced reprogramming to pluripotency. Stem Cell Rep 4:727–743. doi: 10.1016/j.stemcr.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao JY, Shire K, Landry C, Gish GD, Pawson T, Frappier L. 2014. Identification of a novel protein interaction motif in the regulatory subunit of casein kinase 2. Mol Cell Biol 34:246–258. doi: 10.1128/MCB.00968-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marcon E, Ni Z, Pu S, Turinsky AL, Trimble SS, Olsen JB, Silverman-Gavrila R, Silverman-Gavrila L, Phanse S, Guo H, Zhong G, Guo X, Young P, Bailey S, Roudeva D, Zhao D, Hewel J, Li J, Graslund S, Paduch M, Kossiakoff AA, Lupien M, Emili A, Wodak SJ, Greenblatt J. 2014. Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation. Cell Rep 8:297–310. doi: 10.1016/j.celrep.2014.05.050. [DOI] [PubMed] [Google Scholar]
- 62.Liu G, Zhang J, Larsen B, Stark C, Breitkreutz A, Lin ZY, Breitkreutz BJ, Ding Y, Colwill K, Pasculescu A, Pawson T, Wrana JL, Nesvizhskii AI, Raught B, Tyers M, Gingras AC. 2010. ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat Biotechnol 28:1015–1017. doi: 10.1038/nbt1010-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapoor P, Lavoie BD, Frappier L. 2005. EBP2 plays a key role in Epstein-Barr virus mitotic segregation and is regulated by aurora family kinases. Mol Cell Biol 25:4934–4945. doi: 10.1128/MCB.25.12.4934-4945.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 2013. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, Hemsley FM, Jennings JL, Kerr D, Klatt P, Kolar P, Kusada J, Lane DP, Laplace F, Youyong L, Nettekoven G, Ozenberger B, Peterson J, Rao TS, Remacle J, Schafer AJ, Shibata T, Stratton MR, Vockley JG, Watanabe K, Yang H, Yuen MM, Knoppers BM, Bobrow M, Cambon-Thomsen A, Dressler LG, Dyke SO, Joly Y, Kato K, Kennedy KL, Nicolas P, Parker MJ, Rial-Sebbag E, Romeo-Casabona CM, Shaw KM, Wallace S, Wiesner GL, Zeps N, Lichter P, Biankin AV, Chabannon C, Chin L, Clement B, de Alava E, Degos F, Ferguson ML, Geary P, Hayes DN, Johns AL, Kasprzyk A, Nakagawa H, Penny R, Piris MA, Sarin R, Scarpa A, van de Vijver M, Futreal PA, Aburatani H, Bayes M, Botwell DD, Campbell PJ, Estivill X, Grimmond SM, Gut I, Hirst M, Lopez-Otin C, Majumder P, Marra M, McPherson JD, Ning Z, Puente XS, Ruan Y, Stunnenberg HG, Swerdlow H, Velculescu VE, Wilson RK, Xue HH, Yang L, Spellman PT, Bader GD, Boutros PC, Flicek P, Getz G, Guigo R, Guo G, Haussler D, Heath S, Hubbard TJ, Jiang T, Jones SM, Li Q, Lopez-Bigas N, Luo R, Muthuswamy L, Ouellette BF, Pearson JV, Quesada V, Raphael BJ, Sander C, Speed TP, Stein LD, Stuart JM, Teague JW, Totoki Y, Tsunoda T, Valencia A, Wheeler DA, Wu H, Zhao S, Zhou G, Lathrop M, Thomas G, Yoshida T, Axton M, Gunter C, Miller LJ, Zhang J, Haider SA, Wang J, Yung CK, Cros A, Liang Y, Gnaneshan S, Guberman J, Hsu J, Chalmers DR, Hasel KW, Kaan TS, Lowrance WW, Masui T, Rodriguez LL, Vergely C, Bowtell DD, Cloonan N, deFazio A, Eshleman JR, Etemadmoghadam D, Gardiner BB, Kench JG, Sutherland RL, Tempero MA, Waddell NJ, Wilson PJ, Gallinger S, Tsao MS, Shaw PA, Petersen GM, Mukhopadhyay D, DePinho RA, Thayer S, Shazand K, Beck T, Sam M, Timms L, Ballin V, Lu Y, Ji J, Zhang X, Chen F, Hu X, Yang Q, Tian G, Zhang L, Xing X, Li X, Zhu Z, Yu Y, Yu J, Tost J, Brennan P, Holcatova I, Zaridze D, Brazma A, Egevard L, Prokhortchouk E, Banks RE, Uhlen M, Viksna J, Ponten F, Skryabin K, Birney E, Borg A, Borresen-Dale AL, Caldas C, Foekens JA, Martin S, Reis-Filho JS, Richardson AL, Sotiriou C, Thoms G, van't Veer L, Birnbaum D, Blanche H, Boucher P, Boyault S, Masson-Jacquemier JD, Pauporte I, Pivot X, Vincent-Salomon A, Tabone E, Theillet C, Treilleux I, Bioulac-Sage P, Decaens T, Franco D, Gut M, Samuel D, Zucman-Rossi J, Eils R, Brors B, Korbel JO, Korshunov A, Landgraf P, Lehrach H, Pfister S, Radlwimmer B, Reifenberger G, Taylor MD, von Kalle C, Majumder PP, Pederzoli P, Lawlor RA, Delledonne M, Bardelli A, Gress T, Klimstra D, Zamboni G, Nakamura Y, Miyano S, Fujimoto A, Campo E, de Sanjose S, Montserrat E, Gonzalez-Diaz M, Jares P, Himmelbauer H, Bea S, Aparicio S, Easton DF, Collins FS, Compton CC, Lander ES, Burke W, Green AR, Hamilton SR, Kallioniemi OP, Ley TJ, Liu ET, Wainwright BJ. 2010. International network of cancer genome projects. Nature 464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kalla M, Gobel C, Hammerschmidt W. 2012. The lytic phase of Epstein-Barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J Virol 86:447–458. doi: 10.1128/JVI.06314-11. [DOI] [PMC free article] [PubMed] [Google Scholar]