

ABSTRACT
The cellular protein SPOC1 (survival time-associated PHD [plant homeodomain] finger protein in ovarian cancer 1) acts as a regulator of chromatin structure and the DNA damage response. It binds H3K4me2/3-containing chromatin and promotes DNA condensation by recruiting corepressors such as KAP-1 and H3K9 methyltransferases. Previous studies identified SPOC1 as a restriction factor against human adenovirus (HAdV) infection that is antagonized by E1B-55K/E4-orf6-dependent proteasomal degradation. Here, we demonstrate that, in contrast to HAdV-infected cells, SPOC1 is transiently upregulated during the early phase of human cytomegalovirus (HCMV) replication. We show that the expression of immediate early protein 1 (IE1) is sufficient and necessary to induce SPOC1. Additionally, we discovered that during later stages of infection, SPOC1 is downregulated in a glycogen synthase kinase 3β (GSK-3β)-dependent manner. We provide evidence that SPOC1 overexpression severely impairs HCMV replication by repressing the initiation of viral immediate early (IE) gene expression. Consistently, we observed that SPOC1-depleted primary human fibroblasts displayed an augmented initiation of viral IE gene expression. This occurs in a multiplicity of infection (MOI)-dependent manner, a defining hallmark of intrinsic immunity. Interestingly, repression requires the presence of high SPOC1 levels at the start of infection, while later upregulation had no negative impact, suggesting distinct temporal roles of SPOC1 during the HCMV replicative cycle. Mechanistically, we observed a highly specific association of SPOC1 with the major immediate early promoter (MIEP), strongly suggesting that SPOC1 inhibits HCMV replication by MIEP binding and the subsequent recruitment of heterochromatin-building factors. Thus, our data add SPOC1 as a novel factor to the endowment of a host cell to restrict cytomegalovirus infections.
IMPORTANCE Accumulating evidence indicates that during millennia of coevolution, host cells have developed a sophisticated compilation of cellular factors to restrict cytomegalovirus infections. Defining this equipment is important to understand cellular barriers against viral infection and to develop strategies to utilize these factors for antiviral approaches. So far, constituents of PML nuclear bodies and interferon gamma-inducible protein 16 (IFI16) were known to mediate intrinsic immunity against HCMV. In this study, we identify the chromatin modulator SPOC1 as a novel restriction factor against HCMV. We show that preexisting high SPOC1 protein levels mediate a silencing of HCMV gene expression via a specific association with an important viral cis-regulatory element, the major immediate early promoter. Since SPOC1 expression varies between cell types, this factor may play an important role in tissue-specific defense against HCMV.
KEYWORDS: human cytomegalovirus, immediate early, intrinsic immunity, restriction factor
INTRODUCTION
Restriction factors represent a frontline defense against viral infections (1). They constitute the basis of intrinsic antiviral immunity, which is considered either part of the innate immune response or an independent, third branch of the immune system. One characteristic of these factors is their constitutive expression within the host cell, allowing a rapid response to viral infection that precedes the interferon response. The best-characterized restriction factors targeting human cytomegalovirus (HCMV) are cellular constituents of nuclear domain 10 (ND10), also called PML (promyelocytic leukemia protein) nuclear bodies, and interferon gamma-inducible protein 16 (IFI16) (2, 3). Previous studies demonstrated that several ND10 components exert their antiviral activity independently by inducing a transcriptionally inactive chromatin state around the major immediate early (IE) promoter (MIEP), which leads to a silencing of viral IE gene expression (2, 4–12). IFI16 was shown to downregulate the transcription of the viral DNA polymerase pUL54, which results in an inhibition of viral DNA synthesis (3). An additional hallmark of restriction factors is the fact that they are saturable and subject to viral countermeasures. During coevolution with its host, HCMV has evolved regulatory proteins, such as the tegument proteins pp71, pp65, and pUL97 as well as immediate early protein 1 (IE1), to counteract the antiviral activity of ND10 factors and IFI16 in order to efficiently initiate lytic replication (2, 10, 11, 13–19).
SPOC1 (survival time-associated PHD [plant homeodomain] protein in ovarian cancer 1), also known as PHF13 (PHD finger 13), was first described in 2003 as a novel cellular protein with a single PHD domain, and enhanced SPOC1 transcript levels were demonstrated to correlate with a shorter survival time for patients suffering from epithelial ovarian cancers (20). Subsequently, this cellular protein has been associated with several functions in cell biology, such as the modulation of cellular proliferation and spermatogenesis, which was attributed to at least three different mechanisms. One proposed mechanism involves the interaction of SPOC1 with chromatin, which occurs in a multivalent fashion. On the one hand, it harbors a C-terminally located PHD, which serves as a molecular reader of the histone marker H3K4me2/3 (21–23). Upon binding to H3K4me2/3, SPOC1 was proposed to induce chromatin compaction by recruiting histone methyltransferases (HMTs), such as SETDB1, G9A, or GLP, subsequently resulting in an increase in the level of repressive H3K9me3 (24). On the other hand, there is recent evidence that a centrally located domain enables an interaction of SPOC1 with DNA, which contributes to its chromatin binding avidity (23). While SPOC1 is able to bind to chromatin directly, it was also shown to contact chromatin indirectly by interacting with several heterochromatin proteins. In this respect, Chung and colleagues postulated that SPOC1 acts as a transcriptional coregulator, by functioning as a scaffolding protein or bridging factor for the recruitment of RNA polymerase II (RNAPII) complexes as well as Polycomb-repressive complex 2 (PRC2) to distinct chromatin landscapes. Those authors suggested that SPOC1 thereby differentially regulates subsets of target genes that were found to mainly function in DNA binding and chromatin organization as well as transcription, cell cycle regulation, and differentiation (23). Besides transcriptional coregulation and chromatin compaction, SPOC1 was also demonstrated to modulate DNA repair, as it is recruited to DNA double-strand breaks (DSBs) in an ATM-dependent manner and regulates the kinetics of DNA damage repair (DDR). Thereby, SPOC1 is able to shift the balance between nonhomologous end joining (NHEJ) and homologous recombination (HR), favoring HR (24).
Apart from its cellular regulatory functions, SPOC1 was also implicated in contributing to the intrinsic defense against viral infections (25). The first evidence for this was presented by Schreiner and colleagues, who demonstrated that human adenovirus type 5 (HAdV5) infection was diminished at the transcriptional level when SPOC1 was overexpressed, while its depletion resulted in increased virus titers. Furthermore, they showed that the HAdV E3 ubiquitin ligase complex E1B-55K/E4-orf6 efficiently antagonizes SPOC1 by inducing its proteasomal degradation early after infection (25). Interestingly, recent reports suggest that SPOC1 plays a dual role during HIV-1 infection (26). Depending on the time point of HIV-1 replication, high SPOC1 levels either improved HIV-1 integration (SPOC1 expression prior to HIV-1 integration) or suppressed viral gene expression (SPOC1 expression after HIV-1 integration). Furthermore, the authors of that study demonstrated that the viral accessory protein Vpr counteracts SPOC1 by targeted degradation (26).
Here, we analyzed the role of SPOC1 in the context of HCMV infection. Intriguingly, and in contrast to HAdV and HIV-1 infection, we observed that SPOC1 expression is upregulated during infection and peaks during the early phase of the HCMV replicative cycle. Furthermore, we show that at late times postinfection, SPOC1 is degraded in a glycogen synthase kinase 3β (GSK-3β)-dependent manner. In order to elucidate the role of SPOC1 during HCMV infection, we generated SPOC1-overexpressing fibroblasts and observed severely impaired virus growth at a low multiplicity of infection (MOI). Furthermore, we show that the overexpression of SPOC1 restricts the onset of viral IE gene expression, while its depletion results in an increase in the level of IE gene expression. Finally, by chromatin immunoprecipitation (ChIP) coupled with deep sequencing (ChIP-seq), we demonstrate a specific association of SPOC1 with the HCMV MIEP region, strongly arguing for a scenario whereby SPOC1 is able to induce the silencing of viral IE expression via epigenetic modifications.
RESULTS
SPOC1 is transiently upregulated during HCMV infection.
SPOC1 was previously demonstrated to act as a restriction factor against HAdV infection by repressing viral gene expression at the transcriptional level (25). To antagonize this repressive function, HAdV targets SPOC1 for proteasomal degradation immediately upon infection. In order to evaluate the role of SPOC1 during HCMV infection, we inoculated HCMV-permissive primary human foreskin fibroblasts (HFFs) with laboratory strain AD169 at an MOI of 3 and assessed SPOC1 protein levels throughout the HCMV replication cycle (Fig. 1A). In parallel, representatives of the immediate early (IE), early (E), and late (L) phases of HCMV infection were detected to ensure that the replication cycle had fully proceeded. Interestingly, and in contrast to the HAdV-induced depletion of SPOC1, we observed a transient upregulation of SPOC1 during the early phase of HCMV infection, while at late times postinfection, SPOC1 levels declined. In order to investigate whether this upregulation is based on increased SPOC1 transcription, we isolated total RNA at 24 h postinfection (hpi), followed by reverse transcription-quantitative PCR (qRT-PCR) (Fig. 1B, top). This revealed only a mild increase of SPOC1 mRNA levels (2-fold) compared to the 6-fold increase in the SPOC1 protein abundance (Fig. 1B, bottom). Consequently, we assume that the upregulation of SPOC1 takes place at both the transcript and protein levels. Next, we analyzed if the observed upregulation is virus strain or cell type dependent. HFFs and retinal pigment epithelial cells (ARPE-19) were infected with clinical isolate TB40/E, and SPOC1 expression levels were analyzed throughout the replication cycle (Fig. 1C and D, respectively). In both cases, we observed a strong induction of SPOC1 expression culminating at 24 hpi, implying that this event is cell type and virus strain independent. Moreover, it appears to be conserved, since we also detected increased murine SPOC1 levels during murine cytomegalovirus (MCMV) infection beginning at 24 hpi (Fig. 1E). Together, these findings provide evidence that SPOC1 is robustly and specifically upregulated upon CMV infection, raising the question of a pro- or an antiviral function of SPOC1 for viral replication.
FIG 1.

SPOC1 is transiently upregulated during HCMV infection. (A) HFF cells were infected with HCMV laboratory strain AD169 at an MOI of 3 and harvested at the indicated time points postinfection. Total cell extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting with mouse monoclonal antibodies p63-27 (IE1), BS 510 (pUL44), and 28-4 (MCP) and rat monoclonal SPOC1 antibody. (B) HFF cells were infected with HCMV laboratory strain AD169 at an MOI of 3. At 24 hpi, RNA was isolated with TRIzol and subsequently synthesized into cDNA via RT-PCR, and transcript levels were assessed via SYBR green PCR. The relative SPOC1 mRNA levels were calculated by normalization against the housekeeping gene RPL13A (Biomol, Hamburg, Germany). Statistical analysis was performed with Student's t test. Densitometric analysis was performed with AIDA image analyzer v.4.22 software, and SPOC1 band intensities at 24 hpi were normalized against their corresponding β-actin signals. (C and D) HFF (C) or ARPE-19 (D) cells were infected with clinical isolate TB40/E at an MOI of 3 and treated as described above for panel A. (E) Mouse embryonic fibroblasts (MEF) were infected with MCMV at an MOI of 3, and whole-cell lysates were harvested throughout the replication cycle and treated as described above for panel A. Immunoblotting was performed with the rat monoclonal SPOC1 antibody and the monoclonal mouse gB antibody. For all experiments, monoclonal antibody AC-15 (β-actin) served as a loading control.
Elevated SPOC1 protein levels are induced by an IE or E gene product of HCMV.
Next, we set out to investigate whether a viral gene product is responsible for the upregulation of SPOC1 during infection. Since herpesviral gene expression occurs in a cascade fashion with chronological phases termed IE, E, and L, it is possible to confine the responsible viral protein to a specific expression phase during HCMV infection. First, we assessed whether elevated SPOC1 levels are due to stabilization by an HCMV tegument protein. Therefore, we applied cycloheximide (CHX) in parallel with AD169 infection. This inhibits de novo HCMV gene expression but does not affect the abundance of tegument proteins, as confirmed by the detection of IE1 and pp65, respectively (Fig. 2A). Western blot analysis at 12 or 24 hpi revealed that SPOC1 is sensitive to CHX treatment (Fig. 2A), while it can be stabilized by MG132 treatment, as shown in Fig. 2B. These observations are in accordance with data in the literature, where SPOC1 was described to be a labile protein that is tightly regulated by the proteasome (22). Moreover, no stabilizing effect of tegument proteins was detectable (Fig. 2A, compare lanes 3 and 4 and lanes 7 and 8). As an alternative approach, we used UV-inactivated viral supernatants (UV-AD169) for infection of HFFs in parallel with wild-type AD169 (wt-AD169) and harvested the cells at 4 and 24 hpi (Fig. 2C). In contrast to AD169-infected HFFs, cells inoculated with the UV-inactivated virus did not exhibit increased SPOC1 protein levels. Here, too, IE1 as well as pp65 detection served as controls for UV inactivation. Consequently, these data demonstrate that de novo CMV gene expression is necessary to induce the upregulation of SPOC1. In order to determine if a true late viral protein is the decisive factor, we made use of the viral DNA synthesis inhibitor phosphonoformic acid (PFA). However, incubation of the infected cells with PFA did not abolish SPOC1 upregulation, which was assessed at 72 hpi (Fig. 2D). Taken together, these findings indicate that SPOC1 is a labile protein, which is most likely upregulated or stabilized by an IE or E gene product.
FIG 2.

SPOC1 is a labile protein that is not upregulated by a viral tegument or late protein. (A and B) HFF cells were infected with AD169 (MOI of 3) and in parallel treated with 150 μg/ml CHX (A) or with the proteasome inhibitor MG132 at 5 μM (B). The cells were harvested at the indicated time points, and total cell extracts were prepared and subjected to SDS-PAGE and immunoblotting. The antibodies used were directed against SPOC1 (anti-SPOC1 rat), the viral immediate early protein IE1 (mAb p63-27), and the abundant tegument protein pp65 (mAb 65-33) (A). (C) HFF cells were infected with wild-type AD169 and UV-inactivated AD169 at an MOI of 3. Cells were then treated as described above for panel A. The antibodies used were directed against SPOC1, IE1, and pp65. (D) HFFs were infected with wild-type AD169 at an MOI of 3, and in parallel with virus inoculation, 250 μM phosphonoformic acid (PFA) was added as indicated to the infected-cell sample. Cells were harvested at 72 hpi, and total cell extracts were prepared and subjected to SDS-PAGE and immunoblotting. The antibodies used were directed against SPOC1, IE1, and the viral late protein MCP (mAb 28-4), serving as a positive control for the block of true late gene expression. For all experiments, monoclonal antibody AC-15 (β-actin) served as a loading control.
IE1 expression induces the upregulation of SPOC1.
It was previously demonstrated that SPOC1 modulates DNA repair kinetics and is recruited to DNA double-strand breaks in an ATM-dependent manner (24). Since there is increasing evidence that the IE1 protein is able to modulate the DNA damage response during HCMV infection, we assumed that IE1 might be involved in the upregulation of SPOC1 (27–30). In order to investigate this, we utilized IE1-inducible HFF cells (HFF-IE1 Tet-On) and analyzed SPOC1 expression levels in the presence or absence of IE1 (Fig. 3A). As evident from Western blot analyses, SPOC1 expression levels were clearly increased in the presence of IE1. In parallel, PML deSUMOylation was detected to monitor the effect of IE1 on cellular proteins. These data demonstrate that IE1 expression alone is sufficient to induce SPOC1 upregulation. We next set out to investigate whether IE1 is required for the increase in SPOC1 levels during HCMV infection. To this end, we infected HFF cells with a high MOI (MOI of 2) of wild-type AD169 and equivalent genome copy numbers of recombinant HCMV strains that either completely lack IE1 (AD169ΔIE1) or express the mutant IE1 protein IE1-L174P (AD169/IE1-L174P), which exerts a growth defect similar to that of the IE1 deletion virus (31). Subsequently, the cells were harvested at different times postinfection and subjected to Western blot analysis (Fig. 3B). Intriguingly, SPOC1 upregulation at 24 hpi was attenuated in cells infected with the recombinant virus strains, indicating that functional IE1 is necessary to induce increased SPOC1 protein levels.
FIG 3.

The immediate early protein IE1 induces the upregulation of SPOC1. (A) SPOC1 expression levels were analyzed in doxycycline (Dox)-treated and untreated HFF-IE1 cells by Western blotting. IE1 expression was induced by treatment with 500 ng/ml doxycycline for two different time intervals (24 h or 48 h). Cells were harvested, and total cell extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting. The antibodies used were directed against SPOC1, the viral immediate early protein IE1, and the cellular protein PML (pAbs A167 and A168) as a control. (B) HFF cells were infected with wt-AD169, AD169ΔIE1, and AD169/IE1-L174P at an MOI of 2. Cells were harvested at the indicated times postinfection, and total cell extracts were prepared and subjected to SDS-PAGE and immunoblotting. The antibodies used were directed against SPOC1, the viral immediate early proteins IE1 (mAb p63-27) and IE2 (pHM178), and the viral early protein pUL44 (mAb BS510). (C) Determination of the SPOC1 half-life in the presence and absence of IE1 using cycloheximide (CHX) in doxycycline-treated (+Dox) and untreated (−Dox) HFF-IE1 cells. Twenty-four hours after the induction of IE1 expression with doxycycline, cells were treated with 25 μg/ml of CHX and harvested at the indicated times to assess SPOC1 expression levels by Western blotting. (D) Densitometric analysis of data in panel C was performed with AIDA image analyzer v.4.22 software, and SPOC1 band intensities were normalized against their corresponding β-actin signals. For all experiments, monoclonal antibody AC-15 (β-actin) served as a loading control.
Since we already demonstrated that SPOC1 is a labile protein that is rapidly degraded by the proteasome (Fig. 2A and B), we next set out to investigate if IE1 has a favorable effect on SPOC1 protein stability. Therefore, we assessed SPOC1 protein levels in the presence or absence of IE1 while treating HFF-IE1 Tet-On cells with CHX and harvesting them at different times posttreatment (Fig. 3C). While the overall SPOC1 expression level was increased in the presence of IE1, its half-life was not significantly affected (Fig. 3C and D). Taken together, these data allow the assumption that during HCMV infection, IE1 is responsible for the observed upregulation of SPOC1 by mechanisms other than protein stabilization.
The decrease in the SPOC1 level during HCMV infection takes place in a GSK-3β-dependent manner.
We next set out to investigate the observed decline in the SPOC1 abundance at late times of infection in more detail. This decline was particularly prominent in HFF and ARPE-19 cells infected with HCMV strain TB40/E (Fig. 1C and D). Interestingly, Kinkley and colleagues analyzed SPOC1 protein stability in more detail and demonstrated a tight regulation of SPOC1 by the proline-directed serine-threonine kinase GSK-3β (22). In order to confirm this, we first treated HEK293T and HFF cells with inhibitors of GSK-3β (LiCl) and the proteasome (MG132) and analyzed SPOC1 expression levels via Western blotting (Fig. 4A). In line with the findings of Kinkley and colleagues, we observed that SPOC1 was stabilized upon treatment with either LiCl or MG132 and displayed even stronger signal intensities after treatment with both inhibitors. Intriguingly, analysis of GSK-3β expression levels throughout the HCMV replicative cycle revealed an increase at late times postinfection, which correlated with the decline of the SPOC1 abundance (Fig. 4B). Thus, we assumed that GSK-3β is involved in the decrease of SPOC1 levels at late times postinfection. For further investigation, we infected HFFs with HCMV strain TB40/E and applied LiCl at either 1.5 hpi or 48 hpi, followed by harvesting at 96 hpi (Fig. 4C). Regardless of when the inhibitor was added, the decrease of SPOC1 levels was efficiently blocked, even if LiCl was added at late times postinfection and major capsid protein (MCP) levels remained equal. Since the decline in the level of SPOC1 was even more prominent in infected ARPE-19 cells (Fig. 1D), we next added LiCl and two additional GSK-3β inhibitors (SB-216763 and CHIR99021) at 48 hpi and harvested ARPE-19 cells 4 days after infection with HCMV strain TB40/E (Fig. 4D). In line with our above-described findings, we observed a block of the decline in SPOC1 levels with LiCl as well as CHIR99021. However, SB-216763 had no stabilizing effect on SPOC1. Taken together, our data suggest that the observed decrease in SPOC1 levels during the late stage of HCMV infection is in part mediated by GSK-3β.
FIG 4.

The decline in SPOC1 levels at a late stage of HCMV infection is mediated by GSK-3β. (A) HEK293T and HFF cells were treated with the GSK-3β inhibitor LiCl and/or the proteasome inhibitor MG132 for 6 h. Afterwards, cells were harvested, lysed, and subjected to Western blot analysis with subsequent detection of SPOC1 expression levels. (B) Cell lysates from Fig. 1C were analyzed for GSK-3β expression using the polyclonal anti-GSK-3β H-76 antibody. (C) HFF cells were infected with clinical isolate TB40/E at an MOI of 3 and in parallel treated with 40 μM LiCl at 1.5 hpi (*) or 48 hpi. Cells were harvested at 96 hpi, and total cell extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting. (D) ARPE-19 cells were infected with TB40/E (MOI of 3) and treated at 48 hpi with the GSK-3β inhibitors LiCl (40 μM), SB-216763 (3 μM; Sigma-Aldrich, Deisenhofen, Germany), and CHIR99021 (3 μM; Tocris Bioscience, Wiesbaden-Nordenstadt, Germany) and with dimethyl sulfoxide (DMSO) as a control. At 96 hpi, cells were harvested, lysed, and subjected to Western blot analysis, with subsequent detection of SPOC1 expression levels and the viral late protein MCP (MAb 28-4) and pp28 (MAb 41-18) serving as a control for the completion of the HCMV replication cycle. For all experiments, monoclonal antibody AC-15 (β-actin) served as a loading control.
SPOC1 overexpression inhibits the onset of viral IE gene expression.
Next, we wanted to address the relevance of SPOC1 for lytic HCMV replication. First, we generated cells that stably overexpress SPOC1, which was accomplished by lentiviral transduction of primary HFFs using a vector harboring SPOC1. In parallel, the cloning vector served to generate control cells. Subsequent Geneticin selection resulted in cell populations termed HFF/SPOC1 and HFF/control, respectively. These cells were then characterized by Western blotting, which revealed successful SPOC1 overexpression in HFF/SPOC1 cells (Fig. 5A) concurrent with a high transduction efficiency of almost 100%, as assessed by indirect immunofluorescence detection (Fig. 5B). In order to analyze the impact of SPOC1 overexpression on HCMV replication, the generated cells were infected with HCMV laboratory strain AD169 at an MOI of 1 and harvested at different time points of the replication cycle, and expression kinetics of viral immediate early, early, and late proteins were assessed by Western blotting (Fig. 6A). The SPOC1-overexpressing cells showed decreased viral early and late protein expression compared to control cells, which was most prominent for pUL97 and pp71, as revealed by quantification of the Western blot signals (Fig. 6B).
FIG 5.
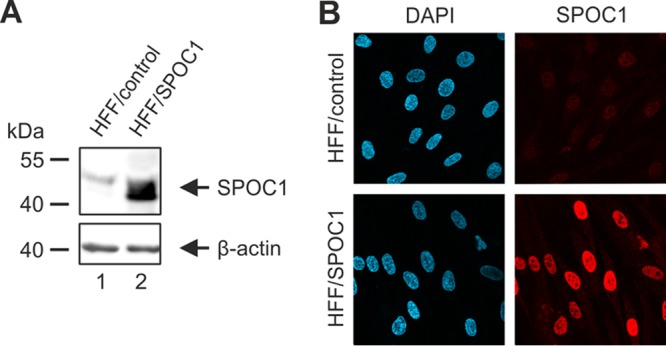
Generation of HFF cells with stable SPOC1 overexpression. (A) Western blot analysis for detection of SPOC1 in lysates of control (HFF/control) and SPOC1-overexpressing (HFF/SPOC1) HFFs using an antibody against endogenous SPOC1 (anti-SPOC1 rat). (B) Immunofluorescence staining of SPOC1 in HFF/control and HFF/SPOC1 cells, using the SPOC1 mAb. Cell nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole).
FIG 6.

SPOC1 overexpression restricts initiation of IE gene expression. (A) HFF/SPOC1 and control HFF cells were either not infected (mock) or infected with AD169 at an MOI of 1 and harvested at the indicated times for Western blotting. Expression kinetics of the viral immediate early protein IE1, viral early proteins (pUL84, pUL44, pUL97, and pp71), and viral late proteins (pp28 and MCP) were compared. (B) Densitometric analysis of the data in panel A was performed with AIDA image analyzer v.4.22 software. (C) Multistep growth curve analyses of AD169 on HFF/control and HFF/SPOC1 cells. Cells were infected with AD169, as indicated, at an MOI of 0.005, 0.001, or 0.0005, and cell supernatants were harvested at the indicated times postinfection and analyzed for genome equivalents by HCMV IE1-specific quantitative real-time PCR. (D) HFF/control and HFF/SPOC1 cells were infected with HCMV laboratory strain AD169 at an MOI of 0.01. At 8 hpi, intracellular DNA was isolated, and genome equivalents were assessed via TaqMan PCR in relation to albumin copy numbers. n.s., not significant. (E) Analysis of IE gene expression in SPOC1-overexpressing cells with AD169 and TB40/E. HFF/control and HFF/SPOC1 cells were grown on coverslips in six-well dishes, infected with 100 IEU/well of either laboratory strain AD169 or clinical isolate TB40/E, and fixed at 24 hpi. The number of IE-expressing cells was determined by indirect immunofluorescence analysis using monoclonal antibody p63-27 against the viral protein IE1. Statistical analysis was performed with Student's t test.
Since cellular restriction is saturable and effects might be masked at a high MOI, we next applied multistep growth curve analyses at low multiplicities of infection. We infected cells with three different MOIs (0.005, 0.001, and 0.0005) and harvested virus-containing cell supernatants at the indicated times postinfection (Fig. 6C). Subsequently, the supernatants were subjected to quantitative real-time PCR to determine HCMV genome equivalents. As evident from Fig. 6C, SPOC1 overexpression strongly impaired virus growth under low-MOI conditions and caused up to a 3-log10 reduction in viral progeny release. SPOC1 did not affect virus uptake and/or genome delivery into the nucleus since the quantification of intranuclear viral genomes did not reveal a significant difference between control and SPOC1-overexpressing cells (Fig. 6D). In order to test if SPOC1 overexpression already acts on the initiation of viral gene expression, HFF/SPOC1 and HFF/control cells were infected in parallel with 100 IE1-forming units (IEU) of HCMV strain AD169 or TB40/E. Twenty-four hours later, the cells were stained for IE1 to determine the number of cells exhibiting an initiation of IE gene expression. Intriguingly, we detected a 4-fold reduction in the number of IE1-positive cells when SPOC1 was overexpressed and infected with laboratory strain AD169 (Fig. 6E, left). An even more pronounced effect was observed after inoculation with clinical strain TB40/E (Fig. 6E, right). Taken together, our data demonstrate that the overexpression of SPOC1 negatively affects virus growth, which could be attributed to a restriction of IE gene expression. This finding, in line with the observed MOI dependency, strongly suggests that SPOC1 acts as a cellular restriction factor for cytomegalovirus infection.
SPOC1 overexpression during the first hours of HCMV infection is critical for efficient restriction.
In order to analyze if high SPOC1 levels prior to infection are necessary for efficient HCMV restriction, we generated HFF cells that were able to induce SPOC1 overexpression upon treatment with doxycycline. In order to accomplish this, we made use of the Lenti-X Tet-On Advanced inducible-expression system (Clontech, Palo Alto, CA, USA). After insertion of the coding sequence for FLAG-tagged SPOC1 into the pLVX Tet-On Advanced vector, lentiviral transduction of HFF cells and selection with puromycin and Geneticin yielded a cell population termed HFF/SPOC1 (Tet-On). These cells showed a strong inducibility already 8 h after treatment with doxycycline (Fig. 7A and B). First, we wanted to know whether high SPOC1 levels are able to restrict the onset of immediate early RNA expression. Thus, cells were either treated with doxycycline for 24 h (+Dox) or not treated (−Dox) and subsequently infected with HCMV strain AD169 (MOI of 0.001), and total RNA was isolated at 8 hpi. Quantification of IE1 transcript levels revealed a strong reduction of IE1 mRNA levels upon the overexpression of SPOC1 (Fig. 7C). Next, we induced SPOC1 overexpression either 24 h prior to, in parallel with, or 8 h after infection with 100 IEU of AD169 and counted the number of IE1-positive cells at 24 h postinfection (Fig. 7D). Interestingly, we found that the restriction of the initiation of IE gene expression was most effective when SPOC1 was preexpressed in the infected cells, leading to an 8-fold decrease in the number of IE-positive cells (Fig. 7D). A significant restriction was still observed when SPOC1 overexpression was induced in parallel with infection, whereas its induction at 8 hpi no longer affected the onset of IE gene expression. Consistent with these results, viral progeny release was inhibited by up to 3 log10 units when SPOC1 overexpression was induced prior to infection (Fig. 7E). From these findings, we conclude that high SPOC1 protein levels prior to the initiation of viral IE gene expression efficiently restrict HCMV replication.
FIG 7.

SPOC1 overexpression during the first hours of HCMV infection is vital for efficient restriction. (A) Western blot analysis of inducible HFF/SPOC1 (Tet-On) cells in the presence (8 h, 16 h, and 24 h) or absence (mock) of doxycycline. (B) Immunofluorescence analysis for detection of SPOC1 in HFF/SPOC1 (Tet-On) cells in the presence (+Dox) (24 h) or absence (−Dox) of doxycycline. (C) HFF/SPOC1 (Tet-On) cells were treated with doxycycline for 24 h or not treated as a control and subsequently infected with HCMV laboratory strain AD169 at an MOI of 0.001. At 8 hpi, total RNA was isolated with TRIzol and synthesized into cDNA via RT-PCR, and IE1 transcript levels were assessed via TaqMan PCR. The relative IE1 mRNA levels were calculated by normalization against albumin. (D) For analysis of IE gene expression, HFF/SPOC1 (Tet-On) cells were grown on coverslips in six-well dishes, while SPOC1 overexpression was induced by the addition of doxycycline at different times prior to (−24 h), in parallel with (0 h), and after (+8 h) infection with 100 IEU/well of AD169 (schematically depicted with the timeline above). Cells were fixed at 24 hpi, and the number of IE-expressing cells was determined by indirect immunofluorescence analysis using monoclonal antibody p63-27 against the viral protein IE1. Statistical analysis was performed with Student's t test. (E) Multistep growth curve analyses of AD169 on HFF/SPOC1 (Tet-On) cells in the presence (24 h prior to infection) and absence of doxycycline. Cells were infected with AD169 at an MOI of 0.001, and cell supernatants were harvested at the indicated times postinfection and analyzed for genome equivalents by HCMV IE1-specific quantitative real-time PCR. dpi, days postinfection.
To further elucidate whether SPOC1 overexpression delays or completely abrogates the onset of viral IE gene expression, we tracked the kinetics of HCMV gene expression in real time using live-cell imaging. HFF/SPOC1 (Tet-On) cells were induced with doxycycline 24 h prior to infection, while untreated cells were used in parallel as a control. For real-time monitoring, we made use of the recombinant reporter virus TB40/E-IE-mNeonGreen, which was used previously by Kasmapour and colleagues to monitor IE protein expression kinetics (32). The dynamics of early viral gene expression (up to 30 hpi) were investigated after infection at an MOI of 0.05, followed by automated whole-frame evaluation and quantification with ImageJ-based software (Fig. 8A). The generated data set provided us with two different pieces of information. On the one hand, detection of the onset of viral IE gene expression was possible, which is characterized by a drop in the mean signal intensity due to the increased area of a true signal compared to autofluorescence. On the other hand, the rate of gene expression could be assessed by measuring the steepness of the slope. The main conclusions were that SPOC1 overexpression appeared to delay the onset of viral IE gene expression and also appeared to suppress gene expression. Within the control cells, the first IE reporter signals were detected at between 5 and 10 hpi (Fig. 8B) and steadily increased with proceeding infection (Fig. 8A). In contrast, SPOC1 overexpression significantly delayed the onset of IE gene expression, starting at between 9 and 15 hpi (Fig. 8B), and showed a reduced increase (Fig. 8A). When comparing images from infected cells with and those without doxycycline treatment, it becomes clear that a significantly lower number of cells expressed IE-mNeonGreen upon the overexpression of SPOC1 (Fig. 8C). To further analyze the heterogeneity of cells exhibiting viral gene expression, we performed manual single-cell tracking. Using this, we assessed the time of onset and the maximal rate of IE gene expression for up to 20 individual HCMV-infected cells (Fig. 8D and E, respectively). Within induced and noninduced cells, we observed high variability regarding the start of de novo viral gene expression, ranging from 4.5 to 20 hpi (Fig. 8D). Although a slight tendency toward SPOC1-mediated inhibition was apparent, the overall values revealed no significant difference in the maximal rates of viral gene expression in SPOC1-positive cells (Fig. 8E). However, single-cell tracking is confined to reporter signal positivity, thus ignoring infected cells that lack de novo viral gene expression, which were taken into consideration by the whole-frame analysis. In summary, this result strongly argues for a scenario whereby high SPOC1 levels are able to induce the complete silencing of viral IE gene expression.
FIG 8.

SPOC1 overexpression completely abrogates the onset of viral IE gene expression. HFF/SPOC1 (Tet-On) cells were seeded in live-cell imaging chamber slides, and SPOC1 overexpression was induced with doxycycline 24 h prior to infection with the recombinant reporter virus TB40/E-IE-mNeonGreen at an MOI of 0.05. In parallel, untreated cells (−Dox) were used as controls. For visualization of cell nuclei, SiR-DNA (Spirochrome AG, Stein am Rhein, Germany) was added 2 h prior to infection. Infection was monitored in real time, while images of 5 fields under each condition were acquired every 15 min for up to 30 h. (A) The acquired stacks of time series for all fields were automatically analyzed by using an ImageJ macro. Solid lines display a three-point average smoothing of the IE-mNeonGreen signal from 5 fields, with standard deviations of the average. a.u., arbitrary units. (B) Scatter dot plots showing the distribution of signal onset in each field of control (−Dox) and SPOC1-overexpressing (+Dox) (−24 h) HFFs. Statistical analysis was performed with an unpaired nonparametric t test (Mann-Whitney test). (C) Representative images taken from the time series (5 to 30 hpi). (D and E) Manual single-cell tracking was performed with ImageJ, and data sets were analyzed by using GraphPad Prism 6. (D) After analysis of the area under curve, the onset of IE gene expression was defined as the first time point when the signal crossed the baseline. (E) The maximal rate of gene expression was assessed by logistic growth curve fitting and subsequently calculating the incline at its inflection point. Statistical analysis was performed with an unpaired nonparametric t test (Mann-Whitney test).
Depletion of endogenous SPOC1 augments the initiation of IE gene expression.
Overall, these findings argue for an antiviral function of SPOC1 when it is present at high levels in HFF cells. In order to investigate whether endogenous SPOC1 is sufficient for the restriction of HCMV, we used two different approaches to deplete SPOC1 from primary HFF cells. On the one hand, we applied small interfering RNA (siRNA)-mediated knockdown by transiently transfecting a mix of siRNAs directed against SPOC1. On the other hand, we used the clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technology to establish stable SPOC1 knockout HFFs. In order to accomplish this, we designed a specific guide RNA (gRNA) directed against SPOC1 and cloned it into the CRISPRv2 lentiviral plasmid, followed by lentiviral transduction of HFF cells (33). Puromycin selection yielded HFF/SPOC1-k.o. cell as well as control cell populations, termed HFF/CRISPR, which were generated with the empty vector CRISPRv2. For both methods, efficient SPOC1 depletion was verified by Western blotting (Fig. 9A and B). Next, SPOC1-depleted HFFs (HFF+siSPOC1 or HFF/SPOC1-k.o.) in parallel with the respective control cells were infected with HCMV (100 IEU/well), followed by immunostaining of IE1 at 24 hpi. Intriguingly, we observed a significant increase in the number of IE1-positive cells in the SPOC1 knockout and knockdown cells compared to the respective control cells, demonstrating that endogenous SPOC1 contributes to the repression of viral IE gene expression (Fig. 9C and D). The IE1 mRNA level was also increased in SPOC1 knockout cells when infection was performed at an MOI of 0.01 (Fig. 9E). In contrast, SPOC1 depletion exerted no significant effect under conditions of high-MOI infection (Fig. 9F). Finally, multistep growth curve analyses confirmed the inhibitory effect of SPOC1 on HCMV replication (Fig. 9G, left). This was even more pronounced when viruses exhibiting a deletion in the region encoding the ND10 antagonistic protein IE1 (AD169/del-IE1) or pp71 (AD169/del-pp71) were used in this analyses (Fig. 9G, middle and right).
FIG 9.

More cells initiate viral IE gene expression in the absence of SPOC1. (A and B) Endogenous SPOC1 levels were diminished either by siRNA-mediated transient transfection using 100 pmol of ON-Target Plus human PHF13 (Dharmacon, Lafayette, CO, USA) with two control siRNAs (siRNAs 1 and 3) (A) or by stable SPOC1 knockout using the CRISPR/Cas9 system, generated via lentiviral transduction and yielding in control (HFF/CRISPR) and SPOC1 knockout HFFs (B). Western blot analyses of endogenous SPOC1 were performed to ensure efficient SPOC1 depletion. (C and D) Analysis of IE gene expression in the absence of SPOC1 with AD169. SPOC1-depleted cells and the respective control cells were grown on coverslips in six-well dishes, infected with 100 IEU/well of either laboratory strain AD169 or clinical isolate TB40/E, and fixed at 24 hpi. The number of IE-expressing cells was determined by indirect immunofluorescence analysis using monoclonal antibody p63-27 against the viral protein IE1. Statistical analysis was performed with Student's t test. (E and F) HFF/CRISPR and HFF/SPOC1-k.o. cells were infected with HCMV laboratory strain AD169 at an MOI of 0.01 (E) or an MOI of 1 (F). At 8 hpi, total RNA was isolated with TRIzol and synthesized into cDNA via RT-PCR, and IE1 transcript levels were assessed via TaqMan PCR. The relative IE1 mRNA levels were calculated by normalization against albumin. (G) Multistep growth curve analyses were conducted in HFF/CRISPR and HFF/SPOC1-k.o. cells with wild-type AD169 (wt-AD169) or recombinant mutant viruses deprived of either IE1 (AD169/del-IE1) or pp71 (AD169/del pp71) (MOI of 0.001). Cell supernatants were harvested at the indicated times postinfection and analyzed for genome equivalents by HCMV gB-specific quantitative real-time PCR. Error bars indicate the standard deviations derived from data from three independent experiments.
SPOC1 specifically associates with the proximal enhancer region of the major immediate early promoter.
The initiation of HCMV IE gene expression is driven by the major immediate early promoter (MIEP). Since SPOC1 is able to directly interact with DNA, a possible mode of action could be the binding of SPOC1 to the MIEP followed by subsequent epigenetic modulation. In order to address this question, we performed SPOC1-specific chromatin immunoprecipitation (ChIP) in parallel with a negative control (hemagglutinin [HA]) and investigated different regions of the CMV promoter by qPCR (Fig. 10A) (23). While SPOC1 showed no significant enrichment over the control antibody HA (Fig. 10A) within regions outside the CMV enhancer (ranges of positions +788 to +732 and −1082 to −1191 relative to the MIEP start site), we found a specific enrichment of SPOC1 within the proximal enhancer region of the MIEP. In order to gain more insight into the binding profile of SPOC1, we performed ChIP coupled with deep sequencing (ChIP-seq) on material isolated from HFF/SPOC1 (Tet-On) cells induced with doxycycline 24 h prior to infection with TB40/E at an MOI of 0.5 and performed cross-linking at 8 hpi. After quality control and alignment against the reference genome, generated by merging the human reference genome (hsGRCh38v87) with the annotated TB40E_BAC4 HCMV genome (GenBank accession number EF999921.1), we utilized the MACS2 irreproducible discovery rate (IDR) pipeline for peak calling in both generated replicates. We had total numbers of 39 million and 51 million reads for replicates 1 and 2, while 0.2% and 0.17% of them mapped to the TB40/E genome, respectively. Among these, the majority (83%) mapped within the region of bp 207200 to 208300 of the HCMV genome, showing a >500-fold enrichment (Fig. 10B). Intriguingly, this region corresponded to the site where the MIEP is located within the HCMV genome (Fig. 10C). Therefore, we conclude that SPOC1 targets the viral genome upon HCMV infection and binds within the CMV enhancer/promoter region, thereby mediating the restriction of viral IE gene expression.
FIG 10.

ChIP experiments reveal specific SPOC1 binding within the proximal enhancer region of the major immediate early promoter. (A) Primary HFF cells were infected with TB40/E at an MOI of 0.5, proteins were cross-linked at 8 hpi, and subsequent ChIP was performed with anti-SPOC1 antibody CR56 or anti-HA as a control. After washing and elution, the samples were subjected to proteinase K digestion and purified with the QIAquick MinElute PCR purification kit (Qiagen, Hilden, Germany). Subsequently, samples were subjected to SYBR green qRT-PCR with primers targeting different regions up- and downstream of the transcription start site of the IE1 and IE2 genes (regions shown relative to position +1 of the transcription start site). (B) HFF/SPOC1 (Tet-On) cells were induced with doxycycline (500 ng/ml) 1 day prior to infection with TB40/E (MOI of 0.5). At 8 h postinfection, proteins were cross-linked, and ChIP was performed with anti-SPOC1 antibody CR56. SPOC1 binding sites within the HCMV genome were mapped onto the reference sequence under GenBank accession number EF999921.1. Reads were deduplicated with SAMtools 1.3 and subjected to peak calling using the MACS2 IDR pipeline. (C) Inset of the peak from panel B depicting open reading frames, the proximal and distal enhancers of the MIEP, and the regions amplified by the primer pairs from panel A.
DISCUSSION
In this report, we investigated the role of the cellular protein SPOC1 as a putative restriction factor against HCMV infection. Previous studies reported an antiviral activity of SPOC1 during human adenovirus infection as well as during HIV-1 replication (25, 26). Both studies demonstrated that SPOC1 levels are rapidly diminished early upon virus infection. Interestingly, and in contrast to HAdV and HIV-1, we identified that SPOC1 is robustly and specifically upregulated by HCMV infection (Fig. 1A). This upregulation appears to be conserved between different HCMV strains and MCMV and could be observed upon infection of various cell types (Fig. 1C to E). SPOC1 was previously described to act as a modulator of DNA double-strand break repair (24). Intriguingly, HCMV differs from HAdV in its requirement for an active DNA damage response (DDR). While HAdV has evolved mechanisms to efficiently inhibit the cellular DDR, HCMV stimulates components of the DDR machinery during the course of its replicative cycle. In particular, the master regulator ATM was demonstrated to be required for efficient HCMV replication (29). Thus, we hypothesize that SPOC1 may undergo upregulation as a component of the virus-induced DDR, while it is rapidly degraded after HAdV5 infection as a mechanism of AdV-mediated DDR countermeasures. Furthermore, we were able to attribute the increase in the SPOC1 abundance to IE1 expression (Fig. 3A and B). Intriguingly, Kulkarni and Fortunato reported that homologous recombination (HR), as one branch of DNA double-strand break repair, is increased upon HCMV infection, which was demonstrated to be specifically stimulated by IE1 expression (27). In line with this, SPOC1 was found to modulate DNA repair kinetics by shifting the balance between nonhomologous end joining (NHEJ) and HR, with the latter being favored (24). After the initial activation of the DDR during the early phase, HCMV inhibits the DDR by protein mislocalization during late stages of infection (34). Hence, the decline of SPOC1 levels that we observed during our studies may correlate with an overall inactivation of the cellular DDR during the late phase of replication. Moreover, we were able to demonstrate that the drop of the SPOC1 abundance is GSK-3β dependent. This fits with recent observations demonstrating that SPOC1 degradation during HIV-1 infection also occurs in a GSK-3β-dependent manner (26). However, whereas during HIV-1 infection, the accessory protein Vpr was identified as the responsible viral factor, the HCMV regulatory proteins inducing SPOC1 downregulation remain to be determined. Since priming phosphorylation of SPOC1 is a prerequisite for subsequent phosphorylation by GSK-3β, a possible candidate factor might be the viral protein kinase pUL97, which will be investigated in future studies (35).
In order to elucidate the role of SPOC1 for HCMV replication, we generated primary human fibroblasts overexpressing SPOC1 and found that high protein levels significantly reduced virus growth (Fig. 6C). In line with this, SPOC1 depletion led to enhanced HCMV infectivity (Fig. 9C and D). We demonstrate that SPOC1 interferes with the initiation of viral IE gene expression, and this occurs in an MOI-dependent manner. MOI dependency is a hallmark of many restriction factors (2, 36, 37). This is due to limiting amounts of preexisting host factors that may be either saturated by incoming viral genomes or antagonized by viral factors. Furthermore, using inducible expression systems, we were able to narrow down the time point of SPOC1 action to very early events of the viral replication cycle: when high SPOC1 levels were present at the time point of HCMV infection, efficient restriction was observed, while later SPOC1 upregulation had no significant effects (Fig. 7D). Moreover, results obtained by live-cell imaging experiments suggest that SPOC1 affects IE gene expression not only by delaying its onset and reducing its rate (Fig. 8A and B) but also by completely abrogating the initiation of viral gene expression (Fig. 8C). Thus, we speculate that high SPOC1 levels constitute an obstacle for the virus that cannot be overcome, leading to a complete shutdown of viral IE gene expression.
Our findings corroborate data from previous studies by Schreiner and colleagues as well as Hofmann and colleagues, which described SPOC1 as an antiviral restriction factor targeting HAdV and in part HIV-1. While SPOC1 appears to play a dual role during HIV-1 infection by promoting HIV-1 integration and repressing viral gene expression, an exclusively repressive role has been described for HAdV infection (25, 26). That group demonstrated that SPOC1 represses HAdV promoter activity and suggested that it targets incoming HAdV genomes immediately upon infection. On the contrary, Komatsu and colleagues were not able to confirm this hypothesis by live-cell imaging techniques. They rather suggested that SPOC1 affects replicating or replicated viral genomes during the late stage of infection (38). Thus, the exact mechanism of how SPOC1 is able to affect viral gene expression requires further investigation.
Interestingly, we observed by ChIP-seq experiments that SPOC1 associates exclusively with a specific gene region of HCMV, namely, the proximal enhancer region of the MIEP (Fig. 10). This regulatory element exerts a critical role for HCMV, and there is increasing evidence that epigenetic modifications of the chromatin structure around the MIEP control the onset of viral gene expression (39, 40). So far, it is not clear whether SPOC1 associates with viral DNA directly or in an indirect manner, for instance, via interactions with H3K4me2/3. Intriguingly, SPOC1 was described to interact with several key regulators of the chromatin structure, e.g., the transcriptional corepressor KAP1 and several lysine methyltransferases (KMTs), such as SETDB1, GLP, and G9A (23, 24). Consequently, we hypothesize that SPOC1 may serve as a recruitment factor for these heterochromatin-building proteins, which may then establish a repressive chromatin structure leading to a shutdown of viral IE gene expression. A promising candidate would be the corepressor protein KAP1, which was recently proposed to act as a critical factor regulating the release of HCMV from latency by a phosphorylation switch (41). In addition, several studies demonstrated that in nonpermissive cells, the MIEP is associated with a number of other heterochromatin-building factors, such as the KMTs Suvar(3-9)H1, SETDB1, and G9A (40, 42–44). Since SPOC1 binds to several of the above-mentioned heterochromatin proteins, we strongly assume that its restrictive function is exerted by the recruitment of at least one of them. Whether these factors act as one large repressor complex or are recruited in a sequential manner needs to be clarified by future studies. Furthermore, since there is a considerable variation of the expression levels between cell types, we assume that restriction by SPOC1 occurs in a cell type-dependent manner. The highest SPOC1 transcript levels were found in testis, more precisely in spermatogonia (21). Therefore, one may speculate that SPOC1 contributes to the intrinsic defense of male germ cells against HCMV infection. In summary, our study adds a novel factor to the host cell endowment contributing to intrinsic immunity against cytomegalovirus infections.
MATERIALS AND METHODS
Oligonucleotides and plasmid construction.
The oligonucleotide primers used for this study were purchased from Biomers GmbH (Ulm, Germany) or Biomol (Hamburg, Germany) and are listed in Table 1. The lentiviral construct pHM4300 for stable SPOC1 overexpression was generated by amplification using the FLAG-tagged SPOC1 plasmid pCMV-Flag-SPOC1 (provided by A. Winterpacht, Erlangen, Germany) and primers SPOC1_PacI_fw and SPOC1_EcoRI_rev, followed by insertion via PacI and EcoRI into a modified pLKO-based lentiviral vector. The modified pLKO-based lentiviral vector was generated as described previously (45). For the inducible expression of SPOC1, FLAG-tagged SPOC1 was generated by PCR amplification with primers F-SPOC1_NotI and SPOC1_EcoRI_rev along with pCMV-Flag-SPOC1 as the template, followed by insertion into the lentiviral pLVX-Tight-Puro vector via NotI and EcoRI. The modified pLVX Tet-On Advanced vector was generated by the replacement of the CMV promoter with an EF1-alpha promoter by the amplification of EF1-alpha promoter sequences with primers EF1-alpha_ClaI_fw and 3′EF1alpha-BamHI, using pHM4300 as the template, followed by insertion into the pLVX Tet-On Advanced vector via ClaI and BamHI. The SPOC1 guide RNA primers SPOC1-fw-gRNA and SPOC1-rev-gRNA were designed with CRISPR Design (see http://crispr.mit.edu/), annealed, and cloned into the pLenti-CRISPR-v2 vector (kindly provided by A. Ensser, Erlangen, Germany) via BsmBI (33).
TABLE 1.
Oligonucleotides used for plasmid construction and qRT-PCR

Cells and viruses.
HEK293T cells were cultivated in Dulbecco's minimal essential medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal calf serum (FCS). Primary human foreskin fibroblasts (HFFs) and human retinal pigment epithelial cells (ARPE-19) were maintained in Eagle's minimal essential medium (MEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 7% fetal calf serum. Mouse embryonic fibroblasts (MEFs) were cultivated with DMEM supplemented with 10% fetal calf serum. HFFs stably overexpressing SPOC1 and the respective control cells were maintained in MEM supplemented with 7% fetal calf serum and 500 μg/ml Geneticin. HFFs with a stable SPOC1 knockout were maintained in MEM supplemented with 10% fetal calf serum and 5 μg/ml puromycin. HFF cells with the inducible overexpression of FLAG-tagged SPOC1 were cultured in MEM supplemented with 10% tetracycline-free fetal bovine serum (Clontech, Palo Alto, CA, USA), 5 μg/ml puromycin, and 500 μg/ml Geneticin. Infection experiments with HFFs were performed with HCMV laboratory strain AD169, the recombinant viruses AD169/IE1-L174P and AD169ΔIE1, and clinical isolate TB40/E (31, 46). UV-inactivated AD169 was generated by the exposure of wild-type AD169 to UV light (0.12 J/cm2). The recombinant reporter HCMV strain used, TB40/E-IE-mNeonGreen, was generated as described previously (32). One day prior to infection, HFF cells were seeded into six-well dishes (3 × 105 cells/well). Virus inoculation was carried out at the specified multiplicities of infection (MOIs), and cells were provided with fresh medium at 1.5 h postinfection (hpi) before being used for subsequent analyses. Viral stocks were titrated as described previously (31). Infection of ARPE-19 cells was performed 1 day after seeding into six-well dishes (4.5 × 105 cells/well). In contrast to HFFs, ARPE-19 cells were inoculated with 2 ml of TB40/E, centrifuged at 1,200 relative centrifugal force (rcf) for 30 min at 37°C, and provided with fresh medium at 1.5 hpi. For MCMV infection, MEF cells were seeded into six-well dishes (1 × 105 cells/well) and 1 day later inoculated with MCMVlucMCK2 (kindly provided by M. Mach, Erlangen, Germany) (47).
Lentivirus transduction and selection of stably transduced cells.
For the generation of HFF/control and HFF/SPOC1 cells, replication-deficient lentiviruses were generated by using pLKO-based expression vectors. For this purpose, HEK293T cells were seeded into 10-cm dishes (5 × 106 cells) cotransfected with an empty pLKO vector or one carrying SPOC1 together with packaging plasmids pLP1, pLP2, and pLP/VSV-G by using the Lipofectamine 2000 reagent (Invitrogen, Karlsruhe, Germany). Viral supernatants were harvested at 48 h posttransfection, cleared by centrifugation, filtered, and stored at −80°C. HFFs of a low passage number were incubated for 24 h with lentiviral supernatants in the presence of 7.5 μg/ml Polybrene (Sigma-Aldrich, St. Louis, MO, USA). Stably transduced HFF cell populations were selected by the addition of 500 μg/ml Geneticin to the cell culture medium. SPOC1 knockout HFFs were generated in an analogous fashion, using the empty pLenti-CRISPR-v2 vector or the SPOC1 gRNA-bearing plasmid pHM4395 for the production of lentiviral supernatants. However, the selection of successfully transduced HFF populations was performed by the addition of 5 μg/ml puromycin to cell culture media. The Lenti-X Tet-On Advanced inducible-expression system (Clontech, Palo Alto, CA, USA) was used to generate HFFs with the inducible overexpression of FLAG-tagged SPOC1. Therefore, replication-deficient lentiviruses expressing the modified tetracycline-controlled transactivator rtTA-Advanced were generated by cotransfecting HEK293T cells with the modified pLVX-Tet-On Advanced vector and the Lenti-X HTX packaging mix (Clontech, Palo Alto, CA, USA), while lentiviruses expressing SPOC1 under the control of the inducible pTight promoter were generated by cotransfection with the pLVX-Tight-Puro vector. The respective viral supernatants were harvested 48 h after transfection, cleared by centrifugation, filtered, and subsequently used for the cotransduction of primary HFFs, followed by selection with 500 μg/ml Geneticin and 5 μg/ml puromycin. HFFs with the inducible expression of IE1 were generated as described previously (31).
Western blotting, indirect immunofluorescence, and antibodies.
Whole-cell lysates from HFF, ARPE-19, HEK293T, or MEF cells were prepared in Roti-Load Laemmli buffer (Roth GmbH, Karlsruhe, Germany) and boiled for 10 min at 95°C (48). Proteins were separated by SDS-PAGE (SDS-polyacrylamide gel electrophoresis) on 8% to 12.5% polyacrylamide gels and transferred onto nitrocellulose membranes (GE Healthcare, Munich, Germany), and subsequent chemiluminescence detection was performed according to the manufacturer's instructions (ECL Western blotting detection kit; Amersham Pharmacia Europe, Freiburg, Germany). For indirect immunofluorescence analysis, the transduced HFFs were fixed with 4% paraformaldehyde, and fluorescence staining was performed as described previously (49). A Leica TCS SP5 confocal microscope with 488-nm and 543-nm laser lines was used for subsequent sample analyses, where each channel was scanned separately under image capture conditions, thereby eliminating channel overlap. The images were then exported, processed with Adobe Photoshop CS5, and assembled by using CorelDraw X5. The following monoclonal antibodies (mAbs) were used for immunofluorescence and Western blot analyses: anti-SPOC1 rat, anti-IE1 p63-27, anti-UL44 BS510 (kindly provided by B. Plachter, Mainz, Germany), anti-UL97 (kindly provided by M. Marschall, Erlangen, Germany), anti-pp65 65-33 (kindly provided by W. Britt, Birmingham, AL, USA), anti-pp28 41-18, anti-MCP 28-4, anti-mouse gB 5F12 (kindly provided by M. Mach, Erlangen, Germany), anti-FLAG 1804, anti-HA clone 7, and anti-β-actin AC-15 (Sigma-Aldrich, Deisenhofen, Germany) (22, 47, 50–53). The following polyclonal antibodies (pAbs) were used for immunofluorescence and Western blot analyses: anti-SPOC1 rabbit CR56, anti-IE2 pHM178 (produced by immunizing rabbits with a prokaryotically expressed protein), anti-UL84, anti-pp71 SA2932, anti-PML A301-167A in combination with α-PML A301-168A (Bethyl Laboratories), and anti-GSK-3β H-76 (Santa Cruz Biotechnology, Heidelberg, Germany) (5, 22, 54). Horseradish peroxidase-conjugated anti-mouse, anti-rabbit, and anti-rat secondary antibodies for Western blot analysis were obtained from Dianova (Hamburg, Germany), and Alexa Fluor 488- and 555-conjugated secondary antibody for indirect immunofluorescence experiments was purchased from Molecular Probes (Karlsruhe, Germany).
siRNA transfection.
HFF cells were seeded into six-well dishes (2.25 × 105 cells/well) and transfected 1 day later with 100 pM of either a mix of 4 specific SPOC1 siRNAs (Dharmacon, Lafayette, CO, USA) or control siRNA 1 or 3 with Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany), using the standard protocol for siRNA transfection provided by the manufacturer.
RNA isolation and quantitative SYBR green real-time PCR.
RNA isolation and subsequent quantitative SYBR green qRT-PCR were performed as described previously (31). SPOC1 transcripts were amplified by utilizing the primer pair 5′SPOC1 and 3′SPOC1 (Table 1) and normalized against the housekeeping gene RPL13A (ribosomal protein L13a) (primer set contained within the HHK-1 set; Biomol GmbH, Hamburg, Germany).
Multistep growth curve analysis and TaqMan real-time PCR.
Multistep growth curve analyses as well as the quantification of viral DNA in supernatants from infected HFF/control, HFF/SPOC1, and HFF/SPOC (Tet-On) cells were performed as described in a previous study (55).
Chromatin immunoprecipitation assays.
For ChIP assays coupled with qRT-PCR (ChIP–qRT-PCR), HFF cells were infected with TB40/E at an MOI of 0.5, and proteins were cross-linked at 8 hpi with 1% formaldehyde for 10 min at room temperature (RT). The reaction was quenched with glycine, and cells were washed with phosphate-buffered saline (PBS) and harvested by scraping. After further washing steps, chromatin preparation and subsequent ChIP were performed according to “chromatin preparation” and “transcription factor ChIP” protocols obtained from the Blueprint website (see http://www.blueprint-epigenome.eu/), with slight alterations. In brief, 1.5 × 107 cells were lysed in 1 ml lysis buffer and fragmented to an average length of 500 to 800 bp by sonication using the Bioruptor NextGene UCD 300 instrument (Diagenode SA, Seraing, Belgium). Subsequently, cell debris was cleared by centrifugation, and ChIP was performed overnight with the fragmented chromatin from 5 × 105 cells, A/G magnetic beads (Merck Chemicals GmbH, Darmstadt, Germany), and anti-SPOC1 CR56 antibody or, alternatively, HA antibody as a control in incubation buffer supplemented with protease inhibitors (Sigma-Aldrich, Deisenhofen, Germany) and 0.1% bovine serum albumin (BSA). After washing the beads with wash buffers I to IV, chromatin was eluted and subjected to proteinase K digestion overnight. Input samples were equally processed in parallel. Finally, chromatin was purified with the QIAquick MinElute PCR purification kit (Qiagen, Hilden, Germany) and subjected to SYBR green PCR, as described previously, with primer pairs +788_+732_fw and +788_+732_rev, +39_−46_fw and +39_−46_rev, and −1082_−1191_fw and −1082_−1191_rev (31). The average threshold cycle (CT) value was determined from triplicate samples and normalized with standard curves for each primer pair. The identities of the obtained products were confirmed by melting-curve analysis.
For ChIP-seq, HFF/SPOC1 (Tet-On) cells were induced with doxycycline (500 ng/ml) 1 day prior to infection with TB40/E (MOI of 0.5). Proteins were cross-linked at 8 hpi with 1% formaldehyde for 10 min at RT, followed by quenching of the reaction and several wash steps. Chromatin preparation and subsequent ChIP were performed as described above, with some alterations. Thereby, cells were lysed and fragmented to an average length of 200 bp using the Covaris S220 AFA ultrasonicator. ChIP was performed overnight with A/G magnetic beads and anti-SPOC1 CR56 antibody in incubation buffer supplemented with protease inhibitors and 0.1% BSA. In order to obtain the highest DNA yield from 1 × 106 cells, four single ChIPs with 2.5 × 105 cells each were performed, which were pooled after elution as one sample, ultimately resulting in two distinct replicates. ChIP-seq libraries were generated by using the TruSeq ChIP kit (Illumina) and sequenced on an Illumina HiSeq2500 platform. Reads were demultiplexed by using Illumina bcl2fastq v2.17.1.14. Fastq files from two flow cells were then concatenated. A reference genome was created by merging the human reference genome (hsGRCh38v87) and the annotated TB40E_BAC4 HCMV genome (GenBank accession number EF999921.1). Alignment to this reference genome was then performed by using bowtie2-2.2.9 with standard parameters. Reads were deduplicated with SAMtools 1.3 and subjected to peak calling using the MACS2 IDR pipeline (outlined at https://sites.google.com/site/anshulkundaje/projects/idr/deprecated). For normalized enrichment calculation, data were mean centered and normalized to the input.
Live-cell imaging.
HFF/SPOC1 (Tet-On) cells were seeded into live-cell imaging chamber slides (4 × 104 cells/chamber) (μ-Slide 8 well; ibidi GmbH, Planegg, Germany), and 24 h prior to infection, SPOC1 overexpression was induced by the addition of doxycycline (500 ng/ml). In parallel, untreated cells were left as controls. SiR-DNA (Spirochrome AG, Stein am Rhein, Germany) was added 2 h prior to infection for tracking of cell nuclei. Subsequently, cells were inoculated with the recombinant reporter virus TB40/E-IE-mNeonGreen at an MOI of 0.05 by centrifugation at 2,000 rpm for 10 min at RT. Afterwards, virus supernatants were removed, and cells were supplemented with fresh medium. In order to monitor the infection in real time, a Leica TCS SP5 confocal microscope with a complete environmental incubation system was used. Thereby, images of 5 fields under each condition were acquired every 15 min for up to 30 h. The acquired stacks of time series for all fields were automatically analyzed by using an ImageJ macro created by B. Kasmapour, Braunschweig, Germany, reporting the mean IE signal associated with the cell nuclei in each frame (32).
ACKNOWLEDGMENTS
We thank M. Mach (Erlangen, Germany), M. Marschall (Erlangen, Germany), A. Ensser (Erlangen, Germany), B. Plachter (Mainz, Germany), and W. Britt (Birmingham, AL, USA) for providing valuable reagents for this study.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB796, TP B3, and STA357/7-1 to T.S. and SFB900 TP B2 to L.C.-S.), the Interdisciplinary Center for Clinical Research Erlangen (IZKF Erlangen) (project A71 to T.S.), and the Wilhelm Sander Stiftung (2016.087.1 to T.S.).
REFERENCES
- 1.Hatziioannou T, Bieniasz PD. 2011. Antiretroviral restriction factors. Curr Opin Virol 1:526–532. doi: 10.1016/j.coviro.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol 80:8006–8018. doi: 10.1128/JVI.00743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gariano GR, Dell'Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2012. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res 229:155–158. doi: 10.1006/excr.1996.0353. [DOI] [PubMed] [Google Scholar]
- 5.Tavalai N, Kraiger M, Kaiser N, Stamminger T. 2008. Insertion of an EYFP-pp71 (UL82) coding sequence into the human cytomegalovirus genome results in a recombinant virus with enhanced viral growth. J Virol 82:10543–10555. doi: 10.1128/JVI.01006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler M, Tavalai N, Muller R, Stamminger T. 2011. Human cytomegalovirus immediate-early gene expression is restricted by the nuclear domain 10 component Sp100. J Gen Virol 92:1532–1538. doi: 10.1099/vir.0.030981-0. [DOI] [PubMed] [Google Scholar]
- 7.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem 281:37652–37660. doi: 10.1074/jbc.M604273200. [DOI] [PubMed] [Google Scholar]
- 10.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J Virol 82:12543–12554. doi: 10.1128/JVI.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn JH. 2011. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol 85:11928–11937. doi: 10.1128/JVI.00758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cantrell SR, Bresnahan WA. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J Virol 79:7792–7802. doi: 10.1128/JVI.79.12.7792-7802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gawn JM, Greaves RF. 2002. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J Virol 76:4441–4455. doi: 10.1128/JVI.76.9.4441-4455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dell'Oste V, Gatti D, Gugliesi F, De Andrea M, Bawadekar M, Lo Cigno I, Biolatti M, Vallino M, Marschall M, Gariglio M, Landolfo S. 2014. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J Virol 88:6970–6982. doi: 10.1128/JVI.00384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biolatti M, Dell'Oste V, Pautasso S, von Einem J, Marschall M, Plachter B, Gariglio M, De Andrea M, Landolfo S. 2016. Regulatory interaction between the cellular restriction factor IFI16 and viral pp65 (pUL83) modulates viral gene expression and IFI16 protein stability. J Virol 90:8238–8250. doi: 10.1128/JVI.00923-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahn JH, Hayward GS. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J Virol 71:4599–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn JH, Hayward GS. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39–55. doi: 10.1006/viro.2000.0448. [DOI] [PubMed] [Google Scholar]
- 19.Kelly C, Van Driel R, Wilkinson GW. 1995. Disruption of PML-associated nuclear bodies during human cytomegalovirus infection. J Gen Virol 76(Part 11):2887–2893. doi: 10.1099/0022-1317-76-11-2887. [DOI] [PubMed] [Google Scholar]
- 20.Mohrmann G, Hengstler JG, Hofmann TG, Endele SU, Lee B, Stelzer C, Zabel B, Brieger J, Hasenclever D, Tanner B, Sagemueller J, Sehouli J, Will H, Winterpacht A. 2005. SPOC1, a novel PHD-finger protein: association with residual disease and survival in ovarian cancer. Int J Cancer 116:547–554. doi: 10.1002/ijc.20912. [DOI] [PubMed] [Google Scholar]
- 21.Bordlein A, Scherthan H, Nelkenbrecher C, Molter T, Bosl MR, Dippold C, Birke K, Kinkley S, Staege H, Will H, Winterpacht A. 2011. SPOC1 (PHF13) is required for spermatogonial stem cell differentiation and sustained spermatogenesis. J Cell Sci 124:3137–3148. doi: 10.1242/jcs.085936. [DOI] [PubMed] [Google Scholar]
- 22.Kinkley S, Staege H, Mohrmann G, Rohaly G, Schaub T, Kremmer E, Winterpacht A, Will H. 2009. SPOC1: a novel PHD-containing protein modulating chromatin structure and mitotic chromosome condensation. J Cell Sci 122:2946–2956. doi: 10.1242/jcs.047365. [DOI] [PubMed] [Google Scholar]
- 23.Chung HR, Xu C, Fuchs A, Mund A, Lange M, Staege H, Schubert T, Bian C, Dunkel I, Eberharter A, Regnard C, Klinker H, Meierhofer D, Cozzuto L, Winterpacht A, Di Croce L, Min J, Will H, Kinkley S. 2016. PHF13 is a molecular reader and transcriptional co-regulator of H3K4me2/3. Elife 5:e10607. doi: 10.7554/eLife.10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mund A, Schubert T, Staege H, Kinkley S, Reumann K, Kriegs M, Fritsch L, Battisti V, Ait-Si-Ali S, Hoffbeck AS, Soutoglou E, Will H. 2012. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res 40:11363–11379. doi: 10.1093/nar/gks868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiner S, Kinkley S, Burck C, Mund A, Wimmer P, Schubert T, Groitl P, Will H, Dobner T. 2013. SPOC1-mediated antiviral host cell response is antagonized early in human adenovirus type 5 infection. PLoS Pathog 9:e1003775. doi: 10.1371/journal.ppat.1003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofmann S, Dehn S, Businger R, Bolduan S, Schneider M, Debyser Z, Brack-Werner R, Schindler M. 2017. Dual role of the chromatin-binding factor PHF13 in the pre- and post-integration phases of HIV-1 replication. Open Biol 7:170115. doi: 10.1098/rsob.170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulkarni AS, Fortunato EA. 2011. Stimulation of homology-directed repair at I-SceI-induced DNA breaks during the permissive life cycle of human cytomegalovirus. J Virol 85:6049–6054. doi: 10.1128/JVI.02514-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castillo JP, Frame FM, Rogoff HA, Pickering MT, Yurochko AD, Kowalik TF. 2005. Human cytomegalovirus IE1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J Virol 79:11467–11475. doi: 10.1128/JVI.79.17.11467-11475.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.E X, Pickering MT, Debatis M, Castillo J, Lagadinos A, Wang S, Lu S, Kowalik TF. 2011. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog 7:e1001342. doi: 10.1371/journal.ppat.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni AS, Fortunato EA. 2014. Modulation of homology-directed repair in T98G glioblastoma cells due to interactions between wildtype p53, Rad51 and HCMV IE1-72. Viruses 6:968–985. doi: 10.3390/v6030968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherer M, Otto V, Stump JD, Klingl S, Muller R, Reuter N, Muller YA, Sticht H, Stamminger T. 2015. Characterization of recombinant human cytomegaloviruses encoding IE1 mutants L174P and 1-382 reveals that viral targeting of PML bodies perturbs both intrinsic and innate immune responses. J Virol 90:1190–1205. doi: 10.1128/JVI.01973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasmapour B, Kubsch T, Rand U, Eiz-Vesper B, Messerle M, Vondran FWR, Wiegmann B, Haverich A, Cicin-Sain L. 2018. Myeloid dendritic cells repress human cytomegalovirus gene expression and spread by releasing interferon-unrelated soluble antiviral factors. J Virol 92:e01138-17. doi: 10.1128/JVI.01138-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaspar M, Shenk T. 2006. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A 103:2821–2826. doi: 10.1073/pnas.0511148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ. 1987. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem 262:14042–14048. [PubMed] [Google Scholar]
- 36.Tavalai N, Stamminger T. 2011. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res 157:128–133. doi: 10.1016/j.virusres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Wagenknecht N, Reuter N, Scherer M, Reichel A, Muller R, Stamminger T. 2015. Contribution of the major ND10 proteins PML, hDaxx and Sp100 to the regulation of human cytomegalovirus latency and lytic replication in the monocytic cell line THP-1. Viruses 7:2884–2907. doi: 10.3390/v7062751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komatsu T, Will H, Nagata K, Wodrich H. 2016. Imaging analysis of nuclear antiviral factors through direct detection of incoming adenovirus genome complexes. Biochem Biophys Res Commun 473:200–205. doi: 10.1016/j.bbrc.2016.03.078. [DOI] [PubMed] [Google Scholar]
- 39.Nevels M, Nitzsche A, Paulus C. 2011. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev Med Virol 21:154–180. doi: 10.1002/rmv.690. [DOI] [PubMed] [Google Scholar]
- 40.Reeves MB. 2011. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res 157:134–143. doi: 10.1016/j.virusres.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D. 2015. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife 4:e06068. doi: 10.7554/eLife.06068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy JC, Fischle W, Verdin E, Sinclair JH. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 21:1112–1120. doi: 10.1093/emboj/21.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91:599–604. doi: 10.1099/vir.0.015602-0. [DOI] [PubMed] [Google Scholar]
- 44.Groves IJ, Reeves MB, Sinclair JH. 2009. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early’ repression of viral gene expression mediated by histone post-translational modification. J Gen Virol 90:2364–2374. doi: 10.1099/vir.0.012526-0. [DOI] [PubMed] [Google Scholar]
- 45.Schilling EM, Scherer M, Reuter N, Schweininger J, Muller YA, Stamminger T. 2017. The human cytomegalovirus IE1 protein antagonizes PML nuclear body-mediated intrinsic immunity via the inhibition of PML de novo SUMOylation. J Virol 91:e2049-16. doi: 10.1128/JVI.02049-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 47.Bootz A, Karbach A, Spindler J, Kropff B, Reuter N, Sticht H, Winkler TH, Britt WJ, Mach M. 2017. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog 13:e1006601. doi: 10.1371/journal.ppat.1006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hofmann H, Floss S, Stamminger T. 2000. Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J Virol 74:2510–2524. doi: 10.1128/JVI.74.6.2510-2524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scherer M, Klingl S, Sevvana M, Otto V, Schilling EM, Stump JD, Muller R, Reuter N, Sticht H, Muller YA, Stamminger T. 2014. Crystal structure of cytomegalovirus IE1 protein reveals targeting of TRIM family member PML via coiled-coil interactions. PLoS Pathog 10:e1004512. doi: 10.1371/journal.ppat.1004512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winkler M, Rice SA, Stamminger T. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J Virol 68:3943–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andreoni M, Faircloth M, Vugler L, Britt WJ. 1989. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J Virol Methods 23:157–167. doi: 10.1016/0166-0934(89)90129-8. [DOI] [PubMed] [Google Scholar]
- 52.Sanchez V, Sztul E, Britt WJ. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-Golgi-intermediate compartment. J Virol 74:3842–3851. doi: 10.1128/JVI.74.8.3842-3851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waldo FB, Britt WJ, Tomana M, Julian BA, Mestecky J. 1989. Non-specific mesangial staining with antibodies against cytomegalovirus in immunoglobulin-A nephropathy. Lancet i:129–131. [DOI] [PubMed] [Google Scholar]
- 54.Gebert S, Schmolke S, Sorg G, Floss S, Plachter B, Stamminger T. 1997. The UL84 protein of human cytomegalovirus acts as a transdominant inhibitor of immediate-early-mediated transactivation that is able to prevent viral replication. J Virol 71:7048–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lorz K, Hofmann H, Berndt A, Tavalai N, Mueller R, Schlotzer-Schrehardt U, Stamminger T. 2006. Deletion of open reading frame UL26 from the human cytomegalovirus genome results in reduced viral growth, which involves impaired stability of viral particles. J Virol 80:5423–5434. doi: 10.1128/JVI.02585-05. [DOI] [PMC free article] [PubMed] [Google Scholar]