

ABSTRACT
Mammalian orthoreovirus attachment to target cells is mediated by the outer capsid protein σ1, which projects from the virion surface. The σ1 protein is a homotrimer consisting of a filamentous tail, which is partly inserted into the virion; a body domain constructed from β-spiral repeats; and a globular head with receptor-binding properties. The σ1 tail is predicted to form an α-helical coiled coil. Although σ1 undergoes a conformational change during cell entry, the nature of this change and its contributions to viral replication are unknown. Electron micrographs of σ1 molecules released from virions identified three regions of flexibility, including one at the midpoint of the molecule, that may be involved in its structural rearrangement. To enable a detailed understanding of essential σ1 tail organization and properties, we determined high-resolution structures of the reovirus type 1 Lang (T1L) and type 3 Dearing (T3D) σ1 tail domains. Both molecules feature extended α-helical coiled coils, with T1L σ1 harboring central chloride ions. Each molecule displays a discontinuity (stutter) within the coiled coil and an unexpectedly seamless transition to the body domain. The transition region features conserved interdomain interactions and appears rigid rather than highly flexible. Functional analyses of reoviruses containing engineered σ1 mutations suggest that conserved residues predicted to stabilize the coiled-coil-to-body junction are essential for σ1 folding and encapsidation, whereas central chloride ion coordination and the stutter are dispensable for efficient replication. Together, these findings enable modeling of full-length reovirus σ1 and provide insight into the stabilization of a multidomain virus attachment protein.
IMPORTANCE While it is established that different conformational states of attachment proteins of enveloped viruses mediate receptor binding and membrane fusion, less is understood about how such proteins mediate attachment and entry of nonenveloped viruses. The filamentous reovirus attachment protein σ1 binds cellular receptors; contains regions of predicted flexibility, including one at the fiber midpoint; and undergoes a conformational change during cell entry. Neither the nature of the structural change nor its contribution to viral infection is understood. We determined crystal structures of large σ1 fragments for two different reovirus serotypes. We observed an unexpectedly tight transition between two domains spanning the fiber midpoint, which allows for little flexibility. Studies of reoviruses with engineered changes near the σ1 midpoint suggest that the stabilization of this region is critical for function. Together with a previously determined structure, we now have a complete model of the full-length, elongated reovirus σ1 attachment protein.
KEYWORDS: fiber protein, reovirus, virus attachment, virus structure
INTRODUCTION
Mammalian orthoreoviruses (reoviruses) cause disease in the very young and show promise as oncolytic agents. These nonenveloped viruses contain a segmented double-stranded RNA (dsRNA) genome, which is encapsidated by a two-layered icosahedral protein shell (1). The outer capsid is composed mainly of heterohexamers of the proteins μ1 and σ3. The pentameric λ2 protein, located at each 5-fold axis, is integral to both protein layers and anchors the trimeric reovirus attachment protein σ1 (2). The σ1 protein is a filamentous molecule that can bind to junctional adhesion molecule A (JAM-A) and sialylated glycans on the host cell surface (3–5). Reovirus cell attachment is thought to follow an adhesion-strengthening mechanism, where the initial low-affinity binding of σ1 to carbohydrate receptors enables lateral virus diffusion along the cell surface, followed by high-affinity binding to JAM-A (6). Reoviruses are internalized by receptor-mediated endocytosis (7). Within endocytic vesicles, the viral outer capsid undergoes stepwise, acid-dependent uncoating, which is catalyzed by cathepsin proteases B, L, and S (8, 9). The first disassembly intermediate, called the infectious subvirion particle (ISVP), is characterized by the loss of σ3, the cleavage of μ1, and a conformational change in the σ1 protein (2, 10). The conversion of the ISVP to the next disassembly intermediate, the ISVP*, involves the release of σ1 and the μ1 cleavage product μ1N, which forms pores within the endosomal membrane and recruits viral particles (11, 12). The contributions of σ1 conformational changes to disassembly and membrane penetration are unclear.
ISVPs are stable particles, which can also be generated in the intestinal lumen or by the in vitro treatment of reovirus with a variety of proteases (13, 14). These particles can be internalized by either endocytosis or direct penetration of the plasma membrane (15). Structural changes of the σ1 protein during virion-to-ISVP conversion have been proposed based on negative-stain electron microscopy (EM) images and cryo-EM reconstructions of both particle types (2, 10). Lower-resolution EM images show filamentous structures frequently protruding from ISVPs but not from virions, and cryo-EM reconstructions reveal electron density corresponding to σ1 that emerges radially from the center of the λ2 pentamer. While the density is knob-like in virions, it is more elongated in ISVPs, suggesting that at least two distinct σ1 conformations exist.
The σ1 protein is thought to consist of four structurally distinct regions based on amino acid sequence analyses and data from structural studies (4, 5, 16, 17). The N-terminal 20 to 25 amino acids anchor the molecule into the viral capsid. The following ∼150 residues, which constitute one-third of the molecule, are predicted to form an uninterrupted α-helical coiled-coil domain, based on the presence of a heptad repeat sequence motif (16, 18). This structural motif, (abcdefg)n, usually incorporates hydrophobic amino acids at positions “a” and “d,” while the other positions are occupied by more-hydrophilic residues (19). A hydrophobic core formed within a helical bundle usually accounts for most intersubunit interactions and provides the largest contribution to overall coiled-coil stability. Residues at positions “g” and “e + 1” are often charged and form interhelical salt bridges that additionally stabilize the oligomer. The predicted coiled-coil domain of the σ1 protein is followed by two domains, termed the body and head. The body encompasses ∼140 amino acids that fold primarily into triple β-spiral repeats (5). This motif has been observed for only a limited number of viral fiber proteins, including adenovirus fiber, avian reovirus σC, and bacteriophage PRD1 (20–22). Each β-spiral repeat is defined by a consensus sequence (“a” through “o”) with nonpolar amino acids at positions “c,” “e,” “g,” “k,” and “m.” This arrangement gives rise to two short antiparallel β-strands, which are connected via a 4-residue β-turn with a proline or glycine at the third position (position “j”). A short loop connects one β-spiral repeat to the next. The C-terminal one-third of the σ1 protein forms the globular head and folds into a compact, eight-stranded β-barrel (23). The head binds the serotype-independent receptor JAM-A within a conserved binding region and can additionally engage carbohydrate receptors, in the case of prototype strain type 1 Lang (T1L) σ1 (4, 24). In contrast to T1L σ1, the carbohydrate-binding epitope of prototype strain type 3 Dearing (T3D) σ1 is located in the body domain between β-spiral repeats β2 and β3 (5).
EM images of σ1 molecules released from virions show long, filamentous structures with three regions of significant flexibility (17). One such region lies near the N terminus, another lies at the midpoint of the fiber (coinciding with the transition of the coiled coil to the body domain), and a third one is located close to the head domain. These flexible regions may facilitate conformational changes in the protein during attachment or entry. Structures of the head and parts of the body domain have been determined for T1L and T3D σ1 proteins and reveal a flexible linker region located just below the head domain (4, 23, 25).
In this study, we investigated structurally uncharacterized regions of T1L and T3D σ1 proteins using X-ray crystallography. Determined at high resolution, our structures include the majority of the tail domain and the coiled-coil-to-body transition, a region at the midpoint of the σ1 molecule predicted to have enhanced flexibility. The structures reveal previously unidentified features, including bound chloride ions at the center of the T1L σ1 coiled-coil domain, a conserved stutter in the coiled coil, and the presence of stabilizing interactions, rather than a flexible linker, at the coiled-coil-to-body junction. To probe the functional relevance of these structural features, we engineered reoviruses containing mutant σ1 proteins and conducted functional assays. Our analyses show that stabilizing interactions at the coiled-coil-to-body domain transition site are crucial for σ1 folding and encapsidation, while bound anions and the heptad repeat stutter are dispensable for efficient viral replication. Moreover, our findings enable modeling of the full-length reovirus σ1 trimer in its fully extended form.
RESULTS
Construct design and structure determination.
To enable structural analyses, we expressed and purified recombinant T1L and T3D σ1 fragments of different lengths encompassing the predicted α-helical coiled-coil tail region (T1L σ1cc [amino acids 29 to 159]) as well as the domain junction and parts of the body domain (T1L σ1cc_body [amino acids 29 to 264] and T3D σ1cc_body [amino acids 25 to 291]). The native crystals of T1L σ1cc diffracted to a resolution of 1.43 Å, and the structure of this fragment was determined with experimental phasing using the anomalous signal of iodide (Table 1). The resulting model was used for molecular replacement to determine the crystal structures of T1L σ1cc_body and T3D σ1cc_body at resolutions of 2.10 Å and 2.15 Å, respectively (Table 1).
TABLE 1.
Data collection and refinement statisticsa
| Parameter | Value |
||||
|---|---|---|---|---|---|
| T1L σ1cc |
T1L σ1cc_body | T3D σ1cc_body | |||
| Chloride | Iodide | Iodide | |||
| Data collection statistics | |||||
| Wavelength (Å) | 0.91841 | 1.00000 | 2.00000 | 1.00003 | 1.00000 |
| Resolution range (Å) | 41.66–1.43 (1.52–1.43) | 48.73–1.35 (1.39–1.35) | 29.76–2.40 (2.46–2.40) | 42.19–2.10 (2.16–2.10) | 49.15–2.15 (2.21–2.15) |
| Space group | P21 | P21 | P21 | P21 | C2 |
| Cell axes (Å) (a, b, c) | 53.4, 37.2, 94.9 | 52.7, 37.6, 89.7 | 52.7, 37.6, 89.7 | 72.7, 34.0, 123.5 | 350.7, 41.5, 63.3 |
| Cell angles (°) (α, γ, β) | 90.0, 102.6 | 90.0, 100.5 | 90.0, 100.5 | 90.0, 101.4 | 90.0, 95.0 |
| Completeness (%) | 99.3 (96.1) | 99.9 (99.9) | 97.1 (80.2) | 99.9 (99.7) | 99.9 (99.8) |
| Total no. of reflections | 445,146 (63,117) | 503,646 (33,260) | 1,039,304 (18,914) | 223,136 (14,337) | 343,646 (24,014) |
| No. of unique reflections | 67,336 (10,431) | 76,273 (5,563) | 25,687 (1,566) | 35,260 (2,552) | 50,259 (3,674) |
| Redundancy | 6.6 (6.1) | 6.6 (6.0) | 40.5 (12.1) | 6.3 (5.6) | 6.8 (6.5) |
| Rmeas (%) | 12.3 (71.4) | 10.9 (87.6) | 7.0 (14.4) | 16.9 (91.7) | 12.7 (99.5) |
| CC1/2 (%) | 99.9 (96.4) | 99.9 (72.3) | 100.0 (99.3) | 99.7 (74.5) | 99.9 (79.7) |
| I/σ | 11.0 (2.1) | 12.0 (1.9) | 50.1 (12.9) | 10.2 (2.3) | 13.7 (2.3) |
| Wilson B (Å2) | 19.2 | 16.2 | 13.5 | 28.8 | 37.5 |
| Anomalous signal (%) | 4.5 (1.4) | ||||
| Refinement statistics | |||||
| Rwork/Rfree (%) | 18.5/22.2 | 17.5/20.3 | 18.9/24.9 | 20.8/24.9 | |
| No. of atoms | |||||
| Protein | 3,100 | 3,104 | 4,921 | 4,771 | |
| Waters | 678 | 817 | 370 | 415 | |
| Chloride/iodide | 2 | 4/3 | 3 | ||
| B factors (Å2) | |||||
| Chain A | 18.8 | 13.5 | 25.4 | 47.8 | |
| Chain B | 19.9 | 14.1 | 27.2 | 49.4 | |
| Chain C | 18.2 | 13.2 | 25.1 | 48.5 | |
| Water | 32.1 | 25.1 | 29.1 | 44.8 | |
| Chloride/iodide | 15.0 | 19.1/8.7 | 31.9 | ||
| RMSD | |||||
| Bond lengths (Å) | 0.015 | 0.015 | 0.011 | 0.01 | |
| Bond angles (°) | 1.15 | 1.24 | 1.07 | 1.03 | |
| PDB accession no. | 6GAK | 6GAJ | 6GAO | 6GAP | |
Values in parentheses are for the highest-resolution shell. RMSD, root mean square deviation; CC1/2, Pearson correlation coefficient calculated between two subsets of unmerged experimental data, each containing a random half of the measurements of each unique reflection (57).
Overall structures of the T1L and T3D σ1 tails connected to adjacent β-spiral repeats.
The structures of T1L σ1cc_body and T3D σ1cc_body reveal an elongated, rodlike homotrimer consisting of two structural domains, a long N-terminal α-helical coiled coil and a C-terminal region formed by β-spiral repeats (Fig. 1). One trimer is present in each asymmetric unit.
FIG 1.

T1L σ1 and T3D σ1 tail structures. The trimeric σ1 protein is shown in blue, red, and yellow. Chloride ions are shown as green spheres, and water molecules are shown as cyan spheres. T1L σ1cc_body (PDB accession number 6GAO) (A) and T3D σ1cc_body (PDB accession number 6GAP) (B) fold into an α-helical coiled-coil domain and a body domain that is formed mostly of β-spiral repeats. The site of transition between the two domains is indicated by black stars. The two structures are aligned based on the heptad repeat register and the positions of their stutter sequences (dark blue). The length of the coiled coil (cc) is indicated.
T1L σ1 residues 29 to 178 form an uninterrupted, left-handed coiled coil with ∼21.5 heptad repeats and a length of ∼220 Å. Three chloride ions are bound at different positions inside the coiled-coil core. Residues 180 to 249 fold into four β-spiral repeats, while the remaining residues of the σ1 fragment (residues 250 to 264) are not defined in the electron density. A sequence alignment with T3D σ1 indicates that these amino acids correspond to the short coiled-coil insertion in the σ1 body domain (5, 16).
The helical bundle of T3D σ1 is formed by ∼20 heptad repeats (residues 27 to 168), resulting in a domain that is approximately 205 Å long. The β-spiral repeats (β1 to β4) and part of the short body domain coiled-coil insertion are present in the electron density map (residues 169 to 244 of chain A) and superimpose well (root mean square deviation [RMSD] of equivalent Cα atoms, 0.38 Å) with those in the previously described structure of a C-terminal region of σ1 (Protein Data Bank [PDB] accession number 3S6X [5]). Residues 245 to 291 are not visible in the electron density map. SDS-PAGE analysis of crystals and protein stored at 4°C revealed that the T3D σ1cc_body protein partly degrades over time and that the crystals consisted of the degraded protein. T3D σ1 is known to be sensitive to trypsin and chymotrypsin cleavage after R245 and L261, respectively (26). Thus, protease cleavage would explain the absence of the C-terminal fragment in the electron density.
The conserved σ1 coiled-coil-to-body transition.
Both σ1cc_body structures show a direct connection of the coiled-coil tail and the body domain, with no intrinsically flexible interdomain region. In both structures, the tight transition of the two domains is stabilized by interhelical salt bridges located at the coiled-coil end and backbone-backbone interactions of valine residues at the domain junction (Fig. 2). An asparagine side chain from the body domain forms hydrogen bonds with carbonyl groups located at the most C-terminal coiled-coil turn. In the case of T1L σ1, a second body domain residue (K189) interacts with a carbonyl group of the coiled-coil end. Although the σ1 proteins of the three reovirus serotypes share limited sequence identity, residues involved in interactions between the two domains are conserved in all three serotypes, suggesting that they have a key function.
FIG 2.

Interactions between the coiled-coil and body domains. The trimeric σ1 protein is shown in blue, red, and yellow. (A) Amino acid sequence alignment of the site of transition between coiled-coil and body domains of the three reovirus serotypes. Conserved residues are highlighted with stars. The heptad repeat consensus sequence is indicated above the alignment. The stutter is highlighted by a yellow box. Residues involved in stabilizing the coiled-coil end and the domain transition are also outlined. (B) T1L σ1 residues N192 and K189 of the body domain, located close to the C-terminal coiled-coil end, form hydrogen bonds with the carbonyl groups of E176 and S178 and the carbonyl group of M179, respectively. An interhelical salt bridge involving R171 and E176 stabilizes the end of the coiled coil. Residue R171 also mediates contacts with S169 and S173 (not shown). Valine residues (V180) at the domain boundary form backbone-backbone interactions, shown along the trimer axis. (C) T3D σ1 residue N182 of the body domain forms hydrogen bonds with the carbonyl groups of E166 and A169, located at the C-terminal coiled-coil end. R161 forms an interhelical salt bridge with E166, stabilizing the coiled coil at the domain boundary. Residue R161 also mediates contacts with D157 and E159 (not shown). Valine residues (V170) of the three T3D σ1 subunits form backbone-backbone interactions, shown along the trimer axis.
Chloride and water coordination in the coiled-coil domain.
The coiled-coil domains of T1L and T3D σ1 proteins are stabilized by a number of intersubunit salt bridges, which are distributed over the length of the helical bundles. In each coiled coil, some hydrophilic residues are located at usually hydrophobic heptad repeat positions (“a” and “d”), with side chains pointing toward the trimer core. T1L σ1 harbors two threonines (T119 and T126) at “a” positions and two asparagines (N38 and N94) at “d” positions. As observed for other trimeric coiled-coil-containing proteins, such as bacterial adhesion proteins or the spike glycoprotein of severe acute respiratory syndrome (SARS) coronavirus, the amide nitrogens of asparagine side chains at position “d” coordinate chloride ions within the central core (Fig. 3A and B) (27, 28). A third chloride ion is located in a hydrophobic pocket formed by three glycine residues (G161) at “d” positions and six isoleucine residues (I158 and I165) at “a” positions above and below the chloride ion (Fig. 3C).
FIG 3.

Chloride ions and water molecules bound in the core of the σ1 coiled coil. The trimeric σ1 protein is shown in blue, red, and yellow. Chloride ions are shown as green spheres, and water molecules are shown as cyan spheres. (A and B) Enlarged views of T1L σ1 showing bound chloride ions complexed with N38 (A) and N94 (B), respectively. (C) A third chloride ion is bound in a hydrophobic pocket inside the core of the structure. (D and E) Enlarged views of T3D σ1 showing bound water molecules in the coiled-coil interior that form hydrogen bonds with H42 (D) and H123 (E).
In the case of T3D σ1, a serine (S39), two histidines (H42 and H123), and an arginine (R67) are present at typically hydrophobic core positions. No asparagines are located at “d” positions, and no chloride ions are present in the interior of the structure. The guanidine group of R67 is shifted to the outside of the core and interacts with a serine (S66) of an adjacent chain. Serines at position “a” (S39) and histidines at position “d” (H42) bind several water molecules inside the coiled-coil core. The histidines (H123) at position “a” also bind a water molecule at the trimer interface. However, H123 lies in a region with elevated temperature factors (amino acids 103 to 151), and the water molecule bound by H123 is therefore not well defined in the electron density.
A heptad repeat stutter in the σ1 tail.
A discontinuity of the heptad repeat pattern in the form of a 4-amino-acid insertion (stutter) is present near the C terminus of the coiled-coil region in all σ1 serotypes. The motif “abcd” (T1L σ1 residues 147 to 150 [VTTE] and T3D σ1 residues 151 to 154 [VTSI]) is present between two “abcdefg” repeats, and our structures show that this stutter leads to local unwinding of the coiled coil, resulting in a d-a layer-type stutter (19, 29). In a d-a layer, the residues at the “d” position of the stutter are shifted outside the core toward the following “a” position, causing a ringlike interaction around the central core (Fig. 4).
FIG 4.

Heptad repeat discontinuity (stutter) of the σ1 coiled coil. The trimeric σ1 protein is shown in blue, red, and yellow. For a sequence comparison of the stutters between different reovirus serotypes, see Fig. 2A. (A) T1L σ1 d-a layer formation between E150 and V151. The view is along the 3-fold axis of the molecule. (B) Side view of the same region. T1L σ1 residue R153 forms a salt bridge with E150 and D155. (C) T3D σ1 d-a layer formation between I154 and Q155. The view is along the 3-fold axis of the molecule. (D) Side view of the same region. T3D σ1 residue Q155 also interacts with R150 of an adjacent chain.
In the case of T1L σ1, the E150 side chains point toward the solvent, leading to hydrophobic interactions of the Cβ and Cγ atoms with V151. The carboxyl group of E150 forms a salt bridge with R153, which is additionally involved in a salt bridge with D155 from an adjacent chain. In the case of T3D σ1, residue I154 at the “d” position of the stutter is shifted toward Q155, which leads to a hydrophobic interaction with the Cβ and Cγ atoms of Q155. Residue Q155 additionally interacts with R150 that also contacts T152.
Interactions at the coiled-coil-to-body domain boundary are required for efficient reovirus replication.
To determine whether the observed structural features (chloride binding of T1L σ1, a heptad repeat stutter, and interactions at the domain transition site) contribute to σ1 function, we engineered reoviruses expressing mutant σ1 molecules. Mutations were engineered into recombinant strain T1L (rsT1L) or rsT3SA+, which contains nine T1L gene segments and a type 3 (T3) σ1-encoding S1 gene segment. We engineered a T1L reovirus expressing σ1 that lacks the capacity to coordinate chloride ions by making valine substitutions at residues N38 and N94. Stutterless T3 σ1 proteins were engineered by the deletion (ΔVTSI) or insertion (+QST) of residues in rsT3SA+. We also engineered rsT3SA+ reoviruses expressing σ1 with alanine substitutions at residue R161 or N182 or both residues. The R161A mutation was designed to prevent the formation of stabilizing salt bridges with D157, E159, and E166, while the N182A mutation removes interactions with carbonyl groups of E166 and A169 at the end of the coiled coil and the beginning of the body domain. The mutant viruses were recovered by using plasmid-based reverse genetics, and multiple clones of each virus were plaque purified and amplified to make working stocks. RNA was extracted from working stocks, and the S1 genes were reverse transcribed and sequenced. The chloride-binding-deficient virus rsT1L σ1 N38V,N94V as well as the stutterless rsT3SA+ viruses (rsT3SA+ σ1 ΔVTSI and rsT3SA+ σ1 +QST) produced titers comparable to those of the parental viruses after two passages in murine L929 fibroblasts (L cells) (Fig. 5A). Following the adsorption of L cells at a low multiplicity of infection (MOI), these mutant viruses replicated with kinetics that were impaired only slightly in comparison to those of the parental rsT1L and rsT3SA+ viruses, respectively (Fig. 5B and C). These observations suggest that T1L σ1 chloride ion coordination and the σ1 heptad repeat stutter are dispensable for efficient reovirus replication in cultured cells. In contrast, titers of the three coiled-coil-to-body transition mutants (rsT3SA+ σ1 R161A, rsT3SA+ σ1 N182A, and rsT3SA+ σ1 R161A,N182A) were substantially lower than those of the parental rsT3SA+ virus after two passages in L cells, with the lowest titers being observed for the N182A and double-mutant viruses (Fig. 5A). Following infection of L cells at a low multiplicity of infection, the σ1 coiled-coil-to-body transition mutants displayed highly impaired replication kinetics in comparison to those of T3SA+, resulting in ∼30- to 100-fold reductions in the titers of rsT3SA+ σ1 R161A and rsT3SA+ σ1 N182A, respectively, and a nearly 1,000-fold reduction in the titer of rsT1L σ1 R161A,N182A by 48 h postinfection (Fig. 5D). These findings indicate that stabilizing interactions at the coiled-coil-to-body domain boundary are required for efficient reovirus replication.
FIG 5.

Mutant reovirus replication in L cells. (A) Average titers from stocks of three independent clones per virus recovered by reverse genetics, plaque purified, and amplified twice in L cells. (B to D) Cells were adsorbed with three independent clones of rsT1L and mutant reoviruses impaired for chloride binding (B), rsT3SA+ and stutter correction mutant reoviruses (C), or rsT3SA+ and coiled-coil-to-body transition mutant reoviruses (D) at an MOI of 0.01 PFU/cell (B and C) or 0.001 PFU/cell (D). Unbound virus was removed, fresh medium was added, and cells were incubated at 37°C for the times shown. Virus titers in cell lysates were determined by a plaque assay using L cells. Error bars represent standard deviations.
Attachment and entry properties of σ1 coiled-coil-to-body transition mutant viruses.
Reoviruses engage both sialylated glycans and JAM-A to mediate cell surface binding (3–5). To compare the receptor utilization of rsT3SA+ to those of coiled-coil-to-body transition mutants, we treated Chinese hamster ovary (CHO) cells expressing human JAM-A (JAM-A-CHO cells) with monoclonal antibody (MAb) J10.4, which blocks reovirus binding to JAM-A, or Arthrobacter ureafaciens neuraminidase (NA), which removes cell surface sialic acid, prior to virus adsorption. We found ∼40% inhibition of rsT3SA+ infection and <20% inhibition of rsT3SA+ σ1 R161A, rsT3SA+ σ1 N182A, and rsT3SA+ σ1 R161A,N182A at the highest NA treatment concentration, with a very modest inhibition of infection by all viruses at lower NA concentrations (Fig. 6A). Following treatment with J10.4, the rsT3SA+ σ1 R161A and rsT3SA+ σ1 R161A,N182A viruses exhibited markedly reduced infectivity, equivalent to that observed for parental rsT3SA+. While the infectivity of the rsT3SA+ σ1 N182A virus was decreased to 30 to 45% in the presence of J10.4, this virus was significantly more infectious following J10.4 treatment than was rsT3SA+ at J10.4 treatment concentrations of 2.5 and 10 μg/ml. These results suggest that mutations destabilizing the coiled-coil-to-body junction do not impair binding to JAM-A and sialic acid. However, the reduced sensitivities of all mutants to NA and of the rsT3SA+ σ1 N182A mutant virus to J10.4 suggest that these viruses may be somewhat less dependent on sialic acid and JAM-A, respectively, for attachment.
FIG 6.

Attachment and entry properties of σ1 coiled-coil-to-body transition mutant reoviruses. (A) JAM-A-CHO cells were treated with the indicated concentrations of NA to remove sialic acid or J10.4 to block binding to JAM-A or left untreated (control) prior to adsorption with equivalently infectious concentrations of rsT3SA+ or σ1 coiled-coil-to-body transition mutant reoviruses. (B) Equivalently infectious concentrations of rsT3SA+ or σ1 coiled-coil-to-body transition mutant reoviruses were incubated with polyclonal antireovirus serum as a positive control, the indicated concentrations of T3 σ1-specific MAb 9BG5 to neutralize the virus or T1 σ1-specific MAb 5C6 as a negative control, or medium only as an additional negative control prior to adsorption on L cells. (C) NH4Cl (25 mM) was added to L cells at the indicated times after adsorption with equivalently infectious concentrations of rsT3SA+ or σ1 coiled-coil-to-body transition mutant reoviruses. Cells were fixed at 16 to 20 h postadsorption and scored for DAPI and reovirus antigen by indirect immunofluorescence. Results are expressed as the mean percentages of virus-infected cells, relative to those under untreated conditions, plus standard deviations (error bars) from four fields of view per well in duplicate wells for three independent experiments. Values that differ significantly from those for rsT3SA+ under each condition by one-way ANOVA with Dunnett's multiple-comparison test are indicated with * (P < 0.01).
MAb 9BG5 binds across the interface of adjacent monomers in the T3 σ1 trimer in the vicinity of the head domain, interferes with binding to JAM-A, and neutralizes reovirus infectivity (30). To determine whether the σ1 proteins of coiled-coil-to-body transition mutant reoviruses fold into functional trimers, we quantified the neutralization of reovirus infectivity using L cells following virus preincubation with a range of concentrations of T3-specific MAb 9BG5 or T1-specific MAb 5C6, as a negative control. We found that the mutant viruses, in nearly all cases, were equivalently sensitive to neutralization by 9BG5 and equivalently resistant to neutralization by 5C6 in comparison to parental rsT3SA+ (Fig. 6B). The rsT3SA+ σ1 N182A virus was less sensitive than rsT3SA+ to neutralization by 9BG5 at the lowest antibody concentrations, but it was still neutralized efficiently. These results provide evidence that the σ1 head domains of the mutant viruses fold into functional trimers.
Ammonium chloride (NH4Cl) prevents endosomal acidification and inhibits reovirus entry. To determine whether mutations affecting the stability of the coiled-coil-to-body junction, which may contribute to σ1 conformational changes, alter the pH sensitivity of reovirus entry, we treated L cells with NH4Cl during a time course and quantified the effects on viral infectivity. We found that rsT3SA+ and all of the coiled-coil-to-body transition mutants were equivalently sensitive to NH4Cl at all times of addition (Fig. 6C). This finding suggests that the mutant viruses have the same requirements for acidic pH to initiate an infectious cycle.
Encapsidation and folding of σ1 coiled-coil-to-body transition mutants.
Reovirus variants and mutants with polymorphisms in the σ1 sequence encapsidate different amounts of σ1 fibers (31, 32). Using quantitative immunoblotting, we found that the mutant viruses encapsidate approximately 50% (rsT3SA+ σ1 R161A and rsT3SA+ σ1 N182A) or 30% (rsT1L σ1 R161A,N182A) of the amount of σ1 encapsidated by the parental rsT3SA+ virus (Fig. 7A). Using agarose gel separation of reovirus particles, we found that the majority of rsT3SA+ particles encapsidate 8 to 12 σ1 trimers (Fig. 7B). In accord with the observed reductions in σ1 encapsidation detected by immunoblotting (Fig. 7A), we found that the majority of rsT3SA+ σ1 R161A and rsT3SA+ σ1 N182A particles encapsidate 0 to 7 σ1 trimers, and the majority of rsT3SA+ σ1 R161A,N182A particles encapsidate 0 to 3 σ1 trimers (Fig. 7B). Together, these observations suggest that altering the stability of the coiled-coil-to-body junction reduces the efficiency of σ1 encapsidation onto reovirus particles. Reduced σ1 encapsidation may explain the observed replication defects of the coiled-coil-to-body transition mutant viruses (Fig. 5C).
FIG 7.

Encapsidation of σ1 by coiled-coil-to-body transition mutant viruses. (A) Purified reovirus particles (1 × 1010) were resolved by SDS-PAGE and immunoblotting using T3D σ1-specific polyclonal antiserum, μ1C-specific monoclonal antibody 8H6, and fluorescently labeled secondary antibodies. Protein band intensities were quantified, and the integrated protein band fluorescence of σ1 was divided by that of μ1C to normalize the results. The percentage of normalized mutant σ1, relative to the amount of T3SA+ σ1, from two independent experiments (1 and 2) is shown. (B) Purified reovirus particles (2.5 × 1011) were resolved by electrophoresis in a 1% agarose gel and visualized by colloidal Coomassie staining. Individual bands on the gel correspond to particles that differ in the numbers (0 to 12) of encapsidated σ1 trimers.
To determine whether the observed reductions in σ1 encapsidation are due to protein misfolding, we engineered the coiled-coil-to-body transition mutations into the T3D σ1cc_body construct and investigated the behavior of the mutant proteins during purification using the protocol established for wild-type T3D σ1cc_body. Expression levels of the R161A and N182A mutants were substantially reduced in comparison to those of the wild-type construct. Less mutant protein was present in the supernatant after cell lysis, and most of the protein eluted in the void volume of the size exclusion chromatography column, suggesting that the proteins form aggregates more readily (data not shown). The tendency for these mutant proteins to aggregate suggests that misfolding of the mutant σ1 proteins mediates their reduced encapsidation into the viral capsid.
Conformational change of encapsidated σ1 for coiled-coil-to-body transition mutant viruses.
While a majority of the σ1 protein containing R161A and N182A mutations may be misfolded, the small amount that is encapsidated appears to be capable of forming trimers, binding to carbohydrate receptors and JAM-A, and mediating pH-dependent entry. To determine whether the encapsidated mutant σ1 molecules also undergo a conformational change during virion-to-ISVP conversion, we generated ISVPs in vitro via incubation with chymotrypsin and conducted hemagglutination (HA) assays with ISVPs and matched virions. Chymotrypsin treatment of T3D increases its capacity to agglutinate red blood cells (26). This phenotype likely results from the increased accessibility of the carbohydrate-binding site in the σ1 body domain following conformational rearrangement, which would enhance red blood cell cross-linking. In accord with this finding, the HA titers of rsT3SA+ ISVPs were higher than those of rsT3SA+ virions (Fig. 8). While no HA was detectable at the highest particle concentration tested for the mutant virions, HA was detected for both rsT3SA+ σ1 R161A and rsT3SA+ σ1 N182A ISVPs. Considering the highly reduced numbers of σ1 trimers present on rsT3SA+ σ1 R161A,N182A particles (Fig. 7B), it is not surprising that neither virions nor ISVPs of this strain were capable of cross-linking red blood cells (Fig. 8). Together, these observations provide indirect evidence that σ1 trimers containing mutations affecting σ1 stability at the coiled-coil-to-body junction can undergo conformational rearrangements during virion-to-ISVP conversion.
FIG 8.
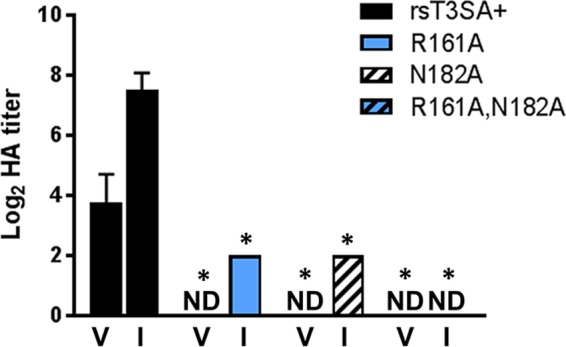
Hemagglutination capacities of rsT3SA+ and σ1 coiled-coil-to-body transition mutant reoviruses. Equal numbers of particles of purified reovirus virions (V) or ISVPs (I) were serially diluted, incubated with a 1% suspension of human erythrocytes at 4°C for 4 h, and scored for HA. Erythrocyte shields indicated HA, and erythrocyte buttons indicated the absence of erythrocyte cross-linking. Results are expressed as mean log2 HA titers for two replicates from each of two independent experiments. The HA titer is defined as 2.5 × 1010 particles divided by the number of particles per HA unit. One HA unit equals the minimum number of particles sufficient to produce HA. ND denotes samples for which HA was not detected at the highest number of particles (2.5 × 1010). Error bars represent standard deviations. Values that differ significantly from those for rsT3SA+ under each condition by one-way ANOVA with Dunnett's multiple-comparison test are indicated with * (P < 0.001).
DISCUSSION
We have structurally and functionally characterized the tail and its connection with the body region of the σ1 proteins of prototype reovirus strains T1L and T3D. Both structures form stable homotrimers that are ∼270 to 280 Å long and comprise an N-terminal α-helical coiled-coil domain and a C-terminal body domain constructed from β-spiral repeats. The structures encompass more than half the length of σ1, which is ∼480 Å in its fully elongated state (17). Both proteins feature an unexpectedly tight transition between the tail and body domains, with direct and conserved interactions linking them near the midpoint of the fiber. The architecture of the transition indicates that it possesses little internal flexibility.
Low-resolution EM images provided the first views of full-length σ1 fibers (17). While some σ1 fibers assume a linear structure, a small fraction feature a kink of up to 90° near the midpoint, at a location that corresponds approximately to the transition between the coiled-coil and body domains. The conformational heterogeneity of the protein was therefore attributed to a high degree of flexibility at the midpoint, with implications for function. The crystal structures reported here define the architecture of this transition at high resolution, and while they do not exclude the possibility of small movements between the two domains, they clearly do not provide any evidence for high flexibility and conformational heterogeneity in this region. As only a minor fraction (2 to 4%) of σ1 molecules analyzed by EM revealed kinks at the midpoint of the fiber, we think that these kinks are most likely caused by the relatively harsh sample preparation, which included heating to 52°C for 30 min to release σ1 from virus particles and negative staining with uranyl formate.
Reovirus σ1 proteins from different serotypes share little sequence identity (16). However, the residues at the coiled-coil-to-body transition are well conserved among all reovirus σ1 proteins, suggesting that their role is essential. The transition is stabilized by a conserved interhelical salt bridge at the C-terminal end of the coiled-coil domain and hydrophilic interactions involving conserved residues of both domains. Mutant T3SA+ viruses lacking the conserved salt bridge or the interactions between body domain residue N182 and carbonyl groups of the coiled coil have defects in viral replication (Fig. 5) and reduced numbers of encapsidated σ1 fibers (Fig. 7). Aggregation during protein purification suggests that the misfolding of R161A and N182A mutant σ1 proteins is responsible for the decreased fiber encapsidation. Thus, our findings indicate that the tight transition between the coiled-coil and body domains is important for σ1 encapsidation into the viral capsid and for protein folding, as even single-amino-acid substitutions in this region are poorly tolerated.
While the significantly impaired replication and σ1 encapsidation observed for coiled-coil-to-body transition mutant reoviruses suggest that the engineered changes can cause misfolding, the sensitivity of these viruses to receptor blockade, neutralizing antibodies, and NH4Cl suggests that the σ1 molecules that are successfully encapsidated enable virions to employ attachment and entry strategies similar to those of the parental virus (Fig. 6). It is unclear why the coiled-coil-to-body transition mutants display increased resistance to NA treatment of L cells or why rsT3SA+ σ1 N182A exhibited J10.4 resistance. It is possible that the mutations increase mobility at the interdomain junction, thereby modestly altering the accessibility of the sialic acid-binding site, which is located in the σ1 body domain, or interactions with JAM-A, which binds the σ1 head. The increase in HA activity observed for ISVPs in comparison to virions suggests that R161 and N182A mutations do not preclude σ1 conformational rearrangement during virion-to-ISVP conversion, at least not when they are introduced individually (Fig. 8). Together, these observations provide evidence that stabilization of the coiled-coil-to-body transition is not required for virus attachment and entry or σ1 conformational rearrangement.
Stutters in α-helical coiled coils introduce instability in coiled-coil structures and may provide sites for the initiation of molecular bending and unfolding (33, 34). The presence of asparagine and the coordination of chloride ions within the coiled-coil interior have been observed for several trimeric bacterial autotransporter adhesins (27). These interactions are thought to reflect special folding requirements in these proteins, such as maintaining protein solubility prior to folding. Likewise, the chloride ions might help to solubilize and properly oligomerize the T1L σ1 tail. It is unclear whether the chloride ions and the conserved stutter contribute to σ1 conformational rearrangements that occur during entry. However, based on a lack of severe replication defects observed for reoviruses with alterations in these features (Fig. 5), they appear to be nonessential for the function of σ1 in viral infection, at least in cell culture.
Together with a previously determined crystal structure that comprises the body and the head domains (PDB accession number 3S6X), we are now able to generate a composite, full-length model of σ1 that has high accuracy (Fig. 9). The two crystal structures contain overlapping segments, which can be superimposed with a low RMSD value of 0.34 Å (Cα atoms), allowing a faithful generation of the composite model. This model corresponds well with averaged EM images of the extended, full-length σ1 protein from strain type 2 Jones (T2J) and likely represents the σ1 conformation of ISVPs.
FIG 9.

Model of the elongated T3D σ1 protein. The trimeric σ1 protein is shown in blue, red, and yellow. N and C termini are indicated. The model was assembled by combining residues 27 to 243 of the T3D σ1cc_body structure (PDB accession number 6GAP) with residues 244 to 455 of a previously determined T3D σ1 structure (PDB accession number 3S6X, comprising the body and the head domain) after alignment of the identical parts of the structures. The main chains of the body domain residues present in both structures align with a low RMSD value of 0.34 Å. The two structures used for the generation of the composite model have resolutions of 2.15 and 2.25 Å, respectively. The T3D σ1 model is placed over a computer-processed electron micrograph of T2J σ1. (Adapted from reference 17).
Elongated fiber proteins are used as attachment moieties by a variety of bacteria and viruses, as exemplified by bacterial adhesins (35) and viral attachment proteins (36, 37). However, structural information about complete fiber proteins is scarce, in part because such proteins are difficult to analyze in their entirety. Often, only sections of the proteins have been resolved structurally, and the structures of the remaining parts are inferred based on sequence homology and modularity. The σ1 structure presented here offers a detailed, high-resolution view of the entire structure of this fiber and lacks only a small number of N-terminal residues. The structure of the avian reovirus functional equivalent to σ1, the σC protein, has been resolved but lacks more than 100 N-terminal residues (20, 38). Our structures of the T1L and T3D σ1 proteins differ from that of σC primarily in the transition between the coiled-coil and β-spiral repeat domains. In contrast to σ1, the coiled coil and the triple β-spiral repeats of σC are separated by a zinc ion-containing flexible linker. The linker of 5 amino acids forms two nested β-turns, and three histidine residues (H158) coordinate a divalent ion that might have a role in stabilizing the extended σC conformation in avian reovirus ISVPs (38). Otherwise, the σC and σ1 proteins have similar overall domain organizations, with a sequence identity of ∼20%. The number of residues that form the σC head domain and the predicted coiled-coil domain is comparable to that for σ1, but the body domain of σC is significantly shorter, containing only two triple β-spirals compared with seven in σ1 (5). Comparisons with other fiber-like proteins show a close structural homology of σ1 to the adenovirus fiber, a fairly rigid attachment protein constructed from β-spiral repeats and a globular head domain. In contrast to σ1, the adenovirus fiber does not contain any α-helical coiled-coil motifs.
The σ1 protein is hypothesized to undergo a large conformational change, from a more contracted to a fully extended trimer, during virus entry (2, 10). For many viruses, attachment proteins exist in a metastable state and refold into more-stable final structures following exposure to an environmental trigger. Perhaps the best-studied example for such a refolding event is influenza virus hemagglutinin, which transitions from a retracted conformation on the particle surface to a fully extended conformation in the low-pH environment encountered during cell entry (37, 39). Refolding of the hemagglutinin exposes the fusion peptide, enabling the fusion of the viral envelope with the host cell membrane. The elongated structure of σ1 defined in this study (Fig. 9) depicts the protein in its fully extended form, which is likely the more stable form, as fragments crystallized under different conditions produce nearly identical structures (4, 5, 23). The structure of contracted σ1 remains a mystery. In light of the evidence that μ1 and its fragments, particularly μ1N, mediate pore formation and the recruitment of viral particles to the endosomal membrane, the functional significance of σ1 conformational rearrangements also remains unknown (11, 12). Nonetheless, there is evidence that the head domain of σ1 is positioned closer to the virus surface in its contracted conformation, as antibodies against the surface capsid protein σ3 inhibit σ1-mediated functions (40). Based on the available structures, we think that a bend at the midpoint of σ1 is unlikely and that a contracted form would require a partial detrimerization of the three chains so that the head domain of each chain can approach the virus surface. There is precedent for such rearrangements in the VP4 attachment protein of rotavirus, which appears to be capable of assuming dimeric and trimeric arrangements (41). Therefore, such dramatic changes in subunit subassemblies during conformational rearrangements may be a common feature of the Reoviridae.
MATERIALS AND METHODS
Protein production and purification. (i) T1L σ1cc.
A cDNA encoding amino acids 29 to 159 of T1L σ1 was cloned into pET16b using the NcoI and BamHI restriction sites. Two additional amino acids (Met and Ala) are present at the N terminus due to the cloning procedure. The T1L σ129–159 protein was produced after induction with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 25°C overnight and purified via anion-exchange chromatography (buffer A, 20 mM HEPES [pH 7.4]; buffer B, 20 mM HEPES [pH 7.4] and 500 mM NaCl), followed by size exclusion chromatography in a solution containing 40 mM HEPES (pH 7.4) and 150 mM NaCl (Superdex 200; GE Healthcare).
(ii) T1L σ1cc_body.
A cDNA encoding amino acids 29 to 455 of T1L σ1 was cloned into pET28b (Novagen) using the NdeI and BamHI restriction sites. A stop codon was introduced at amino acid position 265 via site-directed mutagenesis. The T1L σ129–264 protein was produced at 20°C for 18 to 20 h postinduction (0.2 mM IPTG) in Escherichia coli BL21(DE3). The bacteria were lysed by sonication (Branson 450 sonifier) in a solution containing 50 mM Tris (pH 8.0), 300 mM NaCl, 10 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride (PMSF), and after Ni affinity chromatography, the N-terminal His6 tag was removed by trypsin cleavage in solution. Size exclusion chromatography (Superdex 200; GE Healthcare) with a solution containing 40 mM Tris (pH 8.6) and 150 mM NaCl was used as the last purification step.
(iii) T3D σ1cc_body.
A cDNA encoding amino acids 25 to 291 of T3D σ1 was cloned via the NheI and HindIII restriction sites into pET28b (Novagen), which includes an N-terminal His6 tag. The T3D σ125–291 protein was produced in E. coli Rosetta 2(DE3) by induction with 0.3 mM IPTG at 20°C for 18 to 20 h. Bacteria were resuspended in a solution containing 50 mM Tris (pH 8.0), 300 mM NaCl, 10 mM imidazole, and 1 mM PMSF and lysed by sonication (Branson 450 sonifier). The protein was purified by Ni affinity chromatography (His-Trap FF column; GE Healthcare), followed by size exclusion chromatography (Superdex 200; GE Healthcare) in a solution containing 40 mM HEPES (pH 7.4) and 150 mM NaCl.
(iv) Mutants.
To generate point mutants and stutterless σ1 proteins, plasmids of the T1L σ1cc_body and T3D σ1cc_body genes, respectively, were altered by QuikChange site-directed mutagenesis (Invitrogen) and sequenced to confirm the fidelity of mutagenesis. The primer sequences used are available from the corresponding authors upon request. Mutant proteins were purified via the corresponding purification protocol.
Crystallization. (i) T1L σ1cc.
Well-diffracting crystals of T1L σ1cc were obtained in a solution containing 20% polyethylene glycol 8000 (PEG 8000), 0.1 M N-cyclohexyl-3-aminopropanesulfonic acid (CAPS) (pH 10.5), and 0.2 M NaCl at 4°C by the sitting-drop vapor diffusion method. For an experimental phasing approach, the protein was denatured and refolded in an iodide-containing buffer to exchange (possibly) bound chloride ions of the coiled-coil core. The refolded protein was analyzed by circular dichroism (CD) spectroscopy. The CD spectrum was similar to that of an untreated protein sample (data not shown). Crystals were obtained in a solution containing 18% PEG 8000, 0.1 M CAPS (pH 9.5), and 0.2 mM NaI by seeding with crystals obtained under the original crystallization conditions at 4°C.
(ii) T1L σ1cc_body.
Crystals of T1L σ1cc_body were obtained in 35% (vol/vol) 1,4-dioxane at 4°C by the sitting-drop vapor diffusion method.
(iii) T3D σ1cc_body.
The T3D σ125–291 protein crystallized as small needle clusters in a solution containing 40% 2-methyl-2,4-pentanediol (MPD) and 0.1 M Tris (pH 8.0) at 4°C. An additive screen (Hampton Research) was applied to optimize the crystallization conditions. In addition to the small needlelike clusters that were obtained after 1 day, three-dimensional crystals grew after several weeks in a solution containing 40% MPD, 0.1 M Tris (pH 8.0), and 3% (wt/vol) 1,6-diaminohexane. SDS-PAGE analysis of dissolved crystals and the protein solution stored at 4°C revealed that the protein was partly degraded over time. Matrix-assisted laser desorption ionization (MALDI) mass spectrometry analysis (H. Kalbacher, University of Tübingen) of the gel bands identified protein fragments that covered the T3D σ1 sequence until amino acid R262.
X-ray structure determination.
Crystals of T1L σ1cc, T1L σ1cc_body, and T3D σ1cc_body were frozen in liquid nitrogen with 30% MPD in the mother liquor as a cryoprotectant and used to collect data at beamline X06DA at the Swiss Light Source (Villigen, Switzerland). Diffraction data were processed with the XDS program package (42). The structure of T1L σ1cc was determined by using single-wavelength anomalous dispersion, using the autoSHARP pipeline (43). Molecular replacement with coordinates of T1L σ1cc was used to solve the phase problem of T1L σ1cc_body and T3D σ1cc_body with Phaser (CCP4 suite) (44, 45). Manual model building and structural refinement were conducted in alternating cycles by using COOT (46), PHENIX (47), and autoBUSTER (48).
Cells, viruses, and antibodies.
Spinner-adapted L cells were grown in a suspension culture in Joklik's minimum essential medium (Lonza) supplemented to contain 5% fetal bovine serum (FBS) (Gibco), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). JAM-A-CHO cells (30) were maintained in Ham's F-12 medium (Gibco) supplemented to contain 10% FBS (Gibco), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). These cells were propagated in the presence of 0.8 mg/ml Geneticin during alternate passages.
Laboratory stocks of parental reovirus strains rsT1L and rsT3SA+ and reovirus mutants with altered S1 gene segments were prepared by using plasmid-based reverse genetics (49). rsT1L and rsT3SA+ differ solely in the σ1-encoding S1 gene segment, with strain T3SA+ containing a T3 S1 segment. The pBacT7-S1T1L plasmid was used as a template to engineer pBacT7-S1T1L N38V,N94V by PCR with mutagenic primers (available from the corresponding authors upon request). The pBacT7-S1T3SA+ plasmid was used as a template to engineer pBacT7-S1T3SA+ S1 ΔVTSI, pBacT7-S1T3SA+ S1 +QST, pBacT7-S1T3SA+ S1 R161A, pBacT7-S1T3SA+ S1 N182A, and pBacT7-S1T3SA+ S1 R161A,N182A by QuikChange site-directed mutagenesis (Invitrogen) (primers are available from the corresponding authors upon request). Monolayers of approximately 3 × 106 BHK-T7 cells were cotransfected with 1.8 μg each of nine plasmid constructs representing the T1L reovirus genome (pT7-L1T1L, pT7-L2T1L, pT7-L3T1L, pT7-M1T1L, pT7-M2T1L, pT7-M3T1L, pT-S2T1L, pT7-S3T1L, and pT7-S4T1L) in combination with one of the parental or mutant pBacT7-S1 plasmids by using TransIT-LT1 transfection reagent (Mirus Bio LLC). Following at least 5 days of incubation, recombinant viruses were isolated by plaque purification using L cells. Virus stocks were prepared from at least three plaque-purified clones per recombinant virus strain, as described previously (50). Virus particles were purified from infected L cells by Vertrel XF (DuPont) extraction and CsCl gradient centrifugation (10). Virus titers were determined by a plaque assay using L cells (50). The reovirus particle concentration was determined from the equivalence of one unit of the optical density at 260 nm to 2.1 × 1012 particles and adjusted as appropriate based on SDS-PAGE and Coomassie staining. ISVPs were generated by the treatment of virions with chymotrypsin (Sigma) (51).
Monoclonal antibodies specific for T1L (5C6) (52) and T3D (9BG5) (53) σ1 proteins were obtained and purified as described previously (30). JAM-A MAb J10.4 was obtained and purified as described previously (54, 55).
Multistep virus replication.
L cells were seeded into wells of 24-well plates to achieve a density of ∼4 × 105 cells per well and adsorbed with stocks generated from three plaque-purified clones per reovirus strain diluted in phosphate-buffered saline (PBS) at an MOI of 0.001 or 0.01 PFU/cell at room temperature for 1 h. Inocula were removed, and cells were washed once with PBS to remove unbound virus and incubated in fresh medium at 37°C for 0, 8, 12, 16, 24, or 48 h prior to two rounds of freezing and thawing. Virus titers in inocula and cell lysates were determined by a plaque assay using L cells.
Fluorescent-focus assay.
L cells or JAM-A-CHO cells were seeded into wells of 96-well plates to achieve a density of ∼2 × 104 cells per well. L cells were adsorbed with serial virus dilutions at 37°C for 1 h. Inocula were removed, and cells were washed and incubated in fresh medium at 37°C for 16 to 20 h. Cells were fixed with cold methanol, and reovirus proteins were detected by incubation with polyclonal reovirus antiserum at a 1:500 dilution in PBS containing 0.5% Triton X-100 at 37°C, followed by incubation with Alexa Fluor 488-labeled secondary IgG and 4′,6-diamidino-2-phenylindole (DAPI). Images were captured for four fields of view per well by using an ImageXpress Micro XL automated microscope imager (Molecular Devices). Total numbers and percentages of infected cells were quantified by using MetaXpress high-content image acquisition and analysis software (Molecular Devices). The effect of receptor blockade on reovirus infectivity was tested by incubating JAM-A-CHO cells with various concentrations of Arthrobacter ureafaciens NA (MP Biomedicals) or JAM-A MAb J10.4 (55) at 37°C for 1 h prior to adsorption with virions at particle concentrations that yielded approximately 40% infected cells per well, incubation, fixation, staining, and quantification. The effect of the σ1 blockade was tested by incubating virions at particle concentrations that yielded approximately 80% infected cells per well with medium or medium containing various concentrations of MAb 5C6 or 9BG5 at 37°C for 1 h prior to adsorption onto L cells, incubation, fixation, staining, and quantification. The dependence of reovirus infectivity on endosomal acidification was tested by the addition of 25 mM NH4Cl (EMD Millipore) at various times after the adsorption of L cells with virions at particle concentrations that yielded approximately 80% infected cells per well, incubation, fixation, staining, and quantification. Statistical analyses were performed by using GraphPad Prism 7 (GraphPad). Mean relative values from four fields of view for duplicate wells from three independent experiments were compared by using one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test.
Agarose gel separation of intact virions.
Purified reovirus virions (2.5 × 1011) were diluted into 2.5% glycerol and 0.025% bromophenol blue in TAE buffer (40 mM Tris acetate, 1 mM EDTA [pH 8.0]) and resolved by electrophoresis in 1% agarose gels in TAE buffer at room temperature for 18 h (56). Reovirus particles containing between 0 and 12 σ1 trimers were visualized by staining with PageBlue protein-staining solution (Thermo Scientific).
Immunoblot analysis of σ1 content.
Purified reovirus particles (1 × 1010) were boiled in sample buffer and resolved by SDS-PAGE in a 4 to 20% Mini-Protean TGX precast protein gel (Bio-Rad). Proteins were transferred onto nitrocellulose membranes by using a Trans-Blot SD semidry transfer cell (Bio-Rad) at 15 V for 30 min. Membranes were blocked in Odyssey blocking buffer for >1 h at room temperature and then incubated at 4°C overnight in Odyssey blocking buffer containing rabbit polyclonal anti-T3D σ1 head serum (31) diluted 1:1,000 and mouse μ1C-specific MAb 8H6 (52) diluted 1:1,000. Membranes were washed in PBS-T (PBS plus 0.1% Tween 20) and incubated with anti-rabbit IRDye680LT antibodies and anti-mouse IRDye800CW antibodies (Li-Cor) diluted 1:15,000 in PBS-T plus 5% nonfat dry milk at room temperature for >1 h. After washing with PBS-T, membranes were scanned by using an Odyssey infrared imaging system (Li-Cor). The protein band intensity was quantified by using Image Studio version 5.2. The integrated protein band fluorescence for σ1 was divided by that for μ1C to normalize the results.
Hemagglutination assay.
Purified virions or ISVPs were distributed into wells of 96-well U-bottom microtiter plates (Costar) at an initial concentration of 2.5 × 1010 particles/well and serially diluted 1:2 in 50 μl PBS. Type O human erythrocytes were washed twice with PBS, resuspended as a 1% (vol/vol) suspension in PBS, delivered to virus-containing wells at 50 μl/well, and incubated at 4°C for 4 h. One HA unit is the minimum particle number sufficient to produce a partial or complete shield of erythrocytes in the well. The HA titer is defined as 2.5 × 1010 particles divided by the number of particles per HA unit. Statistical analyses were performed by using GraphPad Prism 7. Mean HA titers from two replicates from each of two independent experiments were compared to the mean for rsT3SA+ virions or ISVPs by using one-way ANOVA with Dunnett's multiple-comparison test.
Accession number(s).
Coordinates and structure factors have been deposited in the Protein Data Bank. PDB accession numbers for T1L σ1cc are 6GAJ (iodide) and 6GAK (chloride). PDB accession numbers for T1L σ1cc_body and T3D σ1cc_body are 6GAO and 6GAP, respectively.
ACKNOWLEDGMENTS
We thank Lisa Kraft, Sophie Stotz, and Hubert Kalbacher at the University of Tübingen for mass spectrometry analysis. We thank members of the Dermody and Stehle laboratories, especially Danica M. Sutherland, Sarah P. Katen, Georg Zocher, and Michael B. Braun, for many helpful discussions during the conduct of this work. We are also grateful to the SLS (PSI, Switzerland) for granting us beam time.
This work was supported by U.S. Public Health Service award R01 AI118887 to Terence S. Dermody and Thilo Stehle. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Dermody TS, Parker JS, Sherry B. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Dryden KA, Wang GJ, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol 122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guglielmi KM, Kirchner E, Holm GH, Stehle T, Dermody TS. 2007. Reovirus binding determinants in junctional adhesion molecule-A. J Biol Chem 282:17930–17940. doi: 10.1074/jbc.M702180200. [DOI] [PubMed] [Google Scholar]
- 4.Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. 2012. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog 8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein sigma 1 in complex with sialylated oligosaccharides. PLoS Pathog 7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 7.Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. 2006. Beta 1 integrin mediates internalization of mammalian reovirus. J Virol 80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson EM, Doyle JD, Wetzel JD, McClung RP, Katunuma N, Chappell JD, Washington MK, Dermody TS. 2009. Genetic and pharmacologic alteration of cathepsin expression influences reovirus pathogenesis. J Virol 83:9630–9640. doi: 10.1128/JVI.01095-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mainou BA, Dermody TS. 2012. In search of cathepsins: how reovirus enters host cells. DNA Cell Biol 31:1646–1649. doi: 10.1089/dna.2012.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furlong DB, Nibert ML, Fields BN. 1988. Sigma-1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein mu 1 mediates membrane disruption. J Virol 76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. 2008. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J 27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. 1990. Intraluminal proteolytic activation plays an important role in replication of type-1 reovirus in the intestines of neonatal mice. J Virol 64:1830–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulz WL, Haj AK, Schiff LA. 2012. Reovirus uses multiple endocytic pathways for cell entry. J Virol 86:12665–12675. doi: 10.1128/JVI.01861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. 1979. Two modes of entry of reovirus particles into L cells. J Gen Virol 45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- 16.Nibert ML, Dermody TS, Fields BN. 1990. Structure of the reovirus cell-attachment protein—a model for the domain organization of sigma-1. J Virol 64:2976–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser RDB, Furlong DB, Trus BL, Nibert ML, Fields BN, Steven AC. 1990. Molecular structure of the cell-attachment protein of reovirus: correlation of computer-processed electron micrographs with sequence-based predictions. J Virol 64:2990–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leone G, Mah DCW, Lee PWK. 1991. The incorporation of reovirus cell attachment protein sigma-1 into virions requires the N-terminal hydrophobic tail and the adjacent heptad repeat region. Virology 182:346–350. doi: 10.1016/0042-6822(91)90678-5. [DOI] [PubMed] [Google Scholar]
- 19.Lupas AN, Gruber M. 2005. The structure of alpha-helical coiled coils. Adv Protein Chem 70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 20.Calvo PG, Fox GC, Parrado XLH, Llamas-Saiz AL, Costas C, Martinez-Costas J, Benavente J, van Raaij MJ. 2005. Structure of the carboxy-terminal receptor-binding domain of avian reovirus fibre SigmaC. J Mol Biol 354:137–149. doi: 10.1016/j.jmb.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 21.Merckel MC, Huiskonen JT, Bamford DH, Goldman A, Tuma R. 2005. The structure of the bacteriophage PRD1 spike sheds light on the evolution of viral capsid architecture. Mol Cell 18:161–170. doi: 10.1016/j.molcel.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 22.van Raaij MJ, Mitraki A, Lavigne G, Cusack S. 1999. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401:935–938. doi: 10.1038/44880. [DOI] [PubMed] [Google Scholar]
- 23.Chappell JD, Prota AE, Dermody TS, Stehle T. 2002. Crystal structure of reovirus attachment protein sigma 1 reveals evolutionary relationship to adenovirus fiber. EMBO J 21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stettner E, Dietrich MH, Reiss K, Dermody TS, Stehle T. 2015. Structure of serotype 1 reovirus attachment protein sigma 1 in complex with junctional adhesion molecule A reveals a conserved serotype-independent binding epitope. J Virol 89:6136–6140. doi: 10.1128/JVI.00433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavalli A, Prota AE, Stehle T, Dermody TS, Recanatini M, Folkers G, Scapozza L. 2004. A molecular dynamics study of reovirus attachment protein sigma 1 reveals conformational changes in sigma 1 structure. Biophys J 86:3423–3431. doi: 10.1529/biophysj.103.030825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nibert ML, Chappell JD, Dermody TS. 1995. Infectious subvirion particles of reovirus type-3 Dearing exhibit a loss in infectivity and contain a cleaved sigma-1 protein. J Virol 69:5057–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartmann MD, Ridderbusch O, Zeth K, Albrecht R, Testa O, Woolfson DN, Sauer G, Dunin-Horkawicz S, Lupas AN, Alvarez BH. 2009. A coiled-coil motif that sequesters ions to the hydrophobic core. Proc Natl Acad Sci U S A 106:16950–16955. doi: 10.1073/pnas.0907256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duquerroy S, Vigouroux AN, Rottier PJM, Rey FA, Bosch BJ. 2005. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology 335:276–285. doi: 10.1016/j.virol.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strelkov SV, Burkhard P. 2002. Analysis of alpha-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J Struct Biol 137:54–64. doi: 10.1006/jsbi.2002.4454. [DOI] [PubMed] [Google Scholar]
- 30.Dietrich MH, Ogden KM, Katen SP, Reiss K, Sutherland DM, Carnahan RH, Goff M, Cooper T, Dermody TS, Stehle T. 2017. Structural insights into reovirus sigma1 interactions with two neutralizing antibodies. J Virol 91:e01621-16. doi: 10.1128/JVI.01621-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bokiej M, Ogden KM, Ikizler M, Reiter DM, Stehle T, Dermody TS. 2012. Optimum length and flexibility of reovirus attachment protein sigma1 are required for efficient viral infection. J Virol 86:10270–10280. doi: 10.1128/JVI.01338-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohamed A, Teicher C, Haefliger S, Shmulevitz M. 2015. Reduction of virion-associated sigma1 fibers on oncolytic reovirus variants promotes adaptation toward tumorigenic cells. J Virol 89:4319–4334. doi: 10.1128/JVI.03651-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arslan M, Qin Z, Buehler MJ. 2011. Coiled-coil intermediate filament stutter instability and molecular unfolding. Comput Methods Biomech Biomed Eng 14:483–489. doi: 10.1080/10255842.2011.560147. [DOI] [PubMed] [Google Scholar]
- 34.Brown JH, Cohen C, Parry DA. 1996. Heptad breaks in alpha-helical coiled coils: stutters and stammers. Proteins 26:134–145. doi:. [DOI] [PubMed] [Google Scholar]
- 35.Bassler J, Hernandez Alvarez B, Hartmann MD, Lupas AN. 2015. A domain dictionary of trimeric autotransporter adhesins. Int J Med Microbiol 305:265–275. doi: 10.1016/j.ijmm.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 36.Zhang YM, Bergelson JM. 2005. Adenovirus receptors. J Virol 79:12125–12131. doi: 10.1128/JVI.79.19.12125-12131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guardado-Calvo P, Fox GC, Llamas-Saiz AL, van Raaij MJ. 2009. Crystallographic structure of the alpha-helical triple coiled-coil domain of avian reovirus S1133 fibre. J Gen Virol 90:672–677. doi: 10.1099/vir.0.008276-0. [DOI] [PubMed] [Google Scholar]
- 39.Schibli DJ, Weissenhorn W. 2004. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion (review). Mol Membr Biol 21:361–371. doi: 10.1080/09687860400017784. [DOI] [PubMed] [Google Scholar]
- 40.Nason EL, Wetzel JD, Mukherjee SK, Barton ES, Prasad BV, Dermody TS. 2001. A monoclonal antibody specific for reovirus outer-capsid protein sigma3 inhibits sigma1-mediated hemagglutination by steric hindrance. J Virol 75:6625–6634. doi: 10.1128/JVI.75.14.6625-6634.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dormitzer PR, Nason EB, Prasad BV, Harrison SC. 2004. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 430:1053–1058. doi: 10.1038/nature02836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vonrhein C, Blanc E, Roversi P, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol Biol 364:215–230. [DOI] [PubMed] [Google Scholar]
- 44.Bailey S. 1994. The Ccp4 suite—programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 48.Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. 2012. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berard A, Coombs KM. 2009. Mammalian reoviruses: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit 15C.1. doi: 10.1002/9780471729259.mc15c01s14. [DOI] [PubMed] [Google Scholar]
- 51.Baer GS, Dermody TS. 1997. Mutations in reovirus outer-capsid protein sigma3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol 71:4921–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Virgin HW IV, Mann MA, Fields BN, Tyler KL. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol 65:6772–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burstin SJ, Spriggs DR, Fields BN. 1982. Evidence for functional domains on the reovirus type 3 hemagglutinin. Virology 117:146–155. doi: 10.1016/0042-6822(82)90514-1. [DOI] [PubMed] [Google Scholar]
- 54.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. 2000. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 113(Part 13):2363–2374. [DOI] [PubMed] [Google Scholar]
- 56.Larson SM, Antczak JB, Joklik WK. 1994. Reovirus exists in the form of 13 particle species that differ in their content of protein sigma 1. Virology 201:303–311. doi: 10.1006/viro.1994.1295. [DOI] [PubMed] [Google Scholar]
- 57.Karplus PA, Diederichs K. 2012. Linking crystallographic model and data quality. Science 336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]