

ABSTRACT
Herpes zoster (HZ) (shingles) is the clinical manifestation of varicella-zoster virus (VZV) reactivation. HZ typically develops as people age, due to decreased cell-mediated immunity. However, the importance of antibodies for immunity against HZ prevention remains to be understood. The goal of this study was to examine the breadth and functionality of VZV-specific antibodies after vaccination with a live attenuated HZ vaccine (Zostavax). Direct enumeration of VZV-specific antibody-secreting cells (ASCs) via enzyme-linked immunosorbent spot assay (ELISPOT assay) showed that Zostavax can induce both IgG and IgA ASCs 7 days after vaccination but not IgM ASCs. The VZV-specific ASCs range from 33 to 55% of the total IgG ASCs. Twenty-five human VZV-specific monoclonal antibodies (MAbs) were cloned and characterized from single-cell-sorted ASCs of five subjects (>60 years old) who received Zostavax. These MAbs had an average of ∼20 somatic hypermutations per VH gene, similar to those seen after seasonal influenza vaccination. Fifteen of the 25 MAbs were gE specific, whereas the remaining MAbs were gB, gH, or gI specific. The most potent neutralizing antibodies were gH specific and were also able to inhibit cell-to-cell spread of the virus in vitro. Most gE-specific MAbs were able to neutralize VZV, but they required the presence of complement and were unable to block cell-to-cell spread. These data indicate that Zostavax induces a memory B cell recall response characterized by anti-gE > anti-gI > anti-gB > anti-gH antibodies. While antibodies to gH could be involved in limiting the spread of VZV upon reactivation, the contribution of anti-gE antibodies toward protective immunity after Zostavax needs further evaluation.
IMPORTANCE Varicella-zoster virus (VZV) is the causative agent of chickenpox and shingles. Following infection with VZV, the virus becomes latent and resides in nerve cells. Age-related declines in immunity/immunosuppression can result in reactivation of this latent virus, causing shingles. It has been shown that waning T cell immunity correlates with an increased incidence of VZV reactivation. Interestingly, serum with high levels of VZV-specific antibodies (VariZIG; IV immunoglobulin) has been administered to high-risk populations, e.g., immunocompromised children, newborns, and pregnant women, after exposure to VZV and has shown some protection against chickenpox. However, the relative contribution of antibodies against individual surface glycoproteins toward protection from shingles in elderly/immunocompromised individuals has not been established. Here, we examined the breadth and functionality of VZV-specific antibodies after vaccination with the live attenuated VZV vaccine Zostavax in humans. This study will add to our understanding of the role of antibodies in protection against shingles.
KEYWORDS: antibody, B cell, cloning, herpes zoster, immunology, shingles, VZV, VZV glycoprotein, Zostavax
INTRODUCTION
Chickenpox (varicella) and herpes zoster (shingles) are both caused by varicella-zoster virus (VZV). VZV is a member of the Herpesviridae family of DNA viruses, which cause both lytic and latent infections. After natural infection with VZV, the virus becomes latent in sensory ganglia. Previous work has shown that immunity induced by natural infection can protect against VZV reactivation (1–3). However, as the immune response to VZV wanes, either in the immunocompromised (i.e., after transplantation, chemotherapy, cancer, HIV/AIDS, or stress) or with age, clinically relevant reactivation of VZV may occur. This can result in the development of shingles, which may cause significant morbidity due to pain associated with postherpetic neuralgia (PHN). Zostavax is a live attenuated viral vaccine licensed for people over the age of 50 and has been shown to significantly decrease the incidence of shingles, by 63.9% in 60- to 69-year-olds and by 37.6% in people >70 years of age. Those who did develop shingles after receiving Zostavax saw a significant decrease in the amount of PHN (66.5% overall) (4). Studies of both Zostavax and the now-FDA-approved VZV vaccine Shingrix (gE protein in AS01B) have demonstrated that boosting the immune response to VZV and/or the gE protein of VZV alone can decrease both the incidence and severity of shingles (4–13).
Earlier studies have shown that the incidence of shingles increases with age, primarily due to a decrease in T cell immunity (9, 14, 15). However, the relative importance of humoral immunity in protection against viral reactivation remains unclear. On one hand, VZV-specific antibodies do not decrease with age, suggesting that antibodies alone are not sufficient for protection against VZV reactivation (9). However, no study to date has evaluated whether neutralizing antibodies to VZV decrease with age; the previous studies measured only total VZV- or glycoprotein E-specific serum antibody levels. On the other hand, antibodies have been shown to be a correlate of vaccine-mediated protection in children receiving the live attenuated varicella vaccine Varivax (16). Moreover, there is evidence that sera from individuals with high titers of antibodies against VZV (VariZIG, VZIG, or ZIG) can protect some immunocompromised children and pregnant women from both subclinical and clinical varicella (17–20).
Four major glycoprotein complexes are located on the surface of VZV viral particles (glycoprotein B [gB], gC, gE/gI, and gH/gL (21)). Glycoprotein E is the most predominant and immunogenic glycoprotein on infected cells and virions (21–24). Glycoproteins E and I form heterodimers and are required for virus replication in vivo (25–27). Inhibition of gE/gI heterodimerization results in decreased gE maturation and surface expression, inhibiting gI incorporation into virions and thus blocking infection of skin and T cells in vivo (27). Glycoprotein B and the gH/gL heterodimer are the second and third most abundant and immunodominant VZV glycoproteins (28). Previous work has shown that gB and gH/gL are necessary and minimally sufficient for virion fusion to the cell, and they have been hypothesized to mediate VZV-induced fusion and virion entry into the cell (27, 29–32). A model of VZV fusion suggests that gH/gL can activate gB to trigger its fusogenic function (27).
Many studies have evaluated the antibody response to VZV in humans. During primary natural varicella infection and after primary varicella vaccination, gE and gB antibodies predominate, followed by gH-specific antibody responses (23, 24). Studies utilizing mouse and mouse-human chimeric hybridomas demonstrated that gE monoclonal antibodies (MAbs) neutralize VZV in vitro in a complement-dependent manner (33, 34). VZV gB-specific antibodies have been shown to neutralize the virus independently of complement and with lower 50% inhibitory concentrations (IC50) than for gE antibodies with complement (33). Interestingly, VZV-specific gH (gH/gL) antibodies have been shown by several groups to neutralize with and without complement as well as to inhibit cell-to-cell spread both in vitro and in a humanized SCID xenograft model (28, 35–44). Structural analysis of two VZV gH/gL monoclonal antibodies isolated from VZV-immune donors via phage display or single-cell antibody-secreting cell cloning IgG-94 (43) and IgG-RC (36) showed that both antibodies target the same site, which is composed of residues in both gH and gL. This epitope is in proximity to the site on gH/gL that activates gB, and thus targeting of this site may represent a potential vulnerability for VZV entry (45).
Using an unbiased approach, we examined the breadth and functionality of VZV-specific antibodies after vaccination with Zostavax in humans. We demonstrated that Zostavax induces VZV-specific IgG and IgA antibody-secreting cells (ASCs) but not IgM ASCs. Analysis of monoclonal antibodies cloned and characterized from single-sorted ASCs revealed that over half of the MAbs generated were gE specific, whereas the other MAbs were gI, gB, and gH specific. Functional analysis of these MAbs demonstrated that antibodies specific for gH displayed the most potent complement-independent neutralization and inhibition of cell-to-cell spread. gE-specific MAbs, as well as a one gB-specific MAb, could neutralize VZV only in the presence of complement and did not inhibit cell-to-cell spread. Overall, these results indicate that gH antibodies may be important in vivo for preventing virus spread, thus potentially limiting the tissue damage/postherpetic neuralgia after reactivation.
RESULTS
Zostavax induces IgG and IgA, but not IgM, ASCs 7 days after vaccination.
In order to examine the breadth and functionality of the antibody response to vaccination by using an unbiased approach, we vaccinated seven subjects (60 to 71 years old) with Zostavax. Serial blood draws were obtained on days 0, 7, and 14 postvaccination. VZV-specific antibody-secreting cells (ASCs) peaked at day 7 postvaccination and returned to baseline by day 14 (Fig. 1A and B). This is consistent with what we have published previously for VZV-specific IgG ASCs following vaccination with Zostavax (46). IgG-producing ASCs dominated among VZV-specific ASCs, followed by IgA ASCs in a few subjects, and no VZV-specific IgM ASCs were detected. The lack of antigen-specific IgM ASCs and the appearance of IgG and IgA ASCs at day 7 are indicative of a true memory B cell recall response. To determine the specificities of the antibodies generated after Zostavax vaccination, we performed single-cell reverse transcription-PCR (RT-PCR) of sorted ASCs 7 days after vaccination to generate monoclonal antibodies as described previously (47–49). CD19+ CD27hi CD38hi ASCs were first sorted in bulk 7 days after vaccination (Fig. 2A). Figure 2B shows that approximately 33 to 55% of sorted bulk IgG-producing ASCs were VZV specific via enzyme-linked immunosorbent spot assay (ELISPOT assay). Next, the ASCs were single-cell sorted into 96-well PCR plates and cloned as described previously (47). Paired heavy- and light-chain plasmids were transiently transfected, and supernatant was collected and screened on a VZV-infected cell lysate via enzyme-linked immunosorbent assay (ELISA). Supernatants that scored positive had antibodies purified and the specificity and functionality tested. Overall, we generated 25 VZV-specific monoclonal antibodies from five subjects. The last two subjects' ASCs were not cloned but were saved for future use. Due to a variable percent frequency of VZV-specific plasmablasts as well as potential primer bias, we cloned variable numbers of antibodies from the 5 subjects (RM, n = 5; ZV301, n = 1; ZV302, n = 4; ZV303, n = 14; and ZV304, n = 1).
FIG 1.

Kinetics of VZV-specific antibody-secreting cells (ASCs) after Zostavax vaccination in older adults. (A) Representative VZV-specific (top row) and total (bottom row) IgG (left), IgM (center), and IgA (right) ASCs were measured on days 0, 7, and 14 after Zostavax vaccination via ELISPOT assay. (B) Cumulative results for VZV-specific IgG, IgM, and IgA ASCs after Zostavax vaccination. *, P < 0.05 by the Student t test.
FIG 2.

ASC sorting and frequency of VZV-specific ASCs. Five older adults were vaccinated with Zostavax, and PBMCs were collected on day 7. (A) ASCs were single-cell sorted into PCR plates using a FACSAria II (left and middle panels, presort; right panel, postsort to assess purity). (B) The frequency of VZV-specific ASCs was calculated via IgG ELISPOT assay. Top row, VZV-specific IgG ASCs; bottom row, total IgG ASCs. The percentages of specific ASCs were measured by taking the number of VZV-specific IgG ASCs divided by the total number of IgG ASCs detected. The number of ASCs plated per well is listed.
Zostavax induces primarily a memory B cell recall response.
Sequence analysis of VZV-specific monoclonal antibodies (Table 1) showed that the median level of somatic hypermutations (SHM) per VH gene was 20, with a range of 7 to 34 (Fig. 3). In contrast, antibody sequences derived from naive B cells averaged approximately zero mutations per VH gene. For reference, in Fig. 3 we also show previously reported data on the number of somatic mutations per VH gene in ASCs generated after seasonal influenza vaccination (50). Levels of SHM in ASC clones responding to Zostavax vaccination are similar to what is seen after influenza vaccination and indicate that Zostavax ASCs are derived from memory B cell precursors. We were able to identify two separate clones (same VDJ junction) with more than one member (different mutations) each for donor 303. The first clone includes antibodies 303-1A8 and 303-1D7 (Table 1). Both antibodies utilized the same variable-region genes (IGHV3-30-3*01 and IGKV1-6*02), had the same CDR3 sequences for both heavy and light chains, had the same number of total mutations in VH and VK, and had the same number of amino acid replacement mutations in VK. The number of replacement mutations in VH differed, as 303-1A8 had 16, compared with 13 for 303-1D7. These data indicate that 303-1A8 and 303-1D7 shared the same parental clone and that 303-1A8 had undergone further affinity maturation. The second clonal family consists of antibodies 303-1A12 and 303-1C6 (Table 1). Both antibodies utilized the same heavy- and light-chain variable region genes (IGHV3-23*04 and IGLV1-44*01) and had identical CDR3 sequences. These antibodies diverged slightly in that 303-1C6 had a higher total number of SHM than 303-1A12 (silent plus replacement mutations). If we look at the number of replacement mutations resulting in an amino acid change, both antibodies had the same number of SHM in IgH (n = 18), but 303-1C6 had more IgL SHM than 303-1A12. These data indicate that 303-1C6 had undergone further affinity maturation.
TABLE 1.
Sequence analysis of VZV-specific monoclonal antibodies
| Antibody | Specificity | Isotype | V region |
J gene | D gene | CDR3 region |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V gene | V % identity | No. of mutations | No. of silent mutations | No. of nonsilent mutations | CDR lengthsa | Amino acid junction | |||||
| RM-1A2 | gE | IgG2 | IGHV1-2*02 | 90 | 30 | 9 | 21 | IGHJ4*02 | IGHD3-9*01 | 8.8.14 | CARIMYFEYDSWSDYW |
| Kappa | IGKV1-39*01 | 96 | 12 | 5 | 7 | IGKJ1*01 | 6.3.9 | CQQSYSLPWTF | |||
| RM-1D1 | gI | IgG1 | IGHV3-11*01 | 97 | 8 | 2 | 6 | IGHJ4*02 | IGHD2-2*02 | 8.8.12 | CAREPPYTSSIDYW |
| Kappa | IGKV1-39*01 | 98 | 7 | 2 | 5 | IGKJ1*01 | 6.3.9 | CQQSYSTPWTF | |||
| RM-2D6 | gH | IgG1 | IGHV3-30*03 | 95 | 16 | 4 | 12 | IGHJ4*02 | IGHD3-3*01 | 8.8.15 | CAKTSWARFLEWTFDYW |
| Kappa | IGKV1-39*01 | 96 | 11 | 6 | 5 | IGKJ5*01 | 6.3.9 | CQQSHSTPTF | |||
| RM-5A2 | gE | IgG1 | IGHV4-59*01 | 94 | 17 | 5 | 12 | IGHJ4*02 | IGHD3-10*02 | 8.7.15 | CARIGGVSFGERPIDYW |
| Kappa | IGKV1-39*01 | 96 | 13 | 5 | 8 | IGKJ4*01 | 6.3.9 | CQQSYSIPLTF | |||
| RM-5B3 | gE | ? | IGHV5-51*01 | 89 | 34 | 13 | 21 | IGHJ4*02 | IGHD6-19*01 | 8.8.12 | CARQRYSSGSFGYW |
| Kappa | IGKV4-1*01 | 94 | 18 | 8 | 10 | IGKJ4*01 | 12.3.9 | CQQYETYPFTF | |||
| 301-1G5 | gE | IgG1 | IGHV1-2*02 | 88 | 25 | 8 | 17 | IGHJ4*02 | IGHD7-27*01 | 8.8.15 | CATLTRETGAPSTFDYW |
| Kappa | IGKV4-1*01 | 91 | 16 | 5 | 11 | IGHJ4*02 | 12.3.9 | CQHYYNTPLTF | |||
| 302-1B12 | gI | IgG1 | IGHV1-69*04 | 83 | 29 | 7 | 22 | IGHJ6*02 | IGHD1-26*01 | 8.8.15 | CARGGSFSLGANGLDVW |
| Kappa | IGKV3-20*01 | 92 | 6 | 1 | 5 | IGKJ1*01 | 7.3.9 | CQQHGSLPWTF | |||
| 302-1C12 | gI | IgA2 | IGHV1-69*04 | 88 | 17 | 7 | 10 | IGHJ3*01 | IGHD4-11*01 | 8.8.16 | CAIRGGMTTLTPEDLDVW |
| Kappa | IGKV2D-29*01 | 93 | 16 | 7 | 9 | IGKJ4*01 | 11.3.9 | CMQSIHLPLTF | |||
| 302-1D11 | gE | IgA1 | IGHV3-9*01 | 89 | 16 | 5 | 11 | IGHJ6*02 | IGHD2-21*02 | 8.8.22 | CAKDQYCGGDCHSKSYYYYGMDVW |
| Lambda | IGLV6-57*02 | 94 | 7 | 6 | 1 | IGLJ2/3*01 | 8.3.10 | CQSYDSQTAVIF | |||
| 302-1G9 | gH | IgA1 | IGHV1-18*01 | 85 | 23 | 5 | 18 | IGHJ4*02 | IGHD4-23*01 | 8.8.16 | CARDRGGSLAMVVGLDHW |
| Kappa | IGKV1(D)-39*01 | 91 | 18 | 8 | 10 | IGKJ4*01 | 6.3.9 | CQQCYSAELTF | |||
| 303-1A8 | gE | IgG1 | IGHV3-30-3*01 | 86 | 23 | 7 | 16 | IGHJ4*02 | IGHD4-23*01 | 8.8.10 | CARDGSFGLDYW |
| Kappa | IGKV1-6*02 | 94 | 8 | 2 | 6 | IGKJ2*01 | 6.3.9 | CLQDYNDPYTF | |||
| 303-1A12 | gB | IgA1 | IGHV3-23*04 | 85 | 21 | 3 | 18 | IGHJ4*02 | IGHD2-21*02 | 8.8.11 | CARDRDWNYFDKW |
| Lambda | IGLV1-44*01 | 83 | 14 | 5 | 9 | IGLJ1*01 | 8.3.11 | CAAWDDRLNGYVF | |||
| 303-1B2 | gI | IgG1 | IGHV4-30-4*01 | 91 | 10 | 2 | 8 | IGHJ5*02 | IGHD3-3*01 | 10.7.15 | CARESIGDDFWSGLGPW |
| Kappa | IGKV2-28*01 | 98 | 2 | 0 | 2 | IGKJ2*01 | 11.3.9 | CMQALQTPYTF | |||
| 303-1C1 | gI | IgG1 | IGHV3-53*01 | 91 | 12 | 2 | 10 | IGHJ4*02 | IGHD6-25*01 | 8.7.14 | CAREAYNSGTYYFDYW |
| Kappa | IGKV1-39*01 | 90 | 12 | 3 | 9 | IGKJ2*01 | 6.3.9 | CQQSHTIPYTF | |||
| 303-1C2 | gB | IgA1 | IGHV3-30*03 | 92 | 9 | 3 | 6 | IGHJ4*02 | IGHD2-21*02 | 8.8.14 | CARDFSGVTTISLDSW |
| Lambda | IGLV1-36*01 | 93 | 5 | 0 | 5 | IGLJ2*01 | 8.3.11 | CAAWDDSLNRGVF | |||
| 303-1C6 | gB | IgA1 | IGHV3-23*01 | 85 | 26 | 8 | 18 | IGHJ4*02 | IGHD2-21*02 | 8.8.11 | CARDRDWNYFDNW |
| Lambda | IGLV1-44*01 | 89 | 16 | 5 | 11 | IGLJ1*01 | 8.3.11 | CAAWDDRLNGYVF | |||
| 303-1C9 | gE | IgG1 | IGHV3-23*01 | 88 | 27 | 9 | 18 | IGHJ5*02 | IGHD5-24*01 | 8.8.15 | CANCPGDSDNCYWFDPW |
| Kappa | IGKV1-12*01 | 88 | 11 | 4 | 7 | IGKJ4*01 | 6.3.9 | CQQAKNFPLTF | |||
| 303-1D7 | gE | IgG1 | IGHV3-30-3*01 | 87 | 23 | 10 | 13 | IGHJ4*02 | IGHD4-23*01 | 8.8.10 | CARDGSFGLDYW |
| Kappa | IGKV1-6*02 | 91 | 8 | 2 | 6 | IGKJ2*01 | 6.3.9 | CLQDYNDPYTF | |||
| 303-1E3 | gE | IgG1 | IGHV4-30-4*01 | 89 | 20 | 7 | 13 | IGHJ4*02 | IGHD2-2*01 | 10.7.15 | CARVRTSSTTSYYFDYW |
| Kappa | IGKV1-39*01 | 93 | 13 | 5 | 8 | IGKJ3*01 | 6.3.8 | CQQSYRTLTF | |||
| 303-1E8 | gE | IgA2 | IGHV4-61*02 | 89 | 18 | 4 | 14 | IGHJ4*02 | IGHD3-22*01 | 10.7.18 | CARAPFYNDFSGYSYYFDYW |
| Lambda | IGLV1-44*01 | 88 | 19 | 7 | 12 | IGLJ1*01 | 8.3.12 | CAAWDDSLNPLYVF | |||
| 303-1E12 | gE | IgG1 | IGHV1-2*02 | 92 | 15 | 3 | 12 | IGHJ4*02 | IGHD2-21*02 | 8.8.13 | CARWEGGDWFAFDFW |
| Lambda | IGLV3-1*01 | 89 | 10 | 3 | 7 | IGLJ1*01 | 6.3.11 | CQAWDSSTASFVF | |||
| 303-1F5 | gE | IgA1 | IGHV3-30*03 | 77 | 32 | 6 | 26 | IGHJ4*02 | IGHD3-9*01 | 8.8.15 | CAKDILTGYYKGNFDYW |
| Kappa | IGKV3-11*01 | 84 | 31 | 11 | 20 | IGKJ4*01 | 6.3.9 | CQLRNGWLFTF | |||
| 303-1F7 | gE | IgG1 | IGHV4-39*07 | 88 | 23 | 8 | 15 | IGHJ4*02 | IGHD3-10*01 | 10.7.11 | CATGALRRPFDSW |
| Kappa | IGKV1-27*01 | 94 | 10 | 4 | 6 | IGKJ1*01 | 6.3.9 | CLKCNTAPWTF | |||
| 303-1G1 | gE | IgA1 | IGHV4-31*03 | 94 | 10 | 3 | 7 | IGHJ5*02 | IGHD2-2*01 | 10.7.19 | CARDSGYCSSTNCPQNWFDPW |
| Kappa | IGKV1-39*01 | 80 | 8 | 0 | 8 | IGKJ1*01 | 6.3.9 | CQQSYTTPQTF | |||
| 304-1A12 | gE | IgA2 | IGHV3-7*01 | 92 | 7 | 1 | 6 | IGHJ6*02 | IGHD3-3*01 | 8.8.18 | CARSPRFLSQDYYYYVMDVW |
| Kappa | IGKV1(D)-39*01 | 91 | 9 | 1 | 8 | IGKJ1*01 | 6.3.10 | CQQSFITRTWTF | |||
Amino acid lengths for CDR1:CDR2:CDR3.
FIG 3.
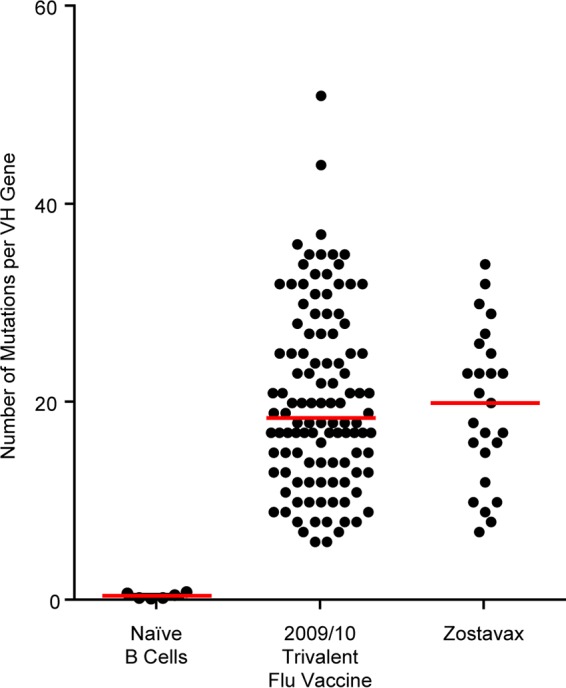
Comparison of the numbers of VH somatic hypermutations in naive B cells, ASCs sorted after vaccination with the 2009/10 influenza trivalent vaccine, and ASCs after Zostavax vaccination. Data from the 2009/10 influenza trivalent vaccine were published by Li et al. (50). IgBLAST was used to determine the number of somatic hypermutations (Fr1-CDR3 heavy chain) as described previously (49).
VZV gE-specific antibodies are predominant in the recall response to Zostavax.
Specificities of the 25 VZV-specific monoclonal antibodies were established as per the scheme depicted in Fig. 4. Briefly, 153 paired heavy- and light-chain genes were transiently transfected for antibody expression. Of the 153 supernatants tested on VZV-infected cell lysates, 54 were shown to be VZV-infected cell lysate positive via ELISA. This indicates that approximately 35% of all antibodies generated were VZV positive, which is similar to the percentage of VZV-specific IgG ASCs shown in Fig. 2B. Next, the supernatants were tested to see whether they bound to a lectin-purified VZV glycoprotein via ELISA. Twenty-five of 54 monoclonal antibodies bound to VZV glycoprotein (46%). Although it was not formally demonstrated, we believe that the remaining antibodies are specific to the other vaccine components, such as the lysate (MRC-5) in which VZV Oka is grown in to make Zostavax. We focused on surface glycoprotein-specific antibodies, as these are most likely to play a relevant role in vivo. Out of the 25 VZV glycoprotein-positive monoclonal antibodies, 15 were determined to bind VZV glycoprotein E via ELISA (60%). The remaining 40% of monoclonal antibodies that were glycoprotein E negative were tested via immunoprecipitation and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to determine their specificity. Lectin-purified VZV glycoproteins from lysate or VZV-infected cell lysate (MAb 302-1G9) were used for immunoprecipitation, followed by protein identification by shotgun proteomics (Fig. 5). We identified 3 VZV-specific gB monoclonal antibodies (303-1A12, 303-1C2, and 303-1C6) (Fig. 5A). As discussed above, 303-1A12 and 303-1C6 are clonally related, and 303-1C6 has additional amino acid changes compared with 303-1A12 (Table 1). We also identified two VZV-specific gH-specific monoclonal antibodies (Fig. 5B). Based on the LC-MS/MS results, we concluded that RM-2D6 precipitates gH by itself, whereas 302-1G9 coprecipitates the gH/gL complex. This could be a result of the fact that an antibody to gH will pull down gL as gH/gL form a complex. Alternatively, this antibody could have an epitope between gH and gL. Epitope mapping was beyond the scope of this study and will be addressed in future studies. Finally, we identified 5 gI-specific monoclonal antibodies (Fig. 5C) (based on predicted molecular weight). Four of these gI-specific monoclonal antibodies, namely, RM-1D1, 302-1C12, 302-1B12, and 303-1C1, coprecipitate the gE/gI complex, as we can detect both VZV-specific glycoprotein E and glycoprotein I using LC-MS/MS. Similar to the rationale for gH and gL, gI-specific antibodies could pull down gE due to the gE/gI complex. Figure 5D demonstrates via Western blotting that these antibodies recognize epitopes on gI, as a glycoprotein-only lysate (including all possible glycoproteins) was run and shows only one band at the predicted gI molecular weight. Additionally, one gI-specific monoclonal antibody, 302-1B2, appears to predominantly precipitate only gI, as the MS signal intensity for gE is 2.5% (data not shown). In total, we were able to identify 15 gE-, 5 gI-, 3 gB-, and 2 gH-specific monoclonal antibodies (Fig. 5E). These results indicate gE is the predominant humoral target upon Zostavax vaccination. This is not surprising, as glycoprotein E is the most abundant glycoprotein present on the surfaces of VZV virions and infected cells (21).
FIG 4.
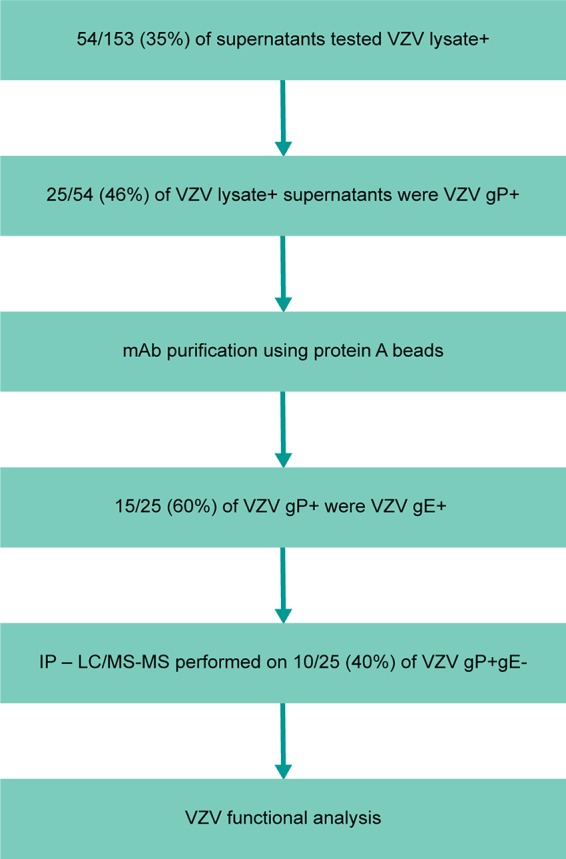
Work flow to identify glycoprotein-specific VZV monoclonal antibodies. Supernatants generated using ASCs single cell sorted, cloned, and expressed in Expi293 cells were tested for VZV lysate binding via ELISA. VZV glycoprotein-positive but non-gE antibodies (10/25) were tested via immunoprecipitation followed by LC/MS to determine the specificity. All glycoprotein-positive antibodies were then tested for neutralization and cell-to-cell spread inhibition in vitro.
FIG 5.

Identification of non-gE-specific monoclonal antibodies. Immunoprecipitation followed by nano-LC-MS/MS was done using the gP-specific monoclonal antibodies and the top three MS hits listed. (A) VZV gB-specific monoclonal antibodies. (B) VZV gH-specific monoclonal antibodies. (C) VZV gI-specific monoclonal antibodies. (D) Western blotting with RM-1D1, 302-1B12, 302-1C12, 303-1C1, and 303-1B2 antibodies was performed using a glycoprotein lysate (including all VZV glycoproteins). The table shows the predicted molecular weights and pIs. (E) Of the 25 VZV glycoprotein-positive antibodies, 15 were gE specific, 5 were gI specific, 3 were gB specific, and 2 were gH specific.
VZV-specific antibodies can neutralize virus in vitro.
After identifying the specificities of all 25 VZV-specific monoclonal antibodies, we tested the ability of these antibodies to neutralize VZV in vitro (Table 2 and Fig. 6). Green fluorescent protein (GFP)-tagged VZV strain Oka was grown in MRC-5 cells in order to make cell-free virus stock. As VZV is highly cell associated in vitro (51), it was critical to make a cell-free virus stock in order to have a reproducible number of input virions in the assay (52). VZV was preincubated with each monoclonal antibody or an influenza virus hemagglutinin head-specific antibody as a control. After 1 h, 10 U/ml of guinea pig complement was added to half of the virus-plus-antibody wells and left for an additional 30 min. ARPE-19 cells were used for VZV infection, as previous groups have shown that they can produce high titers of VZV and show cytopathic effects (53), and the shape of these cells is more suitable for Acumen Cellista imaging. APRE-19 cells were infected for 5 to 7 days at 35°C, and a GFP readout was conducted to quantify infection. Overall, we identified 15 monoclonal antibodies that could neutralize VZV in vitro (Fig. 6A). Twelve of the 15 VZV gE-specific monoclonal antibodies could neutralize VZV only in the presence of complement, with an IC50 range of 0.08 to 0.87 μg/ml (Table 2 and Fig. 6B). Previous work from other groups has shown that gE-specific antibodies require complement to mediate neutralization in vitro (34, 54). None of the gI-specific MAbs could neutralize with or without complement (Fig. 6A). Both gH-specific monoclonal antibodies, RM-2D6 and 302-1G9, had the highest potencies for neutralizing VZV (range, 0.03 to 0.23 μg/ml with or without complement) and did not require the presence of complement. These data are in agreement with previous reports for gH antibodies (28, 35–44). These results confirm previous findings that antibodies directed against gH were the most potent neutralizers in vitro. One out of the three gB-specific monoclonal antibodies neutralized VZV weakly. 303-1C2 neutralized with (3.04 μg/ml) or without (5.27 μg/ml) complement with an IC50 ∼100 times lower than that of the gH antibodies. Interestingly, all of the gE-specific antibodies required complement for in vitro neutralization but were less potent than the gH-specific antibodies (∼3 times less). Representative neutralization curves for VZV preincubated with gH, gE, or gI antibodies are shown in Fig. 6C.
TABLE 2.
In vitro IC50 values
| Antibody | Specificity | IC50 (μg/ml) |
|
|---|---|---|---|
| With complement | Without complement | ||
| RM-1A2 | gE | 0.14 | >10 |
| RM-5A2 | gE | 0.87 | >10 |
| RM-5B3 | gE | 0.52 | >10 |
| 301-1G5 | gE | 0.08 | >10 |
| 302-1D11 | gE | 0.10 | >10 |
| 303-1A8 | gE | 0.11 | >10 |
| 303-1C9 | gE | >10 | >10 |
| 303-1D7 | gE | 0.11 | >10 |
| 303-1E3 | gE | 0.14 | >10 |
| 303-1E8 | gE | 0.10 | >10 |
| 303-1E12 | gE | 0.14 | >10 |
| 303-1F5 | gE | >10 | >10 |
| 303-1F7 | gE | >10 | >10 |
| 303-1G1 | gE | 0.15 | >10 |
| 304-1A12 | gE | 0.17 | >10 |
| RM-1D1 | gI | >10 | >10 |
| 302-1B12 | gI | >10 | >10 |
| 302-1C12 | gI | >10 | >10 |
| 303-1B2 | gI | >10 | >10 |
| 303-1C1 | gI | >10 | >10 |
| RM-2D6 | gH | 0.03 | 0.23 |
| 302-1G9 | gH | 0.06 | 0.09 |
| 303-1A12 | gB | >10 | >10 |
| 303-1C2 | gB | 3.04 | 5.27 |
| 303-1C6 | gB | >10 | >10 |
| Irrelevant | Influenza virus hemagglutinin head | >10 | >10 |
FIG 6.

In vitro VZV neutralization. Purified VZV-specific monoclonal antibodies were tested for in vitro neutralization as described in Materials and Methods. Using an Acumen Cellista, the total number of cells expressing GFP per well was quantified. (A) Cumulative results with and without complement. (B) Cumulative IC50 results (in μg/ml) for all antibodies that showed neutralization with or without the addition of guinea pig complement. (C) Representative neutralization plots with gH-, gE-, and gI-specific monoclonal antibodies added. Red, guinea pig complement added; blue, without guinea pig complement.
VZV anti-gH antibodies inhibit cell-to-cell spread in vitro.
VZV has been shown to be highly cell associated, can induce cell fusion (i.e., formation of syncytia), and transmits through cell-to-cell spread without producing extracellular virions in vitro (55). In order to determine the effects of VZV monoclonal antibodies on cell-to-cell transmission, we performed an in vitro cell spread inhibition assay. Previous reports have shown that gH monoclonal antibodies can inhibit cell-to-cell spread in vitro (28, 35–43) and in a humanized SCID skin xenograft model (44). We tested the ability of all of our monoclonal antibodies to inhibit the cell-to-cell spread of VZV in vitro. Figure 7A shows representative wells with VZV alone or with an irrelevant MAb (against influenza virus hemagglutinin head), 303-1E8 (VZV gE specific), RM-2D6 (VZV gH specific), or 302-1G9 (VZV gH specific). There was significant inhibition of cell spread in a dose-dependent manner using the RM-2D6 (gH) and 302-1G9 (gH) monoclonal antibodies. The cumulative results are shown in Fig. 7B, with all of the gE antibodies shown in the top panel and the non-gE antibodies shown in the bottom panel. Only the gH-specific antibodies could inhibit cell-to-cell spread. Next, we tested whether complement was required for cell spread inhibition. Threefold dilutions of the gH and irrelevant control monoclonal antibodies were performed, starting at 30 μg/ml, with (Fig. 7C, left) or without (Fig. 7C, right) complement. Both 302-1G9 and RM-2D6 could inhibit cell-to-cell spread in vitro independent of complement and with similar IC50 results for each antibody (∼1.4 to 6.2 μg/ml). Overall, our data are consistent with previously published results; only VZV gH-specific monoclonal antibodies have the ability to inhibit cell spread in vitro.
FIG 7.

In vitro cell-to-cell spread inhibition. Cell-to-cell spread inhibition was performed as described in Materials and Methods. (A) Representative well images for samples treated with complement. PGS buffer was used as a negative control. (B) Cumulative data for anti-gE antibodies (top) and non-gE antibodies (bottom). Black bars represent antibodies added at 10 μg/ml, and green bars represent the same antibodies added at 1 μg/ml. (C) Cell spread inhibition curves with IC50 values (μg/ml) with anti-gH or irrelevant MAbs, with (left) or without (right) complement added.
DISCUSSION
In this study, we used molecular techniques to examine the breadth and functionality of the VZV-specific response after Zostavax vaccination in humans. Although decreases in cell-mediated immunity have been shown to result in herpes zoster, we evaluated whether VZV-specific antibodies play a role in in vitro protection. The humoral response induced by Zostavax is predominately a memory B cell recall response: it is composed of IgG and IgA ASCs, with no detectible IgM ASCs, and shows levels of somatic hypermutation similar to what is seen after seasonal influenza vaccination. The majority of antibodies were specific to VZV gE (15/25 = 60%), indicating that gE is the predominant antibody target after vaccination. We could detect 5 gI-specific monoclonal antibodies (5/25 = 20%). These MAbs recognized gI, as determined by SDS-PAGE and Western blotting, but some appeared to coprecipitate gE. Finally, we could detect three gB-specific antibodies (3/25 = 12%) and two gH antibodies (2/25 = 8%) (1 from RM and 1 from ZV303). Functional analyses of these antibodies demonstrated that the gH-specific MAbs were the most potent neutralizers (with or without complement) and could also inhibit cell-to-cell spread of VZV in vitro. Neutralization IC50 for the gH antibodies generated in this study were 0.03 to 0.23 μg/ml (0.2 to 1.5 nM). These potencies are equal to if not better than those previously reported in the literature (range, 0.12 to 8,000 nM [35, 43, 56, 57]). One gB antibody (303-1C2) could also neutralize VZV in vitro with or without complement. However, 303-1C2 was much less potent than the gH MAbs (without complement, 16.6 times less potent; with complement, 73 times less potent). VZV gE-specific MAbs could neutralize in vitro only in the presence of complement but could not inhibit cell spread.
The data in this article are consistent with previous reports in the literature demonstrating that gH MAbs are the most potent neutralizers and can inhibit cell-to-cell spread in vitro (28, 35–44). VZV gB-specific antibodies have also been shown to neutralize in vitro (58). Previously described VZV-specific monoclonal antibodies were generated by (i) hybridoma generation after immunization of mice with VZV (34), (ii) fusion of Ig V genes from mouse hybridomas to human Ig constant regions (59), (iii) in vitro immunization of human lymphocytes with VZV antigens, followed by fusion of the lymphocytes to myeloma cells (42), or (iv) phage display panning of a library of human Ig genes derived from human splenocytes obtained from subjects with idiotypic thrombocytopenic purpura (33). Our approach to isolate ASCs producing anti-VZV antibodies is similar to that used in a previously published study (36). However, we were able to isolate our anti-VZV glycoprotein-specific monoclonal antibodies in an antigen-agnostic manner (from 5 separate subjects), giving more insight into the breadth of glycoprotein-specific ASCs during the response to Zostavax.
Antibodies can act through a variety of mechanisms to prevent virus entry, including blocking receptor engagement, preventing postbinding/prefusion events at the cell surface or inside endosomes, inhibiting release of progeny virus, opsonization, antibody-dependent cell-mediated cytotoxicity (ADCC), and activation of the complement cascade (60). As gH/gL and gB are important for virion fusion (27, 29–32), antibodies directed against these glycoproteins are likely to prevent viral attachment and thus interfere with cell-to-cell spread. VZV-specific gE antibodies have been shown to neutralize in a complement-dependent manner but do not inhibit cell-to-cell spread (34, 54). One possible mechanism for this is that binding of gE-specific antibodies to VZV particles leads to the activation of the complement cascade and inactivation of the virus, while infected cells are protected from complement-mediated lysis due to the presence of complement regulatory proteins on the cell surface and/or low cell surface expression of gE.
As the antibodies that were detected after Zostavax vaccination had a high degree of somatic hypermutations (median, 20; range, 7 to 34), it is unclear whether these mutations were a result of primary infection, exogenous exposure to varicella, or subclinical reactivation over time. There have been some reports that VZV DNA can be detected in the blood of some healthy individuals (61). These data suggest that subclinical reactivation and subsequent boosting of the immune response may occur as people age. However, the importance of endogenous versus exogenous boosting has yet to be fully determined. Since there are similar degrees of somatic hypermutation observed after vaccination with Zostavax and with seasonal influenza vaccine, endogenous/exogenous boosting of the VZV-specific memory B cell compartment could result in increased affinity maturation over time.
The biological importance of VZV-specific antibodies in vivo is unclear. Although patients with agammaglobulinemia are not more susceptible to severe varicella infection (62), treatment of some immunocompromised children and pregnant women with VariZIG (high-titer VZV polyclonal sera) has been shown to prevent severe varicella (17–20). Additionally, VZV-specific total antibody titers do not decrease with age (9). Of note, there are limited data evaluating whether VZV-specific neutralizing antibody titers or gH-specific antibody titers decrease with age. If the neutralizing antibody titers do not decrease with age, this would be further evidence that antibodies probably do not play a protective role against the development of shingles. It could be envisaged that antibodies may play a protective role in preventing disseminated VZV during primary infection when the virus titer in the blood is high. However, the role of antibodies in preventing the development of shingles is probably limited due to the biological transport of the virus from the dorsal root ganglion to the skin through nerves and not through the blood. Evidence in this report demonstrates that gE antibodies can neutralize in the presence of complement in vitro but cannot inhibit cell-to-cell spread. The FDA-approved VZV vaccine Shingrix is a subunit vaccine containing VZV gE adjuvanted with AS01B (7, 8). This vaccine is >90% efficacious, but the correlative mechanism of protection is currently unknown (8). Our data suggest that gE-specific antibodies are not the most functionally potent VZV-specific antibodies (compared to gH-specific antibodies) in terms of neutralization and cell-to-cell spread. Thus, unless the AS01B adjuvant induces a gE antibody qualitatively different from what natural infection/Zostavax vaccination induces (i.e., can inhibit cell-to-cell spread) or acts differently in vivo where there is complement present, the most likely mechanism of protection for Shingrix could be attributed to T cells.
Overall, we identified 25 human VZV-specific antibodies via an unbiased approach from 5 separate subjects 7 days after receiving Zostavax. This is the first study that examined the breadth of the VZV-specific antibody response using molecular biology techniques. All 5 donors tested had gE-specific MAbs, and they were either the only antibody isolated (301 and 304) or the predominant antibody (RM, 302, and 303). Though we see skewing of the results to donor 303, we feel that this distribution as well as the previous literature shows that gE is the predominant antibody target.
Although the majority of antibodies were VZV gE specific, the most potent antibodies in vitro were directed against gH. If antibodies do play a protective role in vivo during chickenpox or shingles, then inclusion of gH (gH/gL) protein would be beneficial in designing a next-generation VZV vaccine. Additionally, gH-specific monoclonal antibody therapy could also be considered an alternative to VariZIG in VZV-negative immunocompromised patients or pregnant women.
MATERIALS AND METHODS
Study subjects.
All studies were approved by the Emory University Institutional Review Board (IRB no. 00045821 [healthy blood subject protocol] and 00050285 [Zostavax-vaccinated subject protocol]). Study subjects provided written informed consent prior to participation. Seven subjects with prior natural immunity to VZV were recruited for collection of peripheral blood mononuclear cells (PBMC) and serum at days 0, 7, 14, and 28. Study demographics are listed in Table 1. Five subjects (RM and ZV301-304) were then used on day 7 for single-cell ASC sorting. The remaining two subjects' ASCs were sorted but not cloned due to limited resources.
PBMC isolation.
PBMCs were isolated using BD Vacutainer CPT tubes, washed, and resuspended in complete FBS medium (RPMI 1640 plus 2% fetal bovine serum [FBS], 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM l-glutamine, and 50 μM 2-mercaptoethanol) for immediate use.
B cell ELISPOT assay.
Millipore Multiscreen-HA 96-well plates (Millipore MAHA N4510, nonsterile) were coated with either 10 μg/ml of goat anti-human IgA+IgG+IgM(H+L) (Jackson, 109-055-064) to measure total IgA or IgM antibody-secreting cells (ASCs), 10 μg/ml of donkey anti-human IgG(H+L) (Jackson, 709-005-149) to measure total IgG ASCs, or 20 to 40 μg/ml of VZV-infected cell lysate (detergent free; Meridian Life Sciences, 7740) to measure VZV-specific ASCs. All were diluted in sterile phosphate-buffered saline (PBS), and 100 μl/well was added and incubated overnight at 4°C. Plates were washed with PBS plus 0.05% Tween 20 and with PBS and blocked with complete medium (RPMI 1640, 5% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM l-glutamine, and 50 μM 2-mercaptoethanol) for >2 h at 37°C. Blocking medium was discarded, and 100 μl/well of complete medium was added followed, by 100 μl/well of 1 × 107 total PBMCs. Twofold serial dilutions were carried out. In some instances, CD19+ CD20lo CD38+ CD27+ cells were bulk sorted and added directly to the blocked ELISPOT assay plate. Figure 2 shows how many ASCs were added per well after sorting. Cells were then incubated for ∼16 h at 37°C with 5% CO2. Plates were washed with PBS followed by PBS plus 0.05% Tween 20, and 100 μl/well of donkey anti-human IgG biotin (Jackson, 709-066-098), goat anti-human IgA biotin (Jackson, 109-065-011), or goat anti-human IgM biotin (Caltag, H15015) diluted 1/1,000 in PBS–0.05% Tween 20–1% FBS was added and left for 2 h at room temperature. Plates were washed with PBS plus 0.05% Tween 20, and 100 μl/well of HRP-avidin D (Vector Laboratories, A-2004) diluted 1/1,000 in PBS–0.05% Tween 20–1% FBS was added and left for 1 h at room temperature. Plates were washed with PBS plus 0.05% Tween 20 followed by PBS, and 100 μl/well of 3-amino-9-ethylcarbazole (AEC) (Sigma, A-5754) in 0.1 M Na-acetate buffer at pH 5.0 (sodium acetate trihydrate [C2H3O2Na · 3H2O] FW 136.1; Sigma, S-8625) was added, left for 5 to 15 min, and then washed thoroughly with distilled water. Plates were scanned on a CTL (Shaker Heights, OH) ImmunoSpot analyzer. Data are representative of the number of antibody-secreting cells (ASCs) per million PBMC.
ASC sorting.
Freshly isolated PBMC from ∼40 ml of blood were stained with anti-human CD19–fluorescein isothiocyanate (FITC) (BD 555412), anti-human CD38–phycoerythrin (PE) (BD 555460), anti-human CD3–PE-Cy7 (BD 557851), anti-human CD20–PE-Cy7 (BD 335793), and anti-human CD27–allophycocyanin (APC) (Ebio, 17-0279) for 30 min on ice. Cells were washed and resuspended in PBS plus 2% FBS. Using a BD FACSAria II, antibody-secreting cells (ASCs) (CD19+ CD38+ CD27+) were either single-cell sorted into a 96-well PCR plate containing 10 mM Tris-HCl with 40 U/μl of RNase inhibitor (Promega) and frozen at −80°C or bulk sorted into complete medium.
Generation of MAbs.
The generation of monoclonal antibodies (MAbs) from single-cell-sorted ASCs was performed as described previously (47, 49, 63). Briefly, IgG and IgA heavy, kappa, and lambda variable regions were amplified by reverse transcription and PCR. cDNA was synthesized from random hexamers and used for a first-round IgGH, IgAH/IgMH, Igκ, and Igλ PCR. The first-round PCR cocktail of primers covered all families of variable (V) and joining (J) genes; this PCR was followed by a nested PCR to identify the sequences of the V and J genes of the IgGH, IgAH/IgMH, Igκ, and Igλ regions. Once the IgGH, IgAH/IgMH, Igκ, and Igλ gene families were known, highly specific primers were used to amplify these regions and put restriction enzyme sites for cloning into IgG1H, Igκ, and Igλ backbones for antibody expression. These antibodies were expressed in HEK293 (donor RM) or Expi293 (Invitrogen) (donors 301, 302, 303, and 304) cells. The antibodies were affinity purified using protein A-agarose beads (GenScript).
V family breadth and somatic hypermutation analysis.
The heavy-chain variable (V) region sequences were analyzed to determine repertoire breadth as described previously (63). Briefly, the International ImmunoGeneTics (IMGT) database information system (www.imgt.org) was used to identify the V and J gene families. To analyze the number of somatic hypermutations in the VH gene, IgBLAST was used to assess the frequency of VH mutations as a combination of both silent and replacement (resulting in a change at the amino acid level) mutations from the framework region 1 to the complementarity-determining region 3 (CDR3). The frequencies of VH mutations in naive B cells and after seasonal trivalent influenza vaccination have been published previously (49, 50).
VZV lysate and gE-specific ELISAs.
The binding specificity of isolated monoclonal antibodies was determined by ELISA. An initial VZV-infected cell lysate ELISA was performed. If the VZV lysate was positive, a VZV total glycoprotein ELISA was used to identify VZV gP-specific MAbs. A follow-up VZV gE ELISA was utilized for those MAbs showing specificity for VZV glycoproteins.
For the VZV-infected cell lysate and gP ELISA, Nunc C96 MaxiSorp Immunoplates (Thermo Scientific) were coated overnight at 4°C with either VZV-infected cell lysate (Meridian Life Sciences, 7740) or lectin-purified VZV glycoproteins (Virusys or in-house reagent) diluted in PBS at a concentration of 1 μg/ml. For the VZV gE ELISA, Pierce nickel-coated plates (Thermo Scientific) were coated overnight at 4°C with His-tagged recombinant VZV gE protein (Oka strain, in-house reagent) diluted in PBS at a concentration of 1 μg/ml. The remainder of the protocol was identical for the VZV lysate, VZV total glycoprotein, and VZV gE ELISAs. Protein-coated plates were washed six times with PBS–0.05% Tween 20 and blocked with blocking buffer (PBS–0.05% Tween 20 with 3% nonfat dry milk) for 1 h at room temperature on a plate rocker. Either supernatant from transiently transfected Expi293 cells diluted 1:2 in blocking buffer (VZV lysate ELISA) or VZV-specific monoclonal antibodies were 5-fold serially diluted in blocking buffer, starting at a concentration of 3 μg/ml, for a total of 4 to 6 dilutions. Blocking buffer was removed from the coated plates, and diluted antibodies were bound to the plate for 1.5 h at room temperature on a plate rocker. Antibody-bound plates were washed six times with PBS–0.05% Tween 20. Goat anti-human IgG–HRP (Southern Biotech) was diluted 1:2,000 in blocking buffer, added to the washed plates, and left for 1 h at room temperature on a plate rocker. Plates were washed six times with PBS–0.05% Tween 20 and developed for 5 min with SuperBlu-Turbo tetramethylbenzidine (TMB) solution (Virolabs) followed by ELISA stop solution for TMB (Virolabs). Absorbance was read at 450 nm on a Victor multilabel counter (Wallac/PerkinElmer), and antibody binding curves were visualized using nonlinear-fit four-parameter variable slope analysis in GraphPad Prism 7 software.
Immunocapture.
Immunocapture experiments were performed with magnetic M-270 Epoxy Dynabeads (Thermo Fisher, 14302D) following the manufacturer's instructions. In brief, 30 μg of MAb was covalently coupled to 1 mg of magnetic beads. The reaction was performed overnight in an incubator shaker at 37°C. Unbound MAb was removed by washing first with PBS and then with PBS with 0.02% bovine serum albumin (BSA) (Pierce, 23209). Twenty-five micrograms of VZV glycoprotein lysate (Virusys) was added to the magnetic beads. Binding of the antigen was allowed by end-over-end rotation of the bead-antigen mixture for 14 h at 4°C. Unbound VZV glycoprotein was removed by three washes with 500 μl PBS. Antigen was released with 25 μl of 8 M urea (ultrapure; Riedel de Haen, 15604) in 100 mM ammonium bicarbonate (Thermo Fisher, BP2413). Control experiments were performed by coupling an irrelevant monoclonal IgG.
MS analysis.
Half the volume (12.5 μl) of the released antigen solution was used for antigen identification. Reduction was performed in an incubator shaker for 20 min at 60°C with 1 μl of 500 mM Tris(2-carboxyethyl)phosphine (TCEP). The sample was then alkylated for 30 min at room temperature in the dark by adding a 1.5-fold molar excess of iodoacetamide (Pierce, 90034). Protein was then digested in an incubator shaker for 4 h at 37°C with 0.1 μg LysC (Roche Diagnostics, 11047825001). A second digest was performed overnight at 37°C with 0.1 μg trypsin (Promega, V5111) after the sample was diluted to 1.5 M urea using 100 mM ammonium bicarbonate. The digestion was stopped by adding 2 μl of formic acid. Samples were analyzed by nano-LC-MS/MS immediately after the digestion or stored at −80°C. Nano-LC-MS/MS analysis was performed on a splitless Ultimate 3000 ultraperformance liquid chromatography (UPLC) system (Dionex) that was directly coupled to an Orbitrap XL mass spectrometer (Thermo Fisher). For the analysis, 50 μl of the protein digest was loaded from the autosampler onto a C18 trapping column (300-μm inner diameter by 5 mm, C18 PepMap100, 5-μm particle size, 100-Å pore size; Thermo Scientific, catalog no. 160454) and then back eluted onto a 75-μm-inner-diameter analytical column (15-cm Acclaim PepMap RSLC; particle size, 2 μm; Thermo Scientific, catalog no. 164534) by gradient elution, increasing the acetonitrile percentage within 50 min from 2% to 35% in 65 min. Peptides were directly eluted into the Orbitrap XL mass spectrometer, and high-mass-accuracy MS spectra were acquired in the Orbitrap mass analyzer at a resolution of 100,000. The top 5 MS/MS spectra were acquired in the ion trap mass analyzer with automated gain control and dynamic exclusion enabled. Sequence identification and label-free quantitation was performed using the software package Protein Discoverer 1.4 (Thermo Scientific), allowing only peptides with less than a 1% false-discovery rate as calculated by the percolator node as positive identifications. Protein sequences of the human as well as the VZV proteome were obtained from the UniProt server (www.uniprot.org).
Western blotting.
VZV glycoprotein preparation (including all glycoproteins, at 2.5 μg per lane) were separated by Bis-Tris 4 to 12% SDS-PAGE (NuPAGE NP0336; Invitrogen) using MES (morpholineethanesulfonic acid) running buffer. Proteins were transferred onto a nitrocellulose membrane following the manufacturer's instructions using the iBlot system from Thermo Fisher (iB301002). Primary anti-VZV antibody and secondary detection antibody were applied using the Snap ID system from Millipore. In brief, the primary antibody was diluted 1:1,000. PBS with 0.1% Tween 20 and 0.5% BSA was used as a blocking buffer. HRP-labeled goat anti-human Fc antibodies (PerkinElmer, NEF802) were used as the detection antibody. The Western blot was developed using chloronaphthol-diaminobenzidine (CN/DAB) substrate (Thermo Scientific, 34000).
Virus.
GFP-VZV (pOKA ORF11-GFP) was kindly provided by Marvin Sommer and Ann Arvin, Stanford University. GFP-tagged VZV Oka strain stocks were grown in MRC-5 cells. MRC-5 cells were cultured 35°C with 5% CO2 in 2% complete EMEM (Eagle minimum essential medium [EMEM] [Corning] supplemented with 2% heat-inactivated fetal bovine serum [HyClone], 1% l-glutamine [Gibco], and 0.5% neomycin [Sigma]). Cell-associated virus was harvested into PGS buffer (in-house buffer) at 40 h postinfection, sonicated to disassociate virus from cells, and centrifuged at 1,500 rpm for 10 min to remove cell debris. Cell-free virus was flash frozen in liquid nitrogen and stored at −70°C.
Neutralization assay.
Purified VZV-specific monoclonal antibodies were 3-fold serially diluted, starting at a concentration of 25 μg/ml, for a total of 10 dilutions. Antibodies were diluted into 2% complete DMEM/F-12 (Dulbecco's modification of Eagle's medium/Ham's F-12 50/50 mix [DMEM/F-12] [Corning] supplemented with 2% heat-inactivated fetal bovine serum [HyClone] and 1% penicillin-streptomycin [Corning]). GFP-tagged VZV Oka strain stocks were quick-thawed in a water bath, diluted to a concentration of 300 PFU/25 μl in PGS buffer, and kept on wet ice. Twenty-five microliters of diluted antibody was thoroughly mixed with 25 μl of diluted VZV-GFP (giving a final starting antibody concentration of 12.5 μg/ml) in a flat-bottom, clear-walled 96-well plate (Costar, 3598) and incubated at 35°C with 5% CO2 for 1 h. Controls included VZV-GFP alone (no antibody) and blank wells (no antibody and no VZV-GFP). All conditions were performed in replicates of 8. After 1 h of incubation, 10 units/well of reconstituted guinea pig complement (Sigma) was added to half of the virus-antibody wells (totaling 4 replicates per condition) and incubated at 35°C with 5% CO2 for an additional 30 min. ARPE-19 target cells were diluted in 2% complete DMEM/F-12, and 20,000 cells, in 50 μl, were added to each well, mixed thoroughly, and incubated at 35°C with 5% CO2 for 1 h with shaking every 15 min. One hundred microliters of 2% complete DMEM/F-12 was then added to each well, and the plate was centrifuged at 1,200 rpm for 10 min to encourage virus-cell interaction and cell settling. The plate was incubated at 35°C with 5% CO2 for 5 to 7 days, and the total number of cells expressing GFP per well was quantified using an Acumen Cellista (TTP LabTech). The percentage of viral neutralization for each well was defined as [(average of 4 replicates of VZV-GFP-only control − condition of interest)/average of 4 replicates of VZV-GFP-only control] × 100. IC50 values were calculated from dilution curves using GraphPad Prism 7 software.
In vitro cell-to-cell spread inhibition assay.
One hundred thousand MRC-5 cells (ATCC) per well were plated in a 12-well plates (Corning 3513) in 2 ml of complete Eagle's minimal essential medium (EMEM) (Sigma) with 10% fetal bovine serum, 1% of l-glutamine, and 0.5% neomycin for 2 to 3 days at 37°C with 5% CO2 until >90% confluence was reached. GFP-labeled cell-free VZV was generated as discussed above (at 7.03 × 104 PFU/ml). Confluent MRC-5 wells were washed once with warm complete EMEM plus 10% FBS, and 100 μl of 1:125-diluted VZV in PGS buffer (Merck in-house) was added per well (∼55 PFU/well). PGS buffer alone was added as a mock control. Plates were rocked back and forth 8 times and incubated for 15 min at 35°C with 5% CO2. This was repeated 3 times for a total infection time of 1 h. After 1 h, 2 ml of EMEM plus 2% FBS plus 1% l-glutamine plus 0.5% neomycin was added and plates placed back at 35°C with 5% CO2 for 16 to 18 h. The next day, wells were washed once with 2 ml of warm EMEM plus 2% FBS plus 1% l-glutamine plus 0.5% neomycin, and antibodies were diluted in EMEM plus 2% FBS plus 1% l-glutamine plus 0.5% neomycin starting at 30 μg/ml. Diluted antibodies (0.5 ml per well) were added and left for 60 min at 35°C with 5% CO2. For the complement-treated wells, 65 μl of guinea pig complement (Sigma, S1639) was added and incubated for 30 min at 35°C with 5% CO2. A 1.5-ml portion of EMEM plus 2% FBS plus 1% l-glutamine plus 0.5% neomycin was added, and plates were placed back at 35°C with 5% CO2 for 4 more days.
To fix and immunostain the cells, wells were washed with PBS, and 2 ml of 90% acetone (Sigma, 534064) diluted in distilled water was added and left for 10 min at room temperature. Wells were washed once with PBS, and 0.5 ml of 1:2,000-diluted VZV glycoprotein E antibody (Abcam, Ab52549) in PBS plus 0.05% Tween 20 added and left for 30 min at 37°C. Wells were gently washed 2 times with PBS, and rabbit anti-mouse IgG peroxidase secondary antibody (Sigma, A9044) diluted 1:1,000 in PBS plus 0.05% Tween 20 was added at 0.5 ml per well and left for 30 min at 37°C. Wells were washed 2 times with PBS, and 150 μl of 1× DAB/Metal in peroxide buffer (Thermo Scientific, 1856090) was added, rocked back and forth 8 times, and incubated at room temperature for 15 min. Plates were then flipped over onto paper towels and dried overnight at room temperature. The plates were scanned on a CTL Immunospot S6 reader (Shaker Heights, OH). The percentage of cell-to-cell spread inhibition for each condition was defined as [(average of 3 replicates of VZV-GFP-only control − condition of interest)/average of 3 replicates of VZV-GFP-only control] × 100. IC50 values were calculated from dilution curves using GraphPad Prism 7 software.
Accession number(s).
Sequences were deposited in GenBank under the following accession numbers (heavy/light chains): 301-1G5, MH259708/MH259733; 302-1B12, MH259709/MH259734; 302-1C12, MH259710/MH259735; 302-1D11, MH259711/MH259736; 302-1G9, MH259712/MH259737; 303-1A8, MH259713/MH259738; 303-1A12, MH259714/MH259739; 303-1B2, MH259715/MH259740; 303-1C1, MH259716/MH259741; 303-1C2, MH259717/MH259742; 303-1C6, MH259718/MH259743; 303-1C9, MH259719/MH259744; 303-1D7, MH259720/MH259745; 303-1E3, MH259721/MH259746; 303-1E8, MH259722/MH259747; 303-1E12, MH259723/MH259748; 303-1F5, MH259724/MH259749; 303-1F7, MH259725/MH259750; 303-1G1, MH259726/MH259751; 304-1A12, MH259727/MH259752; RM-1A2, MH259728/MH259753; RM-1D1, MH259729/MH259754; RM-2D6, MH259730/MH259755; RM-5A2, MH259731/MH259756; and RM-5B3, MH259732/MH259757.
ACKNOWLEDGMENTS
We thank Robert Karaffa and Sommer Durham for sorting antibody-secreting cells and Mary Bower, Wendy Webb Nesheim, and all the staff and subjects at the Hope Clinic of the Emory Vaccine Center.
This study was supported in part by the Emory Flow Cytometry Core (EFCC), one of the Emory Integrated Core Facilities (EICF), and is subsidized by the Emory University School of Medicine. Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000454. GFP-VZV (pOKA ORF11-GFP) was kindly provided by Marvin Sommer and Ann Arvin, Stanford University, with work funded by NIH grant AI20459. This work was funded in part by NIH grant 5U19AI090023, awarded to Bali Pulendran and Rafi Ahmed at Emory University.
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
N.L.S., M.A.R.-M., J.S., E.D., D.K., and K.A.V. are employees of Merck and Co., Inc., Kenilworth, NJ, and receive company stock as a part of their compensation.
REFERENCES
- 1.Arvin AM, Koropchak CM, Williams BR, Grumet FC, Foung SK. 1986. Early immune response in healthy and immunocompromised subjects with primary varicella-zoster virus infection. J Infect Dis 154:422–429. doi: 10.1093/infdis/154.3.422. [DOI] [PubMed] [Google Scholar]
- 2.Kumagai T, Chiba Y, Wataya Y, Hanazono H, Chiba S, Nakao T. 1980. Development and characteristics of the cellular immune response to infection with varicella-zoster virus. J Infect Dis 141:7–13. doi: 10.1093/infdis/141.1.7. [DOI] [PubMed] [Google Scholar]
- 3.Thomas SL, Wheeler JG, Hall AJ. 2002. Contacts with varicella or with children and protection against herpes zoster in adults: a case-control study. Lancet 360:678–682. doi: 10.1016/S0140-6736(02)09837-9. [DOI] [PubMed] [Google Scholar]
- 4.Oxman MN, Levin MJ, Johnson GR, Schmader KE, Straus SE, Gelb LD, Arbeit RD, Simberkoff MS, Gershon AA, Davis LE, Weinberg A, Boardman KD, Williams HM, Zhang JH, Peduzzi PN, Beisel CE, Morrison VA, Guatelli JC, Brooks PA, Kauffman CA, Pachucki CT, Neuzil KM, Betts RF, Wright PF, Griffin MR, Brunell P, Soto NE, Marques AR, Keay SK, Goodman RP, Cotton DJ, Gnann JW Jr, Loutit J, Holodniy M, Keitel WA, Crawford GE, Yeh SS, Lobo Z, Toney JF, Greenberg RN, Keller PM, Harbecke R, Hayward AR, Irwin MR, Kyriakides TC, Chan CY, Chan IS, Wang WW, Annunziato PW, Silber JL, et al. 2005. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 352:2271–2284. doi: 10.1056/NEJMoa051016. [DOI] [PubMed] [Google Scholar]
- 5.Chlibek R, Bayas JM, Collins H, de la Pinta ML, Ledent E, Mols JF, Heineman TC. 2013. Safety and immunogenicity of an AS01-adjuvanted varicella-zoster virus subunit candidate vaccine against herpes zoster in adults ≥50 years of age. J Infect Dis 208:1953–1961. doi: 10.1093/infdis/jit365. [DOI] [PubMed] [Google Scholar]
- 6.Chlibek R, Pauksens K, Rombo L, van Rijckevorsel G, Richardus JH, Plassmann G, Schwarz TF, Catteau G, Lal H, Heineman TC. 2016. Long-term immunogenicity and safety of an investigational herpes zoster subunit vaccine in older adults. Vaccine 34:863–868. doi: 10.1016/j.vaccine.2015.09.073. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham AL, Lal H, Kovac M, Chlibek R, Hwang SJ, Diez-Domingo J, Godeaux O, Levin MJ, McElhaney JE, Puig-Barbera J, Vanden Abeele C, Vesikari T, Watanabe D, Zahaf T, Ahonen A, Athan E, Barba-Gomez JF, Campora L, de Looze F, Downey HJ, Ghesquiere W, Gorfinkel I, Korhonen T, Leung E, McNeil SA, Oostvogels L, Rombo L, Smetana J, Weckx L, Yeo W, Heineman TC, ZOE-70 Study Group. 2016. Efficacy of the herpes zoster subunit vaccine in adults 70 years of age or older. N Engl J Med 375:1019–1032. doi: 10.1056/NEJMoa1603800. [DOI] [PubMed] [Google Scholar]
- 8.Lal H, Cunningham AL, Godeaux O, Chlibek R, Diez-Domingo J, Hwang SJ, Levin MJ, McElhaney JE, Poder A, Puig-Barbera J, Vesikari T, Watanabe D, Weckx L, Zahaf T, Heineman TC, ZOE-50 Study Group. 2015. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 372:2087–2096. doi: 10.1056/NEJMoa1501184. [DOI] [PubMed] [Google Scholar]
- 9.Levin MJ, Oxman MN, Zhang JH, Johnson GR, Stanley H, Hayward AR, Caulfield MJ, Irwin MR, Smith JG, Clair J, Chan IS, Williams H, Harbecke R, Marchese R, Straus SE, Gershon A, Weinberg A, Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. 2008. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis 197:825–835. doi: 10.1086/528696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levin MJ, Schmader KE, Gnann JW, McNeil SA, Vesikari T, Betts RF, Keay S, Stek JE, Bundick ND, Su SC, Zhao Y, Li X, Chan IS, Annunziato PW, Parrino J. 2013. Varicella-zoster virus-specific antibody responses in 50-59-year-old recipients of zoster vaccine. J Infect Dis 208:1386–1390. doi: 10.1093/infdis/jit342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levin MJ, Schmader KE, Pang L, Williams-Diaz A, Zerbe G, Canniff J, Johnson MJ, Caldas Y, Cho A, Lang N, Su SC, Parrino J, Popmihajlov Z, Weinberg A. 2016. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 213:14–22. doi: 10.1093/infdis/jiv480. [DOI] [PubMed] [Google Scholar]
- 12.Morrison VA, Johnson GR, Schmader KE, Levin MJ, Zhang JH, Looney DJ, Betts R, Gelb L, Guatelli JC, Harbecke R, Pachucki C, Keay S, Menzies B, Griffin MR, Kauffman CA, Marques A, Toney J, Boardman K, Su SC, Li X, Chan IS, Parrino J, Annunziato P, Oxman MN, Shingles Prevention Study Group. 2015. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 60:900–909. doi: 10.1093/cid/ciu918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg A, Zhang JH, Oxman MN, Johnson GR, Hayward AR, Caulfield MJ, Irwin MR, Clair J, Smith JG, Stanley H, Marchese RD, Harbecke R, Williams HM, Chan IS, Arbeit RD, Gershon AA, Schodel F, Morrison VA, Kauffman CA, Straus SE, Schmader KE, Davis LE, Levin MJ, US Department of Veterans Affairs (VA) Cooperative Studies Program Shingles Prevention Study Investigators. 2009. Varicella-zoster virus-specific immune responses to herpes zoster in elderly participants in a trial of a clinically effective zoster vaccine. J Infect Dis 200:1068–1077. doi: 10.1086/605611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hope-Simpson RE. 1965. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med 58:9–20. [PMC free article] [PubMed] [Google Scholar]
- 15.Levin MJ, Smith JG, Kaufhold RM, Barber D, Hayward AR, Chan CY, Chan IS, Li DJ, Wang W, Keller PM, Shaw A, Silber JL, Schlienger K, Chalikonda I, Vessey SJ, Caulfield MJ. 2003. Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-dose VZV vaccine. J Infect Dis 188:1336–1344. doi: 10.1086/379048. [DOI] [PubMed] [Google Scholar]
- 16.American Academy of Pediatrics Committee on Infectious Diseases. 2007. Prevention of varicella: recommendations for use of varicella vaccines in children, including a recommendation for a routine 2-dose varicella immunization schedule. Pediatrics 120:221–231. doi: 10.1542/peds.2007-1089. [DOI] [PubMed] [Google Scholar]
- 17.Bapat P, Koren G. 2013. The role of VariZIG in pregnancy. Expert Rev Vaccines 12:1243–1248. doi: 10.1586/14760584.2013.844651. [DOI] [PubMed] [Google Scholar]
- 18.Fisher JP, Bate J, Hambleton S. 2011. Preventing varicella in children with malignancies: what is the evidence? Curr Opin Infect Dis 24:203–211. doi: 10.1097/QCO.0b013e328345d666. [DOI] [PubMed] [Google Scholar]
- 19.Orenstein WA, Heymann DL, Ellis RJ, Rosenberg RL, Nakano J, Halsey NA, Overturf GD, Hayden GF, Witte JJ. 1981. Prophylaxis of varicella in high-risk children: dose-response effect of zoster immune globulin. J Pediatr 98:368–373. doi: 10.1016/S0022-3476(81)80697-X. [DOI] [PubMed] [Google Scholar]
- 20.Zaia JA, Levin MJ, Preblud SR, Leszczynski J, Wright GG, Ellis RJ, Curtis AC, Valerio MA, LeGore J. 1983. Evaluation of varicella-zoster immune globulin: protection of immunosuppressed children after household exposure to varicella. J Infect Dis 147:737–743. doi: 10.1093/infdis/147.4.737. [DOI] [PubMed] [Google Scholar]
- 21.Grose C. 1990. Glycoproteins encoded by varicella-zoster virus: biosynthesis, phosphorylation, and intracellular trafficking. Annu Rev Microbiol 44:59–80. doi: 10.1146/annurev.mi.44.100190.000423. [DOI] [PubMed] [Google Scholar]
- 22.Brunell PA, Novelli VM, Keller PM, Ellis RW. 1987. Antibodies to the three major glycoproteins of varicella-zoster virus: search for the relevant host immune response. J Infect Dis 156:430–435. doi: 10.1093/infdis/156.3.430. [DOI] [PubMed] [Google Scholar]
- 23.Haumont M, Jurdan M, Kangro H, Jacquet A, Massaer M, Deleersnyder V, Garcia L, Bosseloir A, Bruck C, Bollen A, Jacobs P. 1997. Neutralizing antibody responses induced by varicella-zoster virus gE and gB glycoproteins following infection, reactivation or immunization. J Med Virol 53:63–68. doi:. [DOI] [PubMed] [Google Scholar]
- 24.LaRussa PS, Gershon AA, Steinberg SP, Chartrand SA. 1990. Antibodies to varicella-zoster virus glycoproteins I, II, and III in leukemic and healthy children. J Infect Dis 162:627–633. doi: 10.1093/infdis/162.1.627. [DOI] [PubMed] [Google Scholar]
- 25.Arvin AM, Oliver S, Reichelt M, Moffat JF, Sommer M, Zerboni L, Berarducci B. 2010. Analysis of the functions of glycoproteins E and I and their promoters during VZV replication in vitro and in skin and T-cell xenografts in the SCID mouse model of VZV pathogenesis. Curr Top Microbiol Immunol 342:129–146. doi: 10.1007/82_2009_1. [DOI] [PubMed] [Google Scholar]
- 26.Berarducci B, Rajamani J, Zerboni L, Che X, Sommer M, Arvin AM. 2010. Functions of the unique N-terminal region of glycoprotein E in the pathogenesis of varicella-zoster virus infection. Proc Natl Acad Sci U S A 107:282–287. doi: 10.1073/pnas.0912373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerboni L, Sen N, Oliver SL, Arvin AM. 2014. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol 12:197–210. doi: 10.1038/nrmicro3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller PM, Neff BJ, Ellis RW. 1984. Three major glycoprotein genes of varicella-zoster virus whose products have neutralization epitopes. J Virol 52:293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grose C, Carpenter JE, Jackson W, Duus KM. 2010. Overview of varicella-zoster virus glycoproteins gC, gH and gL. Curr Top Microbiol Immunol 342:113–128. doi: 10.1007/82_2009_4. [DOI] [PubMed] [Google Scholar]
- 30.Oliver SL, Sommer M, Zerboni L, Rajamani J, Grose C, Arvin AM. 2009. Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol 83:7495–7506. doi: 10.1128/JVI.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A 108:18412–18417. doi: 10.1073/pnas.1111333108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugano T, Matsumoto Y, Miyamoto C, Masuho Y. 1987. Hybridomas producing human monoclonal antibodies against varicella-zoster virus. Eur J Immunol 17:359–364. doi: 10.1002/eji.1830170309. [DOI] [PubMed] [Google Scholar]
- 34.Wu L, Forghani B. 1997. Characterization of neutralizing domains on varicella-zoster virus glycoprotein E defined by monoclonal antibodies. Arch Virol 142:349–362. doi: 10.1007/s007050050081. [DOI] [PubMed] [Google Scholar]
- 35.Akahori Y, Suzuki K, Daikoku T, Iwai M, Yoshida Y, Asano Y, Kurosawa Y, Shiraki K. 2009. Characterization of neutralizing epitopes of varicella-zoster virus glycoprotein H. J Virol 83:2020–2024. doi: 10.1128/JVI.02097-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birlea M, Owens GP, Eshleman EM, Ritchie A, Traktinskiy I, Bos N, Seitz S, Azarkh Y, Mahalingam R, Gilden D, Cohrs RJ. 2013. Human anti-varicella-zoster virus (VZV) recombinant monoclonal antibody produced after Zostavax immunization recognizes the gH/gL complex and neutralizes VZV infection. J Virol 87:415–421. doi: 10.1128/JVI.02561-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forghani B, Dupuis KW, Schmidt NJ. 1984. Varicella-zoster viral glycoproteins analyzed with monoclonal antibodies. J Virol 52:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grose C, Edwards DP, Friedrichs WE, Weigle KA, McGuire WL. 1983. Monoclonal antibodies against three major glycoproteins of varicella-zoster virus. Infect Immun 40:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montalvo EA, Grose C. 1986. Neutralization epitope of varicella zoster virus on native viral glycoprotein gp118 (VZV glycoprotein gpIII). Virology 149:230–241. doi: 10.1016/0042-6822(86)90124-8. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez JE, Moninger T, Grose C. 1993. Entry and egress of varicella virus blocked by same anti-gH monoclonal antibody. Virology 196:840–844. doi: 10.1006/viro.1993.1543. [DOI] [PubMed] [Google Scholar]
- 41.Shiraki K, Daikoku T, Takemoto M, Yoshida Y, Suzuki K, Akahori Y, Okuno T, Kurosawa Y, Asano Y. 2011. Neutralizing anti-gH antibody of varicella-zoster virus modulates distribution of gH and induces gene regulation, mimicking latency. J Virol 85:8172–8180. doi: 10.1128/JVI.00435-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugano T, Tomiyama T, Matsumoto Y, Sasaki S, Kimura T, Forghani B, Masuho Y. 1991. A human monoclonal antibody against varicella-zoster virus glycoprotein III. J Gen Virol 72:2065–2073. doi: 10.1099/0022-1317-72-9-2065. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki K, Akahori Y, Asano Y, Kurosawa Y, Shiraki K. 2007. Isolation of therapeutic human monoclonal antibodies for varicella-zoster virus and the effect of light chains on the neutralizing activity. J Med Virol 79:852–862. doi: 10.1002/jmv.20838. [DOI] [PubMed] [Google Scholar]
- 44.Vleck SE, Oliver SL, Reichelt M, Rajamani J, Zerboni L, Jones C, Zehnder J, Grose C, Arvin AM. 2010. Anti-glycoprotein H antibody impairs the pathogenicity of varicella-zoster virus in skin xenografts in the SCID mouse model. J Virol 84:141–152. doi: 10.1128/JVI.01338-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, Sullivan NL, Rouphael N, Yu T, Banton S, Maddur MS, McCausland M, Chiu C, Canniff J, Dubey S, Liu K, Tran V, Hagan T, Duraisingham S, Wieland A, Mehta AK, Whitaker JA, Subramaniam S, Jones DP, Sette A, Vora K, Weinberg A, Mulligan MJ, Nakaya HI, Levin M, Ahmed R, Pulendran B. 2017. Metabolic phenotypes of response to vaccination in humans. Cell 169:862–877 e817. doi: 10.1016/j.cell.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. 2009. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc 4:372–384. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, Mehta A, Razavi B, Del Rio C, Zheng NY, Lee JH, Huang M, Ali Z, Kaur K, Andrews S, Amara RR, Wang Y, Das SR, O'Donnell CD, Yewdell JW, Subbarao K, Marasco WA, Mulligan MJ, Compans R, Ahmed R, Wilson PC. 2011. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med 208:181–193. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC. 2008. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li GM, Chiu C, Wrammert J, McCausland M, Andrews SF, Zheng NY, Lee JH, Huang M, Qu X, Edupuganti S, Mulligan M, Das SR, Yewdell JW, Mehta AK, Wilson PC, Ahmed R. 2012. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc Natl Acad Sci U S A 109:9047–9052. doi: 10.1073/pnas.1118979109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harper DR, Mathieu N, Mullarkey J. 1998. High-titre, cryostable cell-free varicella zoster virus. Arch Virol 143:1163–1170. doi: 10.1007/s007050050364. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt-Chanasit J, Bleymehl K, Rabenau HF, Ulrich RG, Cinatl J Jr, Doerr HW. 2008. In vitro replication of varicella-zoster virus in human retinal pigment epithelial cells. J Clin Microbiol 46:2122–2124. doi: 10.1128/JCM.00122-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sloutskin A, Kinchington PR, Goldstein RS. 2013. Productive vs non-productive infection by cell-free varicella zoster virus of human neurons derived from embryonic stem cells is dependent upon infectious viral dose. Virology 443:285–293. doi: 10.1016/j.virol.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kausmally L, Waalen K, Lobersli I, Hvattum E, Berntsen G, Michaelsen TE, Brekke OH. 2004. Neutralizing human antibodies to varicella-zoster virus (VZV) derived from a VZV patient recombinant antibody library. J Gen Virol 85:3493–3500. doi: 10.1099/vir.0.80406-0. [DOI] [PubMed] [Google Scholar]
- 55.Cole NL, Grose C. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev Med Virol 13:207–222. doi: 10.1002/rmv.377. [DOI] [PubMed] [Google Scholar]
- 56.Drew PD, Moss MT, Pasieka TJ, Grose C, Harris WJ, Porter AJ. 2001. Multimeric humanized varicella-zoster virus antibody fragments to gH neutralize virus while monomeric fragments do not. J Gen Virol 82:1959–1963. doi: 10.1099/0022-1317-82-8-1959. [DOI] [PubMed] [Google Scholar]
- 57.Yokoyama T, Ayabe S, Miyagi H, Sugano T, Otsu A, Sato H, Kageyama S, Fujii T, Shiraki K. 2001. Varicella-zoster virus gH:gL contains a structure reactive with the anti-human gamma chain of IgG near the glycosylation site. J Gen Virol 82:331–334. doi: 10.1099/0022-1317-82-2-331. [DOI] [PubMed] [Google Scholar]
- 58.Massaer M, Haumont M, Place M, Bollen A, Jacobs P. 1993. Induction of neutralizing antibodies by varicella-zoster virus gpII glycoprotein expressed from recombinant vaccinia virus. J Gen Virol 74:491–494. doi: 10.1099/0022-1317-74-3-491. [DOI] [PubMed] [Google Scholar]
- 59.Shankar V, Kools JJ, Armour KL, Clark MR. 2005. A chimeric antibody to varicella-zoster virus glycoprotein e. Hybridoma (Larchmt) 24:50–54. doi: 10.1089/hyb.2005.24.50. [DOI] [PubMed] [Google Scholar]
- 60.Marasco WA, Sui J. 2007. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol 25:1421–1434. doi: 10.1038/nbt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toi CS, Lay ML, Lucas R, Chew CB, Taylor J, Ponsonby AL, Dwyer DE, Ausimmune Investigator Group (AIG). 2013. Varicella zoster virus quantitation in blood from symptomatic and asymptomatic individuals. J Med Virol 85:1491–1497. doi: 10.1002/jmv.23605. [DOI] [PubMed] [Google Scholar]
- 62.Nobre FA, Gonzalez IG, de Moraes-Pinto MI, Costa-Carvalho BT. 2015. Protective levels of varicella-zoster antibody did not effectively prevent chickenpox in an X-linked agammaglobulinemia patient. Rev Inst Med Trop Sao Paulo 57:455–457. doi: 10.1590/S0036-46652015000500017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Priyamvada L, Cho A, Onlamoon N, Zheng NY, Huang M, Kovalenkov Y, Chokephaibulkit K, Angkasekwinai N, Pattanapanyasat K, Ahmed R, Wilson PC, Wrammert J. 2016. B cell responses during secondary dengue virus infection are dominated by highly cross-reactive, memory-derived plasmablasts. J Virol 90:5574–5585. doi: 10.1128/JVI.03203-15. [DOI] [PMC free article] [PubMed] [Google Scholar]