

Hepatitis C virus (HCV) is one of the most important human pathogens. Its virion is associated with apolipoprotein E (ApoE), which enhances its infectivity. HCV induces autophagy to enhance its replication. In this report, we demonstrate that autophagy plays an important role in the trafficking of ApoE in HCV-infected cells. This leads to the degradation of ApoE by autophagy. However, if the autophagic protein degradation is inhibited, ApoE is stabilized and colocalized with autophagosomes. This leads to its enhanced colocalization with the HCV E2 envelope protein and increased production of infectious progeny viral particles. If autophagy is inhibited by suppressing the expression of ATG7, a gene essential for the formation of autophagosomes, the colocalization of ApoE with E2 is reduced, resulting in the reduction of progeny viral titers. These results indicate an important role of autophagy in the transport of ApoE to promote the production of infectious HCV particles.
KEYWORDS: apolipoprotein E, autophagosomes, autophagy, hepatitis C virus
ABSTRACT
Apolipoprotein E (ApoE) plays an important role in the maturation and infectivity of hepatitis C virus (HCV). By analyzing the subcellular localization of ApoE in Huh7 hepatoma cells that harbored an HCV subgenomic RNA replicon, we found that ApoE colocalized with autophagosomes. This colocalization was marginally detected in HCV-infected cells, apparently due to the depletion of ApoE by HCV, as treatment with bafilomycin A1 (BafA1), a vacuolar ATPase inhibitor that inhibits autophagic protein degradation, partially restored the ApoE level and enhanced its colocalization with autophagosomes in HCV-infected cells. The role of HCV-induced autophagy in the degradation of ApoE was further supported by the observations that nutrient starvation, which induces autophagic protein degradation, led to the loss of ApoE in HCV subgenomic RNA replicon cells and that the knockdown of ATG7, a protein essential for the formation of autophagic vacuoles, increased the ApoE level in cells with productive HCV replication. Interestingly, the inhibition of autophagy by ATG7 knockdown reduced the colocalization of ApoE with the HCV E2 envelope protein and the HCV titers released from cells. In contrast, the treatment of cells with BafA1 enhanced the colocalization of ApoE and HCV E2 and increased both intracellular and extracellular HCV titers. These results indicated that autophagy played an important role in the trafficking of ApoE in HCV-infected cells. While it led to autophagic degradation of ApoE, it also promoted the interaction between ApoE and HCV E2 to enhance the production of infectious progeny viral particles.
IMPORTANCE Hepatitis C virus (HCV) is one of the most important human pathogens. Its virion is associated with apolipoprotein E (ApoE), which enhances its infectivity. HCV induces autophagy to enhance its replication. In this report, we demonstrate that autophagy plays an important role in the trafficking of ApoE in HCV-infected cells. This leads to the degradation of ApoE by autophagy. However, if the autophagic protein degradation is inhibited, ApoE is stabilized and colocalized with autophagosomes. This leads to its enhanced colocalization with the HCV E2 envelope protein and increased production of infectious progeny viral particles. If autophagy is inhibited by suppressing the expression of ATG7, a gene essential for the formation of autophagosomes, the colocalization of ApoE with E2 is reduced, resulting in the reduction of progeny viral titers. These results indicate an important role of autophagy in the transport of ApoE to promote the production of infectious HCV particles.
INTRODUCTION
Hepatitis C virus (HCV) is one of the most important human pathogens and chronically infects an estimated 71 million people in the world (1). This virus can cause severe liver diseases, including cirrhosis and hepatocellular carcinoma. HCV is an enveloped virus with a positive-stranded RNA genome. It is classified in the Hepacivirus genus of the Flaviviridae family (2). The HCV genome encodes a polyprotein of about 3,000 amino acids. The translation of this polyprotein is mediated by the internal ribosomal entry site (IRES) that consists of most of the 5′ untranslated region (UTR) and the first few codons of the polyprotein-coding sequence (3, 4). This polyprotein is processed by cellular and viral proteases into different viral proteins (5). The structural proteins are the core protein, which forms the viral capsid, and the envelope glycoproteins E1 and E2. The nonstructural proteins NS3, NS4A, NS4B, NS5A, and NS5B are required for viral RNA replication. However, several of the nonstructural proteins, including the ion channel protein p7, NS3, and NS5A, are also involved in viral assembly and release (6–8). The nonstructural protein NS2 also regulates the encapsidation of viral genomic RNA by mediating the interaction between structural and nonstructural proteins (9, 10). Many of the viral proteins also have other regulatory functions.
HCV infection can perturb cellular pathways, including the induction of autophagy (11). Autophagy (i.e., macroautophagy) is a catabolic process that is important for maintaining cellular homeostasis. It begins with the formation of a crescent membranous structure, termed the phagophore or isolation membrane, in the cytoplasm. The membranes of phagophores subsequently expand to form an enclosed double-membrane structure known as autophagosomes. Autophagosomes mature by fusing with lysosomes to form autolysosomes, in which the cargos of autophagosomes are digested by lysosomal enzymes for recycling (12). More than 30 autophagy-related genes (ATG) that regulate the autophagic pathway have been identified (13). The microtubule-associated protein light-chain 3 (LC3), a cytosolic protein, is a mammalian homolog of ATG8 that was initially identified in yeast. During autophagy, LC3 is covalently linked by ATG3, ATG4, and ATG7 to phosphatidylethanolamine (PE) and localized to autophagosomal membranes. For this reason, lipidated LC3 is often used as the marker for autophagosomes. LC3 is either degraded in autolysosomes or delipidated by ATG4 after the maturation of autophagosomes and is released back into the cytosol for recycling. Autophagy can be induced by different stimuli, such as nutrient starvation. It is induced by HCV to enhance its replication (14–18).
Many studies have demonstrated an important role of the very-low-density lipoprotein (VLDL) in the HCV life cycle (19, 20). VLDL released from hepatocytes contains a number of proteins, including apolipoprotein B (ApoB), apolipoprotein E (ApoE), and microsomal triglyceride transfer protein (21). ApoE is known to play an important role in LVDL assembly and lipid transport by binding with high affinity to the LDL receptor (LDLR) (22). It is also detected on the surfaces of HCV particles in ultrastructural analysis (23–28). In addition, ApoE has also been reported to directly interact with HCV NS5A and E2 to enhance the infectivity of virions (29, 30). However, how ApoE is recruited to the sites for HCV assembly remains largely unknown.
To understand the mechanism by which ApoE is associated with and contributes to the assembly and release of HCV particles, we analyzed the subcellular localization of ApoE in Huh7 hepatoma cells that harbored an HCV subgenomic RNA replicon or were infected by HCV. We found that ApoE colocalized with autophagosomes and could be degraded by HCV-induced autophagy. Our further analysis indicated that autophagy was involved in the trafficking of ApoE, which enhanced its interaction with the HCV E2 envelope protein and the production of infectious HCV particles.
RESULTS
Colocalization of ApoE with autophagosomes induced by HCV.
We had previously established a Huh7 hepatoma cell line that stably expressed the green fluorescent protein (GFP)-LC3 fusion protein (i.e., Huh7-GFP-LC3 cells) (16) and used this cell line to produce another stable cell line that contained the HCV genotype 1b subgenomic RNA replicon. This GFP-LC3-replicon cell line was named GLR cells (17). To understand the possible relationship between ApoE and HCV-induced autophagy, we conducted immunofluorescence microscopy using GLR cells. As shown in Fig. 1, the GFP-LC3 signal was diffuse and opaque in the parental Huh7-GFP-LC3 cells, but it became bright and punctate in GLR cells, in agreement with previous reports that HCV replicons could induce the accumulation of autophagosomes in cells (14, 17, 31). Interestingly, although ApoE displayed a reticular staining pattern with enrichment in the perinuclear regions in control cells, consistent with its localization to the endoplasmic reticulum (ER) and Golgi network as previously reported (32, 33), it displayed a bright and punctate staining pattern in GLR cells, largely colocalized with the GFP-LC3 puncta. This result indicated the possible association of ApoE with autophagosomes in GLR cells.
FIG 1.

Colocalization of ApoE with autophagosomes in HCV subgenomic replicon cells. Stable Huh7-GFP-LC3 cells (top panels) or HCV subgenomic RNA replicon (GLR) cells (bottom panels) were fixed and stained for ApoE (red). DAPI was used to stain nuclei.
To determine whether ApoE is also colocalized with autophagosomes in HCV-infected cells, we infected stable GFP-LC3 cells with a cell culture-adapted HCV JFH-1 variant (34). Cells were fixed at 24 and 48 h postinfection for fluorescence microscopy. Curiously, the intensity of the ApoE signal was reduced at both 24 and 48 h postinfection compared with that in the mock-infected cells, and there was only limited colocalization of ApoE with GFP-LC3 puncta (Fig. 2A). To determine whether HCV infection reduced the level of ApoE, we performed immunoblot analysis. As shown in Fig. 2B, the ApoE protein level was not significantly affected by HCV at 6 or 12 h postinfection, but it was decreased at 24 h and further reduced at 48 h postinfection. When the ApoE mRNA was analyzed by real-time reverse transcription-PCR (RT-PCR), no significant difference in ApoE mRNA levels was detected at different time points (Fig. 2C), suggesting the possible depletion of ApoE by HCV via a posttranslational mechanism. We also analyzed LC3, p62, and the HCV core protein by immunoblot analysis. The p62 sequestosome-1 protein mediates autophagic protein degradation and is also degraded by autophagy during the process. It is thus often used as a marker for analyzing the autophagic protein degradation rate (35). As shown in Fig. 2B, lipidated LC3 (i.e., LC3-II), which has a higher electrophoretic mobility than its nonlipidated form (i.e., LC3-I), could be detected as early as 6 h postinfection, but the reduction of the p62 level was not detected until 24 h postinfection, when a slight reduction was detected. The loss of p62 was very pronounced at 48 h postinfection. This result was consistent with our previous finding that HCV temporally regulated the autophagic flux and autophagic protein degradation (36). Interestingly, the peak of ApoE loss coincided with the peak of the autophagic activity (i.e., p62 loss), raising the possibility that ApoE might be degraded by autophagy in HCV-infected cells.
FIG 2.

Effect of HCV infection on ApoE. (A) Stable Huh7 cells that expressed GFP-LC3 were mock infected or infected with HCV, fixed at the time points indicated, and immunofluorescence stained for ApoE (red) and nuclei (blue). Arrows denote the limited colocalization of ApoE with GFP-LC3 puncta. (B and C) Huh7 cells were infected with HCV (MOI = 1) and harvested at the different time points for immunoblot analysis (B) or real-time RT-PCR analysis for ApoE mRNA (C). For panel C, the ApoE mRNA level was normalized against the GAPDH RNA, which served as the internal control. The ApoE mRNA level in mock-infected cells was arbitrarily defined as 1. The experiments were repeated at least three times for statistical analysis.
Degradation of ApoE in an autophagy-dependent manner.
To determine whether autophagy is indeed involved in the degradation of ApoE, we treated HCV-infected cells with bafilomycin A1 (BafA1), a vacuolar ATPase inhibitor that suppresses the maturation of autophagosomes by inhibiting the acidification of lysosomes (37). As shown in Fig. 3A, BafA1 slightly increased the level of ApoE in mock-infected cells. It also increased the levels of LC3-II and p62. This result was as expected, since BafA1 inhibited the degradation of LC3-II and p62 mediated by basal autophagy. More importantly, BafA1 nearly fully restored the ApoE level in HCV-infected cells. BafA1 had no significant effect on the ApoE RNA level (Fig. 3B). These results are consistent with a role of autophagy in the degradation of ApoE in HCV-infected cells. The slight increase of ApoE in mock-infected cells suggested that the basal autophagy might also have a minor effect on ApoE. To confirm the immunoblot results, we also performed immunofluorescence microscopy. As shown in Fig. 3C, the treatment of HCV-infected cells with BafA1 increased both the number of GFP-LC3 puncta (i.e., autophagosomes) and the intensity of the ApoE signal and enhanced their colocalization (Fig. 3D). To determine whether ApoE could also colocalize with endogenous LC3 in HCV-infected cells, we replaced the stable GFP-LC3 cells with naive Huh7 cells for the infection studies. As shown in Fig. 3E, the treatment of HCV-infected Huh7 cells with BafA1 also increased the ApoE signal and enhanced its colocalization with endogenous LC3, in agreement with the results shown in Fig. 3C. As a control, we also treated HCV-infected cells with the proteasome inhibitor MG132 for immunoblot analysis. MG132 did not increase but rather decreased the ApoE level in both mock-infected and HCV-infected cells (Fig. 3F). This result indicated that it was unlikely that ApoE was degraded by proteasomes. The reason why MG132 further reduced the ApoE level is unclear. One possibility is that its inhibition of proteasomes led to the compensatory increase of the autophagic activity for protein degradation, as previously reported (38). This possibility is supported by the observation that MG132 slightly increased the LC3-II level and decreased the p62 level in mock-infected cells. Further studies will be required to confirm this possibility.
FIG 3.

Autophagic degradation of ApoE in HCV-infected cells. (A and B) Huh7 cells were mocked infected or infected with HCV (MOI = 1). At 8 h postinfection, cells were treated with either the vehicle dimethyl sulfoxide (DMSO) or 100 nM bafilomycin A1 (BafA1) for 16 h and then harvested for immunoblot analysis (A) or real-time RT-PCR analysis for quantification of the ApoE mRNA (B), as described in the Fig. 2 legend. (C) Huh7-GFP-LC3 cells were infected with HCV (MOI = 1) and treated with DMSO or BafA1 for 16 h. Cells were fixed at 24 h postinfection and stained for ApoE (red) and DAPI (blue). (D) Percentage of ApoE pixels shown in panel C that were also positive for GFP-LC3 (i.e., yellow/red ratio). The results represent the average for >50 cells. ***, P < 0.001. (E) Huh7 cells were infected with HCV (MOI = 1) and treated with DMSO or BafA1 for 16 h. Cells were fixed at 24 h postinfection and stained for LC3 (green), ApoE (red), and DAPI (blue). (F) Huh7 cells were mock infected or infected with HCV (MOI = 1) for 48 h. Cells treated with MG132 (10 μM) were treated for 6 h before being harvested for immunoblot analysis.
Rubicon is a protein that inhibits the fusion between autophagosomes and lysosomes (39). Due to the induction of Rubicon, the fusion between autophagosomes and lysosomes in HCV subgenomic RNA replicon cells is inefficient (36). To test whether ApoE associated with autophagosomes in HCV replicon cells could also be degraded by autophagy, we nutrient starved the HCV GLR subgenomic RNA replicon cells for 5 h to promote the fusion between autophagosomes and lysosomes. As shown in Fig. 4A, this nutrient starvation led to the loss of LC3-II and p62, indicating the induction of autophagic protein degradation. It also reduced the level of HCV NS5A protein, which is known to be associated with the HCV RNA replication complex on autophagosomes (17, 40). As would be expected if ApoE is degraded by autophagy, it also significantly reduced the ApoE protein level. In a separate experiment, we treated GLR cells that were nutrient starved with the proteasome inhibitor MG132 or the lysosomal protease inhibitors E64d and pepstatin A (35). As shown in Fig. 4B, in agreement with the results shown in Fig. 4A, nutrient starvation reduced ApoE, LC3-II, and p62 in GLR cells. This reduction was not affected by MG132, but it was abolished by E64d and pepstatin A, in agreement with the degradation of ApoE in autolysosomes.
FIG 4.

Analysis of the effect of autophagy on ApoE. (A) HCV GLR replicon cells without treatment or nutrient starved in HBSS for 5 h were lysed for immunoblot analysis. (B) GLR cells were not treated or were treated with HBSS for 5 h in either the absence or presence of 10 μM MG132 or 25 μM E64d and 50 μM pepstatin A (PepA). Cells were then harvested for immunoblot analysis. (C) Huh7 cells were electroporated with JFH-1 or GND RNA and 24 h later were transfected with the control (NC) siRNA or the ATG7 siRNA. Cells were harvested at 48 h after the siRNA transfection for immunoblot analysis. (D) Huh7-GFP-LC3 cells were infected with HCV (MOI = 1) and treated with either DMSO or BafA1. GFP-LC3 and its associated autophagosomes were immunoprecipitated with control beads or anti-GFP antibody-conjugated beads as described in Materials and Methods, followed by immunoblot analysis using the anti-ApoE and anti-GFP antibodies. Total cell lysates were also analyzed by immunoblotting to serve as the input control.
To further confirm the role of autophagy in the degradation of ApoE in HCV-infected cells, we also performed a small interfering RNA (siRNA) knockdown experiment to suppress the expression of ATG7, a protein factor important for autophagy. As the knockdown of ATG7 suppressed HCV infection (16, 41), we chose to introduce the HCV JFH-1 genomic RNA or its replication-defective mutant GND RNA into Huh7 cells by electroporation. Twenty-four hours later, cells were transfected with the ATG7 siRNA (siATG7), and they were lysed 48 h after siRNA transfection for immunoblot analysis. As shown in Fig. 4C, JFH-1 RNA reduced ApoE to an almost undetectable level in Huh7 cells, which was partially restored by ATG7 knockdown. In contrast, ATG7 knockdown did not significantly affect the ApoE level in GND RNA-transfected cells. The effect of ATG7 knockdown on the inhibition of autophagy was confirmed by the analysis of LC3-II and p62, which were decreased and increased, respectively, in JFH-1 RNA-transfected cells. The effect of ATG7 knockdown on LC3-II in GND RNA-transfected cells was inapparent due to the low basal level of LC3-II. However, it also slightly increased the p62 level, in agreement with the suppression of basal autophagy in cells. ATG7 knockdown had little effect on the HCV core protein. This was also consistent with the previous report that the inhibition of autophagy did not affect HCV replication after its replication had been established (14). To confirm the association of ApoE with autophagosomes, we immunoprecipitated GFP-LC3 and its associated autophagosomes from HCV-infected cells using anti-GFP antibody-conjugated magnetic beads. As shown in Fig. 4D, ApoE could be coimmunoprecipitated with GFP-LC3, and their association was enhanced if HCV-infected cells were treated with BafA1 (Fig. 4D). Taken together, the results shown in Fig. 3 and 4 indicate a role of autophagy in the degradation of ApoE.
Regulation of subcellular localization of ApoE by autophagy.
It had previously been demonstrated that ApoE colocalized with the HCV E2 envelope protein in HCV-infected cells (30, 42). To determine whether autophagy is important for the interaction between ApoE and E2, we also conducted the immunofluorescence staining experiment on cells with ATG7 knockdown. As shown in Fig. 5A, in cells electroporated with the GND control RNA, the HCV E2 protein was not detectable. In these cells, ApoE displayed a reticular staining pattern and was enriched in the perinuclear region when the cells were treated with the control siRNA (siNC). This reticular localization pattern was not significantly affected when cells were treated with siATG7, although the enrichment of the ApoE signal in the perinuclear region was slightly reduced, suggesting a possible role of basal autophagy in ApoE trafficking in these cells. In cells electroporated with the JFH-1 genomic RNA, both HCV E2 and ApoE were concentrated in the perinuclear region with a high degree of colocalization with each other when these cells were treated with siNC (Fig. 5A). When these cells were treated with siATG7, the subcellular localization of E2 was not affected, but a significant fraction of ApoE was dispersed from the perinuclear region, resulting in an overall reduction of its colocalization with E2 (Fig. 5B). Thus, the results shown in Fig. 5 indicate a role of autophagy in ApoE trafficking, possibly in GND RNA-transfected cells but clearly in JFH-1 RNA-transfected cells. This trafficking in JFH-1 cells was important to promote the interaction of ApoE with HCV E2.
FIG 5.

Alteration of subcellular localization of ApoE by ATG7 knockdown. (A) Huh7 cells were electroporated with GND or JFH-1 RNA for 24 h and then transfected with control siRNA (siNC) or ATG7 siRNA (siATG7). Cells were fixed at 48 h after siRNA transfection and stained for HCV E2 (green), ApoE (red), or nuclei (blue). (B) Percentage of ApoE puncta that were positive for HCV E2 (i.e., yellow/red ratio). The results represent the average for >50 cells. ***, P < 0.001.
Reduction of infectivity of released HCV particles after the depletion of ATG7.
ApoE has been shown to play important roles in the maturation and infectivity of HCV (23–25, 43). To further determine whether the alteration of subcellular localization of ApoE by autophagy affected the yield of progeny HCV particles, we harvested cells that had been treated with either siNC or siATG7 for quantification of intracellular HCV titers. We also harvested the incubation medium for analysis of extracellular viral titers. Although a slight increase of the intracellular viral titer was observed in cells treated with siATG7, this increase was statistically insignificant (Fig. 6A). However, the treatment with siATG7 significantly reduced the extracellular HCV titer (Fig. 6B). The suppression of ATG7 expression using siATG7 had no effect on the HCV RNA level in cells (Fig. 6C). This result was consistent with the result shown in Fig. 4C, which revealed no significant effect of ATG7 knockdown on the level of the HCV core protein in cells. Interestingly, although ATG7 knockdown increased the intracellular level of ApoE (Fig. 4C), it did not increase the secreted level of ApoE that was reduced by HCV (Fig. 6D). The results shown in Fig. 6 indicate that autophagy was required for the efficient release of infectious HCV particles, which might be due to its role in the transport of ApoE to the HCV assembly site.
FIG 6.

Effect of ATG7 knockdown on progeny HCV titers. (A to C) Huh7 cells electroporated with the JFH-1 RNA or the GND RNA for 24 h were transfected with the control siRNA (siNC) or ATG7 siRNA (siATG7). At 48 h after the siRNA transfection, cells and the incubation media were harvested for determination of intracellular (A) and extracellular (B) HCV titers using the focus formation assay as described in Materials and Methods. A fraction of the cell lysates was also used for real-time RT-PCR analysis for quantification of intracellular HCV RNA levels (C). The results represent the average from at least three independent experiments. (D) Huh7 cells were electroporated with GND or JFH-1 RNA for 24 h and then transfected with control siRNA (siNC) or ATG7 siRNA (siATG7). The incubation media were collected at 48 h after siRNA transfection for quantification of ApoE levels using ELISA (see Materials and Methods). The ApoE level of cells transfected with the GND RNA and siNC was arbitrarily defines as 1. Results represent the average from 3 independent experiments. *, P < 0.05; **, P < 0.01.
Enhanced production of HCV by inhibition of autophagic degradation of ApoE.
As the inhibition of autophagic protein degradation with BafA1 increased the ApoE level in HCV-infected cells, it appeared likely that this increase of ApoE level might enhance the production of infectious HCV particles. To test this possibility, we first determined whether the treatment of HCV-infected cells with BafA1 would enhance the colocalization of ApoE with HCV E2. As shown in Fig. 7A, compared with cells treated with the vehicle dimethyl sulfoxide (DMSO), cells treated with BafA1 had increased levels of ApoE, which remained largely colocalized with HCV E2 in the perinuclear region. We also analyzed the effect of BafA1 on the secretion of ApoE. The same as for ATG7 knockdown, BafA1 did not restore the level of secreted ApoE that was reduced by HCV (Fig. 7B). We next determined whether the increase of E2 and ApoE colocalization would affect intracellular and extracellular HCV titers. As shown in Fig. 7C and D, the treatment with BafA1 increased the intracellular HCV titer approximately 2-fold and extracellular HCV titers approximately 4-fold. BafA1 only slightly increased the level of HCV RNA released from the cells, indicating that it increased primarily the infectivity of HCV released from cells and not the amount of HCV particles produced (Fig. 7E). These results indicated that the inhibition of autophagic degradation of ApoE could indeed enhance the production of infectious HCV particles.
FIG 7.

Effects of ApoE on the colocalization of HCV E2 and ApoE and progeny HCV titers. Huh7 cells were infected with HCV (MOI = 1) and were treated with DMSO or 100 nM BafA1 for 16 h prior to the analysis. (A) Cells were fixed at 24 h postinfection and stained for HCV E2 (green), ApoE (red), and nuclei (blue). (B) The incubation media were collected for analysis of ApoE using ELISA. (C to E) Cells and the incubation media were harvested at 24 h postinfection for quantification of intracellular HCV titers (C), extracellular HCV titers (D), and extracellular HCV RNA (E). **, P < 0.01; ***, P < 0.001.
DISCUSSION
It has been very well documented that ApoE plays an important role in the formation of infectious HCV particles (23–25, 43). The infectivity of HCV can be efficiently neutralized by antibodies targeting ApoE (24), and the depletion of ApoE reduces the production of infectious HCV particles (24). However, the molecular mechanism by which ApoE is recruited to the sites for HCV assembly was unknown. In this work, we studied the relationship between ApoE and HCV and found that ApoE colocalized with autophagosomes in cells harboring an HCV subgenomic RNA replicon (Fig. 1). This colocalization of ApoE with autophagosomes was limited in HCV-infected cells (Fig. 2A). This was apparently due to the depletion of ApoE by HCV (Fig. 2B), because if HCV-infected cells were treated with BafA1, which inhibited autophagic protein degradation and increased the ApoE level (Fig. 3A), the colocalization of ApoE with autophagosomes became apparent (Fig. 3C). The maturation of autophagosomes is inefficient in HCV subgenomic RNA replicon cells, due to the induction of Rubicon, which inhibits the fusion between autophagosomes and lysosomes (36). To test whether ApoE could indeed be degraded by autophagy in replicon cells, we used nutrient starvation to promote the fusion between autophagosomes and lysosomes in these cells. Our results confirmed that nutrient starvation could indeed deplete ApoE from HCV replicon cells (Fig. 4A), and this depletion was abolished by the lysosomal protease inhibitors E64d and pepstatin A (Fig. 4B). Based on these results as well as the observation that the knockdown of ATG7, a gene essential for autophagy, partially restored the ApoE level in cells productively replicating HCV (Fig. 4C), we concluded that ApoE could be degraded by HCV-induced autophagy. Interestingly, in a previous study using lysosomotropic agents to increase lysosomal pH and by conducting light and electron microscopy, it was demonstrated that a substantial amount of newly synthesized ApoE in macrophages was degraded in lysosomes prior to secretion (44). Although the role of autophagy in the degradation of ApoE in that particular study was not addressed, it is conceivable that it was mediated by basal autophagy in macrophages. This speculation is supported by our observations that the inhibition of autophagic protein degradation with BafA1 led to a slight increase of ApoE in mock-infected cells (Fig. 3A), and the knockdown of ATG7 reduced the accumulation of ApoE in the perinuclear region of cells transfected with the GND RNA (Fig. 5A). These observations indicated that basal autophagy might also be involved in the transport and degradation of ApoE in cells, and this degradation is enhanced in HCV-infected cells due to the induction of autophagy by HCV. How ApoE becomes associated with autophagosomes is unclear. As our recent results indicated that autophagosomes induced by HCV originated from the ER (45), it is conceivable that ApoE may be sorted into autophagosomes when they are being formed. Alternatively, transport membranes containing ApoE may fuse with autophagosomes. Our studies also demonstrated that HCV could reduce the level of secreted ApoE (Fig. 6D and 7B). Interestingly, although both ATG7 knockdown and BafA1 could increase the intracellular level of ApoE, they could not restore the level of secreted ApoE reduced by HCV (Fig. 6D and 7B). These results indicated that in addition to enhancing autophagic degradation of ApoE, HCV might also interfere with the secretion of ApoE.
ApoE is important for the production of infectious HCV particles. Thus, the observation that ApoE was degraded by HCV-induced autophagy was curious. To address this question, we studied the possible role of autophagy in the interaction between ApoE and the HCV E2 envelope protein. It had previously been demonstrated that ApoE colocalized and interacted with HCV E2 in the perinuclear region (30, 42). We were able to confirm that previous finding and further demonstrated an important role of autophagy in the transport of ApoE to the perinuclear region, as impairing autophagy by suppressing the expression of ATG7 dispersed ApoE from the perinuclear region and reduced its colocalization with E2 (Fig. 5). This suppression of ATG7 expression did not reduce the HCV RNA level or significantly affect mature HCV particles in cells, but it reduced the HCV titer released from cells (Fig. 6). In contrast, the suppression of the late stage of autophagy using BafA1 increased the level of ApoE in the perinuclear region and enhanced its colocalization with HCV E2 (Fig. 7A). BafA1 also increased both intracellular and extracellular HCV titers (Fig. 7C and D). As BafA1 only slightly increased the amount of HCV RNA released from HCV-infected cells (Fig. 7E), it apparently primarily increased the infectivity of HCV particles. These results indicate that the efficient transport of ApoE to the perinuclear region to interact with E2 required autophagy, which then enhanced the production of infectious HCV particles. A model of the role of autophagy in the trafficking of ApoE is illustrated in Fig. 8. As shown in the figure, ApoE is localized to the ER in hepatocytes and can be transported by autophagosomes to lysosomes for degradation. Upon HCV infection, the association of ApoE with autophagosomes is enhanced by HCV-induced autophagy. This leads to its enhanced transport to the perinuclear region, which may be the ER or Golgi compartment, as previously suggested (30, 46), to interact with the HCV E2 protein and promotes the production of infectious viral particles. As autophagosomes induced by HCV also fuse with lysosomes in the later stage of HCV infection, part of ApoE is also degraded by autophagy during this trafficking process. In this model, the suppression of ATG7 expression inhibits the formation of autophagosomes, resulting in the retention of ApoE in the ER. This prevents ApoE from being delivered to the HCV assembly site and reduces the infectivity of released HCV. In contrast, the suppression of maturation of autophagosomes with BafA1 prevents ApoE from being degraded by autophagy, resulting in its enhanced transport to the HCV assembly site and the increase of the infectivity of both intracellular and extracellular HCV particles.
FIG 8.
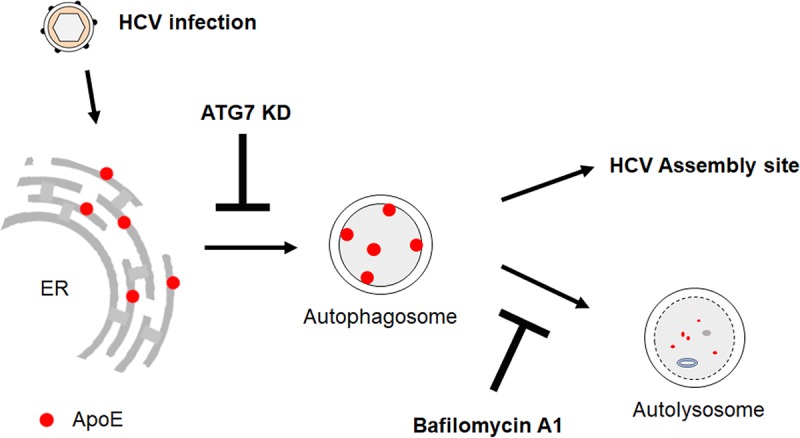
Model of ApoE trafficking by HCV-induced autophagy. See the text for details.
Our observation that ATG7 knockdown could reduce the released HCV titers was consistent with a previous report that ATG7 knockdown could suppress the release of progeny HCV particles (47), although in that previous report, the reduction of HCV release was attributed to the retention of HCV particles in exosomes. It is possible that autophagy may be involved in both the trafficking of ApoE to the HCV assembly site and the viral release via the exosomal pathway. Interestingly, it had also been reported that syntaxin 17 (STX17), a SNARE protein required for the fusion between autophagosomes and lysosomes to form autolysosomes, negatively regulates the release of infectious HCV particles (48). The overexpression of STX17, which favors the maturation of autophagosomes, reduced the HCV titer released from HCV-infected cells, and the suppression of STX17 expression, which suppresses the maturation of autophagosomes, increased it. These results were also consistent with our finding, as the overexpression of STX17 would be expected to enhance the loss of ApoE in autolysosomes and the suppression of STX17 would be expected to increase the ApoE level to enhance its association with progeny HCV particles.
In conclusion, our results indicate that the autophagy induced by HCV plays an important role in ApoE trafficking. Although this process led to the loss of some ApoE due to autophagic degradation, it also increased the infectivity of progeny HCV particles by delivering ApoE to the perinuclear region to promote its interaction with HCV E2.
MATERIALS AND METHODS
Cell cultures and HCV stocks.
Huh7 cells, a human hepatoma cell line, and their derivative Huh7.5 cells were maintained at 37°C in Dulbecco's modified essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Huh7 cells that stably expressed the GFP-LC3 fusion protein have been described before (16). HCV subgenomic RNA replicon cells (i.e., GLR cells) that were established from Huh7-GFP-LC3 cells have also been described previously (17). This replicon was a bicistronic RNA that contained the hygromycin B phosphotransferase-coding sequence and the HCV NS3-NS5B sequence, which was under the expression control of the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES). GLR cells were maintained in DMEM containing 150 μg/ml hygromycin B (17). All infection studies were conducted using a variant of the HCV JFH-1 isolate (34), which replicates robustly in Huh7 cells. For nutrient starvation, cells were incubated in Hanks' balanced salt solution (HBSS) for 5 h before being harvested for the studies.
DNA plasmids and siRNAs.
The DNA plasmids pJFH1 and pJFH1/GND had been described before (49). The HCV genomic RNA was synthesized from pJFH1 using the MEGA-script kit (Ambion) after linearization of the plasmid with the restriction enzyme XbaI. Huh7 cells were then electroporated with the HCV RNA. All the analyses were conducted on cells 3 days after electroporation. For the small interfering RNA (siRNA) knockdown experiment, ATG7 siRNA was purchased from Qiagen and the negative-control siRNA was purchased from Invitrogen. Cells were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen) per the manufacturer's instructions.
Antibodies.
The primary antibodies used in this study included the mouse anti-ApoE antibody (Santa Cruz), goat anti-ApoE antibody (Abcam), rabbit anti-LC3 antibody (Sigma), rabbit anti-p62 antibody (Cell Signaling), rabbit anti-ATG7 antibody (Cell Signaling), rabbit anti-HCV E2 antibody (GeneTex), rabbit anti-HCV core (50), and mouse anti-HCV NS5A monoclonal antibody 9E10 (a gift from Charles Rice, Rockefeller University).
Immunoblot analysis.
Cells were lysed with M-PER mammalian protein extraction reagent (Thermo Fisher). After centrifugation to remove cell debris, cell lysates were subjected for SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% skim milk for 1 h and incubated with the primary antibody overnight at 4°C. After three washes with Tris-buffered saline containing 1% Tween 20 (TBST), the membrane was incubated with the horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h. After the membrane was washed with TBST, chemiluminescent substrates (Pierce) were applied on the membrane, and the image was captured using the LAS-4000 imaging system (Fujifilm).
Immunofluorescence staining and microscopy.
Cells were rinsed with phosphate-buffered saline (PBS) and then fixed with 3.7% formaldehyde. Cells were permeabilized with PBS containing 0.1% saponin, 1% bovine serum albumin (BSA), and 0.05% sodium azide for 5 min and incubated with antibodies for immunofluorescence microscopy. Coverslips were mounted in Vector Shield (Vector) containing DAPI (4′,6′-diamidino-2-phenylindole), which stained the DNA. Images were taken with a Keyence All-in-One fluorescence microscope. The colocalization coefficient, which measures the fraction of Apo E pixels (red) that are also positive for GFP, was determined with randomly selected cells (>50) using the Keyence All-in-One software.
Immunoprecipitation of autophagic membranes.
Huh7-GFP-LC3 cells were seeded in three 100-mm plates and infected with HCV (multiplicity of infection [MOI] = 1). At 8 h postinfection, cells were treated with either the vehicle dimethyl sulfoxide (DMSO) or 100 nM bafilomycin A1 (BafA1) for 16 h. Cells were lysed by passing through a 26-guage needle 20 times in 500 μl hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 5 mM MgCl2). Nuclei and other cell debris were removed by centrifugation at 100 × g for 5 min at 4°C. The supernatant was incubated with 50 μl anti-GFP antibody conjugated-magnetic beads (MBL) or control magnetic beads (Dynabeads ProteinG from Novex by Life Technologies) for 2 h at 4°C. The immune complex was isolated with a magnetic separator and subjected to immunoblot analysis. Total cell lysates were also analyzed by immunoblotting to serve as the input control.
ELISA for ApoE.
Culture media of cells were centrifuged at 1,500 × g for 10 min to remove cellular debris. Levels of released ApoE in culture media were measured by using the apolipoprotein E (APOE) human ELISA kit (Abcam, catalog no. ab108813) according to the manufacturer's protocols. The optical density of the color reactions was read on a plate reader at 450 nm. Standard curves were generated, and concentrations of ApoE were calculated as described in the manufacturer's protocol.
Focus formation assay for HCV titration.
Huh7.5 cells were seeded onto an 8-well chamber slide (3 × 104 cells/well) and inoculated with serially diluted HCV the next day. At 48 h postinoculation, cells were washed with phosphate-buffered saline (PBS) and fixed with 3.7% formaldehyde for 15 min. Cells were stained with the rabbit anticore antibody for 1 h and then with Alexa 488-conjugated goat anti-rabbit secondary antibody for 45 min. After washing, cells were mounted with Vector Shield with DAPI. The HCV core-positive cells were counted under a microscope for titration.
Quantitative RT-PCR.
Total HCV RNA was analyzed by real-time RT-PCR using the TaqMan Gold RT-PCR kit (Applied Biosystems) following the manufacturer's instructions. HCV JFH1 primers 5′-TCTGCGGAACCGGTGAGTA-3′ (sense) and 5′-TCAGGCAGTACCACAAGGC-3′ (antisense) and the probe 5′-CACTCTATGCCCGGCCATTTGG-3′ were used for the real-time RT-PCR. Control glyceraldehyde 3′-phosphate dehydrogenase (GAPDH) primers and their probe were purchased from Applied Biosystems. ApoE mRNA was analyzed by real-time RT-PCR using the SYBR green-based one-step RT-PCR method (Applied Biosciences). Relative RNA levels were determined after normalization against the GAPDH RNA. The forward and reverse primers used for ApoE were 5′-GTTGCTGGTCACATTCCTGG-3′ (sense) and 5′-GCAGGTAATCCCAAAAGCGAC-3′ (antisense), and those for GAPDH were 5′-ACAACTTTGGTATCGTGGAAGG-3′ (sense) and 5′-GCCATCACGCCACAGTTTC-3′ (antisense).
ACKNOWLEDGMENT
This work was supported by NIH grant DK094652.
REFERENCES
- 1.World Health Organization. 2017. Global hepatitis report, 2017. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Robertson B, Myers G, Howard C, Brettin T, Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O, Netesov S, Nishioka K, Shin iT, Simmonds P, Smith D, Stuyver L, Weiner A. 1998. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol 143:2493–2503. [DOI] [PubMed] [Google Scholar]
- 3.Rijnbrand RC, Lemon SM. 2000. Internal ribosome entry site-mediated translation in hepatitis C virus replication. Curr Top Microbiol Immunol 242:85–116. [DOI] [PubMed] [Google Scholar]
- 4.Wang C, Sarnow P, Siddiqui A. 1993. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J Virol 67:3338–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 6.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J Virol 82:7624–7639. doi: 10.1128/JVI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog 6:e1001233. doi: 10.1371/journal.ppat.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol 85:1706–1717. doi: 10.1128/JVI.02268-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Ou JH. 2015. Hepatitis C virus and autophagy. Biol Chem 396:1215–1222. doi: 10.1515/hsz-2015-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reggiori F. 2006. 1. Membrane origin for autophagy. Curr Top Dev Biol 74:1–30. doi: 10.1016/S0070-2153(06)74001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. 2012. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol 86:8705–8712. doi: 10.1128/JVI.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, Machida K, Ou JH. 2012. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem 287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. 2009. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 5:937–945. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]
- 19.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A 104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatters DM, Peters-Libeu CA, Weisgraber KH. 2006. Apolipoprotein E structure: insights into function. Trends Biochem Sci 31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C. 2010. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53. doi: 10.1002/hep.23278. [DOI] [PubMed] [Google Scholar]
- 24.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J Virol 81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuhara T, Wada M, Nakamura S, Ono C, Shiokawa M, Yamamoto S, Motomura T, Okamoto T, Okuzaki D, Yamamoto M, Saito I, Wakita T, Koike K, Matsuura Y. 2014. Amphipathic alpha-helices in apolipoproteins are crucial to the formation of infectious hepatitis C virus particles. PLoS Pathog 10:e1004534. doi: 10.1371/journal.ppat.1004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci U S A 110:9505–9510. doi: 10.1073/pnas.1307527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J Virol 84:11532–11541. doi: 10.1128/JVI.01021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JY, Acosta EG, Stoeck IK, Long G, Hiet MS, Mueller B, Fackler OT, Kallis S, Bartenschlager R. 2014. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J Virol 88:12422–12437. doi: 10.1128/JVI.01660-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. 2008. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol 82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, Pleiss M, McComas C, Hess F, Witter D, Peterson S, Childers M, Goulet M, Liverton N, Hargreaves R, Freedman S, Weisgraber KH, Mahley RW, Huang Y. 2011. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J Biol Chem 286:17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamilton RL, Wong JS, Guo LS, Krisans S, Havel RJ. 1990. Apolipoprotein E localization in rat hepatocytes by immunogold labeling of cryothin sections. J Lipid Res 31:1589–1603. [PubMed] [Google Scholar]
- 34.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algul H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, et al. 2016. Guidelines for the use and interpretation of assays for monitoring autophagy, 3rd ed Autophagy 12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Tian Y, Ou JH. 2015. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog 11:e1004764. doi: 10.1371/journal.ppat.1004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powdrill MH, Bernatchez JA, Gotte M. 2010. Inhibitors of the hepatitis C virus RNA-dependent RNA polymerase NS5B. Viruses 2:2169–2195. doi: 10.3390/v2102169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. 2007. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 39.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. 2009. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 40.Kim JY, Wang L, Lee J, Ou JJ. 2017. Hepatitis C virus induces the localization of lipid rafts to autophagosomes for its RNA replication. J Virol 91:e00541-17. doi: 10.1128/JVI.00541-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim N, Kim MJ, Sung PS, Bae YC, Shin EC, Yoo JY. 2016. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat Commun 7:10631. doi: 10.1038/ncomms10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyer A, Dumans A, Beaumont E, Etienne L, Roingeard P, Meunier JC. 2014. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J Biol Chem 289:18904–18913. doi: 10.1074/jbc.M113.538256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J Virol 84:12048–12057. doi: 10.1128/JVI.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng J, Rudick V, Dory L. 1995. Lysosomal degradation and sorting of apolipoprotein E in macrophages. J Lipid Res 36:2129–2140. [PubMed] [Google Scholar]
- 45.Wang L, Kim JY, Liu HM, Lai MMC, Ou JJ. 2017. HCV-induced autophagosomes are generated via homotypic fusion of phagophores that mediate HCV RNA replication. PLoS Pathog 13:e1006609. doi: 10.1371/journal.ppat.1006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog 8:e1002466. doi: 10.1371/journal.ppat.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shrivastava S, Devhare P, Sujijantarat N, Steele R, Kwon YC, Ray R, Ray RB. 2016. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J Virol 90:1387–1396. doi: 10.1128/JVI.02383-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren H, Elgner F, Jiang B, Himmelsbach K, Medvedev R, Ploen D, Hildt E. 2016. The autophagosomal SNARE protein syntaxin 17 is an essential factor for the hepatitis C virus life cycle. J Virol 90:5989–6000. doi: 10.1128/JVI.00551-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 50.Lo SY, Masiarz F, Hwang SB, Lai MM, Ou JH. 1995. Differential subcellular localization of hepatitis C virus core gene products. Virology 213:455–461. doi: 10.1006/viro.1995.0018. [DOI] [PubMed] [Google Scholar]