

Abstract
Site-selective C–H functionalization is a great challenge in homogeneous transition-metal catalysis. Herein, we present a physical organic approach to delineate the origin of regioselective amidation of N-acylindoles through Ir(III) catalysis. Bulkiness of N-directing groups of indole substrates and electronics of carboxylate additives were identified as two major factors in controlling C2 and C7 selectivity, and their microscopic mechanisms were studied with DFT-based transition state analysis. Computational insights led us to interrogate a linear free energy relationship, and parametrization of molecular determinants enabled the establishment of an intuitive yet robust statistical model that correlates an extensive number of validation data points in high accuracy. This mechanistic investigation eventually allowed the development of a new C2 amidation and alkenylation protocol of indoles, which affords the exclusive functionalization at the C2 position with up to >70:1 selectivity.
Short abstract
Site-selective C−H functionalization is a great challenge in metal catalysis. Herein, we present a physical organic approach to delineate the origin of regioselective indole derivatizations.
Introduction
Discriminating a specific C–H bond and selectively transforming it to valuable functional groups is a formidable challenge in synthetic organic chemistry.1−3 A number of recent studies have led to the consideration of two tools based on substrate-guided4−7 and catalytic system-controlled approaches.8,9 Substrate modification has been widely utilized to drive the reaction toward the desired pathway, but its applications are rather confined to the specially engineered substrates.10−13 In this context, the modulating catalytic system which includes a change of catalyst structure and/or reaction conditions is more ideal for versatile transformations, although identification of effective catalyst systems is much more challenging.14−17 The difficulties in catalyst design mainly arise from a lack of mechanistic understanding behind the desired selectivity.
Given that indoles are widely present in pharmaceutical, natural, and biologically active molecules,18 derivatization of this privileged scaffold in a selective manner could give a great impact in the related research areas.19,20 In recent years, modulation of a N-carbonyl moiety in the indole skeleton has been scrutinized as an effective strategy to direct the C–H bond cleavage at the C2 and/or C7 position in the functionalization of indoles (Scheme 1a). As the C2–H bond is the most acidic in indole compounds,18,21 lithiation of indoles readily takes place at the C2–H bond by applying strong bases.22 Likewise, a number of transition-metal-catalyzed C–H activations of indoles are effective toward the C2 functionalization, and their C2 selectivity is believed to originate from the intrinsic C–H bond strength.23−27 Modification of the directing group to N-pyrimidyl is another effective strategy for the C2 amidation of indoles,28−30 but postmodification of substrates, such as removal of the directing group, often requires harsh conditions.28 On the other hand, when sterically bulky N-acyl groups are installed, C7 metalation is known to be more favored,31 and indeed, this strategy enabled the introduction new C–C and C–N bonds at the C7 position with high selectivity.32−34 Despite these advances, divergent catalytic systems to finely modulate the C2/C7 selectivity are still elusive.
Scheme 1. (a) Substrate-Controlled Approaches in C–H Functionalization of Indoles and (b) Selectivity Reversal Achieved by the Integrated Substrate/Base Effects.

Herein, we report an integrated substrate-guided/base-controlled approach to invert site selectivity in Ir(III)-catalyzed C–H functionalization of N-acylindoles (Scheme 1b). Examination of various reaction parameters led us to reveal microscopic mechanisms associated with site-selective C–H activation steps. Physical organic parametrization of substrates and additives enabled the construction of robust structure-selectivity relationships that can predict an extensive set of reaction outcomes. The series of analyses eventually led to the establishment of a new C2 amidation and alkenylation protocol offering an excellent level of site selectivity.
Results and Discussion
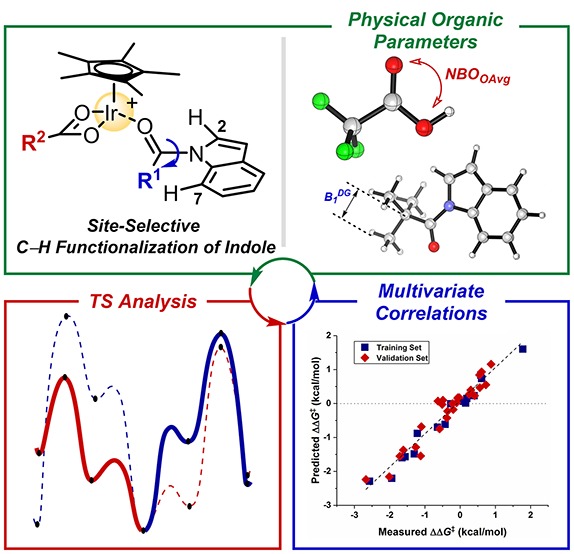
We recently reported versatile C–H amination procedures using organic azides as the amide source35−37 that allow an exclusive C7 functionalization of N-pivaloylindoles through Ir(III) catalysis (Scheme 2a, R1 = tBu).32 A key to achieving high reactivity was to utilize silver acetate as an additive, which is critical in accessing the aromatic C7–H bond by N-pivaloyl-directed C–H activation.38,39 This previous result enabled us to envision that site-selective functionalization of other C–H bonds might also be viable if we can delineate underlying working modes related to the C–H cleavage step. In this context, the C2 position of the indole is an attractive synthetic target because it is proximal to the N-alkylcarbonyl directing group, potentially enabling chelation-assisted C–H bond activation. We sought to interrogate various reaction parameters possibly affecting the selectivity in an anticipation of achieving new C2-amidation protocols. First, four indole substrates having different sizes of directing groups were subjected to the previously optimized conditions by assuming that less bulky substituents might drive the rotational equilibrium toward (E)-isomer (vide supra).31,34,40 As illustrated in Scheme 2a, all substrates were well-converted to the desired amidated products in the presence of silver acetate, but the site selectivity was highly dependent on the N-acyl groups. While exclusive C7 amidation was observed with N-pivaloylindole without any sign of C2 functionalization, subjecting N-isobutyrylindole, which has a smaller alkyl chain, gave rise to C2 amidation in 21% yield in addition to 54% of C7-amidated product. Indeed, a further decrease in the size of the N-substituent increased the portion of C2 products, and an almost nonbiased formation of two regioisomeric products was obtained with N-acetylindole (C7/C2 = 1.4:1).41
Scheme 2. Influence of Reaction Parameters on the Site Selectivity: (a) N-Directing Group Effects and (b) Silver Carboxylate Effects.
Unless otherwise indicated, reactions were run with indole substrate (0.20 mmol), tosyl azide (0.22 mmol), [IrCp*Cl2]2 (5 mol %), silver carboxylate (20 mol %), and AgNTf2 (10 mol %) in 1,2-dichloroethane (0.5 mL) at 40 °C for 12 h. Ratios were determined by 1H NMR analysis of the crude mixture, and averaged over two runs.
Ratios were determined by HPLC analysis of the crude mixture.
Having identified the steric influence of the substrate, we further sought to examine the additive effects on the site selectivity (Scheme 2b). We hypothesized that the stereoelectronic property of the carboxylate additives could be important, since the C–H activation step involves a close interaction between the catalyst and the base in the selectivity-determining deprotonation process.42−45 When more basic carboxylate salt, i.e., silver pivalate, was applied to the reaction in lieu of silver acetate, C7 selectivity was increased to give the ratio of 2.5:1. Interestingly, in sharp contrast, utilizing silver trifluoroacetate significantly suppressed the C7 functionalization, and highly favorably gave C2-amidated product 4 in 62:1 selectivity. These observations highlight that the nature of carboxylates directly affects the relative energy differences between selectivity-determining transition states.46
For a better understanding of the underlying origin of these effects, putative reaction intermediates and their working modes were evaluated with DFT calculation (Figure 1). The most and least basic carboxylates (i.e. pivalate and trifluoroacetate) were representatively chosen for the case study. The pivalate-mediated C–H cleavage, as marked with the blue line, might be initiated from an iridium-substrate adduct species I, which contains κ2 coordination of the pivalate. This adduct species has been frequently proposed in the related C–H functionalization reactions.42,44,45 Upon decoordination of one dative Ir–O bond and subsequent interaction between the metal center and either C2–H or C7–H bond, an agostic complex II or III can be readily formed.39,42 As predicted, replacing the Ir–O bond with the agostic interaction is a thermodynamically uphill process. Complex I was destabilized by 11.2 kcal/mol to furnish complex II, while 7.5 kcal/mol was required for the formation of its C7 isomeric species III. These intermediates subsequently traverse transition states II-TS and III-TS via the concerted metalation–deprotonation (CMD) mechanism to generate iridacycle IV and V, respectively. The reaction barrier for the C2–H cleavage from the agostic complex II was 6.1 kcal/mol, whereas that of the C7–H cleavage was 8.8 kcal/mol. Interestingly, however, the overall activation barrier for the C7–H iridation was still lower in energy than the C2 pathway: the activation barrier from I to II-TS was 17.3 kcal/mol, whereas 16.3 kcal/mol was required to reach III-TS.
Figure 1.

Competitive reaction energy profiles of C2–H and C7–H cleavage steps. Solid lines indicate kinetically favorable pathways. Dashed lines are disfavored ones. Free energy surfaces were constructed at the M06/SDD+6-311+G**/SMD(dichloroethane)//M06/Lanl2dz+6-31G** level of theory.
In sharp contrast, reverted selectivity was computationally estimated when similar reaction pathways were evaluated with trifluoroacetate as the base (red line, Figure 1). The overall activation barrier for C2–H cleavage (I′ to II′-TS) was 2.3 kcal/mol lower in energy than the corresponding C7–H activation (I′ to III′-TS), which is consistent with the experimental observation in Scheme 2b. This dichotomy with two different carboxylates might be explained by considering two aspects: (i) stability of the agostic intermediates, and (ii) deprotonation barriers from them. In general, C7-agostic intermediates are more stable than analogous C2 complexes, mainly because of the higher covalency of less acidic C7–H bonds. However, their relative stability depends on the electron-richness of the iridium metal center: while pivalate-containing III is 3.7 kcal/mol more stable than II, trifluoroacetate complex III′ is slightly more stable than II′ by only 2.2 kcal/mol, suggesting that the electron-deficient metal center favors the agostic complexation with both C2–H and C7–H bonds, and in turn, the stability difference is only marginal with trifluoroacetate complexes. On the other hand, the deprotonation barrier for the C2–H cleavage is generally lower in energy than the C7–H event with both carboxylates, mainly because of the intrinsic acidity of weak C2–H bonds.18,21 The reaction barrier is especially higher when weak trifluoroacetate base is utilized to cleave the strong C7–H bond (III′ to III′-TS), which requires a free energy of 13.4 kcal/mol. We propose that these two influences are convoluted and eventually reflected in C2 and C7 selectivity.
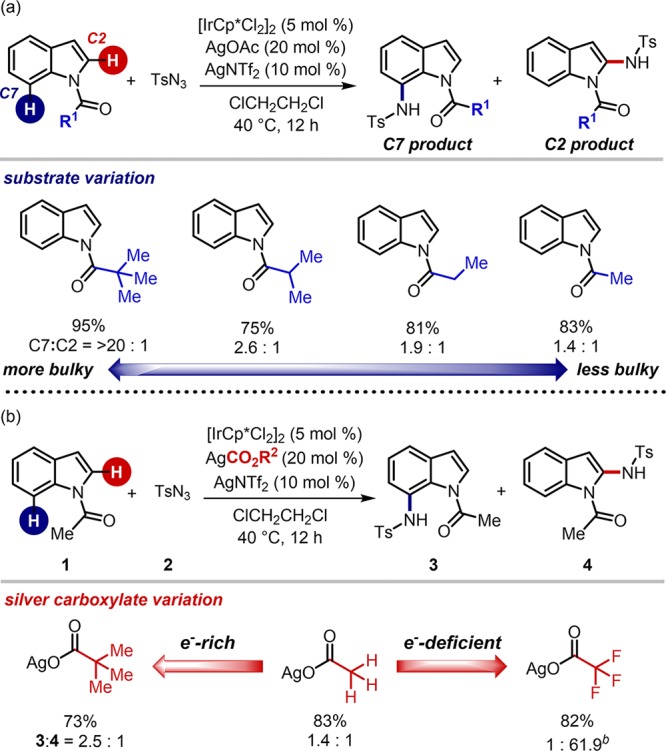
The above transition state analysis revealed that the electron-richness of carboxylate additives is directly associated with the stability of agostic complexes and deprotonation barriers. This implication led us to envision that thermodynamic descriptors of carboxylates might be useful to draw a quantitative relationship that accounts for the measured selectivity. We anticipated that a mathematical model would allow the prediction of the relative free energy differences and provide chemical insights for the selectivity. Among various quantitative physical parameters,47−50 one modern approach is to utilize computational descriptors, which have been recently introduced by Sigman and co-workers.51 This DFT-based approach is particularly effective when experimentally derived parameters are not available.51 For the description of the stereoelectronic features, corresponding carboxylic acids were employed as the carboxylate surrogates, and their structures were computationally optimized with the M06-2X/jun-cc-pVTZ level of theory. From the optimized geometries, a number of stereoelectronic parameters, such as natural bond orbital (NBO) charges52,53 of oxygens, IR frequencies54,55 of carbonyl stretches, and the related Sterimol values,56,57 were enumerated.58
For an extensive evaluation of the structure-selectivity relationship, an additional 20 silver carboxylates were prepared and subjected to the standard reaction condition (Figure 2a). Whereas the use of alkyl carboxylates (entries 1–4) preferred the formation of C7 amidation, phenylacetate derivatives reversed the selectivity (entries 5–7 and 10–13). Significantly, C2 selectivity became more pronounced when halogenated acetates were employed: while chloroacetate gave the C2 product in moderate selectivity (2.6:1), exclusive C2 amidation was observed with pentafluoropropionate (entry 23). These ratios were then converted to ΔΔG‡ in an assumption with the Curtin–Hammett situation and subjected into regression analysis with interrogated molecular descriptors.59 Whereas no notable relationship was found with various steric parameters, such as Sterimol B5 values of carboxylic acids, excellent univariate correlation was found when the average charge of two carboxyl oxygens (NBOO-avg) was considered (Figure 2b).60 In this model, 23 experimental outcomes were well-correlated with NBOO-avg with R2 of 0.91. Cross-validation of the model by the leave-one-out (LOO) method further suggested that the constructed model is highly robust (QL1O2 = 0.89). Remarkable linearity with a negative slope strongly suggests that carboxylic acids having more positive NBOO-avg charge may give rise to higher selectivity in favor of the C2 product.
Figure 2.

(a) Dependence of site selectivity on the type of silver carboxylates (regiomeric ratio was determined by 1H NMR or HPLC analysis from an average of two runs). Measured ΔΔG‡ values were calculated from ΔΔG‡ = −RT ln(4/3) at 40 °C. (b) Plot of measured ΔΔG‡ versus Sterimol B5 value (left) and average NBO charges of two oxygens (NBOO-avg, right) of carboxylic acid surrogates.
Motivated by the remarkable correlation, we further wondered if two identified factors, which are N-directing groups of indole substrates and carboxylate additives, could be simultaneously accounted for in a quantitative manner (Scheme 3a). For this purpose, a series of reactions were examined with different directing groups and additive combinations, as listed in Figure S14. A total of 44 reaction outcomes were obtained and subjected to multivariate analysis. We selected 16 responses to adequately train simultaneous change of the substrates and additives (Figure S14a), and subsequent regression analysis revealed a two-parameter model displaying an excellent level of accuracy and robustness (blue squares, Figure 3b). This normalized model includes a linear combination of NBOO-avg, which was an effective parameter in Figure 2b, and the Sterimol B1 value of N-protecting groups that is a direct illustration of the minimum radius perpendicular to the substituent. The NBO charge displayed a higher contribution to the model, as noticed by a larger absolute value of coefficient (−0.97). Robustness of the relationship was further examined by applying a validation set having an extra 28 responses, and those outcomes are remarkably well-fitted in the developed model (red diamonds, R2 = 0.94). This quantitative relationship clearly suggests that both bulkiness of directing groups and electronic nature of carboxylate additives are directly involved, as predicted by transition state analysis.
Scheme 3. (a) Synthesis of C2 Iridacycle 5, (b) Stoichiometric C–N Coupling with Tosyl Azide, (c) Catalytic Reaction Using 5 as the Catalyst, and (d) Kinetic Isotope Effect (KIE) Experiments.

Figure 3.
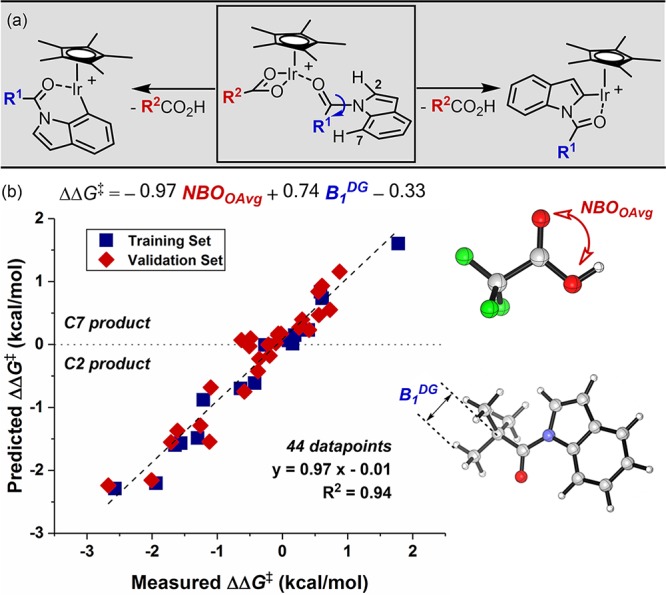
(a) Hypothetical reaction mechanisms on the selectivity determining step. (b) Multivariate regression analysis for the regioselective C–H amidation of indole.
Having understood the selectivity-determining mechanism, experimental attempts to isolate putative reaction intermediates were made to support hypothesized working modes (Scheme 3). Upon treatment of N-acetylindole with a dimeric iridium(III) complex in the presence of AgOTFA, the formation of an iridacycle 5 was observed. The solid state structure of this species was characterized by X-ray analysis, which unambiguously confirmed the selective C2–H activation. When azide 2 was added to 5, C–N coupling smoothly took place at room temperature to afford an amido-inserted complex 6 in high yield, and its structure was also confirmed by X-ray analysis (Scheme 3b). Importantly, an amidation reaction utilizing complex 5 as a catalyst afforded C2-amidated product 4 in an exclusive manner, strongly suggesting that the cyclometalated species 5 is an active intermediate in the catalytic cycle (Scheme 3c). In addition, primary kinetic isotope effect (KIE) values revealed that the C–H bond cleavage is irreversible and turnover-determining (Scheme 3d).61
The substrate generality of this mechanism-driven C2-amidation approach was evaluated with pregenerated Cp*Ir(OTFA)2 as the catalyst (Scheme 4).62 All reactions smoothly took place under mild conditions to furnish C2-amidated indoles in synthetically acceptable yields. While electron-donating substituents facilitated the amidation reactions (4, 7, 8), electron-withdrawing groups provided slightly lower reactivity (9–11). The position of substituents was less critical in the efficiency (12–17). The selective C2-amidation was not impeded even in the presence of C-3 substituents (18–20): the structure of compound 18 was fully characterized including its X-ray structure to confirm the site selectivity. The reaction was compatible with versatile functional groups as representatively demonstrated by the tryptophan derivative (19). In addition, a variety of amide sources were applicable to the present condition. Indeed, sulfonyl azides of benzene derivatives (20–24) and naphthalene (25) were readily reacted under the optimal conditions. Moreover, aliphatic variants also worked well without difficulty (26–28).
Scheme 4. Substrate Scope of Indoles and Sulfonyl Azides.

Unless otherwise indicated, reactions were run with substrates (0.20 mmol), sulfonyl azides (0.22 mmol), IrCp*(OTFA)2 (10 mol %), and NaNTf2 (10 mol %) in 1,2-dichloroethane (0.5 mL) at 40 °C for 16 h; isolated yields.
Run at 80 °C.
To further highlight broad applicability of our strategy, a related C–H functionalization reaction was examined. Inspired by Ma’s approach on C7 alkenylation of N-pivaloylindoles,33 we hypothesized that the site selectivity might be inverted to C2 functionalization on the basis of our mechanistic understanding. Indeed, as illustrated in Scheme 5a, the C2/C7 selectivity was significantly altered by the choice of carboxylate additives: while no preferential formation was observed with copper acetate, the more electron-rich pivalate gave an increase in C7 selectivity (C2:C7 = 1:1.9). Notably, selective C2 functionalization was achieved with electron-deficient copper trifluoroacetate in 13.2:1 selectivity. Ethyl and benzyl acrylates also gave excellent reactivity and selectivity with slightly modified conditions (Scheme 5b). These results clearly demonstrated that our mechanism-guided strategy could be broadly applicable to various C–H functionalization reactions of indoles.
Scheme 5. Application of the Strategy to C–H Alkenylation Reaction: (a) Electronic Effect of the Additive on the Selectivity and (b) C2-Selective C–H Alkenylation with Various Olefin Partners.
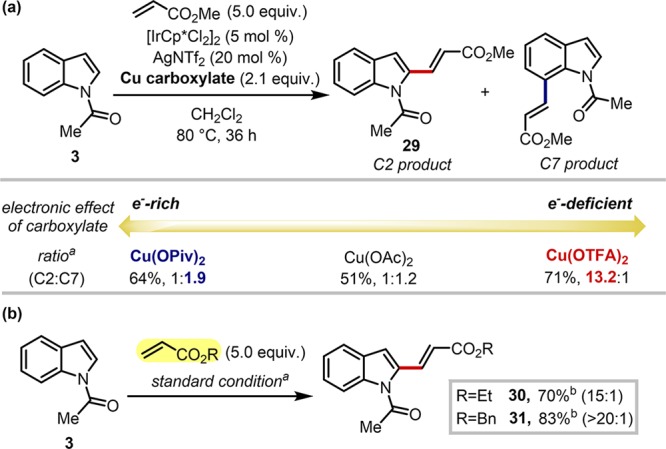
Reactions were run with 3 (0.20 mmol), acrylates (1.0 mmol), [IrCp*Cl2]2 (5 mol %), NaNTf2 (20 mol %), and copper carboxylate (0.42 mmol) in methylene chloride (1.5 mL) at 80 °C for 36 h. Site selectivities were determined by 1H NMR analysis of the crude mixture.
Isolated yields.
Conclusion
In summary, we demonstrated that regioselectivity in the Ir(III)-catalyzed C–H amidation of indoles could be completely switched from the C7 to C2 position by modulating stereoelectronic parameters of substrates and catalysts. Transition state analysis shed light on the origin of the selectivity-determining step, and the understanding was leveraged to construct a quantitative structure-selectivity relationship by utilizing computationally derived descriptors. A robust two-parameter model enabled the correlation of 44 reaction outcomes with a high accuracy. A mechanistic understanding further allowed the establishment of a selective amidation/alkenylation protocol to access C2-amidated indoles in good to excellent yields.
Acknowledgments
This research was supported by the Institute for Basic Science (IBS-R010-D1) in Korea. We thank Dr. Sung-Woo Park (Institute for Basic Science) for helpful discussion.
Supporting Information Available
X-ray crystallographic data (CIF) The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00264.
Author Contributions
† Y.K. and Y.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Neufeldt S. R.; Sanford M. S. Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc. Chem. Res. 2012, 45, 936–946. 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Dong G. Site-selectivity control in organic reactions: A quest to differentiate reactivity among the same kind of functional groups. Acc. Chem. Res. 2017, 50, 465–471. 10.1021/acs.accounts.6b00476. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.; Larsen M. A. Undirected, homogeneous C–H bond functionalization: Challenges and opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückl T.; Baxter R. D.; Ishihara Y.; Baran P. S. Innate and guided C–H functionalization logic. Acc. Chem. Res. 2012, 45, 826–839. 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Wasa M.; Chan K. S. L.; Shao Q.; Yu J.-Q. Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev. 2017, 117, 8754–8786. 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel J. R.; Boerth J. A.; Ellman J. A. Transition-metal-catalyzed C–H bond addition to carbonyls, imines, and related polarized π bonds. Chem. Rev. 2017, 117, 9163–9227. 10.1021/acs.chemrev.6b00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.; Kim Y.; Chang S. Transition metal-catalyzed C–H amination: Scope, mechanism, and applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Beyond directing groups: Transition-metal-catalyzed C-H activation of simple arenes. Angew. Chem., Int. Ed. 2012, 51, 10236–10254. 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Catalyst-controlled site-selective bond activation. Acc. Chem. Res. 2017, 50, 549–555. 10.1021/acs.accounts.6b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J. J.; Rakshit S.; Dröge T.; Glorius F. Palladium-catalyzed amidation of unactivated C(sp3)–H bonds: From anilines to indolines. Angew. Chem., Int. Ed. 2009, 48, 6892–6895. 10.1002/anie.200903035. [DOI] [PubMed] [Google Scholar]
- Kawano T.; Hirano K.; Satoh T.; Miura M. A new entry of amination reagents for heteroaromatic C–H bonds: Copper-catalyzed direct amination of azoles with chloroamines at room temperature. J. Am. Chem. Soc. 2010, 132, 6900–6901. 10.1021/ja101939r. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Ruhl K. E.; Rovis T. A coupling of benzamides and donor/acceptor diazo compounds to form γ-lactams via Rh(III)-catalyzed C–H activation. J. Am. Chem. Soc. 2013, 135, 5364–5367. 10.1021/ja402274g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu L.; Shang M.; Tanaka K.; Chen Q.; Pissarnitski N.; Streckfuss E.; Yu J.-Q. Remote meta C–H activation using a pyridine-based template: Achieving site-selectivity via the recognition of distance and geometry. ACS Cent. Sci. 2015, 1, 394–399. 10.1021/acscentsci.5b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza I. A.; Wagner A. M.; Sanford M. S.; Schoenebeck F. On the role of anionic ligands in the site-selectivity of oxidative C-H functionalization reactions of arenes. Chem. Sci. 2013, 4, 2767–2775. 10.1039/c3sc00017f. [DOI] [Google Scholar]
- Liao K.; Pickel T. C.; Boyarskikh V.; Bacsa J.; Musaev D. G.; Davies H. M. L. Site-selective and stereoselective functionalization of non-activated tertiary C–H bonds. Nature 2017, 551, 609–613. 10.1038/nature24641. [DOI] [PubMed] [Google Scholar]
- Tran G.; Hesp K. D.; Mascitti V.; Ellman J. A. Base-controlled completely selective linear or branched rhodium(I)-catalyzed C–H ortho-alkylation of azines without preactivation. Angew. Chem., Int. Ed. 2017, 56, 5899–5903. 10.1002/anie.201702409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Verma P.; Xia G.; Shi J.; Qiao J. X.; Tao S.; Cheng P. T. W.; Poss M. A.; Farmer M. E.; Yeung K.-S.; et al. Ligand-accelerated non-directed C–H functionalization of arenes. Nature 2017, 551, 489–493. 10.1038/nature24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg R. J.Indoles; Academic Press: San Diego, 1996. [Google Scholar]
- Seregin I. V.; Gevorgyan V. Direct transition metal-catalyzed functionalization of heteroaromatic compounds. Chem. Soc. Rev. 2007, 36, 1173–1193. 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck E. M.; Gaunt M. J. Pd-catalyzed C–H bond functionalization on the indole and pyrrole nucleus. Top. Curr. Chem. 2009, 292, 85–121. 10.1007/128_2009_15. [DOI] [PubMed] [Google Scholar]
- Shen K.; Fu Y.; Li J.-N.; Liu L.; Guo Q.-X. What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO?. Tetrahedron 2007, 63, 1568–1576. 10.1016/j.tet.2006.12.032. [DOI] [Google Scholar]
- Pelkey E. T.Metalation of indole. In Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Gribble G. W., Ed.; Springer: Berlin, Heidelberg, 2010; pp 141–191. [Google Scholar]
- Stuart D. R.; Villemure E.; Fagnou K. Elements of regiocontrol in palladium-catalyzed oxidative arene cross-coupling. J. Am. Chem. Soc. 2007, 129, 12072–12073. 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]
- Campbell A. N.; Meyer E. B.; Stahl S. S. Regiocontrolled aerobic oxidative coupling of indoles and benzene using Pd catalysts with 4,5-diazafluorene ligands. Chem. Commun. 2011, 47, 10257–10259. 10.1039/c1cc13632a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Ryu N.; Shibata T. Ir(I)-Catalyzed C–H bond alkylation of C2-position of indole with alkenes: Selective synthesis of linear or branched 2-alkylindoles. J. Am. Chem. Soc. 2012, 134, 17474–17477. 10.1021/ja308742x. [DOI] [PubMed] [Google Scholar]
- Qin X.; Liu H.; Qin D.; Wu Q.; You J.; Zhao D.; Guo Q.; Huang X.; Lan J. Chelation-assisted Rh(III)-catalyzed C2-selective oxidative C–H/C–H cross-coupling of indoles/pyrroles with heteroarenes. Chem. Sci. 2013, 4, 1964–1969. 10.1039/c3sc22241a. [DOI] [Google Scholar]
- Gandeepan P.; Koeller J.; Ackermann L. Expedient C–H chalcogenation of indolines and indoles by positional-selective copper catalysis. ACS Catal. 2017, 7, 1030–1034. 10.1021/acscatal.6b03236. [DOI] [Google Scholar]
- Shi J.; Zhou B.; Yang Y.; Li Y. Rhodium-catalyzed regioselective amidation of indoles with sulfonyl azides via C-H bond activation. Org. Biomol. Chem. 2012, 10, 8953–8955. 10.1039/c2ob26767e. [DOI] [PubMed] [Google Scholar]
- Sun B.; Yoshino T.; Matsunaga S.; Kanai M. Air-stable carbonyl(pentamethylcyclopentadienyl)cobalt diiodide complex as a precursor for cationic (pentamethylcyclopentadienyl)cobalt(III) catalysis: Application for directed C-2 selective C-H amidation of indoles. Adv. Synth. Catal. 2014, 356, 1491–1495. 10.1002/adsc.201301110. [DOI] [Google Scholar]
- Gensch T.; Klauck F. J. R.; Glorius F. Cobalt-catalyzed C–H thiolation through dehydrogenative cross-coupling. Angew. Chem., Int. Ed. 2016, 55, 11287–11291. 10.1002/anie.201605193. [DOI] [PubMed] [Google Scholar]
- Fukuda T.; Maeda R.; Iwao M. Directed C-7 lithiation of 1-(2,2-diethylbutanoyl)indoles. Tetrahedron 1999, 55, 9151–9162. 10.1016/S0040-4020(99)00487-1. [DOI] [Google Scholar]
- Kim Y.; Park J.; Chang S. A direct access to 7-aminoindoles via iridium-catalyzed mild C–H amidation of N-pivaloylindoles with organic azides. Org. Lett. 2016, 18, 1892–1895. 10.1021/acs.orglett.6b00662. [DOI] [PubMed] [Google Scholar]
- Xu L.; Zhang C.; He Y.; Tan L.; Ma D. Rhodium-catalyzed regioselective C7-functionalization of N-pivaloylindoles. Angew. Chem., Int. Ed. 2016, 55, 321–325. 10.1002/anie.201508117. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Qiu X.; Zhao Y.; Mu Y.; Shi Z. Palladium-catalyzed C–H arylation of indoles at the C7 position. J. Am. Chem. Soc. 2016, 138, 495–498. 10.1021/jacs.5b11569. [DOI] [PubMed] [Google Scholar]
- Ryu J.; Kwak J.; Shin K.; Lee D.; Chang S. Ir(III)-Catalyzed mild C–H amidation of arenes and alkenes: An efficient usage of acyl azides as the nitrogen source. J. Am. Chem. Soc. 2013, 135, 12861–12868. 10.1021/ja406383h. [DOI] [PubMed] [Google Scholar]
- Park S. H.; Kwak J.; Shin K.; Ryu J.; Park Y.; Chang S. Mechanistic studies of the rhodium-catalyzed direct C–H amination reaction using azides as the nitrogen source. J. Am. Chem. Soc. 2014, 136, 2492–2502. 10.1021/ja411072a. [DOI] [PubMed] [Google Scholar]
- Park Y.; Park K. T.; Kim J. G.; Chang S. Mechanistic studies on the Rh(III)-mediated amido transfer process leading to robust C–H amination with a new type of amidating reagent. J. Am. Chem. Soc. 2015, 137, 4534–4542. 10.1021/jacs.5b01324. [DOI] [PubMed] [Google Scholar]
- Ackermann L. Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- Davies D. L.; Macgregor S. A.; McMullin C. L. Computational studies of carboxylate-assisted C–H activation and functionalization at group 8–10 transition metal centers. Chem. Rev. 2017, 117, 8649–8709. 10.1021/acs.chemrev.6b00839. [DOI] [PubMed] [Google Scholar]
- Shukla V. K.; Al-Abdullah E. S.; El-Emam A. A.; Sachan A. K.; Pathak S. K.; Kumar A.; Prasad O.; Bishnoi A.; Sinha L. Spectroscopic (FT-IR, FT-Raman, and UV–visible) and quantum chemical studies on molecular geometry, frontier molecular orbitals, NBO, NLO and thermodynamic properties of 1-acetylindole. Spectrochim. Acta, Part A 2014, 133, 626–638. 10.1016/j.saa.2014.06.043. [DOI] [PubMed] [Google Scholar]
- While the electronic effect of the directing groups on the site selectivity cannot be completely ruled out, a univariate regression analysis with sterimol B1 values gave a quantitative correlation. See Figure S11 for details.
- Boutadla Y.; Davies D. L.; Macgregor S. A.; Poblador-Bahamonde A. I. Computational and synthetic studies on the cyclometallation reaction of dimethylbenzylamine with [IrCl2Cp*]2: Role of the chelating base. Dalton Trans. 2009, 5887–5893. 10.1039/b905469c. [DOI] [PubMed] [Google Scholar]
- Walsh A. P.; Jones W. D. Mechanistic insights of a concerted metalation-deprotonation reaction with [Cp*RhCl2]2. Organometallics 2015, 34, 3400–3407. 10.1021/acs.organomet.5b00369. [DOI] [Google Scholar]
- Liu J.-B.; Sheng X.-H.; Sun C.-Z.; Huang F.; Chen D.-Z. A computational mechanistic study of amidation of quinoline N-oxide: The relative stability of amido insertion intermediates determines the regioselectivity. ACS Catal. 2016, 6, 2452–2461. 10.1021/acscatal.5b02938. [DOI] [Google Scholar]
- Zell D.; Bursch M.; Müller V.; Grimme S.; Ackermann L. Full selectivity control in cobalt(III)-catalyzed C–H alkylations by switching of the C–H activation mechanism. Angew. Chem., Int. Ed. 2017, 56, 10378–10382. 10.1002/anie.201704196. [DOI] [PubMed] [Google Scholar]
- For the amidation reactions giving rise to modest product yields, no other detectable side products were formed
- Hammett L. P. Some relations between reaction rates and equilibrium constants. Chem. Rev. 1935, 17, 125–136. 10.1021/cr60056a010. [DOI] [Google Scholar]
- Taft R. W. Linear steric energy relationships. J. Am. Chem. Soc. 1953, 75, 4538–4539. 10.1021/ja01114a044. [DOI] [Google Scholar]
- Charton M. Steric effects, I. Esterification and acid-catalyzed hydrolysis of esters. J. Am. Chem. Soc. 1975, 97, 1552–1556. 10.1021/ja00839a047. [DOI] [Google Scholar]
- Piou T.; Romanov-Michailidis F.; Romanova-Michaelides M.; Jackson K. E.; Semakul N.; Taggart T. D.; Newell B. S.; Rithner C. D.; Paton R. S.; Rovis T. Correlating reactivity and selectivity to cyclopentadienyl ligand properties in Rh(III)-catalyzed C–H activation reactions: An experimental and computational study. J. Am. Chem. Soc. 2017, 139, 1296–1310. 10.1021/jacs.6b11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman M. S.; Harper K. C.; Bess E. N.; Milo A. The development of multidimensional analysis tools for asymmetric catalysis and beyond. Acc. Chem. Res. 2016, 49, 1292–1301. 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]
- Santiago C. B.; Milo A.; Sigman M. S. Developing a modern approach to account for steric effects in Hammett-type correlations. J. Am. Chem. Soc. 2016, 138, 13424–13430. 10.1021/jacs.6b08799. [DOI] [PubMed] [Google Scholar]
- Park Y.; Niemeyer Z. L.; Yu J.-Q.; Sigman M. S. Quantifying structural effects of amino acid ligands in Pd(II)-catalyzed enantioselective C–H functionalization reactions. Organometallics 2018, 37, 203–210. 10.1021/acs.organomet.7b00751. [DOI] [Google Scholar]
- Milo A.; Bess E. N.; Sigman M. S. Interrogating selectivity in catalysis using molecular vibrations. Nature 2014, 507, 210–214. 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- Bess E. N.; DeLuca R. J.; Tindall D. J.; Oderinde M. S.; Roizen J. L.; Du Bois J.; Sigman M. S. Analyzing site selectivity in Rh2(esp)2-catalyzed intermolecular C–H amination reactions. J. Am. Chem. Soc. 2014, 136, 5783–5789. 10.1021/ja5015508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper K. C.; Bess E. N.; Sigman M. S. Multidimensional steric parameters in the analysis of asymmetric catalytic reactions. Nat. Chem. 2012, 4, 366–374. 10.1038/nchem.1297. [DOI] [PubMed] [Google Scholar]
- Niemeyer Z. L.; Milo A.; Hickey D. P.; Sigman M. S. Parameterization of phosphine ligands reveals mechanistic pathways and predicts reaction outcomes. Nat. Chem. 2016, 8, 610–617. 10.1038/nchem.2501. [DOI] [PubMed] [Google Scholar]
- Please see the Supporting Information for full list of parameters.
- Curtin D. Y. Stereochemical control of organic reactions differences in behaviour of diastereoisomers. Rec. Chem. Prog. 1954, 15, 110–128. [Google Scholar]
- The charge of a single carbonyl oxygen also gave a good correlation. Please see Figures S12 and S13 for detailed discussion.
- When parallel KIE experiments were carried out with 1 and 1-d, essentially the same value (1.5) was obtained. See Figure S10 for detailed information.
- See Table S2 for reaction optimization.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.