

Abstract

Tumors are phenotypically heterogeneous and include subpopulations of cancer cells with stemlike properties. The natural product salinomycin, a K+-selective ionophore, was recently found to exert selectivity against such cancer stem cells. This selective effect is thought to be due to inhibition of the Wnt signaling pathway, but the mechanistic basis remains unclear. Here, we develop a functionally competent fluorescent conjugate of salinomycin to investigate the molecular mechanism of this compound. By subcellular imaging, we demonstrate a rapid cellular uptake of the conjugate and accumulation in the endoplasmic reticulum (ER). This localization is connected to induction of Ca2+ release from the ER into the cytosol. Depletion of Ca2+ from the ER induces the unfolded protein response as shown by global mRNA analysis and Western blot analysis of proteins in the pathway. In particular, salinomycin-induced ER Ca2+ depletion up-regulates C/EBP homologous protein (CHOP), which inhibits Wnt signaling by down-regulating β-catenin. The increased cytosolic Ca2+ also activates protein kinase C, which has been shown to inhibit Wnt signaling. These results reveal that salinomycin acts in the ER membrane of breast cancer cells to cause enhanced Ca2+ release into the cytosol, presumably by mediating a counter-flux of K+ ions. The clarified mechanistic picture highlights the importance of ion fluxes in the ER as an entry to inducing phenotypic effects and should facilitate rational development of cancer treatments.
Short abstract
An active fluorescent salinomycin conjugate reveals rapid cellular uptake and strong localization to the endoplasmic reticulum. Ionophore activity in this organelle is connected to phenotype effects.
Introduction
Phenotypic heterogeneity within tumors presents a significant obstacle toward curative cancer treatment.1 Extensive efforts have therefore been directed at new therapies to eliminate subpopulations of cancer cells with stem-cell-like properties, as these are linked to recurrence and metastasis.2,3 Stemlike cancer cells are particularly problematic as they exhibit up-regulated cellular defense mechanisms and are less susceptible to chemotherapy treatment.4
In 2009, the natural product salinomycin was shown by Gupta et al. to have promise in this regard.2 Salinomycin efficiently and selectively reduced the proportion of breast cancer CD44+/CD24– cells, a phenotype associated with enhanced tumorigenic capacity.2,5 Importantly, pretreatment of cancer cells with salinomycin was also shown to reduce tumor-initiating capacity in animal models. Since the original report, salinomycin has shown activity across numerous cancer cell lines of varying origins.6
Mechanistically, effects on several signal transduction pathways have been invoked to explain the change in phenotype composition after salinomycin treatment. Importantly, Lu et al. showed that salinomycin treatment inhibits the Wnt signaling pathway, a pathway involved in tumorigenesis and embryogenesis, through more than one mechanism.7 Other pathways inhibited by salinomycin include K-Ras8 and Hedgehog signaling.9 However, the molecular origin of the phenotypic effects is unclear, and the mechanistic picture is obscured by the diversity of cellular responses to salinomycin treatment, especially when high μM concentrations are used. Demonstrated effects of salinomycin treatment include impaired mitochondrial function, induction of autophagy, decreased ATP levels, increased reactive oxygen species (ROS) production,10−13 and a recent study suggested sequestration of iron in lysosomes.14 In addition, salinomycin treatment has been shown to induce ER (endoplasmic reticulum) stress in different cell lines.15,16 ER stress is a cellular defense mechanism that can force epithelial stem cells17 and stemlike cancer cells to differentiate.18,19 It is known that this response can be induced by depletion of the ER Ca2+ stores,20 and increases in cytosolic Ca2+ have been observed following salinomycin treatment.10,21
Functionally, salinomycin is a K+-selective ionophore, and accumulating evidence points to its ion transport properties as the origin of phenotype effects.22−25 However, the molecular basis of the changes in phenotype composition, i.e., which ion fluxes in the cell are mediated by salinomycin to induce phenotype effects, is unknown. We reasoned that the action of a small molecule ionophore like salinomycin should be connected to the membrane(s) into which it is inserted, and therefore that subcellular imaging of a fluorescent analog would help to elucidate the basis of its activity.
Here, we report a mechanistic investigation of the molecular origin of the phenotype effects of salinomycin guided by subcellular localization of a fluorescent salinomycin conjugate that retains the biological profile of salinomycin. For this purpose, we utilized synthetic methodologies previously developed in our laboratories for synthesis of improved semisynthetic analogs.23 We show that the conjugate exhibits a rapid cellular uptake in breast cancer cells and a pronounced accumulation in the endoplasmic reticulum (ER) and lipid droplets (LDs). Furthermore, the ER is shown to be the intracellular source of the increased cytosolic Ca2+ resulting from salinomycin treatment. Depletion of Ca2+ in the ER lumen causes ER stress and up-regulation of C/EBP homologous protein (CHOP), which is a known Wnt signaling inhibitor.26 In addition, the concomitant increase in cytosolic Ca2+ activates protein kinase C (PKC), which too is an inhibitor of Wnt signaling.27,28 Moreover, the canonical Wnt/LRP6-mediated signaling pathway is impeded by increased Ca2+ levels.29 Importantly, these data provide a connection between the ionophoric activity of salinomycin in the ER and previously described changes in phenotype composition via inhibition of Wnt signaling.30
Results
Synthesis and Cellular Uptake of Fluorescent Salinomycin Conjugates
To visualize cellular uptake and subcellular localization of salinomycin, we sought a fluorescent conjugate that was functionally equivalent to the native structure. Selective ligation of fluorophores to complex natural products like salinomycin without impairing their properties is nontrivial. A fluorescein derivative of okadic acid ligated at the carboxylate position which is vital to activity31 was reported by Sandler et al.,32 but no fluorescent polyether ionophore conjugate shown to retain activity is known. Whitehouse’s nitrobenzoxadiazole (NBD) reporter33 has previously found use for labeling steroids and phospholipids because of its strong fluorescence in hydrophobic environments.34 It has also been used for uptake studies of amino sugars in mammalian cells.35 We reasoned that the comparatively small size, lack of reactivity (bio-orthogonality), and low polarity of the NBD suggested that it might also be a suitable reporter for subcellular imaging of complex polyoxygenated natural products like salinomycin.
We therefore devised a conjugate of salinomycin bearing an NBD reporter ligated at the C20-hydroxyl group (Figure 1). Esterification at this position was shown by Miazaki et al. to enhance the antibiotic activity.36 We recently demonstrated that salinomycin derivatives such as ethyl carbonate 3 with modifications to this position exhibit both increased basal toxicity and enhanced activity against breast cancer stem cells.24,25
Figure 1.

Design and synthesis of the fluorescent salinomycin NBD conjugates 6 and 7. brsm = based on recovered starting material.
Synthetically, the readily available alcohol 5(37) was reacted with phosgene in pyridine and subsequently with ester 4 in a one-pot procedure to give the corresponding carbonate with a complete selectivity for the C20-hydroxyl group. A fluoride-mediated deprotection of the carboxyl group then provided the targeted conjugate 6, which merged the activity-enhancing structural features of ethyl carbonate 3 with the fluorescent NBD moiety. As a negative control, methyl ester 7, which is incapable of electroneutral alkali metal ion transport, was similarly prepared. The stability of the conjugates was corroborated by treatment with a large excess of the nucleophile glutathione in d6-dimethyl sulfoxide at room temperature. No decomposition was observed by 1H NMR spectroscopy under these conditions, even after several days.
Conjugate 6 was found to be functionally equivalent to salinomycin when evaluated in the JIMT-1 breast cancer cell line, with respect to both basal toxicity and phenotype selectivity. An MTT-based assay showed an IC50 of 0.36 ± 0.04 μM (Figure 2A), which is comparable to that of salinomycin (IC50 = 0.52 μM).23 The methyl ester 7 was shown to be over 2 orders of magnitude less active than salinomycin in this assay. Significantly, conjugate 6 was found equally effective as salinomycin in reducing Wnt signaling, when evaluated both in a Wnt reporter cell line (Figure 2B) and in TOPFlash-transfected JIMT-1 cells (Figure 2C). Conjugate 7 was inactive in these assays, and the compounds did not show FOPFlash activity in transfected JIMT-1 cells (Figure S4). Pretreatment of JIMT-1 cells with 6 at IC50 also gave a reduction in colony-forming efficiency (CFE) in serum-free medium by ∼50% compared to control (Figure 2D). At the protein level, treatment of JIMT-1 cells with 6 gave a similar reduction in the levels of active β-catenin, low-density lipoprotein receptor-related protein 6 (LRP6), and phospho-LRP6 (p-LRP6) as salinomycin (Figure 2E).
Figure 2.
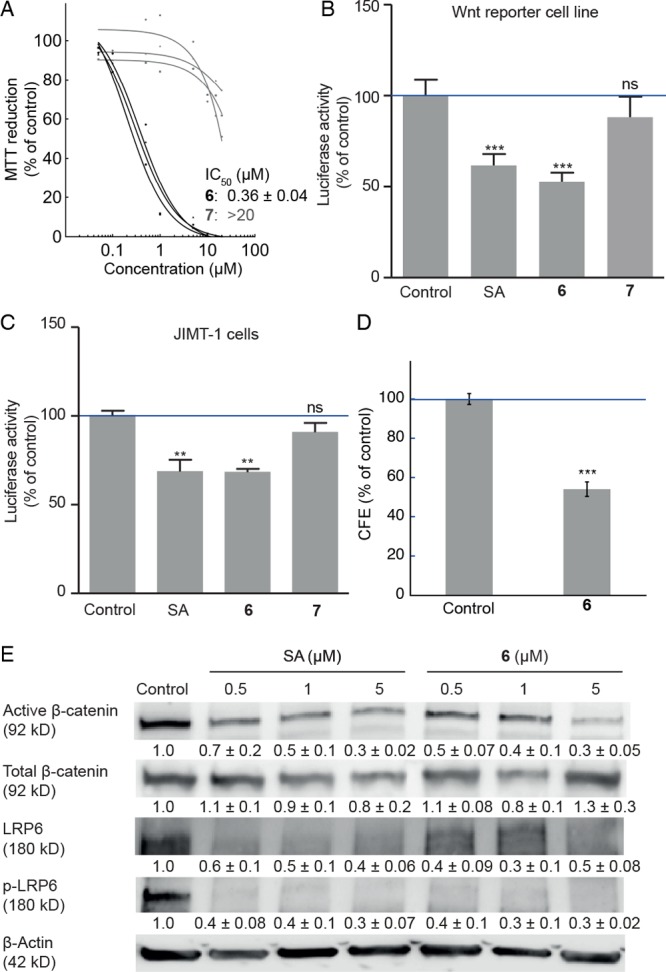
Biological activity of conjugates 6 and 7. (A) Dose response curves and IC50 (mean ± SE, n = 3) of conjugates 6 (black) and 7 (gray) in JIMT-1 cells obtained using an MTT-based assay. (B) Wnt/β-catenin luciferase reporter assay in Leading Light Wnt reporter cells. Columns show mean ± SE (n = 8). DMSO control = 100%. Cells were treated for 24 h. Firefly luciferase levels were normalized to Renilla luciferase levels. (C) Wnt/β-catenin luciferase reporter assay in JIMT-1 cells transfected with TOPFlash and Renilla plasmids. Cells were treated for 24 h. Columns show mean ± SE (n = 3). DMSO control = 100%. Firefly luciferase levels were normalized to Renilla luciferase levels. (D) Colony-forming efficiency (CFE) of JIMT-1 cells following treatment with conjugate 6 at IC50 (0.36 μM) for 72 h. Reported as percentage of colonies formed compared to DMSO control. Columns show mean ± SE (n = 3). (E) Inhibition of Wnt/β-catenin signaling in JIMT-1 cells. Cells were treated with the indicated concentrations for 72 h. Representative Western blots (n ≥ 3) used for densitometric scanning. SA, salinomycin (1). **P < 0.01; ***P < 0.001; ns, not significant.
Conjugate fluorescence was measured with time-resolved fluorescence microscopy imaging using the experimental setup illustrated in Figure 3A.38 The fluorescence properties of N,N-dialkyl NBD’s are reported to be highly sensitive to the local environment;39 however, both conjugates 6 and 7 exhibited quite high fluorescence quantum yield enabling fluorescence imaging of cells containing these molecules. The active conjugate 6 emitted green light in JIMT-1 cells with a λmax of 535 nm. The fluorescence lifetimes of conjugate 6 were similar in different parts of the cell (the bright spots and the less bright regions visible in the images), indicating that fluorescence quantum yields are not very sensitive to differences in cellular environments. In quantitative terms, the fluorescence lifetime was on average 2.1 times longer in cells than in the surrounding buffer solution, and 1.7 times longer in cells than in pure DMSO (Figure 3B, and Figures S1 and S2). In practice this means that the ratio of concentration of 6 in the cell and in buffer is approximately equal to the difference in observed brightness divided by a factor of 2 (the ratio of the lifetimes). Fluorescence can therefore be used to assess the localization of conjugate 6, both within and outside of the cell.
Figure 3.

Cellular uptake and spectroscopic properties of conjugates. (A) Fluorescence microscopy experimental setup. (B) Fluorescence decay of conjugate 6 in various environments: ⟨τ⟩amp: cell = 2.7 ns; buffer = 1.2 ns; DMSO = 1.6 ns. (C) Representative data for real-time accumulation of conjugates 6 (black) and 7 (gray) (20 μM in HBSS with 2% DMSO) in individual JIMT-1 cells as monitored by fluorescence intensity. Fluorescence intensity flashes originating from aggregates of 7 during image capture (seen as gaps in the curve) were removed for clarity. (D) Fluorescence images of representative individual cells recorded during real-time accumulation experiments of conjugate 6 and 7. Scale bar = 20 μm.
The fluorescence microscope also allowed us to monitor the cellular uptake of conjugate 6 and 7 in real time in individual JIMT-1 cells cultured on a microscope coverslip. Prior to addition of compound, an isolated cell covered in buffer was selected for imaging. At time zero, a Hank’s balanced salt solution (HBSS) of conjugate 6 or 7 was added to the buffer outside of the irradiated area, and fluorescence images were continuously recorded with 0.1 s time resolution. Following addition, the conjugate spread rapidly throughout the solution (<1 s), and accumulation in the cell was monitored as an increase in cell fluorescence (Figure 3C; Movie S1, Supporting Information). The time scale of uptake was found to be in the order of tens of seconds, as illustrated by sequences of fluorescence images (Figure 3D and Figure S3). Figure 3C shows how the fluorescence intensity inside and outside the cell is evolving after addition of the conjugates. After approximately 1 min, the fluorescence intensity density in the cells became at least 100 times larger than that in the surrounding buffer indicating strong and rapid accumulation of the conjugates in the cell. This shows a fast passage across the plasma membrane of JIMT-1 cells for both conjugates 6 and 7, and by extension, for salinomycin itself.
Thus, we demonstrated that ligation of an NBD reporter to the C20 hydroxyl of salinomycin gives a fluorescent conjugate that is functionally equivalent to salinomycin and hence suitable for mechanistic investigations. The real-time uptake experiments moreover revealed salinomycin derivatives entering cells on a time scale that supports using such structures for acute cell experiments.
Salinomycin Accumulates in the Endoplasmic Reticulum and in Lipid Droplets
The subcellular localization of salinomycin conjugates in breast cancer cells was investigated using confocal microscopy. Cells treated with a 2 μM solution of conjugate 6 showed a rapid perinuclear accumulation of fluorescence together with the appearance of brightly fluorescent spots (Figure 4A). Costaining with ER-Tracker Red revealed strong colocalization of 6 within the ER in three breast cancer cell lines: JIMT-1, MCF-7, and HCC1937 (Figure 4A and Figure S5A).
Figure 4.

Imaging of fluorescent conjugate 6 in three breast cancer cell lines. Confocal and epifluorescence microscopy was carried out on JIMT-1, MCF-7, and HCC1937 cell lines. (A) Colocalization with ER-Tracker Red. Colocalization shown in orange/yellow. (B) Colocalization with Nile Red in LDs. The cells were incubated with Nile Red and images captured. The Nile Red was then bleached until no fluorescence was observed. Compound 6 was added to the medium of the cells, and images were captured ∼10 min after the imaging of Nile Red. (C, D) Absence of colocalization with Rhodamine 123 in mitochondria. The cells were incubated with Rhodamine 123, and images were captured. The Rhodamine 123 was then bleached until no fluorescence was observed. Compound 6 was added to the medium of the cells, and images were captured ∼10 min after imaging of Rhodamine 123. Panels C and D show maximum intensity projections and single optical planes, respectively. All images in panels A–D were obtained with a laser scanning confocal microscope. Scale bars = 10 μm. (E) DIC and fluorescence images of conjugate 6 in the three breast cancer cell lines were obtained with an epifluorescence microscope. Scale bar = 20 μm.
Imaging of LDs by staining with Nile Red, followed by bleaching and subsequent addition of conjugate 6, revealed a highly similar pattern of bright spots suggesting the presence of 6 also within LDs (Figure 4B and Figure S6). Minor movement of cells and LDs during the course of the experiment explains why the patterns are not fully superimposable. Differential interference contrast (DIC) images of cells treated with conjugate 6 further corroborated the distribution to the ER and LDs (Figure 4E and Figure S9).
The nonactive conjugate 7 showed a similar distribution in cells (Figures S5B and S8).
The effect of salinomycin on cells has generally been associated with interference of mitochondrial function.10,40 Since mitochondria and ER have a similar distribution in cells, we also investigated if salinomycin localized to mitochondria. Imaging of mitochondria in JIMT-1 cells incubated with the mitochondrial stain Rhodamine 123, followed by bleaching and subsequent addition of conjugate 6, revealed markedly different patterns of staining which indicated a low mitochondrial localization (Figure 4C,D and Figure S7).
LDs are lipophilic structures that originate from lipid deposits in the ER phospholipid bilayer and thus share many of its characteristics.41 We interpret the preferential accumulation of the conjugates in the ER and LDs as a reflection of their lipophilic nature. Combined with the observation that only salinomycin derivatives capable of electroneutral alkali metal ion transport induces phenotype effects,42 the localization data indicate that the principal function of salinomycin is ionophore activity in the ER membrane.
Salinomycin Induces ER Ca2+ Release, ER Stress, and PKC Activation
Accumulation of salinomycin in the ER suggests that its ion transport properties may underlie its effect on the cytosolic Ca2+ concentration. The Ca2+ source contributing to the increase in cytosolic Ca2+ caused by salinomycin was thus investigated. JIMT-1 cells were incubated with and without Ca2+ added to the buffer and then treated with 2 μM salinomycin. Significant increases in cytosolic Ca2+ compared to control were found within 10 min in both cases. This implies that the increase originates, at least in part, from intracellular Ca2+ stores (Figure 5A). Elevated levels of cytosolic Ca2+ were found also after 72 h of treatment (Figure 5B). We also investigated the effects on Ca2+ release of a more active analog, ethyl carbonate 3 (Figure 5B). It is noteworthy that carbonate 3 elicited a similar response as salinomycin already at 1/10 of the dose in this assay.
Figure 5.

Salinomycin treatment increases cytosolic Ca2+ and induces ER stress. (A) Acute salinomycin treatment induced Ca2+ release from ER. JIMT-1 cells were labeled with Fluo-4 AM and imaged with confocal microscopy. Salinomycin or salinomycin methyl ester 2 was added at a 2 μM concentration in Ca2+-containing or Ca2+-free medium. Ca2+ release channels in the ER membrane were blocked with 100 μM ryanodine and 50 μM 2-APB before the addition of salinomycin. Data shown are mean ± SE (n = 4). (B) Salinomycin or salinomycin 20-ethyl carbonate 3 treatment increased cytosolic Ca2+. After 72 h of treatment at indicated concentrations, cells were stained with Fluo-3 AM and analyzed with flow cytometry. The relative fluorescence intensity representing the Ca2+ level in the cytosol was calculated. Data shown are mean ± SE (n = 4). (C) Increased PKC activity in cells treated with 0.5 μM salinomycin for 72 h. Data shown are mean ± SE (n ≥ 4). (D) Supervised hierarchical clustering of differentially expressed genes in JIMT-1 cells treated with 50 nM salinomycin or salinomycin 20-ethyl carbonate 3 for 72 h (n = 6). SAM analysis was performed to identify differentially expressed genes between the groups. Genes with q-value ≤1 and an absolute fold change ≥2 were considered to be significantly differentially expressed. Red represents relative up-regulation, and green represents relative down-regulation. (E) Top significantly enriched biological processes up-regulated in carbonate 3-treated cells. Gene ontology (GO) enrichment analysis was performed using the AmiGO database. (F) Activation of the ATF6α pathway by salinomycin or inactive salinomycin methyl ester 2 treatment in JIMT-1 and MCF-7 cells. Cells were treated with the indicated concentrations for 72 h. Representative Western blots (n = 4) used for densitometric scanning showing the expression of UPR-related proteins. SA, salinomycin (1). (G) CHOP-siRNA-treated JIMT-1 cells reduce the level of active β-catenin upon salinomycin treatment. Cells were transfected with CHOP-siRNA and scramble siRNA for 72 h, followed by treatment with salinomycin (5 μM) for 72 h. Representative Western blots (n ≥ 4) used for densitometric scanning showing the expression of active β-catenin and CHOP. SA, salinomycin (1). *P < 0.05; **P < 0.01; ***P < 0.001.
For verification of whether the ER was a significant contributor to the increase in cytosolic Ca2+, the ER Ca2+ release channels, inositol trisphosphate receptor (IP3R) and ryanodine receptor (RyR), were simultaneously inhibited by a combination of 2-aminoethoxydiphenyl borate (2-APB) and ryanodine, respectively. In this experiment, no increase in cytosolic Ca2+ compared to control was seen after treatment with salinomycin which supports that the ER is indeed the primary intracellular source of released Ca2+ (Figure 5A). As expected, methyl ester 2 had no effect on cytosolic Ca2+ concentration in JIMT-1 cells (Figure 5A).
Treatment with ER Ca2+ channel blockers prior to salinomycin treatment gave small changes in the cytosolic Ca2+ level. This suggests that salinomycin-mediated Ca2+ release from other intracellular stores such as mitochondria was small. In agreement with this, no increase in cytosolic Ca2+ was found following salinomycin treatment after depletion of ER calcium by thapsigargin (Figure S10). This suggests that the mitochondrial effects following treatment with salinomycin are secondary effects to increased cytosolic Ca2+. This causes uptake of Ca2+ into the mitochondria with decreased mitochondrial membrane potential and reduced ATP production as a result.
For a broader view of the changes in gene activity following salinomycin treatment, a global mRNA analysis of JIMT-1 cells after treatment with 50 nM of salinomycin or ethyl carbonate 3 was performed. In all, 273 differentially expressed genes were found (Data Set S1). Strikingly, there were a high number of hits with low P-values for genes in pathways involved in the unfolded protein response (UPR) (Figure 5D,E; Data Set S2). In particular, the 78 kDa glucose-regulated protein (GRP78) was significantly up-regulated at the mRNA level (Figure 5E). Increased expression of this protein was also confirmed in both JIMT-1 and MCF-7 cells by Western blot analysis (Figure 5F). Increased GRP78 is important for maintaining cell viability against several kinds of stress including depletion of Ca2+ from the ER.20,43
The gene DNA damage inducible transcript 3 (DDIT3) encoding for CHOP was also significantly up-regulated at the mRNA level in cells treated with carbonate 3 (Figure 5E). CHOP is a multifunctional transcription factor in the ER-stress response and is known to inhibit the Wnt/β-catenin signaling pathway.26 At the protein level, only a weak activation of CHOP was seen; however, CHOP is most efficiently increased when both the protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6) pathways are activated.44 We found activation of ATF6 (Figure 5F) in treated cells but not of PERK or inositol-requiring enzyme (IRE1) (Figure S11), which may explain the small increase in CHOP. These three ER-stress sensors are not always activated in consort during ER stress, and different conditions are known to result in differing patterns of activation.45
As shown in Figure 2C, salinomycin treatment decreased the expression of active β-catenin, but the total β-catenin level was not significantly changed. In cells where CHOP was knocked down by transfection with CHOP-siRNA, salinomycin treatment gave a lower reduction of active β-catenin compared to cells transfected with scrambled siRNA (Figure 5G). These results support that up-regulation of CHOP following salinomycin treatment contributes to inhibition of the Wnt signaling pathway.
The increase in cytosolic Ca2+ is also important for a description of the mechanism of salinomycin. Ca2+ is a vital second messenger that activates conventional (calcium-dependent) PKCs46 to further inhibit the canonical Wnt/β-catenin pathway.28 In line with this, we found that treatment of JIMT-1 cells with salinomycin promotes a significant activation of PKC (Figure 5C).
Combined, the data show that salinomycin causes an increase in the release of Ca2+ from the ER. This release ultimately results in an increase in CHOP expression and activation of calcium-dependent PKC, both known factors contributing to inhibition of Wnt signaling.
Discussion
Salinomycin treatment is known to reduce the proportion of stemlike cancer cells in a cancer cell population via inhibition of the Wnt signaling pathway, but the molecular basis of this effect is unclear. The work described here reveals Ca2+ release from the ER into the cytosol as the molecular initiating event. Our interpretation of the mode of action in the ER is that it derives from a salinomycin-mediated net influx of K+ from the cytosol to the ER lumen (Figure 6). Both release and uptake of Ca2+ into the ER is contingent on a counter-flux of K+ to retain charge-neutrality.47 A K+-selective ionophore like salinomycin, localized in the ER membrane and acting as a passive potassium–hydrogen exchanger, can thus in principle facilitate both uptake and release of Ca2+.
Figure 6.

Schematic representation of the proposed mechanism for Wnt/β-catenin inhibition by salinomycin.
In accordance, nigericin, which is a functionally and structurally related K+-selective polyether ionophore, has previously been shown to both reverse apamin blockage of IP3-mediated Ca2+ release from the ER47 and attenuate propranolol inhibition of Ca2+ reuptake.48,49 Ionophore-mediated K+ flux into the ER also connects to Ca2+ release by causing decomplexation of protein-bound Ca2+ in the ER via K+/Ca2+ exchange. This accentuates Ca2+ release, since increased ER Ca2+ levels are the trigger of Ca2+-induced Ca2+ release.47,50 We thus attribute the increase in cytosolic Ca2+ to a promoted release of Ca2+ from the ER, rather than to an impeded reuptake.
In summary, we developed a fluorescent NBD conjugate of salinomycin that retains the activity profile of the native structure. This conjugate was used to guide a mechanistic investigation of the molecular basis for the activity of salinomycin against stemlike cancer cells. The conjugate was shown to rapidly enter breast cancer cells and localize in the ER and LDs. Uptake of salinomycin into the ER was then shown to result in an enhanced Ca2+ release from this organelle, presumably a result of a counter transport of K+ by salinomycin. Depletion of Ca2+ from the ER led to ER stress and activation of the UPR, which induced up-regulation of CHOP. The concomitant increase in cytosolic Ca2+ caused activation of conventional PKCs. Since both up-regulation of CHOP and activation of PKC inhibit the Wnt signaling pathway, our work connects the mechanism of salinomycin at the molecular level to previously described phenotype effects.
Finally, we anticipate that the simplicity of selective carbonate ligation of NBD fluorophores paired with the favorable chemical, biological, and photophysical properties of such conjugates should find wider use in imaging cellular uptake and mechanistic studies, in the context of both natural products and other complex molecules. In a broader sense, the results of this mechanistic study emphasize the potential importance of K+ flux across the ER membrane as a prospective mechanism to induce and study phenotype effects in cancer cells and possibly also in other cell types.
Acknowledgments
We thank the Swedish Cancer Foundation, Crafoord foundation, the Royal Academy of Sciences, the Royal Physiographical Society, the Percy Falk Foundation, the Mrs Berta Kamprad Foundation, and NanoLund. We thank Ewa Dahlberg and Sebastian Kempengren for expert technical help.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00257.
Synthetic procedures and characterization data for all new compounds and protocols for real-time uptake assay, cell culturing, dose response assay, luciferase reporter assays, colony-forming efficiency assay, subcellular localization, Ca2+ assays, gene expression, RNA interference, Western blot, PKC assay, and statistical analysis (PDF)
Data Set S1: differentially expressed genes between groups identified using SAM analysis (XLSX)
Data Set S2: enriched biological processes up-regulated by treatment with carbonate 3 (XLSX)
Movie S1: representative examples of cellular uptake of conjugates 6 and 7 (AVI)
Author Contributions
○ X.H. and B.B. contributed equally to this study.
The authors declare no competing financial interest.
Supplementary Material
References
- Visvader J. E.; Lindeman G. J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Gupta P. B.; Onder T. T.; Jiang G.; Tao K.; Kuperwasser C.; Weinberg R. A.; Lander E. S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R.; Ozsvari B.; Lisanti C. L.; Tanowitz H. B.; Howell A.; Martinez-Outschboorn U. E.; Sotgia F.; Lisanti M. P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. 10.18632/oncotarget.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Al-Hajj M.; Wicha M. S.; Benito-Hernandez A.; Morrison S. J.; Clarke M. F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3983–3988. 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokat C.; Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol. 2012, 2012, 604076. 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D.; Choi M. Y.; Yu J.; Castro J. E.; Kipps T. J.; Carson D. A. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13253–13257. 10.1073/pnas.1110431108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najumudeen A. K.; Jaiswal A.; Lectez B.; Oetken-Lindholm C.; Guzmán C.; Siljamäki E.; Posada I. M.; Lacey E.; Aittokallo T.; Abankwa D. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene 2016, 35, 5248–5262. 10.1038/onc.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Ma W.; Mao J.; Yu X.; Hou Z.; Fan S.; Song B.; Wang H.; Li J.; Kang L.; Liu P.; Liu Q.; Li L. Salinomycin exerts anticancer effects on human breast carcinoma MCF-7 cancer stem cells via modulation of Hedgehog signaling. Chem.-Biol. Interact. 2015, 228, 100–107. 10.1016/j.cbi.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Jangamreddy J. R.; Ghavami S.; Grabarek J.; Kratz G.; Wiechec E.; Fredriksson B. A.; Rao Pariti R. K.; Cieślar-Pobuda A.; Panigrahi A.; Łos M. J. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: Differences between primary and cancer cells. Biochim. Biophys. Acta, Mol. Cell Res. 2013, 1833, 2057–2069. 10.1016/j.bbamcr.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Choi Y. J.; Kim K. Y.; Yu S. N.; Seo Y. K.; Chun S. S.; Noh K. T.; Suh J. T.; Suh J. T.; Ahn S. C. Salinomycin simultaneously induces apoptosis and autophagy through generation of reactive oxygen species in osteosarcoma U2OS cells. Biochem. Biophys. Res. Commun. 2016, 473, 607–613. 10.1016/j.bbrc.2016.03.132. [DOI] [PubMed] [Google Scholar]
- Boehmerle W.; Endres M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. 10.1038/cddis.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoodt B.; Voght M.; Schmitz I.; Liffers S. T.; Tannapfel A.; Mirmohammadsadegh A. Salinomycin induces autophagy in colon and breast cancer cells with concomitant generation of reactive oxygen species. PLoS One 2012, 7, e44132. 10.1371/journal.pone.0044132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai T. T.; Hamaï A.; Hienzsch A.; Cañeque S.; Müller S.; Wicinski J.; Cabaud O.; Leroy C.; David A.; Acevedo V.; Ryo A.; Ginestier C.; Birnbaum D.; Charafe-Jauffret E.; Codogno P.; Mehrpour M.; Rodriguez R. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. 10.1038/nchem.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Su L.; Zhong N.; Hao X.; Zhong D.; Singhal S.; Liu X. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy 2013, 9, 1057–1068. 10.4161/auto.24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.-Y.; Seo Y.-K.; Yu S.-N.; Kim S.-H.; Suh P.-G.; Ji J.-H.; Yu H.-S.; Park Y.-M.; Ahn S.-C. Gene expression profiling from a prostate cancer PC-3 cell line treated with salinomycin predicts cell cycle arrest and endoplasmic reticulum stress. J. Cancer Sci. Ther. 2013, 5, 023–030. 10.4172/1948-5956.1000180. [DOI] [Google Scholar]
- Heijmans J.; van Lidth de Jeude J. F.; Koo B. K.; Rosekrans S. L.; Wielenga M. C.; van de Wetering M.; Ferrante M.; Lee A. S.; Onderwater J. J.; Paton J. C.; Paton A. W.; Mommaas A. M.; Kodach L. L.; Hardwick J. C.; Hommes D. W.; Clevers H.; Muncan V.; van den Brink G. R. ER Stress Causes Rapid Loss of Intestinal Epithelial Stemness through Activation of the Unfolded Protein Response. Cell Rep. 2013, 3, 1128–1139. 10.1016/j.celrep.2013.02.031. [DOI] [PubMed] [Google Scholar]
- Wielenga M. C. B.; Colak S.; Heijmans J.; van Lidth de Jeude J. F.; Rodermond H. M.; Paton J. C.; Paton A. W.; Vermeulen L.; Medema J. P.; van den Brink G. R. ER-Stress-Induced differentiation sensitizes colon cancer stem cells to chemotherapy. Cell Rep. 2015, 13, 489–494. 10.1016/j.celrep.2015.09.016. [DOI] [PubMed] [Google Scholar]
- Feng Y. X.; Sokol E. S.; Del Vecchio C. A.; Sanduja S.; Claessen J. H.; Proia T. A.; Jin D. X.; Reinhardt F.; Ploegh H. L.; Wang Q.; Gupta P. B. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discovery 2014, 4, 702–715. 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- Mekahli D.; Bultynck G.; Parys J. B.; de Smedt H.; Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harbor Perspect. Biol. 2011, 3, a004317. 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; He L.; Dai W. Q.; Xu Y. P.; Wu D.; Lin C. L.; Wu S. M.; Cheng P.; Zhang Y.; Shen M.; Wang C. F.; Lu J.; Zhou Y. Q.; Xu X. F.; Xu L.; Guo C. Y. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS One 2012, 7, e50638. 10.1371/journal.pone.0050638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huczyński A. Polyether ionophores - Promising bioactive molecules for cancer therapy. Bioorg. Med. Chem. Lett. 2012, 22, 7002–7010. 10.1016/j.bmcl.2012.09.046. [DOI] [PubMed] [Google Scholar]
- Borgström B.; Huang X.; Pošta M.; Hegardt C.; Oredsson S.; Strand D. Synthetic modification of salinomycin: selective O-acylation and biological evaluation. Chem. Commun. 2013, 49, 9944–9946. 10.1039/c3cc45983g. [DOI] [PubMed] [Google Scholar]
- Huang X.; Borgström B.; Kempengren S.; Persson L.; Hegardt C.; Strand D.; Oredsson S. Breast Cancer Stem Cell Selectivity of Synthetic Nanomolar-Active Salinomycin Analogs. BMC Cancer 2016, 16, 145. 10.1186/s12885-016-2142-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgström B.; Huang X.; Hegardt C.; Oredsson S.; Strand D. Structure–Activity Relationships in Salinomycin: Cytotoxicity and Phenotype Selectivity of Semi-synthetic Derivatives. Chem. - Eur. J. 2017, 23, 2077–2083. 10.1002/chem.201603621. [DOI] [PubMed] [Google Scholar]
- Horndasch M.; Lienkamp S.; Springer E.; Schmitt A.; Pavenstädt H.; Walz G.; Gloy J. The C/EBP homologous protein CHOP (GADD153) is an inhibitor of Wnt/TCF signals. Oncogene 2006, 25, 3397–3407. 10.1038/sj.onc.1209380. [DOI] [PubMed] [Google Scholar]
- Luna-Ulloa L. B.; Hernández-Maqueda J. G.; Castañeda-Patlán M. C.; Robles-Flores M. Protein kinase C in Wnt signaling: Implications in cancer initiation and progression. IUBMB Life 2011, 63, 915–921. 10.1002/iub.559. [DOI] [PubMed] [Google Scholar]
- Gwak J.; Cho M.; Gong S. J.; Won J.; Kim D. E.; Kim E. Y.; Lee S. S.; Kim M.; Kim T. K.; Shin J. G.; Oh S. Protein-kinase-C-mediated beta-catenin phosphorylation negatively regulates the Wnt/beta-catenin pathway. J. Cell Sci. 2006, 119, 4702–4709. 10.1242/jcs.03256. [DOI] [PubMed] [Google Scholar]
- Lu D.; Carson D. A. Spiperone enhances intracellular calcium level and inhibits the Wnt signaling pathway. BMC Pharmacol. 2009, 9, 13. 10.1186/1471-2210-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang G. B.; Kim J. Y.; Cho S. D.; Park K. S.; Jung J. Y.; Lee H. Y.; Hong I. S.; Nam J. S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. 10.1038/srep12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwaki S.; Fujiki H.; Suganuma M.; Furuya-Suguri H.; Matsushima R.; Iida Y.; Ojika M.; Yamada K.; Uemura D.; Yasumoto T.; Schmitz F. J.; Sugimura T. Structure-activity relationship within a series of okadaic acid derivatives. Carcinogenesis 1990, 11, 1837–1841. 10.1093/carcin/11.10.1837. [DOI] [PubMed] [Google Scholar]
- Sandler J. S.; Fenical W.; Gulledge B. M.; Chamberlin A. R.; La Clair J. J. Fluorescent profiling of natural product producers. J. Am. Chem. Soc. 2005, 127, 9320–9321. 10.1021/ja042756u. [DOI] [PubMed] [Google Scholar]
- Ghosh P. B.; Whitehouse M. W. 7-Chloro-4-nitrobenzo-2-oxa-1,3-diazole: a new fluorigenic reagent for amino acids and other amines. Biochem. J. 1968, 108, 155–156. 10.1042/bj1080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay A. Chemistry and biology of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: fluorescent probes of biological and model membranes. Chem. Phys. Lipids 1990, 53, 1–15. 10.1016/0009-3084(90)90128-E. [DOI] [PubMed] [Google Scholar]
- Yamada K.; Nakata M.; Horimoto N.; Saito M.; Matsuoka H.; Inagaki N. Measurement of glucose uptake and intracellular calcium concentration in single, living pancreatic β-cells. J. Biol. Chem. 2000, 275, 22278–22283. 10.1074/jbc.M908048199. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y.; Kinashi H.; Otake N.; Mitani M.; Yamanishi T. Chemical modification and structure-activity correlation of salinomycin. Agric. Biol. Chem. 1976, 40, 1633–1640. 10.1080/00021369.1976.10862252. [DOI] [Google Scholar]
- Kupryushkin M. S.; Konevetz D. A.; Vasilyeva S. V.; Kuznetsova A. S.; Stetsenko D. A.; Pyshnyi D. V. Oligonucleotide Functionalization by a Novel Alkyne-Modified Nonnucleosidic Reagent Obtained by Versatile Building Block Chemistry. Nucleosides, Nucleotides Nucleic Acids 2013, 32, 306–319. 10.1080/15257770.2013.787147. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Kuzimenkova M. V.; Xie M.; Meyer M.; Larsson P.-O.; Scheblykin I. G. Watching two conjugated polymer chains breaking each other when colliding in solution. NPG Asia Mater. 2014, 6, e134. 10.1038/am.2014.91. [DOI] [Google Scholar]
- Fery-Forgues S.; Fayet J. P.; Lopez A. Drastic changes in the fluorescence properties of NBD probes with the polarity of the medium: involvement of a TICT state?. J. Photochem. Photobiol., A 1993, 70, 229–243. 10.1016/1010-6030(93)85048-D. [DOI] [Google Scholar]
- Managò A.; Leanza L.; Carraretto L.; Sassi N.; Grancara S.; Quintana-Cabrera R.; Trimarco V.; Toninello A.; Scorrano L.; Trentin L.; Semenzato G.; Gulbins E.; Zoratti M.; Szabò I. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 2015, 6, e1930. 10.1038/cddis.2015.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q.; Goodman J. M. The lipid droplet—a well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 1–12. 10.3389/fcell.2015.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgström B.; Huang X.; Chygorin E.; Oredsson S.; Strand D. Salinomycin Hydroxamic Acids: Synthesis, Structure, and Biological Activity of Polyether Ionophore Hybrids. ACS Med. Chem. Lett. 2016, 7, 635–640. 10.1021/acsmedchemlett.6b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake H.; Hara I.; Arakawa S.; Kamidono S. Stress protein GRP78 prevents apoptosis induced by calcium ionophore, ionomycin, but not by glycosylation inhibitor, tunicamycin, in human prostate cancer cells. J. Cell. Biochem. 2000, 77, 396–408. . [DOI] [PubMed] [Google Scholar]
- Okada T.; Yoshida H.; Akazawa R.; Negishi M.; Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. 10.1042/bj20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuolo J.; Bulotta S.; Verderio C.; Benfante R.; Borgese N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7832–7837. 10.1073/pnas.1101379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg G.; Benedetti L. G.; Abera M. B.; Wang H.; Abba M.; Kazanietz M. G. Protein kinase C and cancer: what we know and what we do not. Oncogene 2014, 33, 5225–5237. 10.1038/onc.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.; Chin W. C.; Verdugo P. Role of Ca2+/K+ ion exchange in intracellular storage and release of Ca2+. Nature 1998, 395, 908–912. 10.1038/27686. [DOI] [PubMed] [Google Scholar]
- Kuum M.; Veksler V.; Kaasik A. Potassium fluxes across the endoplasmic reticulum and their role in endoplasmic reticulum calcium homeostasis. Cell Calcium 2015, 58, 79–85. 10.1016/j.ceca.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Kuum M.; Veksler V.; Liiv J.; Ventura-Clapier R.; Kaasik A. Endoplasmic reticulum potassium-hydrogen exchanger and small conductance calcium-activated potassium channel activities are essential for ER calcium uptake in neurons and cardiomyocytes. J. Cell Sci. 2012, 125, 625–633. 10.1242/jcs.090126. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A.; Shmigol A. Calcium-induced calcium release in neurones. Cell Calcium 1996, 19, 1–14. 10.1016/S0143-4160(96)90009-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.