

Abstract
Cardiac arrest (CA) is sudden loss of heart function and abrupt stop in effective blood flow to the body. The patients who initially achieve return of spontaneous circulation (RoSC) after CA have low survival rate. It has been known that multiorgan dysfunctions after RoSC are associated with high morbidity and mortality. Most previous studies have focused on the heart and brain in RoSC after CA. Therefore, the aim of this research was to perform serological, physiological, and histopathology study in the lung and to determine whether or how pulmonary dysfunction is associated with low survival rate after CA. Experimental animals were divided into sham-operated group (n=14 at each point in time), which was not subjected to CA operation, and CA-operated group (n=14 at each point in time), which was subjected to CA. The rats in each group were sacrificed at 6 hours, 12 hours, 24 hours, and 2 days, respectively, after RoSC. Then, pathological changes of the lungs were analyzed by hematoxylin and eosin staining, Western blot and immunohistochemistry for tumor necrosis factor α (TNF-α). The survival rate after CA was decreased with time past. We found that histopathological score and TNF-α immunoreactivity were significantly increased in the lung after CA. These results indicate that inflammation triggered by ischemia-reperfusion damage after CA leads to pulmonary injury/dysfunctions and contributes to low survival rate. In addition, the finding of increase in TNF-α via inflammation in the lung after CA would be able to utilize therapeutic or diagnostic measures in the future.
Keywords: Asphyxial cardiac arrest, Post-cardiac arrest syndrome, Lung, Tumor necrosis factor α
Introduction
Cardiac arrest (CA) leads to sudden loss of heart function and causes abrupt stop in effective blood flow to the body. In patients who achieve return of spontaneous circulation (RoSC) after CA, morbidity and mortality still remains significantly high because of the development of post-cardiac arrest syndrome (PCAS) [1,2]. In PCAS, multi-organ dysfunctions are involved in and associated with low survival rate. Survival rate in patients at the early period after CA ranges from 4% to 33% [3]. The low survival rate after CA is attributed to a unique pathophysiological process called PCAS [1]. PCAS includes all clinical and biological manifestations such as brain injury, myocardial dysfunction, and persistently precipitating pathology [3]. The pathophysiological process of PCAS could be the results of a combination of whole-body ischemia-reperfusion (IR)–mediated damage and nonspecific activation of systemic inflammatory response triggered by CA and RoSC [4].
Inflammatory response usually occurs due to infection. However, “sterile inflammation” occurs in cases of chemically induced injury, trauma, or ischemia [2]. Systemic IR injury after RoSC triggers the release of inflammatory cytokines and eventually leads to systemic inflammatory response syndrome even in the absence of infection [5]. Ischemia and subsequent reperfusion in multiple organs are unavoidable during CA and RoSC, which commonly results in acute and sterile systemic inflammation, and IR activates and infiltrates innate immune cells [2,6]. Increased pro-inflammatory cytokines have been reported following resuscitation [7]. The inflammatory response after RoSC is characterized by activation of polymorphonuclear leukocytes, expressions of adhesion molecules, production of reactive oxygen species from inducible nitric oxide synthase, and release of cytokines, such as interleukin-6 and tumor necrosis factor α (TNF-α) [8]. TNF-α increases shortly in myocardium after RoSC and is predictive of early death, especially, plasma levels of TNF-α have been shown to be inversely correlated with myocardial function after CA [9]. In addition, cytokines including TNF-α are secreted by alveolar macrophages and amplify the response of endothelial and epithelial cells in the lung [2,6,10]. However, the role of TNF-α in other organs is still unclear after CA.
Recently, Roberts et al. [11] have reported that multiple organ dysfunctions are common in PCAS after RoSC, which is associated with survival rate. However, many studies after RoSC have focused on injuries and dysfunctions of the myocardium (heart) and brain [12,13]. Failures in other vital organs including the lung, liver, and kidney have been ignored [1,11,13]. We had perceived a paucity of serological and histopathological studies regarding pulmonary damage and dysfunction in RoSC. The relationship between survival rate and multiple organ dysfunctions in PCAS remains unclear. Therefore, we aimed to perform serological, physiological, and histopathology study in the lung. Furthermore, we hypothesized that IR following CA causes inflammation response in the lung, which leads to pulmonary dysfunction and alteration of pro-inflammatory factors. To assess these, we designed to induce asphyxial CA model in rats and observed survival rate during post-resuscitation phase. Moreover, we studied the dysfunction of lung by examining histopathological change during the early period after RoSC. In addition, we investigated alterations in TNF-α, as a pro-inflammatory cytokine, by immunohistochemistry as well as western analysis.
Materials and Methods
Experimental animals and groups
Male Sprague-Dawley rats were obtained from the Experimental Animal Center of Kangwon National University (Chuncheon, South Korea). All experimental protocols were approved based on ethical procedures and scientific care by the Kangwon National University-Institutional Animal Care and Use Committee (approval no. KW-151127-1). All subjects gave consent. Animals were divided into (1) sham-operated group (n=14 at each point in time), which was not subjected to CA operation, and (2) CA-operated group (n=14 at each point in time), which was subjected to CA. The rats in each group were sacrificed at 6 hours, 12 hours, 24 hours, and 2 days, respectively, after RoSC.
CA induction and cardiopulmonary resuscitation
CA and cardiopulmonary resuscitation (CPR) were performed using published procedures [14,15] with minor modification. Briefly, rats were anesthetized with 2%–3% isoflurane and mechanically ventilated to maintain respiration using a rodent ventilator (Harvard Apparatus, Holliston, MA, USA). To monitor peripheral oxygen saturation (SpO2), an oxygen saturation probe of pulse oximetry (Nonin Medical Inc., Plymouth, MN, USA) was attached to the left foot. Body temperature was maintained at 37±0.5℃ during and after the CA surgery. For electrocardiogram (ECG), electrocardiographic probes (GE Healthcare, Milwaukee, WI, USA) were placed in limbs and data were monitored continuously. The left femoral artery and right femoral vein were separately cannulated to monitor mean arterial pressure (MAP) (MLT 1050/D, AD Instruments, Bella Vista, Austria) and intravenous injection.
Vecuronium bromide (2 mg/kg, Gensia Sicor Pharmaceuticals, Irvine, CA, USA) was intravenously administered after 5 minutes of stabilization period, anesthesia was stopped, and mechanical ventilation was stopped. MAP below 25 mm Hg and subsequent pulseless electric activity were used to define CA [1,2]. CA was confirmed at 3–4 minutes after vecuronium bromide injection. At 5 minutes after CA, CPR was initiated by intravenously administering a bolus injection of epinephrine (0.005 mg/kg) and sodium bicarbonate (1 mEq/kg) followed by mechanical ventilation with 100% oxygen and mechanical chest compression at a rate of 300/min until MAP reached 60 mm Hg and electrocardiographic activity was observed. Once the animals were hemodynamically stable and spontaneously breathing (usually at 1 hour after RoSC), they were extubated at 2 hours after resuscitation and monitored for outcome evaluation.
Tissue processing
According to general method, the rats were anesthetized with sodium pentobarbital (30 mg/kg, intraperitoneal administration) and perfused transcardially with 4% paraformaldehyde. The lung tissues were isolated, cut, embedded in paraffin, and sectioned (6 µm).
Hematoxylin and eosin staining
Hematoxylin and eosin (H&E) staining was done to examine histopathological changes according to general protocol. Shortly, sections of 6 µm were prepared and stained with H&E, dehydrated by a serial of ethanol and mounted with Canada balsam (Kanto Chemical, Tokyo, Japan). Histopathological analysis for lesion of the lung was semi-quantitatively assessed by a published procedure [16]. In brief, lung injury was scored according to (1) thickness of the alveolar wall; (2) infiltration or aggregation of neutrophils in airspace, the alveolar wall, or the vessel wall; and (3) alveolar congestion, and each item was graded on a four-point scale. Each component ranged from 0 to 3, with higher scores indicating more severe damage.
Western analysis
To examine change in level of TNF-α protein in the lung after CA, western blot analysis (n=7 per group) was performed according to our published method [17]. In short, the lung tissues were homogenized and centrifuged. The protein level in the supernatants was determined using a Micro BCA protein assay kit with bovine serum albumin as a standard (Pierce Chemical, Rockford, IL, USA). The membranes were incubated with 5% non-fat dry milk to reduce background staining and followed by incubation with rabbit anti–TNF-α (diluted 1:500, Abcam, Cambridge, UK), peroxidase conjugated goat anti-rabbit IgG (Sigma-Aldrich, St. Louis, MO, USA) and an enhanced chemiluminescence kit (Pierce Biotechnology). The result of the western blot analysis was scanned, and densitometric analysis for the quantification of the bands was done using Scion Image software (Scion Corp., Frederick, MD, USA), which was used to count relative optical density (ROD). A ratio of the ROD was calibrated as %, with vehiclesham group designated as 100%.
Immunohistochemistry
TNF-α immunohistochemistry was carried out according to our published procedure [17], the sections were incubated with a rabbit anti–TNF-α (diluted 1:500, Abcam), followed with secondary antibody (Vector Laboratories Inc., Burlingame, CA, USA), developed using Vectastain ABC (Vector Laboratories Inc.), and visualized with 3,3′-diaminobenzidine. To quantitatively analyze TNF-α immunoreactivity, the staining intensity of TNF-α–immunoreactive structures was evaluated on the basis of an optical density (OD), which was obtained after the transformation of the mean gray level using the formula: OD=log(256/mean gray level). The OD of background was taken from areas adjacent to the measured area. After the background density was subtracted, a ration of the OD of an image file was calibrated in Adobe Photoshop 8.0 and then analyzed as a percent, with sham-operated-group designated as 100% in NIH Image 1.59.
Statistical analysis
All data were entered into SAS version 9.02 (SAS Institute Inc., Cary, NC, USA). All data were presented as means±standard error of mean. Survival was analyzed using Kaplan-Meier statistic and log-rank test. MAP and peripheral oxygen were compared using one and two-way repeated measures of ANOVA to assess the effect of time. To determine the significance of differences, post-hoc analyses were conducted using Tukey test for all pairwise multiple comparisons. To conduct semiquantitative analysis for histopathology and TNF-α immunoreactivity, differences were considered significant when P-value was less than 0.05.
Results
Physiological variables
There were no significant (P>0.05) differences between the sham operated and CA operated group for baseline characteristics (Table 1). CA was confirmed with isoeletric ECG, SpO2, and MAP. ECG, SpO2, and MAP were changed as expected according to the experimental protocol. Body weight was not changed, and body temperature and heart rate were the same as that at the baseline or after RoSC. The survival rate after CA was 100% at 6 hours after RoSC; the rate was decreased with time past about 64% at 12 hours, 36% at 1 day, and 7% at 2 days after RoSC.
Table 1. Physiologic variables and survival rate after cardiac arrest.
| Baseline | Cardiac arrest | 6-Hour post-CA | 12-Hour post-CA | 1-Day post-CA | 2-Day post-CA | |
|---|---|---|---|---|---|---|
| Body weight (g) | 296.4±22 | - | 293.2±37 | 296.7±38 | 295.2±52 | 295.8±38 |
| Temperature (°C) | 36.7±0.2 | 34.7±0.7 | 36.6±0.4 | 36.7±0.6 | 36.7±0.3 | 37.2±0.1 |
| Asphyxial time to CA (s) | - | - | 186±20 | 192±32 | 188±48 | 189±33 |
| CPR time (min) | - | - | 1.4±0.3 | 1.5±0.7 | 1.6±0.3 | 1.5±0.4 |
| Mean arterial pressure (mm Hg) | 118±12 | - | 109±12 | 113±34 | 114±16 | 117±11 |
| Heart rate (beat/min) | 333±11 | - | 358±43 | 347±45 | 328±56 | 340±56 |
| Survival rate (%) | - | - | 100 | 64.3 | 35.7 | 0 |
CA, cardiac arrest; CPR, cardiopulmonary resuscitation.
Histopathological change
In the CA operated group, histopathology in the lung was significantly increased in a time-dependent manner after CA (P<0.05) (Fig. 1). Damage of the alveolar structure was significantly increased at 12-hour post-CA. The damage was more increased at 1 day after CA and maintained until 2-day post-CA. The histopathological changes involved alveolar septa thickness and infiltration of a number of inflammatory cells. The alveoli were filled with pigment-laden macrophages and red blood cells. The histopathological changes also included bronchointerstitial pneumonia and small caliber bronchioles filled with inflammatory exudates surrounded by zones of interstitial pneumonia. In addition, bronchioles and alveolar spaces contained lymphocytes, plasma cells and neutrophils.
Fig. 1. H&E staining of the lung of the sham (A) and cardiac arrest (CA) operation (B–E) groups. In the CA operated lungs, histopathology is markedly increased at 12-hour post-CA (P<0.05). The histopathological change is more increased at 1- and 2-day post-CA, showing the thickness of alveolar septa and infiltration of a number of inflammatory cells and that alveoli are filled with pigment-laden macrophages and red blood cells. Scale bar=100 µm. (F) Lung lesion score of the sham and CA operation groups (n=7 per group; *P<0.05, significantly different from the sham group). The bars indicated the mean±standard error of mean.

Levels of TNF-α
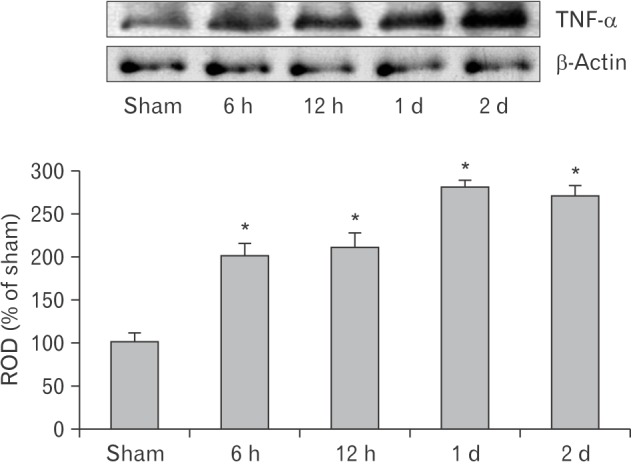
Levels of TNF-α protein in the lung of the CA operated group were altered in a time-dependent manner after CA (Fig. 2). TNF-α levels were significantly increased 6 and 12 hours after CA compared with the sham operated group and peaked 1 day after CA. Two days after CA, TNF-α level was similar to that at 1-day post-CA.
Fig. 2. Western blot analysis of tumor necrosis factor α (TNF-α) in the lung of the sham and cardiac arrest (CA) operated groups. TNF-α protein level is highest 1 day after CA. Relative optical density (ROD) as the mean percentage values of the immunoblot band is represented (n=7 per group; *P<0.05, significantly different from the sham group). The bars indicate the mean±standard error of mean.
TNF-α immunoreactivity
TNF-α reaction was not markedly observed in the lung tissue of the sham operated group (Fig. 3). TNF-α immunoreactivity in the lung of the CA operated group was significantly changed with time after CA (Fig. 3). TNF-α immunoreactivity was increased in the bronchial epithelium at 6-hour post-CA (Fig. 3B). At 12-hour post-CA, the intensity of TNF-α reaction was more increased in the bronchial and bronchiolar epithelium, and many TNF-α immunoreactive cells were found in the parenchyma. At 1-day post-CA, immunohistochemical reaction of TNF-α was observed in numerous cells in the alveolar interseptal region and the epithelium of the bronchi and bronchioles. At 2-day post-CA, the distribution pattern of TNF-α immunoreactivity in the lung was similar to that at 1-day post-CA.
Fig. 3. Immunohistochemistry of tumor necrosis factor α (TNF-α) in the lung of the sham (A) and cardiac arrest (CA) operated (B–E) groups. TNF-α immunoreactivity is shown in the bronchial epithelium from 6-hour post-CA, which is gradually increased with time. TNF-α immunoreactive cells (arrows) are found in the parenchyma from 12-hour post-CA. Scale bar=100 µm. (F) Relative optical density (ROD) of TNF-α immunoreactive structures in the sham and CA operation groups (n=7 per group; *P<0.05, significantly different from the sham group). The bars indicate the mean±standard error of mean.

Discussion
PCAS is known as the main cause of early mortality after resuscitation from CA in patients and animal models [11,18]. The pathophysiology of PCAS is complex and only partially understood [4]. One of the pathophysiological processes of PCAS is known to be attributed to IR-mediated damage and activation of systemic inflammatory response triggered by CA and RoSC [1].
Che et al. [19] reported that the survival rate was about 40% at 2 days after RoSC in a rat model of asphyxial CA. Kida et al. [20] reported that all mice died within 1 day after RoSC in a mouse model of potassium induced CA. In our present study, the survival rate was immediately and significantly decreased after RoSC in the CA operated group and reached about 7% at 2 days after RoSC. As described above, the survival rate in animal models of CA was very low in the early stage of RoSC. Therefore, histopathological evaluation was focused in the early period after CA in this present study.
Multiple organs have been involved in IR damage, and systemic inflammation reaction took place in RoSC after CA [10]. The lung is considered as the most sensitive vital organ to inflammatory response following ischemic injury [10,21]. For instance, cardiopulmonary bypass (CPB) can cause diffuse injury in the lung and leads to post-operative pulmonary edema and abnormal gas exchange [21]. Furthermore, IR after CPB is associated with whole body inflammatory response, and the systemic inflammation is thought to be, in a part, the result of IR injury [10,21]. Asimakopoulos et al. [21] reported that alveolar macrophages were activated at early stage (within 30 minutes) after acute lung injury and produced high levels of TNF-α. They also demonstrated that pulmonary macrophages were found in airways, alveolar spaces, interstitium, pleura space, and pulmonary capillary lumens in the development of inflammation.
This inflammation involves activation of leukocytes, secretions of cytokines, leukocytes adhesions to microvascular endothelium, and leukocytes extravasation, which develop tissue damage in final [2,6,21,22]. In addition, immune cells are rapidly activated upon reperfusion and induce direct tissue injury or augment inflammation via production of pro-inflammatory cytokines. The release of various proinflammatory cytokines such as TNF-α can lead to entrapment of neutrophils in the pulmonary capillaries. Elevated plasma levels of TNF-α, interleukin (IL)-6, IL-8 are strongly correlated with pulmonary dysfunction [10,21,22]. Those cytokines cause generation of polymorphonuclear cells and their entrapment in pulmonary capillaries, and eventually result in cell swelling, plasma and protein extravasation into the interstitial tissue, release of proteolytic enzymes, congestion of alveoli with plasma, erythrocytes and inflammatory debris which alter capillary permeability [6,23]. Finally, these abnormal pathophysiological processes lead to massive interstitial pulmonary edema, pulmonary fibrosis, and acute respiratory distress syndrome (ARDS), which is acute, non-cardiogenic and high-permeability lung injury, and associated with high mortality [21,24]. In addition, inflammatory mediators such as TNF-α, IL-1, IL-6 are increased in plasma and lavage fluid during ARDS, and correlate with adverse outcomes [21].
Generally, ischemic pulmonary injury evokes serious histopathology changes and infiltrated inflammatory cells [24]. TNF-α is produced primarily by macrophages and by a broad variety of cell types including lymphoid cells, mast cells, endothelial cells, myocytes, fibroblasts, and neurons following CA, and implicated in myocardial and brain dysfunctions in early post-CA period [25,26]. Recently, it was reported that TNF-α protein levels were increased in the left ventricle of the heart and in the striatum of the brain from 6 hours after CA [9,27]. Chenoune et al. [28] reported similar histopathological changes 7 days after CA in a rabbit model of CA, which included pulmonary congestion with serous edema or foci of bronchopneumonia [28].
In this study, histopathological score in the CA operated lungs was significantly (P<0.05) increased at 12-hour post-CA, and infiltration of inflammatory cells as well as pulmonary edema were significantly increased at 1 day after CA, which was similar at 2-day post-CA. This indicates that pulmonary injury might be still progressed after 1-day post-CA. Moreover, TNF-immunoreactivity was significantly increased from 6-hour post-CA and highest at 1-day post-CA, which was maintained until 2-day post-CA. Although, there are no reports on causal relationship between lung damage and mortality after RoSC, it has been reported that pulmonary dysfunction following lung injury after cardiac surgery constitutes a major factor influencing perioperative morbidity and mortality [29]. Therefore, our results support the hypothesis that inflammatory response involving TNF-α within lungs is associated with pulmonary dysfunction and could lead to high mortality at early stage of PCAS. In conclusion, future researches should aim to better understand mechanisms of lung IR injury to identify more effective therapeutic targets or diagnostic measures and aim to push the most promising therapeutics toward clinical trials. It is a reasonable strategy to eliminate or reduce the degree of inflammation-induced morbidity and mortality by suppressing secretion and function of various inflammatory mediators.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1D1A1B01011790&NRF-2017R1D1A1B03031998) and by the Ministry of Science, ICT &Future Planning (NRF-2017R1A2B4009079), and by the Bio-Synergy Research Project of the Ministry of Science, ICT and Future Planning through the National Research Foundation (NRF-2015M3A9C4076322).
References
- 1.Girotra S, Chan PS, Bradley SM. Post-resuscitation care following out-of-hospital and in-hospital cardiac arrest. Heart. 2015;101:1943–1949. doi: 10.1136/heartjnl-2015-307450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laubach VE, Sharma AK. Mechanisms of lung ischemia-reperfusion injury. Curr Opin Organ Transplant. 2016;21:246–252. doi: 10.1097/MOT.0000000000000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.López-Herce J, del Castillo J, Matamoros M, Canadas S, Rodriguez-Calvo A, Cecchetti C, Rodríguez-Núnez A, Carrillo Á; Post return of spontaneous circulation factors associated with mortality in pediatric in-hospital cardiac arrest: a prospective multicenter multinational observational study. Crit Care. 2014;18:607. doi: 10.1186/s13054-014-0607-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mongardon N, Dumas F, Ricome S, Grimaldi D, Hissem T, Pène F, Cariou A. Postcardiac arrest syndrome: from immediate resuscitation to long-term outcome. Ann Intensive Care. 2011;1:45. doi: 10.1186/2110-5820-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bro-Jeppesen J, Kjaergaard J, Wanscher M, Nielsen N, Friberg H, Bjerre M, Hassager C. Systemic inflammatory response and potential prognostic implications after out-of-hospital cardiac arrest: a substudy of the target temperature management trial. Crit Care Med. 2015;43:1223–1232. doi: 10.1097/CCM.0000000000000937. [DOI] [PubMed] [Google Scholar]
- 6.Rancan L, Paredes SD, Huerta L, Casanova J, Guzmán J, Garutti I, González-Aragoneses F, Simón C, Vara E. Chemokine involvement in lung injury secondary to ischaemia/reperfusion. Lung. 2017;195:333–340. doi: 10.1007/s00408-017-0001-x. [DOI] [PubMed] [Google Scholar]
- 7.Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, Fraisse F, Dinh-Xuan AT, Carli P, Spaulding C, Dhainaut JF, Cavaillon JM. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation. 2002;106:562–568. doi: 10.1161/01.cir.0000023891.80661.ad. [DOI] [PubMed] [Google Scholar]
- 8.Bro-Jeppesen J, Kjaergaard J, Wanscher M, Nielsen N, Friberg H, Bjerre M, Hassager C. The inflammatory response after out-of-hospital cardiac arrest is not modified by targeted temperature management at 33 °C or 36 °C. Resuscitation. 2014;85:1480–1487. doi: 10.1016/j.resuscitation.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Zhao ZG, Tang ZZ, Zhang WK, Li JG. Protective effects of embelin on myocardial ischemia-reperfusion injury following cardiac arrest in a rabbit model. Inflammation. 2015;38:527–533. doi: 10.1007/s10753-014-9959-1. [DOI] [PubMed] [Google Scholar]
- 10.Apostolakis E, Filos KS, Koletsis E, Dougenis D. Lung dysfunction following cardiopulmonary bypass. J Card Surg. 2010;25:47–55. doi: 10.1111/j.1540-8191.2009.00823.x. [DOI] [PubMed] [Google Scholar]
- 11.Roberts BW, Kilgannon JH, Chansky ME, Mittal N, Wooden J, Parrillo JE, Trzeciak S. Multiple organ dysfunction after return of spontaneous circulation in postcardiac arrest syndrome. Crit Care Med. 2013;41:1492–1501. doi: 10.1097/CCM.0b013e31828a39e9. [DOI] [PubMed] [Google Scholar]
- 12.Laurent I, Monchi M, Chiche JD, Joly LM, Spaulding C, Bourgeois B, Cariou A, Rozenberg A, Carli P, Weber S, Dhainaut JF. Reversible myocardial dysfunction in survivors of out-of-hospital cardiac arrest. J Am Coll Cardiol. 2002;40:2110–2116. doi: 10.1016/s0735-1097(02)02594-9. [DOI] [PubMed] [Google Scholar]
- 13.Madl C, Holzer M. Brain function after resuscitation from cardiac arrest. Curr Opin Crit Care. 2004;10:213–217. doi: 10.1097/01.ccx.0000127542.32890.fa. [DOI] [PubMed] [Google Scholar]
- 14.Drabek T, Foley LM, Janata A, Stezoski J, Hitchens TK, Manole MD, Kochanek PM. Global and regional differences in cerebral blood flow after asphyxial versus ventricular fibrillation cardiac arrest in rats using ASL-MRI. Resuscitation. 2014;85:964–971. doi: 10.1016/j.resuscitation.2014.03.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han F, Boller M, Guo W, Merchant RM, Lampe JW, Smith TM, Becker LB. A rodent model of emergency cardiopulmonary bypass resuscitation with different temperatures after asphyxial cardiac arrest. Resuscitation. 2010;81:93–99. doi: 10.1016/j.resuscitation.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uryu H, Hashimoto D, Kato K, Hayase E, Matsuoka S, Ogasawara R, Takahashi S, Maeda Y, Iwasaki H, Miyamoto T, Saijo S, Iwakura Y, Hill GR, Akashi K, Teshima T. alpha-Mannan induces Th17-mediated pulmonary graft-versus-host disease in mice. Blood. 2015;125:3014–3023. doi: 10.1182/blood-2014-12-615781. [DOI] [PubMed] [Google Scholar]
- 17.Park JH, Park O, Cho JH, Chen BH, Kim IH, Ahn JH, Lee JC, Yan BC, Yoo KY, Lee CH, Hwang IK, Kwon SH, Lee YL, Won MH, Choi JH. Anti-inflammatory effect of tanshinone I in neuroprotection against cerebral ischemia-reperfusion injury in the gerbil hippocampus. Neurochem Res. 2014;39:1300–1312. doi: 10.1007/s11064-014-1312-4. [DOI] [PubMed] [Google Scholar]
- 18.Cour M, Abrial M, Jahandiez V, Loufouat J, Belaïdi E, Gharib A, Varennes A, Monneret G, Thibault H, Ovize M, Argaud L. Ubiquitous protective effects of cyclosporine A in preventing cardiac arrest-induced multiple organ failure. J Appl Physiol (1985) 2014;117:930–936. doi: 10.1152/japplphysiol.00495.2014. [DOI] [PubMed] [Google Scholar]
- 19.Che D, Li L, Kopil CM, Liu Z, Guo W, Neumar RW. Impact of therapeutic hypothermia onset and duration on survival, neurologic function, and neurodegeneration after cardiac arrest. Crit Care Med. 2011;39:1423–1430. doi: 10.1097/CCM.0b013e318212020a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kida K, Shirozu K, Yu B, Mandeville JB, Bloch KD, Ichinose F. Beneficial effects of nitric oxide on outcomes after cardiac arrest and cardiopulmonary resuscitation in hypothermia-treated mice. Anesthesiology. 2014;120:880–889. doi: 10.1097/ALN.0000000000000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asimakopoulos G, Smith PL, Ratnatunga CP, Taylor KM. Lung injury and acute respiratory distress syndrome after cardiopulmonary bypass. Ann Thorac Surg. 1999;68:1107–1115. doi: 10.1016/s0003-4975(99)00781-x. [DOI] [PubMed] [Google Scholar]
- 22.Kang DH, Kim J, Rhee JE, Kim T, Kim K, Jo YH, Lee JH, Lee JH, Kim YJ, Hwang SS. The risk factors and prognostic implication of acute pulmonary edema in resuscitated cardiac arrest patients. Clin Exp Emerg Med. 2015;2:110–116. doi: 10.15441/ceem.14.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei W, Lai SC, Xie Y, Liu BF, He YR, Hu H, Wan Z. The protective effect of target temperature management combined with prostaglandin E(1) on ischemia/teperfusion injury of cerebral micro-vascular endothelium of ROSC rat. Sichuan Da Xue Xue Bao Yi Xue Ban. 2016;47:310–315. [PubMed] [Google Scholar]
- 24.Fang X, Wang L, Shi L, Chen C, Wang Q, Bai C, Wang X. Protective effects of keratinocyte growth factor-2 on ischemia-reperfusion-induced lung injury in rats. Am J Respir Cell Mol Biol. 2014;50:1156–1165. doi: 10.1165/rcmb.2013-0268OC. [DOI] [PubMed] [Google Scholar]
- 25.Youngquist ST, Niemann JT, Heyming TW, Rosborough JP. The central nervous system cytokine response to global ischemia following resuscitation from ventricular fibrillation in a porcine model. Resuscitation. 2009;80:249–252. doi: 10.1016/j.resuscitation.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Youngquist ST, Niemann JT, Shah AP, Thomas JL, Rosborough JP. A comparison of etanercept vs. infliximab for the treatment of post-arrest myocardial dysfunction in a swine model of ventricular fibrillation. Resuscitation. 2013;84:999–1003. doi: 10.1016/j.resuscitation.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drabek T, Janata A, Wilson CD, Stezoski J, Janesko-Feldman K, Tisherman SA, Foley LM, Verrier JD, Kochanek PM. Minocycline attenuates brain tissue levels of TNF-alpha produced by neurons after prolonged hypothermic cardiac arrest in rats. Resuscitation. 2014;85:284–291. doi: 10.1016/j.resuscitation.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chenoune M, Lidouren F, Adam C, Pons S, Darbera L, Bruneval P, Ghaleh B, Zini R, Dubois-Rande JL, Carli P, Vivien B, Ricard JD, Berdeaux A, Tissier R. Ultrafast and whole-body cooling with total liquid ventilation induces favorable neurological and cardiac outcomes after cardiac arrest in rabbits. Circulation. 2011;124:901–911. doi: 10.1161/CIRCULATIONAHA.111.039388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng CS, Wan S, Yim AP, Arifi AA. Pulmonary dysfunction after cardiac surgery. Chest. 2002;121:1269–1277. doi: 10.1378/chest.121.4.1269. [DOI] [PubMed] [Google Scholar]