

Abstract
Pathway complexity in supramolecular polymerization has recently sparked interest as a method to generate complex material behavior. The response of these systems relies on the existence of a metastable, kinetically trapped state. In this work, we show that strong switch-like behavior in supramolecular polymers can also be achieved through the introduction of competing aggregation pathways. This behavior is illustrated with the supramolecular polymerization of a porphyrin-based monomer at various concentrations, solvent compositions, and temperatures. It is found that the monomers aggregate via an isodesmic mechanism in weakly coupled J-type aggregates at intermediate solvent quality and temperature, followed by nucleated H-aggregates at lower solvent qualities and temperatures. At further increased thermodynamic driving forces, such as high concentration and low temperature, the H-aggregates can form hierarchical superhelices. Our mathematical models show that, contrary to a single-pathway polymerization, the existence of the isodesmic aggregation pathway buffers the free monomer pool and renders the nucleation of the H-aggregates insensitive to concentration changes in the limit of high concentrations. We also show that, at a given temperature or solvent quality, the thermodynamically stable aggregate morphology can be selected by controlling the remaining free external parameter. As a result, the judicious application of pathway complexity allows us to synthesize a diverse set of materials from only a single monomer. We envision that the engineering of competing pathways can increase the robustness in a wide variety of supramolecular polymer materials and lead to increasingly versatile applications.
Introduction
Supramolecular interactions of porphyrins have ubiquitous applications in natural systems. For example, allosteric supramolecular host–guest interactions in the heme protein permit the efficient transport of oxygen through the blood plasma,1,2 and the intramolecular orientation of chlorophyll in the light-harvesting complex allows for control over electron transfer.3−5 Their diverse self-assembly properties and chemical functionalities have led researchers to apply these systems in a wide variety of synthetic systems,6 ranging from photovoltaics7,8 to sensing and molecular electronics.9,10
Despite the great insight that has been gained throughout the past decades into the behavior of these molecules, still new and intriguing phenomena are observed in porphyrin-based systems. For example, the group of Ribó and co-workers showed that the direction of stirring induces chirality in supramolecular aggregates of achiral porphyrin monomers.11,12 In another example, Kihara et al.13 reported that coordination control over supramolecular polymers unexpectedly induces responsiveness to static electricity. Previously, we reported that aggregation can be achieved by diluting the system through the addition of coordinating ligands to a supramolecular porphyrin, whereas concentration increases lead to destruction of the polymer.14
Recently, the emerging field of living supramolecular polymerizations has sparked a new interest in supramolecular polymers from porphyrin-based monomers. In seminal examples by the group of Takeuchi and Sugiyasu, chiral porphyrins were shown to assemble into kinetically trapped small J-aggregates. Over time, the kinetically trapped J-aggregate could be converted to other, thermodynamically stable aggregates, which enabled seeded growth of nucleated H-aggregated fibers15,16 or large J-aggregated nanosheets.17 Thus, it was possible to generate several different materials from one monomer by controlling pathway complexity. Due to the kinetic trapping of the initial aggregation state, only the thermodynamically most stable aggregate retained its properties throughout prolonged periods. In another report, the group of Aida showed that addition of small amounts of alcohol as co-solvent to dodecane solutions can successfully compete with the hydrogen bonding that stabilizes supramolecular polymers.18 Through this competition, a temperature interval emerged where the monomeric porphyrin was molecularly dissolved, but above or below this temperature range, polymers were formed. Through the introduction of these competing processes, a thermally bi-signate feature of the polymerization emerged, and a single polymer type was obtained by both increasing and decreasing the temperature.
Competition between several aggregate types is an increasingly observed phenomenon. The groups of Würthner and Lochbrunner recently reported that perylene bis-imides display complex supramolecular polymerization, where the monomers can assemble into nucleated J-aggregates but can also form an off-pathway H-aggregated dimeric structure, which is thermodynamically stable at low concentrations and intermediate temperatures.19 Similar off-pathway dimerization has also been reported for zinc chlorins20 and covalently coupled naphthalene diimide derivatives,21 which indicates that the concepts developed in complex supramolecular polymerizations are not limited to specific molecular platforms. Moreover, these recent findings strongly suggest that pathway complexity in the energy landscape of supramolecular polymerizations can be introduced through the incorporation of large π-cores as well as flexible pendant moieties. Hence, a general description of artificial systems in various complex competing assembly processes is desirable. These descriptions then allow these systems to be compared with their natural counterparts and will lead to a better understanding of supramolecular materials.
In this work, we show that the introduction of competing polymerization pathways in a supramolecular polymerization of a porphyrin-based monomer (Scheme 1) can lead to very responsive transitions between various thermodynamically stable aggregate morphologies. The presence of the competing aggregation pathways increases the responsiveness of the aggregation state of our supramolecular system to changes in temperature and solvent quality. Moreover, the competing pathway not only increases the sensitivity of the system toward external triggers but also dramatically reduces the sensitivity to changes in monomer concentration. These features show that pathway complexity of supramolecular systems can also be used under thermodynamic control and that, contrary to the present understanding, kinetic traps are not a requirement for highly sensitive systems. As a result, the introduction of competing pathways leads to the emergence of increased responsiveness in this class of materials.
Scheme 1. Chiral Amide-Functionalized Zinc Porphyrin (S)-1 and (R)-1.

First, we show that the solvent quality dictates the aggregate morphology of our porphyrin-based monomer. The experimental data are fitted to a thermodynamic model to obtain the energetic parameters that describe the system. Our model shows that the presence of the competing pathways makes the system increasingly insensitive to concentration changes. Atomic force microscopy (AFM) analysis indicates that hierarchical assemblies are present in the system at high thermodynamic driving forces, such as low temperature and high concentration. Kinetic analysis of the polymerization shows that the two aggregation pathways are in competition with each other. Additionally, we show that changing the temperature has a similar effect on the aggregate state as changing the solvent, and these two parameters function as independent handles to obtain the desired material morphology in a wide range of conditions.
Results and Discussion
Solvent Quality Determines the Supramolecular Polymerization of (S)-1
Chiral porphyrin (S)-1 (Scheme 1) was synthesized according to a previously reported procedure.14 Full analytical details are given in the Supporting Information (SI, Figures S1–S3). Upon dissolving (S)-1 in methylcyclohexane (MCH), long supramolecular polymers are formed through hydrogen-bonding interactions along the polymer backbone.14 The presence of the helical aggregates, in which the monomers are co-facially stacked into H-aggregates, is indicated by an absorption band at 392 nm that displays a strong bi-signate Cotton effect. Conversely, solutions of (S)-1 in CHCl3, in which (S)-1 is molecularly dissolved, display a strong absorption around 425 nm and no CD signal.14 To investigate the solvent dependency of the thermodynamic parameters of the supramolecular polymerization of (S)-1, UV–vis and CD spectra of (S)-1 in solutions with varying volume percentages CHCl3 in MCH were measured (Figure 1 and Figure S4). In pure MCH, a CD-active absorption band with a maximum at 392 nm is observed due to the presence of H-aggregates. Upon increasing the volume fraction CHCl3, the absorbance and CD intensity at 392 nm decrease, with a concomitant gradual emergence of a broad, CD-silent absorption band at 425 nm, indicative of the formation of a weakly coupled J-type aggregate or otherwise less ordered aggregate (from here on referred to as J-type aggregates).22,23 Approaching the critical solvent composition, the CD intensity at 392 nm rapidly decreases and the absorption at 425 nm rises sharply, indicating the destruction of cooperatively formed co-facially stacked H-aggregates (Figure 2). At volume percentages above 5–7.5 vol% CHCl3, the CD signal has disappeared due to the destabilization of the H-aggregates, and a further increase in the CHCl3 volume fraction results in a gradual increase in intensity and sharpening of the absorption peak. This gradual increase indicates the presence of the J-type aggregates at intermediate solvent compositions that, upon further increase of the CHCl3 volume fraction, form monomerically dissolved (S)-1. The presence of J-type aggregates is corroborated by a slight red-shift of the Q-bands of (S)-1 at intermediate volume fractions of CHCl3 (Figure S5). The comparably low dependency of the stability of the J-type aggregates on the solvent quality is reflected in the sigmoidal dependency of the absorbance on the solvent composition. This sigmoidal dependency indicates that (S)-1 aggregates into J-type aggregates through an isodesmic aggregation mechanism. Figure 2a illustrates that the onset of isodesmic aggregation shifts to higher CHCl3 fractions as the concentration is increased. Remarkably, the critical solvent composition at which the cooperative pathway is favored over the isodesmic pathway, however, does not shift significantly over the 2 orders of magnitude concentration difference.
Figure 1.

Solvent-dependent (a) UV–vis and (b) CD spectra of 1 μM solutions of (S)-1. In MCH, 1 shows a broad absorption band at 392 nm. The CD spectrum displays a strong bi-signate Cotton effect at those wavelengths, indicating helical H-aggregation of (S)-1 (red line). Upon increasing the volume fraction of CHCl3, the CD intensity and absorbance at 392 nm decrease, while a broad absorption band at 425 nm arises (purple line). This broad band indicates that (S)-1 assembles in J-type aggregates. Further increase in the CHCl3 volume fractions leads to the increase in absorption at 425 nm until (S)-1 is monomerically dissolved (blue line).
Figure 2.

(a) Absorbance at 425 nm and (b) CD intensity at 392 nm at various solvent compositions for [(S)-1] = 1–100 μM and optical path lengths between 1 cm and 0.1 mm. Increasing the CHCl3 leads to a strong decrease in CD intensity at 392 nm and increase in absorbance at 425 nm as the critical solvent composition is approached, indicating the destruction of the cooperatively formed H-aggregates. At CHCl3 fractions above the critical solvent composition, the sigmoidal increase in absorbance at 425 nm indicates the gradual destabilization of the isodesmic J-type aggregates. The symbols indicate experimental data and the lines depict the fits obtained from the equilibrium model. The insets show the sharp change in absorbance and CD intensity at the critical solvent composition.
The presence of two different aggregate types is also confirmed by FT-IR spectroscopy (Figure S6). At low CHCl3 fractions, the amide NH stretch and amide CO stretch frequencies are shifted to lower wavenumbers with respect to molecularly dissolved (S)-1, indicating that at low CHCl3 fractions, the amides of (S)-1 are engaged in hydrogen bonding, indicative of H-aggregates. In intermediate CHCl3 fractions, the IR spectra show vibrational transitions at similar wavenumbers as in pure CHCl3 solutions, but the amide and carbonyl bands have broadened, indicating poorly defined aggregation. The isodesmic aggregates, formed at intermediate CHCl3 fractions, are proposed to be stabilized through π–π-stacking, resulting in J-type aggregates.
It should be noted that the molar ellipticity (Δε) of the H-aggregates that is calculated from the CD signal is not constant over the entire concentration regime. We attribute this variation to subtle changes in atmospheric water content, which has recently been shown to dramatically impact aggregation behavior in supramolecular polymers.24,25
Thermodynamic Parameters Are Obtained with Fits of Experimental Data
In previous works, we have employed numerical models to understand the underlying principles of various types of supramolecular polymerizations, including kinetically trapped states,26 kinetic aspects of competing aggregation pathways,27 sergeants-and-soldiers and majority rules experiments,28,29 racemizing systems,30 and supramolecular copolymers.31 To further understand the pathway complexity in the supramolecular polymerization of (S)-1 and quantify the thermodynamic parameters that govern the aggregation processes, the results obtained from the UV–vis and CD measurements at various CHCl3 volume fractions have been fitted to a thermodynamic equilibrium model (Full details in the SI). In the model, the monomers can assemble into one-dimensional, CD-inactive isodesmic aggregates and CD-active cooperative aggregates, which are both in equilibrium with monomeric (S)-1. This model shows resemblance with the model introduced by the groups of Würthner, Lochbrunner, and co-workers,19 but that model does not account for the observed insensitivity of the critical solvent composition to an increase in concentration.32 The dependency of the aggregate stabilities on the solvent is introduced by33
| 1 |
where ΔG is the Gibbs free energy gain upon addition of monomeric (S)-1 to the aggregate at a given volume fraction of CHCl3, f. The Gibbs free energy of the aggregation process in the absence of CHCl3 is given by ΔG°. The dependence of ΔG on f is introduced with the m-value. In our model, the m-value of the nucleation and elongation phase of the cooperative polymerization are taken to be equal. The obtained fits are given in Figure 2 and the corresponding thermodynamic parameters in Table 1.
Table 1. Values of ΔG° and m for the Cooperative and Isodesmic Pathways, Obtained from the Global Fitting of UV and CD Data for the Various Concentrations of (S)-1.
| 1 μM | 10 μM | 100 μM | |
|---|---|---|---|
| ΔG°coop (kJ/mol) | –46 | –45 | –33 |
| mcoop (kJ/mol) | 194 | 101 | 67 |
| σa | 7.3 × 10–3 | 2.3 × 10–3 | 6.7 × 10–4 |
| ΔG°iso (kJ/mol) | –40 | –41 | –31 |
| miso (kJ/mol) | 91 | 56 | 41 |
The cooperativity parameter σ is determined at 293 K.28
The results show that the Gibbs free energies in MCH of the cooperative pathway (ΔG°coop) range between −46 kJ/mol at [(S)-1] = 1 μM and −33 kJ/mol at [(S)-1] = 100 μM. The Gibbs free energies in MCH of the isodesmic aggregation pathway (ΔG°iso) range between −41 and −31 kJ/mol and are less negative than ΔG°coop at every measured concentration. The solvent dependency parameter of the cooperative polymerization, mcoop, ranges between 194 kJ/mol at [(S)-1] = 1 μM and 67 kJ/mol at [(S)-1] = 100 μM. For the isodesmic pathway, miso, ranges between 91 kJ/mol at [(S)-1] = 1 μM and 41 kJ/mol at [(S)-1] = 100 μM and is lower than mcoop at every measured concentration. Combined, the obtained values of ΔG°coop and ΔG°iso and their respective dependencies on solvent quality, mcoop and miso, lead to a CHCl3 fraction at which ΔG°coop and ΔG°iso are equal and above which the J-type aggregates are the most stable polymer.
The thermodynamic model also allows us to determine the degree of cooperativity of the cooperative pathway, expressed by the cooperativity parameter, σ. Interestingly, despite the strong response observed in the experimental curves (Figure 2b), the resulting cooperativity of the nucleated pathway is not very high, as indicated by the values of σ, ranging between 7.3 × 10–3 and 6.7 × 10–4. This result, together with the observed low sensitivity of the critical solvent composition to increases in CHCl3 fraction prompted us to investigate the competition of isodesmic and cooperative pathways in more detail.
Equilibrium Model Gives Insight into Polymerization Thermodynamics
The observed insensitivity of the critical solvent composition and the low degree of cooperativity obtained from the fits of the experimental data are explained by our theoretical model of a two-pathway supramolecular polymerization. At CHCl3 above the critical solvent fraction, the isodesmic aggregation pathway buffers the monomer concentration, preventing the elongation of nucleated polymers. Increasing the total concentration then leads to an increase in the J-type aggregate concentration and only a marginal increase in the free monomer concentration that is available for cooperative polymerization. Since the free monomer concentration cannot exceed Kiso–1 and the concentration required for the elongation of the cooperative polymers is Ke–1, the H-aggregates can only form when the cooperative polymerization is lower in Gibbs free energy than the isodesmic pathway (see SI for additional details). As a result, there is a fundamental upper solvent fraction above which the formation of the H-aggregates is impossible. The weak concentration dependence of the formation of cooperative polymers in the presence of an isodesmic pathway has previously also been recognized by Bouteiller and Van der Schoot.34,35 This is in strong contrast with a single-pathway nucleated polymerization, in which polymers can theoretically always form at any solvent composition or temperature upon simply increasing the concentration (Figure 3).
Figure 3.

Calculated minimal concentration required for H-aggregate formation in MCH for a system in which the cooperative polymerization is in competition with an isodesmic pathway (a) and a system that can only polymerize via a single cooperative pathway supramolecular polymerization (b). The shaded area indicates the presence of cooperative polymers. In the case of a competitive system (a), the minimum concentration required for the formation of H-aggregates explodes when the solvent composition is such that ΔGcoop= ΔGiso. For the single cooperative pathway, aggregates can form at any solvent composition. The thermodynamic parameters obtained from the denaturation experiment of 1 μM were used in the calculation.
Additionally, the sharp transition in the CD response at the critical solvent composition, despite the moderate cooperativity obtained from the fits, can be explained by our model. At CHCl3 fractions below the critical solvent composition, the H-aggregates are more stable than the J-type aggregates and in this regime, the free monomer concentration follows the behavior similar to a single-pathway cooperative polymerization (Figures S7 and S8). As the solvent quality becomes better, the H-aggregates are increasingly destabilized with respect to the J-type aggregates. The increase in competition between the polymer types upon increasing the solvent quality leads to a mass transfer from the H-aggregates to the J-type aggregates, which does not occur in a single-pathway polymerization, while the free monomer concentration behaves similar to a single-pathway cooperative polymerization. The additional mass transfer from the H-aggregates to the J-type aggregates causes the strong response that is observed at solvent compositions just below the critical solvent fraction.
The aggregation energies of the cooperative and isodesmic pathway in pure MCH do not vary significantly upon increasing the concentration from 1 to 10 μM. However, upon increasing the concentration 1 order of magnitude further, considerable deviations are observed. The solvent-dependency parameter m shows a similar trend: increasing the concentration leads to an apparent decrease in the destabilization by CHCl3 of the cooperative pathway. We propose that this apparent increased stabilization at higher CHCl3 fractions is due to the formation of superhelices of cooperatively formed fibers (vide infra). This hypothesis also explains the observed increase in cooperativity upon increasing the concentration. The formation of hierarchical structures becomes more pronounced at higher concentrations, where the concentration of aggregated material is higher. At these concentrations, the hierarchical structures effectively act as a monomer sink, leading to a more cooperative system.
Atomic Force Microscopy Reveals the Existence of Superhelices
The microscopic morphology of aggregates of (S)-1 was studied using AFM measurements (Figure 4). The AFM images reveal the existence of helical aggregates when (S)-1 is drop-cast from MCH solutions. At low concentrations, fibers with a width of 4.2 nm are observed (for full details, see Table S1). This is in line with the diameter of 4.9 nm of (S)-1 obtained from molecular simulations. The helical pitch of the fibers is estimated at 5.7 nm. AFM images of (R)-1 (Figure S10), which has chiral side chains of opposite configuration, show fibers with similar fiber width and helical pitches as are observed in the AFM images of (S)-1, but the fibers of (R)-1 show opposite helicity. This indicates that the helical features are due to the molecular chirality of the monomers.
Figure 4.

AFM images of helical porphyrin stacks drop-cast from (a) fresh and (b) refrigerated 33 μM MCH solutions of (S)-1. The width of the observed aggregates in micrographs of samples drop-cast from fresh solutions corresponds to the diameter of 1, obtained from molecular models (49 Å), indicating that the helices consist of a single fiber. Micrographs of the refrigerated samples show large aggregates with widths of 25 (green box, B) to 50 nm (red box, A). (c) Cartoon representation of the collapse of a single helical fiber to form a superhelix at high thermodynamic driving forces, which may further assemble to form double and triple helices.
Samples that are drop-cast from a cooled solution, in which the thermodynamic driving force for aggregation is increased, show that single fibers aggregate to form hierarchically assembled double- and triple-helix assemblies with estimated diameters of ca. 25 and 50 nm, respectively, and helical pitches of 23 nm (Figure 4b and Table S1). Strikingly, the handedness of the superhelices is the same as that of the constituent single helices. Formation of hierarchical aggregates from supramolecular polymers usually coincides with inversion of helicity, as has been demonstrated by Nolte,36 Würthner,37 and others.38,39 The observed preservation of helix handedness in our system suggests that the single helices are aggregated into superhelices through a torsional strain mechanism, as observed for peptide amphiphiles by the group of Stupp,40,41 and other systems reported by Yagai42 and Zhu.43 In line with their observations, we observe that the helical pitch of the superhelices increases 4-fold (Table S1). In addition, the bundle width increases considerably. We speculate that the superhelix formation is driven by the collapse of the fiber, maximizing interactions between the aliphatic side chains and causing a release of solvation entropy.44 The collapsed helices can intertwine to form superhelices from multiple individual H-aggregate helices, as observed by the variable superhelix diameter (Figure 4b, boxes A and B).
The observation of the superhelices only at low temperatures indicates that high association constants or concentrations are required to cluster the large number of molecules in one superstructure. Hence, this process will only occur at low temperature, high concentrations and poor solvent quality. Although very different morphologies are observed under AFM, spectroscopic investigations of samples of (S)-1 that are cooled for prolonged periods do not show any appreciable changes in the CD and absorption spectra (Figure S11). Since the handedness of the single helix is retained upon aggregation into superhelices and the molecular environment of the monomers remains almost identical, the helix–superhelix transition is proposed to be not observable in our spectroscopic measurements. Thus, the combination of UV–vis and CD spectroscopy with AFM reveals that (S)-1 can exist in four thermodynamically stable states, but to gain further insight into their mechanism of interconversion, kinetic measurements are necessary.
Kinetic Analysis Indicates the Aggregation Pathways Are in Competition
Not only thermodynamic aspects of supramolecular polymerizations are altered by the solvent quality and other external parameters. Also the kinetic characteristics of the assembly process are strongly dependent on solvent quality. The kinetic traces of single-aggregate supramolecular polymerizations have been reported to equilibrate slowest around the critical solvent composition, resulting from the competition between comparable rates in polymer assembly and disassembly at the critical solvent composition.33 The kinetic traces of the formation of the nucleated H-aggregates of (S)-1 at various solvent compositions is studied with CD spectroscopy. The results are given in Figure 5.
Figure 5.

(a) Kinetic profiles of the CD intensity at 392 nm upon mixing MCH and CHCl3 solutions of (S)-1. The colored labels indicate the volume percentage CHCl3 in the final solution for every trace. (b) The time at which 90% of the final CD signal of the traces depicted in (a) are obtained (t90) shows a maximum around the critical solvent composition. The kinetics of H-aggregate formation is slowest around the critical solvent composition. The behavior of (S)-1 in a dual-pathway energy landscape is qualitatively similar to the kinetics described for OPVs.33
Despite the presence of the isodesmic pathway, the kinetic trace of nucleated H-aggregate formation shows similar behavior as that reported for single-aggregate-type polymerization. At the critical solvent composition, the time at which 90% of the final CD signal is obtained (t90) shows a distinct peak (Figure 5b). The rate of J-type aggregation was too fast to be measured and only equilibrium states could be observed at CHCl3 fractions above the critical solvent composition (Figure S12). Additionally, t90 values did not show a significant dependence on concentration. The absence of a strong concentration dependency of the equilibration time indicates that the two aggregate types do not convert directly from one type to the other, but that aggregate interconversion proceeds through the monomeric state.27
Temperature Variation Gives Morphology Changes Similar to Those Resulting from Changes in Solvent Quality
Thermodynamic considerations dictate that the destabilization by nonspecific interactions of a denaturing co-solvent has a similar effect as an increase in temperature. Taken together, these two external parameters serve as orthogonal handles to control the self-assembly state of a material, potentially giving greater control over the aggregate morphology. To test this hypothesis, we followed the CD intensity and absorbance signals at 392 nm, at which the H-aggregates absorb, and at 425 nm, at which the J-type aggregate and monomers absorb as a function of temperature in MCH. The melting curves obtained from the temperature-dependent CD and UV–vis experiments are given in Figure 6. The melting curves are fit to a thermodynamic model that describes the aggregation of (S)-1 in a similar fashion as the model that is used to fit the denaturation experiments. The temperature dependency of the Gibbs free energy gain upon monomer addition is introduced via
| 2 |
Figure 6.

(a) Absorbance change with respect to the minimum absorbance values of the observed melting curves at 425 nm. (b) CD intensity at 392 nm MCH solutions with varying concentrations of (S)-1 as a function of temperature. The CD-silent J-type aggregation process is not visible in the CD trace but is apparent in the UV signal. The experimental data (symbols) are fitted to the two-pathway thermodynamic model (solid lines). The absorbance traces are corrected for baseline drift between experiments by subtracting the absorbance by the minimum absorbance in the melting curve to obtain ΔAbsorbance.
The thermodynamic parameters obtained from the fitting procedure are given in Table 2. The fitted curves are plotted in Figure 6.
Table 2. Values of ΔH and ΔS for the Cooperative and Isodesmic Pathways, Obtained from the Global Fitting of UV and CD Data for the Various Concentrations of (S)-1 in MCH.
| 1 μM | 10 μM | 100 μM | |
|---|---|---|---|
| ΔHcoop (kJ/mol) | –102 | –66 | –63 |
| ΔScoop (J/mol/K) | –184 | –90 | –107 |
| σa | 2.4 × 10–4 | 2.6 × 10–7 | 8.3 × 10–10 |
| ΔGcoop (kJ/mol)a | –48 | –39 | –32 |
| ΔHiso (kJ/mol) | –72 | –63 | –57 |
| ΔSiso (J·mol–1·K–1) | –97 | –89 | –106 |
| ΔGiso (kJ/mol)a | –43 | –37 | –26 |
The cooperativity parameter σ and Gibbs free energies are determined at 293 K.
The enthalpies of elongation of the nucleated polymers, ΔHcoop, range between −102 kJ/mol at [(S)-1] = 1 μM and −63 kJ/mol at [(S)-1] = 100 μM. Qualitatively, a similar decreasing trend can be seen in the values of the entropy of elongation, which range between −184 J/mol/K at [(S)-1] = 1 μM and −90 and −107 kJ/mol at [(S)-1] = 10 μM and [(S)-1] = 100 μM, respectively. As a result, the Gibbs free energies of elongation of the cooperative polymer at 293 K (ΔGcoop) is in good agreement with the values obtained from the denaturation experiments (Table 1). The enthalpies of the isodesmic polymerization, ΔHiso, range between −72 kJ/mol at [(S)-1] = 1 μM and −57 kJ/mol at [(S)-1] = 100 μM. The entropies of the isodesmic polymerization, ΔSiso, are comparable for all concentrations measured.
Similar relations between the energetic parameters of the cooperative and isodesmic aggregation polymerizations as found in the denaturation studies (Table 1) are found in the melting experiments (Table 2). At all concentrations, the Gibbs free energy of the cooperative polymerization at 293 K is more negative than the Gibbs free energy of the isodesmic aggregation and upon increasing the concentration, a decrease in Gibbs free energy is obtained from the fits. Additionally, the extraction of the enthalpic and entropic contributions in the self-assembly process sheds light on the molecular nature of both aggregation pathways. Upon increasing the concentration, the change in enthalpy upon monomer addition decreases together with the entropic cost of elongation. In addition, the cooperativity parameter σ becomes smaller, indicating a more cooperative formation of H-aggregates. The thermodynamic parameters obtained for the isodesmic pathway are relatively insensitive to a change in concentration. The concentration dependency of the cooperative pathway corroborates the hypothesis that hierarchical structures are formed at increased thermodynamic driving forces for aggregation (i.e., high concentration, low temperature, and low solvent quality). The thermodynamic parameters obtained from the cooling curve at 1 μM give a good description of the two-pathway polymerization process, since hierarchical self-assembly is not significant at these concentrations. Both the enthalpy and entropy of the cooperative pathway are more negative than the values of the isodesmic pathway. This can be rationalized by the greater enthalpy release due to the formation of hydrogen bonds as opposed to weaker π–π interactions that lead to isodesmic aggregation. The additional entropic penalty of the decrease in rotational freedom of the amides and clustering of the aliphatic side chains is reflected in the larger entropic penalty for the cooperative pathway. Upon increasing the concentration, the thermodynamic parameters of the cooperative pathway appear to decrease considerably and the cooperativity of the polymerization appears to be enhanced. These deviations suggest that in the cooling experiments at concentrations of 10 μM and above, hierarchical structures are formed.45
From the results above, we conclude that, qualitatively, changing solvent composition or temperature leads to similar trends in the stability of the supramolecular aggregates. As a next step, we performed cooling experiments of solutions of (S)-1 with varying volume fractions of CHCl3.
Consequences of Competing Pathways for Supramolecular Materials
We envision that selective synthesis of a desired aggregate type by tuning of temperature and solvent composition can be an effective method to harness the different properties of the supramolecular materials under various conditions. To demonstrate this concept, we performed cooling experiments of solutions of (S)-1 in MCH with volume percentages of CHCl3 varying from 0 to 7.5 vol%. The melting curves obtained for 1 μM solutions are given in Figure 7. The increase of the volume fraction of CHCl3 leads to a destabilization of the aggregates, which results in a shift of the elongation temperature to lower temperatures (Figure 7b) and a weaker change in absorbance (Figure 7a). At volume fractions above the critical solvent composition at 293 K (5 vol%), only isodesmic aggregation is observed. It should be noted that Gibbs free energies of isodesmic aggregation obtained from the melting curves with 2.5–7.5 vol% CHCl3 do not show a clear decrease upon increasing the CHCl3 fraction (Table S2). The absence of this trend can be explained by a temperature-dependent m-value of CHCl3. Although the boiling point of the mixtures of CHCl3 in MCH are not reached, the destabilizing effect of CHCl3 will likely become temperature dependent. This renders the enthalpic and entropic contributions temperature dependent, and the model does not give the true thermodynamic values of the aggregation process in these mixtures. Nonetheless, the calculated values of the Gibbs free energies of the nucleated polymer obtained from the denaturation experiments and melting experiments are in good agreement, indicating that melting and denaturation experiments can be used as orthogonal methods to study and synthesize supramolecular materials.
Figure 7.

(a) Absorbance change of 1 μM solutions of (S)-1 with respect to the minimum absorbance values of the observed melting curves at 425 nm. (b) CD intensity at 392 nm as a function of temperature for solutions with varying CHCl3 volume fractions. The experimental data (symbols) are fitted to the two-pathway thermodynamic model (solid lines). Upon increasing the volume fraction CHCl3 the elongation temperature of H-aggregate formation decreases. In solutions with CHCl3 volume fractions above the critical solvent composition (7.5 vol%, green line), no H-aggregation is observed anymore. The absorbance traces are corrected for baseline drift between experiments by subtracting the absorbance by the minimum absorbance in the melting curve to obtain ΔAbsorbance.
Our ability to quantitatively describe the supramolecular polymerization of (S)-1 can be used to predict the polymer morphology, length, and dispersity at any given temperature. Hence, this strategy enables full control over the aggregate morphology and allows the prediction of the composition of the system at arbitrary solvent compositions and temperatures (Figure 8). Because the competing J-type aggregation pathway buffers the monomer concentration, there is an upper limit above which no polymers can be formed. Moreover, the asymptotic behavior of the minimal concentration required to form H-aggregates renders the system insensitive to changes in concentration in the limit of high concentration. As the various porphyrin morphologies, whose aggregation behaviors can be engineered through chemical synthesis,22 are known to exhibit different material properties,17 we envision that the combination of solvent composition and temperature control can be used to synthesize a plethora of functional supramolecular materials from only a very limited number of starting materials in a very easy and controllable manner.
Figure 8.

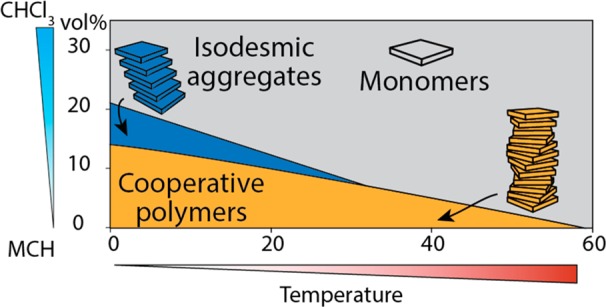
(a) Phase diagram displaying the most prevalent aggregate species of (S)-1 at 100 μM as a function of temperature and solvent composition. In the gray area, 1 is predominantly present as monomer; in the blue area, (S)-1 is predominantly aggregated in J-type aggregates; and in the dark red area, the majority of (S)-1 is aggregated in H-aggregates. The dashed lines indicate the temperatures of the speciation plots to the right. (b) Speciation diagrams of the system at various temperatures as a function of the solvent composition. Note that, although the phase diagram does not indicate J-type aggregates are at any point the most prevalent species at 30 °C, J-type aggregation does take place in a considerable amount, as is evident from the lower speciation diagram.
Outlook and Conclusion
In this work, we show that the supramolecular polymerization of amide-functionalized porphyrin (S)-1 displays pathway complexity. At poor solvent conditions and low temperatures, (S)-1 aggregates into H-aggregated nucleated polymers, whereas at intermediate solvent quality and temperatures, weakly coupled J-type aggregates are formed through an isodesmic aggregation process. Using a two-pathway equilibrium model, a thermodynamic description of the solvent and temperature dependency of the aggregation process has been obtained.
The thermodynamic parameters that are obtained by fitting the two-pathway model to the experimentally obtained curves show a concentration dependency. The curves at higher concentration tend to show decreased Gibbs free energies of aggregation and increased cooperativity in the system. The observation of superhelices in AFM measurements strongly suggests that this concentration dependency is due to the formation of hierarchical structures.
The thermodynamic model revealed that the competition between the cooperative and isodesmic aggregation pathways introduces a fundamental upper solvent composition or temperature at which the nucleated H-aggregates can form. Due to the buffering effect of the isodesmic J-type aggregation at CHCl3 fractions or temperatures above the critical fraction or elongation temperature, H-aggregation is impossible. As a result, formation of the cooperative polymers is very sensitive to solvent composition and temperature, and the system becomes insensitive to changes in concentration. Through the delicate interplay of pathway complexity with the molecular structure of the monomer,22 the presence of competing polymerization pathways in supramolecular polymerizations can be employed to tailor the stability of supramolecular materials in a controllable fashion.
Understanding of the full energy landscape of a supramolecular polymerization is of paramount importance for these materials to be used as functional materials. The full diversity of this pathway complexity can only be achieved through a combination of techniques which are employed at different concentrations that span several orders of magnitude. As the presence of competing aggregation pathways is not restricted to porphyrins, but has also been observed in other systems,27,46 the observed robustness and increased responsiveness are not limited to our porphyrin system, but should be readily observed in other systems. The emergent behavior resulting from the competitive interactions in such systems shows a strong resemblance to supramolecular polymers in nature and holds great promise to further the general design principles of supramolecular materials.
Acknowledgments
M.F.J.M. acknowledges Marçin L. Ślęczkowski for synthesizing the chiral amine precursor for (S)-1 and Ralf A. A. Bovee for MALDI-ToF MS measurements. Tom F. A. de Greef is acknowledged for fruitful discussions. We acknowledge financial support from NWO (TOP-PUNT Grant 10018944) and the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02388.
Additional UV–vis, CD, and IR spectra; details of the thermodynamic model; additional speciation plots, AFM results, results of the fitting procedures, and phase diagrams, including Figures S1–S14 and Tables S1 and S2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Perutz M. F.; Fermi G.; Luisi B.; Shaanan B.; Liddington R. C. Stereochemistry of Cooperative Mechanisms in Hemoglobin High-Spin to Low-Spin Ferrous Iron for the Purpose of Efficient Oxygen Transport: Without Use of That Spin. Acc. Chem. Res. 1987, 20 (9), 309–321. 10.1021/ar00141a001. [DOI] [Google Scholar]
- Rivetti C.; Mozzarelli A.; Rossi G. L.; Henry E. R.; Eaton W. A. Oxygen Binding by Single Crystals of Hemoglobin. Biochemistry 1993, 32 (11), 2888–2906. 10.1021/bi00062a021. [DOI] [PubMed] [Google Scholar]
- Jordan P.; Fromme P.; Witt H. T.; Klukas O.; Saenger W.; Krauß N. Three-Dimensional Structure of Cyanobacterial Photosystem I at 2.5 Å Resolution. Nature 2001, 411 (6840), 909–917. 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- Miyatake T.; Tamiaki H. Self-Aggregates of Bacteriochlorophylls-C, D and E in a Light-Harvesting Antenna System of Green Photosynthetic Bacteria: Effect of Stereochemistry at the Chiral 3-(1-Hydroxyethyl) Group on the Supramolecular Arrangement of Chlorophyllous Pigments. J. Photochem. Photobiol., C 2005, 6, 89–107. 10.1016/j.jphotochemrev.2005.06.001. [DOI] [Google Scholar]
- Sengupta S.; Würthner F. Chlorophyll J-Aggregates: From Bioinspired Dye Stacks to Nanotubes, Liquid Crystals, and Biosupramolecular Electronics. Acc. Chem. Res. 2013, 46 (11), 2498–2512. 10.1021/ar400017u. [DOI] [PubMed] [Google Scholar]
- Elemans J. A. A. W.; van Hameren R.; Nolte R. J. M.; Rowan A. E. Molecular Materials by Self-Assembly of Porphyrins, Phthalocyanines, and Perylenes. Adv. Mater. 2006, 18 (10), 1251–1266. 10.1002/adma.200502498. [DOI] [Google Scholar]
- Van der Boom T.; Hayes R. T.; Zhao Y.; Bushard P. J.; Weiss E. A.; Wasielewski M. R. Charge Transport in Photofunctional Nanoparticles Self-Assembled from Zinc 5,10,15,20-Tetrakis(perylenediimide)porphyrin Building Blocks. J. Am. Chem. Soc. 2002, 124 (32), 9582–9590. 10.1021/ja026286k. [DOI] [PubMed] [Google Scholar]
- D’Souza F.; Smith P. M.; Zandler M. E.; McCarty A. L.; Itou M.; Araki Y.; Ito O. Energy Transfer Followed by Electron Transfer in a Supramolecular Triad Composed of Boron Dipyrrin, Zinc Porphyrin, and Fullerene: A Model for the Photosynthetic Antenna-Reaction Center Complex. J. Am. Chem. Soc. 2004, 126 (25), 7898–7907. 10.1021/ja030647u. [DOI] [PubMed] [Google Scholar]
- Winters M. U.; Dahlstedt E.; Blades H. E.; Wilson C. J.; Frampton M. J.; Anderson H. L.; Albinsson B. Probing the Efficiency of Electron Transfer through Porphyrin-Based Molecular Wires. J. Am. Chem. Soc. 2007, 129 (14), 4291–4297. 10.1021/ja067447d. [DOI] [PubMed] [Google Scholar]
- Anderson H. L. Building Molecular Wires from the Colours of Life: Conjugated Porphyrin Oligomers. Chem. Commun. 1999, 2323–2330. 10.1039/a904209a. [DOI] [Google Scholar]
- Rubires R.; Farrera J.-A.; Ribó J. M. Stirring Effects on the Spontaneous Formation of Chirality in the Homoassociation of Diprotonatedmeso-Tetraphenylsulfonato Porphyrins. Chem. - Eur. J. 2001, 7 (2), 436–446. . [DOI] [PubMed] [Google Scholar]
- Ribó J. M.; Crusats J.; Sagués F.; Claret J.; Rubires R. Chiral Sign Induction by Vortices during the Formation of Mesophases in Stirred Solutions. Science 2001, 292 (5524), 2063–2066. 10.1126/science.1060835. [DOI] [PubMed] [Google Scholar]
- Jintoku H.; Ihara H.; Matsuzawa Y.; Kihara H. Static Electricity-Responsive Supramolecular Assembly. Chem. - Eur. J. 2017, 23 (67), 16961–16965. 10.1002/chem.201704154. [DOI] [PubMed] [Google Scholar]
- Helmich F.; Lee C. C.; Nieuwenhuizen M. M. L.; Gielen J. C.; Christianen P. C. M.; Larsen A.; Fytas G.; Leclère P. E. L. G.; Schenning A. P. H. J.; Meijer E. W. Dilution-Induced Self-Assembly of Porphyrin Aggregates: A Consequence of Coupled Equilibria. Angew. Chem., Int. Ed. 2010, 49 (23), 3939–3942. 10.1002/anie.201000162. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Living Supramolecular Polymerization Realized through a Biomimetic Approach. Nat. Chem. 2014, 6 (3), 188–195. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Würthner F. Mechanism of Self-Assembly Process and Seeded Supramolecular Polymerization of Perylene Bisimide Organogelator. J. Am. Chem. Soc. 2015, 137 (9), 3300–3307. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Fukui T.; Kawai S.; Fujinuma S.; Matsushita Y.; Yasuda T.; Sakurai T.; Seki S.; Takeuchi M.; Sugiyasu K. Control over Differentiation of a Metastable Supramolecular Assembly in One and Two Dimensions. Nat. Chem. 2016, 9 (5), 493–499. 10.1038/nchem.2684. [DOI] [PubMed] [Google Scholar]
- Venkata Rao K.; Miyajima D.; Nihonyanagi A.; Aida T. Thermally Bisignate Supramolecular Polymerization. Nat. Chem. 2017, 9 (11), 1133–1139. 10.1038/nchem.2812. [DOI] [PubMed] [Google Scholar]
- Fennel F.; Wolter S.; Xie Z.; Plötz P.-A.; Kühn O.; Würthner F.; Lochbrunner S. Biphasic Self-Assembly Pathways and Size-Dependent Photophysical Properties of Perylene Bisimide Dye Aggregates. J. Am. Chem. Soc. 2013, 135 (50), 18722–18725. 10.1021/ja409597x. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Grzeszkiewicz C.; Würthner F. Pathway Complexity in the Self-Assembly of a Zinc Chlorin Model System of Natural Bacteriochlorophyll J-Aggregates. Chem. Sci. 2018, 9 (10), 2768–2773. 10.1039/C7SC03725B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K.; Xie J.; Zhang D.; Shi W.; Yan Q.; Zhao D. Concurrent Cooperative J-Aggregates and Anticooperative H-Aggregates. J. Am. Chem. Soc. 2018, 140 (17), 5764–5773. 10.1021/jacs.8b01463. [DOI] [PubMed] [Google Scholar]
- Van der Weegen R.; Teunissen A. J. P.; Meijer E. W. Directing the Self-Assembly Behaviour of Porphyrin-Based Supramolecular Systems. Chem. - Eur. J. 2017, 23 (15), 3773–3783. 10.1002/chem.201605872. [DOI] [PubMed] [Google Scholar]
- The Q-bands of the porphyrin are slightly red-shifted in the isodesmic aggregates with respect to the free monomer (see Figure S5). This suggests a weak J-coupling in the isodesmic aggregate, although the exact geometry of the aggregate remains ambiguous. For a detailed discussion on J- and H-coupled aggregates, see:Hestand N. J.; Spano F. C. Acc. Chem. Res. 2017, 50 (2), 341–350. 10.1021/acs.accounts.6b00576. [DOI] [PubMed] [Google Scholar]; Although the exact orientation of the aggregate is ambiguous, the observed robustness and increased response are not determined by the nature of the coupling in the isodesmic aggregate.
- Van Zee N. J.; Adelizzi A.; Mabesoone M. F. J.; Meng X.; Aloi A.; Zha R. H.; Lutz M.; Filot I. A. W.; Palmans A. R. A.; Meijer E. W. Potential Enthalpic Energy of Water in Oils Exploited to Control Supramolecular Structure. Nature 2018, 558 (7708), 100–103. 10.1038/s41586-018-0169-0. [DOI] [PubMed] [Google Scholar]
- We observed in repeated runs of denaturation and melting experiments that the absolute value of the CD intensity varies from measurement to measurement. Additionally, we observed that sealing the cuvette with a screw cap greatly enhances the reproducibility of the melting curves, which indicates that variations in water content cause this variation. The water dependency of the solutions of (S)-1 illustrates the many local minima that can be encountered and the intricate interactions that play a role in a multidimensional energy landscape of systems with increased pathway complexity.
- Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A. P. H. J.; De Greef T. F. A.; Meijer E. W. Pathway Complexity in Supramolecular Polymerization. Nature 2012, 481 (7382), 492–496. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]
- Van Der Zwaag D.; Pieters P. A.; Korevaar P. A.; Markvoort A. J.; Spiering A. J. H.; De Greef T. F. A.; Meijer E. W. Kinetic Analysis as a Tool to Distinguish Pathway Complexity in Molecular Assembly: An Unexpected Outcome of Structures in Competition. J. Am. Chem. Soc. 2015, 137 (39), 12677–12688. 10.1021/jacs.5b08138. [DOI] [PubMed] [Google Scholar]
- Markvoort A. J.; ten Eikelder H. M. M.; Hilbers P. A. J.; de Greef T. F. A.; Meijer E. W. Theoretical Models of Nonlinear Effects in Two-Component Cooperative Supramolecular Copolymerizations. Nat. Commun. 2011, 2 (4), 509. 10.1038/ncomms1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Eikelder H. M. M.; Markvoort A. J.; De Greef T. F. A.; Hilbers P. A. J. An Equilibrium Model for Chiral Amplification in Supramolecular Polymers. J. Phys. Chem. B 2012, 116 (17), 5291–5301. 10.1021/jp300622m. [DOI] [PubMed] [Google Scholar]
- Cantekin S.; Ten Eikelder H. M. M.; Markvoort A. J.; Veld M. A. J.; Korevaar P. A.; Green M. M.; Palmans A. R. A.; Meijer E. W. Consequences of Cooperativity in Racemizing Supramolecular Systems. Angew. Chem., Int. Ed. 2012, 51 (26), 6426–6431. 10.1002/anie.201201701. [DOI] [PubMed] [Google Scholar]
- Das A.; Vantomme G.; Markvoort A. J.; Ten Eikelder H. M. M.; Garcia-Iglesias M.; Palmans A. R. A.; Meijer E. W. Supramolecular Copolymers: Structure and Composition Revealed by Theoretical Modeling. J. Am. Chem. Soc. 2017, 139 (20), 7036–7044. 10.1021/jacs.7b02835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The model of Würthner and Lochbrunner, in which the monomers can dimerize and form nucleated polymers, shows very similar trends in composition at a set concentration as a function of the solvent composition. However, due to the absence of the isodesmic pathway, the buffering effect is not present, and no robustness is observed. For a more detailed discussion, see the Supporting Information.
- Korevaar P. A.; Schaefer C.; De Greef T. F. A.; Meijer E. W. Controlling Chemical Self-Assembly by Solvent-Dependent Dynamics. J. Am. Chem. Soc. 2012, 134 (32), 13482–13491. 10.1021/ja305512g. [DOI] [PubMed] [Google Scholar]
- Bellot M.; Bouteiller L. Thermodynamic Description of Bis-Urea Self-Assembly: Competition between Two Supramolecular Polymers. Langmuir 2008, 24 (24), 14176–14182. 10.1021/la802367r. [DOI] [PubMed] [Google Scholar]
- Roman M.; Cannizzo C.; Pinault T.; Isare B.; Andrioletti B.; Van Der Schoot P.; Bouteiller L. Supramolecular Balance: Using Cooperativity to Amplify Weak Interactions. J. Am. Chem. Soc. 2010, 132 (47), 16818–16824. 10.1021/ja105717u. [DOI] [PubMed] [Google Scholar]
- Engelkamp H.; Middelbeek S.; Nolte R. J. Self-Assembly of Disk-Shaped Molecules to Coiled-Coil Aggregates with Tunable Helicity. Science 1999, 284 (5415), 785–788. 10.1126/science.284.5415.785. [DOI] [PubMed] [Google Scholar]
- Lohr A.; Lysetska M.; Würthner F. Supramolecular Stereomutation in Kinetic and Thermodynamic Self-Assembly of Helical Merocyanine Dye Nanorods. Angew. Chem., Int. Ed. 2005, 44 (32), 5071–5074. 10.1002/anie.200500640. [DOI] [PubMed] [Google Scholar]
- Aparicio F.; Sánchez L. Thermodynamics of the Helical, Supramolecular Polymerization of Linear Self-Asembling Molecules: Influence of Hydrogen Bonds and π Stacking. Chem. - Eur. J. 2013, 19 (32), 10482–10486. 10.1002/chem.201300553. [DOI] [PubMed] [Google Scholar]
- Hifsudheen M.; Mishra R. K.; Vedhanarayanan B.; Praveen V. K.; Ajayaghosh A. The Helix to Super-Helix Transition in the Self-Assembly of π-Systems: Superseding of Molecular Chirality at Hierarchical Level. Angew. Chem., Int. Ed. 2017, 56 (41), 12634–12638. 10.1002/anie.201707392. [DOI] [PubMed] [Google Scholar]
- Li L. S.; Jiang H.; Messmore B. W.; Bull S. R.; Stupp S. I. A Torsional Strain Mechanism to Tune Pitch in Supramolecular Helices. Angew. Chem., Int. Ed. 2007, 46 (31), 5873–5876. 10.1002/anie.200701328. [DOI] [PubMed] [Google Scholar]
- Tsai W. W.; Li L.; Cui H.; Jiang H.; Stupp S. I. Self-Assembly of Amphiphiles with Terthiophene and Tripeptide Segments into Helical Nanostructures. Tetrahedron 2008, 64 (36), 8504–8514. 10.1016/j.tet.2008.06.033. [DOI] [Google Scholar]
- Yamauchi M.; Ohba T.; Karatsu T.; Yagai S. Photoreactive Helical Nanoaggregates Exhibiting Morphology Transition on Thermal Reconstruction. Nat. Commun. 2015, 6, 8936. 10.1038/ncomms9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Guo Z.; Chen J.; Li W.; Zhong L.; Gao Y.; Jiang L.; Chi L.; Tian H.; Zhu W. H. Enabling Light Work in Helical Self-Assembly for Dynamic Amplification of Chirality with Photoreversibility. J. Am. Chem. Soc. 2016, 138 (7), 2219–2224. 10.1021/jacs.5b11580. [DOI] [PubMed] [Google Scholar]
- Kulkarni C.; Korevaar P. A.; Bejagam K. K.; Palmans A. R. A.; Meijer E. W.; George S. J. Solvent Clathrate Driven Dynamic Stereomutation of a Supramolecular Polymer with Molecular Pockets. J. Am. Chem. Soc. 2017, 139 (39), 13867–13875. 10.1021/jacs.7b07639. [DOI] [PubMed] [Google Scholar]
- We recognize the possibility of incorporating a stack-length-dependent helix–superhelix equilibrium in the model. However, since the superhelices do not have distinct CD signature (Figure S9), spectroscopic investigation into the superhelix formation mechanism is not possible, impairing the unambiguous quantitative implementation of the transition in the model.
- Haedler A. T.; Meskers S. C. J.; Zha R. H.; Kivala M.; Schmidt H. W.; Meijer E. W. Pathway Complexity in the Enantioselective Self-Assembly of Functional Carbonyl-Bridged Triarylamine Trisamides. J. Am. Chem. Soc. 2016, 138 (33), 10539–10545. 10.1021/jacs.6b05184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.