

Abstract
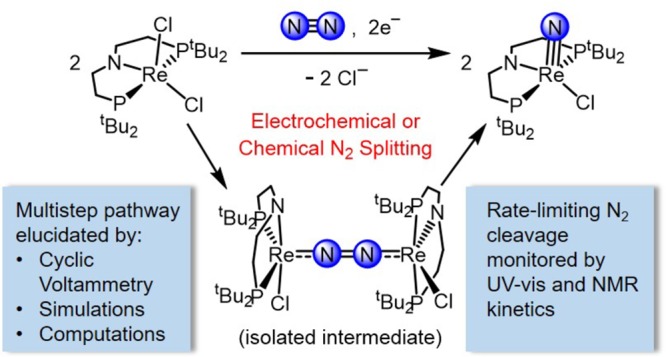
A comprehensive mechanistic study of N2 activation and splitting into terminal nitride ligands upon reduction of the rhenium dichloride complex [ReCl2(PNP)] is presented (PNP– = N(CH2CH2PtBu2)2–). Low-temperature studies using chemical reductants enabled full characterization of the N2-bridged intermediate [{(PNP)ClRe}2(N2)] and kinetic analysis of the N–N bond scission process. Controlled potential electrolysis at room temperature also resulted in formation of the nitride product [Re(N)Cl(PNP)]. This first example of molecular electrochemical N2 splitting into nitride complexes enabled the use of cyclic voltammetry (CV) methods to establish the mechanism of reductive N2 activation to form the N2-bridged intermediate. CV data was acquired under Ar and N2, and with varying chloride concentration, rhenium concentration, and N2 pressure. A series of kinetic models was vetted against the CV data using digital simulations, leading to the assignment of an ECCEC mechanism (where “E” is an electrochemical step and “C” is a chemical step) for N2 activation that proceeds via initial reduction to ReII, N2 binding, chloride dissociation, and further reduction to ReI before formation of the N2-bridged, dinuclear intermediate by comproportionation with the ReIII precursor. Experimental kinetic data for all individual steps could be obtained. The mechanism is supported by density functional theory computations, which provide further insight into the electronic structure requirements for N2 splitting in the tetragonal frameworks enforced by rigid pincer ligands.
1. Introduction
Industrial ammonia synthesis produces more than 150 Mt/a of fixed nitrogen, securing the nutrition of about half of Earth’s population with synthetic fertilizers, and serving as the feedstock for most nitrogen-containing organic chemicals.1 The Haber–Bosch process fixes N2 using the chemical reductant H2, and thus relies on energy-intensive steam reforming of fossil fuels, leading the overall process to consume about 1–2% of the global energy supply. More sustainable approaches to ammonia production, such as electrochemical N2 fixation, would therefore be highly desirable.2 Examples of the use of electrochemical methods to achieve catalysis, or even key individual steps relevant to N2 functionalization, are deeply limited. Heterogeneous electrode materials capable of the nitrogen reduction reaction (NRR) to ammonia have been reported, with recent advances tending toward ambient temperature and pressure conditions.3−5 However, Faradaic yields of the challenging 6e–/6H+ NRR are low, usually due to the competitive hydrogen evolution reaction (HER), a 2e–/2H+ process with a comparable standard reduction potential.6 Molecular catalysts typically exhibit much higher ammonia selectivities,7 but mainly rely on chemical reductants. Importantly, despite some early examples over 30 years ago, electrochemical NRR with molecular (pre)catalysts is still in its infancy.8
Advances in electrochemical nitrogen fixation with molecular catalyst platforms will rely on establishing detailed mechanistic insights. Almost all well-defined molecular catalysts have been proposed to follow sequential N2 protonation/reduction mechanisms9 that are related to that of the [Fe,Mo]-nitrogenase enzyme, which is still under investigation.10 This pathway contrasts with the mechanism of the heterogeneously catalyzed Haber–Bosch process that passes through initial, turnover-limiting dissociative N2 chemisorption to surface-bound nitrides on the way to ammonia formation. A well-defined molecular model for the challenging N2 splitting step was first reported by Laplaza and Cummins in 1995 starting from a three-coordinate molybdenum(III) tris-anilide (Scheme 1).11 This platform binds N2 to form a dinuclear, linearly N2-bridged complex that directly cleaves the extremely strong N–N bond (225 kcal/mol) into molybdenum(VI) nitrides at room temperature.12 Several other examples of N2 splitting into molecular nitrides with group 4–8 transition metals and uranium have since been reported,13,14 and further employed to directly utilize N2 in the synthesis of organic amines, nitriles, or isocyanates.15 However, for most of these systems the reactive intermediates that activate N2 and the mechanistic details of splitting are not known. Furthermore, electrochemically driven N2 cleavage into nitrides remains unknown.16
Scheme 1. N2 Splitting into Terminal Nitrides with Molybdenum Tris-anilides and Transition Metal Pincer Halide Platforms Reported in the Literature11,15d,17,19.
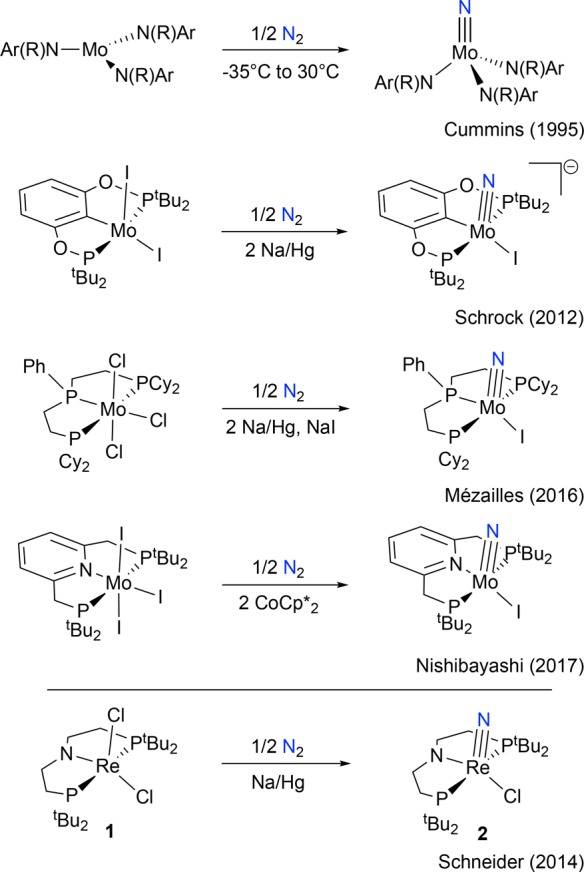
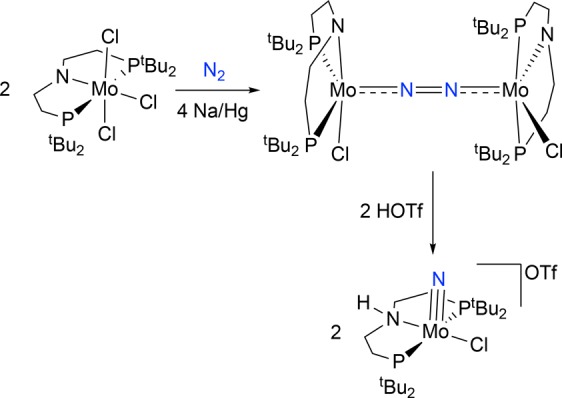
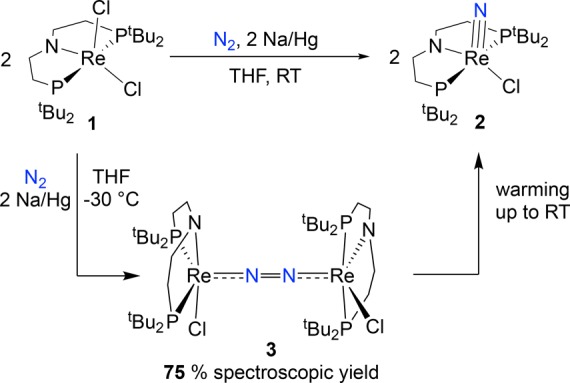
The groups of Schrock, Mézailles, and Nishibayashi independently reported stoichiometric N2 activation and splitting into terminal nitrides upon reduction of pincer supported Mo halide complexes (Scheme 1).15d,17 Nishibayashi’s nitride is also an active catalyst for ammonia synthesis,17b providing early evidence that N2 splitting into nitrides might in fact be relevant to N2 fixation. Experimental information regarding the nature and reactivity patterns of the key intermediates in these pincer supported N2 splitting reactions remains highly limited, particularly for pathways that proceed via in situ reduction of transition metal halide precursors. Dinuclear N2-bridged species are possible intermediates in these reactions, and in fact, starting from a MoIV halide precursor, Schneider and co-workers isolated the N2-bridged complex [{(PNP)ClMo}2(N2)] (PNP– = N(CH2CH2PtBu2)2–), which splits into MoV nitrides upon protonation (Scheme 2).18 This group also expanded N2 splitting reactivity to group 7 precursors.19 Chemical reduction of a rhenium(III) pincer complex [ReCl2(PNP)] (1) under 1 atm N2 affords the terminal nitride [Re(N)Cl(PNP)] (2) in over 80% yield (Scheme 1). The N2-bridged dinuclear complex [{(PNP)ClRe}2(N2)] (3) was proposed as an intermediate based on density functional theory (DFT) computations. Like most of the pincer complexes capable of splitting N2, experimental data regarding the mechanism of formation and structure of polynuclear intermediates remains elusive. In contrast to the molybdenum pincer platforms, reduction of the rhenium system requires only one electron equivalent per metal ion. We therefore chose this system to examine the full mechanism of N2 splitting starting from 1 as an archetypal pincer halide precursor utilized in N2 cleavage. In this article, we present a comprehensive mechanistic picture that evolved from a combined synthetic, spectroscopic, crystallographic, kinetic, electrochemical, and computational study. We also report the first example of electrochemically driven N2 splitting into well-defined molecular nitrides.
Scheme 2. N2 Splitting upon Protonation of an N2-Bridged PNP Pincer Complex18.
2. Results
2.1. Chemical N2 Splitting
The mechanism of N2 splitting was first examined by spectroscopic monitoring of the chemical reduction of 1 under N2. Our previous DFT model predicted a sizable kinetic barrier for the N–N splitting step that forms the final nitride product 2,19 suggesting that an intermediate might be observable. In fact, the presence of a relatively long-lived intermediate was indicated by a temporary red coloration of the mixture during the reduction of violet 1 with Na/Hg to yellow nitride 2 at room temperature.
In an attempt to characterize the intermediate, Na/Hg reduction was carried out in tetrahydrofuran (THF) at −30 °C, leading to a persistent red solution with two new bands at 375 and 533 nm in the electronic absorption spectrum (Figure S4). NMR spectroscopic monitoring at −30 °C gave further insights. The decay of 1 upon reduction was accompanied by the formation of two doublets (δ −120 and +18 ppm) in the 31P{1H} NMR spectrum (Figure 1). The doublets are correlated in a 31P–31P correlation spectroscopy (COSY) experiment (Figure S11) and feature a typical trans P–M–P coupling constant (2JPP = 235 Hz), indicating chemically inequivalent phosphine substituents at a single Re(PNP) fragment. The 1H NMR spectrum of the Re intermediate features sharp resonances covering a wide range of chemical shifts (−16 to +12 ppm, Figure S5) that correlate to the two 31P signals in a 1H–31P heteronuclear multiple-bond correlation spectroscopy (HMBC) experiment (Figure S10). The number and multiplicity of the NMR signals resemble the N2-bridged dimolybdenum complex [{(PNP)ClMo}2(N2)], which also features two doublets in the 31P{1H} NMR spectrum and 12 1H NMR signals for the ligand tert-butyl (4) and backbone ethylene linker protons (8), respectively.18 This NMR pattern is attributed to a rigid, C2-symmetric conformation enforced by the bulky phosphine substituents. Reduction of 1 under 15N2 at −30 °C revealed a single new 15N{1H} NMR signal at 211 ppm (besides the nitride signal of product 2 at 369 ppm; Figure S12), fully consistent with a symmetrically N2-bridged intermediate (and inconsistent with a terminal N2 ligand). The chemical shift lies in the typical range for coordinated N2,20 but the value notably differs from the isostructural Mo complex [{(PNP)ClMo}2(N2)] (δ = 69 ppm). The combined spectroscopic data identify the intermediate as the N2-bridged dimer [{(PNP)ClRe}2(N2)] (3, Scheme 3). The characterization of the dinuclear intermediate 3 (∼75% yield at −30 °C based on 1H NMR integration, Figure S6) substantiates our previous computational proposal.19
Figure 1.
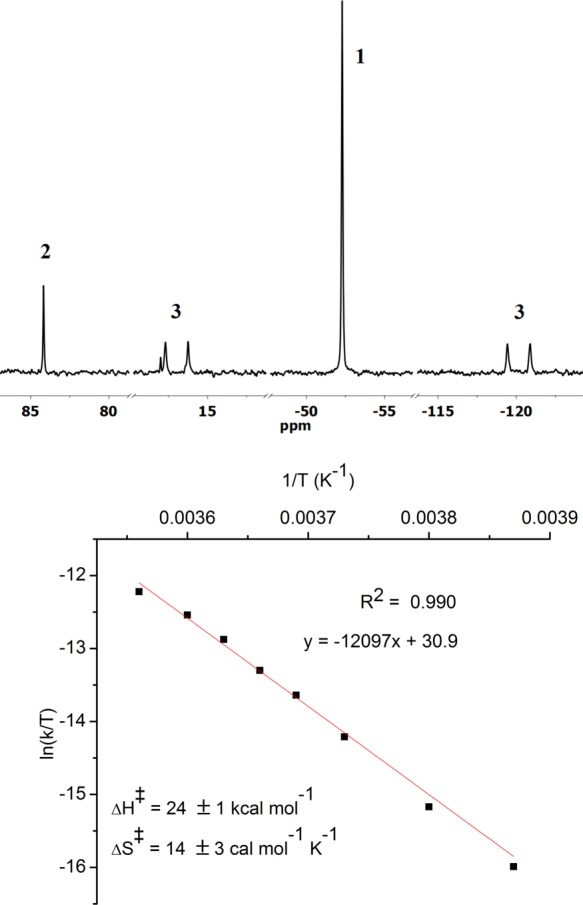
(top) 31P{1H} NMR spectrum during reduction of 1 at −30 °C under N2. (bottom) Eyring plot for the conversion of 3 to 2 in the temperature range −15 to +7.5 °C.
Scheme 3. N2 Splitting upon Chemical Reduction of Rhenium(III) Complex 1 via N2-Bridged Dirhenium Complex 3.
The unusual chemical shift ranges found for the 1H, 31P, and 15N NMR spectra of 3 require an additional comment. These strongly shifted, yet narrow, NMR lines are tentatively attributed to an expression of temperature independent paramagnetism (TIP).21 Unusual chemical shift parameters of rhenium(III) phosphine complexes that were attributed to TIP are well-documented.22 TIP arises from an open-shell electronic structure with a thermally isolated ground state (≫kBT) that results from strong mixing with excited states via spin–orbit coupling, as was found for the related square-planar complex [OsIICl{N(CHCHPtBu2)2}].23 Variable-temperature NMR (VT-NMR) spectra of 3 between −55 and −5 °C feature signals that are almost all temperature independent, supporting TIP. Only the 31P peak at −120 ppm (Δδ = 8 ppm) and one 1H signal of the pincer backbone at 0.4 ppm (Δδ = 1.5 ppm) show some temperature dependence (Figures S13–S15). However, these signals scale linearly with T–1, which would not be expected for the thermal population of a higher lying electronic state (Figures S16 and S17). Furthermore, the tBu 1H NMR signals exhibit significant broadening at lower temperatures, suggesting that an additional dynamic process, such as freezing rotation of the tBu groups at low temperatures, is responsible for the temperature dependence of these two signals. Unfortunately, the low chemical stability of 3 in solution precluded the isolation of samples suitable for SQUID magnetometric characterization to confirm TIP.
The structure of 3 was confirmed through an X-ray diffraction study (Figure 2) of single crystals grown at −80 °C from a THF solution layered with pentane. The asymmetric unit contains one dinuclear molecule of 3 and one mononuclear molecule (comprised of a 50/50 mix of cocrystallized parent 1 and nitride 2). The molecular structure features a μ2:η1:η1-N2 ligand that bridges the apical positions of square-pyramidal rhenium ions (τ5 = 0.18 on Re1 and τ5 = 0.16 on Re2).24 The {ReCl(PNP)} fragments are twisted with respect to each other (Cl1–Re1–Re2–Cl2: 112.1°), which interlocks the bulky PtBu2 groups. This rigid conformation is in agreement with the C2-symmetric structure observed in solution. A moderate degree of activation is indicated by the N2 bond length (1.202(10) Å), which is intermediate between free dinitrogen (1.10 Å)25 and diazene (1.25 Å).26 In turn, the short Re–NN–Re bonds (1.861(6) and 1.886(8) Å) support considerable metal-to-N2 π-backbonding. The conformation and the other structural features are in excellent agreement with the prior computational model.19
Figure 2.
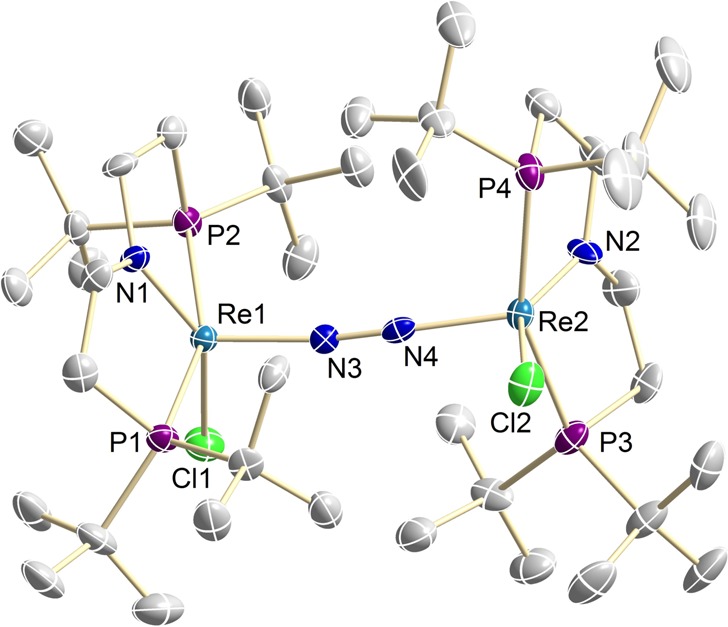
Molecular structure of 3 from single-crystal X-ray diffraction with anisotropic displacement parameters drawn at the 50% probability level. Hydrogen atoms and cocrystallized 1 and 2 are omitted for clarity. Selected bond lengths (Å) and angles (deg): Re1–N3 1.861(8), Re1–N1 1.937(7), N3–N4 1.202(10), Re2–N4 1.886(8), Re2–N4 1.949(8), N1–Re1–Cl2 146.30(2), P1–Re1–P2 157.38(9), P3–Re2–P4 156.28(9), N2–Re2–Cl2 146.6(3), Cl1–Re1–Re2–Cl2 112.0(6).
The direct observation of 3 in high spectroscopic yields at low temperatures allowed for kinetic examination of the N2 splitting reaction. Warming a solution of 3 to room temperature resulted in selective formation of nitride 2 according to 31P and 1H NMR spectroscopies. Monitoring the decay of 3 by 1H NMR spectroscopy at a particular temperature revealed clean first-order kinetics over more than two half-lives (Figure S1). An Eyring analysis within the temperature range −15 to +7.5 °C (Figure 1) provides activation parameters for N2 splitting: ΔH‡ = +24 ± 1 kcal·mol–1; ΔS‡ = 14 ± 3 cal·mol–1·K–1. These experimental activation parameters compare well with the previously computed free energy of activation (+20.2 kcal/mol)19 and the values obtained for a slightly refined computational model (see section 2.3).
After identifying 3 as the pivotal intermediate that splits N2 in the formation of nitride 2, reactivity relevant to the assembly of the N2-bridged species was examined. Dinitrogen binding and chloride dissociation were considered likely initial chemical processes. The possibility of N2 binding to 1 was investigated using infrared (IR) spectroscopy, but a solution of 1 in THF under 1 atm N2 gave no indication of end-on binding of dinitrogen (Figure S25). Furthermore, UV–vis spectra obtained under Ar and N2 were identical, even upon cooling from room temperature to −78 °C (Figures S26 and S27). Finally, 31P{1H} NMR spectra of 1 showed no perceptible difference when recorded under Ar or N2 (1 or 5 atm), even upon cooling to −95 °C (Figures S20–S22). On the basis of these experiments, a limiting binding constant for N2 binding to 1 can be estimated (Keq < 1 M–1, Supporting Information, section 5). The interaction of 1 with chloride, which is released during the N2 splitting reaction, was also assessed. NMR spectroscopic monitoring of a solution of 1 under N2 in the presence of excess [nBu4N]Cl indicated chemical stability and no chloride association over more than 48 h (Figure S24). These binding studies indicate an alternative initial step, such as reduction of rhenium(III). Therefore, electrochemical methods were employed to interrogate formation of 3.
2.2. Electrochemical N2 Splitting
2.2.1. Electrochemical Synthesis of Nitrides from N2
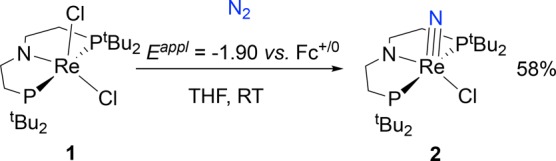
To probe whether N2 splitting can be driven electrochemically, controlled potential electrolysis (CPE) of 1 was explored. Electrolysis of 1 in THF containing 0.2 M [nBu4N][PF6] was carried out at −1.90 V vs Fc+/0 under 1 atm N2,27 based on the CV data for 1 (see section 2.2.4) and the successful use of Co(Cp*)2 (E° = −1.9 V)28 as a chemical reductant for this reaction. During electrolysis at room temperature, the color of the solution changed from purple to yellow as charge amounting to 1.2 electrons per Re was passed. Analysis of the product mixture by 31P{1H} NMR spectroscopy showed that 2 was produced in 58% yield (Scheme 4, Figure S19). An analogous CPE experiment of 1 under 1 atm Ar instead of N2 gave an intractable mixture of unidentified products. Notably, the conversion of 1 to 2 under N2 represents the first synthesis of a nitride complex via electrochemically driven dinitrogen cleavage.
Scheme 4. Electrochemical Conversion of 1 to Rhenium(V) Nitride Complex 2.
2.2.2. UV–Vis Spectroelectrochemical Examination
UV–visible spectroelectrochemistry (SEC) was utilized to probe for the buildup of intermediates during electrolysis. Electrolysis resulted in conversion of 1 (λmax = 530 nm) to a new species with strong absorbances at 384 and 537 nm within 60 s (Figure 3). This intermediate was not observed when the corresponding UV–vis SEC experiment was conducted under Ar (Figure S28), clearly indicating the role of N2 in its formation. The intermediate is assigned as N2-bridged dimer 3, based on the close agreement of the spectral features with a solution of 3 obtained by chemical reduction at −30 °C (λmax = 375 and 533 nm, see section 2.1) and the time-dependent (TD) DFT computed UV–vis spectrum for 3 (see section 2.3 and Figure S4). Upon continued electrolysis, the intermediate converted to product 2 (λmax = 393 nm) over the course of about 2 min (Figure 3, inset). Kinetic data for the N2 splitting chemical reaction was obtained by fitting the decay of 3 in the time period after a minimum in electrolytic current was reached (approximately 100 s) to avoid significant impact by diffusion or competing electrochemical processes. The half-life obtained under these conditions for the conversion of 3 into 2, t1/2 = 28 s at 298 K (Figure S2), is in close agreement with the rate constant derived for splitting of 3 into 2 after chemical reduction of 1 under N2 (k298 K = 0.018 s–1, t1/2 = 38 s, see section 2.1).
Figure 3.
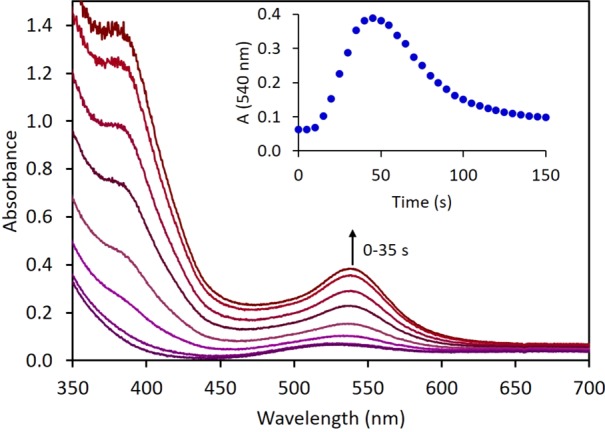
UV–vis spectroelectrochemistry (SEC) electrolysis of 1, showing the absorption profile during conversion of 1 to intermediate 3. (inset) Time course tracking absorbance at 540 nm for formation and decay of 3. Conditions: 2 mM 1, 0.2 M [nBu4N][PF6] in THF at 25 °C, −1.45 V vs Ag wire pseudoreference constant applied potential over 10 min, Au working and counter electrodes, Ag wire pseudoreference electrode in a “honeycomb” cell arrangement.
2.2.3. Cyclic Voltammetry under Ar
Several elementary steps must be involved on the pathway from complex 1 to the N2-bridged dimer and ultimately to the nitride products of N2 cleavage. We therefore turned to cyclic voltammetry (CV) to build a deeper understanding of the initial steps of reductive N2 cleavage. Reduction under Ar was examined first in order to rationalize the redox behavior of the rhenium platform in the absence of N2; the electrochemical behavior under N2 is described in the subsequent section.
CV of 1 under Ar shows two reduction processes at −1.96 and −2.24 V, respectively (Figure S33). The peak current of the first reduction wave (ip,c,1) increases linearly with the square root of the scan rate, indicating a diffusion-controlled process. Increased scan rates lead to a large cathodic shift in the peak potential of the first reduction of 1, which suggests that the first reduction is followed by a chemical reaction. Considering chloride dissociation a plausible reduction-triggered chemical step, the influence of chloride ion concentration on the first reduction process was examined. The peak potential of the first reduction shifts cathodically by ΔE = −0.04 V upon addition of 20 equiv of [nBu4N]Cl (Figure 4). This cathodic shift is accompanied by an increase in reversibility (ip,c,1/ip,a,1 = 1.4 with 20 equiv of Cl– and 0.1 V·s–1) when sweeping through only the initial reduction event. The second reduction process remains irreversible at all examined scan rates and is essentially unaffected by chloride addition. Varying the concentration of 1 from 0.5 to 4 mM has a negligible effect on the overall features of the CV and the peak current ratio of the two reductive features is virtually unaffected. These observations are consistent with an ECE mechanism (electrochemical–chemical–electrochemical reaction sequence). Initial reduction of 1 forms [ReCl2(PNP)]−, which undergoes reversible chloride ligand dissociation forming [ReCl(PNP)]. The second feature is assigned to irreversible reduction of [ReCl(PNP)].
Figure 4.
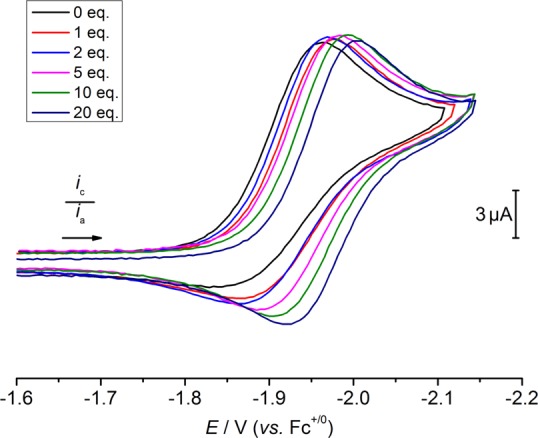
CV data of 1 in THF containing 0.2 M [nBu4N][PF6] under Ar with varying amounts of [nBu4N]Cl, ν = 0.1 V·s–1.
The proposed ECE mechanistic model was probed by digital simulation of the electrochemical data. The simulation parameters (formal potentials, rate constants, and electron transfer parameters) for the best fit are given in Table 1. Figure 5 compares the experimental and simulated CV data with 0–20 equiv of added chloride ion across a wide range of scan rates (see the Supporting Information for more details). The peak current of the second reduction process and the reversibility of the first process cannot be modeled satisfactorily by a simple ECE mechanism (i.e., reduction, chloride ion loss, reduction). In order to achieve good agreement of the simulated and experimental CV data, unimolecular (k2) decay of [ReCl(PNP)] had to be considered in addition to reduction of [ReCl2(PNP)] (E1) and [ReCl(PNP)] (E2), which are connected by reversible chloride dissociation from [ReCl2(PNP)]− (k1, K1; Scheme 5).
Table 1. Thermodynamic and Kinetic Parameters Obtained from Simulation of CV Data under Ar.
| Parameter | Value |
|---|---|
| K1 (M) | 0.05 |
| k1 (s–1) | 1000 |
| k2 (s–1) | 0.11 |
| E1 (V) | –2.00 |
| α1 | 0.5 |
| ks,1 (cm/s) | 0.05 |
| E2 (V) | –2.29 |
| α2 | 0.8 |
| ks,2 (cm/s) | 0.05 |
| E1 – E2 (V) | 0.29 |
Figure 5.
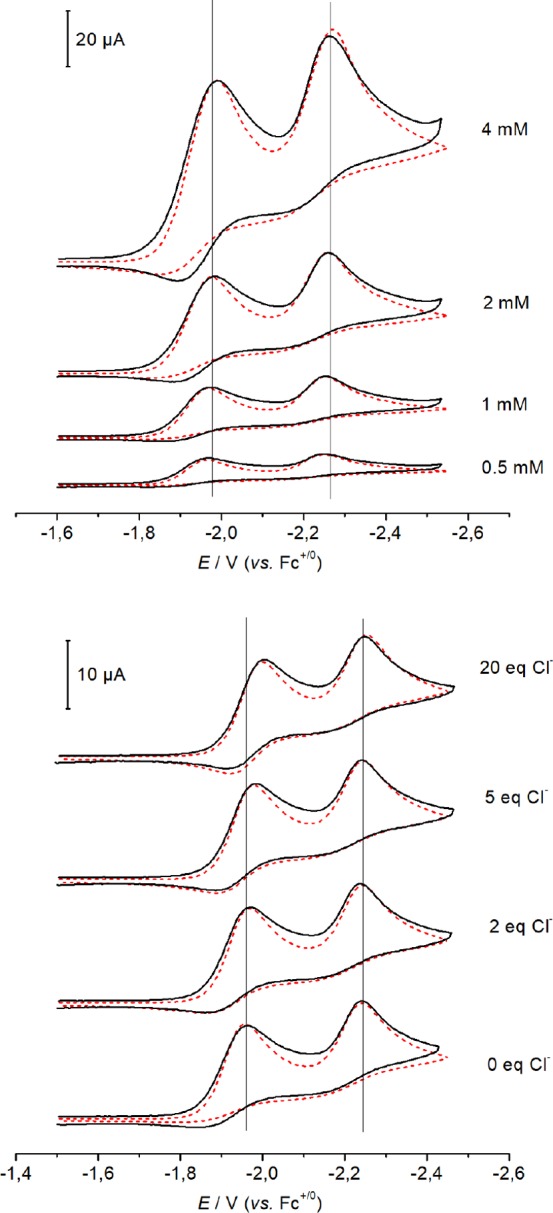
Overlay of experimental (black lines) and simulated (red dashed lines) CV data (I = 0.2 M [Bu4N][PF6] in THF at room temperature, ν = 0.1 V·s–1) for 1 at different concentrations (top) and with various amounts of Cl– ions (bottom). Simulations according to the mechanism in Scheme 5 with the thermodynamic and kinetic parameters given in Table 1.
Scheme 5. Minimum Mechanistic Models for Electrochemical Reduction of 1 under Ar (Orange Box) and under N2 (Gray Box), Respectively.

The PNP ligand is omitted for all species for clarity.
A sensitivity analysis was carried out to provide insight into the accuracy of the parameters derived from the simulation. Doubling/halving of k2 leads to substantially worse fits for the second reduction peak, which cannot be compensated satisfactorily by changing any other rate constant. The rate and equilibrium constants of chloride loss (k1 and K1) were extracted from the chloride titration experiments, and doubling/halving of the values leads to considerably worse fits.
2.2.4. Cyclic Voltammetry under N2
The CV data of 1 under N2 (1 atm) is dramatically different than under Ar (Figure 6). The peak potential of the first reduction process shifts anodically by 60 mV under N2 with respect to Ar (Ep,c = −1.90 V, ν = 0.1 V·s–1) and exhibits a substantial increase in the current response indicative of a multielectron reduction. The feature that is assigned to the reduction of [ReCl(PNP)] near −2.2 V under Ar is absent under N2; a new minor reduction feature is observed near Ep,c = −2.4 V (ν = 0.1 V·s–1).
Figure 6.
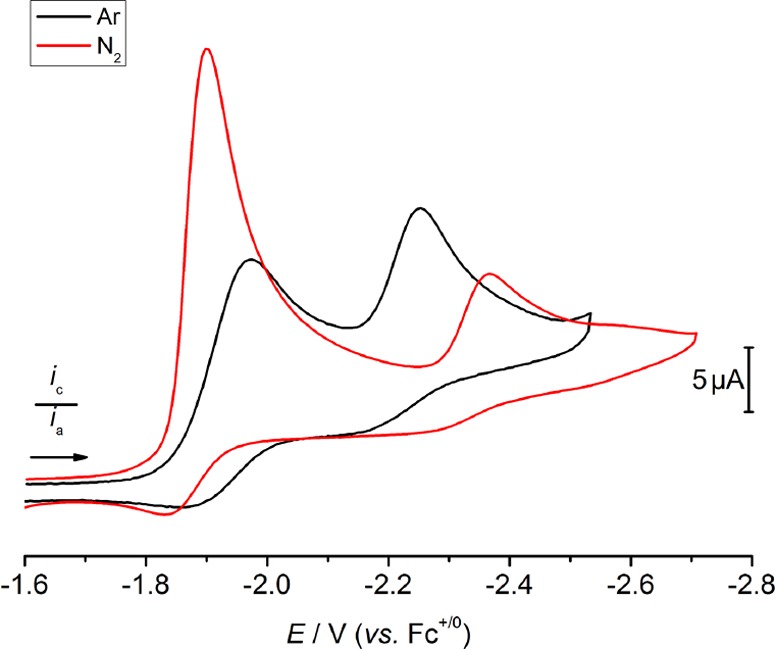
CV data of 1 in THF containing 0.2 M [nBu4N][PF6] under 1 atm N2 (red) and Ar (black). Conditions: 1 (0.8 mM) in 0.2 M [nBu4N][PF6] measured at 0.1 V·s–1.
Upon increasing scan rate, the peak potential of the first reduction process of 1 shifts cathodically and exhibits increasing reversibility, which indicates a coupled chemical reaction following initial reduction (Figure S40). This chemical reaction must involve N2, because the reduction feature assigned to [ReCl(PNP)] in the CV data under Ar is not present. The characteristic reversible ReVI/ReV redox couple (E1/2 = −0.09 V) of the nitride complex 2 is not observed among the several oxidation features in the return sweep, indicating that the nitride is not produced at the electrode in significant quantities within the time scale of a single CV experiment, which is in line with the rate constant for the decay of 3 (see section 2.1). The smaller second reduction feature observed at a 0.1 V·s–1 scan rate decreases in current and then disappears altogether as the scan rate is increased, which is consistent with the formation of an intermediate that is electrochemically detectable by subsequent reduction. It remains unclear whether this species corresponds to the N2-bridged intermediate 3 observed in NMR and SEC experiments. However, the potential of this reduction is far too negative to play a significant role in electrolytic N2 splitting, so further characterization of this feature has not been pursued.
Increasing the concentration of free chloride ions leads to a cathodic shift of the peak potential of the initial reduction of 1, and a slight increase in reversibility (Figure 7). In the presence of 20 equiv of [nBu4N]Cl, Ep,c shifts from −1.90 to −1.94 V (ν = 0.1 V·s–1). The ratio of cathodic and anodic peak currents is 1.9 at a scan rate of 0.1 V·s–1, indicating less reversibility than was achieved under the same conditions but under an Ar atmosphere. More current passes at higher concentrations of 1, as expected, and the peak potential of the initial reduction is essentially unchanged in the range of 0.5–4 mM 1 (Figure S36).
Figure 7.
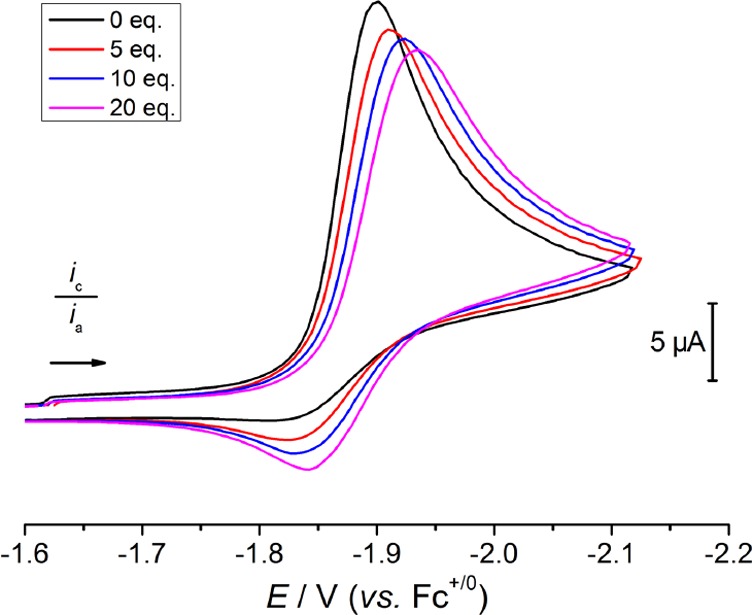
CV of 0.8 mM 1 in THF containing 0.2 M [nBu4N][PF6] and 0–20 equiv [nBu4N]Cl under N2 at 0.1 V·s–1.
Finally, the N2 binding step of the reaction was evaluated by variation of the N2 concentration. Upon increasing the pressure to 10 atm, the first cathodic process rises in current and seems to reach a plateau in this pressure range, while the potential remains unchanged (Figures S37 and S38). The second reduction feature near Ep,c = −2.4 V (ν = 0.1 V·s–1) decreases in intensity with increasing N2 pressure. CV data measured at 1 atm before and after pressurization are almost unchanged.
Simulation of the CV data was pursued to identify the N2 activating species and derive a comprehensive, quantitative picture of the formation of the N2-bridged intermediate that splits into nitrides. Several mechanisms that involve the necessary N2 binding (CN2), chloride ion dissociation (CCl), and dimerization (Cdim) chemical steps were considered (Scheme S1). The multielectron transfer character of the reduction peak is consistent with rapid 2e– reduction of complex 1 to a rhenium(I) species before formation of a bridging N2 intermediate with parent 1 in the diffusion layer. In contrast, lower current similar to a 1e– reduction would be expected for a pathway involving a mixed-valent N2-bridged rhenium(II)/rhenium(III) intermediate, as confirmed by digital simulations (Figure S45). The pronounced peak potential shift and current increase of the first cathodic process observed moving from Ar to N2 suggest fast chemical reactions and a potential inversion of the subsequent reduction process. The increased reversibility in the presence of added chloride suggests reversible chloride ion dissociation (CCl). Irreversible dimerization (Cdim) is taken to be the last step of the reaction sequence based on the SEC studies above in which N2-bridged dimer 3 builds up before N2 splitting and the multielectron nature of the first reduction process.
Three conceivable pathways involve pre-equilibrium N2 binding (Scheme S1). The N2 coordination step could be followed by (a) two-electron reduction, chloride ion loss, and dimerization (CN2E2eCClCdim), (b) two successive one-electron reductions, chloride ion loss, and dimerization (CN2EECClCdim), or (c) successive steps of reduction, chloride ion loss, reduction, and dimerization (CN2ECClECdim). Such pre-equilibrium pathways are unlikely due to the lack of NMR, UV–vis, and IR spectroscopic evidence for a dinitrogen adduct of 1 under various conditions (see section 2.1). These pathways were further excluded by electrochemical simulations (Figures S45–S47) using the spectroscopically estimated upper limit for initial N2 binding (Keq < 1 M–1, Supporting Information, section 5). Most importantly, a pre-equilibrium should lead to a dramatic shift of the peak potential with increasing N2 pressure, which has not been observed experimentally.
Alternative pathways all involve the initial electrochemical reduction of 1, followed by various orders of the N2 binding, chloride ion loss, and second reduction preceding the final dimerization step. An ECClCN2ECdim sequence would follow the kinetics of reduction and chloride ion loss, just as observed under argon, with subsequent N2 binding to the monochloride intermediate [ReCl(PNP)]. In this situation, the formal potential of the first reduction and the chemical kinetics of chloride dissociation must be the same under N2 and Ar. The observed anodic shift of the peak potential moving from Ar to N2 immediately casts doubt on [ReCl(PNP)] being involved in N2 binding. Indeed, even with diffusion-controlled N2 binding, the simulation could not account for the large peak potential shift observed under N2 (Figure S48). The ECN2ECClCdim scenario features initial reduction of 1 followed by N2 binding to form [ReCl2(N2)(PNP)]−, which would be subsequently reduced. It would be surprising for the reduction potential of this anionic dinitrogen complex to be in a similar range to neutral complex 1. In fact, for this species, a reduction potential E = −3.26 V was computed by DFT (see section 2.3). Even if taking an unrealistic reduction potential of −1.89 V for the intermediate, the current dependence on chloride addition could not be sufficiently modeled for this pathway, which makes the ECN2ECClCdim scenario also unlikely (Figure S49).
The minimum scenario that could adequately simulate the CV data is the ECN2CClECdim pathway (Scheme 5): reduction of 1 is followed by N2 binding to form [ReCl2(N2)(PNP)]−. This rhenium(II) species undergoes chloride loss to neutral [ReCl(N2)(PNP)], which can be reduced at a less negative potential (ΔE ≈ 0.4 V) than [ReCl(PNP)] due to the π-acceptor ligand N2. Resultant rhenium(I) species [ReCl(N2)(PNP)]− ultimately comproportionates with parent 1. Subsequent or simultaneous chloride loss would form N2-bridged dimer 3, but for simplicity this process was not included in the simulations. The simulated and experimental CV data are in excellent agreement over a wide scan rate and chloride ion concentration range (Figure 8 and Figures S50 and S51) for this highly complex reaction sequence. The simulation parameters were chosen to maximize conserved parameters from the Ar data (E1, E2, k1, k2), and to minimize the number of variables in the simulation; other processes such as redox disproportionation and competing concurrent pathways were not considered for the sake of simplicity.29 The numerical ranges of the fit parameters under N2 (E3, k3, K3, k4, K4, k5) lead to fits within the experimental error of the measurements as judged by the standard deviations of the peak currents and potential shifts obtained from multiple experiments (Tables S1 and S2; see the Supporting Information for further information).
Figure 8.
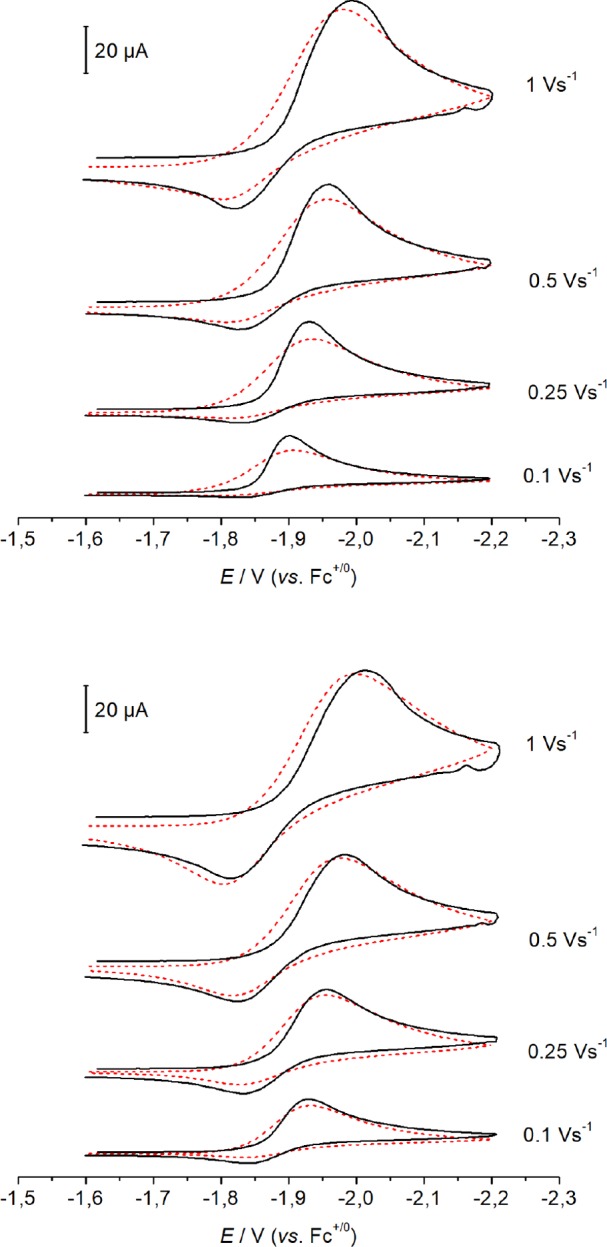
Overlay of experimental (black lines) and simulated (red dashed lines) CV data (I = 0.2 M [nBu4N][PF6] in THF at room temperature) for 1 (top) and 1 with 10 equiv [nBu4N]Cl (bottom). Simulation according to Scheme 5 with the thermodynamic and kinetic parameters given in Tables 1 and 2.
Key results of the simulations (Table 2) include the following: (1) trapping of the electron-rich and coordinatively unsaturated rhenium(II) species [ReCl2(PNP)]− by the π-acceptor ligand N2 is both thermodynamically favorable (K3 ≈ 104 M–1) and kinetically rapid (k3 > 5 × 107 M–1 s–1); (2) chloride ion loss from [ReIICl2(N2)(PNP)]− is thermodynamically not favored (K4 ≈ 10–2 M) but rapid (k4 > 5 × 102 s–1); (3) reduction of [ReIICl(N2)(PNP)] to rhenium(I) (E3 = −1.86 V) is at a more positive potential than parent 1, resulting in the characteristic peak shape. The potential value E3 is somewhat less well-defined by the simulations, given that changes of ±20 mV did not have a dramatic influence on the simulation quality. This simple minimum model does not reproduce the increasing peak current with N2 pressure (Figure S52). The conversion of [ReICl(N2)(PNP)]− to bis-dinitrogen complex [ReICl(N2)2(PNP)]− through an additional N2 binding equilibrium could account for this trend. Stabilization of Re(I) might slow the rate of dimerization to 3 and therefore the depletion of parent 1 in the diffusion layer resulting in higher currents at higher N2 pressure. Furthermore, computational analysis suggests reversible formation of a rhenium(I) bis-dinitrogen compound (see section 2.3). With K6 ∼ 50 M–1 and k6 ∼ 1000 M–1 s–1 for this equilibrium, the simulations for ambient pressure are virtually unchanged while the current increase for the high pressure data is well resembled (Figure S53).
Table 2. Estimated Ranges for Rate and Equilibrium Constants of the Best Model for Electrochemical Reduction of 1 under N2 Atmospherea.
| Parameter | Range | Value |
|---|---|---|
| K3 (M–1) | 5 × 103–5 × 104 | (1 × 104) |
| k3 (M–1 s–1) | >5 × 107 | (5 × 109) |
| K4 (M) | 2 × 10–2–2 × 10–3 | (5 × 10–3) |
| k4 (s–1) | >5 × 102 | (1 × 105) |
| k5 (M–1 s–1) | 4 × 103–1 × 104 | (7 × 103) |
| E3 (V) | –1.84 to −1.88 | (−1.86 V) |
The values in parentheses were used to plot the simulation in Figures 8 and Figures S50–S53.
2.3. Computational Analysis
Density functional theory (DFT) computations were carried out to evaluate the N2 splitting mechanism presented in the preceding sections. The calculations were mainly restricted to reaction steps directly supported by experimental data. This study expands upon the previous report that focused on the splitting of dinuclear complex 3 into nitrides.19 This N2 splitting step was also revisited in light of the additional experimental results presented above that include ample structural, spectroscopic, electrochemical, and kinetic data for benchmarking.
Electrochemical potentials were anchored with the experimental [Re(N)Cl(PNP)] (2+/2) redox couple (E1/2 = −0.086 V).30,31 Several functionals were evaluated by comparison with (a) the experimental/simulated redox potentials and equilibrium constants E1, E2, E3, K1, K3, and K4 and (b) DLPNO-CCSD(T)32 calculations of truncated model complexes (see Supporting Information, section 7, and Tables S6–S8). Best agreement was obtained with the M06 functional33 for the thermochemistry of both redox (E°M06 – E°exp = 0.1–0.2 V) and chemical steps (Scheme 6; see also the Supporting Information, section 7). Reaction free energies in THF were estimated by application of Truhlar’s SMD solvation model,34,35 including structural relaxation of gas-phase optimized geometries in solution.
Scheme 6. Computed (M06/def2-TZVP(SMD(THF))//D3BJ-RIJ-PBE/def2-SVP) Formal Reduction Potentials and Reaction Free Energies (kcal·mol–1) of Electrochemical and Chemical Steps That Connect 1 with Nitride 2.

The proposed mechanism is highlighted in gray. Experimentally derived potentials and free energies are given in parentheses. Irreversible decomposition routes under Ar were not considered in the computational model.
Binding of N2 (ΔG°M06 = +2.8 kcal·mol–1) or chloride (ΔG°M06 = +1.8 kcal·mol–1) to parent 1 are both calculated to be endergonic, in accord with the experimental stability of 1 in the presence of N2 and chloride. An initial reduction process from 1 to [ReIICl2(PNP)]− was considered next, with the computed potential in excellent agreement with experiment (E°M06 = −2.1 V; E°exp = −2.0 V).27 Further reduction of all anionic intermediates to dianionic species is computed at reduction potentials well beyond −3.0 V, and can therefore be safely discarded. In the absence of N2, chloride dissociation, which is slightly overestimated in endergonicity (ΔG°M06 = +5.8 kcal·mol–1; ΔG°exp = +1.8 kcal·mol–1), and subsequent reduction of [ReIICl(PNP)] (E°M06 = −2.4 V; E°exp = −2.3 V) represent the preferred pathway, fully supporting the experimental CV simulations. For both redox couples [ReCl2(PNP)]0/– and [ReCl(PNP)]0/–, respectively, small structural rearrangements were computed suggesting fast and electrochemically reversible electron transfer.
Coordination of N2 to the initial reduction product [ReIICl2(PNP)]− is considerably more favorable than chloride dissociation (ΔΔG°M06 = 6.6 kcal·mol–1; Scheme 6). The product of N2 binding is predicted to be most stable as the cis-dichloro isomer. The chloride ligand in octahedral cis-[ReIICl2(N2)(PNP)]− was computed to be labilized relative to [ReIICl2(PNP)]− (ΔΔG°M06 = 6.6 kcal·mol–1), rendering Cl– dissociation slightly exergonic. Electrochemical simulations actually point to slightly less favorable Cl– dissociation from the N2 adduct, which might be explained by computational underestimation of the stability of [ReIICl2(N2)(PNP)]−. Reduction is reflected by a shift of the computed N2 stretching vibration and slight elongation of the N–N bond relative to hypothetical cis-[ReIIICl2(N2)(PNP)] (Table 3), indicating a moderate degree of N2 activation at rhenium(II)36 with less than 20% of the computed spin density located at the N2 ligand.
Table 3. Computed Structural and Electronic Parameters of Selected Dinitrogen Intermediates in the Most Stable Configuration.
| [ReIIICl2(N2)(PNP)] | [ReIICl2(N2)(PNP)]− | [ReIICl(N2)(PNP)] | [ReICl(N2)(PNP)]− | 3 | |
|---|---|---|---|---|---|
| dRe–Na (Å) | 1.969 | 1.892 | 1.892 | 1.877 | 1.875 |
| dN–Na (Å) | 1.141 | 1.162 | 1.158 | 1.170 | 1.200 |
| υN–Na (cm–1) | 2105 | 1975 | 1955 | 1935 | 1771 |
| ρReb | 0.36 | 0.24 (0.50) | 0.42 (0.59) | 0.13 | 0.37 (0.91) |
| ρNαb | –0.05 | –0.09 (−0.01) | –0.13 (−0.03) | –0.13 | –0.22 (0.00) |
| ρNβb | –0.08 | –0.28 (0.18) | –0.15 (0.30) | –0.42 | – |
Gas phase structures.
Calculated NPA charges with natural spin densities in parentheses.
Subsequent reduction of [ReIICl(N2)(PNP)] after chloride loss was computed to occur at E° = −2.0 V, i.e., about the same potential as the initial reduction and thus consistent with an ECCE-type mechanism. A closely related pathway, in which N2 binding is followed by reduction of the bis-dinitrogen complex to rhenium(I) at slightly less negative reduction potential, cannot be ruled out computationally (Scheme 6). For each of these pathways, the key finding of potential inversion upon N2 addition and chloride loss is preserved. As expected, reduction of rhenium(II) is accompanied by an additional bathochromic shift of the N2 stretching vibration and N–N bond elongation (Table 3). Furthermore, natural population analysis (NPA) shows an accumulation of negative charge at the terminal N atom of [ReICl(N2)(PNP)]−. However, natural bond orbital (NBO) analysis (Figure S57) and the computed N2 stretching vibration (1935 cm–1) clearly favor formulation as a ReI dinitrogen over ReIII diazenido complex. In comparison, Cummins’ anionic diazenido complex [Mo(N2)(NtBuAr)3]− (Ar = C6H3-3,5-Me2) exhibits an N2 stretching vibration at 1761 cm–1.37
Structural comparison of the range of computed mononuclear rhenium structures (Supporting Information, section 7.7) shows that the octahedral rhenium(II) and rhenium(I) species exhibit distorted minimum conformations of the pincer backbone that involve either a considerable tilt around the Re–NPNP bond or pyramidalization of the pincer nitrogen atom. Both structural distortions would help minimize a π-mismatch of filled rhenium d-orbitals with the amide lone pair. This observation suggests that the strong π-donating ability of the dialkylamide ligand works in concert with the steric bulk to facilitate halide loss.
Comproportionation of the rhenium(I) dinitrogen intermediate with parent rhenium(III) complex 1 and concomitant chloride loss to form the dinuclear compound 3 was computed to be exergonic (ΔG°M06 = −12.0 kcal·mol–1). Consequently, the formation of 3 is the first irreversible reaction within the lowest energy ECN2CClECdim pathway, in support of the electrochemical model. In the previous model, using the PBE functional, an antiferromagnetically coupled electronic singlet ground state was computed to be energetically almost degenerate with a triplet state for 3.19 In contrast, the M06 functional favors the triplet state by 4.6 kcal·mol–1, with the spin density mainly centered on the Re ions (Table 3). However, the shifted, yet sharp NMR signals of 3 (see section 2.1) indicate an energetically well-separated ground state presumably due to further splitting by spin–orbit coupling. Despite the obvious inability of DFT to properly describe the multireference character of 3, the molecular structure is excellently reproduced. Furthermore, TD-DFT computations predict an absorption spectrum for 3 (Figure S4) that is in excellent agreement with the spectra obtained in SEC experiments. In comparison with the mononuclear N2 complexes discussed above, the computed N–N bond distance (1.20 Å) and stretching frequency (1771 cm–1) range between typical values for a moderately activated N2 ligand and a diazenido (N22–) bridge.36 Within a localized description of the core, the rhenium-centered spin density is in agreement with a ReII(N2)ReII formulation or alternatively ReIII(N22–)ReIII with intermediate-spin rhenium(III) (S = 1) antiferromagnetically coupled to (triplet) N22–. However, oxidation states can be physically meaningless in the case of highly covalent metal–ligand multiple bonding as was pointed out, e.g., for nitrosyl or nitride complexes.38 Within a more covalent bonding picture, diazenido (formal N–N double bonding) character of the N2 bridge is reflected by the occupation of two Re–N–N–Re πReN–π*NN–πReN molecular orbitals (MO236/237; Figure 9) with four electrons and two electrons in the π*ReN–πNN–π*ReN MOs (MO240/241), respectively.
Figure 9.
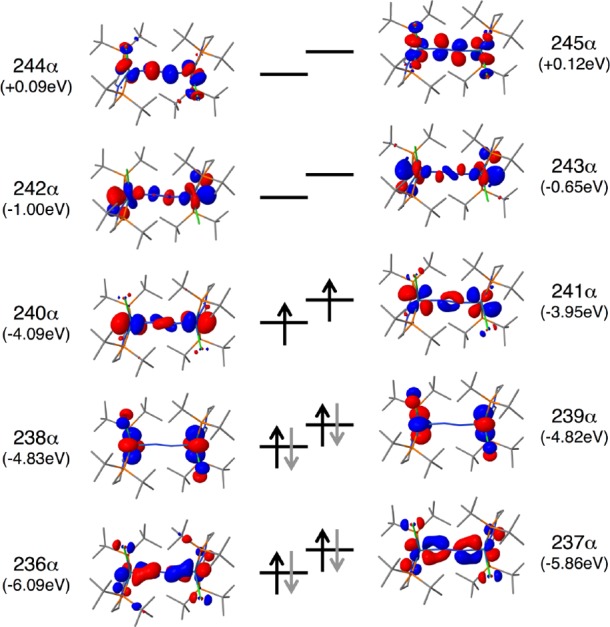
DFT computed frontier molecular orbital (FMO) diagram of 3.
As in the previous computational model,19 the transition state (TS) for splitting of 3 into nitrides exhibits a Re–N–N–Re in-plane zigzag structure39 with an N–N distance (1.59 Å) that is considerable longer than the N–N single bond in hydrazine (1.45 Å). Upon approaching the TS, MO242 (Figure 9), which is essentially Re–N–N–Re nonbonding, gains considerable σRe–N and σ*N–N character and is gradually stabilized with respect to the π-MO manifold (Figure S59). Population of this MO from the π*ReN–πNN–π*ReN level leads to both N–N bond weakening and Re–N strengthening and ultimately full N–N bond cleavage. The computed free energy of activation at 298 K (ΔG‡M06 = +26.9 kcal/mol) is slightly higher than the experimental value (ΔG‡exp = +19.8 kcal/mol) and the previously computed values (ΔG‡PBE = +20.2 kcal/mol). However, the enthalpy of activation (ΔH‡M06 = 26.2 kcal·mol–1) is in excellent agreement with experiment (ΔH‡exp = 24 kcal·mol–1). The considerable deviation of the entropy of activation (ΔS‡M06 = −3 cal·mol–1 K–1; ΔS‡exp = 14 cal·mol–1 K–1) can in part be attributed to an underestimated contribution of the electronic partition function, which arises from the inability of DFT to describe the multireference character of 3. With that notion, the better agreement of ΔG‡PBE with experiment might be a consequence of error compensation.
3. Discussion
3.1. Chemical vs Electrochemical N2 Splitting
The transition metal mediated reduction of N2 to two nitrides formally requires the transfer of six electrons and the cooperative action of at least two metal ions. As the closest analogues of our rhenium platform, Schrock, Mézailles, and Nishibayashi have reported N2 splitting upon 2e– reduction of either MoIII or MoIV pincer halides under dinitrogen.15d,17 For all of these systems, experimental information about the nature of the key species and kinetics of the elementary steps remains unknown. A complex reaction mechanism can be expected that is composed of several steps, such as reduction, N2 binding, halide dissociation, formation of N2-bridged species, and N–N bond cleavage. Consequently, the kinetics of the elementary steps need to be aligned to obtain the desired reactivity, specifically if reactive intermediates with short lifetimes under the experimental conditions are involved.
In many systems that were shown to fully split N2, the nature of the chemical reductant has a strong influence on the nitride yield. For example, Holland and co-workers have shown that alkali metal reductants facilitate N2 splitting in Fe(nacnac) complexes by producing alkali metal cations that stabilize the products of N2 activation and cleavage through contact ion-pair formation.14,40 However, in most cases the origin of reductant effects is not well understood. In the case of parent 1, the N2 splitting yield is slightly higher with Na/Hg (>85%) than with Co(Cp*)2 (up to 75%).19 A productive role for Na+ in removing chloride after dissociation (K4 = 10–3 M, Table 2) could be envisioned in this case. Our mechanistic model also reveals that the rhenium(II) species formed upon initial reduction (E1 = −2.0 V) is the N2 binding state, followed by reduction to rhenium(I) at a slightly less negative potential (E3 = −1.9 V) and ReI/ReIII comproportionation leading to productive nitride synthesis (Scheme 5). If N2 does not bind, the rhenium(II) intermediate can undergo reductive decomposition (E2 = −2.3 V), as seen in the Ar CV data. Very strong reductants (E° < – 2.3 V) could introduce the possibility of over-reduction of the rhenium(II) species competing with productive N2 binding, which might lead to unproductive decomposition pathways.
As judged from the complexity of our mechanistic model, it is obvious that molecular platforms for N2 splitting with chemical reductants are not necessarily transferable to electrochemical approaches. Electrochemical methods also have inherent speciation and concentration gradients from the electrode surface through the diffusion/reaction layer to the bulk solution, which give rise to different kinetic profiles compared with both molecular and solid chemical reductants, respectively. However, in contrast to the other pincer halide precursors that have been used for N2 splitting, our rhenium platform 1 only requires one reduction equivalent per metal ion. It therefore is the simplest candidate to directly initiate N2 activation by electrochemical electron transfer and obtain mechanistic information from the voltammetric response. In fact, the rhenium pincer system represents the first example of electrochemical N2 splitting into well-defined molecular nitride complexes. Other electrochemical nitride syntheses have started from ammonia, such as Meyer and co-workers’ electrochemical oxidation of osmium(III) ammine complexes to terminal osmium(VI) nitrides.41 In the present reductive N2 splitting reaction, the same N2-bridged intermediate is formed in both chemical and electrochemical reductions, as evidenced by UV–vis absorption spectroscopy (λmax = 533 nm). This mechanistic connection and the relatively high Faradaic yield offer, for the first time, the possibility of electrochemical examination of a full N2 splitting sequence triggered by reduction of a transition metal halide complex.
3.2. Formation of the N2-Bridged Intermediate
The accumulation of N2-bridged complex 3 as the only observable intermediate, within seconds of electrolysis, indicates rapid kinetics for all elementary steps prior to dinitrogen cleavage. Simulations of the electrochemical data under a broad range of experimental conditions and scan rates are supported by DFT computations to provide compelling evidence for a ECN2CClECdim mechanism. The process begins with electrochemical reduction immediately followed by coordination of N2. The kinetic model reveals two key findings about the initial steps: (1) the N2 binding step occurs in the Re(II) formal oxidation state; (2) N2 binding occurs before chloride dissociation. According to experimental data, N2 binding to [ReIICl2(PNP)]− is strongly favorable (K3 > 5 × 103 M–1) and rapid (k3 > 5 × 107 M–1 s–1), in sharp contrast to the unobserved binding to the rhenium(III) complex 1 (Keq < 1 M–1). This increased affinity for the π-acid N2 is anticipated for the more electron-rich, anionic rhenium(II) complex. The DFT computations qualitatively align with this trend, as reflected by the large computed bathochromic shift of the N2 stretching vibration for [ReCl2(N2)(PNP)]0/– (Δν = 130 cm–1). Peters and co-workers observed an increase in N2 binding affinity by 6 orders of magnitude upon reduction of a low valent bimetallic iron complex.42 The rate of N2 binding for this Fe system (7 × 108 M–1 s–1) is similar to that found for rhenium(II) intermediate [ReCl2(PNP)]−. An alternative pathway involving N2 activation by a rhenium(I) species (ECClE-type mechanism) was irreconcilable with the distinct current increase observed for the first reduction feature under N2 relative to Ar and the missing reduction feature E2 of [ReCl(PNP)] under N2. The rapid N2 binding to rhenium(II) is a prerequisite to avoid unimolecular decay after chloride loss (k2). We therefore hypothesize that the bulky pincer ligand, which enforces a five-coordinate geometry in the starting complex 1, might be instrumental for productive N2 activation and splitting.
The next step is chloride dissociation from the anionic dinitrogen complex, [ReIICl2(N2)(PNP)]−. The rate constant for chloride dissociation from [ReIICl2(N2)(PNP)]− (k4 > 5 × 102 s–1) is comparable to or faster than that of [ReIICl2(PNP)]− under Ar (k1 = 1 × 103 s–1). Chloride dissociation is likely facilitated by the bulky tert-butyl substituents and the strong amide π-donating ability of the PNP ligand framework. In comparison, chloride loss from another rhenium(II) complex, [ReII(PMe2Ph)3Cl3]−, was reported to be several orders of magnitude slower (k = 0.91 s–1).43 On the other hand, chloride dissociation from isoelectronic [MoICl2(dppe)2]− (k = 2.5 × 102 s–1; dppe = bis(diphenylphosphino)ethane) proceeds with a comparable rate constant to the present system.44 The net result of the two chemical steps that follow reduction is an associative substitution at five-coordinate [ReIICl2(PNP)]−.
Substantial evidence has been collected that the neutral dinitrogen complex [ReIICl(N2)(PNP)] undergoes reduction to the corresponding anionic ReI complex at a potential less negative than the reduction potential of parent 1. Compared with the CV under argon, the current amplification and the potential shift of the first reduction feature under N2 is indicative of potential inversion; i.e., reduction of this intermediate is more facile than reduction of 1. The presence of a π-accepting N2 ligand should shift the reduction of [ReIICl(N2)(PNP)] to more anodic potentials relative to 1, and computations bear out this hypothesis. Similar potential inversion has been observed with [ReIIICl2(dppe)2]+, which is reduced at a more cathodic potential relative to the reduction of [ReIICl(N2)(dppe)2]+ (−0.81 and −0.27 V, respectively), despite the lower formal oxidation state for the latter.45 [ReIICl2(N2)(PNP)]− was also considered a candidate for this second electron transfer process, but as expected the reduction potential was computed at a much more negative potential.
Formation of the bridging dinitrogen complex 3 requires a bimolecular reaction step. Since the transformation involves one electron per rhenium, and ReII is easily reduced to ReI, a reasonable hypothesis involves the comproportionation of ReIII and ReI through the reaction of 1 with [ReICl(N2)(PNP)]−. The two species would react as the anionic Re(I) complex diffuses away from the electrode and 1 diffuses from the bulk solution toward the electrode (Figure S54). This is taken to be the rate-limiting process in the electrochemical reduction of 1, on the basis of the observed accumulation of N2-bridged 3 in UV–visible SEC experiments. For the sake of simplicity, CV simulations only include the bimetallic dimer formation process, and chloride dissociation is not directly considered. Chloride dissociation does occur, either in a concerted fashion or in a separate dissociation process, to form 3. DFT calculations suggest that the net dimerization and chloride dissociation process is in fact strongly exergonic (ΔG° = −12.0 kcal·mol–1) and thus irreversible.
At least five steps occur during the sweep of a cyclic voltammogram to form the N2-bridged intermediate 3 from molecular nitrogen. The complexity of the ECCEC-type reaction sequence adds some degree of uncertainty to the quantitative rate and equilibrium constants, and a sensitivity analysis provides a range of viable values. However, DFT calculations provide strong support for the proposed mechanism. As an additional key finding, the computational results indicate that considerable N2 activation, as judged by the stretching vibrations, is not obtained prior to ReI/ReIII comproportionation to dinuclear complex 3, thereby avoiding the unfavorable one-electron reduction of N2.46
3.3. Splitting of the N2-Bridged Intermediate into Nitrides
The final step of nitride formation is the rate-determining cleavage of dirhenium complex 3. Dinitrogen splitting with molecular platforms has recently been comprehensively reviewed.13 Within a simple, covalent bonding model, the degree of N2 activation in linearly N2-bridged dinuclear complexes can be predicted based on the occupation of the π-manifold that is constructed by linear combination of metal d-orbitals and nitrogen p-orbitals of the {M–N–N–M} core.47 Splitting of such compounds into terminal nitride complexes has been associated with π10–{M–N–N–M} electronic configurations to reach stable nitrides with filled M–N π and vacant π* MOs.13,48,49 These simple considerations are showcased by Cummins’ S6-symmetric intermediate [{(tBuArN)3Mo}2(N2)]12,50 (Scheme 7) and other examples.18,51 The electronic structure computations for 3 are in full agreement with this picture (Figure 9). In the current case, the square-pyramidal ligand field renders nonbonding, metal d-orbital based MOs with δ-symmetry (Figure 9, MO238/239) energetically accessible to host the four additional valence electrons in the dirhenium complex, i.e., {ReN2Re}4+ (π10δ4) vs {MoN2Mo}6+ (π10) (Scheme 7). The similar degree of N2 activation is expressed by the N–N bond distances found for 3 (1.202(10) Å) and [{(tBuArN)3Mo}2(N2)] (1.212(2) Å),50 respectively. These values are considerably shorter than related compounds with π8 ([{(tBuArN)3Mo}2(N2)]2+, 1.265(5) Å)) or π8δ4 ([{(PNP)ClMo}2(N2)], 1.258(9) Å) configurations.18,50
Scheme 7. Qualitative Comparison of the Electronic Configurations of L3MoIII(N2)MoIIIL3 in S6-Symmetric (left) and of L4ReII(N2)ReIIL4 in D4h-Symmetric (right) Ligand Fields, Respectively.

The simple analogy of the trigonal- and square-pyramidal platforms suggests that the success with group 7 metal rhenium is facilitated by the square-pyramidal coordination geometry with apical N2 coordination, enforced by the bulky pincer ligand. Schneider and co-workers showed that the stable (π8δ4)-complex [{(PNP)ClMo}2(N2)] (S = 0) also splits into nitrides upon protonation of the pincer backbone, which is associated with a transition to a π10δ2 (S = 2) configuration.18 Hence, besides coordination geometry, the spin state seems to have a subtle influence on N2 splitting. In this context, the Mo pincer halides that are overall reduced by 2e–/Mo are particularly interesting (Scheme 1). There, several redox and spin states are conceivable for binuclear N2-bridged intermediates that split into nitrides. Applying the simple considerations discussed above, e.g., to Schrock’s POCOP complex,17a low-spin [{(POCOP)(I)Mo}2(N2)]2– (π10δ4, S = 0; POCOP = C6H3-2,6-(OPtBu2)2) or high-spin [{(POCOP)(I)Mo}2(N2)] (π10δ2, S = 2) might be the preferred states for N2 splitting.
DFT studies for the splitting of [{(RR′N)3Mo}2(N2)] suggest a distortion of the {Mo–N–N–Mo}-core from linearity toward a zigzag shaped transition state prior to full cleavage into nitrides.52 Krewald and González recently examined the photochemical splitting of [{(H3N)5Os}2(N2)]5+ using multireference computations with perturbative treatment of spin–orbit splittings (SO-CASPT2) for a defined linear trajectory of the two separating {(H3N)5OsN}n+ fragments.53 However, as a general picture that evolves for the different pathways, N2 bond cleavage can be correlated with the population of N–N σ* orbitals at the expense of M–N π*-MOs. This electronic rearrangement weakens the N–N and strengthens the M–N bonds. These considerations also hold true for the computational model for splitting of 3, which nicely reproduces the experimental activation enthalpy. In the transition state, the MO with σ*N–N character (Figure 9, MO242; Scheme 7, a2u) is populated. Notably, this MO would be destabilized by an additional ligand (e.g., N2) trans to the bridging N2-ligand. For example, in contrast to Cummins’ tris-anilides with trigonal-pyramidally coordinated Mo, Schrock’s five-coordinate triamidoamine complex [{(tBuMe2SiNCH2CH2)3NMo}2(N2)], which exhibits the same spin states, formal oxidation states, and similar degrees of N2 activation, does not split into nitrides. In this context, it is tempting to speculate that the square-pyramidal (rather than octahedral) geometries enforced by the bulky PtBu2-containing pincer ligands utilized in Re- and Mo-mediated N2 splitting are a prerequisite for facile kinetics.
4. Concluding Remarks
The pincer-supported rhenium complex described here is, to date, the only group 7 transition metal complex reported to split N2 into nitrides. It is also a representative example of a high-valent metal halide that produces nitrides upon reduction under N2 at ambient conditions. The nitride is formed rapidly and in high yield, both using chemical reductants and, for the first time, electrochemical reduction. These traits are prerequisites for successful incorporation into future (electro)catalytic cycles. Furthermore, this system is amenable to detailed mechanistic analysis that has provided unprecedented insight into the elementary steps from reduction and N2 activation all the way to splitting. The electrochemical data, chemical kinetics, and supporting computational treatment give a comprehensive mechanistic picture that involves initial, extremely rapid assembly of the dinuclear intermediate 3, followed by slightly slower N–N bond scission to furnish nitrides. Key findings for this sequence are the following: (1) the N2binding state is a mononuclear ReIIspecies with dramatically increased dinitrogen affinity after reduction of precursor 1, (2) binding of the π-accepting N2 ligand leads to immediate further reduction of the ReIIN2adduct to ReIdue to potential inversion, followed by comproportionation with the ReIIIprecursor to reach the N2-bridged dimer, and (3) considerable N2activation is not achieved prior to formation and subsequent splitting of this dimer. It is reasonable to assume that the accessibility of more stable ReI is instrumental to avoiding ReII decay pathways. Furthermore, it allows for storage of two reduction equivalents at the metal, thereby avoiding thermodynamically unfavorable one-electron N2 reduction. This chain of events is facilitated by the bulky, π-donating pincer ligand, which stabilizes the coordinatively unsaturated precursor and low-valent, transient intermediates.
This ReI/ReIII comproportionation defines the first irreversible step in the electrochemically driven reaction sequence. The steric bulk of the pincer ligand might also be advantageous in stabilizing coordinatively unsaturated 1 in this step. Trapping of the ReI dinitrogen intermediate in the diffusion layer by parent 1, which is fully depleted at the cathode surface, emphasizes the importance of mass transport for electrochemical N2 functionalization. The electronic configuration of N2-bridged 3 resembles that of Cummins’ prototypical N2 splitting intermediate [{(RR′N)3Mo}2(N2)], with the square-pyramidal geometry proving crucial to accommodating the additional electrons of ReII relative to MoIII. The mechanistic insight gained in this Re system offers guidance for the future development of additional platforms for chemical and electrochemical N2 splitting.
Acknowledgments
This article is dedicated to the scientific legacy of Frances P. Venable (1856–1934), who received his Ph.D. at the University of Goettingen before becoming professor of chemistry and president of the University of North Carolina, on the occasion of the 200th anniversary of the Department of Chemistry at the University of North Carolina at Chapel Hill. B.M.L. and A.J.M.M. gratefully acknowledge support from the National Science Foundation under Grant CHE-1665135. A.J.M.M. is an Alfred P. Sloan Fellow. R.S.v.A. and S.S. thank the European Research Council for financial support (ERC Grant Agreement 646747, grant holder S.S.). I.S. thanks the DFG (Emmy Noether Program: SI 1577/2) and the Fonds der Chemischen Industrie for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03755.
Author Contributions
∥ B.M.L. and R.S.v.A.: These authors contributed equally.
The authors declare no competing financial interest.
Notes
CCDC-1832925 (3·0.51·0.52·0.5C5H12) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/products/csd/request/ (or from Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, U.K. Fax: + 44-1223-336-033. E-mail: deposit@ccdc.cam.ac.uk).
Supplementary Material
References
- Patil B. S.; Hessel V.; Seefeldt L. C.; Dean D. R.; Hoffman B. M.; Cook B. J.; Murray L. J.. Nitrogen Fixation. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: 2017. [Google Scholar]
- a van der Ham C. J. M.; Koper M. T. M.; Hetterscheid D. G. H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. 10.1039/C4CS00085D. [DOI] [PubMed] [Google Scholar]; b Sustainable Ammonia Production. DOE Roundtable Report; 2016. https://science.energy.gov/~/media/bes/pdf/reports/2016/SustainableAmmoniaReport.pdf.
- a Marnellos G.; Stoukides M. Ammonia Synthesis at Atmospheric Pressure. Science 1998, 282, 98–100. 10.1126/science.282.5386.98. [DOI] [PubMed] [Google Scholar]; Licht S.; Cui B.; Wang B.; Li F.-F.; Lau J.; Liu S. Ammonia Synthesis by N2 and Steam Electrolysis in Molten Hydroxide Suspensions of Nanoscale Fe2O3. Science 2014, 345, 637–640. 10.1126/science.1254234. [DOI] [PubMed] [Google Scholar]
- Garagounis I.; Kyriakou V.; Skodra A.; Vasileiou E.; Stoukides M. Electrochemical Synthesis of Ammonia in Solid Electrolyte Cells. Front. Energy Res. 2014, 10.3389/fenrg.2014.00001. [DOI] [Google Scholar]
- a Lan R.; Irvine J. T. S.; Tao S. Synthesis of Ammonia Directly from Air and Water at Ambient Temperature and Pressure. Sci. Rep. 2013, 3, 1145. 10.1038/srep01145. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen S.; Perathoner S.; Ampelli C.; Mebrahtu C.; Su D.; Centi G. Electrocatalytic Synthesis of Ammonia at Room Temperature and Atmospheric Pressure from Water and Nitrogen on a Carbon-Nanotube-Based Electrocatalyst. Angew. Chem., Int. Ed. 2017, 56, 2699–2703. 10.1002/anie.201609533. [DOI] [PubMed] [Google Scholar]; c Shi M.-M.; Bao D.; Wulan B.-R.; Li Y.-H.; Zhang Y.-F.; Yan J.-M.; Jiang Q. Au Sub-Nanoclusters on TiO2 toward Highly Efficient and Selective Electrocatalyst for N2 Conversion to NH3 at Ambient Conditions. Adv. Mater. 2017, 29, 1606550. 10.1002/adma.201606550. [DOI] [PubMed] [Google Scholar]
- Lindley B. M.; Appel A. M.; Krogh-Jespersen K.; Mayer J. M.; Miller A. J. M. Evaluating the Thermodynamics of Electrocatalytic N2 Reduction in Acetonitrile. ACS Energy Lett. 2016, 1, 698–704. 10.1021/acsenergylett.6b00319. [DOI] [Google Scholar]
- Eizawa A.; Arashiba K.; Tanaka H.; Kuriyama S.; Matsuo Y.; Nakajima K.; Yoshizawa K.; Nishibayashi Y. Remarkable Catalytic Activity of Dinitrogen-bridged Dimolybdenum Complexes Bearing NHC-based PCP-pincer Ligands Toward Nitrogen Fixation. Nat. Commun. 2017, 8, 14874. 10.1038/ncomms14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pickett C. J.; Talarmin J. Electrosynthesis of Ammonia. Nature 1985, 317, 652–653. 10.1038/317652a0. [DOI] [Google Scholar]; b Becker J. Y.; Posin B. Nitrogen Reduction by Electrochemically Generated Vanadium(II) Promoted by Various Organic Ligands in Basic Methanol. J. Electroanal. Chem. Interfacial Electrochem. 1988, 250, 385–397. 10.1016/0022-0728(88)85178-7. [DOI] [Google Scholar]; c Becker J. Y.; Avraham S.; Posin B. Electrochemical Reduction of Titanium Compounds in the Presence of Catechol and N2 in MeOH or THF. J. Electroanal. Chem. Interfacial Electrochem. 1987, 230, 143–153. 10.1016/0022-0728(87)80138-9. [DOI] [Google Scholar]; d Luneva N. P.; Mironova S. A.; Shilov A. E.; Antipin M. Y.; Struchkov Y. T. A Novel Mixed-Valence Tetranuclear VII/VIII Complex Isolated from 3,5-Di-tert-Butylcatechol/Vanadium(II)/Sodium Methoxide System for Dinitrogen Reduction. Angew. Chem., Int. Ed. Engl. 1993, 32, 1178–1179. 10.1002/anie.199311781. [DOI] [Google Scholar]; e Del Castillo T. J.; Thompson N. B.; Peters J. C. A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc. 2016, 138, 5341–5350. 10.1021/jacs.6b01706. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Munisamy T.; Schrock R. R. An Electrochemical Investigation of Intermediates and Processes Involved in the Catalytic Reduction of Dinitrogen by [HIPTN3N]Mo (HIPTN3N = (3,5-(2,4,6-i-Pr3C6H2)2C6H3NCH2CH2)3N). Dalton Trans. 2012, 41, 130–137. 10.1039/C1DT11287B. [DOI] [PubMed] [Google Scholar]; g Chalkley M. J.; Del Castillo T. J.; Matson B. D.; Peters J. C. Fe-Mediated Nitrogen Fixation with a Metallocene Mediator: Exploring pKa Effects and Demonstrating Electrocatalysis. J. Am. Chem. Soc. 2018, 140, 6122–6129. 10.1021/jacs.8b02335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schrock R. Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum: Theory versus Experiment. Angew. Chem., Int. Ed. 2008, 47, 5512–5522. 10.1002/anie.200705246. [DOI] [PubMed] [Google Scholar]; b Tanaka H.; Nishibayashi Y.; Yoshizawa K. Interplay between Theory and Experiment for Ammonia Synthesis Catalyzed by Transition Metal Complexes. Acc. Chem. Res. 2016, 49, 987–995. 10.1021/acs.accounts.6b00033. [DOI] [PubMed] [Google Scholar]; c Thompson N.; Green M.; Peters J. Nitrogen Fixation via a Terminal Fe(IV) Nitride. J. Am. Chem. Soc. 2017, 139, 15312–15315. 10.1021/jacs.7b09364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114, 4041–4062. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplaza C. E.; Cummins C. C. Dinitrogen Cleavage by a Three-Coordinate Molybdenum(III) Complex. Science 1995, 268, 861–863. 10.1126/science.268.5212.861. [DOI] [PubMed] [Google Scholar]
- Laplaza C. E.; Johnson M. J. A.; Peters J. C.; Odom A. L.; Kim E.; Cummins C. C.; George G. N.; Pickering I. J. Dinitrogen Cleavage by Three-Coordinate Molybdenum(III) Complexes: Mechanistic and Structural Data. J. Am. Chem. Soc. 1996, 118, 8623–8638. 10.1021/ja960574x. [DOI] [Google Scholar]
- Klopsch I.; Yuzik-Klimova E. Y.; Schneider S. Functionalization of N2 by Mid to Late Transition Metals via N–N Bond Cleavage. Top. Organomet. Chem. 2017, 60, 71–112. 10.1007/3418_2016_12. [DOI] [Google Scholar]
- Representative examples:; a Pool J. A.; Lobkovsky E.; Chirik P. J. Hydrogenation and Cleavage of Dinitrogen to Ammonia with a Zirconium Complex. Nature 2004, 427, 527–530. 10.1038/nature02274. [DOI] [PubMed] [Google Scholar]; b Akagi F.; Matsuo T.; Kawaguchi H. Dinitrogen Cleavage by a Diniobium Tetrahydride Complex: Formation of a Nitride and Its Conversion into Imide Species. Angew. Chem., Int. Ed. 2007, 46, 8778–8781. 10.1002/anie.200703336. [DOI] [PubMed] [Google Scholar]; c Rodriguez M. M.; Bill E.; Brennessel W. W.; Holland P. L. N2 Reduction and Hydrogenation to Ammonia by a Molecular Iron-Potassium Complex. Science 2011, 334, 780–783. 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shima T.; Hu S.; Luo G.; Kang X.; Luo Y.; Hou Z. Dinitrogen Cleavage and Hydrogenation by a Trinuclear Titanium Polyhydride Complex. Science 2013, 340, 1549–1552. 10.1126/science.1238663. [DOI] [PubMed] [Google Scholar]; e Keane A. J.; Farrell W. S.; Yonke B. L.; Zavalij P. Y.; Sita L. R. Metal-Mediated Production of Isocyanates, R3EN = C=O from Dinitrogen, Carbon Dioxide, and R3ECl. Angew. Chem., Int. Ed. 2015, 54, 10220–10224. 10.1002/anie.201502293. [DOI] [PubMed] [Google Scholar]; f Falcone M.; Chatelain L.; Scopelliti R.; Zivkovic I.; Mazzanti M. Nitrogen Reduction and Functionalization by a Multimetallic Uranium Nitride Complex. Nature 2017, 547, 332–337. 10.1038/nature23279. [DOI] [PubMed] [Google Scholar]
- Representative examples:; a Curley J. J.; Sceats E. L.; Cummins C. C. A Cycle for Organic Nitrile Synthesis via Dinitrogen Cleavage. J. Am. Chem. Soc. 2006, 128, 14036–14037. 10.1021/ja066090a. [DOI] [PubMed] [Google Scholar]; b Ishida Y.; Kawaguchi H. Nitrogen Atom Transfer from a Dinitrogen-Derived Vanadium Nitride Complex to Carbon Monoxide and Isocyanide. J. Am. Chem. Soc. 2014, 136, 16990–16993. 10.1021/ja510317h. [DOI] [PubMed] [Google Scholar]; c Klopsch I.; Kinauer M.; Finger M.; Würtele C.; Schneider S. Conversion of Dinitrogen into Acetonitrile under Ambient Conditions. Angew. Chem., Int. Ed. 2016, 55, 4786–4789. 10.1002/anie.201600790. [DOI] [PubMed] [Google Scholar]; d Liao Q.; Cavaille A.; Saffon-Merceron N.; Mézailles N. Direct Synthesis of Silylamine from N and a Silane: Mediated by a Tridentate Phosphine Molybdenum Fragment. Angew. Chem., Int. Ed. 2016, 55, 11212–11216. 10.1002/anie.201604812. [DOI] [PubMed] [Google Scholar]; e Guru M. M.; Shima T.; Hou Z. Conversion of Dinitrogen to Nitriles at a Multinuclear Titanium Framework. Angew. Chem., Int. Ed. 2016, 55, 12316–12320. 10.1002/anie.201607426. [DOI] [PubMed] [Google Scholar]
- For a DFT study of electrochemical N2 splitting to form surface nitrides, see:; Abghoui Y.; Garden A. L.; Howalt J. G.; Vegge T.; Skúlason E. Electroreduction of N2 to Ammonia at Ambient Conditions on Mononitrides of Zr, Nb, Cr, and V: A DFT Guide for Experiments. ACS Catal. 2016, 6, 635–646. 10.1021/acscatal.5b01918. [DOI] [Google Scholar]
- a Hebden T. J.; Schrock R. R.; Takase M. K.; Müller P. Cleavage of Dinitrogen to Yield a (t-BuPOCOP)molybdenum(iv) Nitride. Chem. Commun. 2012, 48, 1851–1853. 10.1039/c2cc17634c. [DOI] [PubMed] [Google Scholar]; b Arashiba K.; Eizawa A.; Tanaka H.; Nakajima K.; Yoshizawa K.; Nishibayashi Y. Catalytic Nitrogen Fixation via Direct Cleavage of Nitrogen–Nitrogen Triple Bond of Molecular Dinitrogen under Ambient Reaction Conditions. Bull. Chem. Soc. Jpn. 2017, 90, 1111–1118. 10.1246/bcsj.20170197. [DOI] [Google Scholar]
- Silantyev G. A.; Förster M.; Schluschaß B.; Abbenseth J.; Würtele C.; Volkmann C.; Holthausen M. C.; Schneider S. Dinitrogen Splitting Coupled to Protonation. Angew. Chem., Int. Ed. 2017, 56, 5872–5876. 10.1002/anie.201701504. [DOI] [PubMed] [Google Scholar]
- Klopsch I.; Finger M.; Würtele C.; Milde B.; Werz D. B.; Schneider S. Dinitrogen Splitting and Functionalization in the Coordination Sphere of Rhenium. J. Am. Chem. Soc. 2014, 136, 6881–6883. 10.1021/ja502759d. [DOI] [PubMed] [Google Scholar]
- Mason J. Nitrogen Nuclear Magnetic Resonance Spectroscopy in Inorganic, Organometallic, and Bioinorganic Chemistry. Chem. Rev. 1981, 81, 205–227. 10.1021/cr00043a001. [DOI] [Google Scholar]
- Soncini A.; Van den Heuvel W. Communication: Paramagnetic NMR Chemical Shift in a Spin State Subject to Zero-field Splitting. J. Chem. Phys. 2013, 138, 021103. 10.1063/1.4775809. [DOI] [PubMed] [Google Scholar]
- Chatt J.; Leigh G. J.; Mingos D. M. P. Configurations of Some Complexes of Rhenium, Ruthenium, Osmium, Rhodium, Iridium, and Platinum Halides with Mono(tertiary phosphines) and Mono(tertiary arsines). J. Chem. Soc. A 1969, 1674–1680. 10.1039/j19690001674. [DOI] [Google Scholar]
- Abbenseth J.; Diefenbach M.; Bete S. C.; Würtele C.; Volkmann C.; Demeshko S.; Holthausen M. C.; Schneider S. A Square-Planar Osmium(II) Complex. Chem. Commun. 2017, 53, 5511–5514. 10.1039/C7CC01569K. [DOI] [PubMed] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) Perchlorate. J. Chem. Soc., Dalton Trans. 1984, 1349–1356. 10.1039/DT9840001349. [DOI] [Google Scholar]
- Stoicheff B. High Resolution Raman Spectroscopy of Gases: III. Raman Spectrum of Nitrogen. Can. J. Phys. 1954, 32, 630–634. 10.1139/p54-066. [DOI] [Google Scholar]
- a Carlotti M.; Johns J. W. C.; Trombetti A. The ν5 Fundamental Bands of N2H2 and N2D2. Can. J. Phys. 1974, 52, 340–344. 10.1139/p74-048. [DOI] [Google Scholar]; b Craig N. C.; Levin I. W. Vibrational assignment and potential function for trans-diazene (diimide): Predictions for cis-diazene. J. Chem. Phys. 1979, 71, 400–407. 10.1063/1.438084. [DOI] [Google Scholar]
- All potentials in this article are internally referenced to the ferrocenium/ferrocene reduction potential, Fc+/0.
- Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- a Mastragostino M.; Nadjo J. M.; Saveant J. Disproportionation and ECE Mechanisms – I. Theoretical analysis. Relationships for Linear Sweep Voltammetry. Electrochim. Acta 1968, 13, 721–762. 10.1016/0013-4686(68)85007-8. [DOI] [Google Scholar]; Amatore C.; Saveant J. ECE and Disproportionation: Part V. Stationary State General Solution Application to Linear Sweep Voltammetry. J. Electroanal. Chem. Interfacial Electrochem. 1977, 85, 27–46. 10.1016/S0022-0728(77)80150-2. [DOI] [Google Scholar]
- The potential of the 2+/2 redox couple in CH2Cl2 was reported at −0.13 V (ref (19)).
- Konezny S. J.; Doherty M. D.; Luca O. R.; Crabtree R. H.; Soloveichik G. L.; Batista V. S. Reduction of Systematic Uncertainty in DFT Redox Potentials of Transition-Metal Complexes. J. Phys. Chem. C 2012, 116, 6349–6356. 10.1021/jp300485t. [DOI] [Google Scholar]
- a Pinski P.; Riplinger C.; Valeev E. F.; Neese F. Sparse Maps – A Systematic Infrastructure for Reduced-scaling Electronic Structure Methods. I. An Efficient and Simple Linear Scaling Local MP2Method that Uses an Intermediate Basis of Pair Natural Orbitals. J. Chem. Phys. 2015, 143, 034108. 10.1063/1.4926879. [DOI] [PubMed] [Google Scholar]; b Riplinger C.; Sandhoefer B.; Hansen A.; Neese F. Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Natural Orbitals. J. Chem. Phys. 2013, 139, 134101. 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]; c Neese F.; Hansen A.; Liakos D. G. Efficient and Accurate Approximations to the Local Coupled Cluster Singles Doubles Method using a Truncated Pair Natural Orbital Basis. J. Chem. Phys. 2009, 131, 064103. 10.1063/1.3173827. [DOI] [PubMed] [Google Scholar]; d Saitow M.; Becker U.; Riplinger C.; Valeev E. F.; Neese F. A New Near-Linear Scaling, Efficient and Accurate, Open-Shell Domain-Based Local Pair Natural Orbital Coupled Cluster Singles and Doubles Theory. J. Chem. Phys. 2017, 146, 164105. 10.1063/1.4981521. [DOI] [PubMed] [Google Scholar]; e Riplinger C.; Neese F. An Efficient and Near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem. Phys. 2013, 138, 034106. 10.1063/1.4773581. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of four M06-Class Functionals and 12 other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Marenich A. V.; Ho J.; Coote M. L.; Cramer C. J.; Truhlar D. G. Computational Electrochemistry: Prediction of Liquid-Phase Reduction Potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. 10.1039/C4CP01572J. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- a Holland P. L. Metal-Dioxygen and Metal-Dinitrogen Complexes: Where are the Electrons?. Dalton Trans 2010, 39, 5415–5425. 10.1039/c001397h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Studt F.; Tuczek F. Theoretical, Spectroscopic, and Mechanistic Studies on Transition-Metal Dinitrogen Complexes: Implications to Reactivity and Relevance to the Nitrogenase Problem. J. Comput. Chem. 2006, 27, 1278–1291. 10.1002/jcc.20413. [DOI] [PubMed] [Google Scholar]
- Peters J. C.; Cherry J.-P. F.; Thomas J. C.; Baraldo L.; Mindiola D. J.; Davis W. M.; Cummins C. C. Redox-Catalyzed Binding of Dinitrogen by Molybdenum N-tert-Hydrocarbylanilide Complexes: Implications for Dinitrogen Functionalization and Reductive Cleavage. J. Am. Chem. Soc. 1999, 121, 10053–10067. 10.1021/ja991435t. [DOI] [Google Scholar]
- a Enemark J. H.; Feltham R. D. Principles of Structure, Bonding and Reactivity for Metal Nitrosyl Complexes. Coord. Chem. Rev. 1974, 13, 339–406. 10.1016/S0010-8545(00)80259-3. [DOI] [Google Scholar]; b Bendix J.; Meyer K.; Weyhermüller T.; Bill E.; Metzler-Nolte N.; Wieghardt K. Nitridocyanometalates of Cr(V), Mn(V), and Mn(VI). Inorg. Chem. 1998, 37, 1767–1775. 10.1021/ic971377h. [DOI] [Google Scholar]
- Abbenseth J.; Finger M.; Würtele C.; Kasanmascheff M.; Schneider S. Coupling of Terminal Iridium Nitrido Complexes. Inorg. Chem. Front. 2016, 3, 469–477. 10.1039/C5QI00267B. [DOI] [Google Scholar]
- a Figg T. M.; Holland P. L.; Cundari T. R. Cooperativity Between Low-Valent Iron and Potassium Promoters in Dinitrogen Fixation. Inorg. Chem. 2012, 51, 7546–7550. 10.1021/ic300150u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b MacLeod K. C.; Menges F. S.; McWilliams S. F.; Craig S. M.; Mercado B. Q.; Johnson M. A.; Holland P. L. The Mechanism of N-N Double Bond Cleavage by an Iron(II) Hydride. J. Am. Chem. Soc. 2016, 138, 11185–11191. 10.1021/jacs.6b04984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pipes D. W.; Bakir M.; Vitols S. E.; Hodgson D. J.; Meyer T. J. Reversible Interconversion between a Nitrido Complex of Os(VI) and an Ammino Complex of Osmium(II). J. Am. Chem. Soc. 1990, 112, 5507–5514. 10.1021/ja00170a014. [DOI] [Google Scholar]; b El-Samanody E.-S.; Demadis K. D.; Meyer T. J.; White P. S. Reversible Osmiun(VI) Nitrido to Osmium(II) Ammine Interconversion in Complexes Containing Polypyrazolyl Ligands. Inorg. Chem. 2001, 40, 3677–3686. 10.1021/ic000764f. [DOI] [PubMed] [Google Scholar]
- Rittle J.; McCrory C. C. L.; Peters J. C. Catalytic Reduction of N2 to NH3 by an Fe-N2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc. 2014, 136, 13853–13862. 10.1021/ja507217v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncari E.; Mazzi U.; Seeber R.; Zanello P. J. Electrochemical Investigation on the Phosphine Rhenium Complexes [ReCl3(PMe2Ph)3)] and [ReCl4(PMe2Ph)2)] in an Aprotic Medium. J. Electroanal. Chem. Interfacial Electrochem. 1982, 132, 221–231. 10.1016/0022-0728(82)85020-1. [DOI] [Google Scholar]
- Martins N. C. T.; Guedes da Silva M. F. C.; Wanke R.; Pombeiro A. J. L. Electrocatalytic Reduction of Organohalides Mediated by the Dihalo-Molybdenum Phosphinic Complexes trans-[MoX2(Ph2PCH2CH2PPh2)2)] (X = I, Br) – A Mechanistical Study by Cyclic Voltammetry Digital Simulation. Dalton Trans. 2009, 4772–4777. 10.1039/b811393a. [DOI] [PubMed] [Google Scholar]
- a Kirchhoff J. R.; Heineman W. R.; Deutsch E. Technetium Electrochemistry. 4. Electrochemical and Spectroelectrochemical Studies on the bis(Tertiary Phosphine or Arsine (D))Rhenium(III) Complexes, trans-[ReD2X2]+ (X = Cl, Br). Comparison with Technetium(III) Analogues. Inorg. Chem. 1987, 26, 3108–3113. 10.1021/ic00266a010. [DOI] [Google Scholar]; b Lever A. P. B. Electrochemical Parametrization of Rhenium Redox Couples. Inorg. Chem. 1991, 30, 1980–1985. 10.1021/ic00009a008. [DOI] [Google Scholar]
- Bazhenova T. A.; Shilov A. E. Nitrogen Fixation in Solution. Coord. Chem. Rev. 1995, 144, 69–145. 10.1016/0010-8545(95)01139-G. [DOI] [Google Scholar]
- a Treitel I. M.; Flood M. T.; Marsh R. E.; Gray H. B. Molecular and Electronic Structure of μ-Nitrogen-decaamminediruthenium(II). J. Am. Chem. Soc. 1969, 91, 6512–6513. 10.1021/ja01051a070. [DOI] [Google Scholar]; b Chatt J.; Fay R. C.; Richards R. L. Preparation and Characterisation of the Dinuclear Dinitrogen Complex, Trichloro-μ-Dinitrogen-Bis(tetrahydrofuran){Chlorotetrakis[dimethyl-(phenyl)phosphine]Rhenium(I)}Chromium(III)[(PMe2Ph)4ClReN2CrCl3(thf)2]. J. Chem. Soc. A 1971, 0, 702–704. 10.1039/J19710000702. [DOI] [Google Scholar]
- Rebreyend C.; de Bruin B. Photolytic N2-Splitting: A Road to Sustainable NH3 Production?. Angew. Chem., Int. Ed. 2015, 54, 42–44. 10.1002/anie.201409727. [DOI] [PubMed] [Google Scholar]
- Scheibel M. G.; Abbenseth J.; Kinauer M.; Heinemann F. W.; Würtele C.; de Bruin B.; Schneider S. Homolytic N–H Activation of Ammonia: Hydrogen Transfer of Parent Iridium Ammine, Amide, Imide, and Nitride Species. Inorg. Chem. 2015, 54, 9290–9302. 10.1021/acs.inorgchem.5b00829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curley J. J.; Cook T. R.; Reece S. Y.; Müller P.; Cummins C. C. Shining Light on Dinitrogen Cleavage: Structural Features, Redox Chemistry, and Photochemistry of the Key Intermediate Bridging Dinitrogen Complex. J. Am. Chem. Soc. 2008, 130, 9394–9405. 10.1021/ja8002638. [DOI] [PubMed] [Google Scholar]
- Miyazaki T.; Tanaka H.; Tanabe Y.; Yuki M.; Nakajima K.; Yoshizawa K.; Nishibayashi Y. Cleavage and Formation of Molecular Dinitrogen in a Single System Assisted by Molybdenum Complexes bearing Ferrocenyldiphosphine. Angew. Chem., Int. Ed. 2014, 53, 11488–11492. 10.1002/anie.201405673. [DOI] [PubMed] [Google Scholar]
- a Cui Q.; Musaev D. G.; Svensson M.; Sieber S.; Morokuma K. N2 Cleavage by Three-Coordinate Group 6 Complexes. W(III) Complexes would be better than Mo(III) Complexes. J. Am. Chem. Soc. 1995, 117, 12366–12367. 10.1021/ja00154a052. [DOI] [Google Scholar]; b Neyman K. M.; Nasluzov V. A.; Hahn J.; Landis C. R.; Rösch N. Density Functional Study of N2 Activation by Molybdenum(III) Complexes. Unusually Strong Relativistic Effects in 4d Metal Compounds. Organometallics 1997, 16, 995–1000. 10.1021/om9607863. [DOI] [Google Scholar]; c Graham D. C.; Beran G. J. O.; Head-Gordon M.; Christian G.; Stranger R.; Yates B. F. Nitrogen Activation via Three-Coordinate Molybdenum Complexes: Comparison of Density Functional Theory Performance with Wave Function Based Methods. J. Phys. Chem. A 2005, 109, 6762–6772. 10.1021/jp044217h. [DOI] [PubMed] [Google Scholar]
- Krewald V.; González L. A Valence-Delocalised Osmium Dimer capable of Dinitrogen Photocleavage: Ab Initio Insights into Its Electronic Structure. Chem. - Eur. J. 2018, 24, 5112–5123. 10.1002/chem.201704651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.