

Summary
The human gastrointestinal tract hosts a diverse network of microorganisms, collectively known as the microbiota that plays an important role in health and disease. For instance, the intestinal microbiota can prevent invading microbes from colonizing the gastrointestinal tract, a phenomenon known as colonization resistance. Perturbations to the microbiota, such as antibiotic administration, can alter microbial composition and result in the loss of colonization resistance. Consequently, the host may be rendered susceptible to colonization by a pathogen. This is a particularly relevant concern in the hospital setting, where antibiotic use and antibiotic-resistant pathogen exposure are more frequent. Many nosocomial infections arise from gastrointestinal colonization. Due to their resistance to antibiotics, treatment is often very challenging. However, recent studies have demonstrated that manipulating the commensal microbiota can prevent and treat various infections in the intestine. In this review, we discuss the members of the microbiota, as well as the mechanisms, that govern colonization resistance against specific pathogens. We also review the effects of antibiotics on the microbiota, as well as the unique epidemiology of immunocompromised patients that renders them a particularly high-risk population to intestinal nosocomial infections.
Keywords: antibiotic, colonization resistance, gut, infection, microbiota
1 | INTRODUCTION
The intestinal microbiota has important implications in human health and disease. One of its clearest roles has been in providing protection against enteric bacterial pathogens. This is particularly important in the hospital setting to prevent nosocomial (i.e. hospital-acquired) infections originating from the gastrointestinal tract. The patient’s commensal microbiota can exclude these pathogens from colonizing the intestinal tract. However, under certain circumstances the patient may develop a compromised microbiota that can no longer protect against colonization by exogenous bacteria. Consequently, these patients can become colonized with a pathogen that can then proliferate to high densities; the gastrointestinal tract can thus serve as an important reservoir for a variety of bacterial pathogens. This poses two important concerns. First, densely colonized patients serve as reservoirs for patient-to-patient transmission, contributing to the endemic persistence of nosocomial infections in hospitals. Second, the pathogen can cause potentially life-threatening disease, especially in immunocompromised patients. Therefore, studying the intestinal microbiota can guide strategies to defend against infectious enteric pathogens, and thereby prevent these complications. Although the microbiota has been implicated in preventing pathogen colonization, the specific contributions and mechanisms that mediate this protection are incompletely defined and continue to be an active area of investigation. This review will explore our current knowledge of the role of the intestinal microbiota in providing protection against enteric infectious diseases, and the strategies that are being developed to enhance resistance against intestinal pathogens.
2 | AN OVERVIEW OF THE MICROBIOTA
2.1 | Overall community structure of the microbiota
Microorganisms have coevolved within the intestines of animal hosts to form a complex ecosystem that is known as the intestinal microbiota. This rich microbial community is densely populated by trillions of bacteria belonging to several hundreds of different species.1 In addition to the diversity within an individual’s microbiota (alpha diversity), there is great diversity in the microbial composition between individuals (beta diversity). Despite this diversity, most of the microbiota members predominantly belong to just four phyla—the Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria. The Firmicutes and Bacteroidetes phyla account for greater than 90% of the bacterial population in the colon, where bacterial density is greatest. On the other hand, Actinobacteria and Proteobacteria are regularly present, but in low abundance.2,3 Below, we will organize and discuss these phyla and highlight important members to better understand the community structure of the microbiota.
2.2 | Bacteroidetes
The Bacteroidetes phylum is composed of both anaerobic and aerobic, non-spore-forming, Gram-negative, rod-shaped bacteria that colonize all of the different regions of the intestinal tract. Within this phylum, the Bacteroides genus is one of the most predominant groups in the intestine.1 These bacteria are well known to digest complex polysaccharides that are resistant to the host’s digestive enzymes. For instance, Bacteroides thetaiotamicron primarily forages on complex O-glycans found in mucin.4 The degradation of these complex carbohydrates yields the release of volatile short-chain fatty acids (SCFAs) like acetate, propionate, and butyrate that are reabsorbed by the host for energy. Additionally, SCFAs have been implicated in regulating intestinal epithelial cell growth,5 and differentiation and stimulation of the immune system6 among various other important biological processes.
Although the Bacteroides genus contributes important metabolic functions and generally maintains a beneficial relationship with the host when retained in the intestinal lumen, certain members within this genus can become pathogenic if they disseminate. For instance, Bacteroides fragilis is typically found in the lower gastro-intestinal tract lumen, and has been shown to have beneficial effects, such as stimulating host immune development.7 However, it is also the most commonly isolated species from infections caused by an obligate anaerobic pathogen. It commonly causes abdominal abscesses and bloodstream infections if the intestinal mucosa is perforated and subsequently traversed.8 Most Bacteroides that remain in the intestinal lumen do not cause intestinal disease, with one exception: Enterotoxigenic B. fragilis (ETBF) produces a toxin termed B. fragilis toxin (BFT) that causes colitis and has been associated with promoting colon tumorigenesis.9,10 In addition, Bacteroides spp. have the most antibiotic resistance mechanisms, andthe highest resistance rates of all obligate anaerobic pathogens.11
2.3 | Firmicutes
The Firmicutes phylum is composed of both obligate and facultative anaerobic bacteria. Most members are Gram-positive, and most can form endospores, providing an ecological advantage for survival under adverse conditions. Endospores are dormant, non-reproductive structures that the bacterium can reduce itself to when encountering environmental stress. Endospores can survive in the absence of nutrients and are remarkably resistant to oxygen, ultraviolet radiation, desiccation, extreme temperatures, and chemical disinfectants. This enables bacteria to lie dormant for extended periods of time, and reactivate into its metabolically active state when the environment becomes favorable.
The Clostridia class within this phylum contains a highly heterogeneous group of bacteria that can further be separated into clusters. Members of Clostridium cluster XIVa and IV represent a majority of the organisms residing in the intestinal tract and have been implicated in a number of beneficial roles. These bacteria are thought to preferentially colonize between mucosal folds to establish a close relationship with the intestinal epithelial cells to help maintain them.12 Species within these clusters are well known to release butyrate as an end-product of fermentation, which like the SCFAs produced by Bacteroides spp., promote intestinal epithelial health. Additionally, members of these clusters promote host immune homeostasis in the intestine by inducing colonic T regulatory cells.13 Other clusters of the Clostridia class, however, include important pathogens that cause human disease including members of cluster I, C. perfringens and C. tetani, as well as C. difficile, a member of cluster XI.14
Finally, another class within the Firmicutes phylum is the Bacilli class. Notable members of this class are the clinically relevant, oxygen-tolerant pathobionts Enterococcus spp. and Streptococcus spp. that are normally found in low abundance, but can undergo pathogenic expansion during intestinal dysbiosis.15,16
2.4 | Actinobacteria
The Actinobacteria phylum is composed of aerobic and anaerobic bacteria. Like the Firmicutes, this phylum is also composed of Gram-positive bacteria, but differs in that they have a high G+C content in their DNA. Within this phylum, Bifidobacteria spp. are one of the major genera of bacteria residing in the intestinal tract.1 Certain species within this genus, including B. longum, have been considered to have probiotic effects. They have been implicated to protect against pathogens through various processes including competitive exclusion, bile salt hydrolase activity, immune modulation, and the ability to adhere to the mucus or intestinal epithelium.17,18
2.5 | Proteobacteria
The Proteobacteria phylum encompasses a wide variety of Gram-negative bacteria. While most of the microbes in the intestinal tract are obligate anaerobes, members of the Proteobacteria phylum are facultative anaerobes. Although Proteobacteria are natural inhabitants of the intestine, they normally comprise a small minority in a healthy, homeostatic microbiota. Some research suggests the increased prevalence of Proteobacteria in the microbial community can serve as a potential diagnostic signature of dysbiosis and risk of disease.19 Specifically, the Enterobacteriaceae family within the Gammaproteobacteria class contains pathogens including Escherichia coli and Klebsiella spp. that are normally in low abundance, but have the potential for overgrowth and intestinal domination during dysbiosis.15,16
2.6 | Anatomic distribution
The gastrointestinal tract is comprised of the stomach, small intestine, cecum, large intestine, and rectum. There are several changes that occur in the environment along this tract. For instance, the pH and oxygen concentration exist on a gradient beginning in the acidic and aerobic stomach and continuing to the more neutral and anaerobic colon. Additionally, nutrient sources shift as resident microbes digest dietary components.20 Particularly, most simple sugars are absorbed in the terminal ileum, and therefore most carbohydrate sources beyond the ileocecal valve are host-derived mucins or dietary carbohydrates indigestible by host enzymes. As a result of several factors, bacteria have a specific distribution in the intestine. The total density of bacteria is greater in the colon than the small intestine. Generally, members belonging to Lactobacillales or Proteobacteria are the major residents of the small intestine. However, in the colon bacteria from Bacteroides and Clostridiales become the dominating members.21
2.7 | The microbiome
The collective genes that an individual’s intestinal microbiota encompasses are known as the microbiome. It overwhelmingly surpasses the coding capacity of the human genome with more than 3 million genes.22 Although there is large inter-individual variability in the bacterial species comprising the host’s microbiota, many microbial genes share functions, resulting in high functional redundancy between microbiomes. These overlapping functions can be considered the core microbiome shared among healthy individuals.23,24 This includes genes involved in the biodegradation of dietary complex sugars and glycans that are indigestible by the host, synthesis of essential amino acids and vitamins, and detoxification of xenobiotics.25,26
3 | ANTIBIOTICS CAN COMPROMISE THE MICROBIOTA
3.1 | Antibiotic selection has implications for the microbiota
Antibiotics are used for the prophylactic and curative treatment of a vast range of potentially life-threatening bacterial infections. Countless lives are saved by antibiotics in the fight against infectious diseases. In recent years, microbiome studies have shown that overuse, prolonged use, incorrect use and the mechanistic properties of many antibiotics may lead to unanticipated and undesirable consequences. Sequelae include antibiotic resistance, intestinal domination by pathogenic bacteria, transient or profound loss of microbial diversity, transient or profound loss of the number of microbial species, increased and prolonged susceptibility to infection and the risk of reoccurring infection.27–34 While these studies have examined the commensal microbiota and its nuanced relationship with antibiotic therapy, careful consideration of the pharmacokinetic implications of antibiotics can allow us to mitigate the damaging effects of antibiotic therapy (Table 1).
TABLE 1.
Pharmacodynamic and pharmacokinetic overview of select antibiotics and their effect on the microbiota28–30,32,33,35,37
| Ampicillin | Clindamycin | Metronidazole | Neomycin | Vancomycin | |
|---|---|---|---|---|---|
| Classification | Aminopenicillin | Lincosamide | Nitroimidazole | Aminoglycoside | Glycopeptide |
| Route of administration | Intramuscular Intravenous Oral |
Intramuscular Intravenous Oral Topical Vaginal |
Intravenous Oral Topical Vaginal |
Intravenous Intramuscular Oral Topical |
Intraocular Intraperitoneal Intrathecal Intravenous Intraventricular Oral |
| Spectrum | (1) Gram + (2) Gram – (3) Anaerobes |
(1) Gram + (2) Anaerobes |
(1) Anaerobes | (1) Gram – (2) Aerobes |
(1) Gram + (2) Aerobes |
| Intestinal absorption by oral administration | Moderate absorption | High absorption | High absorption | Minimal absorption | Minimal absorption |
| Site of absorption | Small Intestine | Small Intestine | Small Intestine | — | — |
| Clearance mechanism | Renala | Biliary | Renala Biliary | Renal | Renala Minimal Biliary |
| Microbiota diversity with oral administration | Long-term changes | Long-term changes | Short-term changes | Long-term changes | Long-term changes |
| Microbiota diversity with systemic administration | Long-term changes | Long-term changes | Undetermined | Minimal changes | Minimal changes |
The pharmacological properties of select antibiotics and their effect on the microbiota are shown. Possible routes of administration—not just the methods commonly used in the clinical setting—are described. Various bacterial exceptions are excluded from spectrum; and moderate absorption and high absorption are defined as 40%-60% and 61%-100%, respectively. Antibiotics may be excreted unchanged in the urine, bile, and/or feces. These antibiotics, depending upon route of administration, may also affect the diversity of species in the intestinal microbiota. These changes may be short-term (less than 2 weeks) or long-term (greater than 2 weeks).
Primary excretion mechanism.
3.2 | Antibiotic pharmacokinetics
Pharmacokinetics is the study of how a drug moves into, through, and out of the body. Considering the pharmacokinetics of an antibiotic can be helpful to understand its effects on the intestinal microbiota. Theoretically, an antibiotic can only alter the composition of the intestinal microbiota by direct exposure, which is only achieved if it reaches the intestinal lumen. While oral administrations are directly delivered to the intestinal lumen, systemic administrations can also indirectly reach the intestine. Systemic antibiotics circulating in the blood eventually reach the liver where they may undergo modifications; then depending upon the antibiotic, they may either be concentrated and excreted into the bile or returned to the blood as waste products for renal clearance. Importantly, bile is delivered to the gastrointestinal tract and excreted in the feces, while blood is filtered by the kidneys and passed through the genitourinary tract for excretion in the urine.35 Generally, hydrophilic agents (e.g. β-lactam antibiotics, aminoglycosides, glycopeptides, lipopeptides) are predominantly eliminated by renal mechanisms. By contrast, hepatic mechanisms of clearance are more common in lipophilic agents (e.g. fluoroquinolones, glycylcyclines, lincosamides, macrolides, metronidazole, streptogramins, tetracyclines).36
Upon reaching the gastrointestinal tract, it is also important to consider the antibiotic’s intestinal absorption. Intestinal absorption depends upon several factors, including the structure and integrity of the intestinal membrane, the specific transport mechanisms involved, and the specific properties of an antibiotic. Antibiotics that are readily absorbed will result in a lower final concentration in the intestinal lumen, reducing exposure to the microbiota. For example, orally administered metronidazole is almost entirely absorbed in the small intestine35 and, as a result, intestinal concentrations of metronidazole significantly decrease along the gastrointestinal tract, with minimal amounts reaching the colon. In contrast, poorly absorbed antibiotics, such as vancomycin, upon oral administration, maintain high concentrations throughout the length of the GI tract. This suggests that along the gastrointestinal tract, oral metronidazole may have less of an impact on the microbiota than oral vancomycin, at least in part due to its absorption by the intestine.
Therefore, considering if an antibiotic’s route of administration, mechanism of excretion, and intestinal permeability deliver the antibiotic, either directly or indirectly, to the intestinal lumen can help to predict whether it will have an effect on the microbiota. This has recently been emphasized by a study that compared the oral and parenteral administration of two antibiotics that differed in their excretion routes, and their consequent effects on the intestinal microbiota. Reporter bacteria resistant to either tetracycline or ampicillin were orally inoculated into conventionally raised mice. Following antibiotic administration, changes in 16S microbial composition and antibiotic resistance (AR) gene pool expression in the intestine were monitored. Oral administrations of either antibiotic resulted in the expansion of the antibiotic-resistant bacteria and expression of AR gene pools. However, intravenous administration of ampicillin, which is predominantly excreted by renal clearance, had no effect on microbial composition and no detected expression of AR gene pools compared to untreated mice. Meanwhile tetracycline, which is excreted by both biliary and renal mechanisms, had a delayed intermediate expansion of tetracycline-resistant bacteria and expression of AR gene pools compared to oral administration.34 These findings are consistent with an antibiotic’s expected effects on the intestinal microbiota when considering its pharmacokinetics. Therefore, in the absence of experimentally defining an antibiotic’s potential to alter the intestinal microbiota, classic pharmacokinetic studies can provide a logical approximation.
3.3 | An antibiotic’s effect on the composition of the microbiota
Recent studies have uncovered the specific changes that occur over time in the intestinal microbiota’s composition during antibiotic-mediated dysbiosis and recovery. These studies serially collected stool specimens from mice or humans treated with an antibiotic to study the composition of bacterial taxa over time after stopping treatment. Different antibiotics that reached the intestine were found to have different effects on the density and diversity of the microbiota.
For instance, two antibiotics used to treat C. difficile, metronidazole and vancomycin, have been compared in mice and demonstrated to exhibit different effects.29 Vancomycin selectively kills Gram-positive bacteria, while metronidazole predominantly targets anaerobic bacteria. The total bacterial density of the microbiota in metronidazole-treated mice was not reduced. However, vancomycin-treated mice had a decrease that returned to pretreatment levels by approximately 2 weeks after stopping treatment. Moreover, mice treated with metronidazole only experienced a relatively transient disruption in their microbiota composition, recovering to a diverse baseline composition within 1-2 weeks. However, studies examining the microbiota of mice29,33 or patients33 treated with vancomycin experienced a profound, long-lasting shift. The overall diversity significantly decreased, with the permanent disappearance of certain bacterial taxa that were abundant prior to treatment. This was accompanied by the expansion of bacterial taxa that were normally present in low or undetectable levels.
Similar patterns of long-term changes to the composition and diversity of the microbiota have been observed with the use of ampicillin and clindamycin.32,37 The expanding bacterial populations during early recovery of the microbiota varied across studies and largely depended on the initial commensal bacteria present after discontinuing antibiotic treatment. Generally, expansion of members from the Proteobacteria phylum was common. Furthermore, the length of time that an antibiotic reduced the microbiota diversity correlated with the window of susceptibility to intestinal colonization by various nosocomial pathogens.29,32,33,37
Upon reaching the intestine, an antibiotic’s effect on the composition depends upon which members of the microbiota it targets as well as its intestinal absorption. For instance, metronidazole and clindamycin both target anaerobic bacteria and are readily absorbed in the small intestine, but clindamycin additionally targets Gram-positive bacteria. The broader spectrum of activity may explain clindamycin’s more pronounced effect in reducing microbial diversity in the long term. Additionally, both vancomycin and ampicillin have broader spectra of activity and are more poorly absorbed than metronidazole in the intestine35; this may similarly explain why both cause greater perturbations to the microbiota.
An antibiotic’s spectrum of activity can also play an important role in which pathogenic bacteria can consequently colonize and expand within the intestine. For instance, metronidazole treatment results in a threefold increased risk of intestinal enterococcal expansion, whereas intravenous vancomycin and beta-lactam administration did not increase this risk.15 Metronidazole narrowly targets obligate anaerobes, and therefore oxygen-tolerant enterococcal species are spared. Vancomycin and beta-lactams, however, have broader spectra that can target antibiotic-sensitive enterococcal species and thereby restrict their growth.
Thus, an antibiotic’s spectrum of activity and intestinal absorption, in part, determine its impact on the microbiota composition and a host’s susceptibility to colonization by pathogens. These can be important aspects, among various other factors, to consider when choosing an antibiotic and predicting its effects on the intestinal microbiota.
4 | NOSOCOMIAL INFECTIONS OF THE GASTROINTESTINAL TRACT
4.1 | Immunocompromised patients are a high-risk population
The microbiota and its role in protecting against infectious diseases is particularly important among hospitalized immunocompromised patients. These individuals with immune suppression, particularly those receiving chemotherapy or undergoing hematopoietic stem cell transplantation, are at an increased risk of infection originating from the gastrointestinal tract.38,39 Prophylactic administration of antibiotics can prevent these infections. However, antibiotics can compromise the intestinal microbiota while simultaneously giving rise to antibiotic-resistant bacteria. There are predominantly two general complications that typically result as a consequence. Colonization with C. difficile can cause intestinal disease,40 whereas other common nosocomial bacterial pathogens can expand in the gastrointestinal tract and translocate into the bloodstream to cause systemic disease.41 We will review the pathogenesis of both of these types of infections that can arise from colonizing the gastrointestinal tract.
4.2 | Bloodstream infections originating from the gastrointestinal tract
Bloodstream infections originating from the gastrointestinal tract that are encountered in immunocompromised patients can result from changes in the microbiota and damage to the mucosal barrier in the setting of an impaired immune system. This is mediated by the combination of chemotherapy, irradiation, and antibiotics that ultimately enable intestinal bacteria to disseminate systemically. The most common bacteria to translocate are oxygen-tolerant pathobionts including vancomycin-resistant Enterococcus, Enterobacteriaceae such as E. coli and Klebsiella spp., and viridans streptococci.41–44
Cytotoxic chemotherapy remains one of the most common treatment options for a variety of cancers, and a common cause of immune suppression in patients. It encompasses a variety of drugs that inhibit different steps involved in cellular mitosis, thereby preferentially affecting rapidly dividing cells like cancer cells. However, specialized stem cells in the gastrointestinal tract,45 and hematopoietic stem cells in the bone marrow46 are two important cell populations that also rapidly divide under healthy homeostatic conditions. These cells become susceptible to the anti-mitotic effects of chemotherapy, collaterally disrupting the normal turnover of these cells.
The specialized stem cells in the gastrointestinal tract normally replenish the epithelial cells of the mucosal barrier to maintain its integrity. However, cytotoxic chemotherapy inhibits the replacement of aging, and damaged epithelial cells lining the intestine. Damaged cells generate reactive oxygen species and initiate a repair response that activates the transcription factor, nuclear factor-κB (NF-κB), not only in epithelial cells, but in all of the surrounding cells and tissue within the mucosa. Downstream pro-inflammatory cytokine production causes inflammatory damage to the tissue and provides a positive-feedback loop to amplify the inflammatory response at the primary site of damage by further activating NF-κB. A combination of inflammatory damage and apoptosis of the cells in the gastrointestinal mucosa often leads to the development of painful lesions and a compromised mucosal barrier. This condition, known as mucositis, is a common complication of chemotherapy.47
Chemotherapy concurrently targets hematopoietic stem cells in the bone marrow. These cells give rise to all blood cell types, with the highest turnover of neutrophils. Neutrophils are the most abundant, but short-lived white blood cells that act as an immediate primary defense against infections.46 The earliest recognizable precursor of neutrophils is the promyelocytes, which actively synthesize DNA and are vulnerable to the anti-mitotic effects of chemotherapy. Their progeny become myelocytes, which are the most numerous proliferating neutrophil precursors, and are consequently the population of cells that are most severely affected by chemotherapy. All of the succeeding precursor cells following myelocytes are non-dividing. Therefore, the loss of cells from the myelocyte compartment has the greatest downstream impact on the degree of neutrophil deficiency (i.e. neutropenia), and the recovery of cells from this population largely determines the duration of neutropenia.48 Chemotherapy-induced neutropenia can render the patient susceptible to certain infections. Therefore, prophylactic broad-spectrum antibiotics are commonly administered to prevent opportunistic infections in the immunocompromised patient.
Due to these changes, immunocompromised patients are specifically susceptible to infections originating from the gastrointestinal tract. First, the patient can develop neutropenia which impairs the patient’s ability to limit bacterial invasion. Second, the patient can be administered broad-spectrum antibiotics that can, together with chemotherapy, promote intestinal dysbiosis and expansion of antibiotic-resistant pathogens. Third, the patient can develop mucositis, creating a portal for intestinal bacteria to enter systemic circulation. Under these circumstances, the colonized pathogen can traverse across the epithelium and enter the bloodstream to establish infection. The pathogen can thereby reside in an intestinal reservoir but continuously seed bloodstream infections when host defenses are impaired.
4.3 | C. difficile pathogenesis
Clostridium difficile infection in immunocompromised patients, like HSCT patients, is far more frequent than in the general patient population.49–54 A steady increase in the incidence of C. difficile infection has been continuing for decades, partially attributed to the rise in emerging antibiotic-resistant strains.55,56 Hosts are rendered susceptible to opportunistic CDI by antibiotic-induced perturbations of the intestinal microbiota that result in the loss of colonization resistance.57 This poses a concern, particularly in the hospital setting, where C. difficile is easily communicable from patient reservoirs. Although an obligate anaerobe, C. difficile persists in the environment and is readily transmitted from individuals due to its ability to form an endospore. The C. difficile spores are most commonly ingested orally by susceptible hosts, whereupon they can germinate into their metabolically active, vegetative state and proliferate in the colon where environmental conditions are favorable again. They then begin to produce toxins A and B, which are internalized by colonic epithelial cells, and consequently glucosylate Rho GTPases that lead to the disruption of tight junctions in the intestinal epithelial barrier.40,57 This establishes C. difficile infection, which can initiate the onset of disease in the host ranging from mild diarrhea to pseudomembranous colitis with life-threatening toxic megacolon.58
5 | COLONIZATION RESISTANCE
5.1 | Discovery of colonization resistance
The intestinal microbiota plays a critical role in excluding invading bacteria and inhibiting the overgrowth of indigenous minority bacteria within the intestinal tract. The microbiota’s role in host defense against enteric pathogens was initially discovered by Bohnhoff et al.59 in the early 1950s that investigated why patients undergoing treatment with antibiotics commonly developed secondary infections. They demonstrated that mice orally administered streptomycin dramatically reduced the density of the intestinal microbiota and increased susceptibility to Salmonella enterica subsp. enteritidis infection by almost six orders of magnitude. Several subsequent studies used various permutations of different animal models, pathogens, and antibiotic regimens that similarly resulted in increased susceptibility to infection following antibiotic-mediated depletion of the microbiota within the intestine. The term ‘colonization resistance’ was thus coined to describe the now widely recognized protection conferred by the intestinal microbiota.60
5.2 | Fecal microbiota transplants restore colonization resistance
The finding that an antibiotic-naive microbiota is resistant to colonization by various exogenous pathogens has led to the investigation of manipulating microbial populations as a potential therapy. An initial approach has been by transferring feces from a healthy donor to the intestinal tract of a recipient patient by fecal microbiota transplantation (FMT). Clear evidence for the benefits of reconstituting the microbiota by FMT was demonstrated by a randomized clinical trial that found duodenal infusion of a fecal solution derived from a healthy donor was significantly more effective than conventional antibiotic therapy in curing recurrent C. difficile infection.61 Additional experimental studies demonstrated that antibiotic-treated mice that were densely colonized with other opportunistic pathogens, including vancomycin-resistant Enterococcus and Klebsiella pneumonieae, similarly fell below the limit of detection following FMT therapy from an antibiotic-naive donor mouse.62,63
However, despite its proven efficacy, FMT therapy still remains a cumbersome treatment that is not commonly performed. The composition of feces is variable, incompletely defined, and complex. While it contains protective species, it simultaneously carries the potential for risks that cannot be reliably detected by current technology. These risks depend upon the microbiota composition and the recipient’s genotype and include the uncertainty of transmitting undetected pathogens and other undefined microbes associated with the development of various disorders including obesity, metabolic syndrome, and autoimmune diseases.64 Therefore, identifying and comprehensively characterizing the precise bacterial species that mediate colonization resistance can standardize therapeutic input and reduce unpredictable outcomes.
5.3 | Key members of the intestinal microbiota mediate colonization resistance
It has been known for over 50 years that commensal anaerobes confer protection against exogenous pathogens.65 However, the specific bacterial species contributing to colonization resistance against various enteric pathogens are incompletely defined. While it has been argued that the recovery of a complex microbiota is important for reestablishing colonization resistance,66 a growing number of studies that analyzed the microbiome of antibiotic-treated mice has led to a compelling argument for, and the subsequent identification of, a few key members that are sufficient to protect against specific pathogens, including C. difficile, VRE, and Listeria monocytogenes.67–69 These studies demonstrated that in mouse cohorts treated with antibiotics, there is a wide distribution in the degree of susceptibility to intestinal pathogen colonization following partial microbiota recovery. Comparing mice that are highly susceptible to pathogen colonization with resistant mice revealed that both groups can have a microbiota with equally low diversity, but that the specific bacterial taxa compositions varied.67,69 These results indicated that the contributions of specific bacterial species, in the absence of a complex microbiota, are sufficient to restore varying degrees of colonization resistance. Subsequent correlation analyses between pathogen susceptibility and bacterial taxa abundance can identify species that correlate with protection against a pathogen. This strategy led to the recent identification of a four-member bacterial consortia that, upon colonizing germ-free mice, confer in vivo resistance to Listeria monocytogenes infection.69
Another study identified a small consortium of protective bacteria against VRE by simplifying the analysis of colonization resistance using a mouse model involving the transplantation of an antibiotic-resistant microbiota. Researchers from this study discovered that a mouse strain had evolved an ampicillin-resistant intestinal microbiota from two decades of chronic oral ampicillin exposure. Subsequently, this ampicillin-resistant microbiota (ARM) and its isolates were studied in a mouse model. Recipient mice with an ampicillin-sensitive microbiota were transplanted with ARM isolates while ampicillin was administered throughout the course of the experiment. Continuous ampicillin treatment minimized the recovery of indigenous ampicillin-sensitive bacteria, but allowed efficient colonization of ARM isolates. This enabled the researchers to dissect the impact of ARM-derived strains on VRE colonization with minimal interference from a recovering indigenous microbiota. This approach led to the discovery of a four-member bacterial consortium that cooperates to eliminate dense intestinal VRE colonization from mice.68
Additionally, several studies have demonstrated that a small consortium of commensal bacteria can treat patients colonized with C. difficile. The first of these studies discovered in the late 1980s that 10 commensal bacterial species cured patients with recurrent C. difficile infection.70 A following study used mice infected with C. difficile to show a consortium of six phylogenetically diverse bacteria can clear C. difficile infection from the intestine,71 while another study used germ-free mice to show that a single Lachnospiraceae isolate can reduce the severity of C. difficile infection.72
6 | DIRECT MECHANISMS OF COLONIZATION RESISTANCE
6.1 | Defining direct colonization resistance
Direct mechanisms of colonization resistance are characterized by the commensal microbiota’s ability to restrict exogenous microbial colonization or prevent pathogenic overgrowth of indigenous microbial members strictly through bacterial factors, independent of any interaction with the host (Figure 1).
FIGURE 1.
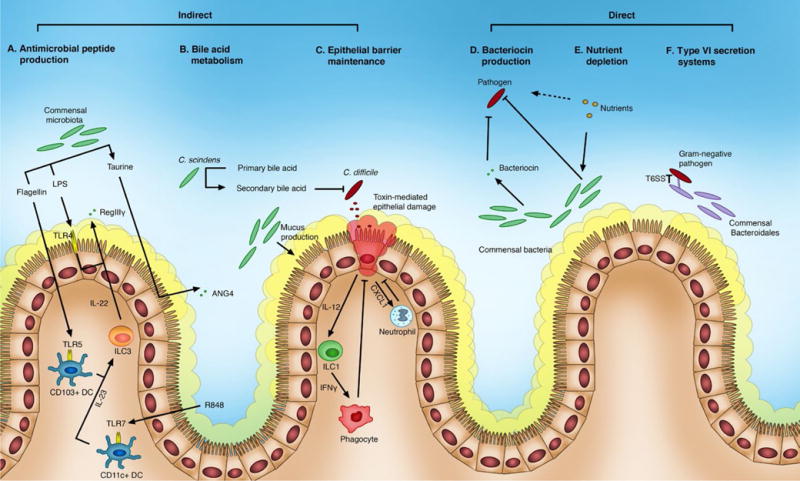
The intestinal microbiota mediates colonization resistance against pathogens by both direct and indirect mechanisms of action. Commensal bacterial species and their products interact with host factors to provide indirect colonization resistance by producing antimicrobial peptides, maintaining the epithelial barrier, and modulating bile acids (A-C). Regenerating islet-derived protein IIIγ (RegIIIγ) and angiogenin-4 (ANG4) are antimicrobial proteins produced by the host that are regulated by the microbiota (A). The microbiota can enhance expression of RegIIIγ by stimulating Toll-like receptors (TLRs). Lipopolysaccharide (LPS) stimulates TLR-4, most likely on intestinal epithelial cells, which results n RegIIIγ production by Paneth cells. Flagellin stimulates TLR-5 on TLR5+CD103+ dendritic cells (DCs), and the TLR-7 agonist resiquimod (R484) stimulates TLR-7 on TLR7+CD11c+ DCs. Activated DCs release IL-23 that stimulates group 3 innate lymphoid cells (ILC3) to secrete IL-22, which subsequently results in Paneth cells producing RegIIIγ. The microbial metabolite, taurine, can signal though an inflammasome complex n the intestinal epithelial cell. The inflammasome contains the nucleotide-binding oligomerization domain (NOD), leucine-rich-repeat (LRR)-containing protein (NLR) family member NLRP6, and caspase-1. Signaling through the inflammasome results in the downstream production of proinflammatory cytokines including IL-18. IL-18 enhances the production of antimicrobial peptides, including ANG4. Certain commensal bacteria, like Clostridium scindens, can dehydroxylate primary bile acids into secondary bile acids using the enzyme 7α-hydroxysteroid deyhydrogenase, which inhibits the vegetative growth of C. difficile (B). The microbiota also maintains the epithelial barrier by inducing mucus production, and transcriptional activation of the nuclear factor-κB (NF-κB) within epithelial cells to delay apoptosis and repair tissue. Pathogens are restricted at sites of epithelial damage by the epithelial cell’s downstream expression of the chemokine CXCL1 that recruits neutrophils, as well as expression of IL-12 that activates ILC1s to release IFNγ for phagocyte recruitment (C). Direct mechanisms of colonization resistance include bacteriocin production, nutrient depletion, and type VI secretion systems (D-F). Several commensal bacteria have been identified to produce bacteriocins with a narrow spectrum of activity that inhibit specific pathogens with minimal impact to the indigenous microbiota (D). Additionally, the microbiota competes with pathogens for various nutrients, including dietary and host-derived carbohydrates, as well as microbial metabolites like succinate, which drives a reduction in pathogen colonization (E). Gram-negative bacteria in the microbiota, like the Bacteroidales order, can deliver toxins to pathogens in a contact-dependent manner via the type VI secretion system. IL, interleukin
6.2 | Nutrient depletion
The availability of host and dietary carbohydrates in the intestine is an important source of energy for members of the microbiota, and can influence its composition.73–76 Dietary carbohydrates can contain both simple monosaccharides and complex polysaccharides. A study identified that certain pathogens, like C. rodentium, are predominantly restricted to utilizing monosaccharides. Additionally, certain members of the intestinal microbiota, like an isolated commensal E. coli species, were found to have similar glycan restrictions. However, other commensal bacterial species, like B. thetaiotaomicron, exhibit broad abilities to readily catabolize both monosaccharides and polysaccharides. A series of mouse experiments investigated the effect of these two types of commensal bacteria on C. rodentium colonization and demonstrated two important concepts.77 First, commensal bacteria and pathogens that share similar glycan preferences compete for the same metabolic niche, which can drive a reduction in pathogen colonization. Second, commensal bacteria that are capable of broad sugar utilization can be manipulated to compete for a targeted metabolic niche by restricting specific dietary carbohydrates. Together, these experiments demonstrated that a pathogen can be antagonized by members of the microbiota with similar metabolic niches, and dietary shifts can influence which members occupy this niche.
Another study similarly demonstrated that a duo of commensal E. coli species together competitively excluded the closely related pathogen enterohemorrhagic E. coli (EHEC) from its metabolic niche, and thereby prevented its colonization.78 A variety of different sugars are available in the mucus layer of the intestinal tract for bacteria to consume. EHEC can use five of these sugars, which defined its distinct niche.79 Additionally, two commensal E. coli strains, E. coli Nissle 1917 and E. coli HS, were capable of utilizing distinct but overlapping sets of defined sugars.78 Neither of the commensal E. coli strains, on their own, utilized all five of the sugars that EHEC was capable of using. However, the combination of the two commensal E. coli strains utilized all five of these sugars, and thereby completely occupied EHEC’s nutrient-defined niche. Mice colonized with only one of the commensal E. coli strains could not resist EHEC colonization. However, co-colonization with both commensal E. coli strains successfully prevented EHEC col-onization.78 This suggests that the distinct but overlapping nutritional profiles of related species in the intestinal microbiota can together saturate a metabolically related pathogen’s niche and outcompete its colonization.
A similar competition for nutrients between a pathogen and the microbiota was demonstrated with host-derived glycans. The mucus layer lining the gastrointestinal tract is primarily composed of mucins, which are heavily O-glycosylated glycoproteins. The O-glycan structures present in mucin are diverse and frequently modified by the sugars fucose and sialic acid.80 Many commensal and pathogenic bacteria utilize sialic acid as a source of energy, carbon, and nitrogen.81 However, sialic acid must be cleaved and released from mucin for access. Some commensal bacteria, like Bacteroides thetaiotaomicron express sialidase which liberates the sialic acid residues from mucin, and thereby provides an important nutrient source for the surrounding microbial community.82,83 Commensal bacteria can antagonize pathogens by competing for the same niche and acquiring nutrients, like sialic acid, more efficiently. C. difficile growth within the intestine of mice was determined to be, at least in part, dependent upon the concentration of free sialic acid. A complex microbiota colonizing the intestine of mice resulted in low levels of free sialic acid, which suggests that this metabolic niche is already efficiently occupied by the indigenous microbial community. Importantly, antibiotic treatment compromises the microbial community occupying this niche. Antibiotic-treated mice had an elevation of free sialic acid levels within the intestine, which promoted C. difficile expansion.84 Together, these experiments suggest the intestinal microbiota likely outcompete C. difficile for available sialic acid, resulting in restricted C. difficile growth.
Additionally, studies have demonstrated that C. difficile competes for microbiota-associated metabolites as an important nutrient source in the intestine. The commensal bacterium, B. thetaiotaomircon, is a primary fermenter that produces high levels of succinate as a by-product in carbohydrate metabolism. There are various secondary fermenters, including C. difficile, that compete for succinate consumption.85 The reduction of succinate to butyrate can be coupled to the oxidation of NADH to NAD+, thereby replenishing the pool of available NAD+.86,87 Subsequently, NAD+ can be utilized in various redox reactions involved in the catabolism of dietary carbohydrates. In an intact microbiota, succinate concentrations were found to be low; this is likely due to succinate consumption by secondary fermenters in the microbiota. However, antibiotic treatment increased succinate concentrations, probably due to the consequent antibiotic-mediated elimination of secondary fermenters. The increased succinate enabled C. difficile to proliferate to higher densities in the intestine.88 This demonstrates that the competing secondary fermenters reduce the pool of available succinate for C. difficile, thereby restricting C. difficile growth.
6.3 | Bacteriocin production
Bacteriocins are ribosomally synthesized peptides that are active against other bacteria and against which the producer has a specific immunity mechanism. This encompasses a large, heterogeneous group of peptides that are usually classified into post-translationally modified peptides (class I) and unmodified peptides (class II).89 Recent analysis of the metagenomic samples from the NIH Human Microbiome Project revealed that small-molecule biosynthetic gene clusters, including many classes of bacteriocins, were widely distributed in genomes and metagenomes of the human microbiota.90 This suggests that these antimicrobial peptides may have a significant influence on the composition, maintenance, and stability of microbial populations.
A commensal bacterium isolated from the human intestinal microbiota, Bacillus thuringiensis DPC 6431, produced a post-translationally modified bacteriocin, thuricin CD.91 This is a class I bacteriocin, and more specifically a type of sactibiotic, which is defined to be a post-translationally modified peptide containing sulfur-α-carbon linkages.92 It was found to have a narrow spectrum of activity in vitro, mainly targeting spore-forming Gram-positive bacteria, and was specifically demonstrated to inhibit several C. difficile isolates. Inhibition of C. difficile in an ex vivo model of the distal colon by thuricin CD was comparable to vancomycin and metronidazole, the current treatments for C. difficile infection. Moreover, unlike metronidazole and vancomycin, thuricin CD appeared to have little impact in the composition of the commensal microbiota.93
Another study screened bacterial strains isolated from resected human terminal ilea, which identified Lactobacillus salivarius strain UCC118 as a probiotic candidate.94 Further studies revealed this bacterium harbored a megaplasmid that encoded the genes for the class II bacteriocin Abp118 that inhibits L. monocytogenes.95 Oral administration of L. salivarius strain UCC118 reduced intestinal L. monocytogenes colonization in mice compared to a L. salivarius strain that does not produce bacteriocins. Moreover, this protection was lost when mice had been infected with a strain of L. monocytogenes that heterologously expressed the cognate Abp118 immunity protein, AbpIM.96 Together, these experiments determined that L. salivarius directly inhibits L. monocytogenes in the intestinal tract by the production of the Abp118 bacteriocin.
Bacteriocins have also been implicated to protect against VRE. Pediococcus acidilactici MM33 is a commensal bacterium isolated from human stool that produces the class II bacteriocin, pediocin PA-1.97,98 P. acidilacticii MM33 had an associated trend toward reducing intestinal VRE colonization, but further study is warranted to assess the statistical significance of these observations.99 A similar study identified Enterococcus faecalis strain CK135, a research strain derived from the commonly studied OGR1F strain, protected against VRE. The strain harbored a plasmid, pPD1, that encoded the enterococcal bacteriocin, bacteriocin 21 (bac-21).100 The expression of this bacteriocin conferred a colonization advantage by antagonizing and displacing related indigenous species. E. faecalis CK135 was shown to specifically compete and ultimately reduce VRE colonization in mice.101
Bacteriocins produced by Gram-negative bacteria are collectively called microcins, despite representing different classes of bacteriocins. Microcins usually exhibit a narrow spectrum of activity, predominantly against other Gram-negative bacteria.89 E. coli Nissle 1917 is a well-studied probiotic that produces two microcins.102 Both are implicated in limiting Enterobacteriaceae overgrowth during dysbiosis.103 Some microcins target cells that express the same nutrient receptors, and hijack these transporters to get internalized into the cell where it can exert its inhibitory effect.104 Specifically, EcN microcins target siderophore receptors expressed by other competing bacteria.105 Siderophores are synthesized and secreted by certain commensal bacteria and pathogens, and act to retrieve iron, an essential nutrient, from the environment. Eventual reuptake of the iron-bound siderophore occurs through a specific membrane receptor that microcins strategically target.106 EcN microcins are suggested to have a post-translationally modified siderophore moiety at the C terminus; this enables them to intercept cells expressing siderophore receptors, and subsequently antagonize them.102,104 EcN administration led to the reduced colonization of E. coli or Salmonella enterica in infected mice.103
6.4 | Type VI secretion system
Type VI secretion systems (T6SSs) have only been identified in Gram-negative bacteria, and most often mediate interactions between Gram-negative bacterial species that are in close proximity to each other. It involves the contact-dependent transport of proteins from a donor cell to a recipient cell.107 This system is composed of two main complexes that assemble with additional bridging and cytoplasmic components. The first complex is membrane-associated, and includes the assembly of two proteins that are homologous to other bacterial secretion systems. The second complex is loaded with the effector protein in the cytoplasm and assembles into a needle-like structure resembling the T4 contractile bacteriophage tail.108,109 These two complexes organize together and coordinate the delivery of effector proteins across the envelope of the donor cell and through the outer membrane into the periplasmic space of the recipient cell. The effector protein is often an antimicrobial toxin, and commonly used as a contact-dependent mechanism of bacterial antagonism.110,111
Several enteric pathogens have T6SSs, including Salmonella enter-ica,112 C. rodentium,113 Aeromonas hydrophila,114 and enteroaggregative E. coli,115 which suggests that there may be an adaptive role for the system in the intestine. Since Gram-positive organisms are not known to be targeted by T6SSs, the two major Gram-negative phyla in the intestinal microbiota, Bacteroidetes and Proteobacteria, can potentially be involved in T6SSs. A comprehensive bioinformatics analysis of human intestinal Bacteroidales genomes revealed that more than half of these Bacteroidales species encode T6SS genes.116 These T6SS loci of human intestinal Bacteroidales species segregated into three evolutionarily distinct genetic architectures (GA) designated GA1, GA2, and GA3. GA1 and GA2 T6SS loci were located on conserved integrative conjugative elements (ICE). This readily enabled transfer between strains and explained the finding that GA1 and GA2 T6SSs are present in diverse Bacteroidales species within the human intestinal microbiota.116 These findings revealed that the microbiota harbors several Bacteroidales strains with numerous different T6SS loci. Communal sharing of these antagonistic systems suggests they function as a defense strategy against a common competitor, such as an invading pathogen.
The GA3 T6SS loci, however, are only found in B. fragilis and are not contained on ICE and therefore do not readily transfer to other Bacteroidales species. Studies in vitro demonstrated that these T6SSs were able to inhibit most Bacteroidales species that did not have the cognate immunity proteins, whereas other Gram-negative E. coli strains were not inhibited.117 This demonstrates that these computationally predicted T6SSs identified from analyzing Bacteroidales genomes are expressed and functional in the human intestinal micro-biota. Moreover, this suggests that T6SSs may also play a role in localized competition between indigenous species, possibly contributing to ecological homeostasis by preventing the overgrowth of certain species.
7 | INDIRECT MECHANISMS OF COLONIZATION RESISTANCE
7.1 | Defining indirect colonization resistance
Indirect mechanisms of colonization resistance are characterized by the commensal microbiota’s dependence upon host-derived factors in order to provide protection against an exogenous pathogen (Figure 1).
7.2 | Antimicrobial peptide production
The microbiota can stimulate the host innate immune receptors, and can consequently result in the production of host-derived antimicrobial peptides. Antimicrobial peptides are produced in the intestine by Paneth cells and epithelial cells.118 Many antimicrobial peptides target bacterial cell wall structures including the membrane and peptidoglycan layer. There is an important compositional difference between bacterial and eukaryotic cell membranes that some antimicrobial peptides exploit to selectively target bacteria. Bacterial cell membranes are predominantly composed of acidic phospholipids, like phosphatidylglycerol and cardiolipin, that results in a net negative charge.119 In contrast, eukaryotic membranes only have negatively charged phospholipids in the inner leaflet of the cell membrane, which faces the cytoplasm. Therefore, many antimicrobial peptides are positively charged and electrostatically interact with the negative charges on the bacterial membrane to disrupt these structures. This leads to membrane potential dysregulation, the loss of ions and metabolites, and eventual osmotic lysis.120
The most well-studied example is the antimicrobial protein RegIIIγ. Germ-free mice reconstituted with an intestinal microbiota demonstrated that the microbiota drives the expression of RegIIIγ.121 RegIIIγ is a C-type lectin produced by Paneth cells in the crypts of the small intestine that mediates Gram-positive bacterial killing by binding peptidoglycan of the bacterial cell wall and forming a hexameric pore that permeabilizes the bacterial membrane.122 This antimicrobial peptide effectively inhibits intestinal pathogens like VRE123 and L. monocytogenes124 in the intestine.
Studies have demonstrated that innate immune signaling through specific toll-like receptors (TLRs) and subsequent recruitment of the adapter, myeloid differentiation primary response protein 88 (MyD88), can substitute for the microbiota to induce RegIIIγ expression. Oral administrations of the TLR-4 ligand lipopolysaccharide (LPS), activate TLR-4-expressing non-hematopoietic-derived cells, likely intestinal epithelial cells. This leads to MyD88-dependent RegIIIγ expression that confers resistance to VRE colonization.123
Additionally, RegIIIγ induction can be mediated by innate immune activation that originates in deeper subepithelial tissues of the GI tract, possibly as a mechanism of alerting epithelial cells to the loss of mucosal integrity. Systemic administration of the TLR-5 ligand, flagellin, activates TLR-5-expressing CD103+CD11b+ dendritic cells residing in the intestinal lamina propria to rapidly produce IL-23; this stimulates innate lymphoid cells (ILCs) to produce IL-22, resulting in IL-22-dependent expression of RegIIIγ in intestinal epithelial cells, which markedly reduced VRE colonization.125,126 These findings suggest bacterial-derived ligands, like LPS or flagellin, can target specific innate immune receptors in lieu of the microbiota to mediate an antibacterial response.
Interestingly, antiviral signaling through innate immune receptors can also augment microbiota-driven colonization resistance by enhancing RegIIIγ expression. This was exemplified using antibiotic-treated mice infected with murine norovirus, which activates TLR-7, or by orally administering resiquimod (R848)—a synthetic TLR-7 agonist. Both activate TLR-7-expressing CD11c+ dendritic cells that respond by secreting IL-23. ILCs release IL-22 upon activation by IL-23, resulting in the IL-22-dependent induction of RegIIIγ and restoration of colonization resistance against VRE.127
Although RegIIIγ is the most well-studied example of the micro-biota inducing the host to produce antimicrobial peptides, various others have been identified. Defensins, also known as cryptdins, are a class of small cationic peptides produced by Paneth cells that similarly disrupt cell membranes. Alpha-defensins are expressed exclusively by Paneth cells in the small intestine and are among the most highly expressed antimicrobial peptides in the intestine.128 Expression of these antimicrobial peptides has been implicated to be regulated by the intracellular pattern-recognition receptor NOD2. Studies have demonstrated that NOD2−/− mice have impaired α-defensin expression in the intestine, and are consequently more susceptible to oral infection with L. monocytogenes.129,130 Subsequent studies revealed that α-defensin expression can be independent of NOD2 when the genetic background of the mouse strain used in experiments is different.131 Studies with germ-free mice also suggest that α-defensin expression may be independent of microbial signals.132
Angiogenin-4 (ANG4) is another antimicrobial peptide exclusively expressed by Paneth cells. It is a protein belonging to a family of ribo-nucleases with antimicrobial functions that target both Gram-negative and Gram-positive bacteria. Colonizing germ-free mice with a conventional microbiota, or specifically with Bacteroides thetaiotaomicron, was found to enhance the production of ANG4 by Paneth cells.133 Additionally, ANG4 expression has been determined to be regulated, at least in part, by the activation of an intestinal inflammasome complex. An inflammasome is a cytoplasmic multiprotein complex that is composed of one or several nucleotide-binding oligomerization domain protein-like receptors (NLRs). The complex senses molecular patterns, including microbial products. Specifically, NLRP6 is a member of the NLR family of proteins that interacts with the adapter protein, apoptosis-associated speck-like protein containing a CARD (ASC), to form an inflammasome complex that has an important role in regulating the colonic microbiota.5 Activation of the NLRP6 inflammasome results in the downstream activation of caspase-1 and subsequent cleavage of effector proinflammatory cytokines including IL-18. The consequent induction of IL-18 leads to the production of antimicrobial peptides, including ANG4. Moreover, the microbiota produces metabolites that modulate the NLRP6 inflammasome and subsequent antimicrobial peptide expression. For instance, taurine is a bile acid conjugate produced by certain members of the microbiota that has been shown to activate NLRP6 inflammasome signaling and specifically enhance ANG4 production.133
7.3 | Epithelial barrier maintenance
Mucus covers and protects the intestinal epithelium by maintaining the spatial separation between lumenal bacterial colonization and the host epithelial barrier.134 The thickness of the mucus is dependent upon the microbiota. Germ-free mice have thinner mucus layers that can grow to the thickness of conventionally raised mice by the administration of microbial products like lipopolysaccharide or peptidoglycan.135
However, upon mucosal adherence, pathogens like C. difficile disrupt the epithelial barrier by damaging epithelial cells to establish an infection. NF-κB is an important signaling pathway for epithelial tissue repair. Although it has been implicated in the pathogenesis of various inflammatory diseases when signaled in immune cells,136 NF-κB-mediated transcriptional activation within intestinal epithelial cells has been shown to reduce epithelial cell death and promote tissue repair.137,138 The microbiota engages innate immune receptors that can lead to the activation of NF-κB transcription in intestinal epithelial cells, and thereby maintain the integrity of the epithelial barrier.139,140 NF-κB leads to the downstream production of proinflammatory cytokines and induces anti-apoptotic factors, stimulates proliferation, and stabilizes tight junctions.137 Innate immune activation via TLR-5 stimulation improved survival and reduced severe weight loss during acute C. difficile infection in mice, potentially due to downstream transcriptional activation of NF-κB. This was demonstrated with the systemic administration of flagellin, a TLR-5 agonist, which significantly reduced C. difficile colonization and toxin production in antibiotic-treated mice. Furthermore, examining the cecum and colon revealed that flagellin treatment protected against C. difficile-mediated epithelial cell loss, thereby maintaining the structural integrity of the epithelial barrier.141
This preserved barrier reduces pathogen invasion while host immune cells are recruited to the site of damage. The majority of TLR signaling is mediated through the MyD88 adapter protein. MyD88 signaling also induces the expression of a potent neutrophil-recruiting chemokine, CXCL1, in the colon. The CXCR2 chemokine receptor expressed by neutrophils allows them to migrate to the colon to respond to CXCL1, where they subsequently associate closely with the intestinal epithelium, especially at sites of damage. This MyD88-dependent recruitment of neutrophils plays a critical role in creating a barrier to contain luminal bacteria and prevent systemic bacterial dissemination, and is required for recovery from an acute C. difficile infection.142
The downstream activation of proinflammatory cytokines also activates ILCs that reside within the epithelial layer and underlying lamina propria. These cells are important early responders to acute C. difficile infection by maintaining tissue integrity. More specifically, two CD90+CD127+ ILC subsets, T-bet-expressing ILC1s and retinoic acid-related orphan receptor-γt (Rorγt)-expressing ILC3s are activated following C. difficile-mediated epithelial damage. ILC1s are activated by IL-12 and produce IFNγ, and activate phagocyte-mediated clearance of bacteria that enter the lamina propria. In addition, ILC3s produce IL-22 upon activation by IL-23 or IL-1 β, and contribute a minor role in eliminating bacteria that do disseminate by activating the complement pathway via IL-22.143
Together, these findings suggest that the microbiota provides baseline stimulation to innate immune receptors that function to keep both epithelial and immune cells alert. These host cells readily prevent and respond to pathogen-mediated epithelial damage through the production of mucus, preservation of epithelial cells, and recruitment of immune cells.
7.4 | Bile acid metabolism
In addition to interacting with the host’s innate immune system, the microbiota interacts with host-derived molecules, like bile acids, to convert them into toxic products that inhibit C. difficile growth. Bile acids are amphipathic molecules that are produced in the liver and secreted by the gallbladder into the intestinal tract to aid in emulsifying dietary fats for digestion. Most are reabsorbed in the terminal ileum, but a small remaining fraction continues to the large intestine where a subset of bacteria in the colon can convert them into secondary bile acids. Primary bile acids are deconjugated by a wide range of bacterial species that express bile salt hydrolases. Then, a smaller subset of bacteria in the colon dehydroxylates the unconjugated bile acid to form secondary bile acids.144 Importantly, different bile acids have different effects on promoting germination and vegetative growth. While the primary bile acid taurocholic acid induces germination of C. difficile spores, secondary bile acids have been found to inhibit the growth of vegetative, toxin-producing cells.145
A recent study used mathematical modeling to identify commensal bacterial species involved in secondary bile acid conversion correlated with resistance to the development of C. difficile colitis in both mouse models and in hospitalized patients. The commensal anaerobe, Clostridium scindens, demonstrated the strongest association with resistance to C. difficile infection in both mice and humans. C. scindens converts endogenous primary bile acids into secondary bile acids, particularly into deoxycholate acid and lithocholate acid. These secondary bile acids inhibit C. difficile growth in a dose-dependent manner. The enzyme 7α-hydroxysteroid dehydrogenase is responsible for conversion of primary bile acids into secondary bile acids, and is part of a bile acid inducible operon that was identified in C. scinden’s genome. Additionally, sequestering bile acids by the addition of cholestyr-amine eliminates C. scinden’s inhibitory effect on C. difficile growth.67 Together, these experiments suggest the commensal bacterium, C. scindens, mediates C. difficile inhibition through the conversion of host-derived primary bile acids into secondary bile acids.
8 | CONCLUSION
Recent studies have provided insight into the mechanisms mediating colonization resistance against pathogens. However, research in this field is still in an early phase. Considerable research efforts are still in the screening phase to identify important protective species from the microbiota. These recent advances in identifying small consortia of protective microbial members are promising, and establish a catalogue of important species implicated in human health. However, while this simplifies the complexity of the microbiota to a minimum, our understanding of colonization resistance remains incomplete. The recently discovered four-member consortium that can decolonize VRE from the gastrointestinal tract serves as an example. Although the consortium demonstrates impressive efficacy in eradicating VRE colonization, it operates through an undefined mechanism of bacterial cooperation. The future direction of research is in exploring these dynamic relationships between bacteria within the microbiota; dissecting these complex interactive networks will help to isolate the precise mechanisms mediating colonization resistance and provide insight into the foundations that govern specific bacterial interdependencies.
Subsequently, strategies targeted to enhance these mechanisms can provide an alternative to the use of current antibiotics in treating challenging infections. This is particularly relevant to addressing the rise of antibiotic-resistant infections. VRE and several Enterobacteriaceae species are major causes of intestinal nosocomial infection that exhibit concerning degrees of AR. Patients rendered susceptible to intestinal infections because of the loss of microbiota-mediated colonization resistance can harbor high concentrations of these pathogens and contribute to their persistence and transmission. However, the recent discoveries of specific bacteria capable of defending against certain pathogens suggest that reestablishing precise, key components of the microbiota has promising therapeutic potential. A more comprehensive understanding of the microbiota’s mechanisms of colonization resistance can guide future strategies for identifying susceptible patients, as well as preventing and treating infections originating from the gastrointestinal tract.
Acknowledgments
S. K. is supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences, NIH (award T32GM07739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program). E. G. P. has received funding from NIH grants R01 AI042135, AI095706, U01 AI124275, and P30 CA008748.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
References
- 1.Rajilic-Stojanovic M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. doi: 10.1111/1574-6976.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley R, Walter J. The human gut microbiome: Ecology and recent evolutionary changes. Annu Rev Microbiol. 2011;65:411–429. doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- 3.Tap J, Mondot S, Levenez F, et al. Towards the human intestinal micro-biota phylogenetic core. Environ Microbiol. 2009;10:2574–2584. doi: 10.1111/j.1462-2920.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- 4.Thomas F, Hehemann J, Rebuffet E, Czjzek M, Michel G. Environmental and gut bacteroidetes: The food connection. Front Micobiol. 2011;2:93. doi: 10.3389/fmicb.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy M, Thaiss CA, Zeevi D, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163:1428–1443. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazmanian SK, Kasper DL. The love-hate relationship between bacterial polysaccharides and the host immune system. Nat Rev Immunol. 2006;6:849–858. doi: 10.1038/nri1956. [DOI] [PubMed] [Google Scholar]
- 8.Wexler H. Bacteroides: The good, the bad, and the nittygritty. Clin Microbiol Rev. 2007;4:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sears CL. Enterotoxigenic Bacteroides fragilis: A rogue among symbiotes. Clin Microbiol Rev. 2009;22:349–369. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via actiation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salyers AA, Gupta A, Wang Y. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol. 2004;12:412–416. doi: 10.1016/j.tim.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011;5:627–638. doi: 10.1038/ismej.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. Commensal Clostridia: Leading players in the maintenance of gut homeostasis. Gut Pathog. 2013;5:23. doi: 10.1186/1757-4749-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins MD, Lawson PA, Willems A, et al. The phylogeny of the genus Clostridium: Proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994;4:812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- 15.Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55:905–914. doi: 10.1093/cid/cis580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taur Y, Pamer EG. The intestinal microbiota and susceptibility to infection in immunocompromised patients. Curr Opin Infect Dis. 2013;26:332–337. doi: 10.1097/QCO.0b013e3283630dd3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Picard C, Fioramonti J, Francois A, Robinson T, Neant F, Matuchansky C. Review article: Bifidobacteria as probiotic agents— Physiological effects and clinical benefits. Aliment Pharmacol Ther. 2005;22:495–512. doi: 10.1111/j.1365-2036.2005.02615.x. [DOI] [PubMed] [Google Scholar]
- 18.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin NR, Whon TW, Bae JW. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Koropatkin NM, Cameron EA, Martens EC. How glycans metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2009;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lozupone C, Stombaugh J, Gordon J, Jansson J, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurokawa K, Itoh T, Kuwahara T, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friefeld AG, Bow EJ, Sepkowitz KA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the infectious diseases society of America. Clin Infect Dis. 2011;52:e56–e93. doi: 10.1093/cid/cir073. [DOI] [PubMed] [Google Scholar]
- 28.Gilbert DN, Moellering RC, Eliopoulous GM, Chambers HF, Saag MS. The Sanford Guide to Antimicrobial Therapy 2013. 43rd. Virginia: Antimicrobial Therapy; 2013. pp. 71–76. [Google Scholar]
- 29.Lewis BB, Buffie CG, Carter RA, et al. Loss of microbiota-mediated colonization resistance to Clostridium difficile infection with oral vancomycin compared with metronidazole. J Infect Dis. 2015;212:1656–1665. doi: 10.1093/infdis/jiv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandell GL, Bennet JE, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. Seventh. Philadelphia: Churchill Livingstone; 2009. pp. 309–467. [Google Scholar]
- 31.Taur Y, Jenq RR, Ubeda C, van den Brink M, Pamer EG. Role of intestinal microbiota in transplantation outcomes. Best Pract Res Clin Haematol. 2015;28(2–3):155–161. doi: 10.1016/j.beha.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buffie CG, Jarchum I, Equinda M, et al. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun. 2012;80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isaac S, Scher JU, Djukovic A, et al. Short-and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother. 2017;72:128–136. doi: 10.1093/jac/dkw383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Huang Y, Zhou Y, Buckley T, Wang H. Antibiotic administration routes significantly influence the levels of antibiotic resistance in gut microbiota. Animicrob Agents Chemother. 2013;57:3659–3666. doi: 10.1128/AAC.00670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levison ME, Levison JH. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect Dis Clin North Am. 2009;23:791–vii. doi: 10.1016/j.idc.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vincent JL, Bassetti M, Francois B, et al. Advances in antibiotic therapy in the critically ill. Crit Care. 2016;20:133. doi: 10.1186/s13054-016-1285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ubeda C, Taur Y, Jenq R, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. Journal of Clinical Investigation. 2010;120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Velasco E, Byington R, Martins CAS, Schirmer M, Dias LMC, Goncalves VMSC. Comparative study of clinical characteristics of neutropenic and non-neutropenic adult cancer patients with bloodstream infections. Eur J Clin Microbiol Infect Dis. 2006;25:1–7. doi: 10.1007/s10096-005-0077-8. [DOI] [PubMed] [Google Scholar]
- 39.Koll BS, Brown AE. The changing epidemiology of infections at cancer hospitals. Clin Infect Dis. 1993;17:S322–S328. doi: 10.1093/clinids/17.supplement_2.s322. [DOI] [PubMed] [Google Scholar]
- 40.Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: Pathogenesis and host defence. Nat Rev Microbiol. 2016;14:609–620. doi: 10.1038/nrmicro.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taur Y, Pamer EG. Microbiome mediation of infections in the cancer setting. Microbiome mediation of infections in the cancer setting. Genome Med. 2016;8:40. doi: 10.1186/s13073-016-0306-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamboj M, Chung D, Seo SK, et al. The changing epidemiology of vancomycin-resistant Enterococcus (VRE) bacteremia in allogeneic hematopoietic stem cell transplant (HSCT) recipients. Biol Blood Marrow Transplant. 2010;16:1571–1581. doi: 10.1016/j.bbmt.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almyroudis N, Fuller A, Jakubowski A, et al. Pre-and post-engraftment bloodstream infection rates and associated mortality in allogeneic hematopoietic stem cell transplant receipients. Tranplant Infect Dis. 2005;7:11–17. doi: 10.1111/j.1399-3062.2005.00088.x. [DOI] [PubMed] [Google Scholar]
- 44.Weinstock DM, Conlon M, Iovino C, et al. Colonization, bloodstream infection, and mortality caused by vancomycin-resistant enterococcus early after allogeneic hematopoietic stem cell transplant. Biol Blood Marrow Transplant. 2007;13:615–621. doi: 10.1016/j.bbmt.2007.01.078. [DOI] [PubMed] [Google Scholar]
- 45.Van Vliet MJ, Harmsen HJ, de Bont ES, Tissing WJ. The role of intestinal microbiota in the development and severity of chemotherapy-induced mucositis. PLoS Pathog. 2010;6:e1000879. doi: 10.1371/journal.ppat.1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia: Risks, consequences, and new directions for its management. Cancer. 2004;100:228–237. doi: 10.1002/cncr.11882. [DOI] [PubMed] [Google Scholar]
- 47.Sonis ST. The pathobiology of mucositis. Nat Rev Cancer. 2004;4:277–284. doi: 10.1038/nrc1318. [DOI] [PubMed] [Google Scholar]
- 48.Dale DC. Myelosuppresion. In: Sonis ST, Keefe DM, editors. Pathobiology of Cancer Regimen-Related Toxicities. New York, NY: Springer; 2013. [Google Scholar]
- 49.Alonso CD, Treadway SB, Hanna DB, et al. Epidemiology and outcomes of Clostridium difficile infections in hematopoietic stem cell transplant recipients. Clin Infect Dis. 2012;54:1053–1063. doi: 10.1093/cid/cir1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leung S, Metzger BS, Currie BP. Incidence of Clostridium difficile infection in patients with acute leukemia and lymphoma after allogeneic hematopoietic stem cell transplantation. Infect Control Hosp Epidemiol. 2010;31:313–315. doi: 10.1086/651066. [DOI] [PubMed] [Google Scholar]
- 51.Chopra T, Chandrasekar P, Salimnia H, Heilbrun LK, Smith D, Alangaden GJ. Recent epidemiology of Clostridium difficile infection during hematopoietic stem cell transplantation. Clin Transplant. 2011;25:E82–E87. doi: 10.1111/j.1399-0012.2010.01331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chakrabarti S, Lees A, Jones S, Milligan D. Clostridium difficile infection in allogeneic stem cell transplant recipients is associated with severe graft-versus-host disease and non-relapse mortality. Bone Marrow Transplant. 2000;26:871–876. doi: 10.1038/sj.bmt.1702627. [DOI] [PubMed] [Google Scholar]
- 53.Willems L, Porcher R, Laufaurie M, et al. Clostridium difficile infection after allogeneic hematopoietic stem cell transplantation: Incidence, risk factors, and outcome. Biol Blood Marrow Transplant. 2012;18:1295–1301. doi: 10.1016/j.bbmt.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 54.Lessa FC, Gould CV, McDonald LC. Current status of Clostridium difficile infection epidemiology. Clin Infect Dis. 2012;55:S65–S70. doi: 10.1093/cid/cis319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 56.Owens RC, Jr, Donskey CJ, Gaynes RP, Loo VG, Muto CA. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S19–S31. doi: 10.1086/521859. [DOI] [PubMed] [Google Scholar]
- 57.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 58.Johnson S, Gerding DN. Clostridium difficile-associated diarrhea. Clin Infect Dis. 1998;26:1027–1036. doi: 10.1086/520276. [DOI] [PubMed] [Google Scholar]
- 59.Bohnhoff M, Drake BL, Miller CP. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc Soc Exp Biol Med. 1954;86:132–137. doi: 10.3181/00379727-86-21030. [DOI] [PubMed] [Google Scholar]
- 60.van der Waaij D, Berghuis de Vries JM, Lekkerkerk V. Colonization resistance of the digestive tract in conventional and antibiotic-treated mice. J Hyg (Lond) 1971;69:405–411. doi: 10.1017/s0022172400021653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 62.Caballero S, Carter R, Ke X, et al. Distinct but spatially overlapping intestinal niches for vancomycin-resistant Enterococcus faecium and carbapenem-resistant Klebsiella pneumoniae. PLoS Pathog. 2015;11:e1005132. doi: 10.1371/journal.ppat.1005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ubeda C, Bucci V, Caballero S, et al. Intestinal microbiota containing barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect Immun. 2013;81:965–973. doi: 10.1128/IAI.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pamer EG. Fecal microbiota transplantation: Effectiveness, complexities, and lingering concerns. Mucosal Immunol. 2014;7:210–214. doi: 10.1038/mi.2013.117. [DOI] [PubMed] [Google Scholar]
- 65.Bohnhoff M, Miller CP, Martin WR. Resistance of the mouse’s intestinal tract to experimental salmonella infection. I. Factors which interfere with the initiation of infection by oral inoculation. J Exp Med. 1964;120:805–816. doi: 10.1084/jem.120.5.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang JY, Antonopoulos DA, Kalra A, et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 67.Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caballero S, Kim S, Carter RA, et al. Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enter-ococcus faecium. Cell Host Microbe. 2017;21:592–602. doi: 10.1016/j.chom.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Becattini S, Littmann E, Carter RA, et al. Commensal microbes provide first line defense against Listeria monocytogenes infection. J Exp Med. 2017;214 doi: 10.1084/jem.20170495. https://doi.org/10.1084/jem.20170495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tvede M, Rask-Madsen J. Bacteriotherapy for chronic relapsing Clostridium difficile diarrhoea in six patients. Lancet. 1989;1:1156–1160. doi: 10.1016/s0140-6736(89)92749-9. [DOI] [PubMed] [Google Scholar]
- 71.Lawley TD, Clares S, Walker AW, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun. 2012;80:3786–3794. doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walker AW, Ince J, Duncan SH, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salonen A, Lahti L, Salojarvi J, et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014;8:2218–2230. doi: 10.1038/ismej.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez I, Lattimer JM, Hubach KL, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013;7:269–280. doi: 10.1038/ismej.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamada N, Kim YG, Sham HP, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maltby R, Leatham-Jensen MP, Gibson T, Cohen PS, Conway T. Nutritional basis for colonization resistance by human commensal Escherichia coli strains HS and Nissle 1917 against E. coli O157:H7 in the mouse intestine. PLoS ONE. 2013;8:e53957. doi: 10.1371/journal.pone.0053957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fabich AJ, Jones SA, Chowdhury FZ, et al. Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76:1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brockhausen I, Schachter H, Stanley P. Chapter 9 O-GalNAc glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al., editors. Essentials of Glycobiology. 2nd. New York, NY: Cold Spring Harbor; 2009. [PubMed] [Google Scholar]
- 81.Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev. 2004;68:132–153. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcobal A, Barboza M, Froehlich JW, et al. Consumption of human milk oligosaccharides by gut-related microbes. J Agric Food Chem. 2010;58:5334–5340. doi: 10.1021/jf9044205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ng KM, Ferreyra JA, Higginbottom SK, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fischbach MA, Sonnenburg JL. Eating for two: How metabolism establishes interspecies interactions in the gut. Cell Host Microbe. 2011;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kenealy WR, Waselefsky DM. Studies on the substrate range of Clostridium kluyveri: The use of propanol and succinate. Arch Microbiol. 1985;141:187–194. [Google Scholar]
- 87.Wolff RA, Urben GW, O’Herrin SM, Kenealy WR. Dehydrogenases involved in the conversion of succinate to 4-hydroxybutanoate by Clostridium kluyveri. Appl Environ Microbiol. 1993;59:1876–1882. doi: 10.1128/aem.59.6.1876-1882.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe. 2014;16:770–777. doi: 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cotter PD, Ross PR, Hill C. Bacteriocins—A viable alternatiave to antibiotics? Nat Rev Microbiol. 2013;11:95–105. doi: 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- 90.Donia MS, Cimermancic P, Schulze CJ, et al. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158:1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rea MC, Sit CS, Clayton E, et al. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci USA. 2010;107:9352–9357. doi: 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mathur H, Rea MC, Cotter PD, Hill C, Ross RP. The sactibiotic subclass of bacteriocins: An update. Curr Protein Pept Sci. 2015;16:549–558. doi: 10.2174/1389203716666150515124831. [DOI] [PubMed] [Google Scholar]
- 93.Rea MC, Dobson A, O’Sullivan O, et al. Effect of broad-and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4639–4644. doi: 10.1073/pnas.1001224107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Collins JK, Thornton G, Sullivan GO. Selection of probiotic strains for human applications. Int Dairy J. 1998;8:487–490. [Google Scholar]
- 95.Claesson MJ, Li Y, Leahy S, Canchaya C, van Pijkeren JP, Cerdeno-Tarraga AM. Multireplicon genome architecture of Lactobacillussalivarius. Proc Natl Acad Sci USA. 2006;103:6718–6723. doi: 10.1073/pnas.0511060103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corr SC, Li Y, Riedel CU, O’Toole PW, Hill C, Gahan CGM. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci USA. 2007;104:7617–7621. doi: 10.1073/pnas.0700440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Millette M, Dupont C, Archambault D, Lacroix M. Partial characterization of bacteriocins produced by human Lactococcus lactis and Pediococcus acidilactici isolates. J Appl Microbiol. 2007;102:274–282. doi: 10.1111/j.1365-2672.2006.03044.x. [DOI] [PubMed] [Google Scholar]
- 98.Millette M, Dupont C, Shareck F, Ruiz MT, Archambault D, Lacroix M. Purification and identification of the pediocin produced by Pediococcus acidilactici MM33, a new human intestinal strain. J Appl Microbiol. 2008;104:269–275. doi: 10.1111/j.1365-2672.2007.03583.x. [DOI] [PubMed] [Google Scholar]
- 99.Millette M, Cornut G, Dupont C, Shareck F, Archambault D, Lacroix M. Capacity of human nisin-and pediocin-producing lactic acid bacteria to reduce intestinal colonization by vancomycin-resistant Enterococci. Appl Environ Microbiol. 2008;74:1997–2003. doi: 10.1128/AEM.02150-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tomita H, Fujimoto S, Tanimoto K, Ike Y. Cloning and genetic and sequence analyses of the bacteriocin 21 determinant encoded on the Enterococcus faecalis pheromone-responsive conugative plasmid pPD1. J Bacteriol. 1997;179:7843–7855. doi: 10.1128/jb.179.24.7843-7855.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kommineni S, Bretl DJ, Lam V, et al. Bacteriocin production augments niche competition by enterococci in the mammalian GI tract. Nature. 2015;526:719–722. doi: 10.1038/nature15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vassiliadis G, Destoumieux-Garzon D, Lombard C, Rebuffat S, Peduzzi J. Isolation and characterization of two members of the siderophore-microcin family, microcins M and H47. Antimicrob Agents Chemother. 2010;54:288–297. doi: 10.1128/AAC.00744-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sassone-Corsi M, Nuccio SP, Liu H, et al. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature. 2016;540:280–283. doi: 10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Patzer SI, Baquero MR, Bravo D, Moreno F, Hantke K. The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IroN. Microbiology. 2003;149:2557–2570. doi: 10.1099/mic.0.26396-0. [DOI] [PubMed] [Google Scholar]
- 105.Nolan EM, Walsh CT. Investigations of the MceIJ-catalyzed post-translational modification of the microcin E492 C-terminus: Linkage of ribosomal and nonribosomal peptides to form “Trojan horse” antibiotics. Biochemistry. 2008;47:9289–9299. doi: 10.1021/bi800826j. [DOI] [PubMed] [Google Scholar]
- 106.Fischbach MA, Lin H, Liu DR, Walsh CT. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol. 2006;22:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 107.Russell AB, Peterson SB, Mougous JD. Type VI secretion system effectors: Poisons with a purpose. Nat Rev Microbiol. 2014;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leiman PG, Basler M, Ramagopal UA, et al. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci USA. 2009;106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature. 2012;483:182–186. doi: 10.1038/nature10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011;475:343–347. doi: 10.1038/nature10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 112.Blondel CJ, Jimenez JC, Contreras I, Santiviago CA. Comparative genomic analysis uncovers 3 novel loci encoding type six secretion systems differentially distributed in Salmonella serotypes. BMC Genom. 2009;10:354. doi: 10.1186/1471-2164-10-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gueguen E, Cascales E. Promoter swapping unveils the role of the Citrobacter rodentium CTS1 type VI secretion system in interbacterial competition. Appl Environ Microbiol. 2013;79:32–38. doi: 10.1128/AEM.02504-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suarez G, Sierra JC, Sha J, Wang S, Erova TE, Fadl AA. Molecular characterization of a functional type VI secretion system from a clinical isolate of Aeromonas hydrophila. Microb Pathog. 2008;44:344–361. doi: 10.1016/j.micpath.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brunet YR, Espinosa L, Harchouni S, Mignot T, Cascales E. Imaging type VI secretion-mediated bacterial killing. Cell Rep. 2013;3:36–41. doi: 10.1016/j.celrep.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 116.Coyne MJ, Roelofs KG, Comstock LE. Type VI secretion systems of human gut Bacteroidales segregate into three genetic architectures, two of which are contained on mobile genetic elements. BMC Genom. 2016;17:58. doi: 10.1186/s12864-016-2377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chatzidaki-Livanis M, Geva-Zatorsky N, Comstock LE. Bacteroides fragilis type VI secretion systems use novel effector and immunity proteins to antagonize human gut Bacteroidales species. Proc Natl Acad Sci USA. 2016;113:3627–3632. doi: 10.1073/pnas.1522510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 119.Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim Biophys Acta. 1999;1462:1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 120.Brogden KA. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 121.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mukherjee S, Zheng H, Derebe MG, et al. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature. 2014;505:103–107. doi: 10.1038/nature12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brandl K, Plitas G, Mihu CN, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455:804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. 2007;204:1891–1900. doi: 10.1084/jem.20070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kinnebrew MA, Buffie CG, Diehl GE, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kinnebrew MA, Ubeda C, Zenewicz LA, Smith N, Flavell RA, Pamer EG. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. J Infect Dis. 2010;201:534–543. doi: 10.1086/650203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abt MC, Buffie CG, Susac B, et al. TLR-7 activation enhances IL-22-mediated colonization resistance against vancomycin-resistant enterococcus. Sci Transl Med. 2016;8:327ra25. doi: 10.1126/scitranslmed.aad6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 129.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 130.Petnicki-Ocwieja T, Hrncir T, Liu YJ, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shanahan MT, Carroll IM, Grossniklaus E, et al. Mouse Paneth cell antimicrobial function is independent of Nod2. Gut. 2014;63:903–910. doi: 10.1136/gutjnl-2012-304190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Putsep K, Axelsson LG, Boman A, et al. Germ-free and colonized mice generate the same products from enteric prodefensins. J Biol Chem. 2000;275:40478–40482. doi: 10.1074/jbc.M007816200. [DOI] [PubMed] [Google Scholar]
- 133.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 134.Johansson ME, Ambort D, Pelaseyed T, et al. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci. 2011;68:3635–3641. doi: 10.1007/s00018-011-0822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Petersson J, Schreiber O, Hansson GC, et al. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G327–G333. doi: 10.1152/ajpgi.00422.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eckmann L, Nebelsiek T, Fingerle AA, et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: Implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 138.Ben-Neriah Y, Schmidt-Supprian M. Epithelial NF-kappaB maintains host gust microflora homeostasis. Nat Immunol. 2007;8:479–481. doi: 10.1038/ni0507-479. [DOI] [PubMed] [Google Scholar]
- 139.Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 140.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 141.Jarchum I, Liu M, Lipuma L, Pamer EG. Toll-like receptor 5 stimulation protects mice from acute Clostridium difficile colitis. Infect Immun. 2011;79:1498–1503. doi: 10.1128/IAI.01196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun. 2012;80:2989–2996. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abt MC, Lewis BB, Caballero S, et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe. 2015;18:27–37. doi: 10.1016/j.chom.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 145.Sorg JA, Sonenshein LA. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]