

Abstract
Biological processes are naturally regulated with high spatial and temporal control, as is perhaps most evident in metazoan embryogenesis. Chemical tools have been extensively utilized in cell and developmental biology to investigate cellular processes, and conditional control methods have expanded applications of these technologies toward resolving complex biological questions. Light represents an excellent external trigger since it can be controlled with very high spatial and temporal precision. To this end, several optically regulated tools have been developed and applied to living systems. In this review we discuss recent developments of optochemical tools, including small molecules, peptides, proteins, and nucleic acids that can be irreversibly or reversibly controlled through light irradiation, with a focus on applications in cells and animals.
Keywords: caged compounds, chemical biology, optochemical tools, photochemistry, photoswitches
Graphical abstract

1. Introduction
Achieving precise conditional control of biological function represents a crucial tool for studying the mechanisms of cellular processes. Naturally, these processes occur in a strict spatially and temporally regulated fashion on a cell and organism level. In order to study these processes, the utilized tools must operate with the same level of spatiotemporal resolution. To this end, the use of light as a conditional stimulus has found extensive applications for the activation and deactivation of small molecules, proteins, peptides, and oligonucleotides. Harnessing light has enabled significant advances in biological studies and holds promise toward use in clinical settings. This Review showcases many of the most recent applications and methodology developments in optochemical control of biology. In contrast to excellent previous reviews that focus on the chemistry of controlling biological function with light,[1] this Review focuses on the manipulation of cell and animal biology using photochemical approaches. Purely optogenetic approaches that do not utilize chemical operations have been reviewed elsewhere.[2]
2. Optical Control of Small Molecules
Small-molecule probes have been essential tools to perturb and control cellular processes, thereby providing a detailed understanding of biological function. Optical control of small-molecule function provides an additional layer of precision to the molecular toolbox by enabling temporal and spatial control with high resolution, as discussed in the examples below. Small-molecule protein dimerizers, inhibitors, metabolites, and metal ions have all been rendered light-responsive through the chemical installation of caging groups and have been applied to the investigation of living systems. In contrast to photocaged nucleic acids, peptides, and proteins, small molecules have the added benefits of being synthetically more accessible and capable of being readily delivered into cells and animals.
2.1. Optical Activation of Rapamycin-Induced Protein Dimerization
Chemical inducers of dimerization (CIDs) are prominent tools for chemical biologists to place biological processes under conditional control.[3] The most commonly utilized CID is rapamycin, which binds to FK506-binding protein (FKBP) and the FKBP-rapamycin binding (FRB) domain of mTOR to form a ternary complex. Due to the small size of FKBP and FRB, these domains have been fused to numerous proteins, and subsequent heterodimerization is induced by rapamycin. Processes that have been placed under rapamycin control include Golgi/endoplasmic reticulum association to study mitosis,[4] phosphoinositide control of endocytic trafficking,[5] and inactivation of proteins by rerouting them to the mitochondria.[6] Caged rapamycin analogues allow these processes to be placed under optical control in order to enhance temporal and spatial resolution.
The photocaged rapamycin analogue 1 was generated through installation of a nitrobenzyl caging group at the C-40 position (Figure 1a).[7] When applied to cells, 1 still induces FKBP–FRB dimerization, thus indicating that the caging group alone is not sufficient to abrogate protein/small-molecule interactions, which is consistent with previous modifications at C-40.[8] However, work by the Hahn group has shown that a truncated FKBP, termed iFKBP,[9] exhibits increased flexibility in the loop that interacts with the C-40 position of rapamycin. This flexibility increases contacts for interaction with 1 and results in a distorted binding conformation that prevents formation of the ternary complex consisting of iFKBP, FRB, and 1. This system was then applied to the optical activation of FAK (focal adhesion kinase), using an engineered iFKBP-FAK fusion that rendered the kinase inactive until UV irradiation led to rapamycin decaging, FAK activation, and an expected cell ruffling phenotype (Figure 1b). While 1 was readily synthesized, the need for FKBP truncation required some protein engineering.
Figure 1.
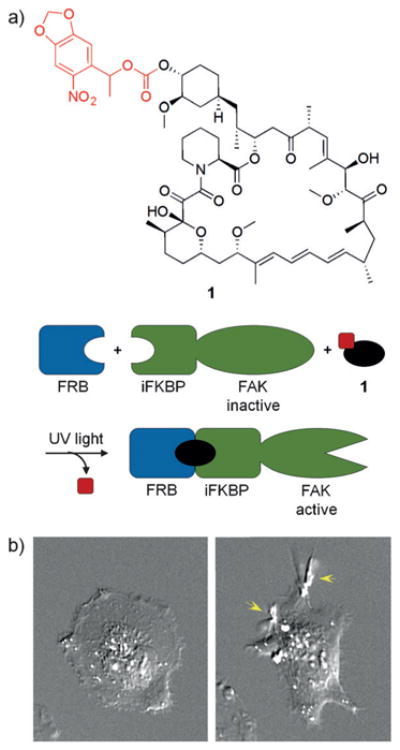
a) The structure of 1 is shown with the caging group in red. In this system, FAK activity was monitored in the presence of 1 with and without UV exposure. b) Cells treated with 1 alone did not display active FAK (left); however, UV irradiation led to activation of FAK and subsequent cell ruffling (right). Adapted with permission from Ref. [7]. Copyright 2011 American Chemical Society.
A concurrent approach by Inoue and co-workers addressed this limitation with the photocleavable rapamycin construct 2, which contains a biotin linked through a photo-cleavable caging group to the C-40 position of rapamycin.[10] Similar to 1, the biotin moiety itself is too small to block FKBP interaction; however, when bound to the avidin protein, steric demand is significantly increased and cell permeability is diminished, thus sequestering 2 outside of the cell (Figure 2a). A related strategy of using a ligand/protein complex as a caging group, rather than a small synthetic chromophore, was previously reported by Miller.[11] Upon irradiation, the avidin-biotin moiety is removed to generate the cell-permeable hydroxyethyl rapamycin 3, which leads to intracellular dimerization of FKBP and FRB and, for example, membrane recruitment of a protein of interest, such as Tiam, a Rac1 activator. The latter induces cell migration and ruffling at the edge of cells (Figure 2b). Overall, 2 enables the spatiotemporal activation of protein dimerization and can be readily interfaced with a range of biological systems that have already been placed under control by rapamycin.
Figure 2.

a) The complex of 2 and streptavidin is unable to enter cells and does not induce protein dimerization until UV irradiation generates 3. b) Rac-FKBP was used together with membrane localized FRB. In the absence of UV light, cells displayed normal cell edges (left); however, upon irradiation and Rac localization, cell ruffling is apparent (right). Adapted with permission from Ref. [10]. Copyright 2011 American Chemical Society.
In order to alleviate the need for an external avidin protein while still capitalizing on the dramatic steric demand provided through recruitment of a protein to the caging group, a second rapamycin molecule was linked to generate the symmetric dimer 4 (Figure 3a).[12] Computational and experimental studies confirmed that the formation of a FKBP–4–FKBP homodimer sterically blocked binding of FRB. Since 4 does not require the use of an engineered iFKBP or the use of an exogenous protein, such as avidin, this represents a more practical engineering approach for light-triggered dimerization. The broad applicability of light-activated 4 was demonstrated by optical control of mTOR kinase, TEV protease, and Cre recombinase function through protein dimerization (Figure 3b,c).
Figure 3.
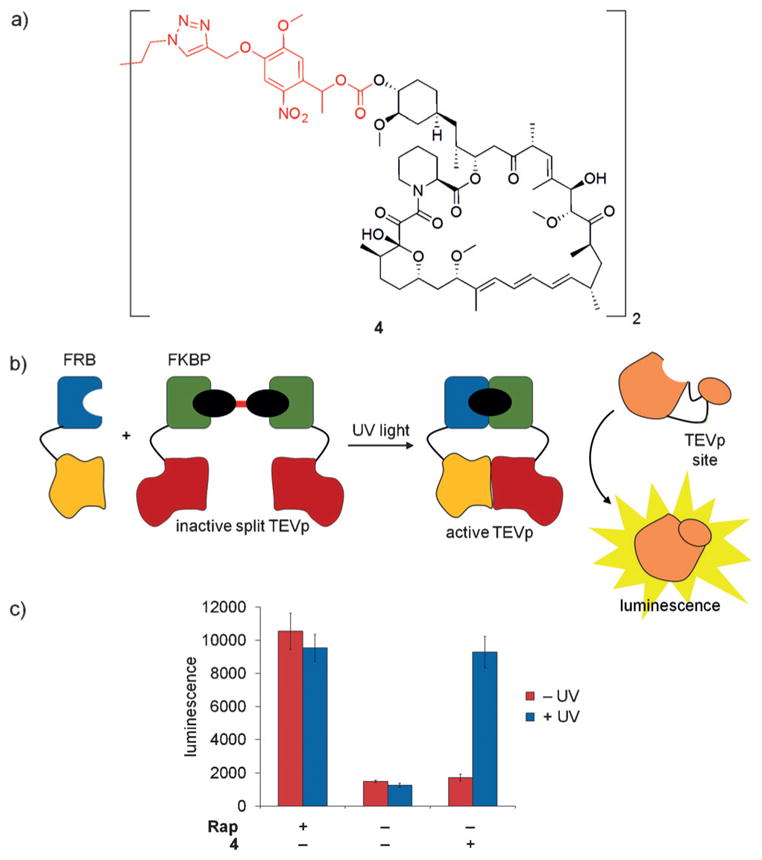
a) The structure of 4 is shown with the caging group highlighted in red. b,c) The caged rapamycin dimer 4 was applied to a split TEV protease system consisting of FRB-TEVp (N-terminus) and FKBP-TEVp (C-terminus) to demonstrate optical control. In the presence of 4, no protease activity is detected. However, after UV irradiation and dimerization of split TEV, a luciferase reporter is proteolytically cleaved and luminescence is generated. Adapted with permission from Ref. [12]. Copyright 2015 Royal Society of Chemistry.
2.2. Optical Activation of Other Chemical Inducers of Protein Dimerization
While rapamycin remains the best-studied CID, other dimerizer systems have been developed and placed under optical control. One limitation of the caged rapamycin system is the generation of free and diffusible rapamycin following UV irradiation, which limits spatial resolution. To address this concern, Chenoweth and co-workers developed the photo-activatable covalent protein dimerizer 5, which contains a chloro-alkyl moiety for bioconjugation to the HaloTag enzyme, and a trimethoprim (TMP) group that binds to E. coli dihydrofolate reductase (DHFR).[13] The caging group blocks the TMP/DHFR interaction until irradiation and subsequent ternary complex formation (Figure 4a,b). This interaction can be reversed through addition of an excess of TMP, which outcompetes the dimerizer ligand. Additionally, the modularity of this approach allows easy exchange of components, such as caging groups and binding proteins (e.g., SNAP-tag or cutinase).[14] The caged TMP-HaloTag ligand has enabled spatial control of protein dimerization in a variety of cellular compartments, including kinetochores, centromeres, and centrosomes (Figure 4c). Unlike the rapamycin dimerization system, where protein binding and ternary complex formation occur following irradiation, this approach overcomes the diffusion limitation through covalent tethering of TMP to the HaloTag protein, followed by ternary complex formation after light activation to yield tight spatial control of dimerization.
Figure 4.
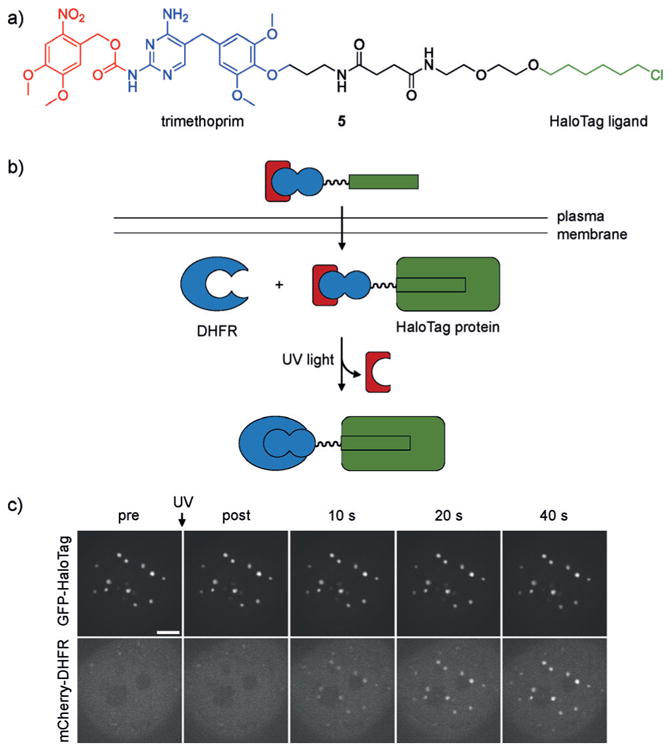
a) The structure of 5 is shown with the photolabile group highlighted in red, the trimethoprim group that interacts with DHFR is shown in blue, and the alkyl chloride is shown in green. b) Compound 5 enters cells and covalently labels HaloTag protein fusions. The removal of the caging group with UV light allows dimerization with the DHFR protein fusion. c) Recruitment of mCherry to centromere-localized GFP following 387 nm light exposure. Adapted with permission from Ref. [13a]. Copyright 2014 Nature Publishing Group.
In order to complement light-activated CIDs, Wymann and co-workers developed the light-cleavable CID 6, which enables the deactivation of protein dimerization.[15] This approach also utilizes the HaloTag protein and its chloro-alkyl ligand, as well as SNAP-tag technology (Figure 5a). The SNAP protein is an engineered mutant of the DNA repair enzyme O6-alkylguanine-DNA alkyltransferase and reacts specifically and covalently with benzylguanine analogues. The chloro-alkyl group and the benzylguanine are linked through a photocleavable linker. Delivery into cells induces covalent dimerization of SNAP-tag and HaloTag fusion proteins until the linker is cleaved through UV irradiation (Figure 5b,c). Cells expressing NLS-CFP-SNAP (nuclear) and Halo-RFP-giantin (Golgi) were treated with 6, which induced dimerization and translocation of CFP from the nucleus to the Golgi. Upon UV irradiation and subsequent linker cleavage, CFP translocated back to the nucleus. This approach may enable the deactivation of proteins through sequestration to a non-native location until photoactivation triggers protein transport to its native compartment.
Figure 5.
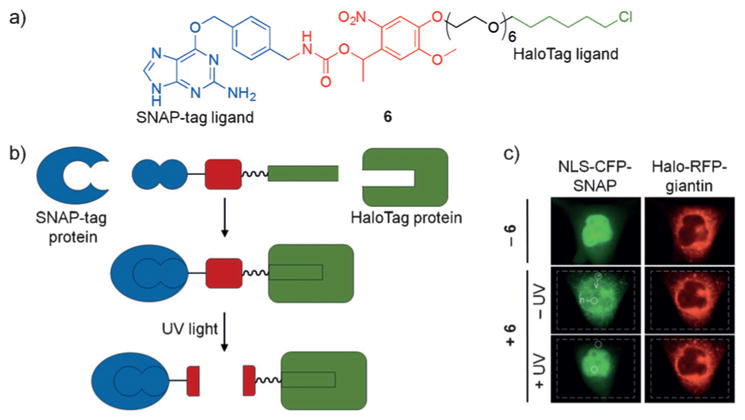
a) The structure of 6 is shown with the SNAP-tag reactive region in blue, the caging group in red, and the HaloTag ligand in green. b) Upon addition of 6 to cells, the SNAP-tag and HaloTag moieties react with their respective protein binding partners, thus dimerizing the two proteins. Upon UV irradiation, the linker is cleaved to generate free proteins again. c) Hela cells co-expressing NLS-CFP-SNAP and Halo-RFP-giantin show nuclear and mitochondrial localized CFP and RFP, respectively in the absence of 6 (row 1). Following addition of 6, the SNAP-tag and HaloTag ligands react with their protein partners to form a covalent complex, which results in export of NLS-CFP-SNAP from the nucleus to the Golgi (row 2). Upon UV irradiation, nuclear localization of NLS-CFP-SNAP is obtained again (row 3). Adapted with permission from Ref. [15]. Copyright 2014 Wiley-VCH.
Additionally, two other natural product inspired, caged CIDs have been developed based on abscisic acid[16] and gibberellic acid.[17] These methods present alternative CID approaches that do not require protein engineering for efficient optical control and have no endogenous off-target interactions when applied in mammalian cells since the dimerization components are native to plants.
Overall, several photo-controlled CIDs have been reported and enable the activation or deactivation of protein–protein dimerization. The tools directly interface with well-established and commercially available systems, such as FKBP/FRB, HaloTag, and SNAP-tag, and thus have the potential to provide optical control for a wide range of cellular processes.
2.3. Engineering Pharmacophores for Reversible Photoswitching of Cellular Processes
While optical control of most small molecules, including the examples above, involves an irreversible photolysis step, pharmacologically active compounds have also been rendered reversibly light-switchable to provide enhanced spatial and temporal regulation. Specifically, azobenzene isosteres (“azosteres”) have been identified (Figure 6a)[18] including stilbenes, N-aryl benzamides, benzyl phenyl (thio)ethers, benzyl anilines, and 1,2-diaryl ethanes. Replacement of these structural motifs with a photoswitchable azobenzene is expected to yield comparable efficacy as the parent compound, while optical switching to the other azobenzene configuration should abrogate or diminish biological activity. This direct substitution of a similar structural motif with an azobenzene has been termed “azologization” (Figure 6b), whereas the addition of an azobenzene group to an existing fragment has been referred to as “azoextension” (Figure 6c).[19] Unlike traditional caging approaches, this method enables highly controlled on/off/on cycles for the precise and dynamic activation and deactivation of biological processes.
Figure 6.
a) Isosteres of azobenzene (“azosteres”), for example stilbenes and arylamides, are common structural motifs found in existing pharmaceutical compounds. b) An example of azologization is shown wherein an arylamide is replaced with an azobenzene. c) Replacement of the morpholine ring in ciprofloxacin with an azobenzene functionality demonstrates an “azoextension approach.
In pioneering work by Trauner and co-workers, photo-switchable small molecules have been successfully applied for controlling endogenous G-protein-coupled receptors,[19a] bacterial growth,[19b,20] ion channels,[21] GABAA receptors,[22] nicotinic acetylcholine receptors,[23] insulin release,[24] histone deacetylases,[25] amidohydrolases,[26] and proteasomal function.[27]
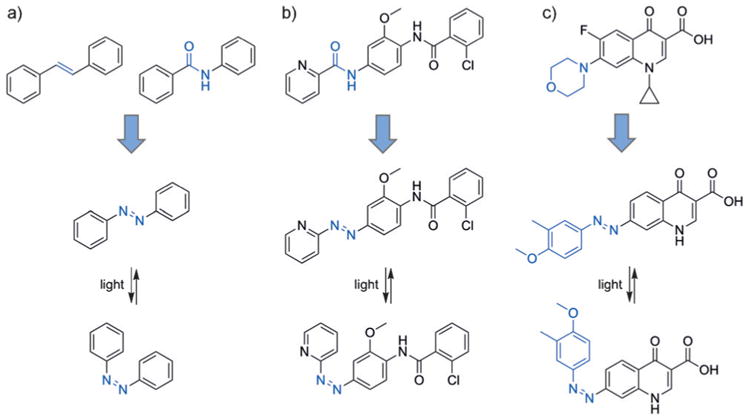
Microtubules are cytoskeletal proteins that play a role in cell proliferation and migration, as well as signaling and trafficking pathways.[28] They are attractive targets for cancer therapeutics, and several small-molecule ligands have been discovered. Colchicine-like inhibitors typically bind to the soluble α/β-tubulin dimers to prevent microtubule polymerization; however, they can also bind to functional micro-tubules to induce depolymerization.[29] Unfortunately, existing small-molecule inhibitors cannot be controlled spatially or temporally, and in order to address this challenge Trauner and co-workers structurally analyzed colchicine (7) and combretastatin A-4 (8). The photoswitchable analogue 9 was derived by replacing the central stilbene motif with an azobenzene (Figure 7a).[30] While in the cis configuration, the azobenzene displays a set of pharmacologically essential methoxy groups in the same arrangement as 7 and 8; however upon photo-chemical or thermal reversion to the trans isomer, the pharmacophore is lost. Cells were treated with 9 and stained for the presence of tubulin (absence of tubulin indicates an active inhibitor). After optical switching of 9 to the active cis configuration, minimal tubulin formation was observed and a 250-fold increase in cytotoxicity was detected compared to cells that were kept in the dark (Figure 7b). Additionally, the function of 9 can be reversibly regulated, which addresses the limitations of photocaged small-molecules that can only be activated once.
Figure 7.
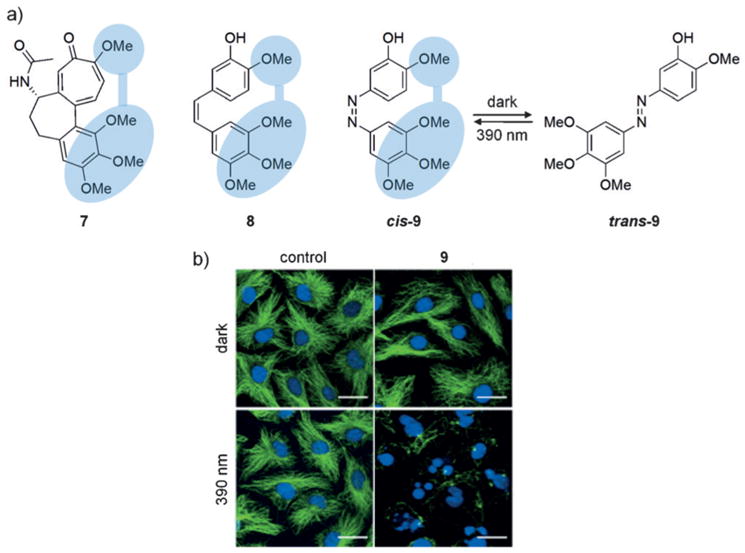
a) The structures of 7 and 8 are shown with the pharmacophore indicated in blue (left). Replacement of the stilbene moiety in combrestatin A-4 with an azobenzene group produces cis-9 which can be reversibly converted into trans-9. b) MDA-MB-231 cells treated with 9 (1.5 μM) and maintained in the dark (predominantly containing the thermodynamically more stable trans isomer) exhibit no microtubule inhibition as seen from the tubulin (green) and DNA (blue) staining. However, upon irradiation and formation of the cis isomer, microtubule inhibition is observed. Adapted with permission from Ref. [30]. Copyright 2015 Elsevier Inc.
Diacylglycerols (DAGs) are components of the phospholipid bilayer and play a key role as second messengers in signaling.[31] In addition, they are involved in membrane recruitment of kinases and nucleotide-exchange factors through their C1 domain, a cysteine-rich region found at the N-terminus of several proteins.[32] Although C1 domains are found in a variety of kinases, only a specific subset is recruited to the membrane by DAG, thereby providing an opportunity for controlled recruitment and dissociation of protein–membrane interactions. Inspired by previous work on mimicking arachidonic acid with photoswitchable fatty acids,[33] a photo-switchable DAG was engineered by Trauner and co-workers.[34] The molecule mimics the natural glycerol 10 when photoisomerized from the more stable trans configuration to the cis configuration (Figure 8). Thus, introduction of trans-11 does not lead to membrane recruitment of a C1 domain GFP fusion protein until UV-induced switching to cis-11 takes place. Thermal reversion resulted in dissociation from the membrane. In these experiments, recombinantly overexpressed C1 fusion proteins were utilized, possibly due to the necessity of outcompeting endogenous proteins bearing C1 domains. Further applications of photoswitchable DAGs include regulating intracellular Ca2+ levels and controlling synaptic transmission in mammalian cells and C. elegans. Additionally, reversibly controlled protein kinase C (PKC) localization and activation at the membrane was demonstrated. Previously, PKC and C1 domains have been controlled through the introduction of nitrobenzyl and coumarin caging groups on dioctanoyl glycerol[35] or through bromohydroxycoumarin-caged DAG lactones;[36] however these methods only allow off-to-on activation. The azobenzene-based system described above provides multiple cycles of recruitment/activation and dissociation/inactivation in an innovative approach (see Section 3.3 for a further discussion of the advantages of reversible photoswitching of cellular processes).
Figure 8.
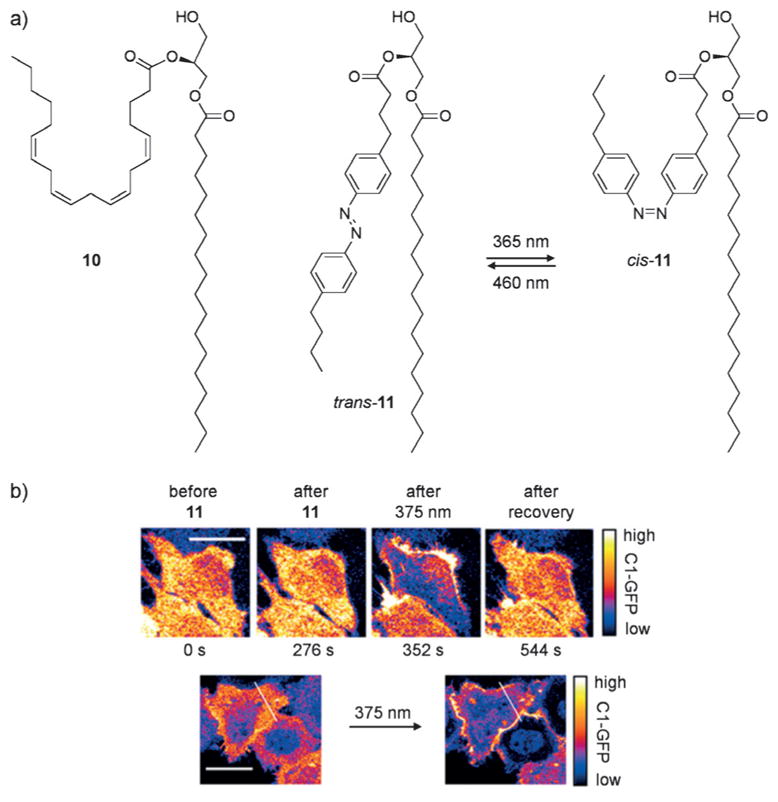
a) The structures of 10, trans-11, and cis-11 are shown. b) Hela cells were transfected with C1-GFP, then treated with trans-11. Treatment with trans-11 does not induce membrane recruitment; however, following UV-induced conversion to cis-11, C1-GFP rapidly localizes to the membrane. Adapted with permission from Ref. [34]. Copyright 2016 Nature Publishing Group.
In addition to the reversible control of signaling lipids, fatty acids have been caged through traditional approaches. Lysophosphatidic acid (LPA) has been implicated in the mobility and invasion of cancer cells.[37] The biological effects of LPA are heavily dependent on localization and timing, and Schultz and co-workers applied a coumarin caging group to the phosphate head group in 12 to generate a concentration gradient of active LPA following localized irradiation (Figure 9).[38] When A375M cells (a cell line with known chemotactic behavior toward LPA) were treated with caged LPA in the dark, no cell migration was observed. However, upon pulsed irradiation (20 ms every 40 s) for 4 hours, cells noticeably migrated to the point of irradiation as the LPA concentration was locally increased (Figure 9). This demonstrates that localized release of a small, diffusible signaling molecule through spatially controlled repetitive decaging enables the establishment of a concentration gradient due to the diffusion of the caged effector into the irradiation area and the rapid dilution of small quantities of decaged compound through diffusion into the surrounding area.
Figure 9.
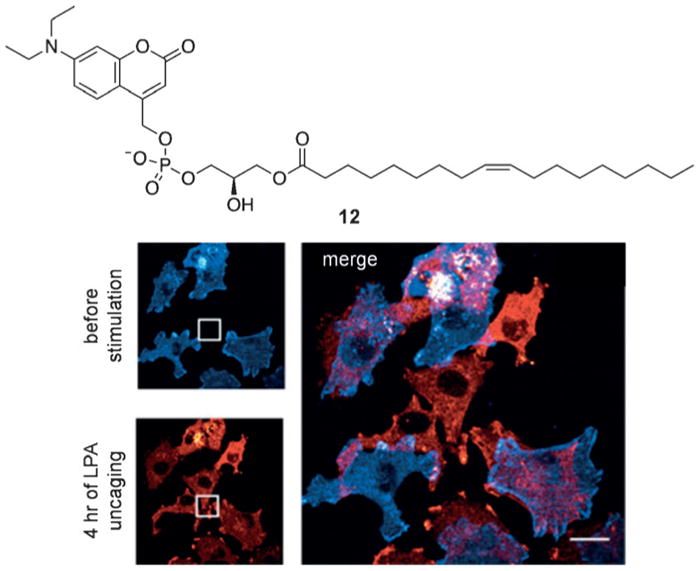
The caged lysophosphatidic acid 12 was used in A375M cells to demonstrate optotaxis through cell movement toward an LPA gradient established by repetitive, localized decaging. Adapted with permission from Ref. [38]. Copyright 2016 Elsevier Inc.
2.4. Multiwavelength Activation of Small Molecules
Although optochemical probes enable precise spatial and temporal activation and perturbation of cellular processes, this is typically restricted to the light-triggering of one biomolecule using a single wavelength (often 365 or 405 nm). In order to sequentially control multiple molecules, wavelength-selective caging groups have been utilized.[39]
Microorganisms commonly exist in mixed populations, making treatment/removal of a specific bacterial species challenging in the context of an infection.[40] The ability to remove one strain in the presence of another by using an external trigger would enable controlled selection and bacterial patterning. Feringa and co-workers utilized the complementary antibiotics fluoroquinolone 13 and benzylpenicillin 14, which specifically target two different bacterial species, E. coli and S. aureus, and rendered both light-activatable using wavelength-selective caging groups (Figure 10). The 7-dialkylaminocoumarin (in 15) and 7-alkoxycoumarin (in 16) have absorption maxima at 381 nm and 322 nm, respectively, and thus can be triggered independently (or at least sequentially).[41] In the absence of light, both bacterial strains grow on agar plates; however, irradiation at 312 nm removed all S. aureus leaving the E. coli untouched, while treatment with white light (>400 nm) showed the opposite effect. This demonstrates the ability to optically activate different antibiotics for the control of bacterial growth. The development of additional complementary antibiotics and additional light-orthogonal caging groups will enable the selective removal of specific bacteria within complex microbial communities.
Figure 10.
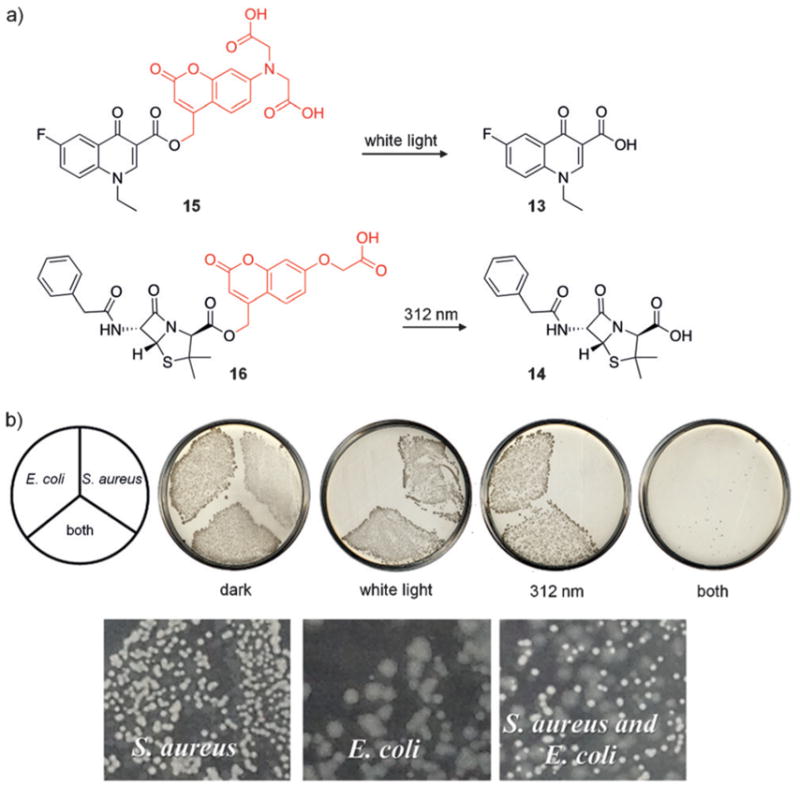
a) The structures of 13–16 are shown, with caging groups indicated in red. b) Plates containing both caged antibiotics in the dark allowed growth of both strains. Plates subjected to visible and 312 nm light show no bacterial growth, whereas individual strains were able to grow in the presence of only one light exposure. Adapted with permission from Ref. [41]. Copyright 2014 American Chemical Society.
Wavelength-selective photoactivation was also applied to the control of protein kinase activity through caged-cyclic AMP and caged protein kinase G.[42] Utilization of nitro-benzyl and coumarin caging groups in concert allowed sequential activation in human cells.
Multiwavelength probing of neuronal functions has been performed in mouse brain slices. Two neurotransmitters, γ-aminobutyric acid (GABA) and glutamate, have been extensively studied and rendered light-responsive through caging. The work of Ellis-Davies and others has combined multiwavelength, orthogonal caging groups in order to achieve the activation of both neurotransmitters in a single experiment,[43] thereby enabling complex analysis of synaptic integration upon simultaneous or sequential activation of glutamate and GABA in living systems.
While wavelength-selective activation and deactivation of small molecules, oligonucleotides (see Figure 23, Figure 29, and Figure 30), and proteins has provided unique tools, these methods offer only off-to-on or on-to-off capabilities. In order to transition reversible control of biological function to multiwavelength control, Feringa and co-workers developed photoswitching groups that provide selective control in the <300 or >500 nm range, thereby enabling pairing with classical azobenzenes that are switched with 300–500 nm light. However, these switches have not been applied in cells or animals to date (also see Figure 18c for orthogonal photoswitching).[44]
Figure 23.

a) Sequential activation of cMOs targeting the genes flh (purple) or spt (blue). Structures of the NB (red) and DEACM (orange) caging groups are shown. b) Representative images of myod1 expression patterns from control (wildtype), flh knockdown (aberrant expression), and spt knockdown (no expression) phenotypes after irradiation. c) Quantification of phenotypes is shown for embryos injected with the cMOs and irradiation conditions denoted in the graphs. Adapted with permission from Ref. [48]. Copyright 2014 Wiley-VCH.
Figure 29.

a) Structures of caging groups used for two-photon-triggered decaging of DNA. DEACM (yellow box) and ANBP (red box) were incorporated into DNA1 and DNA2, respectively. b) Caging groups prevent DNA duplex formation until irradiation and subsequent toehold-mediated displacement of a quencher-modified strand. c) Schematic of decaging strategies and their expected DNA duplex outcomes for the demonstration of spatial control. d) Images of the irradiated area of the hydrogel after decaging. The ATTO 565 channel is shown in blue and the ATTO Rho14 channel in pink. Selective activation of either DNA1 or DNA2 results in the images on the left and center, respectively. The picture on the right is an overlay of the other two images. Adapted with permission from Ref. [205]. Copyright 2016 Wiley-VCH.
Figure 30.
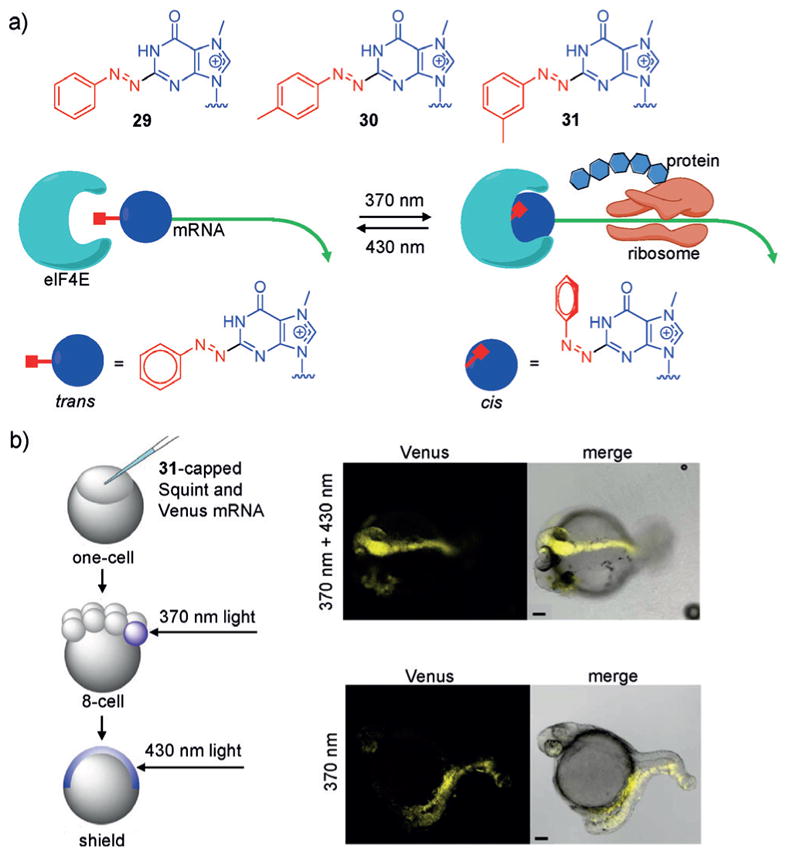
a) Structures of the 2-phenylazo caps 29, 30, and 31 are shown with the nucleobase in blue and the photoswitchable groups in red. In the trans form, the 2-phenylazo caps inhibit binding of elF4E and translation; however, upon photoisomerization (370 nm) to the cis form, elF4E can bind and initiate protein expression. b) mRNAs capped with 31 were injected into zebrafish embryos at the one-cell stage and irradiated at the 8-cell and shield stages with 370 nm or 430 nm light, respectively. After photoswitching, development of a second midline with head structures was observed. Adapted with permission from Ref. [213]. Copyright 2017 American Chemical Society.
Figure 18.
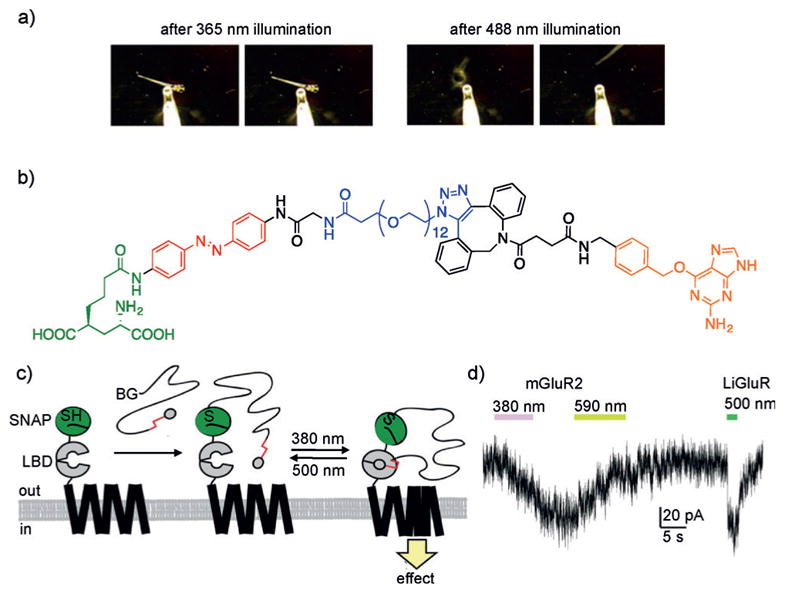
a) Reversible optical control of the zebrafish larvae fast escape response. b) Structure of a PORTL comprised of a glutamate ligand (green) connected to an azobenzene (red), a flexible PEG linker (blue), and a benzylguanine (orange). c) Scheme depicting the mechanism of PORTL-mediated reversible photocontrol of a SNAP-mGluR2 receptor using two different wavelengths of light. The ligand binding domain (LBD) binds to glutamate. d) Dual optical control of mGluR2 and LiGluR in HEK239T cells enables independent activation of receptors, as demonstrated by a whole-cell patch-clamp electrophysiology assay. Adapted with permission from Ref. [118] (Copyright 2015 American Chemical Society) and Ref. [117] (Copyright 2007 Elsevier Inc).
Zebrafish and frog oocytes are two frequently used model organisms in developmental biology. Jullien combined the photocontrol of 13-cis-retinoic acid (RA) with a caged inducer of the transcription factor En2 that can be activated with blue light.[45] Previous efforts had successfully demonstrated that the En2 inducer cyclofen, which results in a distinct reduction in eye size or eye loss in the zebrafish model, could be successfully caged to optically control eye development.[46] Additional work by Jullien demonstrated that 13-cis-RA could be photoisomerized to all-trans-RA using UV light, which allowed rescue of hindbrain formation in embryos whose trans-retinoic acid pathway had been blocked by a small-molecule inhibitor.[47] A transgenic zebrafish line in which the third and fifth rhombomeres were fluorescently labeled with GFP was used as a reporter for hindbrain rescue (presence of GFP at the fifth rhombomere indicated hindbrain rescue). An orthogonal system for photo-activating 13-cis-RA (365 nm) with a thiocoumarin caged cyclofen (488 nm) was developed, thereby enabling independent activation of eye loss or hindbrain rescue when applied in vivo. Additionally, multiwavelength activation has been applied for the control of antisense oligonucleotide function in zebrafish, as presented below (Figure 23).[48]
2.5. Optical Activation of Small-Molecule Drug Release
Antibody–drug conjugates (ADCs) have emerged as innovative therapeutics to enhance the specificity of existing drugs.[49] One of the limitations of traditional ADCs is their inability for controlled release of a small molecule upon localization to the desired target. Linkers that undergo proteolytic or reductive cleavage are commonly used; however, they are dependent on the endogenous cellular environment, provide no temporal control, and may lead to off-target effects due to premature cleavage. Schnermann and coworkers developed a new linker approach that utilizes near-IR light to cleave the drug from the antibody.[50] Panitumumab, a monoclonal antibody for the human epidermal growth factor receptor, was conjugated to the microtubule inhibitor combrestatin 8 through a previously reported cyanine photocage[51] that is activated at 690 nm (Figure 11a). The novel caging group capitalizes on the propensity of cyanine dyes to undergo photobleaching as a result of carbon–carbon double bond photooxidation (Figure 11b). Upon irradiation at 690 nm, photooxidation of the C=C bond results in bond cleavage to generate an enone. This cleavage event alters the π-conjugation of the dye, thus making the C–N bond more susceptible to hydrolysis. Hydrolysis generates a secondary amine, which cyclizes and releases the drug. When applied to a xenograft tumor model, the injected conjugate demonstrated full localization to the implanted tumors. Following irradiation at 690 nm, only the light-exposed areas showed a loss of fluorescence as a result of cyanine dye cleavage and concomitant release of 8 (Figure 11c).
Figure 11.
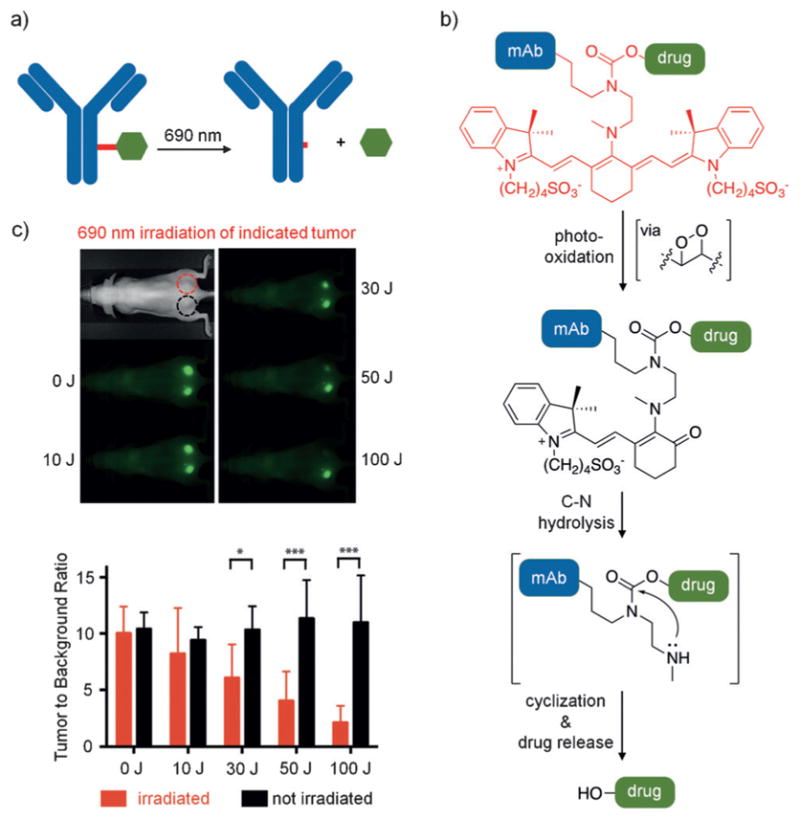
a) The monoclonal antibody (mAb) panitumumab is shown in blue with the photocleavable cyanine linker in red conjugated to 8 in green. b) The decaging mechanism of the cyanine dye is shown in an abbreviated form. c) Mice were implanted with A431 cells on both sides of the dorsal region. They were then injected with panitumumab-8, which localized to the developed tumors. The top tumor was irradiated with 690 nm light, while the bottom one was shielded from light. Only the tumor treated with light showed a decrease in fluorescence, which is indicative of the release of 8. The bar graph indicates that tumors maintained in the dark show minimal change in fluorescence (black bars), while the irradiated sample shows a light-dependent decrease in fluorescence (red bars). Adapted with permission from Ref. [50]. Copyright 2015 Wiley-VCH.
This innovative approach provides spatiotemporal drug release through illumination with significantly red-shifted light due to the absorbance spectrum of the cyanine dye, thereby enabling good tissue penetration and minimal toxicity in live mice. In addition, it enables tracking of the location of the ADC using the inherent fluorescent nature of the cyanine caging group and drug release can be visualized through a decrease in fluorescence following IR-light treatment. While this initial work provides a proof of concept in the release of combrestatin A-4, the biological potency of the drug delivery was not tested. Future work in the field may focus on measuring potency and cell viability in response to spatiotemporal drug release.
2.6. Caged Fluorophores for Super-Resolution Microscopy
Photoactivation localization microscopy (PALM) has become a common super-resolution microscopy technique, and can resolve structural features below the diffraction limit. In this approach, a small molecule or protein fluorophore is stochastically activated, imaged, and then bleached over thousands of micrographs.[52] Increased resolution is achieved through the higher photon yield obtained from synthetic fluorophores compared to that of fluorescent proteins. Caged fluorophores that are non-fluorescent prior to irradiation have been developed for PALM applications. This enables fluorophore activation in specified locations and elucidation of the exact location where the photons were emitted, which contrasts with traditional imaging in which an emission of an excess of photons hampers the generation of highly resolved images. In the last few years, the development and optimization of photocaged fluorophores has provided researchers with several options.[53] Johnsson and co-workers developed an O6-benzylguanine-modified rhodamine 110, which was caged with a nitrobenzyl group (17, Figure 12a).[54] Various SNAP-modified proteins were used to demonstrate localization to the membrane (SNAP-β-adrenergic receptor), the nucleus (SNAP-NLS), and within the cytoplasm (SNAP-MEK1) of fixed mammalian cells (Figure 12b). Following 488 nm irradiation to bleach any previously decaged molecules, and irradiation at 405 nm to decage the fluorophores, PALM imaging was performed to generate super-resolution images. However, one limitation of this approach is the necessity to fix cells prior to imaging unless a cell-surface target is used due to the impermeability of 17. Similarly, Moerner and co-workers developed the HaloTag-modified dicyanomethylenedihydrofuran 18, which bears an azide that is converted into an amine upon blue-light illumination (594 nm), thereby activating the fluorophore (Figure 12c).[55] In this particular example, a nearby computer monitor provided sufficient light to maintain a steady-state of photo-activated fluorophores, thus eliminating the need for laser or LED activation and minimizing photodamage. This photo-activatable fluorophore was used for labeling of α-tubulin in fixed mammalian cells and for high-resolution imaging of swarmer-pole-localized PopZ in live Caulobacter crecentus (Figure 12d). More recent advancements have been made to generate photoactivatable fluorophores with different emission wavelengths for imaging.[56]
Figure 12.
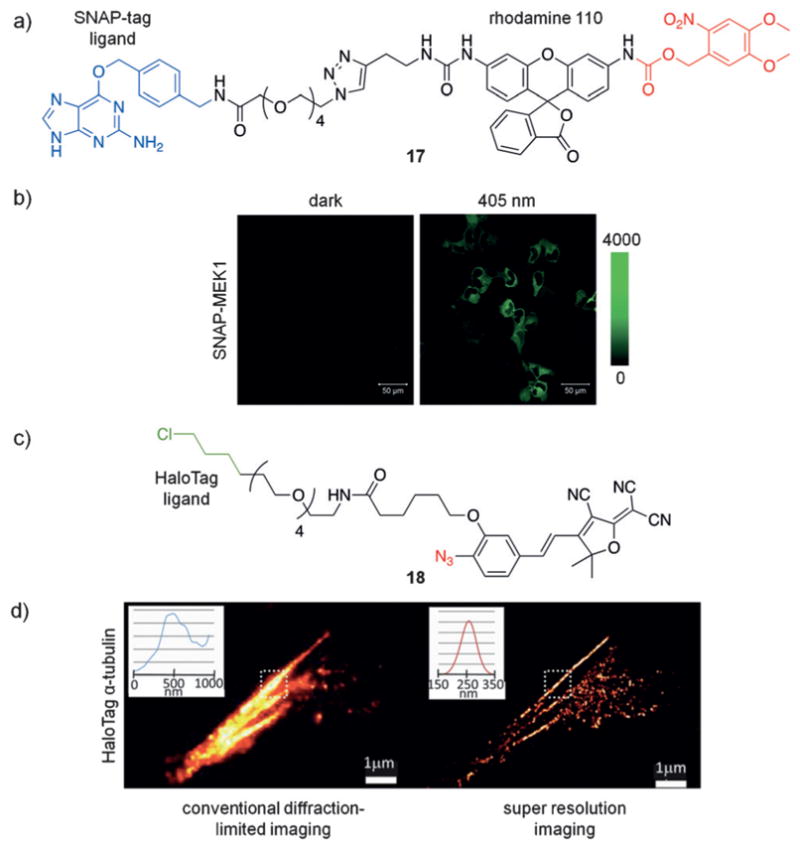
a) The structure of 17 is shown with the SNAP-tag ligand highlighted in blue and the caging group in red. b) U2OS cells expressing cytoplasmic SNAP-MEK1 were fixed and treated with 17. UV irradiation leads to photoactivation of 17. c) The structure of 18 is shown with the HaloTag ligand in green and the photoactivatable azide in red. d) Fixed BS-C-1 cells expressing HaloTag-α-tubulin, were treated with 18 and then subjected to diffraction-limited imaging (left) or super-resolution imaging (right). Fluorophore activation occurred under ambient light. Adapted with permission from Ref. [54] (Copyright 2011 American Chemical Society) and Ref. [55] (Copyright 2010 American Chemical Society).
The photoactivatable fluorophores described above have advanced the field of super-resolution microscopy and have aided in the observation of protein localization and organization. This is especially relevant in bacterial cells, which due to their small size often only show diffuse fluorescence in traditional diffraction-limited imaging. Additionally, the modularity of these photoactivatable fluorophores allows facile swapping of ligands, fluorophores, or caging groups, thus making this a highly adaptable tool for imaging. Future work in the field should focus on developing cell-permeable compounds to enable live-cell imaging.
2.7. Optical Activation of Metal Ion Release
Divalent metal ions play critical roles in biological processes, including as second messengers in signaling cascades (e.g., Ca2+) and as coordination sites in metalloproteins (e.g., Zn2+ or Cu2+).[57] Photocaging these ions has enabled temporal control in live cells. Ellis-Davies and co-workers applied a nitrobenzyl derivative, as well as other chromophores, for the regulation of calcium function.[58] Recently, the calcium chelator 19 was developed and was activated with 810 nm (two-photon) or 405 nm light (Figure 13a).[59] When cardiac myocytes bearing a fluorescent Ca2+ reporter were treated with 19, no change in calcium levels was observed. However, upon illumination, a calcium wave was detected in both directions from the light source (Figure 13b,c). This blue-light-activated caging group may enable orthogonal calcium release together with other light-activated metal ion chelators or signaling elements. Similar to calcium, optical release of Pt2+,[60] Zn2+,[61] and other ions[57,62] has recently been reported. Advancements in caging group technology in conjunction with two-photon excitation have enabled red-shifted ion release, which is important for adapting the approach for future applications in living systems. While the existing caging approaches have enabled the investigation of Ca2+, Zn2+, and Cu2+ release, there are many other biologically relevant and disease-linked metal ions (e.g., Cu+, Fe2+/Fe3+, or Mg2+) that would benefit from optically controlled release.
Figure 13.
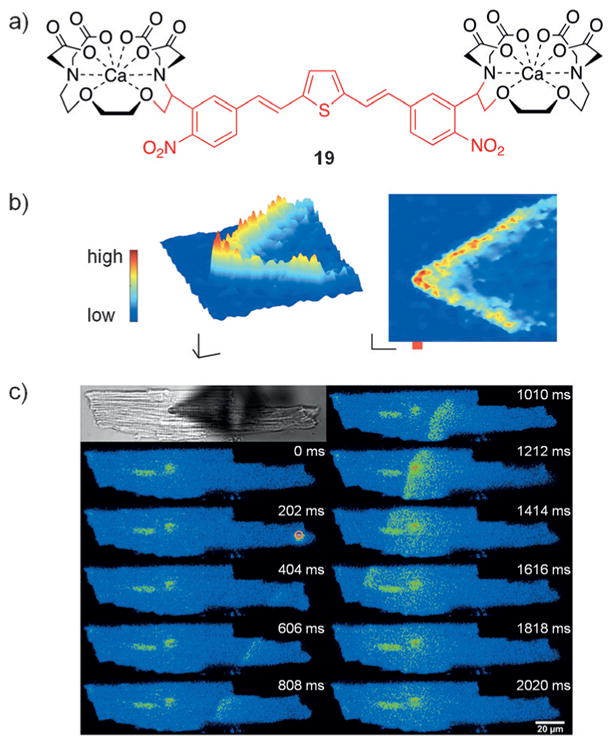
a) The structure of the light-triggered chelator 19 is shown. b) Myocytes containing a fluorescent reporter for calcium were treated with 19 and then irradiated with 810 nm light at the point indicated in red, which propagated a wave of calcium in both directions from the spot of illumination. c) A similar experiment was conducted as in (b); however, a 405 nm light source was used. Adapted with permission from Ref. [59]. Copyright 2016 American Chemical Society.
3. Optical Control of Proteins and Peptides
Proteins and peptides play significant roles in the function of many cellular processes crucial to maintaining homeostasis at the molecular, cellular, and organism levels. Not surprisingly, the development of light-controlled tools to interrogate protein and peptide function has proven highly valuable.[63] Optical control of protein and peptide function has been achieved recently through the development of unnatural amino acid mutagenesis, enabling the site-specific incorporation of photoactivatable caging groups and photoswitchable azobenzene groups. Alternative methodologies provide complementary photodeactivation of proteins. These advances have not only proven useful in probing numerous biological processes, but have also been applied to a variety of drug delivery systems. Recent applications of optical control of proteins and peptides are discussed below and exemplify the significant potential that light-controllable systems afford.
3.1. Optical Activation of Protein Function
Optical control of peptide and protein function using chemical means has been achieved through the introduction of photolabile caging groups. Traditionally, this is accomplished through chemical modification of fully (or partially) synthetic proteins or through post-translational modification of isolated protein samples.[64] However, labor-intensive synthesis approaches and the need for peptide/protein delivery have limited their application in cells and organisms. These limitations with regard to caging group installation were addressed through the development of photocaged, genetically encoded unnatural amino acids (UAAs) that can be incorporated into proteins and peptides in living cells. This was accomplished through the engineering of orthogonal tRNA synthetases and their cognate tRNAs to create biosynthetic machinery that enables site-specific installation of UAAs in response to an amber stop codon (TAG) within a gene of interest. Using this method, nitrobenzyl and coumarin caging groups have been installed on Tyr, Lys, Ser, and Cys residues.[63] Strategic insertion of the caged amino acid at a critical residue on the protein of interest can perturb function by masking an important chemical functionality or by generating steric hindrance. Following photolysis of the caging group, the amino acids are returned to their native state, thereby restoring wild-type protein structure and function.
Numerous applications of optical control of protein function using UAA mutagenesis have been demonstrated, including control of protein localization,[65] DNA transcription,[66] DNA recombination,[67] intein splicing,[68] protease activity,[69] epigenetic events,[70] and protein phosphorylation.[71]
A precise understanding of protein phosphorylation events in kinase signal transduction pathways requires 1) a spatiotemporal understanding of kinase activity within the connectivity of a given network and 2) a detailed model of how individual components within a pathway interact with other nodes within the same network or other networks. Optochemical tools provide the necessary temporal and spatial control to meet these requirements since genetic perturbations (e.g., gene knockdown or overexpression) are too slow to prevent network adaptation, and since pharmacological perturbations (e.g., small-molecule inhibitors) often lack the required level of specificity.[72] By employing the genetically encoded caged lysine 20 (Figure 14a), a light-activated kinase MEK1 was developed in which a universally conserved lysine residue within the ATP binding pocket is photocaged (Figure 14b).[73] MEK1 is a critical component of the Raf/MEK/ERK signaling pathway and is involved in many essential cell functions including cell growth, adhesion, survival, and differentiation.[74] MEK1 phosphorylates ERK, and employing an EGFP-ERK2 reporter demonstrated that expression of the caged inactive MEK1 results in cytosolic localization of EGFP-ERK2. Following illumination and decaging of MEK1, rapid phosphorylation and sustained translocation of EGFP-ERK2 into the nucleus was observed. In contrast, cell-surface stimulation of the Raf/MEK/ERK pathway with epidermal growth factor leads to network adaptation and nuclear export of EGFP-ERK2, thus revealing that adaptation of the network to a persistent stimulus may occur upstream of MEK1 and not downstream as previously hypothesized (Figure 14c).[75] While MAPK phosphatase activity alone is not sufficient to prevent nuclear localization of ERK2, these results suggest that MAPK phosphatase (MKP) activity is capable of maintaining an apparent steady-state of nuclear ERK2 (Figure 14d). Overall, site-specific installation of 20 at a conserved lysine residue in an active site has the potential to be broadly applicable to a wide range of kinases, and ATP-dependent enzymes in general.[76] The limitation of requiring UV light for activation can be overcome by using a coumarin-caged lysine.[77]
Figure 14.
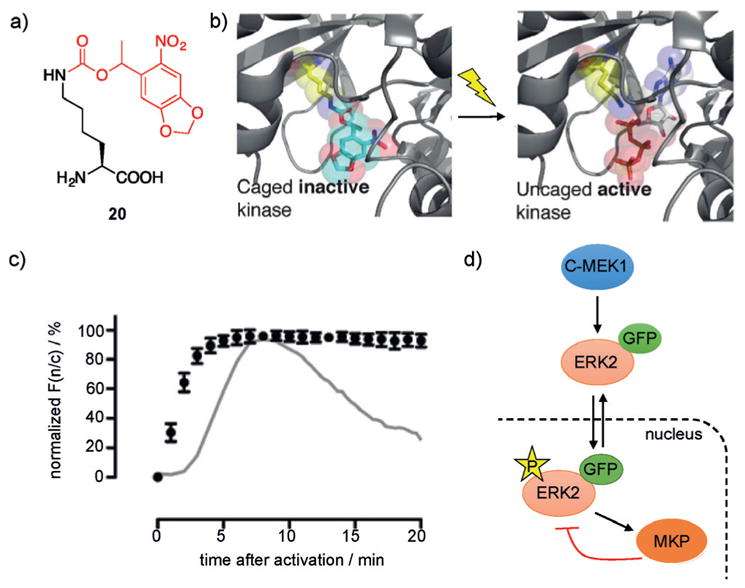
a) Structure of photocaged lysine 20 with the caging group shown in red. b) Caging of a conserved lysine in the MEK1 active site blocks ATP binding and renders it inactive. c) Nuclear translocation of EGFP-ERK2 following phosphorylation after photoactivation of caged MEK1 depicted as normalized F(n/c) (ratio of nuclear to cytoplasmic EGFP fluorescence signal) as a function of time after photoactivation. The gray line represents normalized F(n/c) observed when cells are induced with EGF. d) Schematic representation of the sub-network and phosphorylation/dephosphorylation of ERK downstream of MEK. Adapted with permission from Ref. [73]. Copyright 2011 American Chemical Society.
Gene-editing tools, such as zinc-finger nucleases, TALENs, and CRISPR/Cas nucleases have been extensively used in the modification of cell lines and model organisms for research purposes[78] and represent highly promising technologies for the treatment of a wide range of diseases.[79] In order to enable spatiotemporal studies of gene function and to potentially reduce off-target effects, different approaches to optically control gene-editing activity have been reported. A photocaged zinc-finger nuclease was developed to enable conditional generation of gene modifications with optical control by caging a tyrosine residue critical for protein–DNA interaction through genetic code expansion.[80] Following irradiation and subsequent decaging, gene-editing activity was rescued. Similar light-triggered approaches have been applied to the spatiotemporal control of other genome-modifying enzymes, such as Cre recombinase[67a,b] and CRISPR/Cas9.[81] The development of CRISPR/Cas9 as a biological tool has led to significant advances in genome editing, since the sequence-specific targeting of the nuclease through a guide RNA (gRNA) greatly simplifies the experimental effort and readily enables high-throughput and multiplexing studies.[82] The first optically activated Cas9 protein was generated through UAA mutagenesis, thereby providing photocontrol over gene editing (Figure 15a).[81] Based on structural data, several lysine residues in close proximity to the Cas9–gRNA binding interface and the nuclease domains were screened through UAA scanning, and ultimately K866 (Figure 15b,c) was found to lead to an inactive enzyme when replaced with the caged lysine 20. UV exposure fully restored Cas9 activity in cells, as demonstrated by a fluorescent reporter assay. Furthermore, light-triggered silencing of the transmembrane transferrin receptor CD71 was used to showcase the applicability of this optical gene editing system to endogenous targets. Additional approaches to optically control Cas9 function have employed light-inducible split-protein systems or light-induced protein homodimerizers.[83] The same approach has been applied to catalytically inactive dCas9.[84] Moreover, optical control of gRNA function has been achieved, as discussed below (Figure 27).[85]
Figure 15.
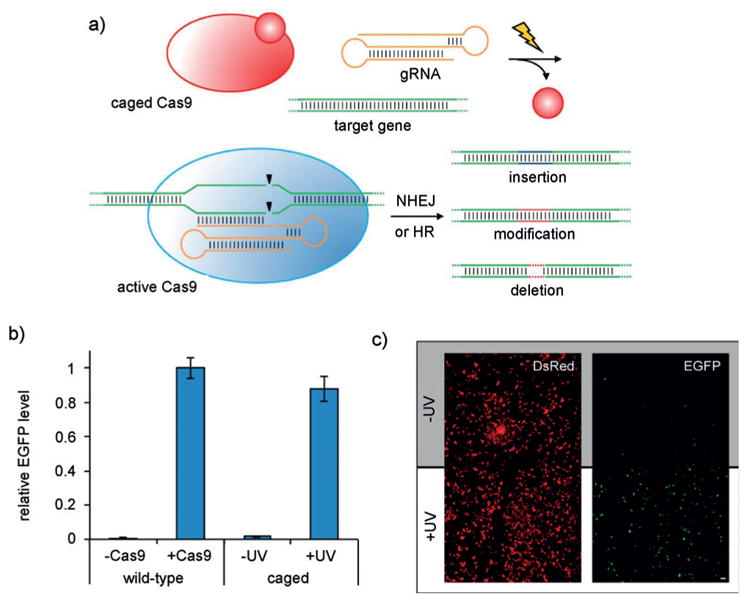
a) Photoactivation of caged Cas9 (K866→20 mutant) affords optochemical control of gene editing. Site-specific incorporation of a nitrobenzyl caging group at the critical lysine residue K866 renders Cas9 inactive until the caging group is removed by illumination, which generates wild-type Cas9 and rescues gene-editing functions, such as non-homologous end-joining (NHEJ) or homology-directed repair (HR). b) Activation of EGFP expression in a dual-reporter assay by Cas9 following irradiation, showed gene-editing levels similar to wild-type Cas9. c) Spatial control of Cas9 gene editing through patterned illumination through a mask. Adapted with permission from Ref. [81]. Copyright 2015 American Chemical Society.
Figure 27.
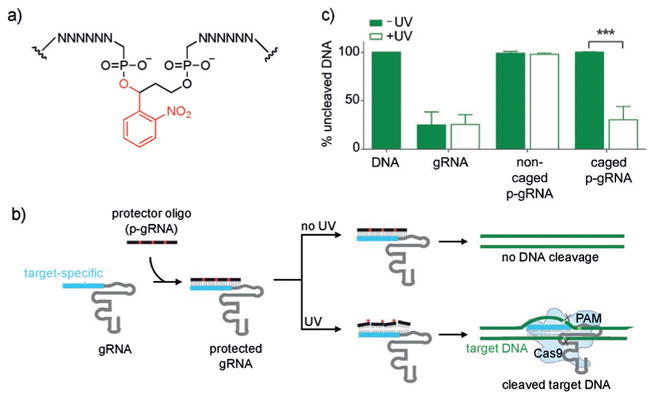
a) Structure of the photocaged backbone for optical control of gRNA function and gene editing. b) Caged complementary ssDNA protectors hybridize to the gRNA to prevent binding to the DNA target. Following UV irradiation, the photolabile groups are removed from the protector, which allows subsequent Cas9 cleavage. c) Percentage of non-cleaved DNA targeted in vitro by a corresponding gRNA. Irradiation with UV light results in simultaneous Cas9-mediated DNA cleavage. Adapted with permission from Ref. [85]. Copyright 2016 Wiley-VCH.
In addition to genetically encoded light activation of proteins, synthetic photocaged peptides have been generated and applied to the optical control of biological processes. Lawrence and co-workers adapted a highly potent and selective bivalent peptide inhibitor of Src tyrosine kinase into a light-triggered molecular probe by strategically inserting a photocleavable group within the linker of the two inhibitor domains.[86] In the absence of light, the peptide potently inhibited kinase activity. However, upon light exposure and cleavage of the bivalent inhibitor, 90% of Src kinase activity was restored. While this method avoids the need for genetic manipulation, it has been limited to in vitro and cell lysate applications thus far. Other applications of peptides containing light-cleavable groups include optical control of cell signaling,[87] protease activity,[88] and cell motility.[89] Simultaneous applications of two different caging groups (see Section 5.1) has enabled multiwavelength control of protein or peptide function.[90]
In summary, the development of photocaged amino acids has enabled precise optical control of a wide range of protein and peptide targets. These approaches offer several key advantages over other optogenetic approaches: 1) irradiation leads to the generation of the wild-type protein structure; 2) the small size of the caging groups minimizes non-specific perturbation of the protein structure, while providing precise blocking of essential amino acid functions; and 3) sites for caging-group installation can be predicted using structural and mechanistic data. However, this technology still faces difficult challenges for in vivo engineering, and the irreversible nature of decaging events may limit dynamic control of biological processes. The continued development of novel photocaged amino acids with distinct photochemical properties promises to expand the capabilities and scope of this technology to increasingly more complex models, thereby enabling a more detailed understanding of intricate cellular processes.
3.2. Optical Deactivation of Protein Function
While examples of photoactivation of proteins and peptides through optical removal of caging groups are prevalent, photodeactivation approaches are much less common but constitute valuable tools for generating knockout phenotypes with spatiotemporal control. One method that has been successfully applied is chromophore-assisted light inactivation (CALI).[91] CALI utilizes a chromophore in close proximity to the target protein that, when irradiated with light, produces reactive oxygen species (ROS) that react with and deactivate the target protein. Early methods relied on conjugation of photosensitizing dyes to antibodies that bind to specific target proteins;[92] however, they were limited by the need for microinjection into cells, the large size of the antibodies, and the risk of perturbing the native function of the target protein prior to irradiation.[93] To overcome these limitations, small-molecule ligands have been explored and applied to proteins in cells.[94] Recently, the Kodadek group developed ruthenium-conjugated peptoids, which are chemically stabilized peptide analogues capable of generating singlet-oxygen species to target and deactivate cell-surface and cytoplasmic proteins (Figure 16a).[95] Using a peptoid targeting the VEGF receptor 2 (VEGFR2), a growth factor receptor important in vasculogenesis and angiogenesis, a Ru(tris-bipyridyl)2+ ROS generator was appended to both the C- and N-termini. Following illumination, highly efficient and selective VEGFR2 inhibition was observed (Figure 16b). Similar constructs were designed to successfully deactivate the ATPase Rpt4 and the serine hydrolase RBBP9.[96] Thus, peptoid-ruthenium CALI conjugates represent a selective and potent method to conditionally deactivate native proteins in cells. One non-trivial limitation of this method is the necessity for a targeting ligand that has reasonable binding affinity for the protein of interest without significantly inhibiting protein function prior to illumination.
Figure 16.
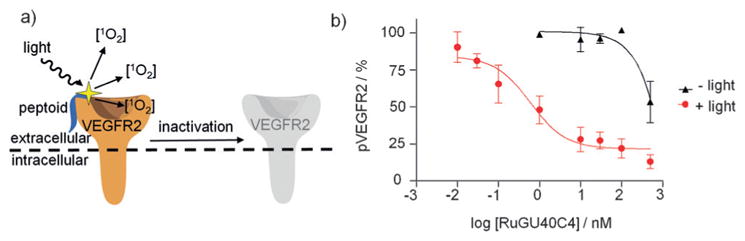
a) Scheme depicting inactivation of VEGFR2 with a ruthenium-conjugated, bound peptoid. Binding of the peptoid to VEGFR2 followed by illumination results in localized generation of singlet-oxygen (1O2) species and inactivation of VEGFR2. b) Dose-dependent inhibition of VEGF-induced autophosphorylation of VEGFR2 following irradiation for 10 min. Adapted with permission from Ref. [95]. Copyright 2010 Nature Publishing Group.
CALI systems based on genetically encoded target-specific labeling with photosensitizing dyes through SNAP-tag and HaloTag systems have also been developed.[97,98] Other genetically encoded CALI agents have been based on ROS-generating proteins. While fluorescent proteins such as GFP have been widely utilized for fluorescence imaging, they are also capable of generating ROS, albeit very inefficiently. Optimized forms of GFP have been developed to boost ROS generation, such as KillerRed.[99] Due to the size of KillerRed (27 kDa) and tendency to dimerize, limitations on the folding, function, and cellular localization of KillerRed fusion proteins have represented a challenge and are being addressed through second-generation versions.[100] To continue to improve upon current CALI techniques and to reduce off-target damage through ROS generation, new technologies should seek to enable improved proximity of the chromophore to the target protein and minimize perturbation of native protein function prior to illumination.[101]
Taken together, the development of CALI has enabled photodeactivation in cells and provides an attractive complementary system to traditional photoactivation techniques. Continued improvement of CALI systems will look to reducing off-target ROS generation and improving the general applicability of the approach to cell biological studies.
3.3. Reversible Optical Switching of Protein Function
The development of genetically encoded photo-caging techniques has provided an exquisite level of target specificity while also granting precise spatio-temporal control; however, photoactivation (or deactivation) is largely an irreversible event. In contrast, reversible photoswitching of protein function may offer the following advantages: 1) close mimicking of nature’s often reversible control of protein activity, and 2) enhanced spatial resolution due to protein activity only in illuminated areas since diffusion outside of the light beam can lead to reversible off switching.[102] Purely optogenetic approaches to reversible control have been developed using naturally light-sensitive fusion proteins. In response to light, these proteins (e.g., LOV domains, channelrhodopsins, or Cry/Phy protein dimerizers) undergo conformational changes or changes in aggregation state and have been engineered to optically control kinase activity,[103] protein localization,[104] transcription,[105] DNA recombination,[106] and motor function.[107]
To circumvent the use of bulky protein fusion constructs, several chemistry-based approaches have been developed that enable reversible and robust photoswitching of proteins and peptides. An early report utilized solid-phase peptide synthesis to incorporate a photoisomerizable azobenzene motif at specific residues within RNAse S, however only modest changes in activity were observed.[108] Toward incorporating azobenzene motifs into larger proteins, Maruta synthesized a cysteine-reactive azobenzene maleimide that was bioconjugated to different kinesin motor domain cysteine mutants.[109] Upon trans-to-cis photoisomerization, kinesin exhibited a 2-fold increase in ATPase activity. Further taking advantage of the selective reactivity of surface-exposed cysteine residues, several groups have developed bis-reactive azobenzene reagents that enable the installation of a photo-isomerizable bridging moiety between two proximal cysteine residues.[110] A noteworthy advantage of bridging two residues on a peptide/protein is the enhanced induction of large-scale conformational changes following photoisomerization. While general bifunctional thiol-reactive azobenzene derivatives have proven successful in the photocontrol of protein conformation, due to their high reactivity toward thiols and diminished selectivity, they have limited utility in cellular experiments.
In efforts to site-specifically introduce a reversibly photo-switchable azobenzene motif into proteins in live cells, Schultz and co-workers genetically encoded the azophenylalanine 21 in E. coli using an engineered M. jannaschii tyrosyl-tRNA synthetase/tRNA pair, and applied it to the reversible control of protein–DNA interactions (Figure 17a).[111] However, the lack of a reactive functional group in 21 prohibits the formation of a photoswitchable azobenzene bridge, thus limiting photoisomerization to impose localized structural changes rather than large-scale conformational alterations. To address this limitation, Wang and co-workers genetically incorporated the para-methylene chloride-modified azophenylalanine 22, which is capable of reacting with a nearby cysteine residue to form a photoswitchable bridge on the modified protein.[112] They applied it to the calcium-binding messenger protein calmodulin. Incorporation of 22 in close proximity to an engineered cysteine residue resulted in the formation of a covalent azobenzene bridge, and photoisomerization through irradiation at 365 nm induced a significant shift in the protein conformation. To improve upon the design while maintaining the covalent bridging capability, Wang and co-workers introduced five fluorine atoms onto the benzene ring to generate 23, affording visible (blue/green)-light photoswitching (Figure 17b).[113] Conformational switching allowed photocontrol of calmodulin binding to neuronal nitric oxide synthase (NOS1) in vitro. This represents a very interesting approach that overcomes the non-specific labeling limitations of previous azobenzene bridges, while simultaneously translating small structural changes in an azobenzene chromophore into large structural changes in protein conformation.
Figure 17.
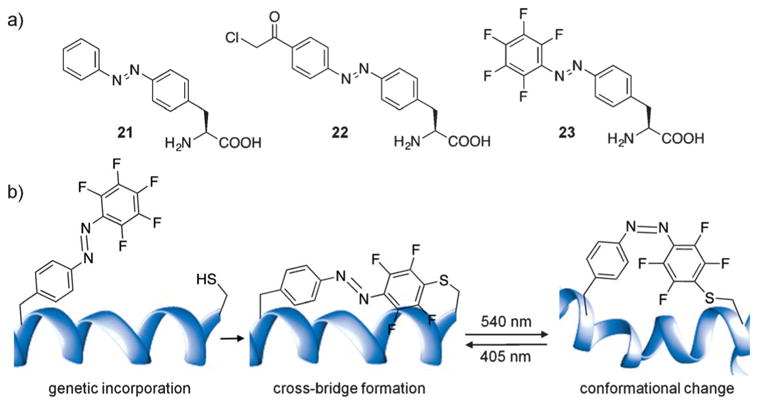
a) Structures of genetically incorporated photoswitchable azobenzene amino acids. b) Genetic encoding of 23 enables bridge formation through proximity-induced reaction with a nearby cysteine residue. Illumination with green or blue light allows photoswitching between the cis/trans isomers and leads to significant conformational changes in the protein structure.
Rather than relying on photoinduced protein conformation changes, other methods have utilized photoswitchable tethered ligands (PTLs) to perturb protein function.[114] PTLs consist of three primary components: 1) a reactive moiety for bioconjugation to target proteins; 2) a molecular photoswitch (e.g., an azobenzene); and 3) a ligand capable of modulating the activity of the target protein. While efforts have been made to directly incorporate molecular photoswitches into ligand structures, this requires the ligand to be amenable to appropriate chemical modifications while also retaining high affinity and specificity for the protein of interest (see Section 2.3 for a more detailed discussion of this concept). In contrast, PTLs only require small-molecule ligands of low affinity because covalent tethering places the ligand in close proximity to the specific protein, thereby increasing its localized concentration.[115] Isomerization of the tethered ligand is typically efficient and does not rely on conformational changes to the protein structure to perturb protein function. Furthermore, with traditional azobenzene bridging techniques (discussed above), one must take into careful consideration the effect the peptide–protein structure imposes on the isomerization and photoequilibrium properties of the azobenzene motif. PTLs do not face this challenge and thus are more readily adaptable to a variety of applications. Several cysteine-reactive PTLs have been successfully applied to various receptors and ion channels.[116] Trauner and co-workers successfully applied a cysteine-reactive PTL for control of the ionotropic glutamate receptor LiGluR in a live animal, thus demonstrating photoswitchable control over neuronal activity and discreet dissection of zebrafish neural circuitry.[117] Larvae expressing the PTL-functionalized LiGluR were exposed to 365 nm light and exhibited a loss of their fast escape response following mechanical stimulation. However, following irradiation with 488 nm light, the zebrafish larvae regained the touch response, thus demonstrating optical neuronal control in vivo (Figure 18a). Recently, the Trauner group developed a photoswitchable orthogonal remotely tethered ligand (PORTL), based on SNAP-tag labeling, thereby significantly improving selectivity and enabling multiplexed optical control.[118] As a proof of principle, PORTL was applied to a G-protein-coupled metabotropic glutamate receptor (mGluR2), where glutamate acts as the ligand. Synthetic constructs consisted of a glutamate ligand, an azobenzene moiety to enable photoswitching, and a PEG linker containing the terminal benzylguanine moiety for conjugation to the SNAP-tag fusion protein (Figure 18b). Expression of a SNAP-mGluR2 fusion construct produced efficient labeling and robust photoswitchable activation of SNAP-mGluR2 receptors in HEK293T cells (Figure 18c). Due to the bioorthogonal nature of the SNAP-tag technology,[119] Trauner and coworkers were also able to demonstrate simultaneous optical control of two independent glutamate receptors: mGluR2 and LiGluR. By employing two orthogonal azobenzene moieties harboring distinctive spectral properties, they achieved independent and sequential activation of the receptors, thus showcasing the ability to multiplex PORTL (Figure 18d). Monitoring receptor activity using a whole-cell patch-clamp electrophysiology assay, treatment of cells with 380 nm light resulted in a slow response, as expected by activation of the slow mGluR2 receptor. Treatment with 590 nm reversed mGluR2 activation, while subsequent exposure to 500 nm light activated a fast LiGluR-mediated response. Multiplexing multiple receptor populations provides an opportunity to probe crosstalk between proteins, enabling complex investigations at the molecular, circuit, or cellular levels.
To capitalize on specific bioconjugation reactions in live cells, while removing the requirement of a fusion protein for PTL bioconjugations, Chin and co-workers developed a genetically encoded photoswitchable bioorthogonal ligand tethering (photo-BOLT) technique that takes advantage of the rapid and specific conjugation of genetically encoded strained alkenes and alkynes with tetrazines through an inverse electron demand Diels–Alder reaction.[120] Covalent modification effectively increases the local concentration of the inhibitor for the mutant protein, while inclusion of an azobenzene moiety provides photoswitchable control over the ability of the inhibitor to bind (Figure 19a). As a proof of principle, this approach was successfully applied to the kinase MEK1 in HEK293T cells expressing a mutant MEK1 protein containing the strained alkyne UAA 24 (Figure 19d), thereby complementing previous photocaging efforts (Figure 14).[73] Upon treatment with 25, a non-selective MEK1/MEK2 inhibitor modified with an azobenzenetetrazine moiety (Figure 19d), significant inhibition of mutant MEK1 activity at concentrations as low as 100 nM was observed. In contrast, treatment of cells with the tetrazine-modified inhibitor alone at concentrations of up to 10 μM (the solubility limit) showed no inhibition of wild-type kinase, thus demonstrating the ability of BOLT to confer protein selectivity and high efficacy to otherwise non-selective, low-affinity inhibitors. Following illumination at 360 nm and photoisomerization to the cis isomer, the activity of mutant MEK1 was restored, since the inhibitor could not access the binding pocket anymore. Subsequent switching to the trans isomer through illumination at 440 nm led to inhibition of the mutant MEK1 (Figure 19b,c), thus demonstrating the reversible photo-switching of kinase activity. Alternatively, thermal relaxation from the cis isomer to the trans isomer also results in inhibition of mutant MEK1 activity. Capitalizing on the enhanced specificity and potency associated with tethering, this approach is widely applicable to all kinases, even those for which no selective inhibitors exist. This crucial advantage may also extend the potential of photo-BOLT to other protein families in which selective inhibition with small molecules is challenging.
Figure 19.
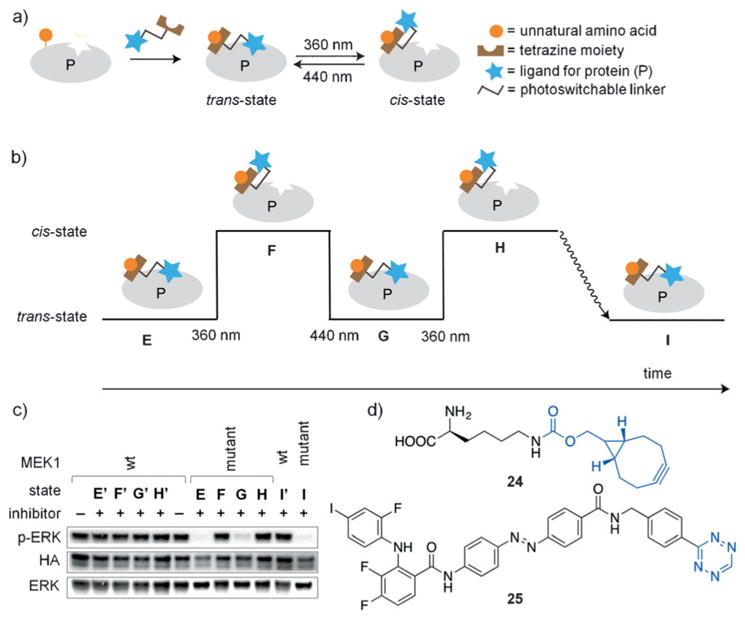
a) General scheme of photo-BOLT for reversible toggling of protein activity using light. Following incubation of target proteins with tetrazine-modified ligand, protein activity is inhibited. Due to the presence of a photoswitchable linker, illumination at 360 nm induces photoisomerization (cis state) and rescues protein activity. Additional illumination at 440 nm or thermal relaxation results in reversion back to the original state (trans state). b) Graphical representation of the sequential illuminations conducted in (c). c) HEK293T cells expressing wild-type MEK1 (E′–I′) or mutant MEK1 (E–I) were incubated with the tetrazine-modified inhibitor to achieve state E (inactive MEK1). Illumination at 360 nm results in state F (active MEK1). Subsequent illumination at 440 nm achieves state G (inactive MEK1). Further illumination (360 nm) achieves state H (active MEK1). Finally, incubation for 3 h without illumination induces thermal relaxation to state I (inactive MEK1). d) Structures of the strained alkyne-modified UAA 24 and the azobenzene-tetrazine-modified MEK1 inhibitor 25. Adapted with permission from Ref. [120]. Copyright 2015 Nature Publishing Group.
Photoswitchable technologies have significantly improved the options for optical control of systems, providing improved regulation of protein and peptide activity. Recent advances in azobenzene photoswitches have been focused on the chemical properties of the chromophore in the hope of tuning thermal relaxation rates, enhancing isomer conversion stoichiometry, and allowing red-shifted isomerization for improved in vivo efficiency.[121] Reduction of azobenzenes by thiols (e.g., glutathione) remains a concern for the application of these photoswitches in cells and organisms and should be taken into consideration when utilizing azobenzene derivatives.[122] Continued development of systems that are easier to design and less invasive holds promise for generating new approaches for improving the implementation of reversible photochemical control in living organisms.
3.4. Photocaging Groups for Peptide and Protein Delivery
Precise analysis of suitable biomarkers is one prerequisite to the development of personalized medicine approaches. Methods to quantify oncogenic kinase activity in human primary cells has been challenging due to the small quantity of cells available in clinical specimens and the low throughput of current techniques.[123] To achieve a feasible clinical assay for kinase activity, the assay must be 1) specific for the kinase of interest; 2) resistant to phosphorylation until the desired assay start time; and 3) able to deliver statistically significant results from single (or few) cells. To meet these requirements, a cell-permeable light-activated peptide sensor system (Figure 20a) was developed by Allbritton and Lawrence to assess phosphorylation activity of the protein kinase Akt, a key player in tumor formation and progression.[124] The threonine phosphorylation site within a short fluorophore-labeled native peptide substrate was caged with a nitrobenzyl group, thereby blocking phosphorylation by Akt and silencing the reporter as a kinase substrate (26) until UV exposure decages it to 27, followed by phosphorylation to 28. Phosphorylation was analyzed by capillary electrophoresis (CE; Figure 20b,c). The ability to optically control the assay start time enables precise detection of reaction kinetics and prevents the sensor from being acted upon by kinases during cellular uptake and distribution. Most peptide reporters have relied on micro-injection or cell-penetrating peptides (CPPs) to be delivered into cells, which limits efficiency and throughput of these approaches.[125] Interestingly, incorporation of a single nitro-benzyl caging group into the peptide substrate provided improved cell permeability of the peptide, thereby overcoming a major limitation of previous kinase sensors, which suggests that incorporation of the caging group may sufficiently increase the hydrophobicity of similar peptides. In a pilot study, the sensor was applied to an automated single-cell CE system. Treatment of pancreatic cancer cells with the sensor enabled the measurement of Akt kinase activity with substantially increased throughput (7.2 cellsh−1) compared to previous methods (0.5 cellsh−1).[126] This sensor design has the potential to be developed for additional kinases and may enable rapid evaluation of aberrant kinase activities in small clinical samples, ideally in a multiplexed fashion in order to use oncogenic patterns of hyper-phosphorylation as cancer biomarkers.[127]
Figure 20.
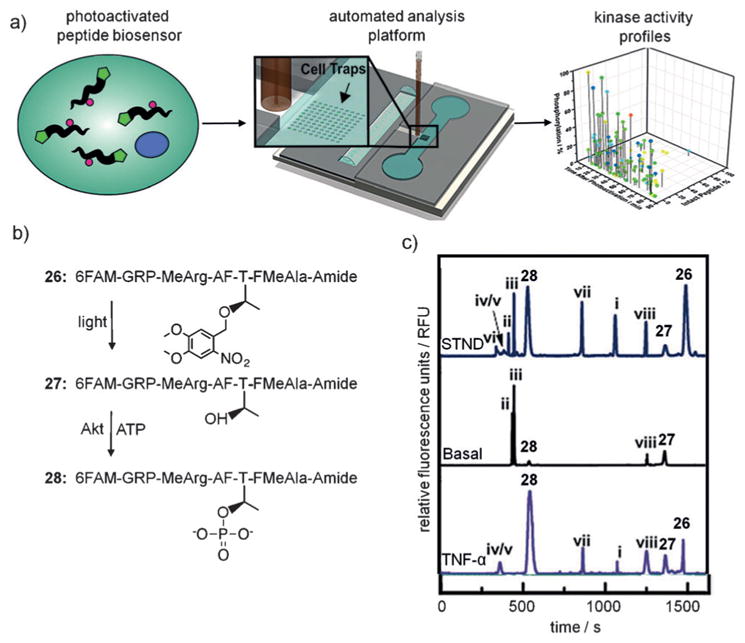
a) Schematic representation of the kinase activity profiling technique using a photoactivated peptide biosensor and single-cell capillary electrophoresis. b) The native substrate is modified with a nitrobenzyl caging group to generate the photocaged substrate 26, which upon illumination is decaged to 27 and subsequently phosphorylated to 28 by Akt. c) Single-cell Akt activity measured by CE, with the peaks 26–28 corresponding to structures 26–28. STND =standard solution of peptides, Basal =serum starved PANC-1 cells, TNF-α=cells stimulated with TNF-α. Proteolytic products of 26 are labeled as i–viii. Adapted with permission from Ref. [124]. Copyright 2016 Wiley-VCH.
The field of drug delivery has also harnessed light to improve the spatiotemporal control of various systems. As discussed earlier, photocleavable antibody–drug conjugates have been developed that enable cell-targeted and light-controlled delivery of therapeutics. Conjugation of combrestatin to an EGFR monoclonal antibody via a photolabile linker was shown to facilitate targeted release at tumor sites (Figure 11). The field of RNA/DNA therapeutics has extensively incorporated light-controlled mechanisms to improve spatiotemporal control and delivery. In contrast to the caged DNA and RNA nucleobases discussed below, one approach pioneered by the Ohtsuki lab utilizes a protein carrier to deliver RNA cargo (i.e., siRNA or shRNA) into cells through the conjugation of photosensitizer dyes to cell-permeable RNA-binding proteins (RBPs).[128] In the absence of light, a significant portion of the RNA/RBP complex remains trapped in endosomes and is eventually degraded. However, illumination with light affords improved endosomal escape and subsequent release of the RNA cargo into the cytosol. Another strategy for the delivery of DNA/RNA involves conjugation of CPPs derived from the HIV TAT protein to caged nucleobases.[129] This technology was applied to DNA antisense reagents and following efficient cellular uptake mediated by the CPP, light exposure resulted in removal of the caging group/CPP conjugate and activation of the antisense agent. Importantly, by installing the CPP onto the caging groups, multiple CPPs were conjugated and light-triggered decaging yielded the antisense product in its native state with no potentially deleterious modifications that could perturb activity. This technology proved to be highly modular, and conjugation of the caged nucleobases to folic acid enabled targeted cell delivery through cell-surface folate receptors. In an alternative approach, Mei and co-workers made use of conjugating caged CPPs to liposomes, where the caging group inhibits interaction of the CPPs with the cell membrane, thereby preventing liposomal delivery. Following decaging, the CPP is permitted to interact with the cell membrane, which results in efficient cellular uptake and delivery of cargo. This approach was successfully applied for the delivery of siRNA,[130] antimicrobial peptides,[131] and small-molecule therapeutics.[132] The Friedman group successfully developed a photoactivated insulin depot for the treatment of diabetes.[133] Conjugating insulin proteins to a biodegradable insoluble matrix via a photolabile linker enabled light-controlled release of insulin in an in vivo mouse model.[134] This application of light-controlled release has the potential to be applied to numerous other drugs in which spatiotemporal delivery is advantageous for treatment.
Finally, spatiotemporal release of therapeutic small molecules and proteins from modified hydrogels has been demonstrated using a light-controlled system. To this end, Anseth and co-workers employed two wavelength-orthogonal photolabile linkers covalently attached to bone morphogenic proteins 2 and 7, thereby enabling sequential release and delivery into mesenchymal stem cells for improved osteogenic differentiation.[90b] Importantly, the extent of each respective protein release was tunable by adjusting the duration of light exposure. This work has implications for the design of biomaterials capable of delivering therapeutics to tissues for regeneration, wound healing, and disease treatment.
In summary, optical control of protein and peptide function is not limited to the perturbation of biological systems, but also has potential to further improve biological sensors. Moreover, even small structural changes imparted through caging-group installation on peptides can have beneficial effects on the properties of the molecules, such as greatly improved cellular delivery. Finally, various drug delivery systems have benefited from the enhanced spatio-temporal control through the incorporation of photorelease technologies to afford improved targeting and controlled dosing. While the incorporation of caging groups directly into proteins has enabled the investigation of many biological processes, light-responsive motifs cannot be genetically inserted into nucleic acids and thus require synthetic (or semi-synthetic/enzymatic) approaches.
4. Optical Control of Oligonucleotides
Nucleic acids have been extensively used as biological probes, with promising developments into therapeutics already underway.[135] Oligonucleotides can be ideal for studying biological pathways because they can affect processes at the DNA, RNA, and protein level in a sequence-specific and fully programmable fashion. Optical control of nucleic acid function has allowed the spatiotemporal control of RNAi, transcription, translation, gene editing, and nucleic acid detection.[1f,136]
Photocleavable groups have been synthetically incorporated into oligonucleotides in a variety of different ways to allow precise activation or deactivation of nucleic acid function with spatiotemporal control (Figure 21). A straightforward approach consists of utilizing a photocleavable linker, for example, in an antisense agent,[137,138] thereby enabling optical activation of gene expression (Figure 21a). Photocleavable linkers have also been utilized for tethering of antagomirs,[139] peptide nucleic acids,[140] and morpholino oligonucleotides[141] directly to short inhibitor strands that block activity through hairpin formation. The inhibitory strand is removed with light, leading to activation of the antisense strand (Figure 21b). Recently, this approach was further advanced by synthesizing circular nucleic acids in which the two termini are linked via a photocleavable moiety. The resulting cyclic structure cannot efficiently hybridize to its target due to the induced curvature until it is linearized through photochemical cleavage of the linker (Figure 21c).[142] Other approaches to optical control include the introduction of chemical modifications such as caged phosphate backbones,[143] caged 2′-hydroxy groups,[144] and caged nucleobases.[145] Caged nucleobases have demonstrated particularly broad applicability since they block Watson–Crick hydrogen bonding, thereby rendering the oligonucleotide inactive until irradiation. This has been employed for the light-mediated regulation of antisense agents,[146] antagomirs,[147] splice-switching oligonucleotides,[148] PCR primers,[149] and many other oligonucleotides (Figure 21d).[150] The same approach can also be used for the optical deactivation of oligonucleotide function through light-triggered hairpin formation, thereby sequestering the active nucleic acid sequence (Figure 21e), as demonstrated for antisense agents,[151] DNAzymes,[151] and triplex-forming oligonucleotides.[150c]
Figure 21.
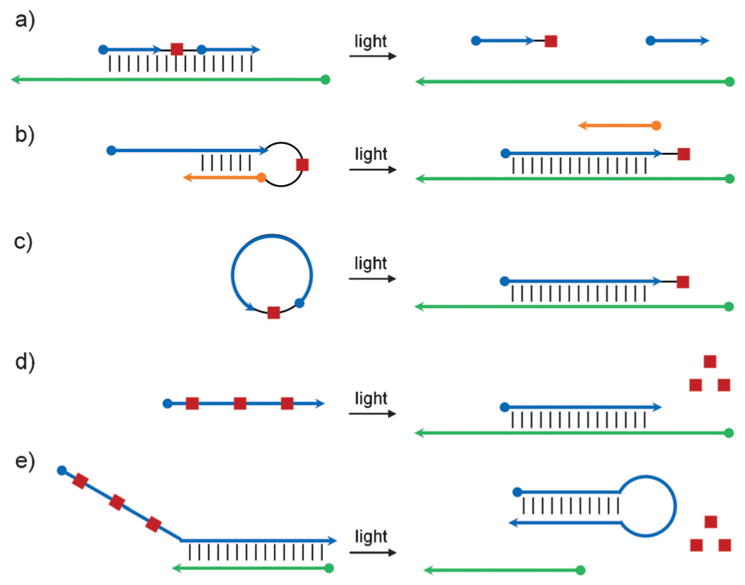
Approaches to the regulation of oligonucleotide hybridization using light-cleavable groups. a) An oligonucleotide sequence containing a photocleavable linker is able to bind its target sequence, inhibiting activity, until light-induced cleavage. b) Hairpin formation of a short inhibitory strand mediated by a photocleavable linker blocks oligonucleotide function until irradiation. c) Formation of a cyclic oligonucleotide via a photocleavable linker inhibits function due to induced curvature until light exposure. d) Caged nucleobases inhibit oligonucleotide function until photo-deprotection. e) Deprotection of caged nucleobases results in hairpin formation, which inhibits oligonucleotide activity. Adapted with permission from Ref. [1f]. Copyright 2013 American Chemical Society.
4.1. Optical Activation of RNA Interference
Sequence-specific gene silencing through RNA interference (RNAi) has been extensively used as a research tool and is being evaluated in clinical studies.[152] Several strategies have been developed to optically control short-interfering RNAs (siRNA duplexes of 21–23 nt),[153] with the first report utilizing the incorporation of nitrobenzyl caging groups at random nucleobases along the phosphate backbone.[143c] The McMaster and Friedman groups installed caging groups at the 5′ phosphate of an siRNA antisense strand to block binding to the RNA-induced silencing complex (RISC), however background activity of the caged siRNA was observed and attributed to some level of tolerance for 5′-blocked phosphates.[154] Friedman and co-workers then developed a more sterically demanding cyclododecyl-containing nitrobenzyl caging group to improve blocking of RNAi function through installation at the 3′ and 5′ termini.[155] Not only did this new photolabile group improve the dynamic range of caged siRNAs, but also the site-specific installation allowed a more facile synthesis of the reagents.
This design was further simplified by the Tang group, who recognized that a single vitamin E modification on the 5′ terminus of siRNAs results in loss of RNAi activity.[156] Thus, by placing a photolabile linker between a vitamin E moiety and the 5′ terminus of the antisense strand of siRNA reagents, they were able to reduce the number of required terminal phosphate caging groups from four to one, thereby enabling activation with a single photolysis step (Figure 22a,b). Efficient off-to-on switching of gene silencing was detected after UV exposure, and spatial control of GFP-targeting siRNA was demonstrated through patterned irradiation of a monolayer of mammalian cells using a mask (Figure 22c). The ability to achieve virtually complete off-to-on switching through UV irradiation with a single chemical modification further facilitates the synthesis and application of these light-activated gene-silencing tools.
Figure 22.
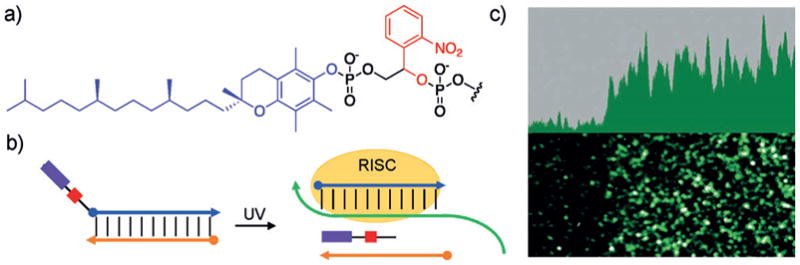
a) Structures of vitamin E (purple) and the photocleavable linker (red). b) siRNA duplexes targeting GFP were modified at the 5′ terminus on the sense (blue) strand with a photocleavable linker and a vitamin E moiety. This modification blocks loading into RISC until irradiation with UV light. c) Spatial control was demonstrated in HEK293T cells using an EGFP reporter. Adapted with permission from Ref. [156]. Copyright 2016 Wiley-VCH.
In a nucleobase-caging approach, Heckel and co-workers first incorporated guanidine and thymidine deoxynucleotides carrying nitrobenzyl groups at the O6 and O4 positions into a siRNA reagent, specifically into nucleotides in close proximity to the mRNA cleavage site.[157] The disruption of hydrogen bonding and introduction of steric hindrance resulted in up to 90% inactivation of a caged siRNA targeting EGFP, however, after 28 hours, fluorescence began to decrease in the absence of light, possibly due to instability of the caging groups. Subsequently, a series of siRNA reagents were synthesized in which nitrobenzyl-caged uridine or guanosine residues were incorporated into the mRNA cleavage site or seed region of an siRNA duplex.[158] The photolabile groups were installed at the N1 or N3 position of guanosine or uridine, providing enhanced stability of the caged siRNAs for at least 48 hours and resulting in excellent off-to-on photoswitching. Moreover, optical control of siRNAs through the installation of caged nucleobases within the seed region directly translates to optical activation of microRNA (miRNA) function.
Overall, development of siRNA duplexes containing bulky caging groups at the 5′ terminus provides a method for facile synthesis of RNAi reagents capable of spatiotemporal gene control. Incorporation of caged nucleobases during oligonucleotide synthesis eliminates the need for post-synthetic installation of bulky caging groups and provides excellent optical activation, thus potentially extending the applicability of these approaches to the control of miRNA function. Continued development of these reagents through the incorporation of photoswitchable groups may enable reversible optical control of siRNA function.
4.2. Optical Activation of Antisense Function
In a complementary fashion, miRNAs have commonly been silenced using antagomirs, synthetic phosphorothioate- and 2′OCH3-modified oligonucleotides,[135b,c,159] which have been placed under optical control through a nucleobase-caging approach (Figure 21d).[147b] This enabled the spatio-temporal investigation of miR-22 and miR-124 function in migrating neurons.[160] Alternatively, inhibition of antagomir function through temporary hairpin formation mediated by photocleavable linkers (Figure 21e) has also been utilized for optical deactivation of miRNA function.[139a]
While DNA/RNA-based oligonucleotides have been extensively used as antisense agents, other synthetic oligomers, such as morpholinos (MOs),[161] have advantages in specific applications; in particular, experiments in aquatic embryos such as those of frogs and fish. MOs are modified nucleic acids containing morpholine rings and phosphorodiamidate backbones instead of sugars and phosphodiesters, respectively, which renders them nuclease-resistant, less likely to interact non-specifically with cellular proteins, and still capable of binding to complementary sequences via Watson–Crick base pairing.[162] As such, MOs have been utilized as antisense agents for the inhibition of miRNA function, mRNA translation, and mRNA splicing.[163] Caged morpholinos (cMOs) have been developed as molecular probes of gene function in cells and animals because they provide precise spatiotemporal control of gene expression.[138b,141,146b,164] In early developments, the Chen group used the light-cleavable hairpin approach,[141a] including the two-photon activation of cMOs containing a bromohydroxyquinoline-based linker (Figure 21b).[141b]
However, this approach requires careful inhibitor sequence design to minimize background activity while still retaining rapid photoactivation. Additionally, release of the inhibitory oligonucleotide may create potential for off-target effects. Similarly, Washbourne and co-workers utilized a MO inhibitor that contained a photocleavable linker (Figure 21b).[138b] In contrast, optical control of MO function in cells and animals has also been demonstrated by incorporating caged thymidine nucleobases.[146b] This approach avoids the necessity for an inhibitory oligonucleotide; however, multiple caging groups are needed to fully block MO–mRNA hybridization.
Recently, conformationally gated MOs[48,142d,165] and DNA oligomers[142b,166] were developed in which the introduction of curvature reduces antisense oligonucleotide–mRNA interactions, thereby inhibiting gene silencing until photochemical cleavage of a linker linearizes the nucleic acid and fully restores mRNA hybridization. This nicely addresses both limitations discussed above, since only a single photolysis step is needed for activation and no inhibitory oligonucleotide is released.[142d] Moreover, the ability to insert a wide range of chromophores into the circular MO enables wavelength-specific gene silencing by sequentially irradiating zebrafish embryos with 405–470 nm (cleaving a diethylaminocoumarin (DEACM) group) and 365 nm (cleaving a 2-nitrobenzyl (NB) group; Figure 23a).[48] Cyclic MOs were designed to target the T-box transcription factor spadetail (spt/tbx16) and the homeobox-containing repressor flh. Spt controls cell differentiation during embryogenesis and its transcription within the embryo midline is inhibited by flh. Mutation of spt also corresponds with the absence of myogenic differentiation 1 (myod1), while flh silencing leads to aberrant myod1 expression in early stages of development.[167] Upon irradiation with 365 nm light, zebrafish injected with the NB cyclic spt cMO displayed a drastic loss in myod1 expression, but showed minimal response to irradiation with 405 or 470 nm light, whereas embryos injected with the DEACM cyclic flh cMO exhibited abnormal myod1 expression when irradiated with either 470, 405, or 365 nm light, as expected (Figure 23b,c). Co-injection of both cMOs led to zebrafish embryos that displayed the spt mutant phenotype only after irradiation at 405/470 nm followed by 365 nm light exposure. Sequential irradiation at defined time points during embryonic development demonstrated that spt acts during gastrulation in embryos bearing flh mutations to direct muscle cell precursors toward muscle cell fates, thus showcasing the utility of sequentially activated cMOs for the investigation of gene interactions with precise temporal control.
In summary, caged oligonucleotides, including circular MOs, have been established for optical control of gene function through blocking translation. The development of circular MOs that are subject to photoactivation using wavelength-specific chromophores enables the investigation of genetic networks through sequential knockdown of different gene targets. However, while nuclease-resistant MOs are attractive antisense agents, delivery into mammalian cells requires additional modifications.[168] Furthermore, depending on the mRNA sequence, background antisense activity of the circular MO may be observed.
4.3. Optical Control of Transcription
Several approaches to optically control gene expression at the transcriptional level have been developed, including light activation of triplex-forming oligonucleotides (TFOs).[169] TFOs are single-stranded oligonucleotides that bind to the major groove of DNA promoter regions through Hoogsteen hydrogen bonding (Figure 24a), thereby preventing the association of transcription factors.[170] Contrary to antisense agents, which regulate gene translation by targeting mRNAs, for which thousands of copies may be present in cells, TFOs can target genomic DNA.[171] Optical control of transcription through photoactivation of a TFO containing caged thymidine bases has been demonstrated (Figure 24b).[150c]
Figure 24.

a) Control of both Watson–Crick base pairing and Hoogsteen base pairing through photolysis of nucleobase-caged thymidines. b) Optical activation of a triplex-forming oligonucleotide (TFO; yellow) through nucleobase decaging and resulting DNA triplex formation through Hoogsteen base pairing. The TFO blocks binding of the transcription factor (brown) to the promoter region (blue/green), thereby inhibiting gene expression. Photoactivation of the caged TFO led to reduction in luminescence similar to a non-caged TFO. c) Optical activation of a DNA decoy through light-induced dumbbell formation, leading to sequestration of the transcription factor targeting the decoy promoter region and inhibition of gene expression. UV irradiation of the caged DNA decoy led to a decrease in luciferase activity similar to that of a non-caged decoy. Adapted with permission from Ref. [150c] (Copyright 2012 American Chemical Society) and Ref. [150d] (Copyright 2011 American Chemical Society).
Besides TFOs, DNA decoys have also been employed for transcriptional control. DNA decoys inhibit gene expression by sequestering transcription factors rather than relying on the binding to and blocking of promoter sequences.[172] Three caged thymidine bases were incorporated into a DNA decoy targeting NF-κB (Figure 24c) to optically regulate transcription factor binding by initially preventing formation of the DNA decoy dumbbell structure.[150d] Following UV irradiation, the decoy is formed, resulting in sequestration of the transcription factor and deactivation of gene expression (Figure 24c). Complementary optochemical transcriptional activation was demonstrated using NF-κB DNA decoys[173] containing up to three 7-nitroindole moieties, a nucleobase mimic known to undergo elimination upon irradiation with UV light. Here, the decoy/transcription factor complex inhibits gene expression, until exposure to UV light depurinates the photoresponsive nucleotides, resulting in release of the transcription factor and activation of gene expression.
Plasmids containing a gene of interest under control of the cytomegalovirus (CMV) promoter are one of the most common expression platforms in mammalian cells.[174] Transcription is initiated when the TATA box binding protein binds to the TATA box sequence within the CMV promoter and recruits additional transcriptional machinery.[175] In order to achieve spatial and temporal activation of gene expression, caged thymidine nucleobases were site-specifically introduced into the TATA box (Figure 25a).[176] The caged plasmid is inactive until brief UV exposure removes the caging groups, forming the wild-type CMV promoter, and completely restoring gene expression, as demonstrated for EGFP and polo-like kinase 3. In order to apply this method in an animal model, the caged TATA box plasmid was injected into zebrafish embryos (Figure 25b). Following microinjection at the 1-cell stage, only embryos that were exposed to UV irradiation expressed EGFP. The ability to activate transcription using a caged expression plasmid at defined time points and locations in cells and animals has broad implications for investigating many biological pathways, including those involved in development, and it complements the previously discussed light-triggered inhibition of transcription and translation using light-activated nucleic acids. With recent advances in caging groups with different spectral properties, the activation and deactivation of multiple genes using light of different wavelengths will enable the study of increasingly complex genetic systems. Also, the caged plasmid system is assembled in a very modular fashion and thus should allow optical control of any gene of interest.
Figure 25.
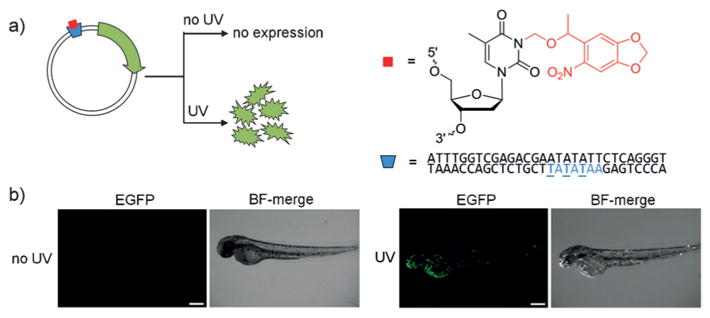
a) Optical control of transcription using a caged promoter. EGFP is only expressed following removal of the nitrobenzyl caging group (red) upon irradiation with UV light. Three caged thymidine residues (underlined) were incorporated into the TATA box sequence (blue) of the CMV promoter. b) Optical activation of EGFP expression in zebrafish embryos. Following injection of the caged plasmid at the one-cell stage, embryos were either irradiated with 365 nm light or left in the dark. EGFP fluorescence and brightfield (BF)-merge micrographs are shown. Adapted with permission from Ref. [176]. Copyright 2014 American Chemical Society.
Taken together, optical control of TFOs and DNA decoys through the incorporation of caged nucleobases enables spatial and temporal regulation of transcription. Continued expansion of light-triggered DNA decoys to include alternative caging groups may enable orthogonal control of multiple transcription factors and investigation of the corresponding genetic networks. Development of an optochemically regulated plasmid through caging of the promoter region provides a modular approach to controlling exogenous gene expression in mammalian cells as well as zebrafish. The latter may benefit from enhanced three-dimensional precision through the application of two-photon excitation in future iterations of this methodology.
4.4. Optical Activation of RNA Editing and Gene Editing
In addition to transcriptional and translational control using synthetic nucleic acids, caged oligoribonucleotides have been applied to the conditional editing of RNA sequences. The Stafforst group established a method for introducing a single adenosine-to-inosine point mutation in an mRNA sequence by using a modified targeting RNA that recruits the ADAR1 enzyme (adenosine deaminases acting on RNA).[177] The editing machinery was assembled by fusing a SNAP-tag domain to ADAR1 and incorporating a benzylguanine (BG) ligand on the targeting RNA for covalent attachment. In order to achieve precise optical control, a photolabile group was installed at the N7 position of the BG moiety (Figure 26a).[178] As a proof of concept, a caged RNA targeting a premature amber stop codon was used to activate an EGFP reporter after optically triggered adenosine-to-inosine conversion in both mammalian cells and the worm Platynereis dumerilii (Figure 26b). Minor background activity of RNA editing was observed in the worms in the absence of light. This technology is highly adaptable for the optochemical control of translation of protein isoforms or investigation of developmental processes with precise spatial control in model organisms, simply by changing the targeting RNA sequence. Additionally, similar approaches could be used in the optical control of nucleic acid editing machinery, such as that for pseudouridylation[179] or 2′-O-methylation,[180] if directed by a small guide RNA.
Figure 26.
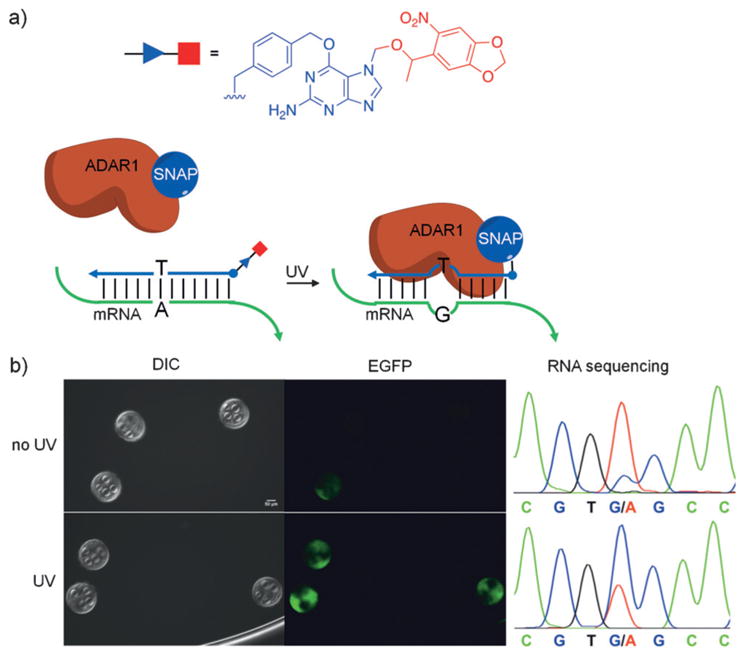
a) Light-activated site-directed RNA editing. Irradiation with UV light removes the photolabile group (red), exposing the benzylguanine moiety (purple) for conjugation to the SNAP-ADAR1 protein. Once the RNA–protein conjugate forms, site-specific editing is performed. b) The RNA–protein conjugate activates EGFP fluorescence in worms only after UV irradiation. RNA sequencing indicated an increase in editing yield from 10% to 60% after irradiation as shown in the chromatogram. Adapted with permission from Ref. [178]. Copyright 2015 American Chemical Society.
Recently, Bhatia and co-workers developed a light-activated CRISPR/Cas9 gene-editing system by blocking the gRNA with a complementary single-stranded DNA molecule that contains several photocleavable groups in its backbone. In an approach that is similar to the optical activation of antisense oligonucleotide function discussed above,[137,138,181] the inhibitory DNA strand binds to the target region of the gRNA and prevents Cas9 function until it is fragmented through UV-induced photolysis (Figure 27a,b).[85] As a proof of concept, this system was delivered into HeLa cells stably expressing a destabilized GFP reporter, and UVactivation led to reporter DNA cleavage and subsequent reduction in GFP expression. It was determined that longer protectors (24 nt) with three photolabile groups spaced six bases apart are optimal for inhibiting Cas9-mediated DNA cleavage (Figure 27c). The approach was also applied to the optical editing of an endogenous genomic target, and the gRNA/protector duplexes were stable for up to five days in live cells. However, compared to other optochemically controlled Cas9 systems (e.g., see Figure 15),[81] the use of a light-triggered DNA inhibitor strand seems to display a lower dynamic range.
All in all, light-triggered RNA editing was achieved by utilizing endogenous machinery in conjunction with a modular guide oligonucleotide. While incorporation of the SNAP-tag enables site-directed RNA editing, it also requires the guide RNA to be chemically modified prior to delivery into cells (i.e., it cannot be genetically encoded). Delivery of the guide RNAs and their cellular stability will be an important consideration for applying these tools more broadly for the investigation of biological systems. Application of a caged gRNA protector allows optical control of the CRISPR/Cas9 system without the need for protein engineering. However, depending on the sequence, the use of protector oligonucleotides may lead to potential background activity or toxicity.
4.5. Optical Activation of Oligonucleotide-Based Sensors
Caged oligonucleotides have also been utilized for various detection systems. DNA computation is an emerging method for the analysis of oligonucleotide patterns in which sequence-specific hybridization of target input nucleic acids to a DNA-based scaffold triggers a detectable output signal based on toehold-mediated strand-exchange reactions.[182] In a strand-exchange reaction, the incoming oligonucleotide hybridizes to a short single-stranded domain (referred to as a toehold) of a duplex, resulting in branch migration and subsequent strand displacement. Optical control of DNA logic gates using caged nucleobases[150a] and their application in live cells has been demonstrated.[183] Similarly, light-controlled amplification circuits relying on toehold-mediated strand-displacement reactions have also been developed.[184] Optical control of DNA computation devices provides many opportunities for spatiotemporal detection of oligonucleotides involved in the development of diseases and other biological processes.
Molecular beacons are short oligonucleotide hairpins that maintain an “off” state until a complementary DNA or RNA molecule hybridizes to the loop, separates the stem, and turns fluorescence “on”.[185] Two approaches to achieve optical control over molecular beacons have been reported: 1) installation of caging groups into the stem region to prevent formation of the hairpin[186] and 2) installation of caging groups into the loop region to prevent target hybridization,[187] with the latter showing reduced background. With a system using a fused aptamer against a cell-surface protein for nucleic acid uptake, the Tan group developed optical control of a molecular beacon in breast cancer cells.[188] Because biological processes occur at specific times and locations, this technology could easily be applied to the investigation of transcriptional events with enhanced precision relative to previous methods.
Aptamers are RNA- or DNA-based oligonucleotides that bind to proteins with high specificity and affinity.[189] They have been rendered responsive to light using photocleavable linkers,[142e,190] caged nucleobases,[191] caged backbones,[192] and photoswitchable motifs.[193] For example, Mayer and Heckel demonstrated optochemically induced deactivation of an anti-thrombin aptamer through the incorporation of caged nucleobases.[191a] The aptamer was shown to inhibit thrombin-mediated blood clotting in human plasma in the absence of light; however, upon UV irradiation the aptamer was deactivated, effectively applying light as an antidote to an anticoagulant.[194] Recently, caging groups were applied to Apta-PCR[195] as a sensitive technique for quantification of target proteins.[196]
DNAzymes are enzyme composed of DNA and have been engineered to catalyze sequence-specific RNA cleavage.[197] They have been used as gene regulatory tools and as cellular sensors, but issues remain due to activity during cellular delivery and uptake. To circumvent this concern and achieve spatiotemporal control over activity, several strategies have been developed to optochemically regulate DNAzymes.[144a,151,198] Caged adenosine bases were incorporated into the scissile position of the substrate strand for DNA-zymes selective for Zn2+ or Pb2+ ions, thus demonstrating a modular approach for sensing metal ions in mammalian cells.[199] Recently, Xiang and co-workers reported the development of a DNAzyme that was caged post-synthetically in order to circumvent the addition of caged phosphoramidites during solid-phase synthesis.[200] Following optimization through in vitro experiments, the fluorescently labeled DNA-zyme was delivered into HeLa cells along with the substrate and the Zn2+ cofactor. After a brief UV irradiation, the DNAzyme was activated, resulting in substrate cleavage and subsequent enhancement of the fluorescence signal, thus demonstrating temporal control of DNAzyme function.
While several methods to detect mRNA expression in cells have been developed,[201] single-cell analysis techniques remain complex and challenging.[202] To address these limitations, a transcriptome in vivo analysis (TIVA)-tag was developed to capture mRNA from single cells in live tissue upon photoactivation,[203] thereby providing transcriptome analysis with spatial and temporal control. The TIVA-tag was delivered into cells using a reductively cleaved CPP and contained a light-cleavable oligonucleotide hairpin. After photolysis, the poly-2′-deoxyfluorouridine sequence hybridizes to poly-A tails and thus captures mRNAs. A biotin tag was used for affinity purification and a Cy3-Cy5 FRET pair was added to monitor uptake and light-mediated activation (Figure 28a). Targeted irradiation of a single neuron in mouse or human brain slices activated the TIVA-tag and the mRNA/TIVA-tag complexes were isolated and analyzed through RNA-seq (Figure 28b). This approach revealed that cells in dispersed cultures express 30% more genes than those in live brain slices, thus suggesting that the microenvironment of a cell plays an important role in determining its transcriptome. Additionally, while all of the studied cells expressed characteristic neuronal markers, many also expressed markers indicative of other cell types, thus illustrating the utility of this technology for investigating cell heterogeneity. This study successfully demonstrated that the TIVA-tag can be used to isolate RNA from live cells with spatial and temporal resolution, thereby creating snapshots of the transcriptome that otherwise cannot be obtained.
Figure 28.
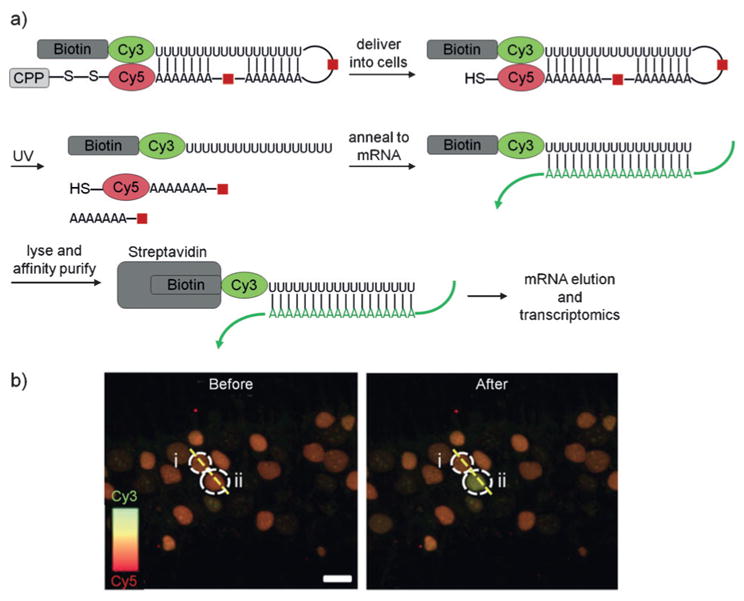
a) Schematic showing optical control of the TIVA-tag for single-cell transcriptome analysis. b) Decaging of the TIVA-tag in single cells in live hippocampal slices from mice. Neuron ii was irradiated, leading to activation of the TIVA-tag in a single cell but not in adjacent cells. The fluorescence signal of Cy5 at 647–704 nm generated from exciting Cy3 at 514 nm was recorded in the two neurons outlined by dotted white lines and labeled “i” and “ii”. Adapted with permission from Ref. [203]. Copyright 2014 Nature Publishing Group.
Overall, several methods for light-activated nucleic acid based sensors have been developed, including DNA logic gates, molecular beacons, aptamers, DNAzymes, and the TIVA-tag. These optochemical tools allow the precise spatiotemporal detection of oligonucleotide targets and metal ions in living systems. Moreover, the TIVA-tag enabled analysis of mRNA transcripts in single neurons. Future development of the TIVA-tag may include sequence elements that enable the detection of other RNA molecules, such as non-coding RNAs.
4.6. Two-Photon Activation of Oligonucleotide Function
In addition to the two-photon activation of morpholino oligonucleotides described above,[141b] Heckel and co-workers utilized coumarin (DEACM) and nitrobiphenyl (ANBP) caging groups,[204] which can be removed through orthogonal two-photon activation, to nucleobase-protect deoxythymidine and deoxyguanosine bases in two oligonucleotides, DNA1 and DNA2 (Figure 29a).[205] Decaging of DNA1 and DNA2, embedded in a hydrogel to prevent diffusion, initiated sequence-specific hybridizations of complement strands labeled with the fluorophores ATTO 565 and ATTO Rho14 respectively (Figure 29c), as part of a light-triggered toehold-mediated strand exchange (Figure 29b). Irradiation with 980 nm light resulted in exclusive decaging of DNA2 (Figure 29d center), while irradiation with 840 nm light (at reduced laser power) selectively decaged DNA1 (Figure 29d left). However, upon irradiation with 840 nm light at increased laser power, it was possible to decage both DNA1 and DNA2, resulting in the detection of both fluorophores (Figure 29d right). The strand-exchange reaction triggered by DNA1 was also demonstrated in hippocampal neurons. Because two-photon imaging has been routinely used in live tissues,[206] this approach delivers an additional level of precision for probing biological pathways and nucleic acid function in model organisms with complex three-dimensional structures.
Thus, wavelength-selective spatially controlled two-photon activation of oligonucleotides has been achieved in hydrogels. This methodology may provide an opportunity for multiplexable nucleic acid sensors and gene-control agents in living organisms due to the enhanced tissue penetration and three-dimensional resolution of two-photon decaging.
4.7. Reversible Optical Switching of Oligonucleotide Function
Recent advances have been made in the development of photoisomerizable nucleic acids.[136a,193,207] Photoswitchable oligonucleotides have enabled precise reversible control of nucleic acid based processes such as DNA crosslinking[208] and DNA–RNA hybridization.[209] More complex photoswitching tools have also been reported, such as azobenzene-modified DNAzymes and ribozymes[210] to reversibly control RNA cleavage. Diarylethene photoswitches incorporated at the C5 position of uracil were installed in the T7 promoter region of a double-stranded DNA template, thereby allowing photo-switching of transcription.[211] However, these technologies have not yet been applied in vivo and achieving reversible control of oligonucleotide function in living organisms remains a challenge. Ogasawara demonstrated reversible optical control of translation in cells by installing a styryl-modified 2′-deoxyguanosine residue as the 5′-cap of mRNA encoding a constitutively active H-Ras gene.[212] In the trans form, the styryl group inhibits translation by blocking the interaction between translation initiation factor elF4E and the 5′-cap. Delivery of the styryl-capped H-Ras mRNA into rat brain cells led to expansion and contraction of cells upon irradiation with 405 nm and 310 nm light, respectively. One major limitation of this tool is the requirement for potentially damaging UV-B light for photoswitching. More recently, 2-phenylazo groups were incorporated at the C2 amine of a guanosine residue and subsequently installed as the 5′-cap of a mRNA sequence through in vitro transcription, thereby allowing photoisomerization at longer wavelengths.[213] In addition to a 2-phenylazo cap (29), 2-para-methyl- (30) and 2-meta-methyl- (31) phenylazo caps were also investigated for their ability to alter affinity for elF4E through steric hindrance (Figure 30a). Phenylazo cap-modified mRNA encoding a fluorescent protein was injected into zebrafish embryos at the one-cell stage and photoisomerized to the cis form using 370 nm light. The cap configurations were reversed to the trans form through thermal isomerization over 1–2.5 h. The switch 31 exhibited the highest photo-modulation efficiency, while both 30 and 31 elicited the greatest translational inhibition in the trans state. To further demonstrate the utility of this technology, 31 was incorporated as the 5′-cap of mRNA encoding the squint protein in zebrafish embryos. Squint protein plays a major signaling role between the eight-cell and shield stages of zebrafish development and its overexpression beyond these stages in a specific region of the embryo leads to the formation of a second midline without defined head features such as eyes. Following injection of 31-capped squint protein mRNA and Venus fluorescent lineage tracer mRNA, embryos were irradiated with 370 nm light at the 8-cell stage and 430 nm light at the shield stage. After photoswitching, most of the embryos expressed a second midline with defined head features (Figure 30b). In contrast, embryos continuously exposed to 370 nm light but not 430 nm light expressed a second midline but no defined head features, thus suggesting that squint protein expression promotes midline development in early stages of embryogenesis and inhibits the formation of head features at later stages. Overall, the induction rate of the second midline was low due to difficulty in differentiating specific regions of the embryos at the 8-cell stage, thus highlighting a potential limitation to the use of optochemical tools for investigating developmental processes in very early stages of development. However, photoswitchable 2-phenyl-azo caps provide precise on/off/on cycles of translational control in cells and animals with minimal toxicity, which allows reversible temporal control of gene function.
In summary, reversible optical control of translation has been demonstrated with 5′-capped mRNAs in mammalian cells and in zebrafish embryos, and enable precise temporal control over reversible activation/inactivation of gene expression, thereby mimicking nature’s gene regulatory abilities. Future development and incorporation of red-shifted photo-switchable caps may enable deeper tissue penetration, thereby expanding the utility of this technology to other model organisms.
5. Selected Advances in the Development of Photo-activatable Caging Groups
Nitrobenzene- and coumarin-derived caging groups have been the most extensively used photocleavable groups, and recent advances in shifting activation wavelengths into the red and far-red region of the spectrum have been made. Nitrobenzene derivatives remain important caging groups, due to minimal leaving-group pKa requirements, in contrast to coumarin-based groups.[1e,214] Notable examples for red-shifted nitrobenzene derivatives are bisstyrylthiophene (see Figure 13), ANBP (see Figure 29), and nitrodibenzofuran (NDBF).[215] Long-wavelength activated coumarin groups include thiocoumarin,[45] dicyanocoumarin,[216] and 3-acrylamide-modified aminocoumarin, for example, 32 (see Figure 32).[217]
Figure 32.
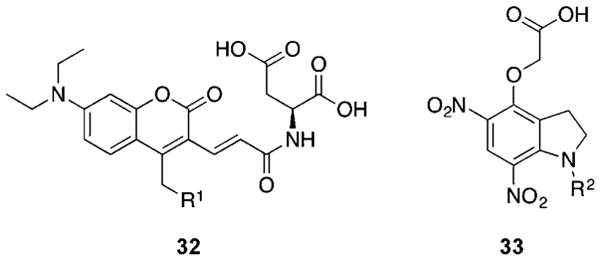
Structures of the caging groups 32 and 33. Examples of caged molecules include cyclic-AMP (R1) and γ-aminobutyric acid (R2).
The Lawrence group utilized naturally occurring vitamin B12, which contains a corrin ring coordinated to a central cobalt atom, as a unique caging group (Figure 31a).[218] The weak cobalt–carbon bond is photocleaved at 330–580 nm, releasing the R group (e.g., a drug or a fluorophore).[219] Additional chromophores were conjugated to vitamin B12, such as Cy5, Cy7, or Alexa700, to act as antennas to allow further red-shifted photocleavage with 640–780 nm light.[220] A related approach for low energy, near-IR light activation utilizes photosensitizers to generate singlet-oxygen species that react with an electron-rich alkene (e.g., ~ SHC=CHS ~ or anthracene) through [2+2] or [4+2] cycloaddition, respectively. Rapid decomposition generates two carbonyl-bearing fragments (Figure 31b), in a similar fashion as the photo-bleaching of Cy dyes (see Figure 11). This approach has been applied to zinc release,[221] DNA hybridization,[222] regulation of PCR,[223] anti-miRNAs,[224] and siRNAs.[225] Moreover, ruthenium complexes have been cleverly used for the caging of cyclic morpholinos[142a] and chemotherapeutic agents.[226]
Figure 31.
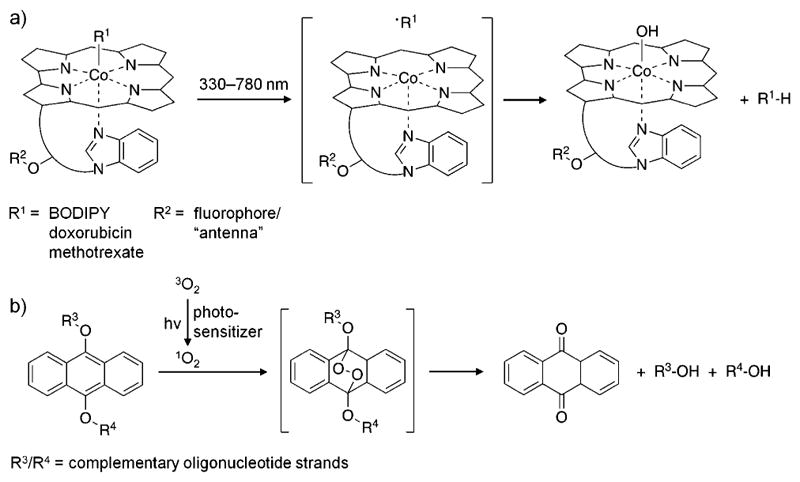
a) The vitamin B12 core structure with the photocleavable group as R1 and the antenna/fluorophore as R2. b) The dialkoxyanthracene reacts with singlet oxygen to form an endoperoxide intermediate that rapidly fragments into an anthraquinone, thereby releasing R3 and R4 as alcohols. Adapted with permission from Ref. [218] (Copyright 2015 American Chemical Society) and Ref. [222] (Copyright 2011 American Chemical Society).
Several of the red-shifted caging groups mentioned previously have been applied to the multiwavelength activation of biological processes, since they can be paired with UV-triggered nitrobenzyl groups.[39] For example, Heckel and coworkers utilized DEACM and ANBP groups for the sequential activation of oligonucleotides with 840 and 980 nm light (see Figure 29 and Section 4.7 for a detailed discussion).[205] In another example, the coumarin caging group 32 was applied in combination with carboxydinitroindole 33 for selective control of neuronal activation (Figure 32).[227] Overall, the caging groups applied in these examples could be exchanged for other nitrobenzyl or coumarin moieties with similar absorption maxima to enable the caging of other functional groups.
6. Summary and Outlook
The optical control of biological processes has dramatically advanced in recent years, showcasing innovative combinations of synthetic chromophores with biologically active molecules, including biological macromolecules such as nucleic acids and proteins. For example, direct control of protein function can be achieved through the site-specific installation of light-removable caging groups using fully genetically encoded tools in mammalian cells and model organisms. The chromophores are chemically tunable (in contrast to classic optogenetic approaches), and an extensive number of biological activities has been optically triggered, including post-translational protein modifications, cell signaling, sub-cellular protein translocation, gene editing, and RNA polymerization. The optical control of nucleic acid function has also seen significant advances, and distinct chemical approaches have been further developed, ranging from the installation of nucleobase-caging groups to the generation of circular oligonucleotides through photocleavable linkers, all with the goal of light-triggered activation or deactivation of nucleic acid hybridization. Thus, virtually any nucleic acid based cellular process can be placed under precise spatial and temporal control, most importantly transcription and translation. Recent examples in the context of rendering small molecules light-controllable are small-molecule dimerizers of proteins, for example, the natural product rapamycin or other chimeric molecules, through the installation of light-removable caging groups. Despite rapid diffusion, spatial control of small-molecule function in tissue culture has been achieved through repeated, pulsed decaging in a specific location. In order to place small-molecule function under reversible optical control, an azostere approach has been developed, which replaces common pharmacophores with light-switchable azobenzene motifs. Azobenzene chromophores have also been linked directly to the control of protein function, either through bioconjugation approaches that convert low-potency inhibitors into highly specific and light-regulated molecules, or through the direct genetic encoding of amino acids containing azobenzene moieties. Similarly, diazo-modified mRNA caps have been developed for reversibly photo-switchable control of translation. It can be expected that other cellular processes involving nucleic acids will soon be controlled in a reversible fashion to provide enhanced spatial control, even in long-term experiments. Wavelength-specific and two-photon activation of oligonucleotides has been demonstrated, including optical control in model organisms, such as fully transparent zebrafish embryos.
The methods discussed herein represent recent key developments; however, in order to further increase their relevance, it will be essential to apply these very precise tools to the study of important biological questions. Thus, close collaborations between chemists and biologists will be essential and applications beyond proof-of-principle experiments will be necessary in order to convince scientists in fields outside of chemistry and chemical biology of the utility of optochemical methods. The feedback generated in a collaborative environment will motivate further technological developments to expand the capabilities of these light-controlled reagents.
Furthermore, chemists must strive to develop optical tools that are adaptable to a variety of biological systems and that are readily available to cellular and developmental biologists, who often do not have synthetic chemistry expertise. Reagent stability, including the ability to handle optically triggered compounds and biological molecules under ambient light, and ease of delivery into cells and animals should remain of critical consideration when advancing these technologies. Finally, commercialization of photochemically controlled reagents will greatly facilitate applications by lowering the barrier to enter this field for biologists. Caged neurotransmitters have been extensively utilized in neurobiology not only due to their very precise temporal control, but also due to their commercial availability. The sharing of biological reagents and model systems is facilitated by non-profit repositories (e.g., Addgene or ZFIN), while similar services do not exist for (photocaged) synthetic agents, thus making it more difficult for biologists to test and apply these tools. While recent investigations into genetic networks, neuronal networks, and cell-signaling networks have capitalized on the temporal and spatial control provided by the approaches discussed here, the field is still only in its infancy and holds great potential for the coming years.
Acknowledgments
T.C. was supported by a National Science Foundation Graduate Research Fellowship and Y.N. was supported by a Mellon Graduate Fellowship. A.D. acknowledges support from the National Institutes of Health (GM112728, GM108952, and HD085206) and the National Science Foundation (CHE-1404836, CBET-1603930, and MCB-1330746). Since the community of chemical biologists who are developing and applying optochemical and optobiological tools is continuously growing, we apologize to all colleagues whose work we were unable to discuss due to space limitations.
Biographies

Nicholas Ankenbruck received his BS and MS in Chemistry from Ball State University under the direction of Prof. J. Ribblett. Since 2013, he has been pursuing his Ph.D. at the University of Pittsburgh in the development of synthetic molecular probes for the perturbation and detection of microRNA function under the guidance of Prof. Deiters.

Yuta Naro received his BS in Health Sciences from Ithaca College and started pursuing his Ph.D. at the University of Pittsburgh in 2012. His current research under the guidance of Prof. Deiters pertains to the discovery of small-molecule inhibitors of micro-RNA function and the development of conditionally controlled chemical probes for studying biological processes.

Taylor Courtney received her BA in Chemistry and BS in Molecular Biology from North Carolina State University in 2015 and started pursuing her Ph.D. at the University of Pittsburgh. Under the mentorship of Prof. Deiters, her research focuses on the development and application of optically controlled chemical tools to study protein function.

Alexander Deiters received his Ph.D. from the University of Münster (with D. Hoppe) in 2000 and subsequently conducted postdoctoral studies at the University of Texas (with S. F. Martin) and The Scripps Research Institute (with P. G. Schultz). He is currently a professor at the University of Pittsburgh, where his group conducts research at the interface of chemistry and biology, with an emphasis on optical control of oligonucleotide and protein function in cells and animals.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201700171.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.a) Brieke C, Rohrbach F, Gottschalk A, Mayer G, Heckel A. Angew Chem Int Ed. 2012;51:8446–8476. doi: 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2012;124:8572–8604. [Google Scholar]; b) Mayer G, Heckel A. Angew Chem Int Ed. 2006;45:4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:5020–5042. [Google Scholar]; c) Lee HM, Larson DR, Lawrence DS. ACS Chem Biol. 2009;4:409–427. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gautier A, Gauron C, Volovitch M, Bensimon D, Jullien L, Vriz S. Nat Chem Biol. 2014;10:533–541. doi: 10.1038/nchembio.1534. [DOI] [PubMed] [Google Scholar]; e) Klán P, Šolomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J. Chem Rev. 2013;113:119–191. doi: 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liu Q, Deiters A. Acc Chem Res. 2014;47:45. doi: 10.1021/ar400036a. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Mart RJ, Allemann RK. Chem Commun. 2016;52:12262–12277. doi: 10.1039/c6cc04004g. [DOI] [PubMed] [Google Scholar]; h) Ouyang X, Chen JK. Chem Biol. 2010;17:590–606. doi: 10.1016/j.chembiol.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Niu J, Johny MB, Dick IE, Inoue T. Biophys J. 2016;111:1132–1140. doi: 10.1016/j.bpj.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang K, Cui B. Trends Biotechnol. 2015;33:92–100. doi: 10.1016/j.tibtech.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deisseroth K. Nat Neurosci. 2015;18:1213–1225. doi: 10.1038/nn.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Weitzman M, Hahn KM. Curr Opin Cell Biol. 2014;30:112–120. doi: 10.1016/j.ceb.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Pathak GP, Vrana JD, Tucker CL. Biol Cell. 2013;105:59–72. doi: 10.1111/boc.201200056. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Repina N, Rosenbloom A, Mukherjee A, Schaffer DV, Kane RS. Annu Rev Chem Biomol Eg. 2017;8(1):13–39. doi: 10.1146/annurev-chembioeng-060816-101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Putyrski M, Schultz C. FEBS Lett. 2012;586:2097–2105. doi: 10.1016/j.febslet.2012.04.061. [DOI] [PubMed] [Google Scholar]; b) Voss S, Klewer L, Wu YW. Curr Opin Chem Biol. 2015;28:194–201. doi: 10.1016/j.cbpa.2015.09.003. [DOI] [PubMed] [Google Scholar]; c) Fegan A, White B, Carlson JC, Wagner CR. Chem Rev. 2010;110:3315–3336. doi: 10.1021/cr8002888. [DOI] [PubMed] [Google Scholar]; d) DeRose R, Miyamoto T, Inoue T. Pflugers Archiv: Eur J Physiol. 2013;465:409–417. doi: 10.1007/s00424-012-1208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pecot MY, Malhotra V. Cell. 2004;116:99–107. doi: 10.1016/s0092-8674(03)01068-7. [DOI] [PubMed] [Google Scholar]
- 5.Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P. Cell. 2009;136:1110–1121. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson MS, Sahlender DA, Foster SD. Dev Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karginov AV, Zou Y, Shirvanyants D, Kota P, Dokholyan NV, Young DD, Hahn KM, Deiters A. J Am Chem Soc. 2011;133:420–423. doi: 10.1021/ja109630v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banaszynski LA, Liu CW, Wandless TJ. J Am Chem Soc. 2005;127:4715–4721. doi: 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- 9.Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM. Nat Biotechnol. 2010;28:743–747. doi: 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umeda N, Ueno T, Pohlmeyer C, Nagano T, Inoue T. J Am Chem Soc. 2011;133:12–14. doi: 10.1021/ja108258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harwood KR, Miller SC. ChemBioChem. 2009;10:2855–2857. doi: 10.1002/cbic.200900546. [DOI] [PubMed] [Google Scholar]
- 12.Brown KA, Zou Y, Shirvanyants D, Zhang J, Samanta S, Mantravadi PK, Dokholyan NV, Deiters A. Chem Commun. 2015;51:5702–5705. doi: 10.1039/c4cc09442e. [DOI] [PubMed] [Google Scholar]
- 13.a) Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, Chenoweth DM. Nat Commun. 2014;5:5475. doi: 10.1038/ncomms6475. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ballister ER, Ayloo S, Chenoweth DM, Lampson MA, Holzbaur ELF. Curr Biol. 2015;25:R407–R408. doi: 10.1016/j.cub.2015.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jing C, Cornish VW. Acc Chem Res. 2011;44:784–792. doi: 10.1021/ar200099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmermann M, Cal R, Janett E, Hoffmann V, Bochet CG, Constable E, Beaufils F, Wymann MP. Angew Chem Int Ed. 2014;53:4717–4720. doi: 10.1002/anie.201310969. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:4808–4812. [Google Scholar]
- 16.Wright CW, Guo ZF, Liang FS. ChemBioChem. 2015;16:254–261. doi: 10.1002/cbic.201402576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schelkle KM, Griesbaum T, Ollech D, Becht S, Buckup T, Hamburger M, Wombacher R. Angew Chem Int Ed. 2015;54:2825–2829. doi: 10.1002/anie.201409196. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:2867–2871. [Google Scholar]
- 18.Broichhagen J, Frank JA, Trauner D. Acc Chem Res. 2015;48:1947–1960. doi: 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- 19.a) Pittolo S, Gomez-Santacana X, Eckelt K, Rovira X, Dalton J, Goudet C, Pin JP, Llobet A, Giraldo J, Llebaria A, Gorostiza P. Nat Chem Biol. 2014;10:813–815. doi: 10.1038/nchembio.1612. [DOI] [PubMed] [Google Scholar]; b) Velema WA, van der Berg JP, Hansen MJ, Szymanski W, Driessen AJ, Feringa BL. Nat Chem. 2013;5:924–928. doi: 10.1038/nchem.1750. [DOI] [PubMed] [Google Scholar]
- 20.Velema WA, Hansen MJ, Lerch MM, Driessen AJ, Szymanski W, Feringa BL. Bioconjugate Chem. 2015;26:2592–2597. doi: 10.1021/acs.bioconjchem.5b00591. [DOI] [PubMed] [Google Scholar]
- 21.Schoenberger M, Damijonaitis A, Zhang Z, Nagel D, Trauner D. ACS Chem Neurosci. 2014;5:514–518. doi: 10.1021/cn500070w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Huckvale R, Mortensen M, Pryde D, Smart TG, Baker JR. Org Biomol Chem. 2016;14:6676–6678. doi: 10.1039/c6ob01101b. [DOI] [PubMed] [Google Scholar]; b) Stein M, Middendorp SJ, Carta V, Pejo E, Raines DE, Forman SA, Sigel E, Trauner D. Angew Chem Int Ed. 2012;51:10500–10504. doi: 10.1002/anie.201205475. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:10652–10656. [Google Scholar]
- 23.Damijonaitis A, Broichhagen J, Urushima T, Hull K, Nagpal J, Laprell L, Schonberger M, Woodmansee DH, Rafiq A, Sumser MP, Kummer W, Gottschalk A, Trauner D. ACS Chem Neurosci. 2015;6:701–707. doi: 10.1021/acschemneuro.5b00030. [DOI] [PubMed] [Google Scholar]
- 24.Broichhagen J, Schonberger M, Cork SC, Frank JA, Marchetti P, Bugliani M, Shapiro AM, Trapp S, Rutter GA, Hodson DJ, Trauner D. Nat Commun. 2014;5:5116. doi: 10.1038/ncomms6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Szymanski W, Ourailidou ME, Velema WA, Dekker FJ, Feringa BL. Chemistry. 2015;21:16517–16524. doi: 10.1002/chem.201502809. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reis SA, Ghosh B, Hendricks JA, Szantai-Kis DM, Tork L, Ross KN, Lamb J, Read-Button W, Zheng B, Wang H, Salthouse C, Haggarty SJ, Mazitschek R. Nat Chem Biol. 2016;12:317–323. doi: 10.1038/nchembio.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weston CE, Kramer A, Colin F, Yildiz O, Baud MG, Meyer-Almes FJ, Fuchter MJ. ACS Infect Dis. 2017;3:152–161. doi: 10.1021/acsinfecdis.6b00148. [DOI] [PubMed] [Google Scholar]
- 27.Hansen MJ, Velema WA, de Bruin G, Overkleeft HS, Szymanski W, Feringa BL. ChemBioChem. 2014;15:2053–2057. doi: 10.1002/cbic.201402237. [DOI] [PubMed] [Google Scholar]
- 28.Dumontet C, Jordan MA. Nat Rev Drug Discovery. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griggs J, Metcalfe JC, Hesketh R. Lancet Oncol. 2001;2:82–87. doi: 10.1016/S1470-2045(00)00224-2. [DOI] [PubMed] [Google Scholar]
- 30.Borowiak M, Nahaboo W, Reynders M, Nekolla K, Jalinot P, Hasserodt J, Rehberg M, Delattre M, Zahler S, Vollmar A, Trauner D, Thorn-Seshold O. Cell. 2015;162:403–411. doi: 10.1016/j.cell.2015.06.049. [DOI] [PubMed] [Google Scholar]
- 31.Almena M, Merida I. Trends Biochem Sci. 2011;36:593–603. doi: 10.1016/j.tibs.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Colon-Gonzalez F, Kazanietz MG. Biochim Biophys Acta Mol Cell Biol Lipids. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Frank JA, Moroni M, Moshourab R, Sumser M, Lewin GR, Trauner D. Nat Commun. 2015;6:7118. doi: 10.1038/ncomms8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frank JA, Yushchenko DA, Hodson DJ, Lipstein N, Nagpal J, Rutter GA, Rhee JS, Gottschalk A, Brose N, Schultz C, Trauner D. Nat Chem Biol. 2016;12:755–762. doi: 10.1038/nchembio.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadler A, Reither G, Feng S, Stein F, Reither S, Muller R, Schultz C. Angew Chem Int Ed. 2013;52:6330–6334. doi: 10.1002/anie.201301716. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:6455–6459. [Google Scholar]
- 36.Nomura W, Narumi T, Ohashi N, Serizawa Y, Lewin NE, Blumberg PM, Furuta T, Tamamura H. ChemBioChem. 2011;12:535–539. doi: 10.1002/cbic.201000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imamura F, Horai T, Mukai M, Shinkai K, Sawada M, Akedo H. Biochem Biophys Res Commun. 1993;193:497–503. doi: 10.1006/bbrc.1993.1651. [DOI] [PubMed] [Google Scholar]
- 38.Hövelmann F, Kedziora KM, Nadler A, Müller R, Jalink K, Schultz C. Cell Chem Biol. 2016;23:629–634. doi: 10.1016/j.chembiol.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 39.Hansen MJ, Velema WA, Lerch MM, Szymanski W, Feringa BL. Chem Soc Rev. 2015;44:3358–3377. doi: 10.1039/c5cs00118h. [DOI] [PubMed] [Google Scholar]
- 40.Eyre DW, Cule ML, Griffiths D, Crook DW, Peto TE, Walker AS, Wilson DJ. PLoS Comput Biol. 2013;9:e1003059. doi: 10.1371/journal.pcbi.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Velema WA, van der Berg JP, Szymanski W, Driessen AJ, Feringa BL. ACS Chem Biol. 2014;9:1969–1974. doi: 10.1021/cb500313f. [DOI] [PubMed] [Google Scholar]
- 42.Priestman MA, Sun L, Lawrence DS. ACS Chem Biol. 2011;6:377–384. doi: 10.1021/cb100398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.a) Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Nat Neurosci. 2012;15:423–430. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]; Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Nat Neurosci. 2012;15:s421–s423. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]; b) Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GC, Higley MJ. Science. 2013;340:759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, Kasai H. Nat Neurosci. 2013;16:1409–1416. doi: 10.1038/nn.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Amatrudo JM, Olson JP, Agarwal HK, Ellis-Davies GCR. Eur J Neurosci. 2015;41:5–16. doi: 10.1111/ejn.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lerch MM, Hansen MJ, Velema WA, Szymanski W, Feringa BL. Nat Commun. 2016;7:12054. doi: 10.1038/ncomms12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fournier L, Gauron C, Xu L, Aujard I, Le Saux T, Gagey-Eilstein N, Maurin S, Dubruille S, Baudin JB, Bensimon D, Volovitch M, Vriz S, Jullien L. ACS Chem Biol. 2013;8:1528–1536. doi: 10.1021/cb400178m. [DOI] [PubMed] [Google Scholar]
- 46.a) Sinha DK, Neveu P, Gagey N, Aujard I, Le Saux T, Rampon C, Gauron C, Kawakami K, Leucht C, Bally-Cuif L, Volovitch M, Bensimon D, Jullien L, Vriz S. Zebrafish. 2010;7:199–204. doi: 10.1089/zeb.2009.0632. [DOI] [PubMed] [Google Scholar]; b) Sinha DK, Neveu P, Gagey N, Aujard I, Benbrahim-Bouzidi C, Le Saux T, Rampon C, Gauron C, Goetz B, Dubruille S, Baaden M, Volovitch M, Bensimon D, Vriz S, Jullien L. ChemBioChem. 2010;11:653–663. doi: 10.1002/cbic.201000008. [DOI] [PubMed] [Google Scholar]
- 47.Neveu P, Aujard I, Benbrahim C, Le Saux T, Allemand JF, Vriz S, Bensimon D, Jullien L. Angew Chem Int Ed. 2008;47:3744–3746. doi: 10.1002/anie.200800037. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:3804–3806. [Google Scholar]
- 48.Yamazoe S, Liu Q, McQuade LE, Deiters A, Chen JK. Angew Chem Int Ed. 2014;53:10114–10118. doi: 10.1002/anie.201405355. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:10278–10282. [Google Scholar]
- 49.a) Casi G, Neri D. J Controlled Release. 2012;161:422–428. doi: 10.1016/j.jconrel.2012.01.026. [DOI] [PubMed] [Google Scholar]; b) Iyer U, Kadambi VJ. J Pharmacol Toxicol Methods. 2011;64:207–212. doi: 10.1016/j.vascn.2011.07.005. [DOI] [PubMed] [Google Scholar]; c) Senter PD. Curr Opin Chem Biol. 2009;13:235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]; d) Jerjian TV, Glode AE, Thompson LA, O’Bryant CL. Pharmacotherapy. 2016;36:99–116. doi: 10.1002/phar.1687. [DOI] [PubMed] [Google Scholar]
- 50.Nani RR, Gorka AP, Nagaya T, Kobayashi H, Schnermann MJ. Angew Chem Int Ed. 2015;54:13635–13638. doi: 10.1002/anie.201507391. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2015;127:13839–13842. [Google Scholar]
- 51.Gorka AP, Nani RR, Zhu J, Mackem S, Schnermann MJ. J Am Chem Soc. 2014;136:14153–14159. doi: 10.1021/ja5065203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 53.Li WH, Zheng G. Photochem Photobiol Sci. 2012;11:460–471. doi: 10.1039/c2pp05342j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banala S, Maurel D, Manley S, Johnsson K. ACS Chem Biol. 2012;7:289–293. doi: 10.1021/cb2002889. [DOI] [PubMed] [Google Scholar]
- 55.Lee HL, Lord SJ, Iwanaga S, Zhan K, Xie H, Williams JC, Wang H, Bowman GR, Goley ED, Shapiro L, Twieg RJ, Rao J, Moerner WE. J Am Chem Soc. 2010;132:15099–15101. doi: 10.1021/ja1044192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.a) Anzalone AV, Chen Z, Cornish VW. Chem Commun. 2016;52:9442–9445. doi: 10.1039/c6cc04882j. [DOI] [PubMed] [Google Scholar]; b) Vaughan JC, Jia S, Zhuang X. Nat Methods. 2012;9:1181–1184. doi: 10.1038/nmeth.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mbatia HW, Burdette SC. Biochemistry. 2012;51:7212–7224. doi: 10.1021/bi3001769. [DOI] [PubMed] [Google Scholar]
- 58.Kaplan JH, Ellis-Davies GC. Proc Natl Acad Sci USA. 1988;85:6571–6575. doi: 10.1073/pnas.85.17.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agarwal HK, Janicek R, Chi SH, Perry JW, Niggli E, Ellis-Davies GC. J Am Chem Soc. 2016;138:3687–3693. doi: 10.1021/jacs.5b11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ciesienski KL, Hyman LM, Yang DT, Haas KL, Dickens MG, Holbrook RJ, Franz KJ. Eur J Inorg Chem. 2010:2224–2228. [Google Scholar]
- 61.Basa PN, Antala S, Dempski RE, Burdette SC. Angew Chem Int Ed. 2015;54:13027–13031. doi: 10.1002/anie.201505778. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2015;127:13219–13223. [Google Scholar]
- 62.Gwizdala C, Burdette SC. Curr Opin Chem Biol. 2013;17:137–142. doi: 10.1016/j.cbpa.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 63.Baker AS, Deiters A. ACS Chem Biol. 2014;9:1398–1407. doi: 10.1021/cb500176x. [DOI] [PubMed] [Google Scholar]
- 64.a) Lawrence DS. Curr Opin Chem Biol. 2005;9:570–575. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]; b) Hahn ME, Muir TW. Trends Biochem Sci. 2005;30:26–34. doi: 10.1016/j.tibs.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 65.Gautier A, Nguyen DP, Lusic H, An W, Deiters A, Chin JW. J Am Chem Soc. 2010;132:4086–4088. doi: 10.1021/ja910688s. [DOI] [PubMed] [Google Scholar]
- 66.a) Hemphill J, Chou C, Chin JW, Deiters A. J Am Chem Soc. 2013;135:13433–13439. doi: 10.1021/ja4051026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chou C, Young DD, Deiters A. ChemBioChem. 2010;11:972–977. doi: 10.1002/cbic.201000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.a) Edwards WF, Young DD, Deiters A. ACS Chem Biol. 2009;4:441–445. doi: 10.1021/cb900041s. [DOI] [PubMed] [Google Scholar]; b) Luo J, Arbely E, Zhang J, Chou C, Uprety R, Chin JW, Deiters A. Chem Commun. 2016;52:8529–8532. doi: 10.1039/c6cc03934k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ren W, Ji A, Ai H-w. J Am Chem Soc. 2015;137:2155–2158. doi: 10.1021/ja508597d. [DOI] [PubMed] [Google Scholar]
- 69.Nguyen DP, Mahesh M, Elsässer SJ, Hancock SM, Uttamapinant C, Chin JW. J Am Chem Soc. 2014;136:2240–2243. doi: 10.1021/ja412191m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walker OS, Elsässer SJ, Mahesh M, Bachman M, Balasubramanian S, Chin JW. J Am Chem Soc. 2016;138:718–721. doi: 10.1021/jacs.5b07627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.a) Arbely E, Torres-Kolbus J, Deiters A, Chin JW. J Am Chem Soc. 2012;134:11912–11915. doi: 10.1021/ja3046958. [DOI] [PubMed] [Google Scholar]; b) Lahiri S, Seidel R, Engelhard M, Becker CFW. Mol BioSyst. 2010;6:2423–2429. doi: 10.1039/c0mb00019a. [DOI] [PubMed] [Google Scholar]
- 72.a) Dar AC, Shokat KM. Annu Rev Biochem. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]; b) Knight ZA, Shokat KM. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 73.Gautier A, Deiters A, Chin JW. J Am Chem Soc. 2011;133:2124–2127. doi: 10.1021/ja1109979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roberts PJ, Der CJ. [Google Scholar]
- 75.Cirit M, Wang C-C, Haugh JM. J Biol Chem. 2010;285:36736–36744. doi: 10.1074/jbc.M110.148759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.a) Luo J, Kong M, Liu L, Samanta S, Van Houten B, Deiters A. ChemBioChem. 2017;18:466–469. doi: 10.1002/cbic.201600624. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liaunardy-Jopeace A, Murton BL, Mahesh M, Chin JW, James JR. Nature Struct Mol Biol. 2017;24:1155–1163. doi: 10.1038/nsmb.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luo J, Uprety R, Naro Y, Chou C, Nguyen DP, Chin JW, Deiters A. J Am Chem Soc. 2014;136:15551–15558. doi: 10.1021/ja5055862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.a) Gaj T, Gersbach CA, Barbas CF., III Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM. Acta Naturae. 2014;6:19–40. [PMC free article] [PubMed] [Google Scholar]
- 79.a) Maeder ML, Gersbach CA. Mol Ther. 2016;24:430–446. doi: 10.1038/mt.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cox DBT, Platt RJ, Zhang F. Nat Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chou C, Deiters A. Angew Chem Int Ed. 2011;50:6839–6842. doi: 10.1002/anie.201101157. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2011;123:6971–6974. [Google Scholar]
- 81.Hemphill J, Borchardt EK, Brown K, Asokan A, Deiters A. J Am Chem Soc. 2015;137:5642–5645. doi: 10.1021/ja512664v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.a) Sternberg SH, Doudna JA. Mol Cell. 2015;58:568–574. doi: 10.1016/j.molcel.2015.02.032. [DOI] [PubMed] [Google Scholar]; b) Shalem O, Sanjana NE, Zhang F. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.a) Nihongaki Y, Kawano F, Nakajima T, Sato M. Nat Biotechnol. 2015;33:755–760. doi: 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]; b) Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M. Chem Biol. 2015;22:169–174. doi: 10.1016/j.chembiol.2014.12.011. [DOI] [PubMed] [Google Scholar]; c) Zhou XX, Zou X, Chung HK, Gao Y, Liu Y, Qi LS, Lin MZ. ACS Chem Biol. 2017 doi: 10.1021/acschem-bio.7b00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polstein LR, Gersbach CA. Nat Chem Biol. 2015;11:198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jain PK, Ramanan V, Schepers AG, Dalvie NS, Panda A, Fleming HE, Bhatia SN. Angew Chem Int Ed. 2016;55:12440–12444. doi: 10.1002/anie.201606123. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2016;128:12628–12632. [Google Scholar]
- 86.Li H, Hah J-M, Lawrence DS. J Am Chem Soc. 2008;130:10474–10475. doi: 10.1021/ja803395d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.a) Janett E, Bernardinelli Y, Müller D, Bochet CG. Bioconjugate Chem. 2015;26:2408–2418. doi: 10.1021/acs.bioconjchem.5b00473. [DOI] [PubMed] [Google Scholar]; b) Lee HM, Xu W, Lawrence DS. J Am Chem Soc. 2011;133:2331–2333. doi: 10.1021/ja108950q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sainlos M, Iskenderian-Epps WS, Olivier NB, Choquet D, Imperiali B. J Am Chem Soc. 2013;135:4580–4583. doi: 10.1021/ja309870q. [DOI] [PubMed] [Google Scholar]; d) Mancini RJ, Stutts L, Moore T, Esser-Kahn AP. Angew Chem Int Ed. 2015;54:5962–5965. doi: 10.1002/anie.201500416. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) O’Banion CP, Nguyen LT, Wang Q, Priestman MA, Holly SP, Parise LV, Lawrence DS. Angew Chem Int Ed. 2016;55:950–954. doi: 10.1002/anie.201508767. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2016;128:962–966. [Google Scholar]
- 88.Decaneto E, Abbruzzetti S, Heise I, Lubitz W, Viappiani C, Knipp M. Photochem Photobiol Sci. 2015;14:300–307. doi: 10.1039/c4pp00297k. [DOI] [PubMed] [Google Scholar]
- 89.a) Goubko CA, Basak A, Majumdar S, Jarrell H, Khieu NH, Cao X. J Biomed Mater Res Part A. 2013;101A:787–796. doi: 10.1002/jbm.a.34381. [DOI] [PubMed] [Google Scholar]; b) Wirkner M, Weis S, Miguel VS, Álvarez M, Gropeanu RA, Salierno M, Sartoris A, Unger RE, Kirkpatrick CJ, del Campo A. ChemBioChem. 2011;12:2623–2629. doi: 10.1002/cbic.201100437. [DOI] [PubMed] [Google Scholar]; c) Miller DS, Chirayil S, Ball HL, Luebke KJ. ChemBio-Chem. 2009;10:577–584. doi: 10.1002/cbic.200800679. [DOI] [PubMed] [Google Scholar]; d) Petersen S, Alonso JM, Specht A, Duodu P, Goeldner M, del Campo A. Angew Chem Int Ed. 2008;47:3192–3195. doi: 10.1002/anie.200704857. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:3236–3239. [Google Scholar]
- 90.a) Goguen BN, Aemissegger A, Imperiali B. J Am Chem Soc. 2011;133:11038–11041. doi: 10.1021/ja2028074. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Azagarsamy MA, Anseth KS. Angew Chem Int Ed. 2013;52:13803–13807. doi: 10.1002/anie.201308174. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:14048–14052. [Google Scholar]
- 91.Jacobson K, Rajfur Z, Vitriol E, Hahn K. Trends Cell Biol. 2008;18:443–450. doi: 10.1016/j.tcb.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.a) Müller BK, Jay DG, Bonhoeffer F. Curr Biol. 1996;6:1497–1502. doi: 10.1016/s0960-9822(96)00754-3. [DOI] [PubMed] [Google Scholar]; b) Schröder R, Jay DG, Tautz D. Mech Dev. 1999;80:191–195. doi: 10.1016/s0925-4773(98)00211-1. [DOI] [PubMed] [Google Scholar]; c) Schröder R, Tautz D, Jay DG. Dev Genes Evol. 1996;206:86–88. doi: 10.1007/s004270050034. [DOI] [PubMed] [Google Scholar]
- 93.Sano Y, Watanabe W, Matsunaga S. J Cell Sci. 2014;127:1621. doi: 10.1242/jcs.144527. [DOI] [PubMed] [Google Scholar]
- 94.a) Yogo T, Urano Y, Mizushima A, Sunahara H, Inoue T, Hirose K, Iino M, Kikuchi K, Nagano T. Proc Natl Acad Sci USA. 2008;105:28–32. doi: 10.1073/pnas.0611717105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sato S, Morita K, Nakamura H. Bioconjugate Chem. 2015;26:250–256. doi: 10.1021/bc500518t. [DOI] [PubMed] [Google Scholar]; c) Joyner JC, Hocharoen L, Cowan JA. J Am Chem Soc. 2012;134:3396–3410. doi: 10.1021/ja208791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee J, Udugamasooriya DG, Lim H-S, Kodadek T. Nat Chem Biol. 2010;6:258–260. doi: 10.1038/nchembio.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu X, Dix M, Speers AE, Bachovchin DA, Zuhl AM, Cravatt BF, Kodadek TJ. ChemBioChem. 2012;13:2082–2093. doi: 10.1002/cbic.201200445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keppler A, Ellenberg J. ACS Chem Biol. 2009;4:127–138. doi: 10.1021/cb800298u. [DOI] [PubMed] [Google Scholar]
- 98.Takemoto K, Matsuda T, McDougall M, Klaubert DH, Hasegawa A, Los GV, Wood KV, Miyawaki A, Nagai T. ACS Chem Biol. 2011;6:401–406. doi: 10.1021/cb100431e. [DOI] [PubMed] [Google Scholar]
- 99.Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, Lukyanov KA. Nat Biotechnol. 2006;24:95–99. doi: 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- 100.a) Takemoto K, Matsuda T, Sakai N, Fu D, Noda M, Uchiyama S, Kotera I, Arai Y, Horiuchi M, Fukui K, Ayabe T, Inagaki F, Suzuki H, Nagai T. Sci Rep. 2013;3:2629. doi: 10.1038/srep02629. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wojtovich AP, Foster TH. Redox Biol. 2014;2:368–376. doi: 10.1016/j.redox.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toettcher JE, Voigt CA, Weiner OD, Lim WA. Nat Methods. 2011;8:35–38. doi: 10.1038/nmeth.f.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yi JJ, Wang H, Vilela M, Danuser G, Hahn KM. ACS Synth Biol. 2014;3:788–795. doi: 10.1021/sb5001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.a) Yumerefendi H, Lerner AM, Zimmerman SP, Hahn K, Bear JE, Strahl BD, Kuhlman B. Nat Chem Biol. 2016;12:399–401. doi: 10.1038/nchembio.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Levskaya A, Weiner OD, Lim WA, Voigt CA. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Spiltoir JI, Strickland D, Glotzer M, Tucker CL. ACS Synth Biol. 2016;5:554–560. doi: 10.1021/acssynbio.5b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.a) Konermann S, Brigham MD, Trevino A, Hsu PD, Heidenreich M, Le C, Platt RJ, Scott DA, Church GM, Zhang F. Nature. 2013 doi: 10.1038/nature12466. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Polstein LR, Gersbach CA. J Am Chem Soc. 2012;134:16480–16483. doi: 10.1021/ja3065667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kawano F, Okazaki R, Yazawa M, Sato M. Nat Chem Biol. 2016;12:1059–1064. doi: 10.1038/nchembio.2205. [DOI] [PubMed] [Google Scholar]
- 107.Kravitz AV, Freeze BS, Parker PRL, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.James DA, Burns DC, Woolley GA. Protein Eng. 2001;14:983–991. doi: 10.1093/protein/14.12.983. [DOI] [PubMed] [Google Scholar]
- 109.Yamada MD, Nakajima Y, Maeda H, Maruta S. J Biochem. 2007;142:691–698. doi: 10.1093/jb/mvm183. [DOI] [PubMed] [Google Scholar]
- 110.a) Schierling B, Noël AJ, Wende W, Volkov E, Kubareva E, Oretskaya T, Kokkinidis M, Römpp A, Spengler B, Pingoud A. Proc Natl Acad Sci USA. 2010;107:1361–1366. doi: 10.1073/pnas.0909444107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Umeki N, Yoshizawa T, Sugimoto Y, Mitsui T, Wakabayashi K, Maruta S. J Biochem. 2004;136:839–846. doi: 10.1093/jb/mvh194. [DOI] [PubMed] [Google Scholar]; c) Caamaño AM, Vázquez ME, Martínez-Costas J, Castedo L, Mascareñas JL. Angew Chem Int Ed. 2000;39:3104–3107. doi: 10.1002/1521-3773(20000901)39:17<3104::aid-anie3104>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2000;112:3234–3237. [Google Scholar]; d) Zhang F, Zarrine-Afsar A, Al-Abdul-Wahid MS, Prosser RS, Davidson AR, Woolley GA. J Am Chem Soc. 2009;131:2283–2289. doi: 10.1021/ja807938v. [DOI] [PubMed] [Google Scholar]; e) Blacklock FKM, Yachnin BJ, Woolley GA, Khare SD. J Am Chem Soc. 2018;140:14–17. doi: 10.1021/jacs.7b08709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bose M, Groff D, Xie J, Brustad E, Schultz PG. J Am Chem Soc. 2006;128:388–389. doi: 10.1021/ja055467u. [DOI] [PubMed] [Google Scholar]
- 112.Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, Wang L. Angew Chem Int Ed. 2014;53:3932–3936. doi: 10.1002/anie.201400001. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:4013–4017. [Google Scholar]
- 113.Hoppmann C, Maslennikov I, Choe S, Wang L. J Am Chem Soc. 2015;137:11218–11221. doi: 10.1021/jacs.5b06234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reiner A, Isacoff EY. In: Photoswitching Proteins: Methods and Protocols. Cambridge S, editor. Springer; New York: 2014. pp. 45–68. [Google Scholar]
- 115.Broichhagen J, Trauner D. Curr Opin Chem Biol. 2014;21:121–127. doi: 10.1016/j.cbpa.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 116.a) Bartels E, Wassermann NH, Erlanger BF. Proc Natl Acad Sci USA. 1971;68:1820–1823. doi: 10.1073/pnas.68.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sandoz G, Levitz J, Kramer RH, Isacoff EY. Neuron. 2012;74:1005–1014. doi: 10.1016/j.neuron.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Volgraf M, Gorostiza P, Numano R, Kramer RH, Isacoff EY, Trauner D. Nat Chem Biol. 2006;2:47–52. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH. Nat Neurosci. 2004;7:1381–1386. doi: 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Szobota S, Gorostiza P, Del Bene F, Wyart C, Fortin DL, Kolstad KD, Tulyathan O, Volgraf M, Numano R, Aaron HL, Scott EK, Kramer RH, Flannery J, Baier H, Trauner D, Isacoff EY. Neuron. 2007;54:535–545. doi: 10.1016/j.neuron.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 118.Broichhagen J, Damijonaitis A, Levitz J, Sokol KR, Leippe P, Konrad D, Isacoff EY, Trauner D. ACS Cent Sci. 2015;1:383–393. doi: 10.1021/acscentsci.5b00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hinner MJ, Johnsson K. Curr Opin Biotechnol. 2010;21:766–776. doi: 10.1016/j.copbio.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 120.a) Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Nat Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tsai YH, Essig S, James JR, Lang K, Chin JW. Nat Chem. 2015;7:554–561. doi: 10.1038/nchem.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.a) Knie C, Utecht M, Zhao F, Kulla H, Kovalenko S, Brouwer AM, Saalfrank P, Hecht S, Bléger D. Chem Eur J. 2014;20:16492–16501. doi: 10.1002/chem.201404649. [DOI] [PubMed] [Google Scholar]; b) Bléger D, Schwarz J, Brouwer AM, Hecht S. J Am Chem Soc. 2012;134:20597–20600. doi: 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]; c) Beharry AA, Sadovski O, Woolley GA. J Am Chem Soc. 2011;133:19684–19687. doi: 10.1021/ja209239m. [DOI] [PubMed] [Google Scholar]
- 122.Boulégue C, Löweneck M, Renner C, Moroder L. Chem-BioChem. 2007;8:591–594. doi: 10.1002/cbic.200600495. [DOI] [PubMed] [Google Scholar]
- 123.Turner NC, Reis-Filho JS. [Google Scholar]
- 124.Mainz ER, Wang Q, Lawrence DS, Allbritton NL. Angew Chem. 2016;128:13289–13292. doi: 10.1002/anie.201606914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.González-Vera JA, Morris MC. Proteomes. 2015;3:369–410. doi: 10.3390/proteomes3040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Proctor A, Herrera-Loeza SG, Wang Q, Lawrence DS, Yeh JJ, Allbritton NL. Anal Chem. 2014;86:4573–4580. doi: 10.1021/ac500616q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.a) Kolch W, Pitt A. Nat Rev Cancer. 2010;10:618–629. doi: 10.1038/nrc2900. [DOI] [PubMed] [Google Scholar]; b) Fleuren EDG, Zhang L, Wu J, Daly RJ. Nat Rev Cancer. 2016;16:83–98. doi: 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]
- 128.a) Endoh T, Sisido M, Ohtsuki T. Nucleic Acids Symp Ser. 2007;51:127–128. doi: 10.1093/nass/nrm064. [DOI] [PubMed] [Google Scholar]; b) Endoh T, Sisido M, Ohtsuki T. Bioconjugate Chem. 2008;19:1017–1024. doi: 10.1021/bc800020n. [DOI] [PubMed] [Google Scholar]; c) Endoh T, Sisido M, Ohtsuki T. J Controlled Release. 2009;137:241–245. doi: 10.1016/j.jconrel.2009.04.015. [DOI] [PubMed] [Google Scholar]; d) Matsushita-Ishiodori Y, Kuwabara R, Sakakoshi H, Endoh T, Ohtsuki T. Bioconjugate Chem. 2011;22:2222–2226. doi: 10.1021/bc200095a. [DOI] [PubMed] [Google Scholar]
- 129.Govan JM, Uprety R, Thomas M, Lusic H, Lively MO, Deiters A. ACS Chem Biol. 2013;8:2272–2282. doi: 10.1021/cb400293e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.a) Yang Y, Yang Y, Xie X, Wang Z, Gong W, Zhang H, Li Y, Yu F, Li Z, Mei X. Biomaterials. 2015;48:84–96. doi: 10.1016/j.biomaterials.2015.01.030. [DOI] [PubMed] [Google Scholar]; b) Xie X, Yang Y, Yang Y, Mei X. J Drug Targeting. 2015;23:789–799. doi: 10.3109/1061186X.2015.1009077. [DOI] [PubMed] [Google Scholar]
- 131.Mizukami S, Hosoda M, Satake T, Okada S, Hori Y, Furuta T, Kikuchi K. J Am Chem Soc. 2010;132:9524–9525. doi: 10.1021/ja102167m. [DOI] [PubMed] [Google Scholar]
- 132.a) Yang Y, Yang Y, Xie X, Cai X, Wang Z, Gong W, Zhang H, Li Y, Mei X. Colloids Surf B. 2015;128:427–438. doi: 10.1016/j.colsurfb.2015.02.041. [DOI] [PubMed] [Google Scholar]; b) Xie X, Yang Y, Yang Y, Zhang H, Li Y, Mei X. Drug Delivery. 2016;23:2445–2456. doi: 10.3109/10717544.2015.1008707. [DOI] [PubMed] [Google Scholar]
- 133.Jain PK, Karunakaran D, Friedman SH. Angew Chem Int Ed. 2013;52:1404–1409. doi: 10.1002/anie.201207264. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:1444–1449. [Google Scholar]
- 134.Sarode BR, Kover K, Tong PY, Zhang C, Friedman SH. Mol Pharmaceutics. 2016;13:3835–3841. doi: 10.1021/acs.molpharmaceut.6b00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.a) Sharma VK, Rungta P, Prasad AK. RSC Adv. 2014;4:16618–16631. [Google Scholar]; b) Reautschnig P, Vogel P, Stafforst T. RNA Biol. 2017;14:651–668. doi: 10.1080/15476286.2016.1208323. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bennett CF, Swayze EE. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 136.a) Lubbe AS, Szymanski W, Feringa BL. Chem Soc Rev. 2017;46:1052–1079. doi: 10.1039/c6cs00461j. [DOI] [PubMed] [Google Scholar]; b) Ruble BK, Yeldell SB, Dmochowski IJ. J Inorg Biochem. 2015;150:182–188. doi: 10.1016/j.jinorgbio.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Richards JL, Tang X, Turetsky A, Dmochowski IJ. Bioorg Med Chem Lett. 2008;18:6255–6258. doi: 10.1016/j.bmcl.2008.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.a) Tomasini AJ, Schuler AD, Zebala JA, Mayer AN. Genesis. 2009;47:736–743. doi: 10.1002/dvg.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tallafuss A, Gibson D, Morcos P, Li Y, Seredick S, Eisen J, Washbourne P. Development. 2012;139:1691–1699. doi: 10.1242/dev.072702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.a) Zheng G, Cochella L, Liu J, Hobert O, Li W-h. ACS Chem Biol. 2011;6:1332–1338. doi: 10.1021/cb200290e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Griepenburg JC, Ruble BK, Dmochowski IJ. Bioorg Med Chem. 2013;21:6198–6204. doi: 10.1016/j.bmc.2013.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tang X, Swaminathan J, Gewirtz AM, Dmochowski IJ. Nucleic Acids Res. 2008;36:559–569. doi: 10.1093/nar/gkm1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.a) Shestopalov IA, Sinha S, Chen JK. Nat Chem Biol. 2007;3:650–651. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]; b) Ouyang X, Shestopalov IA, Sinha S, Zheng G, Pitt CLW, Li WH, Olson AJ, Chen JK. J Am Chem Soc. 2009;131:13255–13269. doi: 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.a) Griepenburg JC, Rapp TL, Carroll PJ, Eberwine J, Dmochowski IJ. Chem Sci. 2015;6:2342–2346. doi: 10.1039/c4sc03990d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu L, Wang Y, Wu J, Lv C, Wang J, Tang X. Nucleic Acids Res. 2013;41:677–686. doi: 10.1093/nar/gks996. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang Y, Wu L, Wang P, Lv C, Yang Z, Tang X. Nucleic Acids Res. 2012;40:11155–11162. doi: 10.1093/nar/gks840. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yamazoe S, Shestopalov IA, Provost E, Leach SD, Chen JK. Angew Chem Int Ed. 2012;51:6908–6911. doi: 10.1002/anie.201201690. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:7014–7017. [Google Scholar]; e) Seyfried P, Eiden L, Grebenovsky N, Mayer G, Heckel A. Angew Chem Int Ed. 2017;56:359–363. doi: 10.1002/anie.201610025. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2017;129:365–369. [Google Scholar]
- 143.a) Monroe WT, McQuain MM, Chang MS, Alexander JS, Haselton FR. J Biol Chem. 1999;274:20895–20900. doi: 10.1074/jbc.274.30.20895. [DOI] [PubMed] [Google Scholar]; b) Ando H, Furuta T, Tsien RY, Okamoto H. Nat Genet. 2001;28:317–325. doi: 10.1038/ng583. [DOI] [PubMed] [Google Scholar]; c) Shah S, Rangarajan S, Friedman SH. Angew Chem Int Ed. 2005;44:1328–1332. doi: 10.1002/anie.200461458. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2005;117:1352–1356. [Google Scholar]
- 144.a) Chaulk SG, MacMillan AM. Nucleic Acids Res. 1998;26:3173–3178. doi: 10.1093/nar/26.13.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chaulk SG, MacMillan AM. Angew Chem Int Ed. 2001;40:2149–2152. [PubMed] [Google Scholar]; Angew Chem. 2001;113:2207–2210. [Google Scholar]
- 145.a) Usui K, Aso M, Fukuda M, Suemune H. J Org Chem. 2008;73:241–248. doi: 10.1021/jo702080r. [DOI] [PubMed] [Google Scholar]; b) Anhäuser L, Muttach F, Rentmeister A. Chem Commun. 2018 doi: 10.1039/c7cc08300a. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 146.a) Young DD, Lusic H, Lively MO, Yoder JA, Deiters A. ChemBioChem. 2008;9:2937–2940. doi: 10.1002/cbic.200800627. [DOI] [PubMed] [Google Scholar]; b) Deiters A, Garner RA, Lusic H, Govan JM, Dush M, Nascone-Yoder NM, Yoder JA. J Am Chem Soc. 2010;132:15644–15650. doi: 10.1021/ja1053863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.a) Schäfer F, Wagner J, Knau A, Dimmeler S, Heckel A. Angew Chem Int Ed. 2013;52:13558–13561. doi: 10.1002/anie.201307502. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:13801–13805. [Google Scholar]; b) Connelly CM, Deiters A. In: Cancer Cell Signaling: Methods and Protocols. Robles-Flores M, editor. Springer; New York: 2014. pp. 99–114. [Google Scholar]; c) Connelly CM, Uprety R, Hemphill J, Deiters A. Mol BioSyst. 2012;8:2987–2993. doi: 10.1039/c2mb25175b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hemphill J, Liu Q, Uprety R, Samanta S, Tsang M, Juliano RL, Deiters A. J Am Chem Soc. 2015;137:3656–3662. doi: 10.1021/jacs.5b00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.a) Young DD, Edwards WF, Lusic H, Lively MO, Deiters A. Chem Commun. 2008:462–464. doi: 10.1039/b715152g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanaka K, Katada H, Shigi N, Kuzuya A, Komiyama M. ChemBioChem. 2008;9:2120–2126. doi: 10.1002/cbic.200800285. [DOI] [PubMed] [Google Scholar]
- 150.a) Prokup A, Hemphill J, Deiters A. J Am Chem Soc. 2012;134:3810–3815. doi: 10.1021/ja210050s. [DOI] [PubMed] [Google Scholar]; b) Govan JM, Young DD, Lively MO, Deiters A. Tetrahedron Lett. 2015;56:3639–3642. doi: 10.1016/j.tetlet.2015.01.165. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Govan JM, Uprety R, Hemphill J, Lively MO, Deiters A. ACS Chem Biol. 2012;7:1247–1256. doi: 10.1021/cb300161r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Govan JM, Lively MO, Deiters A. J Am Chem Soc. 2011;133:13176–13182. doi: 10.1021/ja204980v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Young DD, Lively MO, Deiters A. J Am Chem Soc. 2010;132:6183–6193. doi: 10.1021/ja100710j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bobbin ML, Rossi JJ. Annu Rev Pharmacol Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- 153.Matsushita-Ishiodori Y, Ohtsuki T. Acc Chem Res. 2012;45:1039–1047. doi: 10.1021/ar200227n. [DOI] [PubMed] [Google Scholar]
- 154.a) Nguyen QN, Chavli RV, Marques JT, Conrad PG, II, Wang D, He W, Belisle BE, Zhang A, Pastor LM, Witney FR, Morris M, Heitz F, Divita G, Williams BRG, McMaster GK. Biochim Biophys Acta Biomembr. 2006;1758:394–403. doi: 10.1016/j.bbamem.2006.01.003. [DOI] [PubMed] [Google Scholar]; b) Shah S, Friedman SH. Oligonucleotides. 2007;17:35– 43. doi: 10.1089/oli.2006.0067. [DOI] [PubMed] [Google Scholar]
- 155.Jain PK, Shah S, Friedman SH. J Am Chem Soc. 2011;133:440–446. doi: 10.1021/ja107226e. [DOI] [PubMed] [Google Scholar]
- 156.Ji Y, Yang J, Wu L, Yu L, Tang X. Angew Chem Int Ed. 2016;55:2152–2156. doi: 10.1002/anie.201510921. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2016;128:2192–2196. [Google Scholar]
- 157.Mikat V, Heckel A. RNA. 2007;13:2341–2347. doi: 10.1261/rna.753407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Govan JM, Young DD, Lusic H, Liu Q, Lively MO, Deiters A. Nucleic Acids Res. 2013;41:10518–10528. doi: 10.1093/nar/gkt806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 160.Volvert M-L, Prévot P-P, Close P, Laguesse S, Pirotte S, Hemphill J, Rogister F, Kruzy N, Sacheli R, Moonen G, Deiters A, Merkenschlager M, Chariot A, Malgrange B, Godin JD, Nguyen L. Cell Rep. 2014;7:1168–1183. doi: 10.1016/j.celrep.2014.03.075. [DOI] [PubMed] [Google Scholar]
- 161.a) Watanabe T, Hoshida T, Sakyo J, Kishi M, Tanabe S, Matsuura J, Akiyama S, Nakata M, Tanabe Y, Suzuki AZ, Watanabe S, Furuta T. Org Biomol Chem. 2014;12:5089–5093. doi: 10.1039/c4ob00418c. [DOI] [PubMed] [Google Scholar]; b) Guha S, Graf J, Göricke B, Diederichsen U. J Pept Sci. 2013;19:415–422. doi: 10.1002/psc.2514. [DOI] [PubMed] [Google Scholar]; c) Stafforst T, Hilvert D. Angew Chem Int Ed. 2010;49:9998–10001. doi: 10.1002/anie.201004548. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2010;122:10195–10198. [Google Scholar]
- 162.Summerton J, Weller D. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 163.Eisen JS, Smith JC. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- 164.a) Shestopalov IA, Chen JK. Methods Cell Biol. 2011;104:151–172. doi: 10.1016/B978-0-12-374814-0.00009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Payumo AY, McQuade LE, Walker WJ, Yamazoe S, Chen JK. Nat Chem Biol. 2016;12:694–701. doi: 10.1038/nchembio.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yamazoe S, McQuade LE, Chen JK. ACS Chem Biol. 2014;9:1985–1990. doi: 10.1021/cb500429u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tang X, Su M, Yu L, Lv C, Wang J, Li Z. Nucleic Acids Res. 2010;38:3848–3855. doi: 10.1093/nar/gkq079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.a) Weinberg ES, Allende ML, Kelly CS, Abdelhamid A, Murakami T, Andermann P, Doerre OG, Grunwald DJ, Riggleman B. Development. 1996;122:271. doi: 10.1242/dev.122.1.271. [DOI] [PubMed] [Google Scholar]; b) Amacher SL, Kimmel CB. Development. 1998;125:1397. doi: 10.1242/dev.125.8.1397. [DOI] [PubMed] [Google Scholar]
- 168.Ferguson DP, Schmitt EE, Lightfoot JT. PLoS One. 2013;8:e61472. doi: 10.1371/journal.pone.0061472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jain A, Magistri M, Napoli S, Carbone GM, Catapano CV. Biochimie. 2010;92:317–320. doi: 10.1016/j.biochi.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 170.Schleifman EB, Chin JY, Glazer PM. In: Chromosomal Mutagenesis. Davis GD, Kayser KJ, editors. Humana; Totowa: 2008. pp. 175–190. [Google Scholar]
- 171.Yang N, Singh S, Mahato RI. J Controlled Release. 2011;155:326–330. doi: 10.1016/j.jconrel.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kemme CA, Nguyen D, Chattopadhyay A, Iwahara J. Transcription. 2016;7:115–120. doi: 10.1080/21541264.2016.1188873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Struntz NB, Harki DA. ACS Chem Biol. 2016;11:1631–1638. doi: 10.1021/acschembio.6b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kamensek U, Sersa G, Vidic S, Tevz G, Kranjc S, Cemazar M. Mol Imaging Biol. 2011;13:43–52. doi: 10.1007/s11307-010-0300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Alberini CM. Physiol Rev. 2009:89. doi: 10.1152/physrev.00017.2008. https://doi.org/10.1152/physrev.00017.02008. [DOI] [PMC free article] [PubMed]
- 176.Hemphill J, Govan J, Uprety R, Tsang M, Deiters A. J Am Chem Soc. 2014;136:7152–7158. doi: 10.1021/ja500327g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vogel P, Schneider MF, Wettengel J, Stafforst T. Angew Chem Int Ed. 2014;53:6267–6271. doi: 10.1002/anie.201402634. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:6382–6386. [Google Scholar]
- 178.Hanswillemenke A, Kuzdere T, Vogel P, Jékely G, Stafforst T. J Am Chem Soc. 2015;137:15875–15881. doi: 10.1021/jacs.5b10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Huang C, Wu G, Yu Y-T. Nat Protoc. 2012;7:789–800. doi: 10.1038/nprot.2012.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhao X, Yu Y-T. Nat Methods. 2008;5:95–100. doi: 10.1038/nmeth1142. [DOI] [PubMed] [Google Scholar]
- 181.Ordoukhanian P, Taylor J-S. J Am Chem Soc. 1995;117:9570–9571. [Google Scholar]
- 182.Zhang DY, Winfree E. J Am Chem Soc. 2009;131:17303–17314. doi: 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- 183.Hemphill J, Deiters A. J Am Chem Soc. 2013;135:10512–10518. doi: 10.1021/ja404350s. [DOI] [PubMed] [Google Scholar]
- 184.a) Huang F, You M, Han D, Xiong X, Liang H, Tan W. J Am Chem Soc. 2013;135:7967–7973. doi: 10.1021/ja4018495. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prokup A, Hemphill J, Liu Q, Deiters A. ACS Synth Biol. 2015;4:1064–1069. doi: 10.1021/sb500279w. [DOI] [PubMed] [Google Scholar]
- 185.Tan W, Wang K, Drake TJ. Curr Opin Chem Biol. 2004;8:547–553. doi: 10.1016/j.cbpa.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 186.Wang C, Zhu Z, Song Y, Lin H, Yang CJ, Tan W. Chem Commun. 2011;47:5708–5710. doi: 10.1039/c1cc10481k. [DOI] [PubMed] [Google Scholar]
- 187.Joshi KB, Vlachos A, Mikat V, Deller T, Heckel A. Chem Commun. 2012;48:2746–2748. doi: 10.1039/c2cc16654b. [DOI] [PubMed] [Google Scholar]
- 188.Qiu L, Wu C, You M, Han D, Chen T, Zhu G, Jiang J, Yu R, Tan W. J Am Chem Soc. 2013;135:12952–12955. doi: 10.1021/ja406252w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Mol Ther Nucleic Acids. 2014;3:e182. doi: 10.1038/mtna.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tan Z, Feagin TA, Heemstra JM. J Am Chem Soc. 2016;138:6328–6331. doi: 10.1021/jacs.6b00934. [DOI] [PubMed] [Google Scholar]
- 191.a) Heckel A, Mayer G. J Am Chem Soc. 2005;127:822–823. doi: 10.1021/ja043285e. [DOI] [PubMed] [Google Scholar]; b) Pinto A, Lennarz S, Rodrigues-Correia A, Heckel A, O’Sullivan CK, Mayer G. ACS Chem Biol. 2012;7:360–366. doi: 10.1021/cb2003835. [DOI] [PubMed] [Google Scholar]; c) Mayer G, Lohberger A, Butzen S, Pofahl M, Blind M, Heckel A. Bioorg Med Chem Lett. 2009;19:6561–6564. doi: 10.1016/j.bmcl.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 192.a) Li Y, Shi J, Luo Z, Jiang H, Chen X, Wang F, Wu X, Guo Q. Bioorg Med Chem Lett. 2009;19:5368–5371. doi: 10.1016/j.bmcl.2009.07.128. [DOI] [PubMed] [Google Scholar]; b) Li YM, Shi J, Cai R, Chen X, Luo ZF, Guo QX. J Photochem Photobiol A. 2010;211:129–134. [Google Scholar]
- 193.Kim Y, Phillips JA, Liu H, Kang H, Tan W. Proc Natl Acad Sci USA. 2009;106:6489–6494. doi: 10.1073/pnas.0812402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Buff MCR, Schäfer F, Wulffen B, Müller J, Pötzsch B, Heckel A, Mayer G. Nucleic Acids Res. 2010;38:2111–2118. doi: 10.1093/nar/gkp1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pinto A, Polo PN, Rubio MJ, Svobodova M, Lerga TM, O’Sullivan CK. In: Nucleic Acid Aptamers: Selection, Characterization, and Application. Mayer G, editor. Springer; New York: 2016. pp. 171–177. [Google Scholar]
- 196.Civit L, Pinto A, Rodrigues-Correia A, Heckel A, O’Sullivan CK, Mayer G. Methods. 2016;97:104–109. doi: 10.1016/j.ymeth.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 197.Fokina AA, Stetsenko DA, François J-C. Expert Opin Biol Ther. 2015;15:689–711. doi: 10.1517/14712598.2015.1025048. [DOI] [PubMed] [Google Scholar]
- 198.a) Richards JL, Seward GK, Wang Y-H, Dmochowski IJ. ChemBioChem. 2010;11:320–324. doi: 10.1002/cbic.200900702. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lusic H, Young DD, Lively MO, Deiters A. Org Lett. 2007;9:1903–1906. doi: 10.1021/ol070455u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hwang K, Wu P, Kim T, Lei L, Tian S, Wang Y, Lu Y. Angew Chem Int Ed. 2014;53:13798–13802. doi: 10.1002/anie.201408333. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:14018–14022. [Google Scholar]
- 200.Wang X, Feng M, Xiao L, Tong A, Xiang Y. ACS Chem Biol. 2016;11:444–451. doi: 10.1021/acschembio.5b00867. [DOI] [PubMed] [Google Scholar]
- 201.a) Ozsolak F, Milos PM. Nat Rev Genet. 2011;12:87. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou CY, Alexander SC, Devaraj NK. Chem Sci. 2017;8:7169–7173. doi: 10.1039/c7sc03150e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Saliba A-E, Westermann AJ, Gorski SA, Vogel J. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, Francis C, Spaethling JM, Wolf JA, Grady MS, Ulyanova AV, Yeldell SB, Griepenburg JC, Buckley PT, Kim J, Sul J-Y, Dmochowski IJ, Eberwine J. Nat Methods. 2014;11:190–196. doi: 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Donato L, Mourot A, Davenport CM, Herbivo C, Warther D, Léonard J, Bolze F, Nicoud J-F, Kramer RH, Goeldner M, Specht A. Angew Chem Int Ed. 2012;51:1840–1843. doi: 10.1002/anie.201106559. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:1876–1879. [Google Scholar]
- 205.Fichte MAH, Weyel XMM, Junek S, Schäfer F, Herbivo C, Goeldner M, Specht A, Wachtveitl J, Heckel A. Angew Chem Int Ed. 2016;55:8948–8952. doi: 10.1002/anie.201603281. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2016;128:9094–9098. [Google Scholar]
- 206.a) Benninger RKP, Piston DW. Current protocols in cell biology/editorial board, Juan S. Bonifacino [et al.] 2013;0 4(Unit-4.1124) [Google Scholar]; b) Miller MJ, Wei SH, Parker I, Cahalan MD. Science. 2002;296:1869. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]; c) Belluscio L. Current Protocols in Neuroscience. Wiley; Chichester: 2001. [Google Scholar]
- 207.Wang F, Liu X, Willner I. Angew Chem Int Ed. 2015;54:1098–1129. doi: 10.1002/anie.201404652. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:1112–1144. [Google Scholar]
- 208.a) Kashida H, Doi T, Sakakibara T, Hayashi T, Asanuma H. J Am Chem Soc. 2013;135:7960–7966. doi: 10.1021/ja401835j. [DOI] [PubMed] [Google Scholar]; b) Kawai H, Murayama K, Kashida H, Asanuma H. Chem Eur J. 2016;22:10533–10538. doi: 10.1002/chem.201602006. [DOI] [PubMed] [Google Scholar]; c) Sakamoto T, Fujimoto K. Modified Nucleic Acids. Springer; Cham: 2016. pp. 145–157. [Google Scholar]
- 209.a) Goldau T, Murayama K, Brieke C, Asanuma H, Heckel A. Chem Eur J. 2015;21:17870–17876. doi: 10.1002/chem.201503303. [DOI] [PubMed] [Google Scholar]; b) Goldau T, Murayama K, Brieke C, Steinwand S, Mondal P, Biswas M, Burghardt I, Wachtveitl J, Asanuma H, Heckel A. Chem Eur J. 2015;21:2845–2854. doi: 10.1002/chem.201405840. [DOI] [PubMed] [Google Scholar]; c) Ito H, Liang X, Nishioka H, Asanuma H. Org Biomol Chem. 2010;8:5519–5524. doi: 10.1039/c0ob00432d. [DOI] [PubMed] [Google Scholar]; d) Nishioka H, Liang X, Asanuma H. Chem Eur J. 2010;16:2054–2062. doi: 10.1002/chem.200902789. [DOI] [PubMed] [Google Scholar]; e) Nakasone Y, Ooi H, Kamiya Y, Asanuma H, Terazima M. J Am Chem Soc. 2016;138:9001–9004. doi: 10.1021/jacs.6b02525. [DOI] [PubMed] [Google Scholar]
- 210.a) Zhou M, Liang X, Mochizuki T, Asanuma H. Angew Chem Int Ed. 2010;49:2167–2170. doi: 10.1002/anie.200907082. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2010;122:2213–2216. [Google Scholar]; b) Liang X, Zhou M, Kato K, Asanuma H. ACS Synth Biol. 2013;2:194–202. doi: 10.1021/sb300120n. [DOI] [PubMed] [Google Scholar]
- 211.Cahová H, Jäschke A. Angew Chem Int Ed. 2013;52:3186–3190. doi: 10.1002/anie.201209943. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:3268–3272. [Google Scholar]
- 212.Ogasawara S. ChemBioChem. 2014;15:2652–2655. doi: 10.1002/cbic.201402495. [DOI] [PubMed] [Google Scholar]
- 213.Ogasawara S. ACS Chem Biol. 2017;12:351–356. doi: 10.1021/acschembio.6b00684. [DOI] [PubMed] [Google Scholar]
- 214.Schmidt R, Geissler D, Hagen V, Bendig J. J Phys Chem A. 2007;111:5768–5774. doi: 10.1021/jp071521c. [DOI] [PubMed] [Google Scholar]
- 215.Momotake A, Lindegger N, Niggli E, Barsotti RJ, Ellis-Davies GC. Nat Methods. 2006;3:35–40. doi: 10.1038/nmeth821. [DOI] [PubMed] [Google Scholar]
- 216.Fournier L, Aujard I, Le Saux T, Maurin S, Beaupierre S, Baudin JB, Jullien L. Chemistry. 2013;19:17494–17507. doi: 10.1002/chem.201302630. [DOI] [PubMed] [Google Scholar]
- 217.Olson JP, Kwon H-B, Takasaki KT, Chiu CQ, Higley MJ, Sabatini BL, Ellis-Davies GCR. J Am Chem Soc. 2013;135:5954–5957. doi: 10.1021/ja4019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shell TA, Lawrence DS. Acc Chem Res. 2015;48:2866–2874. doi: 10.1021/acs.accounts.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Shell TA, Shell JR, Rodgers ZL, Lawrence DS. Angew Chem Int Ed. 2014;53:875–878. doi: 10.1002/anie.201308816. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:894–897. [Google Scholar]
- 220.Smith WJ, Oien NP, Hughes RM, Marvin CM, Rodgers ZL, Lee J, Lawrence DS. Angew Chem Int Ed. 2014;53:10945–10948. doi: 10.1002/anie.201406216. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:11125–11128. [Google Scholar]
- 221.Atilgan A, Ecik ET, Guliyev R, Uyar TB, Erbas-Cakmak S, Akkaya EU. Angew Chem Int Ed. 2014;53:10678–10681. doi: 10.1002/anie.201405462. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:10854–10857. [Google Scholar]
- 222.Arian D, Kovbasyuk L, Mokhir A. J Am Chem Soc. 2011;133:3972–3980. doi: 10.1021/ja108819c. [DOI] [PubMed] [Google Scholar]
- 223.Meyer A, Schikora M, Mokhir A. Chem Commun. 2015;51:13324–13326. doi: 10.1039/c5cc01805f. [DOI] [PubMed] [Google Scholar]
- 224.König SG, Mokhir A. Bioorg Med Chem Lett. 2013;23:6544–6548. doi: 10.1016/j.bmcl.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 225.Meyer A, Mokhir A. Angew Chem Int Ed. 2014;53:12840–12843. doi: 10.1002/anie.201405885. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:13054–13057. [Google Scholar]
- 226.Sgambellone MA, David A, Garner RN, Dunbar KR, Turro C. J Am Chem Soc. 2013;135:11274–11282. doi: 10.1021/ja4045604. [DOI] [PubMed] [Google Scholar]
- 227.Olson JP, Banghart MR, Sabatini BL, Ellis-Davies GCR. J Am Chem Soc. 2013;135:15948–15954. doi: 10.1021/ja408225k. [DOI] [PMC free article] [PubMed] [Google Scholar]