

Abstract
SALL2 is a poorly characterized transcription factor that belongs to the Spalt‐like family involved in development. Mutations on SALL2 have been associated with ocular coloboma and cancer. In cancers, SALL2 is deregulated and is proposed as a tumor suppressor in ovarian cancer. SALL2 has been implicated in stemness, cell death, proliferation, and quiescence. However, mechanisms underlying roles of SALL2 related to cancer remain largely unknown. Here, we investigated the role of SALL2 in cell proliferation using mouse embryo fibroblasts (MEFs) derived from Sall2 −/− mice. Compared to Sall2 +/+ MEFs, Sall2 −/− MEFs exhibit enhanced cell proliferation and faster postmitotic progression through G1 and S phases. Accordingly, Sall2 −/− MEFs exhibit higher mRNA and protein levels of cyclins D1 and E1. Chromatin immunoprecipitation and promoter reporter assays showed that SALL2 binds and represses CCND1 and CCNE1 promoters, identifying a novel mechanism by which SALL2 may control cell cycle. In addition, the analysis of tissues from Sall2 +/+ and Sall2 −/− mice confirmed the inverse correlation between expression of SALL2 and G1‐S cyclins. Consistent with an antiproliferative function of SALL2, immortalized Sall2 −/− MEFs showed enhanced growth rate, foci formation, and anchorage‐independent growth, confirming tumor suppressor properties for SALL2. Finally, cancer data analyses show negative correlations between SALL2 and G1‐S cyclins’ mRNA levels in several cancers. Altogether, our results demonstrated that SALL2 is a negative regulator of cell proliferation, an effect mediated in part by repression of G1‐S cyclins’ expression. Our results have implications for the understanding and significance of SALL2 role under physiological and pathological conditions.
Keywords: cell cycle, cell proliferation, cyclin D1, cyclin E1, SALL2, tumorigenesis
Abbreviations
- AP1
activator protein 1
- ATF3
activating transcription factor 3
- BrdU
5‐bromo‐2′‐deoxyuridine
- CCND1
cyclin D1 gene
- CCNE1
cyclin E1 gene
- CDK2
cyclin‐dependent kinase 2
- CDK4/6
cyclin‐dependent kinase 4/6
- CDK
cyclin‐dependent kinase
- CDKN1A
cyclin‐dependent kinase inhibitor 1A gene
- CDKN2A
cyclin‐dependent kinase inhibitor 2A gene
- ChIP
chromatin immunoprecipitation
- c‐MYC
v‐Myc avian myelocytomatosis viral oncogene homolog
- E1A
exon 1A
- E1
exon 1
- G1/S
Gap1/synthesis
- G1
Gap1
- G2‐M
Gap2‐mitosis
- HDAC
histone deacetylase
- HOSE
human ovarian epithelial cells
- HPV16 E6
human papillomavirus type 16 E6
- iMEFs
immortalized mouse embryonic fibroblasts
- LOH
loss of heterozygocity
- MEFs
mouse embryonic fibroblasts
- NFAT
nuclear factor of activated T cells
- p16INK4a
cyclin‐dependent kinase inhibitor 2A
- P1
promoter 1
- p21WAF/CIP
cyclin‐dependent kinase inhibitor 1 or CDK interacting protein 1
- P2
promoter 2
- p53
p53 tumor suppressor protein
- Ppib
peptidylprolyl isomerase B
- pRb
retinoblastoma protein
- SALL2
Spalt‐like 2
- SALL
Spalt‐like
- SKOV3
SK‐ovary adenocarcinoma cells
- SV40
simian virus 40
- SWI/SNF
SwItch/sucrose nonfermenting
- WT1
Wilms tumor 1
- ZO‐2
zonula occludens‐2
1. Introduction
SALL2 belongs to the Spalt‐like family of transcription factors conserved from nematodes to humans (de Celis and Barrio, 2009; Sweetman and Munsterberg, 2006). The gene contains two alternative promoters (P1 and P2), which originate two protein isoforms, named SALL2‐E1 and SALL2‐E1A (Ma et al., 2001). These two isoforms differ in the first 25 amino acids (Ma et al., 2001), and those amino acids are particularly relevant as SALL2‐E1 contains a 12 amino acids conserved motif involved in transcriptional repression through its interaction with the NuRD complex (Lauberth and Rauchman, 2006). Both SALL2 isoforms contain several C2H2 zinc finger motifs along the structure, and several glutamine‐, serine‐, and proline‐rich regions, which are typically present in transcription factors (Hermosilla et al., 2017). E1 is restricted to certain tissues such as thymus, testis, and colon, while E1A has ubiquitous expression. Both isoforms are highly expressed in the brain (Kohlhase et al., 1996, 2000; Ma et al., 2001).
SALL2 has been associated with neurogenesis, neuronal differentiation, and eye development. Consequently, SALL2/Sall2 deficiency associates with neural tube defects in mice, and with coloboma, a congenital eye disease in humans and mice (Böhm et al., 2008; Kelberman et al., 2014). Importantly, SALL2 is deregulated and/or mutated in various cancers, suggesting a role for SALL2 in the disease (Hermosilla et al., 2017). In this context, most findings suggest that SALL2 behaves as a tumor suppressor (Hermosilla et al., 2017; Sung and Yim, 2017). Like the retinoblastoma protein (pRb) and the p53 tumor suppressors (Yim and Park, 2005), SALL2 interacts with viral oncogenic proteins. These include the mouse polyomavirus large T antigen (Li et al., 2001) and the human papillomavirus type 16 E6 (HPV16 E6) protein (Parroche et al., 2011), but fails to interact with the large T antigen of simian virus 40 (SV40) (Li et al., 2004)
SALL2 has also been associated with induction of cellular quiescence in human fibroblasts (Liu et al., 2007) and with cellular apoptosis in mouse embryonic fibroblast and human leukemia cells exposed to genotoxic stress (Escobar et al., 2015). Clinical evidence showed loss of heterozygosity (LOH) at the SALL2 locus in 30% of ovarian cancer patients (Bandera et al., 1997), and recent studies demonstrated that the P2 promoter of SALL2 is susceptible to silencing by methylation (Sung et al., 2013). This epigenetic modification was confirmed in the majority of primary tumors and correlated with negative SALL2 expression in ovarian carcinomas of various histological types. Other studies indicate that lost or reduced SALL2 expression may be involved in leukemogenesis (Chai, 2011) and breast cancer (Liu et al., 2014; Zuo et al., 2017). However, SALL2 is found upregulated in Wilms tumor (Li et al., 2002), synovial sarcoma (Nielsen et al., 2003), and oral (Estilo et al., 2009) and testicular cancer (Alagaratnam et al., 2011), indicating that SALL2 role in cancer is yet controversial.
Evidence of the role of SALL2 in proliferation came mainly from overexpression experiments. Initial studies showed that forced expression of SALL2 in SKOV3 ovarian carcinoma cells inhibits DNA synthesis and increases p21WAF/CIP mRNA and protein levels (Li et al., 2004). Similarly, a microarray analysis indicates that the cyclin‐dependent kinase 4 inhibitor A (p16INK4a) gene (CDKN2A) is upregulated in SALL2‐expressing versus SALL2‐null SKOV3 ovarian carcinoma cells (Wu et al., 2015). Together, these data suggest that SALL2 inhibits cell cycle progression at the G1‐ to S‐phase transition by upregulation of cyclin‐dependent kinase inhibitors. However, few direct transcriptional target genes of SALL2 related to proliferation have been identified.
To further investigate the role of SALL2 in cell proliferation, we used primary and immortalized mouse embryonic fibroblasts (MEFs) derived from previously characterized Sall2‐deficient mice (Sato et al., 2003). Our studies showed that SALL2 is highly expressed in mitotic cells. After cell division, SALL2 protein levels are slightly reduced and then are maintained constant during progression through G1. We demonstrated that SALL2 exerts a negative regulatory role in cell proliferation associated with the regulation of cell cycle progression. We identified a novel mechanism involving the transcriptional repression of G1‐S cyclins, CCND1 and CCNE1, by SALL2. Accordingly, we observed inverse correlation between SALL2 and G1‐S cyclins levels in specific tissues, supporting their negative regulation by SALL2 in vivo. Considering that Sall2 −/− MEFs displayed transformation properties and data from R2 platform show a negative correlation between SALL2 and G1‐S cyclins mRNA expression in various cancers, our studies further support a tumor suppressor role for SALL2.
2. Materials and methods
2.1. Reagents
Propidium iodide, nocodazole (#M1404), SALL2 (#HPA004162) polyclonal antibody, protease inhibitor cocktail I (# P8340), phosphatase inhibitor cocktail II (P5726), and 5‐bromo‐2′‐deoxyuridine (# B5002) were purchased from Sigma‐Aldrich Chemicals (St. Louis, MO, USA). SALL2 antibody used for ChIP experiments was obtained from Bethyl Lab (Montgomery, TX, USA). Cyclin A (C‐19, #SC‐596) polyclonal antibody and cyclin B1 (GNS1, #SC‐245), cyclin D1 (DCS‐6, #SC‐20044), cyclin E1 (E‐4, #SC‐377100), p21 (F‐5, #6246), Myc (9E10, #SC‐40), and β‐actin (AC‐15, #SC‐69879) monoclonal antibodies were obtained from Santa Cruz Biotechnology (San Diego, CA, USA). The SV40 large T antigen expression pBSSVD2005 plasmid was a gift from David Ron (Addgene plasmid # 21826), the plasmid containing the CCNE1 promoter was a gift from Bob Weinberg (Addgene plasmid # 8458) (Geng et al., 1996), and the CCND1 promoter pGL3Basic was a gift from Frank McCormick (Addgene plasmid # 32726) (McCormick and Tetsu, 1999). pcDNA3‐SALL2 plasmid was described elsewhere (Escobar et al., 2015). Alexa Fluor 488‐conjugated phalloidin and Alexa Fluor 488‐conjugated goat anti‐rabbit secondary antibodies were purchased from Invitrogen (Carlsbad, CA, USA). Horseradish peroxidase‐conjugated secondary antibodies and Hoechst 33342 were from Bio‐Rad (Hercules, CA, USA).
2.2. Isolation of primary MEFs and genotyping
Sall2 knockout mice (Sato et al., 2003) were obtained by collaboration with Dr. Ruichi Nishinakamura (Kumamoto University, MTA (2010) to RP, Universidad de Concepción). Mice were group‐housed under standard conditions with food and water available ad libitum and were maintained on a 12‐h light/dark cycle. Mice were fed with a standard chow diet (ProLab, LabDiet, St. Louis, MO, USA) containing no less than 5% crude fat and were treated in compliance with the US National Institutes of Health guidelines for animal care and use. Studies were reviewed and approved by the Animal Ethics Committee of the Chile's National Commission for Scientific and Technological Research (CONICYT, protocol for projects # 1110821 and # 1151031).
Sall2 +/+ and Sall2 −/− fibroblasts were prepared from embryos at 13.5 days postcoitum as previously described (Escobar et al., 2015). Briefly, embryos, whose head and other red organs were removed, were smashed into pieces using a razor blade in a 100‐mm dish with 5 mL trypsin (GE Healthcare HyClone, Logan, UT, USA). The smashed embryo was incubated in trypsin for 15 min at 37 °C followed by dilution in 10 mL DMEM (GE Healthcare HyClone) by pipetting up and down. Cells were centrifuged and seeded in 100‐mm culture dishes (passage 0). MEFs were generated from independent embryos and routinely cultured as described below.
Mice were routinely genotyped by isolating tail DNA as previously reported (Escobar et al., 2015). One microliter of genomic DNA was used for PCR analysis. Sall2 PCR was performed as previously (Escobar et al., 2015) with the following oligonucleotides: forward, 5′‐CACATTTCGTGGGCTACAAG‐3′, and reverse, 5′‐CTCAGAGCTGTTTTCCTGGG‐3,′ and Neo, 5′‐GCGTTGGCTACCCGTGATAT‐3′. The sizes of the PCR products are 188 bp for the wild‐type (WT) and 380 bp for the null mutant.
2.3. Cell culture
Sall2 +/+ and Sall2 −/− primary and immortalized MEFs were cultured in DMEM supplemented with 10% heat‐inactivated fetal bovine serum (FBS, GE Healthcare HyClone), 1% glutamine (Invitrogen), and 0.5% penicillin/streptomycin (Invitrogen). Experiments with primary Sall2 +/+ and Sall2 −/− MEFs were performed with early passages (passages 3–4). Human embryonic kidney epithelial HEK293 cells (American Type Culture Collection CRL‐1573™) used for promoter reporter assays and chromatin immunoprecipitation were cultured in DMEM supplemented with 10% FBS, 1% glutamine, and 0.5 % penicillin/streptomycin.
2.4. 3T3 assays
Primary MEFs from passages 3–4 were seeded at 3 × 105 cells/60 mm dish, cell numbers were determined after 3 days, and cells were reseeded for the next passage at the starting density. This protocol was repeated between 15 and 18 times.
2.5. MEFs immortalization
Primary Sall2 +/+ and Sall2 −/− MEFs (passage 4) were immortalized using SV40 large T antigen based on modified protocol from Zhu et al. (1991). For transfection, we used Lipofectamine 2000 (Invitrogen) and 2 μg SV40 large T antigen expression vector. After cell transfection, we proceeded to select for low density. To complete the immortalization process, 5–6 post‐transfection passages were carried out.
2.6. CRISPR/Cas 9‐mediated gene targeting
HEK293 cells were electroporated with a vector encoding CRISPR/Cas9 coupled to Paprika‐RFP and harboring the following guide RNA against exon 2 of SALL2 5′ GGCTCCTTAGGCCAGACGGT 3′. Cas 9 and Paprika‐RFP genes are linked by the 2A oligopeptide sequence, allowing efficient production of the two proteins by ribosome skipping translation (Provost et al., 2007). After 16 h postelectroporation, the top 2% of the brightest cells were sorted by RFP channel and plated as individual clones. The clones were grown for 2 weeks, and western blot against SALL2 was performed in each clone for knockout identification. After selection of positive clones, genomic PCR and further sequencing confirmed CRISPR/Cas9 cut on the SALL2 locus.
2.7. Proliferation assays
Primary MEFs were seeded at 2 × 105 cells/35‐mm dish in triplicate, and cells were counted daily for 6 days. Media were replaced every second day. The immortalized Sall2 +/+ and Sall2 −/− MEFs were seeded at 1 × 104 cells/well in 6‐well dishes in triplicate, and cells were counted daily for 6 days.
2.8. Cell synchronization
Exponentially growing immortalized MEFs (iMEFs) were treated with 125 ng mL−1 nocodazole for 16 h. Except for flow cytometry (FACS) analyses where the whole cell population was collected to avoid morphological disruption, mitotic cell population was enriched by mechanic detachment (shake‐off) as described by Schorl and Sedivy (2007). Cells were released by a single rinse and subsequent incubation with fresh nocodazole‐free complete media. After nocodazole release, cells were harvested at selected times for western blot, qRT/PCR, and FACS analyses.
2.9. Flow cytometry
Approximately, 1.5 × 106 iMEFs/100‐mm dish were seeded the day before synchronization. At the moment of harvest, cells were washed in phosphate‐buffered saline (PBS). Both culture media and PBS used for rinsing the cells were collected. After PBS wash, cells were detached in 0.25% trypsin and collected in the same tube. Following centrifugation, cells were washed again with PBS, suspended in 300 μL of ice‐cold PBS, and fixed by adding 700 μL of ice‐cold 70% ethanol drop wise to the sample while vortexing, and kept at 4 °C. After fixation, cells were washed twice with PBS and incubated with 0.4 mg mL−1 RNAse (Thermo Fisher Scientific, Waltham, MA, USA) in PBS at 37 °C during 40 min. Propidium iodide was added to a final concentration of 10 μg mL−1. Cells were filtered and analyzed for DNA content on a Becton Dickinson FACSCanto II. Cell population was quantified with modfit lt 5.0.9 software (Verity Software House, Inc., Topsham, ME, USA).
2.10. BrdU incorporation experiments
Cells were synchronized as described above. After mitotic detachment, floating cells were seeded on poly‐l‐lysine‐coated coverslips and grown in nocodazole‐free medium. Cells were cultured in 50 μm BrdU‐supplemented media for 45 min prior to fixation in cold‐fixing solution (70% ethanol, 15 mm glycine; pH 2.0). Cells were rinsed twice with PBS 1X and blocked in 3% BSA‐PBS 1X solution. Coverslips were incubated with 1:20 anti‐BrdU antibody or 1 : 100 anti‐cyclin D1 (M20, #SC‐718) in incubation buffer (Roche Applied Science, Indianapolis, IN, USA) for 40 min at 37 °C. Next, cells were incubated with 1 : 500 fluorophore‐conjugated secondary antibody (Molecular Probes, Invitrogen) in 1% BSA‐PBS 1X solution. Cells were PBS‐rinsed again, and nuclei were stained with Hoechst (Bio‐Rad).
2.11. Western blot analysis
Cells were lysed in 50 mm Tris/HCl pH 7.4, 200 mm NaCl, 2.5 mm MgCl2, 1% Triton X‐100, and 10% glycerol, supplemented with protease and phosphatase inhibitor cocktails. Proteins from cell lysates (50–70 μg total protein) were fractionated by SDS/PAGE and transferred overnight at 30 mA to PVDF membrane (Immobilon, Merck, Kenilworth, NJ, USA) using a wet transfer apparatus. The PVDF membranes were blocked for 1 h at room temperature in 5% nonfat milk in TBS‐T (TBS with 0.1% Tween) and incubated overnight with an appropriate dilution of primary antibody at 4 °C. After washing, the membranes were incubated with horseradish peroxidase‐conjugated secondary antibody (Bio‐Rad) diluted in 5% nonfat milk in TBS‐T for 1 h at room temperature. Immunolabeled proteins were visualized by ECL (Pierce, Thermo Scientific, Waltham, MA, USA). For the study of protein expression in vivo, proteins were extracted from several tissues (kidney, spleen, brain, cerebellum, and liver) of 6‐week‐old Sall2 +/+ and Sall2 −/− mice. Tissues were lysed in 50 mm Tris/HCl pH 7.5, 150 mm NaCl, 2.5 mm MgCl2, 1% NP‐40, 0.1% deoxycholate, and 10% glycerol, supplemented with protease and phosphatase inhibitor cocktails. Lysates were analyzed by western blotting as described above.
2.12. Focus formation assay
Sall2 +/+ and Sall2 −/− iMEFs were seeded at 1 x 104/well in 6‐well dishes and cultured in complete medium with 10% FBS, without splitting, for 14–21 days. Media were replaced every 2 days. Confluent monolayer cultures with foci were rinsed with PBS and stained with 4 mg mL−1 crystal violet in 10% methanol. Experiments were performed in triplicate.
2.13. Anchorage‐independent growth by soft agar assay
Sall2 +/+ and Sall2 −/− iMEFs were seeded at 1.5 × 104/60‐mm dish in 1.8 % Bacto agar in DMEM supplemented with 10% FBS. Every 2 days, 0.5 mL of growth media was applied to the surface of the agar to prevent it from drying out. After incubating for 4 weeks at 37 °C in the 5% CO2 incubator, colonies were counted. Experiments were performed in triplicate.
2.14. Transient transfections and reporter gene assays
To evaluate transcriptional activity of CCNE1 and CCND1 promoters, HEK293 cells were transiently co‐transfected with 0.5 μg of each promoter cyclin (CCNx‐luc), 0.125 μg of RSV‐β‐galactosidase (β‐Gal), and 1 μg of SALL2 (pcDNA3SALL2 E1 and pcDNA3SALL2 E1A) or control vector per well. After 24 h, the transfected cells were washed with PBS, lysed with reporter assay lysis buffer (Promega, Madison, WI, USA), and spun at 14 000 × g to pellet cell debris. The supernatant was then assayed for luciferase and β‐Gal activity using the manufacturer's suggested protocols. Luminescent reporter activity was measured using a Luminometer (Victor3; Perkin‐Elmer, Waltham, MA, USA). All transfections were normalized to β‐Gal activity and performed in triplicate. Luciferase values were expressed as fold induction relative to luciferase.
2.15. ChIP assay
The assay was carried out as previously (Henriquez et al., 2011) with the following modifications: HEK293 cells (2 × 106 cells/100 mm plate) were transfected with pFLAG‐CMV2‐SALL2E1 or pFLAG‐CMV2‐SALL2E1A. To shear DNA, nuclei were sonicated in 300 μL of sonication buffer using a Misonix sonicator (model 3000) (16 times, 15 s on/20 s off each time, 6 W potency), obtaining DNA lengths between 300 and 500 bp. Immunoprecipitations were carried out overnight at 4 °C using 5 μg anti‐SALL2 (Bethyl) or 5 μg normal mouse IgG antibodies, and 40 μg of chromatin. DNA was analyzed by real‐time PCR directed to SALL2‐specific proximal regions of CCNE1 (−70/−242) and CCND1 (−22/−201) promoters. Primer sequences were forward, 5′ CTGATTCCCCGTCCCTGCG 3′, and reverse, 5′ GACATTTAAAATCCCTGCGCGC 3′, for CCCNE1; forward, 5′ TCTATGAAAACCGGACTACAGG 3′, and reverse, 5′ AAAGATCAAAGCCCGGCAGA 3′, for CCND1. In addition, a previously reported region of Pmaip1 promoter (−869/−756) (Escobar et al., 2015) was used as negative control of SALL2 binding. Primer sequences for this unrelated region (URR) were forward, 5′ TGAAGCGGCTCTCAGTAACC 3′, and reverse, 5′ AGCTACCTGGGAACGTGAAA 3′. All PCRs (KAPA SYBR FAST qPCR; Kappa Biosystems, Wilmington, MA, USA) contained 1 μL of input and 3 μL of IP samples.
2.16. Real‐time quantitative reverse transcription/PCR
Total RNA was extracted from cells with TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. RNA was treated with Turbo DNase (Invitrogen Ambion, Thermo Fisher Scientific) to eliminate any residual DNA from the preparation. Total RNA (1 μg) was reverse‐transcribed using the Maloney murine leukemia virus reverse transcriptase (Invitrogen) and 0.25 μg of Anchored Oligo(dT) 20 Primer (Invitrogen; 12577‐011). To control specificity of the amplified product, a melting curve analysis was carried out. No amplification of unspecific product was observed. Amplification of cyclophilin B (Ppib) was carried out for each sample as an endogenous control. Primer sequences were forward, 5′ GATCTCCTCCGCAGTCTGG 3′, and reverse, 5′ ACACAATGGGTATCCGGTCT 3′, for mouse Sall2 E1A; forward, 5′AACGGAGACCCCAACAGTTA 3′, and reverse, 5′ TGGGTCAGTGCAACATGAGT 3′, for mouse Sall2E1; forward, 5′ GTGCTGGGAATGCAAGCCATATCT 3′, and reverse, 5′ AAGCGGCTGGAAATGGCTTAGT 3′, for Ccne1; forward, 5′ AGGAAGCGGTCCAGGTAGTT 3′, and reverse, 5′ AGTGCGTGCAGAAGGAGATT 3′, for Ccnd1; forward, 5′ TTGTGGCCTTAGCTACAGGA 3′, and reverse, 5′ GCTCACCGTAGATGCTCTTT3′, for Ppib.
The relative expression of the Ccne1, Ccnd1, and Sall2 genes was calculated using the standard curve method, and all mRNA expressions were relative to Ppib.
3. Data accessibility
Publicly available databases from R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl) were used as materials for this study.
4. Results
4.1. Enhanced cell proliferation of primary Sall2‐deficient MEFs
To further investigate the role of SALL2 in cell proliferation, we used primary Sall2‐null mouse embryo fibroblasts (MEFs) derived from previously characterized Sall2‐deficient (Sall2 −/− ) mice (Sato et al., 2003). Because Sall2 gene could rise two isoforms of similar molecular weight (Ma et al., 2001), we first evaluated isoforms’ expression in wild‐type (Sall2 +/+) MEFs by RT/PCR. P19 mouse cells, which express both isoforms, were used as positive control. We found that Sall2‐E1A (162‐bp band) is the predominant isoform in MEFs under normal cell culture conditions, while Sall2‐E1 isoform (299‐bp band) is barely detected (Fig. 1A). Quantitative RT/PCR (qPCR) also showed that Sall2 E1A is abundant in MEFs, while E1 was undetectable (Fig. 1B). The low/absent expression of E1 isoform was additionally confirmed by analyzing transcriptome sequencing of MEFs available from public datasets (http://www.ebi.ac.uk/ena, SRX610348). A Sashimi plot depicts preferential usage of isoform E1A from E1 in MEFs (Fig. 1C).
Figure 1.
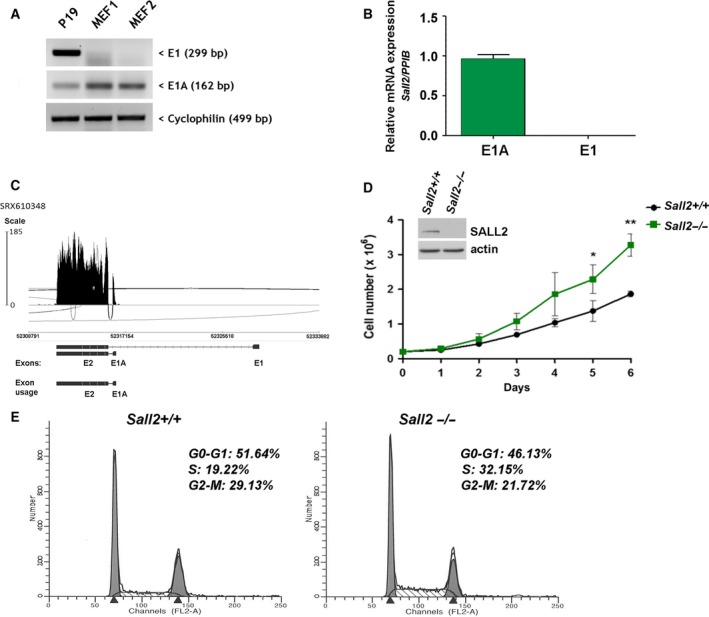
Increased proliferation of Sall2‐deficient cells. (A,B) Expression of Sall2 mRNA isoforms was evaluated in Sall2 wild‐type (Sall2 +/+) MEFs by RT/PCR (A) and qPCR (B) using Ppib as normalizer. P19 cells were used as positive control for SALL2 isoforms expression. (C) Sashimi plot (http://software.broadinstitute.org/software/igv/Sashimi) for alternatively spliced exon and flanking exon of Sall2 from deep RNA‐sequencing data of MEFs (SRX610348). Per‐base expression is plotted on y‐axis of Sashimi plot, genomic coordinates on x‐axis, and mRNA isoforms are represented on the bottom (exons in black, introns as lines). (D) Proliferation curves of Sall2 −/− and Sall2 +/+ MEFs. Equal number of MEFs (passage 3) was plated in triplicate and counted every day for 6 days. Data are expressed as mean ± SD from three independent experiments. *P ≤ 0.05, **P ≤ 0.01, Student's t‐test. (E) Cell cycle analysis of Sall2 −/− and Sall2 +/+ MEFs by flow cytometry. Asynchronous cells (passage 4) were collected, fixed, and stained with propidium iodide (PI) before cell cycle analysis. G0‐G1, S and G2‐M populations are indicated as percentages of the whole population. Figure is representative of two independent experiments of isogenic MEFs performed in triplicate.
We then compared proliferation rate of primary Sall2 +/+ and Sall2 −/− MEFs. Under normal growth conditions, Sall2 −/− cells proliferate significantly faster than Sall2 +/+ cells (Fig. 1D). Similar results were obtained from three independent isogenic Sall2 +/+ and Sall2 −/− MEFs cultures (data not shown). Furthermore, flow cytometry data analysis indicated that asynchronous Sall2 −/− MEFs present higher proportion of cells in S phase (32.2% vs. 19.2%) and lower proportion of cells in G2‐M (21.7% vs. 29.1%) than Sall2 +/+ cells (Fig. 1E).
Because of the limited lifespan of primary MEFs, we attempted to generate immortal MEFs using the 3T3 protocol (Xu, 2005); however, Sall2 deficiency did not result in spontaneous immortalization of fibroblasts. In fact, our study showed that Sall2 −/− MEFs enter senescence and have a lifespan similar to Sall2 +/+ MEFs (Fig. S1A). At passage 8, we noticed that both Sall2 −/− and Sall2 +/+ MEFs present the typical senescence phenotype consisting of enlarged cell with an increased SA‐β‐galactosidase activity (Fig. S1B). Thus, considering that SV40 large T antigen fails to interact with SALL2 (Li et al., 2004), we immortalized MEFs using this antigen‐mediated transformation (Zhu et al., 1991). Primary cells were transfected with SV40 T antigen, and after five passages using cell density selection, immortal Sall2 +/+ and Sall2 −/− MEFs were obtained. There were no obvious phenotypic differences between Sall2 +/+ and Sall2 −/− iMEFs grown for 24 h in complete medium (Fig. S2A,B). According to previous data on the role of SALL2 in the proliferation of cancer cells (Li et al., 2004), and with our data on primary MEFs (Fig. 1), Sall2 −/− iMEFs also showed higher rate of proliferation compared to the Sall2 +/+ counterpart (Fig. S2C). Together, these results indicate that Sall2‐deficiency increases cell proliferation and suggest that SALL2 E1A negatively regulates cell proliferation in embryonic fibroblasts.
4.2. Sall2 deficiency accelerates postmitotic progression into G1 and S phases
Because of the proliferative advantage of Sall2‐deficient MEFs, we investigated the expression profile of SALL2 during cell cycle progression. iMEFs were synchronized in mitosis using nocodazole. Mitotic iMEFs were harvested and then reseeded in complete medium for cell cycle re‐entry into G1. Mitotic synchronization and progression into G1 and S phases were monitored by FACS analysis and expression of specific cell cycle markers (cyclins B, D, E, and A). About 80% of Sall2 +/+ and Sall2 −/− iMEFs are arrested in mitosis after nocodazole treatment (t = 0), consistent with the high levels of cyclin B1 protein (M‐phase marker) (Fig. 2A–C). After nocodazole release (2–6 h), the percentage of mitotic Sall2 −/− and Sall2 +/+ iMEFs progressively decreases to about 20% and 50%, while the percentage of cells in G1 increases to about 50% and 20%, respectively, showing accelerated postmitotic progression of Sall2 −/− iMEF into G1 phase. Consequently, progression beyond G1/S phase transition is evident in Sall2 −/− iMEFs by 4–12 h. At 12 h, the fraction of Sall2 −/− cells in S phase increases to 60%, whereas Sall2 +/+ iMEFs only reach to 40% (Fig. 2A,B). The increase in the percentage of Sall2 −/− iMEFs in S phase was also confirmed by BrdU incorporation experiments (Fig. S3).
Figure 2.
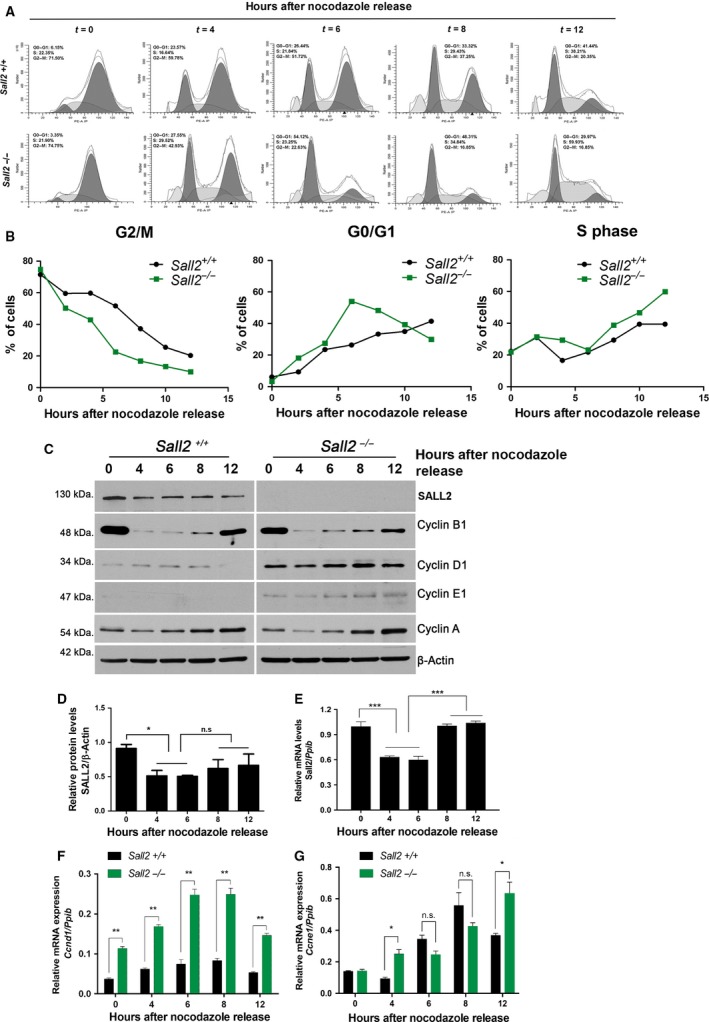
Sall2 deficiency accelerates G1‐/S‐phase transition and correlates with increased levels of cyclins D1 and E1. Sall2 +/+ and Sall2 −/− iMEFs were synchronized at G2‐M by nocodazole as described in materials and methods, and then released into the cell cycle. (A) Cell cycle analysis of Sall2 +/+ and Sall2 −/− iMEFs by flow cytometry. The graphs show representative cell cycle profiles of each iMEFs over time (hours) after release from nocodazole. (B) Progression of Sall2 +/+ iMEFs through the cell cycle was compared with that of Sall2 −/− iMEFs every 2 h for a total of 12 h. Data are presented as the percentage of cells at G2‐M, G1, and S phases over time after nocodazole release and are representative of four independent experiments. (C) Sall2 +/+ and Sall2 −/− iMEFs were harvested at various times after release to evaluate SALL2 protein expression and compare the expression of the indicated cyclins by western blot. β‐actin was used as normalizer. Figure is representative of four independent experiments. (D) Graph shows relative SALL2 protein level in Sall2 +/+ iMEFs after various hours from nocodazole release. SALL2 protein level was determined by densitometric analysis and normalized to the corresponding β‐actin level. Data are expressed as mean ± SD from four independent experiments. *P ≤ 0.05, one‐way ANOVA; n.s, not significant. (E) Sall2 mRNA expression was evaluated by qPCR analysis using Ppib as normalizer. Data are expressed as mean ± SD from three independent experiments performed in triplicate. ***P ≤ 0.001, one‐way ANOVA. (F,G) RNA was isolated from Sall2 +/+ and Sall2 −/− iMEFs and mRNA levels of Ccnd1 (F) and Ccne1 (G) were measured by qPCR. Numbers are relative to Ppib. Data are expressed as mean ± SD from three independent experiments performed in triplicate. *P ≤ 0.05; **P ≤ 0.01, Student's t‐test; n.s, not significant.
Interestingly, SALL2 protein is highly expressed in mitotic Sall2 +/+ iMEFs (Fig. 2C,D). After cell division (4 h), SALL2 protein levels are slightly reduced and then are maintained constant between 6 and 12 h (Fig. 2C,D). Quantification of Sall2 mRNA showed a similar decrease between 4 and 6 h, but it significantly increases over time (8–12 h) (Fig. 2E), suggesting differences between the behavior of SALL2 protein and mRNA at latter times of nocodazole release. Together, these results suggest that SALL2 regulates postmitotic progression into G1 as well as progression from G1 to S phases.
4.3. Cyclin D1 and E1 transcripts are increased in Sall2‐deficient cells
To understand the mechanism underlying SALL2 regulatory role in cell cycle progression, we compared the expression of specific cell cycle markers associated with the control of postmitotic progression into G1 and S phases by western blot (Fig. 2C). Whereas not obvious difference for the expression of cyclin B1 (mitotic marker) and cyclin A (S‐phase marker) was noticed between Sall2 +/+ and Sall2 −/− iMEFs, a consistent upregulation of cyclin D1 (G1 phase) and cyclin E1 (S phase entry) was evident in Sall2 −/− compared to Sall2 +/+ iMEFs (Fig. 2C).
Considering that protein expression of cyclins during cell cycle is regulated at transcriptional level (Klein and Assoian, 2008; Möröy and Geisen, 2004; Suryadinata et al., 2010), we evaluated changes in mRNA levels of cyclins D1 (Ccnd1) and E1 (Ccne1) during postmitotic progression through G1 and S phase, associated with the expression of SALL2. In agreement with protein levels of cyclins D1 and E1 in both genotypes, the levels of Ccnd1 and Ccne1 mRNA were significantly higher in Sall2 −/− iMEFs compared to those in Sall2 +/+ iMEFs (Fig. 2F,G), suggesting that SALL2 could regulate expression at the transcriptional level of key factors (cyclins D1 and E1) controlling G1‐/S‐phase transition and promoting entry into S phase. These results indicate that accelerated postmitotic progression of Sall2 −/− iMEFs into G1/S phases may be explained by a functional disruption of SALL2 on transcriptional repression of cyclins D1 and E1.
To confirm SALL2 regulation of cyclins expression, we approached rescue of SALL2 expression in Sall2‐deficient cells. Because of extremely low efficiency of transfection of iMEFs, we used HEK293 cells. We knocked out SALL2 using CRISPR/Cas 9‐mediated gene targeting and then rescued SALL2 expression. Similar to the Sall2 −/− MEFs, HEK‐SALL2KO cells showed upregulation of cyclin D1 levels (Fig. S4A,B). On the other hand, rescue of SALL2 significantly decreased the levels of cyclin D1 (Fig. S4C,D). The levels of cyclin E1 were already high at time 0 and were maintained constant after nocodazole release. We were unable to detect any significant difference in the levels of cyclin E1 between the SALL2WT and SALL2KO cells (Fig. S4A,B), or by the rescue of SALL2 (Fig. S4C,D). This later result suggests that cyclin E1 protein expression is mainly controlled by other factors in these cells. Altogether, these results suggest a regulatory role for SALL2 in cell cycle progression by downregulating the expression of G1 cyclins in both mouse fibroblast and human HEK293 cells.
4.4. SALL2 transcriptionally regulates G1‐S cyclins
To investigate whether SALL2 transcriptionally regulates cyclins D1 and E1, we initially performed bioinformatic analyses of mouse and human cyclin promoters to identify putative SALL2 binding sites (Fig. 3), using a previously reported binding site matrix (consensus sequence GGG(T/C)GGG) (Gu et al., 2011) in Transcriptional Regulatory Element Database (TRED) (https://cb.utdallas.edu/cgi-bin/TRED/tred.cgi?process=home) (Jiang et al., 2007). Conservation analysis using CLUSTAL‐O indicates 64% (CCND1) and 56% (CCNE1) identities between mouse and human promoters [−2000 bp from transcription start site (+1)], with the highest conservation in the proximal promoter region (−500 bp to +1). Identity between species in the proximal region is 69% for pCCND1 and 63% for pCCNE (Fig. S5). Thus, we focused on SALL2 binding sites at the proximal promoter because those could allow the binding of SALL2 protein to interfere with the transcriptional start site. Figure 3A shows a schematic representation of human promoters. Two putative SALL2 sites are present in CCND1 promoter at positions −76 and −198 bp from transcription start site, while six putative sites are present in the proximal region of CCNE1 promoter at positions −57, −68, −123, −140, −152, and −492 bp from transcription start site (+1) (Fig. S5).
Figure 3.
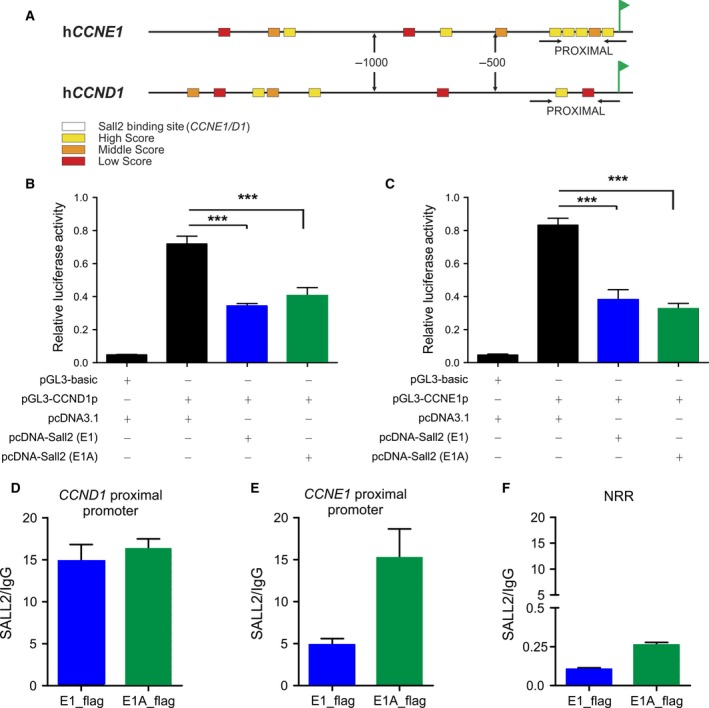
SALL2 binds and represses CCND1 and CCNE1 promoters. Bioinformatic analyses of cyclin promoters to identify putative SALL2 sites were performed using a previously reported binding site matrix (consensus sequence GGG(T/C)GGG) (Gu et al., 2011) in Transcriptional Regulatory Element Database (TRED) (https://cb.utdallas.edu/cgi-bin/TRED/tred.cgi?process=home) (Jiang et al., 2007). Sequences analyzed [−2000 bp from transcription start site (+1)] were obtained from Eukaryotic Promoter Database (EPD) and included mouse Ccnd1 (NM 007631), mouse Ccne1 (NM 007633), human CCND1 (NM 053056), and human CCNE1 (NM 001238). A. Schematic representation of human CCND1 (NM 053056) and CCNE1 (NM 001238) gene promoters. SALL2 putative binding sites are represented by square symbols. Putative SALL2 binding sites are classified as high, middle, and low scores according to their identity with the SALL2 consensus matrix (Gu et al., 2011) The transcription start site (+1) is represented by a flag. (B,C) Repression of CCND1 (B) and CCNE1 (C) promoters’ activities by SALL2. Transient co‐transfections of pGL3‐CCND1 or pGL3‐CCNE1 reporter with or without SALL2 E1 (blue bars) (or E1A, green bars) into HEK293 cells were performed as described under ‘Materials and Methods’. Luciferase activity was measured from cell lysates and normalized to β‐galactosidase activity, and promoter activity was expressed as relative luciferase units (R.L.U). pGL3 vector served as control. Data are expressed as mean ± SD from three independent experiments performed in triplicate. ***P ≤ 0.001, one‐way ANOVA. (D–F) HEK293 cells were transfected with pcDNA3‐SALL2 (E1, blue) or pcDNA3‐SALL2 (E1A, green) vector. Chromatin was immunoprecipitated 24 h after transfection using SALL2 antibody or normal rabbit IgG (control antibody), and specific genomic regions in the human CCND1 (D), CCNE1 (E), proximal promoters, and a nonrelated promoter region (NRR) were analyzed by real‐time PCR. Graphs show quantification of the amplified DNA for each immunoprecipitation relative to IgG. Results are representative of two assays performed in triplicate.
Next, we evaluated responsiveness of human cyclin promoters to SALL2 using reporters previously described: hCCND1 (McCormick and Tetsu, 1999) and hCCNE1 (Geng et al., 1996). Expression of SALL2 (E1 or E1A isoform) significantly decreased the activity of CCND1 and CCNE1 promoters (Fig. 3B,C), indicating that both SALL2 isoforms repress CCND1 and CCNE1 promoter activity.
Finally, to demonstrate in vivo the interaction of SALL2 with CCND1 and CCNE1 promoters, we performed chromatin immunoprecipitation (ChIP) assays in HEK293 cells transfected with SALL2E1 or SALL2E1A. Figure 3D–E shows that both SALL2 isoforms bind to the CCND1 and CCNE1 proximal promoters. In contrast, no binding of SALL2 is observed to a nonrelated promoter region (Fig. 3F; NRR). Together, these results demonstrated that SALL2 binds to and regulates promoters of cyclins controlling cell cycle progression from G1 to S phase.
4.5. SALL2 and G1‐S cyclins’ expression inversely correlate in vivo
It has been reported that in adult organism, Sall2 mRNA is highly expressed in the brain and to a lesser extent in heart, kidney, lung, pancreas, and ovary (Kohlhase et al., 1996, 2000; Ma et al., 2001). To evaluate the significance of SALL2‐dependent regulation of G1‐S cyclins in vivo, we compared the expression of cyclin D1 and E1 in several tissues from 6‐ to 8‐week‐old isogenic Sall2 +/+ and Sall2 −/− mice by western blot and densitometric analysis. Figure 4 shows that SALL2 levels vary between tissues. Consistent with previous reports, the high levels of SALL2 were found in the brain and cerebellum, while it was poorly expressed in other tissues. As positive control of a SALL2‐dependent target, we evaluated p21 expression (Li et al., 2004). We noticed that only the spleen showed the expected positive correlation between p21 expression and the Sall2 genotype (Fig. 4A,B). Consistent with a negative regulation of cyclins by SALL2, the levels of cyclin D1 were significantly upregulated in the liver (Fig. 4A,C) and the levels of cyclin E1 were significantly upregulated in the brain of Sall2 −/− mice (Fig. 4A,D). No significant inverse correlation was found in other tissues analyzed, suggesting that SALL2‐dependent transcriptional regulation of cyclins D1 and E1 is tissue‐specific.
Figure 4.
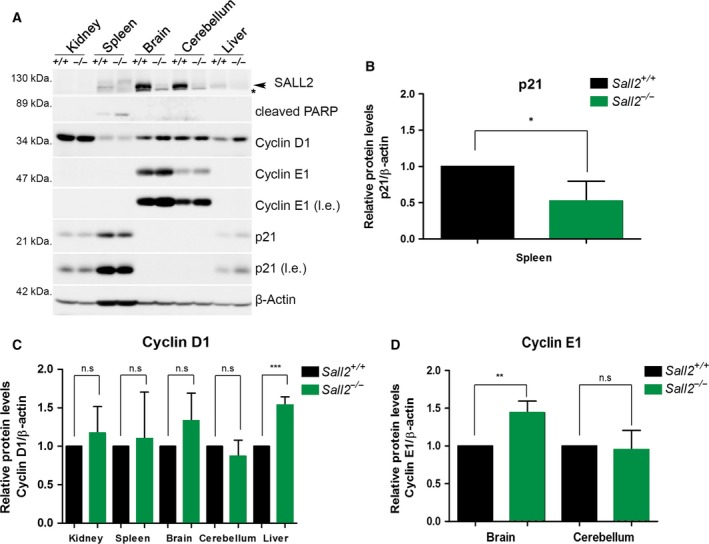
Increased expression of cyclins D1/E1 in tissues from Sall2‐deficient mice. Tissues from 6‐ to 8‐week‐old Sall2 +/+ and Sall2 −/− mice were isolated and lysed to evaluate SALL2, cyclin D1 and cyclin E1 levels by western blot analysis (A). Representative western blot of tissues analyzed. An arrow indicates SALL2, and the asterisk corresponds to nonspecific band. p21—a protein positively regulated by SALL2 (Li et al., 2004)—was used as positive control; β‐actin was used as normalizer. l.e; long exposure. (B–D) Densitometric data from western blots of p21, cyclins D1 and E1 from five isogenic mouse/genotype. Data are expressed as mean ± SD from five independent mouse tissues per genotype. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, Student′s t–test; n.s, nonsignificant.
4.6. Sall2‐deficient iMEFs possess transformation capability
Cyclin D1 and/or E1 overexpression has been associated with cancer (Hwang and Clurman, 2005; Kim and Diehl, 2009; Möröy and Geisen, 2004; Qie and Diehl, 2016). Because of the increased growth rate and expression of cyclins D1 and E1 of Sall2 −/− iMEFs, we evaluated whether these cells present enhanced transformation properties. Loss of contact inhibition was measured by focus‐forming assay. We detected transforming foci in the Sall2 −/− iMEFs within 10–12 days of culture, but not in the Sall2 +/+ iMEFs. Figure 5A shows three representative colonies (focus) of morphologically transformed Sall2 −/− iMEFs in relation to the Sall2 +/+ phenotype. Transformed cells were highly retractile and grew in irregular patterns with occasional balls or stellate patterns, particularly after prolonged incubation. After 16–18 days of culture, cells were fixed and stained to score the number of foci. Sall2 −/− iMEFs presented an average of 33 foci/plate versus the 10 foci/plate observed in Sall2 +/+ iMEFs (Fig. 5B). We also performed soft agar colony formation assay to monitor anchorage‐independent growth. Primary Sall2 +/+ MEFs were used as negative control, and colony formation was compared between Sall2 −/− and Sall2 +/+ iMEFs. Sall2 +/+ iMEFs grew few colonies, while Sall2 −/− iMEFs significantly increased anchorage‐independent growth (Fig. 5C). These results confirm the malignant transformation properties of Sall2‐deficient cells and support a tumor suppressor role for SALL2.
Figure 5.
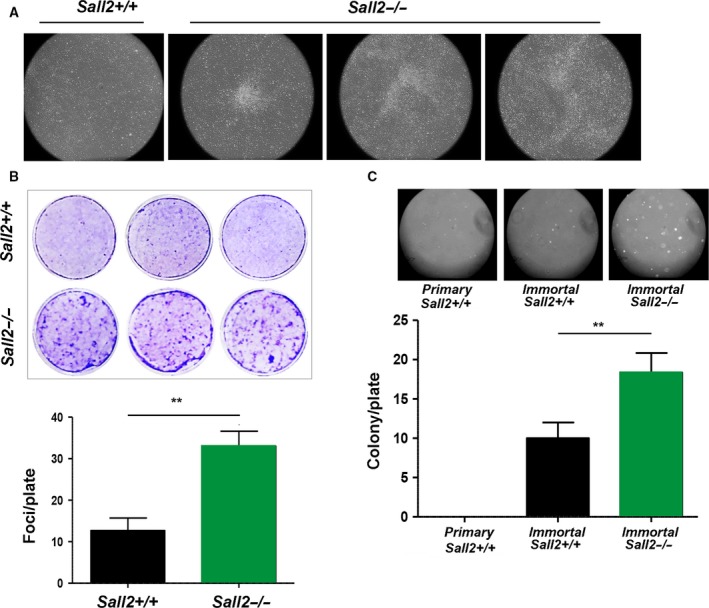
Immortalized Sall2‐deficient cells possess transforming ability. (A,B) Foci formation assay. iMEFs were grown in regular culture medium for 12–18 days prior to staining with crystal violet. (A) Microscopic visualization of three individual focus from Sall2 −/− iMEFs photographed at 4× magnification. The appearance of the Sall2 +/+ iMEFs culture (left) is shown for comparison.(B) Top, crystal violet staining of Sall2 +/+ and Sall2 −/− iMEFs. Bottom, quantification of number of foci per plate. Results are representative of three independent experiments performed in triplicate (**P ≤ 0.01, Student's t‐test). (C) Sall2 −/− MEFs showed increased anchorage‐independent growth. Sall2 +/+ and Sall2 −/− iMEFs were grown in soft agar for 3–4 weeks. Top, colonies were photographed at 4× magnification. Primary Sall2 +/+ MEFs were used as negative control. Bottom, quantification of number of colonies per plate. Results are representative of three independent experiments performed in triplicate (**P ≤ 0.01, Student's t‐test).
4.7. SALL2 and CCND1/E1 genes’ expression inversely correlate in cancer
To assess correlation between SALL2 and CCND1/E1 genes’ expression in cancer, we used R2: Genomic Analysis and Visualization Platform (http://r2.amc.nl). The R2 platform allows analysis of multiple gene expression microarrays from various pathological conditions. Datasets from different types of cancer were used to investigate correlation between SALL2 and CCND1/E1 genes’ expression (Table S1). Tumor types were selected based on previous studies reporting deregulation of SALL2 in cancer (Liu et al., 2014; Ma et al., 2001). In addition, we selected studies performed in samples from patients without chemotherapy treatment.
The microarray analyses showed significant inverse correlation between SALL2 and CCNE1 mRNA expression in various cancers, including glioblastoma, lymphoma, cervix, pancreas, breast, colon, and lung cancer (Table S1). As an example, Fig. 6 shows a representative graph from breast (A), lung (B), colon (C), glioblastoma (D), and lymphoma (E) cancer studies. Noteworthy, in breast cancer, four independent studies showed inverse correlations of similar magnitude, with Pearson's coefficients (r) ranging from −0.322 (GSE2109) to −0.494 (GSE3494) and P values ranging from 1.3e‐09 to 8.0e‐17 (Table S1). However, the correlation between SALL2 and CCND1 expression is puzzling. Although there is an inverse correlation in endometrial cancer (Table S1, GSE11869), tumor bladder (Table S1, GSE3167), pancreatic (Table S1, GSE17891), and colon cancer (Table S1, GSE41258), a positive correlation is found in breast cancer (Table S1, GSE3494, GSE2109, GSE21653, GSE2034) and sarcoma (Table S1; GSE17679). In some cancer studies (Table S1; GSE4536, GSE17891, GSE41258) SALL2 levels inversely correlated with the expression levels of both CCNE1 and CCND1. Altogether, the R2 data analysis showed more consistent inverse correlation between SALL2 and CCNE1 expression in breast cancer tissues. Likely, SALL2 regulation of cyclin D1 and E1 expression is genetic context‐dependent, which could explain the observed inverse correlations in only a subset of cancers.
Figure 6.
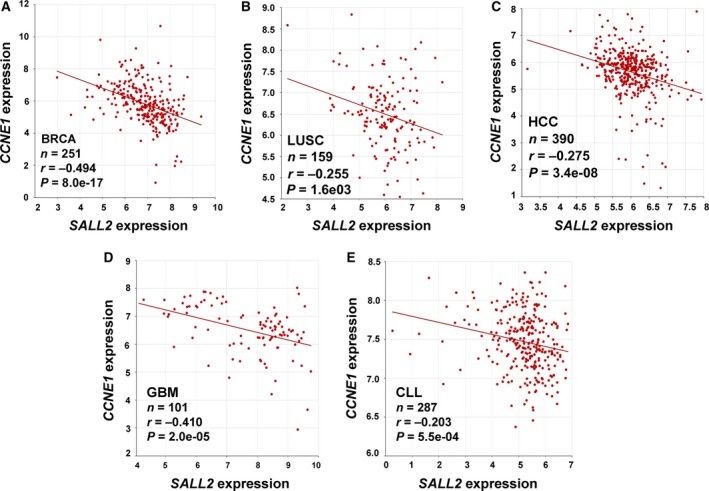
Correlation between SALL2 and CCND1/CCNE1 expression in cancer. Scatter plots of SALL2 by CCND1 and CCNE1 were generated using publicly available databases and software's from R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). Pearson's correlation coefficients (r) and associated P values (p) were calculated using default HugoOnce algorithm and ANOVA statistical test. (A) BRCA, breast invasive carcinoma. (B) LUSC, lung squamous cell carcinoma, and (C) HCC, human colorectal cancer. (D) GBM, glioblastoma multiforme, (E) CLL, chronic lymphocytic leukemia, n = number of samples.
5. Discussion
Increasing studies indicate that SALL2 plays a role in cancer. Because it is downregulated in ovarian cancer as well as in other cancer types, SALL2 has been proposed as a tumor suppressor. However, SALL2 is also upregulated in some cancers and was recently identified as a key factor for glioblastoma propagation (Suvà et al., 2014). Therefore, how SALL2 is associated with cancer is controversial. In addition, mechanisms and targets of SALL2 that could explain its role in disease are yet scarce (Hermosilla et al., 2017; Sung and Yim, 2017).
Here, we demonstrated that Sall2 deficiency, in normal and immortal fibroblasts, triggers uncontrolled cell proliferation, which correlates with cell cycle alteration and increased tumorigenic potential of immortal cells in vitro. Our data are consistent with a role of SALL2 in cell cycle arrest and with previous studies in other cell types. Indeed, depletion of SALL2 (silencing) in human ovarian surface epithelial HOSE (Sall2 expressing) cells increased DNA synthesis measured by BrdU incorporation (Li et al., 2004). On the other hand, gain of SALL2 function (overexpression) decreased DNA synthesis in SKOV3 (Sall2‐deficient) ovarian cancer cells (Li et al., 2004). SALL2 was also identified as a key factor for cellular quiescence of human foreskin fibroblasts, showing early upregulation of SALL2 upon serum deprivation (Liu et al., 2007). Silencing of SALL2 blocked the ability of cells to arrest in G0–G1 after growth factor deprivation, leading to inappropriate progression through S and G2‐M phases (Liu et al., 2007). In agreement, we found that asynchronous primary Sall2 −/− MEFs present higher proportion of cells in S phase. However, we did not observe a notorious difference in the G2‐M phases between asynchronous (Fig. 1E 29.13% vs 21.72%) or nocodazole synchronized Sall2 +/+ and Sall2 −/− MEFs (t = 0). Still, SALL2 levels are high in mitotic synchronized MEFs. These results suggest that SALL2 is also involved in regulation of mitotic exit, which could in part explain the accelerated progression of Sall2 −/− cells into G1. As previous studies used siRNA pools and serum deprivation in human foreskin fibroblasts (Liu et al., 2007), or overexpression of SALL2 in—p16‐deficient—SKOV3 cells (Wu et al., 2015), the difference in the effect of SALL2 at G2‐M might relate to experimental conditions, cell‐type specificity or the genetic context. Whether the role of SALL2 in G2‐M is cell‐type dependent is out of the focus of the present study and should be analyzed in future studies. Nevertheless, nocodazole‐synchronized Sall2 −/− iMEFs inappropriately progressed through the cell cycle, entering earlier than wild‐type iMEFs into G1 and S phases, which is associated with the increased proliferation of the Sall2‐deficient cells detected by FACs and BrdU incorporation studies, and consistent with the negative regulation of G1/S cyclins by SALL2.
Our current study shows for the first time that SALL2 represses cyclin D1 and cyclin E1 expression, further supporting a role of SALL2 during G1/S transition. Cyclins are sequentially expressed during the cell cycle, forming complexes with cyclin‐dependent kinases that phosphorylate target proteins required for progression through the cell cycle (Suryadinata et al., 2010). Cyclin types D and E play major roles at the G1‐ to S‐phase transition; specifically, the induction of cyclin D1 is a rate‐limiting event for cyclin‐dependent activation of CDK4 kinase and the subsequent transcriptional activation of cyclin E gene for progression beyond G1/S transition (Kim and Diehl, 2009). Type E cyclins express during late G1 phase until the end of the S phase and are limiting for the passage of cells through the restriction point ‘R’ from a resting state, or passing from G1 to S phase (Möröy and Geisen, 2004; Siu et al., 2012). Because of the relevance of cyclin D1 and cyclin E1 expression during cell cycle progression, both cyclins are highly regulated at specific times through transcriptional, post‐transcriptional, and post‐translational mechanisms (Kim and Diehl, 2009; Klein and Assoian, 2008; Möröy and Geisen, 2004; Qie and Diehl, 2016; Siu et al., 2012). Consistent with transcriptional repression, ectopic SALL2 (E1 or E1A isoform) repressed cyclin D1 and E1 promoter's activity. In addition, chromatin immunoprecipitation experiments showed specific binding of each SALL2 isoform to the proximal region of CCND1 and CCNE1 genes’ promoters. Of note, even though most of our studies were carried out in MEFs, which express mainly E1A isoform, ectopic expression of either E1 or E1A represses the transcriptional activities of cyclin promoters, suggesting that CCND1 and CCNE1 are not isoform‐specific but rather common targets.
SALL2 isoforms differ in the first exon but share exon 2, which contains most of the protein sequence including the DNA binding and transactivation domains (Hermosilla et al., 2017). The repressor function of E1 could be explained by a conserved repressor domain present at its N‐terminal region, which binds to the NuRD complex (Lauberth and Rauchman, 2006). E1A does not contain this domain neither interacts with the NuRD complex (Lauberth and Rauchman, 2006); however, it has been reported that similar to the effect of SALL2 E1, the expression of SALL2 E1A represses several gene promoters activity, including TK, hTERT, c‐Myc, and Sp1. (Wu et al., 2015). How SALL2 E1A represses the cyclins promoter is at the present unknown, but might result from direct repression of promoter's activity. Similar to repression mediated by p53, E1A could bind to an element that overlaps—or are similar—to sites of activator/coactivator molecules, directly bind to promoter elements and disrupt the pre‐initiation complex assembly, or assemble a multiprotein repressor complex (van Bodegom et al., 2010; Bohlig and Rother, 2011; Ho and Benchimol, 2003; Zaky et al., 2008). Our ChIP studies suggest a direct repression of cyclin D1/E1 by SALL2, although they did not rule out an indirect effect through participation of other transcription factors or cofactors in the process. For instance, SALL2 transcriptionally represses c‐Myc (Sung et al., 2012). As c‐Myc regulates CCND1 and CCNE1 expression and p21CIF/WAF (Jansen‐Dürr et al., 1993; Obaya et al., 1999; Coller et al., 2000; Gartel et al., 2001), it could be indirectly responsible for some of the effects of SALL2. However, our results argue against this possibility because we were unable to find SALL2‐dependent changes of c‐Myc expression in HEK293 cells (Fig. S4). On the other hand, cyclin D–Cdk4/6 complexes could lead to partial activation of E2F transcription factor, allowing the transcription of CCNE1 gene (Suryadinata et al., 2010), suggesting that SALL2‐dependent cyclin D repression could lead to repression of cyclin E (Fig. 7). Although this pathway is clearly not involved in HEK293 cells where we failed to detect any change on cyclin E1 expression, it could contribute to SALL2‐dependent repression of cyclin E1 in MEFs. Nevertheless, our transcriptional studies strongly suggest that SALL2 directly regulates cyclin E1 expression.
Figure 7.
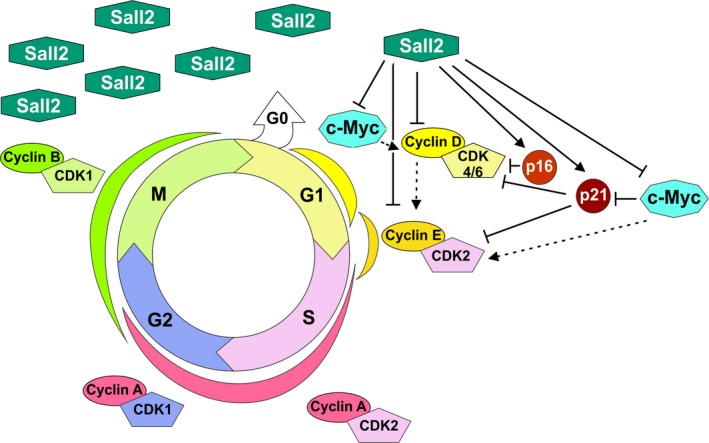
Model of SALL2‐dependent regulation of the cell cycle. SALL2 directly inhibits G1‐S phase progression by repressing cyclin D1 and cyclin E1 expression (this article). Additionally, SALL2 induces p21CIF / WAF (p21) and p16INK 4a (p16) and represses c‐Myc. As c‐Myc represses p21, SALL2‐mediated c‐Myc repression could indirectly increase p21 levels. As Myc could trigger cyclin E1 and D1 expression, SALL2 could indirectly downregulate cyclins by repressing c‐Myc. Dotted arrow indicates indirect effects.
Identification of other SALL2 transcriptional targets related to the control of cell proliferation comes mainly from overexpression experiments, which identified that SALL2 induces the expression of two cell cycle inhibitory proteins: cyclin‐dependent kinase inhibitor I (CDKN1A, p21CIF/WAF) (Li et al., 2004) and cyclin‐dependent kinase 4 inhibitor A (CDKN2A, p16INK4a) (Wu et al., 2015) (Fig. 7). As p16INK4a blocks cyclin D‐Cdk4/6 complexes during G1 phase (Serrano et al., 1993), and p21 blocks cyclin E/Cdk2 complex at the G1/S check point (Harada and Ogden, 2000; Harper et al., 1995), SALL2 regulation of previously reported targets also supports its role during G1‐/S‐phase transition.
We showed that Sall2 deficiency associates with increased expression of Cyclin D1 and E1 (at mRNA and protein levels), but does not affect the expression and/or kinetics of cyclin A and B1 through the cell cycle progression. Therefore, the deregulation of cyclins D1 and E1 is likely involved in the proliferative advantage of the Sall2 −/− MEFs. Relevant is that the inverse correlation between SALL2 and cyclin D1/E1 expression in MEFs is also confirmed in tissues from wild‐type and Sall2 knockout mice, suggesting that SALL2 repression of cyclin D1 and E1 expression occurs in vivo. These results open the possibility of SALL2 association with other functions of cyclins. In particular, in addition to its role as a CDK‐dependent regulator of the cell cycle, cyclin D1 has also CDK‐independent functions associated with the regulation of cellular metabolism, cell differentiation, and cellular migration (Dai et al., 2013; Fu et al., 2005; Pestell, 2013). It is particularly intriguing that SALL2 and cyclin D1 have both been associated with neurogenesis (Bohm et al., 2007; Pincheira and Donner, 2008; Pogoriler et al., 2006).
Considering only the transcriptional aspect of the regulation of cyclin D1 expression, our results suggest that SALL2 controls the steady‐state levels of cyclin D1, but changes in the activity of other transcriptional repressors and/or activators surely contributes in the increase in cyclin D1 (Klein and Assoian, 2008). Several positive and negative regulators of cyclin D1 expression are known, and these include transcription factors that directly bind and repress or activate CCND1 promoter, such as ATF3, a member of the AP1 family that represses cyclin D1 expression (Lu et al., 2006). Besides ATF3, other factor such as SMAR1 inhibits cyclin D1 transcription by recruiting to the CCND1 promoter a repressor complex containing Sin3, HDAC1, and the pocket proteins p107 and p130 (Rampalli et al., 2005). Other identified CCND1 repressors include ZO‐2 and p19ARF (D'Amico et al., 2004; Huerta et al., 2007). In relation to cyclin E1 transcriptional repression less information is available. Repression of CCNE1 gene during G2‐M and the early G1 phases of the cell cycle is mediated through the assembly of a multiprotein complex containing hypophosphorylated pRb, HDAC, and SWI/SNF, which is recruited to E2F transcription factors to the CCNE1 promoter to silence transcription (Möröy and Geisen, 2004; Suryadinata et al., 2010; Zhang et al., 2000). In addition, studies identified Wilms tumor 1 (WT1) (Loeb et al., 2002), NF‐kB (p65/RelA) (Janbandhu et al., 2010), and NFAT (Teixeira et al., 2016) transcription factors as negative regulators of CCNE1 gene expression. How all these factors are coordinated with the regulation of cyclins by SALL2 as well as the underlying mechanism of SALL2 transcriptional repression will require further investigation.
Interesting for cancer research, it is the fact that Sall2 deficiency increases the growth rate, foci formation, and ability of immortalized MEFs to form colonies in soft agar, similar to the effect of knocking out ATF3, an inhibitor of cyclin D1 (Lu et al., 2006). In addition, we found a significant inverse correlation between CCND1/E1 and SALL2 expression in various cancers, but most notoriously between CCNE1 and SALL2 in breast cancer. Analysis of four independent breast cancer datasets showed that SALL2 mRNA inversely correlates with CCNE1 mRNA levels. Although further studies are needed to address whether these correlations are consequence of a functional deficiency of SALL2, it is intriguing that upregulation of cyclin E1 has been reported in many cancer types, and most carefully investigated in breast cancer (Barton et al., 2006; Hwang and Clurman, 2005; Inoue and Fry, 2016; Keyomarsi et al., 2002; Möröy and Geisen, 2004). The mechanisms associated with cyclin E1 overexpression include deregulation of RB pathway by mutations on its regulators, which increased E2F activity, CCNE1 gene amplification, and disrupted proteolysis (Siu et al., 2012). Cyclin E1 (full length and short form) over expression correlates with poor clinical outcome and greatest risk of recurrence (Hunt et al., 2017; Keyomarsi et al., 2002). In relation to SALL2 and breast cancer, two independent bioinformatic studies identified SALL2 as a highly relevant breast cancer biomarker (Liu et al., 2014; Zuo et al., 2017). The studies suggest SALL2 as a putative shared target gene of MYB and ARNT2 transcription factors and as putative suppressor of epithelial–mesenchymal transition activities. SALL2 is identified as one of the candidate drivers for attenuating histological grade promotion, and in preventing cancer progression (Liu et al., 2014; Zuo et al., 2017). Our findings suggest a novel mechanism through CCND1/E1 promoter derepression by loss/deficiency of SALL2 tumor suppressor function. However, it is intriguing the inverse correlation between SALL2/CCNE1 in glioblastoma, as SALL2 was identified as a factor that promotes glioblastoma propagation (Suvà et al., 2014). In addition, the correlation between SALL2 and CCND1 expression was ambiguous, finding positive and negative correlations depending on the cancer type. This suggests that other factors, genetic context or tissue specificity, are involved in the regulation of CCND1/E1 by SALL2. This possibility is consistent with Fig. 4A,C and D that shows inverse correlation only in a subset of normal mouse tissues. Additional clinical and basic studies are needed to support the role of SALL2 as a tumor suppressor in breast cancer, lymphoma, cervix, pancreas, colon, and/or lung cancer.
In summary, we presented evidence of a role of SALL2 in the inhibition of the cell cycle during G1‐ and G1‐ to S‐phase transition. Together with the results showing increased tumorigenic potential of Sall2 −/− cells, our studies indicate a potential mechanistic association of SALL2 deficiency with cancer, through loss of a tumor suppressor function. In further support of this function, there are the previous studies demonstrating that SALL2 transcriptionally increases p21 and p16 Cdk inhibitors’ expression and that SALL2 represses the proto‐oncogene c‐Myc. We also previously demonstrated that SALL2 induces apoptosis of MEFs and human leukemia cells exposed to genotoxic stress (Escobar et al., 2015). Still, SALL2 upregulation in some types of cancer seems to be inconsistent with a tumor suppressor role (Alagaratnam et al., 2011; Estilo et al., 2009; Li et al., 2002; Nielsen et al., 2003; Suvà et al., 2014). However, the SALL2 gene status, subcellular localization, and/or SALL2 isoform expression in those cancers are unknown, which could shed light on the apparently conflicting results. Also, we cannot rule out other SALL2 functions associated with other cellular contexts, targets, and/or cell types. In this sense, there is evidence indicating that SALL2 is involved in stemness (Hermosilla et al., 2017), a function that may explain its role in reprogramming differentiated glioblastoma cells into those with ability to propagate a tumor in vivo. Nevertheless, the revealed function of SALL2 on the repression of cyclins D1 and E1 opens a new perspective for understanding not only SALL2 association with disease, but also its normal function.
6. Conclusions
SALL2 inhibits cell proliferation by repressing G1‐ to S‐phase cell cycle transition. The effect of SALL2 on cell cycle progression is associated with the transcriptional repression of cyclins D1 and E1 expression. Accordingly, SALL2 behaves as a tumor suppressor.
Author contributions
RP conceived and coordinated the project. VEH, GS, ER, DE, MIH, and CF performed experiments and acquisition of data. VEH and RP performed overall data interpretation. MG, VM, MAG, and AFC helped with data interpretation and critically reviewed the manuscript, and VEH, MG, AFC, and RP wrote the manuscript. All authors read and approved the final version of the manuscript.
Supporting information
Fig. S1 Sall2 deficiency does not contribute to immortalization of MEFs.
Fig. S2. Characterization of iMEFs.
Fig S3. Increased BrdU incorporation of Sall2‐deficient iMEFs.
Fig S4. Loss/gain of SALL2 function inversely correlated with levels of cyclin D1 in HEK293 cells.
Fig. S5. DNA sequences of human and mouse proximal promoter regions of CCND1 and CCNE1.
Table S1. Inverse correlation between SALL2 and CCNE1/D1 expression in various cancers.
Acknowledgements
We thank Carolina Benítez and Jocelin González for animal technical support. We thank Patricio Ordenes (Department of Cell Biology, Universidad de Concepción) and Daniela Peña (Departamento de Bioquímica y Biología Molecular, Universidad de Concepción) for experimental technical support; Mr. Germán Osorio (CMA Bio Bio) for microscopy analysis support; and Dr. Mauricio Chandía, Sergio Medina, and Cristofer Palma (Laboratorio Anatomía Patológica, Universidad de Concepción) for FACS analysis support. This work was supported by Fondecyt #1110821 and Fondecyt #1151031 to RP; Fondecyt Grant #1160731 to AC; and Fondecyt #3160129 to MIH. ER, VEH, GS, DE, and CF were supported by the Fondecyt Grant #1151031. This work was also supported by Millennium Science Initiative (ICM Chile) of the Ministry of Economy, Development and Tourism, Grant Number P09/016‐F to MG.
References
- Alagaratnam S, Lind GE, Kraggerud SM, Lothe RA and Skotheim RI (2011) The testicular germ cell tumour transcriptome. Int J Androl 34, e133‐50–1. [DOI] [PubMed] [Google Scholar]
- Bandera CA, Takahashi H, Behbakht K, Liu PC, LiVolsi VA, Benjamin I, Morgan MA, King SA, Rubin SC and Boyd J (1997) Deletion mapping of two potential chromosome 14 tumor suppressor gene loci in ovarian carcinoma. Cancer Res 57, 513–515. [PubMed] [Google Scholar]
- Barton MC, Akli S and Keyomarsi K (2006) Deregulation of cyclin E meets dysfunction in p53: closing the escape hatch on breast cancer. J Cell Physiol 209, 686–694. [DOI] [PubMed] [Google Scholar]
- van Bodegom D, Roessingh W, Pridjian A and El Dahr SS (2010) Mechanisms of p53‐mediated repression of the human polycystic kidney disease‐1 promoter. Biochim Biophys Acta 1799, 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlig L and Rother K (2011) One function–multiple mechanisms: the manifold activities of p53 as a transcriptional repressor. J Biomed Biotechnol 2011, 464916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm J, Buck A, Borozdin W, Mannan AU, Matysiak‐Scholze U, Adham I, Schulz‐Schaeffer W, Floss T, Wurst W, Kohlhase J et al (2008) Sall1, sall2, and sall4 are required for neural tube closure in mice. Am J Pathol 173, 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm J, Kaiser FJ, Borozdin W, Depping R and Kohlhase J (2007) Synergistic cooperation of Sall4 and Cyclin D1 in transcriptional repression. Biochem Biophys Res Commun 356, 773–779. [DOI] [PubMed] [Google Scholar]
- de Celis JF and Barrio R (2009) Regulation and function of Spalt proteins during animal development. Int J Dev Biol 53, 1385–1398. [DOI] [PubMed] [Google Scholar]
- Chai L (2011) The role of HSAL (SALL) genes in proliferation and differentiation in normal hematopoiesis and leukemogenesis. Transfusion 51, 87S–93S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN and Golub TR (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA 97, 3260–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Al‐Odaini AA, Fils‐Aimé N, Villatoro MA, Guo J, Arakelian A, Rabbani SA, Ali S and Lebrun JJ (2013) Cyclin D1 cooperates with p21 to regulate TGFβ‐mediated breast cancer cell migration and tumor local invasion. Breast Cancer Res 15, 3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico M, Wu K, Fu M, Rao M, Albanese C, Russell RG, Lian H, Bregman D, White MA and Pestell RG (2004) The inhibitor of cyclin‐dependent kinase 4a/alternative reading frame (INK4a/ARF) locus encoded proteins p16INK4a and p19ARF repress cyclin D1 transcription through distinct cis elements. Cancer Res 64, 4122–4130. [DOI] [PubMed] [Google Scholar]
- Escobar D, Hepp MI, Farkas C, Campos T, Sodir NM, Morales M, Álvarez CI, Swigart L, Evan GI, Gutiérrez JL et al (2015). Sall2 is required for proapoptotic Noxa expression and genotoxic stress‐induced apoptosis by doxorubicin. Cell Death Dis 6, e1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estilo CL, O‐charoenrat P, Talbot S, Socci ND, Carlson DL, Ghossein R, Williams T, Yonekawa Y, Ramanathan Y, Boyle JO et al (2009) Oral tongue cancer gene expression profiling: Identification of novel potential prognosticators by oligonucleotide microarray analysis. BMC Cancer 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Wang C, Rao M, Wu X, Bouras T, Zhang X, Li Z, Jiao X, Yang J, Li A et al (2005) Cyclin D1 represses p300 transactivation through a cyclin‐dependent kinase‐independent mechanism. J Biol Chem 280, 29728–29742. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F and Tyner AL (2001) Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA 98, 4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picón M, Roberts JM, Lundberg AS, Gifford A, Sardet C and Weinberg RA (1996) Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 12, 1173–1180. [PubMed] [Google Scholar]
- Gu HC, Li DW, Sung CK, Yim H, Troke P and Benjamin T (2011) DNA‐binding and regulatory properties of the transcription factor and putative tumor suppressor p150(Sal2). Biochim Biophys Acta‐Gene Regul Mech 1809, 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K and Ogden GR (2000) An overview of the cell cycle arrest protein, p21(WAF1). Oral Oncol 36, 3–7. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell‐Crowley L and Swindell E (1995) Inhibition of cyclin‐dependent kinases by p21. Mol Biol Cell 6, 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriquez B, Hepp M, Merino P, Sepulveda H, van Wijnen AJ, Lian JB, Stein GS, Stein JL and Montecino M (2011) C/EBPβ binds the P1 promoter of the Runx2 gene and up‐regulates Runx2 transcription in osteoblastic cells. J Cell Physiol 226, 3043–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermosilla VE, Hepp MI, Escobar D, Farkas C, Riffo EN, Castro AF and Pincheira R (2017) Developmental SALL2 transcription factor: a new player in cancer. Carcinogenesis. 38, 680–690. [DOI] [PubMed] [Google Scholar]
- Ho J and Benchimol S (2003) Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ 10, 404–408. [DOI] [PubMed] [Google Scholar]
- Huerta M, Muñoz R, Tapia R, Soto‐Reyes E, Ramírez L, Recillas‐Targa F, González‐Mariscal L and López‐Bayghen E (2007) Cyclin D1 is transcriptionally down‐regulated by ZO‐2 via an E box and the transcription factor c‐Myc. Mol Biol Cell 18, 4826–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt KK, Karakas C, Ha MJ, Biernacka A, Yi M, Sahin AA, Adjapong O, Hortobagyi GN, Bondy ML, Thompson PA et al (2017) Cytoplasmic cyclin E predicts recurrence in patients with breast cancer. Clin Cancer Res, 23, 2991–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HC and Clurman BE (2005) Cyclin E in normal and neoplastic cell cycles. Oncogene 24, 2776–2786. [DOI] [PubMed] [Google Scholar]
- Inoue K and Fry E (2016) Novel molecular markers for breast cancer. Biomark. Cancer 8, 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janbandhu VC, Singh AK, Mukherji A and Kumar V (2010) p65 negatively regulates transcription of the cyclin E gene. J Biol Chem 285, 17453–17464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen‐Dürr P, Meichle A, Steiner P, Pagano M, Finke K, Botz J, Wessbecher J, Draetta G and Eilers M (1993) Differential modulation of cyclin gene expression by MYC. Proc Natl Acad Sci USA 90, 3685–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xuan Z, Zhao F and Zhang MQ (2007) TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res 35, D137–D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, Islam L, Lakowski J, Bacchelli C, Chanudet E, Lescai F, Patel A, Stupka E, Buck A, Wolf S et al (2014) Mutation of SALL2 causes recessive ocular coloboma in humans and mice. Hum Mol Genet 23, 2511–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M et al (2002) Cyclin E and survival in patients with breast cancer. N Engl J Med 347, 1566–1575. [DOI] [PubMed] [Google Scholar]
- Kim JK and Diehl JA (2009) Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol 220, 292–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA and Assoian RK (2008) Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci 121, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase J, Altmann M, Archangelo L, Dixkens C and Engel W (2000) Genomic cloning, chromosomal mapping, and expression analysis of msal‐2. Mamm Genome 11, 64–68. [DOI] [PubMed] [Google Scholar]
- Kohlhase J, Schuh R, Dowe G, Kuhnlein RP, Jackle H, Schroeder B, Schulz‐Schaeffer W, Kretzschmar HA, Kohler A, Muller U et al (1996) Isolation, characterization, and organ‐specific expression of two novel human zinc finger genes related to the Drosophila gene spalt. Genomics 38, 291–298. [DOI] [PubMed] [Google Scholar]
- Lauberth SM and Rauchman M (2006) A conserved 12‐amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J Biol Chem 281, 23922–23931. [DOI] [PubMed] [Google Scholar]
- Li D, Dower K, Ma Y, Tian Y and Benjamin TL (2001) A tumor host range selection procedure identifies p150(sal2) as a target of polyoma virus large T antigen. Proc Natl Acad Sci USA 98, 14619–14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C‐M, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, Friedman R, Klein U and Tycko B (2002) Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal‐epithelial transition. Am J Pathol 160, 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Tian Y, Ma Y and Benjamin T (2004) p150(Sal2) is a p53‐independent regulator of p21(WAF1/CIP). Mol Cell Biol 24, 3885–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Adler AS, Segal E and Chang HY (2007) A transcriptional program mediating entry into cellular quiescence. PLoS Genet 3, 0996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LYD, Chang LY, Kuo WH, Hwa HL, Chang KJ and Hsieh FJ (2014) A supervised network analysis on gene expression profiles of breast tumors predicts a 41‐gene prognostic signature of the transcription factor MYB across molecular subtypes. Comput Math Methods Med 2014, 813067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb DM, Korz D, Katsnelson M, Burwell EA, Friedman AD and Sukumar S (2002) Cyclin E is a target of WT1 transcriptional repression. J Biol Chem 277, 19627–19632. [DOI] [PubMed] [Google Scholar]
- Lu D, Wolfgang CD and Hai T (2006) Activating transcription factor 3, a stress‐inducible gene, suppresses Ras‐stimulated tumorigenesis. J Biol Chem 281, 10473–10481. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li D, Chai L, Luciani AM, Ford D, Morgan J and Maizel AL (2001) Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J Biol Chem 276, 48223–48230. [DOI] [PubMed] [Google Scholar]
- McCormick F and Tetsu O (1999) Beta‐catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426. [DOI] [PubMed] [Google Scholar]
- Möröy T and Geisen C (2004) Cyclin E. Int J Biochem Cell Biol 36, 1424–1439. [DOI] [PubMed] [Google Scholar]
- Nielsen TO, Hsu FD, O'Connell JX, Gilks CB, Sorensen PHB, Linn S, West RB, Liu CL, Botstein D, Brown PO et al (2003) Tissue microarray validation of epidermal growth factor receptor and SALL2 in synovial sarcoma with comparison to tumors of similar histology. Am J Pathol 163, 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obaya AJ, Mateyak MK and Sedivy JM (1999) Mysterious liaisons: the relationship between c‐Myc and the cell cycle. Oncogene 18, 2934–2941. [DOI] [PubMed] [Google Scholar]
- Parroche P, Touka M, Mansour M, Bouvard V, Thépot A, Accardi R, Carreira C, Roblot GG, Sylla BS, Hasan U et al (2011) Human papillomavirus type 16 E6 inhibits p21 WAF1 transcription independently of p53 by inactivating p150Sal2. Virology 417, 443–448. [DOI] [PubMed] [Google Scholar]
- Pestell RG (2013) New roles of Cyclin D1. Am J Pathol 183, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincheira R and Donner DB (2008) The Sall2 transcription factor is a novel p75NTR binding protein that promotes the development and function of neurons. Ann N Y Acad Sci 1144, 53–55. [DOI] [PubMed] [Google Scholar]
- Pogoriler J, Millen K, Utset M and Du W (2006) Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development 133, 3929–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost E, Rhee J and Leach SD (2007) Viral A2 peptide allow expression of multiple proteins from a single ORF in transgenic zebrafish embryos. Genesis 45, 625–629. [DOI] [PubMed] [Google Scholar]
- Qie S and Diehl JA (2016) Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med 94, 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampalli S, Pavithra L, Bhatt A, Kundu TK and Chattopadhyay S (2005) Tumor suppressor SMAR1 mediates cyclin D1 repression by recruitment of the SIN3/histone deacetylase 1 complex. Mol Cell Biol 25, 8415–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Matsumoto Y, Koide U, Kataoka Y, Yoshida N, Yokota T, Asashima M and Nishinakamura R (2003) Zinc finger protein sall2 is not essential for embryonic and kidney development. Mol Cell Biol 23, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorl C and Sedivy JM (2007) Analysis of cell cycle phases and progression in cultured mammalian cells. Methods 41, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ and Beach D (1993) A new regulatory motif in cell‐cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707. [DOI] [PubMed] [Google Scholar]
- Siu KT, Rosner MR and Minella AC (2012) An integrated view of cyclin E function and regulation. Cell Cycle 11, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CK, Li D, Andrews E, Drapkin R and Benjamin T (2013) Promoter methylation of the SALL2 tumor suppressor gene in ovarian cancers. Mol Oncol 7, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CK and Yim H (2017) Roles of SALL2 in tumorigenesis. Arch Pharm Res, 40, 146–151. [DOI] [PubMed] [Google Scholar]
- Sung CK, Yim H, Gu H, Li D, Andrews E, Duraisamy S, Li C, Drapkin R and Benjamin T (2012) The polyoma virus large T binding protein p150 is a transcriptional repressor of c‐MYC. PLoS One 7, e46486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryadinata R, Sadowski M and Sarcevic B (2010) Control of cell cycle progression by phosphorylation of cyclin‐dependent kinase (CDK) substrates. Biosci Rep 30, 243–255. [DOI] [PubMed] [Google Scholar]
- Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV et al (2014) Reconstructing and reprogramming the tumor‐propagating potential of glioblastoma stem‐like cells. Cell 157, 580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetman D and Munsterberg A (2006) The vertebrate spalt genes in development and disease. Dev Biol 293, 285–293. [DOI] [PubMed] [Google Scholar]
- Teixeira LK, Carrossini N, Sécca C, Kroll JE, DaCunha DC, Faget DV, Carvalho LDS, de Souza SJ and Viola JPB (2016) NFAT1 transcription factor regulates cell cycle progression and cyclin E expression in B lymphocytes. Cell Cycle 15, 2346–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Cheng K, Shi L, Li Z, Negi H, Gao G, Kamle S and Li D (2015) Sal‐like protein 2 upregulates p16 expression through a proximal promoter element. Cancer Sci 106, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J (2005) Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol Chapter 28, Unit 28.1. [DOI] [PubMed] [Google Scholar]
- Yim E‐K and Park J‐S (2005) The role of HPV E6 and E7 oncoproteins in HPV‐associated cervical carcinogenesis. Cancer Res Treat 37, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S and Bhakat KK (2008) Regulation of the human AP‐endonuclease (APE1/Ref‐1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res 36, 1555–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW and Dean DC (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC‐Rb‐hSWI/SNF and Rb‐hSWI/SNF. Cell 101, 79–89. [DOI] [PubMed] [Google Scholar]
- Zhu JY, Abate M, Rice PW and Cole CN (1991) The ability of simian virus 40 large T antigen to immortalize primary mouse embryo fibroblasts cosegregates with its ability to bind to p53. J Virol 65, 6872–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Cui Y, Yu G, Li R and Ressom HW (2017) Incorporating prior biological knowledge for network‐based differential gene expression analysis using differentially weighted graphical LASSO. BMC Bioinformatics 18, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Sall2 deficiency does not contribute to immortalization of MEFs.
Fig. S2. Characterization of iMEFs.
Fig S3. Increased BrdU incorporation of Sall2‐deficient iMEFs.
Fig S4. Loss/gain of SALL2 function inversely correlated with levels of cyclin D1 in HEK293 cells.
Fig. S5. DNA sequences of human and mouse proximal promoter regions of CCND1 and CCNE1.
Table S1. Inverse correlation between SALL2 and CCNE1/D1 expression in various cancers.
Data Availability Statement
Publicly available databases from R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl) were used as materials for this study.