

 This work is licensed under a
This work is licensed under a Abstract
Context
Von Hippel–Lindau (VHL) disease manifests as a variety of benign and malignant neoplasms. Previous studies of VHL disease have documented several genotype–phenotype correlations; however, many such correlations are still unknown. Increased identification of new mutations and patients with previously described mutations will allow us to better understand how VHL mutations influence disease phenotypes.
Patients and design
A total of 45 individuals from five unrelated families were evaluated, of which 21 patients were either diagnosed with VHL disease or showed strong evidence related to this disease. We compared the patients’ gene sequencing results with their medical records including CT or MRI scans, eye examinations and laboratory/pathological examinations. Patients were also interviewed to obtain information regarding their family history.
Results
We identified four missense mutations: c.239G>T (p.Ser80Ile), linked with VHL Type 2B, was associated with renal cell carcinoma, pheochromocytoma and hemangioma in the cerebellum; c.232A>T (p.Asn78Tyr) manifested as RCC alone and likely caused VHL Type 1; c.500G>A (p.Arg167Gln) mutation was more likely to cause VHL Type 2 than Type 1 as it preferentially induced Pheo and HB in the retina, cerebellum and spinal cord; c.293A>G (p.Try98Cys) was associated with Pheo and thus likely induced VHL Type 2.
Conclusions
Characterizing VHL disease genotype–phenotype correlations can enhance the ability to predict the risk of individual patients developing different VHL-related phenotypes. Ultimately, such insight will improve the diagnostics, surveillance and treatment of VHL patients.
Precis
Four missense mutations in VHL have been identified in 21 individuals when five unrelated Chinese families with VHL disease were analyzed; VHL mutations are highly associated with unique disease phenotypes.
Keywords: VHL disease, renal cell carcinoma, hemangioma, pheochromocytoma
Introduction
Von Hippel–Lindau (VHL) disease is a rare autosomal dominant disease that manifests as a variety of benign and malignant neoplasms, including retinal hemangioma (HB), HB of the central nervous system (CNS), pancreatic neuroendocrine tumor (NET) or cystadenoma, pheochromocytoma (Pheo) and clear cell renal cell carcinoma (RCC) (1, 2). VHL disease is primarily caused by inactivation of the VHL tumor-suppressive protein. A dysfunctional VHL leads to reduced ubiquitylation and proteasomal degradation of the hypoxia-inducible factors (HIFs), HIF-1α and HIF-2α. Elevated levels of HIFs subsequently result in overactivation of the downstream pathways involved in vascular endothelial growth factor, platelet-derived growth factor and transforming growth factor-α (3). Patients with VHL disease exhibit diverse phenotypes. For example, some patients manifest either RCC or Pheo, and some with both. VHL disease is generally classified into two subtypes: Type 1 (without Pheo) and Type 2 (with Pheo) (4). Type 2 disease can be further divided into three sub-categories: Types 2A, 2B and 2C. Patients with Pheo and HB in the CNS, but without RCC were considered as Type 2A; patients with Pheo, RCC and other CNS tumors were known as Type 2B, while those with Pheo alone were categorized as Type 2C.
VHL mutation-induced RCC typically develops between the ages of 20 and 50 years but rarely in patients younger than 20 years old (5). This type of RCCs is often multi-centric and bilateral, arising in conjunction with both cysts and de novo from the non-cystic renal parenchyma. In addition, they usually develop at multiple sites in the kidneys with approximately two-thirds of such patients showing multiple renal cysts and RCCs (5). Pancreatic abnormalities are also common in patients with VHL disease, as about 77% of these patients have different pancreatic lesions, such as cysts (47%), serous cystadenomas (11%) and NETs (15%) (5). VHL mutation-induced Pheo usually develops in the adrenal gland or paraganglia at different ages ranging from <10 years to >40 years with about 18% of them at a median age of 30 years (6). Pheos are often multiple, bilateral or extra-adrenal and are usually asymptomatic. HBs are rare indolent vascular tumors that may occur sporadically (75%) or in association with VHL disease (25%). VHL-associated HBs are frequently multiple and develop during childhood (<10 years) or in the early teens to thirties. HB most commonly developed in the cerebellum (5–30%) and spinal cord (80%) (6).
Variations in disease penetrance have long been recognized in close relation to genotypes. Previous studies have documented that specific VHL mutations are associated with distinct patterns of tumor formation (7). Since some data suggest that the same mutation might cause different VHL types in different races (8, 9), it is critical to fully characterize genotype–phenotype correlations of this disease in different patient populations. In this study, we reported five unrelated Chinese families with VHL disease and analyzed the genotype–phenotype correlations in this population.
Subjects and methods
Patients
A total of 45 individuals from five unrelated families were evaluated. Among them, 21 patients were either diagnosed with VHL disease or showed strong evidence related to this disease. Based on one of the recent review articles (2), VHL is the only gene in which mutations are known to cause VHL disease and these patients have the mutations resulting in loss-of-function of VHL protein (the genetic criteria). The clinical criteria for VHL disease include (i) a positive family history of HB, RCC, Pheo or pancreatic tumors or cysts and epididymal cystadenoma; (ii) HB in combination with other tumors including RCC, Pheo, pancreatic tumors or cysts or epididymal cystadenoma without a positive family history of VHL disease (2). The patients’ gene sequencing results were compared with their medical records including CT or MRI scans, eye examinations and laboratory/pathological examinations. Additional information regarding to their family history was obtained by further interviewing.
Compliance with ethical standards
All procedures involving human participants were performed in accordance with the ethical standards of the Institutional Research Committee, the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The Daping Hospital of Third Military Medical University waived Institutional Review Board approval for the study. However, written informed consent for the use of medical records and related images was obtained from all patients.
Genetic analysis
Total DNA isolated from the peripheral blood of all five probands was screened for potential mutations in the following susceptibility genes of Pheos and/or RCC: SDHAF2 (succinate dehydrogenase complex assembly factor 2), SDHB (succinate dehydrogenase subunit B), SDHC (succinate dehydrogenase subunit C), SDHD (succinate dehydrogenase subunit D), MAX (MYC-associated factor X), NF1 (neurofibromin 1), RET (rearranged during transfection), VHL, FH (fumarate hydratase), FLCN (folliculin), and MET using Target Capture-Based Deep Sequencing (BGI Health, Shenzhen, Guangdong, China). Upon identification of mutations in the probands, the potential mutations in most of the respective family members were further screened by Sanger sequencing.
Results
Cases
Patient 1
A 42-year-old female was admitted to our hospital in May 2015 due to masses in her right kidney and left adrenal gland. The CT scan showed a mass of 2.5 × 2.1 cm and multiple cysts in her right kidney (Fig. 1A, upper image), and a mass of 6.1 × 3.9 cm in her left adrenal gland (Fig. 1A, middle image), while a MRI of her head showed a lesion in her right cerebellum (1.0 × 1.4 cm). In addition, two large masses (>6 cm) were found in her bilateral adrenal glands when she was 29 years old, for which she repeatedly underwent adrenal Pheo resection. At 38 years of age, she underwent pancreatic mass resection and pathological analysis of the mass showed a pancreatic NET. Considering her recurrent and multiple lesions, surgery was not considered suitable and therefore sunitinib was prescribed. A few months later, she experienced dizziness, headache and emesis. In February 2016, she underwent surgery for the mass in her right cerebellum (Fig. 1A, bottom image), and HB was confirmed pathologically. In addition, her eldest brother had RCCs in both kidneys, HBs in his bilateral cerebellums and Pheo in his left adrenal gland. Her second brother also had RCC, while her daughter had Pheos in her bilateral adrenal glands and HB in her left cerebellum. The patient’s father died from brain tumors. Although without pathological confirmation, he was most likely suffered HBs based on the family history. Therefore, we conducted gene sequencing of her DNA and a pathogenic VHL mutation (c.239G>T; p.Ser80Ile) (Fig. 2A) was confirmed.
Figure 1.

Representative CT and MRI scans from the probands. (A) CT and MRI scans showing lesions in proband 1, including a right renal cell carcinoma (RCC) (upper image) and left pheochromocytoma (Pheo) (middle image) by CT scan, and a hemangioma (HB) in the right cerebellum (bottom image) by MRI scan. (B) A CT scan showing RCCs in proband 2. (C) CT scans of Pheos (upper image) and a renal cyst (bottom image) in proband 3. (D) CT and MRI scans showing the Pheos (upper image) and HB (bottom image) in proband 4. (E) CT scans showing Pheos in proband 5. Red arrows indicate the relevant features.
Figure 2.
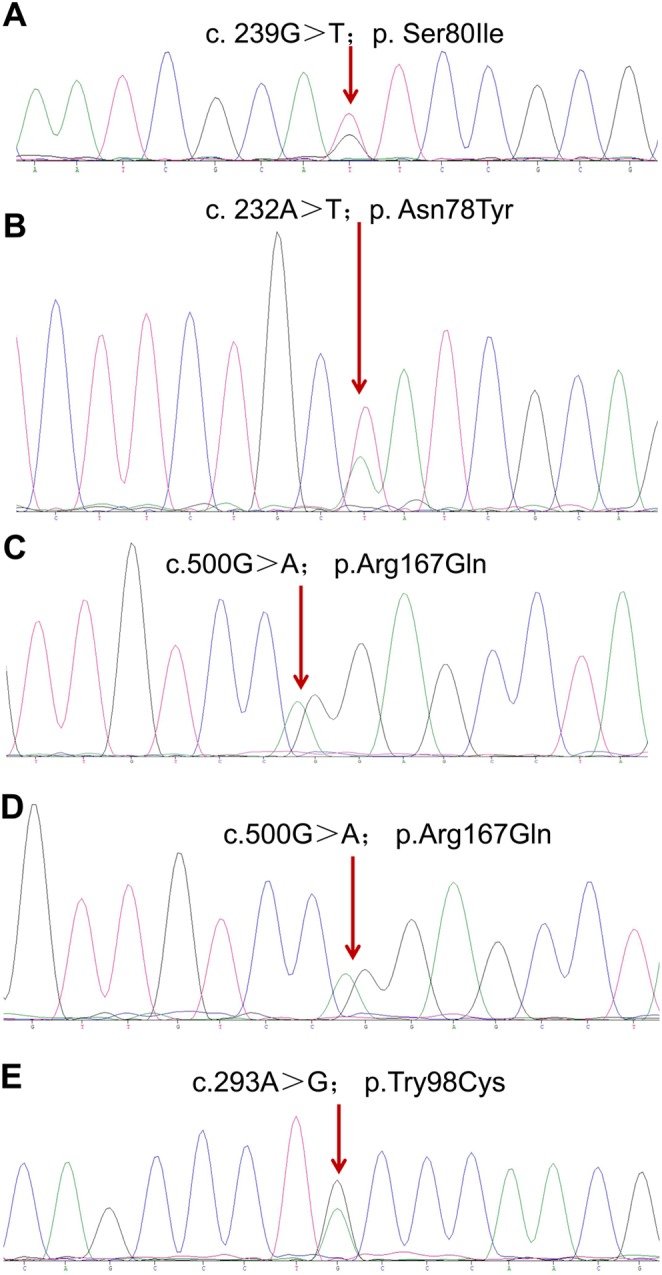
Gene sequencing data reveal VHL mutations for Families 1 (A), 2 (B), 3 (C), 4 (D), and 5 (E), respectively.
Patient 2
A 50-year-old female was referred to our hospital in August 2011 for masses in her left kidney (Fig. 1B). The CT scan showed several masses in her left kidney and the largest one was about 3.1 × 2.4 cm. She underwent a radical nephrectomy 5 years ago for the tumor in her right kidney. Given the fact that she had already lost one kidney, sunitinib instead of surgery was prescribed. Sanger sequencing for potential VHL mutation was conducted for her and her family members and c.232A>T (p.Asn78Tyr) mutation was identified in her, her mother, sister, brother and nephew (Fig. 2B). In fact, her sister was diagnosed with a pancreatic NET and died of hemorrhagic stroke at the age of 40 years, and her mother died of stroke at 50 years of age. Her brother and nephew refused further examinations for VHL-related lesions.
Patient 3
The patient was a 24-year-old male who presented with masses in his bilateral adrenals. In February 2013, he underwent an abdominal CT scan that revealed masses in his bilateral adrenals (Fig. 1C, upper image) and a cyst in his left kidney (Fig. 1C, bottom image). The masses were subsequently resected by laparoscopic surgery and pathological analysis confirmed the presence of Pheos. His mother and aunt died from HBs in their brain, while his sister and cousin had Pheos in their bilateral adrenals and right adrenal, respectively. We decide to sequence the relevant susceptible genes in the proband and confirmed that he had a heterozygous missense VHL mutation c.500G>A (p.Arg167Gln) (Fig. 2C).
Patient 4
A 21-year-old male reported blurry vision and metamorphopsia in his left eye for 10 days beginning on November 9, 2015. An eye examination discovered a macular hole for which he underwent an operation. Tissue pathology revealed an HB in his left retina. After 8 months, he was again referred to our hospital for masses in his bilateral adrenals (Fig. 1D, upper image). These masses were subsequently resected and the pathological report confirmed the presence of Pheos. Additionally, the MRI scan showed a mass in his basal ganglia region (Fig. 1D, bottom image), a mass in his left frontal lobe, as well as a mass in his cervical cord, which were most likely CNS HBs. Sequencing identified the same heterozygous missense mutation as the family of patient 3, p.Arg167Gln (Fig. 2D). Additionally, he revealed that his father also had Pheos in his bilateral adrenals.
Patient 5
The patient was a 10-year-old male who presented with dizziness and headache for 1 month and blurry vision for 2 weeks. An abdominal CT scan revealed two masses in his bilateral adrenals (Fig. 1E) and Pheos were confirmed pathologically. Sequencing screening identified a heterozygous missense mutation (c.293A>G; p.Try98Cys) in VHL (Fig. 2E). Additionally, both his grandmother and mother had Pheos in their bilateral and left adrenals.
Clinical characteristics of patients
In this study, we analyzed a total of 21 patients from five unrelated families who were either diagnosed with VHL disease or showed strong evidence support the conclusion of this disease (Table 1) and their family pedigrees were shown in Fig. 3. Among all the VHL disease-associated tumors, Pheo was the most common one (n = 10, 5 men and 5 women), followed by HB (n = 7, 3 men and 4 women) and RCC (n = 3, 1 men and 2 women). RCC, Pheo and HB frequently manifested bilaterally or as multiple lesions. Among this particular cohort, CNS HB is the leading cause of death due to HB-related hemorrhagic stroke. Routine eye examinations might be critical for identifying early manifestations of VHL pathogenesis so patient care can be managed at earlier points in disease progression. Two probands were admitted to hospital because of blurry vision. Wang et al. have described that HB in CNS and retina represent the most prevalent VHL-associated tumors. By conditional inactivating VHL in a hemangioblast population using a Scl-Cre-ERT2 transgenic mouse line, they found that affected VHL-mutant mice demonstrated retinal vascular lesions associated with prominent vasculature, anomalous capillary networks, hemorrhage, exudates and localized fibrosis (10).
Table 1.
Demographic and disease characteristics of patients with VHL disease.
| Family | Family history | Gender | VHL gene mutation | VHL type | Phenotypes | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RCC | Pheo | HB | Pancreatic NET | Renal cyst | Pancreatic cyst | |||||
| 1 | Proband | Female | c.239G>T; p.Ser80Ile | 2B | Right | Bilateral | Right cerebellum | Yes | Bilateral | — |
| Eldest brother | Male | Bilateral | Left | Bilateral cerebellums | — | Bilateral | — | |||
| Daughter | Female | — | Bilateral | Left cerebellum | — | — | — | |||
| Father | Male | — | — | Suggested HB | — | — | — | |||
| 2 | Proband | Female | c.232A>T; p.Asn78Tyr | 1 | Bilateral | — | — | Yes | Bilateral | — |
| Mother | Female | — | — | — | — | — | — | |||
| Sister | Female | — | — | — | Yes | — | — | |||
| Brother | Male | — | — | — | — | — | — | |||
| Nephew | Male | — | — | — | — | — | — | |||
| 3 | Proband | Male | c.500G>A; p.Arg167Gln | 2B | — | Bilateral | — | — | Left | — |
| Sister | Female | — | Bilateral | — | — | — | — | |||
| Mother | Female | — | — | Suggested HB | — | — | — | |||
| Grandmother | Female | — | ? | — | — | — | — | |||
| Uncle | Male | — | — | — | — | — | — | |||
| Aunt | Female | — | — | Suggested HB | Yes | — | — | |||
| Cousin | Female | — | Right | — | — | — | — | |||
| 4 | Proband | Male | c.500G>A; p.Arg167Gln | 2B | — | Bilateral | In the retina, cerebellum and spinal cord | — | — | — |
| Father | Male | — | Bilateral | — | — | — | — | |||
| 5 | Proband | Male | c.293A>G; p.Try98Cys | — | Bilateral | — | — | — | — | |
| Mother | Female | — | Left | — | — | — | — | |||
| Grandmother | Female | — | Bilateral | — | — | — | — | |||
HB, hemangioma; NET, neuroendocrine tumor; Pheo, pheochromocytoma; RCC, renal cell carcinoma; VHL, Von Hippel-Lindau.
Figure 3.

Family pedigrees for probands 1 (A), 2 (B), 3 (C), 4 (D), and 5 (E), respectively. ‘I, II, III’ represent generations, ‘□, ○’ indicate normal males and females, ‘●, ■’ represent male and female carriers of the corresponding VHL mutation, dashes between symbols ‘□–○’ indicate a couple and ‘↗’ indicates the probands.
Genotype–phenotype correlations
Four missense mutations were identified in these five unrelated Chinese families, specifically, c.239G>T (p.Ser80Ile) in family 1, c.232A>T (p.Asn78Tyr) in family 2, c.500G>A (p.Arg167Gln) in both family 3 and 4 and c.293A>G (p.Try98Cys) in family 5. All affected members in Family 1 presented with RCC, Pheo and HB in their cerebellum, indicating that the mutation (c.239G>T; p.Ser80Ile) is likely linked to VHL Type 2B. In Family 2, only RCC developed in the affected individuals, suggesting that this specific mutation (c.232A>T; p.Asn78Tyr) leads to VHL Type 1. Pheos and HBs developed more frequently in the members of families 3 and 4 who bore the same mutation (c.500G>A; p.Arg167Gln), indicating that this mutation is likely responsible for VHL Type 2 rather than Type 1. The proband in family 5 presented with both Pheo and HB, suggesting that the c.293A>G (p.Try98Cys) mutation is also linked to VHL Type 2. Although both mutations in families 3/4 and 5 resulted in Pheos, the tumors carrying different mutations might have diverse characteristics. The hormone expression profiles of proband 4 and 5 (Table 2) showed that the Pheo in proband 4 (c.500G>A; p.Arg167Gln) might be non-functional, while the Pheo in proband 5 (c.293A>G; p.Try98Cys) could cause hormonal change and result in the hypertension.
Table 2.
Patients’ hormone expression profiles.
| Hormone | Reference range | Proband 4 | Proband 5 |
|---|---|---|---|
| Cortisol 8 (ng/mL) | 50–280 | 241 | 312 |
| Cortisol 16 (ng/mL) | 20–140 | 135 | 381 |
| Cortisol 24 (ng/mL) | 10–120 | 81 | 128 |
| ACTH 8 (pg/mL) | 5.08–32.8 | 16.9 | — |
| ACTH 16 (pg/mL) | 10.7–30.5 | 24.0 | — |
| ACTH 24 (pg/mL) | 5–15 | 12.5 | — |
| Vanillylmandelic acid (μmol/24 h urine) | 9.6–50.0 | 28.1 | 116.8 |
| 17-Hydroxycorticosteroid (μmol/24 h urine) | 5.5–33.2 | 16.4 | 11.3 |
| 17-Ketosteroid (μmol/24 h) | 13.9–76.3 | 67.2 | 40 |
| Orthostatic aldosterone (ng/mL) | 0.1–0.3 | 0.1 | 0.1 |
| Clinostatic aldosterone (ng/mL) | 0.1–0.2 | 0.1 | 0.2 |
| Orthostatic renin activity (ng/mL/h) | 0.3–5.2 | 3.4 | 17.9 |
| Clinostatic renin activity (ng/mL/h) | 0.1–1.5 | 1.0 | 17.1 |
| Orthostatic angiotensin II (pg/mL) | 19–115 | 79 | 322 |
| Orthostatic angiotensin II (pg/mL) | 15–97 | 55 | 306 |
| Hypertension | No | Yes |
ACTH, adrenocorticotropic hormone.
Discussion
In this study, we identified four missense mutations in five unrelated families with VHL disease and analyzed the genotype–phenotype correlation in this small cohort. Based on the genetic mutations and the clinical diagnoses, we inclined to make the following conclusions: the mutation c.239G>T; p.Ser80Ile is associated with RCC, Pheo and HB in the cerebellum, while the mutation c.232A>T; p.Asn78Tyr tended to cause RCC alone; the mutation c.500G>A; p.Arg167Gln preferentially caused Pheo and HB in the retina, cerebellum and spinal cord; the mutation c.293A>G; p.Try98Cys is associated with Pheo alone.
All four mutations identified in this study have been reported previously. The mutation c.239G>T; p.Ser80Ile has been reported to be associated with both VHL Types 1 (11) and Type 2 (12, 13). However, this particular mutation in the Chinese family specifically caused VHL Type 2B. Using molecular modeling of the VHL–ElonginC–HIF–1alpha complex, the mutation c.232A>T; p.Asn78Tyr identified in the current study was previously predicted to be highly associated with RCC, and therefore, to cause VHL Type 1 (14). We found that the mutation c.500G>A; p.Arg167Gln in two unrelated families. This mutation has been first reported by Hes et al. in three families, of which four patients had RCCs and three had renal cysts (15) and later by Ciotti et al. in another two patients (16). In the latter report, one patient presented with bilateral RCCs while the other suffered RCCs and both pancreatic and renal cysts. Therefore, this mutation-associated disease is classified as VHL Type 1 due to the absence of Pheo. In the current study, this mutation was associated with both Pheo and HB, and therefore, we suggest that in Chinese population, this mutation-caused VHL disease should be classified as Type 2. However, we are unable to further determine if it is specific for Type 2A or Type 2B due to the limited size of cohort. The mutation c.293A>G, p.Try98Cys was previously described in two Chinese patients, one of them presented with HB and Pheo, while the other only showed HB. In the current study, the affected patients in family 5 experienced Pheo only. Therefore, this mutation should be classified as VHL Type 2 in the Chinese population.
Patients with VHL disease exhibit diverse phenotypes. Many factors may underlie this phenotypic diversity, including the kind or site of the VHL mutation, the type of amino acid substitutions, modifier genes or other environmental factors. Of note, different kinds of mutations can produce remarkably diverse phenotypes. In VHL patients from non-Chinese population, VHL deletions or truncations are associated with VHL Type 1, while missense mutations are more likely to cause VHL Type 2 (7). This is because VHL missense mutations confer a high risk in development of Pheos (17), while VHL deletions and truncations confer a higher risk of CNS HB relative to missense mutations (18). The findings from a cohort of a large Chinese population also support the notion that missense mutation confers a high risk of Pheos development in Chinese VHL patients (19). All four mutations identified in our study were missenses and only one of them caused VHL Type 1, while the other three resulted in VHL Type 2. These findings also support the notion that missense mutations intend to lead to Pheo.
Mutations at different sites are associated with diverse risks of VHL-related lesions. For instance, mutations in the Missense Cluster Regions MCR-1 (residues 74–90) and MCR-2 (residues 130–136) increase the risk of renal lesions (20). The mutations p.Tyr98His and p.Tyr112His confer a low risk for RCC, while p.Tyr98His, p.Ser111Cys, p.Arg161Gln, p.Leu178Gln and p.Leu188Val confer a high risk of Pheo (7). It has been reported that c.241C4T (p.Pro81Ser) missense mutation increases the risk of RCC and CNS HB (21), while the p.Pro86Leu mutation preferentially induces HB (22). Our findings further substantiated this notion. More importantly, we found that the same tumor (Pheo) resulted from different mutations manifest different characteristics. The mutation c.500G>A; p.Arg167Gln in proband 4 caused non-functional Pheo, while mutation c.293A>G; p.Try98Cys in proband 5 resulted in hormonal changes and subsequent hypertension. This finding need to be further substantiated in a large cohort study.
Different mutations of the same codon can also produce variable phenotypes. Bradley et al. suggest that missense mutation c.334T>A (p.Tyr112Asn) caused VHL Type 1 since most (53.8%) of the affected family members presented with RCC and only one with Pheo (23). On the other hand, substitution of histidine for asparagine (c.334T>C) increases the risk of VHL Type 2A as all the members in two American families (22 patients) carrying this mutation had Pheo and none with RCC (24). It has been speculated that such large phenotypic variation may resulted from different stabilities of the VHL protein produced by different amino acid substitutions. By studying mice carrying VHL-null and Type 2B mutant, Alexandra et al. demonstrated that diverse phenotypes are likely resulted from differential effects of specific mutation on vascular development via downstream perturbations in Notch signal and in conjunction with other pathway misregulation (25).
Although most mutations lead to similar genotype–phenotype correlations across races (26), the same mutation in patients of different races may also manifest diverse phenotypes. Crossey et al. showed that the mutations p.Arg113Ter, p.Gln132Ter, p.Leu158Val and p.Cys162Tyr are associated with VHL Type 1 in western cultures (8), whereas a later report by Yoshida et al. suggested that these mutations are associated with VHL Type 2 in Japanese (10). Similarly, previous studies showed that the mutation c.500G>A (p.Arg167Gln) is associated with RCC and renal cysts in western populations, suggesting that this mutation causes VHL Type 1 (15, 16, 27), while we found that this mutation in Families 3 and 4 frequently caused Pheo, indicating that this mutation (c.500G>A; p.Arg167Gln) is likely linked to VHL Type 2 in the Chinese population.
Different family members bearing the same mutation could also show distinct phenotypes. It has been reported that members in the same family with a c.203C>G (Ser68Trp) mutation demonstrated variable penetrance of isolated Pheos (28). The proband did not manifest Pheo until he was age 63 years. By contrast, one of the proband’s six children presented with an isolated Pheo at age 23 years, and the proband’s sister developed a Pheo at the age of 24 years. In addition, two of her three children are carriers of the mutation and one of them was still asymptomatic at the age of 35, whilst the other had a Pheo when she was 12 years old. Taken together, these data indicate that many other factors, possibly environment, may affect the phenotypes induced by specific mutations. Therefore, it is necessary to characterize genotype-phenotype correlation between different populations. In conclusion, better understanding genotype–phenotype correlation can not only advance our prediction of VHL disease-associated risks but also improve the diagnosis, surveillance and treatment of VHL.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Acknowledgements
The author would like to thank all the patients and the families in this study for their collaboration.
References
- 1.Gossage L, Eisen T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nature Reviews Clinical Oncology 2010. 7 277–288. ( 10.1038/nrclinonc.2010.42) [DOI] [PubMed] [Google Scholar]
- 2.Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Japanese Journal of Clinical Oncology 2006. 36 337–343. ( 10.1093/jjco/hyl052) [DOI] [PubMed] [Google Scholar]
- 3.Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nature Reviews Cancer 2015. 15 55–64. ( 10.1038/nrc3844) [DOI] [PubMed] [Google Scholar]
- 4.Kaelin WG., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nature Reviews Cancer 2002. 2 673–682. ( 10.1038/nrc885) [DOI] [PubMed] [Google Scholar]
- 5.Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature. Journal of Gastrointestinal Surgery 2012. 16 1422–1428. ( 10.1007/s11605-012-1847-0) [DOI] [PubMed] [Google Scholar]
- 6.Wanebo JE, Lonser RR, Glenn GM, Oldfield EH. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. Journal of Neurosurgery 2003. 98 82–94. ( 10.3171/jns.2003.98.1.0082) [DOI] [PubMed] [Google Scholar]
- 7.Nordstrom-O'Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, van Brussel A, Voest EE, Giles RH. Genetic analysis of von Hippel-Lindau disease. Human Mutation 2010. 31 521–537. ( 10.1002/humu.21219) [DOI] [PubMed] [Google Scholar]
- 8.Crossey PA, Foster K, Richards FM, Phipps ME, Latif F, Tory K, Jones MH, Bentley E, Kumar R, Lerman MI, et al Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Human Genetics 1994. 93 53–58. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida M, Ashida S, Kondo K, Kobayashi K, Kanno H, Shinohara N, Shitara N, Kishida T, Kawakami S, Baba M, et al Germ-line mutation analysis in patients with von Hippel-Lindau disease in Japan: an extended study of 77 families. Japanese Journal of Cancer Research: Gann 2000. 91 204–212. ( 10.1111/j.1349-7006.2000.tb00933.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Shepard MJ, Zhang C, Dong L, Walker D, Guedez L, Park S, Wang Y, Chen S, Pang Y, et al Deletion of the von Hippel-Lindau gene in hemangioblasts causes hemangioblastoma-like lesions in murine retina. Cancer Research 2018. 78 1266–1274. ( 10.1158/0008-5472.CAN-17-1718) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, et al Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Human Mutation 1996. 8 348–357. () [DOI] [PubMed] [Google Scholar]
- 12.Patocs A, Gergics P, Balogh K, Toth M, Fazakas F, Liko I, Racz K. Ser80Ile mutation and a concurrent Pro25Leu variant of the VHL gene in an extended Hungarian von Hippel-Lindau family. BMC Medical Genetics 2008. 9 29 ( 10.1186/1471-2350-9-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Huang Y, Pan J, Liu D, Zhou L, Xue W, Chen Q, Dong B, Xuan H. Germline mutations in the von Hippel-Lindau disease (VHL) gene in mainland Chinese families. Journal of Cancer Research and Clinical Oncology 2008. 134 1211–1218. ( 10.1007/s00432-008-0399-x) [DOI] [PubMed] [Google Scholar]
- 14.Losonczy G, Fazakas F, Pfliegler G, Komaromi I, Balazs E, Penzes K, Berta A. Three novel germ-line VHL mutations in Hungarian von Hippel-Lindau patients, including a nonsense mutation in a fifteen-year-old boy with renal cell carcinoma. BMC Medical Genetics 2013. 14 3 ( 10.1186/1471-2350-14-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hes FJ, van der Luijt RB, Janssen AL, Zewald RA, de Jong GJ, Lenders JW, Links TP, Luyten GP, Sijmons RH, Eussen HJ, et al Frequency of Von Hippel-Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, Southern blot analysis and multiplex ligation-dependent probe amplification. Clinical Genetics 2007. 72 122–129. ( 10.1111/j.1399-0004.2007.00827.x) [DOI] [PubMed] [Google Scholar]
- 16.Ciotti P, Garuti A, Gulli R, Ballestrero A, Bellone E, Mandich P. Germline mutations in the von Hippel-Lindau gene in Italian patients. European Journal of Medical Genetics 2009. 52 311–314. ( 10.1016/j.ejmg.2009.05.007) [DOI] [PubMed] [Google Scholar]
- 17.Cybulski C, Krzystolik K, Murgia A, Gorski B, Debniak T, Jakubowska A, Martella M, Kurzawski G, Prost M, Kojder I, et al Germline mutations in the von Hippel-Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. Journal of Medical Genetics 2002. 39 E38 ( 10.1136/jmg.39.7.e38) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franke G, Bausch B, Hoffmann MM, Cybulla M, Wilhelm C, Kohlhase J, Scherer G, Neumann HP. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: molecular characterization and genotype-phenotype correlations in VHL patients. Human Mutation 2009. 30 776–786. ( 10.1002/humu.20948) [DOI] [PubMed] [Google Scholar]
- 19.Peng S, Shepard MJ, Wang J, Li T, Ning X, Cai L, Zhuang Z, Gong K. Genotype-phenotype correlations in Chinese von Hippel-Lindau disease patients. Oncotarget 2017. 8 38456–38465. ( 10.18632/oncotarget.16594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallou C, Chauveau D, Richard S, Joly D, Giraud S, Olschwang S, Martin N, Saquet C, Chretien Y, Mejean A, et al Genotype-phenotype correlation in von Hippel-Lindau families with renal lesions. Human Mutation 2004. 24 215–224. ( 10.1002/humu.20082) [DOI] [PubMed] [Google Scholar]
- 21.Weirich G, Klein B, Wohl T, Engelhardt D, Brauch H. VHL2C phenotype in a German von Hippel-Lindau family with concurrent VHL germline mutations P81S and L188V. Journal of Clinical Endocrinology and Metabolism 2002. 87 5241–5246. ( 10.1210/jc.2002-020651) [DOI] [PubMed] [Google Scholar]
- 22.Fukino K, Teramoto A, Adachi K, Takahashi H, Emi M. A family with hydrocephalus as a complication of cerebellar hemangioblastoma: identification of Pro157Leu mutation in the VHL gene. Journal of Human Genetics 2000. 45 47–51. ( 10.1007/s100380050009) [DOI] [PubMed] [Google Scholar]
- 23.Bradley JF, Collins DL, Schimke RN, Parrott HN, Rothberg PG. Two distinct phenotypes caused by two different missense mutations in the same codon of the VHL gene. American Journal of Medical Genetics 1999. 87 163–167. () [DOI] [PubMed] [Google Scholar]
- 24.Chen F, Slife L, Kishida T, Mulvihill J, Tisherman SE, Zbar B. Genotype-phenotype correlation in von Hippel-Lindau disease: identification of a mutation associated with VHL type 2A. Journal of Medical Genetics 1996. 33 716–717. ( 10.1136/jmg.33.8.716) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arreola A, Payne LB, Julian MH, de Cubas AA, Daniels AB, Taylor S, Zhao H, Darden J, Bautch VL, Rathmell WK, et al Von Hippel-Lindau mutations disrupt vascular patterning and maturation via Notch. JCI Insight 2018. 3 92193 ( 10.1172/jci.insight.92193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rocha JC, Silva RL, Mendonca BB, Marui S, Simpson AJ, Camargo AA. High frequency of novel germline mutations in the VHL gene in the heterogeneous population of Brazil. Journal of Medical Genetics 2003. 40 e31 ( 10.1136/jmg.40.3.e31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JS, Lee JH, Lee KE, Kim JH, Hong JM, Ra EK, Seo SH, Lee SJ, Kim MJ, Park SS, et al Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-alpha binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Medical Genetics 2016. 17 48 ( 10.1186/s12881-016-0306-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin RL, Walpole I, Goldblatt J. Identification of two sporadically derived mutations in the Von Hippel-Lindau gene. Human Mutation 1996. 7 185 () [DOI] [PubMed] [Google Scholar]