

Abstract
Crohn’s disease (CD), a form of inflammatory bowel disease, has a higher prevalence in Ashkenazi Jewish than in non-Jewish European populations. To define the role of non-synonymous mutations, we performed exome sequencing of Ashkenazi Jewish patients with CD, followed by array-based genotyping and association analysis in 2,066 CD cases and 3,633 healthy controls. We detected association signals in the LRRK2 gene that conferred CD risk (N2081D variant, P=9.5×10−10) or protection (N551K variant, tagging R1398H-associated haplotype, P=3.3×10−8). These variants affected CD age of onset, disease location, LRRK2 activity, and autophagy. Bayesian network analysis of CD patient intestinal tissue further implicated LRRK2 in CD pathogenesis. Analysis of the extended LRRK2 locus in 24,570 CD cases, patients with Parkinson’s disease (PD), and healthy controls revealed extensive pleiotropy, with similar genetic effects between CD and PD in both Ashkenazi Jewish and non-Jewish cohorts. The LRRK2 N2081D CD risk allele is located in the same kinase domain as G2019S, a mutation that is the major genetic cause of familial and sporadic PD. Like the G2019S mutation, the N2081D variant is associated with increased kinase activity, whereas neither N551K nor R1398H on the protective haplotype altered kinase activity. R1398H, but not N551K, increased GTPase activity, thereby deactivating LRRK2. The presence of shared LRRK2 alleles in CD and PD provides refined insight into disease mechanisms and may have major implications for the treatment of these two seemingly unrelated diseases.
INTRODUCTION
The inflammatory bowel diseases (IBD) are comprised of two major subtypes, Crohn’s disease (CD) and ulcerative colitis (UC), which are distinguished by the distribution of chronic inflammatory changes. In UC, the inflammation is relatively superficial and is confined to the colon. CD most commonly affects the terminal ileum (last part of the small intestine) and colon, and is frequently associated with deep, transmural inflammation, often resulting in obstruction and abscess formation requiring resectional surgery.
Approved medical therapies for moderate to severe IBD are the same for CD and UC, and include monoclonal antibodies against the pro-inflammatory TNF cytokine and, more recently, antibodies against the α4β7 integrin, which blocks leukocyte trafficking to the intestine. However, present therapies provide prolonged deep remission in only a minority of IBD patients. Consequently, there is a substantial unmet need for more effective medical therapies, especially for CD patients. Genome-wide association studies (GWAS) have identified over 200 loci associated with IBD (1, 2), providing many new potential therapeutic targets. The large majority of these loci are common to CD and UC, implicating numerous pathways, notably the pro-inflammatory interleukin (IL)-23 pathway. In particular, R381Q within the interleukin 23 receptor (IL23R) is a loss-of-function allele that confers protection against developing IBD (3). Importantly, monoclonal antibodies blocking the IL-23 pathway have demonstrated efficacy in IBD, as well as a favorable safety profile (4). CD-predominant loci include nucleotide-binding oligomerization domain-containing protein 2 NOD2 and a number of autophagy genes (e.g. ATG16L1, IRGM). NOD2 is an intracellular receptor for bacterial peptidoglycan and is expressed in a wide variety of cells including plasma cells, innate immune leukocytes (e.g. monocytes, macrophages, dendritic cells) and Paneth cells, which are located at the base of the small intestinal (but not typically colonic) crypts and produce potent antimicrobial peptides. Loss-of-function NOD2 risk alleles are associated with inflammation in the ileum rather than colon and an earlier age of onset with an earlier need for resectional surgery. Among the autophagy-associated signals are the ATG16L1 T300A allele that results in ATG16L1 degradation through caspase-3 activation (5) and multiple polymorphisms in the 5q33.1 region that cause tissue-specific variation in immunity-related GTPase family M protein IRGM expression (6, 7).
However, a fundamental limitation of common variant-predominant GWAS is the imprecise definition of genes, specific alleles and mechanisms driving most association signals identified thus far, with the large majority of independent GWAS signals driven by common variants of modest statistical and functional effects. Furthermore, common variation in composite is predicted to contribute only a modest fraction of expected heritability for many diseases. For these reasons, major sequencing efforts to identify rare variants of potentially higher statistical and functional effects are of importance for refining the pathways associated with disease pathogenesis and designing new therapies.
We hypothesized that uncommon CD susceptibility alleles with higher effects (i.e. odds ratios), which had eluded analysis in common variant-predominant GWAS, play an important role in genetic predisposition to CD and can elucidate new insights into CD pathogenesis. In this study, we sought to identify the strongest functionally relevant associations and to characterize their biological implications. Given that a major epidemiological feature of IBD is its several-fold higher prevalence in Ashkenazi Jewish cohorts (8, 9) compared to non-Jewish Europeans, we performed exome sequencing of Ashkenazi Jewish CD cases followed by custom array-based genotyping in a large case-control cohort. We identified independent coding CD risk and protective alleles in LRRK2, a large multifunctional gene that confers the greatest genetic effects reported thus far in Parkinson’s disease (PD), a neurodegenerative movement disorder affecting the basal ganglia and characterized by resting tremor, bradykinesia, rigidity and postural instability (10). The presence of shared alleles in CD and PD provides refined insight into disease mechanisms and may have major implications for the treatment of these two seemingly unrelated diseases.
RESULTS
Exome sequencing and HumanExome chip study design
We first performed exome sequencing of 50 Ashkenazi Jewish individuals with CD, randomly selected from high quality DNA samples and confirmed using prior chip data (11) to have 100% Ashkenazi Jewish ancestry, in order to optimize cataloguing of new variants (Fig. S1, Table S1). From these results, we selected 4,277 putatively high-yield new mutations, adding these to the HumanExome beadchip (Fig. S2, Table S2). We next performed discovery-phase genotyping and association analyses in individuals with full genetic Ashkenazi Jewish ancestry (11) (Fig. S3, Table S3).
Top coding-region associations in CD
In the discovery-phase cohort of 1,477 unrelated CD cases and 2,614 independent healthy controls, non-synonymous variants at three loci on chromosomes 1, 12, and 16 demonstrated associations that reached a chip-wide significance (Table 1). Importantly, in addition to the previously reported NOD2 and IL23R alleles, non-synonymous variants, N2081D in LRRK2 and S6N in SLC2A13, in strong linkage disequilibrium (LD) with each other (r2=0.91), were identified to be associated with CD risk (minor allele frequency in CD [MAFCD]=8.1%, odds ratio [OR]=1.73, P=2.56×10−9 and MAFCD=8.1%, OR=1.73, P=2.68×10−9, respectively). The LRRK2 N551K variant was also associated with CD protection (MAFCD=6.6%, OR=0.65, P=7.06×10−7; Table 1, Fig. 1A, Fig. S4). We then evaluated the evidence for CD association in an independent Ashkenazi Jewish cohort of 589 CD and 1019 controls (Table S3). This replicated the association signals at LRRK2 N2081D (MAFCD=7.4%, OR=1.34, P=4.40×10−2), at SLC2A13 S6N (MAFCD=7.7%, OR=1.46, P=9.58×10−3), and at LRRK2 N551K (MAFCD=7.0%, OR=0.72, P=1.27×10−2). Meta-analysis revealed genome-wide significant CD risk at LRRK2 N2081D (P=9.51×10−10) and at SLC2A13 S6N (P=1.39×10−10), and protection at LRRK2 N551K (P=3.28×10−8). A list of all coding variants with discovery-phase association P-values<2×10−5 is provided in Table S4. Notably, R1398H (MAFCD=6.6%, OR=0.71, P=7.33×10−5) and K1423K (MAFCD=5.9%, OR=0.66, P=4.4×10−6) in the LRRK2 gene, which previously have been reported to combine with N551K to form a protective haplotype in PD (12–15), were found to show weaker associations in CD (Table S4).
Table 1.
List of the top variants that reached genome-wide significance in meta-analysis
| Ref SNP ID | Chr | Coordinate | Gene | Substitution | Discovery (N=1477 cases, 2614 control) | Replication (N=589 cases, 1019 control) | Meta-analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| MAFCD (%) | MAFCTRL (%) | P-value | OR | MAFCD (%) | MAFCTRL (%) | P-value | OR | P-value | |||||
| rs11209026 | 1 | 67705958 | IL23R | R381Q | 3.22 | 8.03 | 6.79 × 10−18 | 0.38 | 3.15 | 8.05 | 3.36 × 10−8 | 0.37 | 1.38 × 10−24 |
|
| |||||||||||||
| rs139518863 | 12 | 40499594 | SLC2A13 | S6N | 8.10 | 4.84 | 2.68 × 10−9 | 1.73 | 7.65 | 5.36 | 9.58 × 10−3 | 1.46 | 1.39 × 10−10 |
|
| |||||||||||||
| rs7308720 | 12 | 40657700 | LRRK2 | N551K | 6.64 | 9.85 | 7.06 × 10−7 | 0.65 | 7.78 | 10.45 | 1.27 × 10−2 | 0.72 | 3.28 × 10−8 |
| rs33995883 | 12 | 40740686 | LRRK2 | N2081D | 8.13 | 4.86 | 2.56 × 10−9 | 1.73 | 7.40 | 5.61 | 4.40 × 10−2 | 1.34 | 9.51 × 10−10 |
|
| |||||||||||||
| rs141326733 | 16 | 50138853 | HEATR3 | R642S | 2.78 | 1.03 | 3.16 × 10−9 | 2.74 | 1.87 | 0.93 | 2.29 × 10−2 | 2.02 | 4.76 × 10−10 |
|
| |||||||||||||
| rs2066842 | 16 | 50744624 | NOD2 | P268S | 32.42 | 23.03 | 2.25 × 10−20 | 1.60 | 32.44 | 20.07 | 4.21 × 10−15 | 1.91 | 3.31 × 10−33 |
| rs2066844 | 16 | 50745926 | NOD2 | R702W | 3.63 | 1.88 | 1.19 × 10−6 | 1.97 | 3.82 | 2.11 | 4.25 × 10−3 | 1.84 | 1.76 × 10−8 |
| rs104895447 | 16 | 50750842 | NOD2 | M863V | 4.06 | 1.05 | 1.57 × 10−19 | 3.98 | 3.57 | 1.08 | 1.15 × 10−6 | 3.39 | 1.28 × 10−24 |
| rs2066845 | 16 | 50756540 | NOD2 | G908R | 8.73 | 4.21 | 5.14 × 10−17 | 2.18 | 7.99 | 3.29 | 4.12 × 10−9 | 2.55 | 1.52 × 10−24 |
| rs2066847 | 16 | 50763781 | NOD2 | L1007fs | 8.33 | 2.75 | 6.27 × 10−30 | 3.21 | 7.47 | 2.40 | 7.09 × 10−12 | 3.28 | 3.43 × 10−40 |
MAFCD, minor allele frequency in Crohn’s disease cases; MAFCTRL, minor allele frequency in controls; OR, odds ratio. P-values for discovery and replication cohorts calculated using X2 testing. Meta-analysis performed using METAL default method. Chr, chromosome
Figure 1. Crohn’s disease association within the LRRK2 locus.
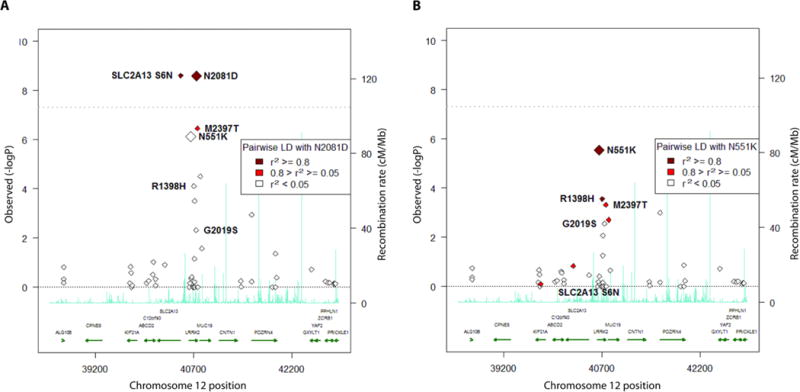
(A)Single-point association without covariates, using Exome chip-genotyped variants only. (B)Association conditioned on N2081D genotypes, using Exome chip-genotyped variants only.
Prior studies have implicated distinct common alleles in the LRRK2 region as being associated with CD (1, 16, 17). To further elucidate the genetic structure of the LRRK2 signal, we conducted a conditional analysis using the discovery cohort, which demonstrated that this broad association peak was entirely dependent on the coding mutation at N2081D in LRRK2 (Fig. 1B); SLC2A13 S6N, as well as the association signal from the previously reported GWAS hits, including non-synonymous variant rs3761863 (M2397T) (16, 18), were substantially attenuated. Conditioning on N2081D genotypes verified the independence of the protective association signal at LRRK2 N551K linked to lower CD risk (OR=0.67, P=1.4×10−6; Fig. 1B). Conditioning on N551K or R1398H genotypes from the protective haplotype as a covariate had minimal effect on the association signal. Interestingly, in phased haplotype association analysis (Table S5), the 2081D risk variant occurred completely on the background of the protein-destabilizing allele M2397(18) (MAFCD=45%; pairwise D’=1.0, r2=0.09), whereas the 551K protective variant co-resided with the stabilizing 2397T(18) allele (pairwise D’=0.94, r2=0.06). Conditioning on both N551K and N2081D together effectively eliminated the association signal at M2397T (conditioned P=0.015; unconditioned P=5.9×10−7).
The multi-functional kinase, LRRK2, has attracted considerable attention given that variants in this gene have been recognized as major risk factors for PD (19). Notably, the G2019S mutation in LRRK2, the best known genetic cause of familial and sporadic PD worldwide and located in the same kinase domain as N2081D, showed suggestive, but not genome-wide significant, association with CD (unconditioned OR=1.9, P=4.8×10−3) and no LD with N2081D (r2=0.0) in the Ashkenazi Jewish cohort.
Further replication and validation of the shared CD and PD risk allele within the LRRK2 locus
To replicate our findings in the non-Jewish cohorts and to explore the pleiotropic effect of LRRK2 variation on CD and PD risk, we expanded our analysis to include a total of 8,314 independent Ashkenazi Jewish and 16,401 independent non-Jewish participants comprising 6,538 CD cases, 5,570 PD cases, and 12,607 healthy controls genotyped in previous studies (Table S3). After performing imputation and quality control measures, we conducted association testing on the set of LRRK2 variants in these datasets (see Supplementary Material and Methods). As in the discovery cohort, in both Ashkenazi Jewish and non-Jewish validation cohorts, we observed a multi-marker CD-associated signal within the LRRK2 gene (Table S6) that was fully conditioned on N2081D (Fig. S5A-B). Also, conditioning on N551K or R1398H as a covariate had minimal effect on the broad association peak. Importantly, in the non-Jewish dataset, association results showed similar marginal effects for N2081D (ORAJ=1.7 [1.4-2.0] vs. ORNJ=1.6 [1.3-2.0]) and N551K (ORAJ=0.67 [0.57-0.79] vs. ORNJ=0.89 [0.79-1.0]) or R1398H (ORAJ=0.71 [0.60-0.84] vs. ORNJ=0.88 [0.78-0.99]) but with substantially lower MAF’s, especially for N2081D (MAFAJ_CD=8.0% vs. MAFNJ_CD=2.9%; Table 2). Notably, G2019S did not have nominally significant CD association (P = 0.12), likely due to subtle stochastic fluctuations in allele frequencies during imputation.
Table 2.
Allele frequencies and association statistics for LRRK2 non-synonymous variants in imputed datasets
| N551K Variant
| |||||||
| CD vs. control association | PD vs. control association | ||||||
| MAFCD(%) | MAFPD(%) | MAFctrl(%)a | Odds ratio (95% CI) | P-value | Odds ratio (95% CI) | P-value | |
|
|
|||||||
| Ashkenazi Jewish | 6.8 | 7.7 | 9.8 | 0.67 (0.57 - 0.79) | 1.4×10−6 | 0.77 (0.67 - 0.90) | 3.9×10−4 |
| Non-Jewish | 6.0 | 6.2 | 6.9 | 0.89 (0.79 - 1.0) | 5.1×10−2 | 0.87 (0.77 - 1.0) | 4.4×10−2 |
|
| |||||||
| R1398H Variant
| |||||||
| CD vs. control association | PD vs. control association | ||||||
| MAFCD(%) | MAFPD(%) | MAFctrl(%)a | Odds ratio (95% CI) | P-value | Odds ratio (95% CI) | P-value | |
|
|
|||||||
| Ashkenazi Jewish | 6.8 | 7.6 | 9.1 | 0.71 (0.60 - 0.84) | 5.0×10−5 | 0.84 (0.72 - 0.98) | 1.6×10−2 |
| Non-Jewish | 6.1 | 6.2 | 6.9 | 0.88 (0.78 - 0.99) | 4.0×10−2 | 0.88 (0.77 - 1.0) | 5.6×10−2 |
|
| |||||||
| N2081D Variant
| |||||||
| CD vs. control association | PD vs. control association | ||||||
| MAFCD(%) | MAFPD(%) | MAFctrl(%)a | Odds ratio (95% CI) | P-value | Odds ratio (95% CI) | P-value | |
|
|
|||||||
| Ashkenazi Jewish | 8.0 | 5.9 | 5.4 | 1.7 (1.4 - 2.0) | 4.3×10−8 | 1.1 (1.0 - 1.4) | 3.6×10−2 |
| Non-Jewish | 2.9 | 2.4 | 1.8 | 1.6 (1.3 - 2.0) | 2.1×10−6 | 1.3 (1.0 - 1.6) | 1.7×10−2 |
Combined control minor allele frequency (MAF). Each healthy control was randomly assigned to only one disease association analysis to ensure independence. P-values calculated using logistic regression.
To examine the genetic link between CD and PD, we then assessed PD association with LRRK2 N2081D and N551K/R1398H in Ashkenazi Jewish and non-Jewish cohorts, observing association signals for all polymorphisms (Table 2). Specifically, the OR estimates of the protective variants, 551K and R1398H, were similar between CD and PD with slight differences between Ashkenazi Jewish and non-Jewish cohorts (N551K: ORAJ_CD=0.67 [0.57–0.79] and ORAJ_PD=0. 77 [0.67-0.90]; ORNJ_CD=0.89 [0.79-1.0] and ORNJ_PD=0.87 [0.77-1.0], and R1398H: ORAJ_CD=0.71 [0.60–0.84] and ORAJ_PD=0.84 [0.72-0.98]; ORNJ_CD=0.88 [0.78-0.99] and ORNJ_PD=0.88 [0.77-1.0]). However, in both populations, the risk allele, N2081D, showed higher ORs in association with CD (ORAJ_CD=1.7 [1.4–2.0], ORNJ_CD=1.6 [1.3–2.0]) than with PD (ORAJ_PD=1.1 [1.0–1.4], ORNJ_PD=1.3 [CI 1.0–1.6]). Conditioning on N2081D or N551K demonstrated no difference, with G2019S remaining the dominant PD signal (Fig. S5C-D).
To determine the degree of pleiotropy in the LRRK2 locus, we selected variants at least nominally (P < 0.05) associated with both CD and PD and assessed their direction and magnitude of effect across diseases. Following LD pruning (i.e. removal of correlated mutations with pairwise r2>0.8, thus ensuring statistical independence among the remaining mutations), we detected a consistent pattern of correlated effect sizes, with 23 of 26 independent variants (88%) exhibiting effects in the same direction for both diseases in the Ashkenazi Jewish dataset (binomial P=5.2×10−6) and, similarly, 25 of 29 variants (86%) in the non-Jewish dataset (P=7.6×10−6; Fig. 2). Taken together, our findings suggest extended pleiotropy between CD and PD throughout the LRRK2 locus.
Figure 2. Odds ratios for Crohn’s disease (CD) and Parkinson’s disease (PD) analysis.
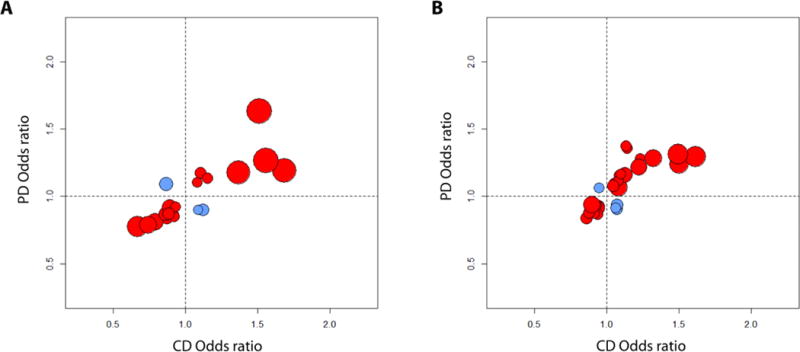
(A)Ashkenazi Jewish cohort odds ratios: 23 of 26 independent variants (88%) exhibited effects in the same direction for both diseases (binomial test P=5.2×10−6). (B)Non-Jewish cohort odds ratios: 25 of 29 variants (86%) exhibited effects in the same direction for both diseases (P=7.6×10−6). Red indicates LRRK2 variants for which both diseases have the same direction of effect; blue indicates opposite-direction effects. Only the variants with at least nominal significance (P<0.05) in both CD and PD analysis after linkage disequilibrium pruning are shown. Circle sizes correspond inversely to the significance (P-value) of CD association at each variant.
Network analysis of IBD patient tissues further implicates LRRK2 in CD
Given strong LD within the LRRK2 locus containing several plausible candidate genes, including SLC2A13 and MUC19 (Table S6), we conducted network analysis to explore which of these genes participate in biological pathways involved in CD pathogenesis. We constructed an IBD Bayesian network using previously described methodology (20), from gene expression data for 8,382 genes. The expression data were collected in 203 intestinal biopsies that included ileum, ascending colon, descending colon and transverse colon, and inflamed and non-inflamed sigmoid and rectum, all collected at baseline from 54 anti-TNFα resistant CD patients enrolled in the Ustekinumab (anti-IL12/IL23) clinical trial (21, 22). Among the full set of genes, we defined a specific subset, located within IBD-associated loci previously defined in an Immunochip-based large-scale genetic analysis (1) with the goal of projecting these genes onto the intestinal network and identifying co-expressed genes that act together. We then excluded genes previously associated with PD (23), including LRRK2, as well as genes within 1 Megabase (Mb) of LRRK2 to see whether either LRRK2 or other genes would be “recovered” by the network as being co-expressed with the IBD-associated genes. We found that the largest connected sub-network of genes, which represents a set of co-expressed IBD-associated genes, contained LRRK2, but no other genes in the genomic neighborhood of LRRK2 (Fig. 3), thus implicating LRRK2 in particular in IBD pathogenesis. Notably, of the 622 genes in this sub-network, there were 102 (16.4%) IBD-associated genes, a 2.5-fold enrichment compared to the full intestinal network (hypergeometric P=7.6×10−8). Importantly, LRRK2 was closely connected to GPR65, a proton-sensing G-protein coupled receptor associated with IBD and altered lysosomal function (24) and to HLA-DPA1, an α-subunit of the major histocompatibility complex protein/peptide-antigen receptor and a graft-versus-host disease antigen complex linked to both IBD(25) and PD (26).
Figure 3. A LRRK2-focused sub-network within the inflammatory bowel disease-associated gene network.
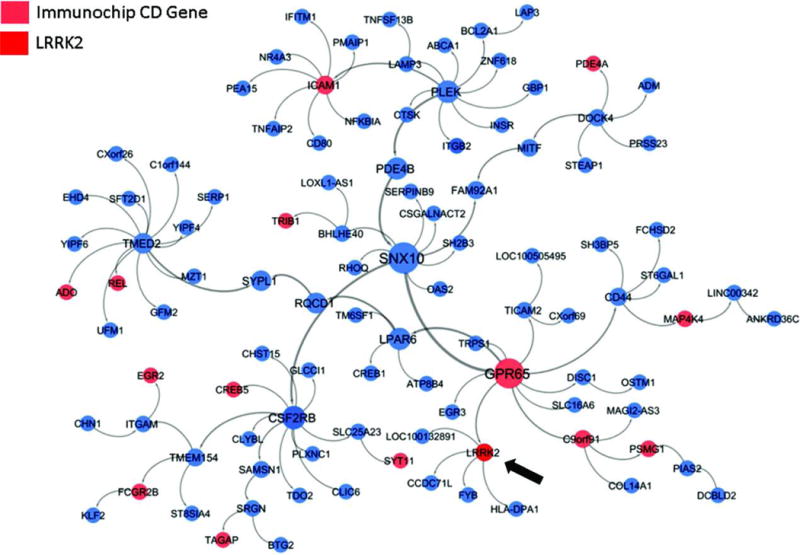
The full intestinal Bayesian network was comprised of 8,382 genes, 551 (6.6%) of them were IBD-associated. From the intestinal network, the largest connected sub-network of genes that were within a path length of two IBD-associated genes was identified; this portion of the network that includes LRRK2 is shown.
Effect of LRRK2 mutations on protein kinase and GTPase activity
Prior studies in PD suggest a central role for increased LRRK2 kinase activity in disease risk resulting from gain-of-function mutations in the LRRK2 kinase domain. Given that both PD-risk G2019S and CD-risk N2081D are located in the kinase domain (Fig. 4A), we investigated the effect of CD-associated LRRK2 mutations on kinase activity. Specifically, we quantified phosphorylation of a newly identified LRRK2 substrate, Rab10 (27), by wildtype LRRK2 protein and mutant LRRK2 proteins bearing G2019S, R1398H, N551K, N551K+R1398H or N2081D mutations that were expressed and purified from HEK293T cells (Fig. 4B). We demonstrated a ~30% increase in phosphorylated Rab10 (pRab10) in the presence of the LRRK2 N2081D mutation compared to wildtype LRRK2 (Fig. 4B) and also confirmed a previous report that the G2019S mutation increased pRab10 (27). In contrast, no change was observed in pRab10 in the R1398H, N551K, or N551K+R1398H carrier cells. Roc, a Ras/GTPase domain in complex proteins, is also a common site of PD-linked LRRK2 mutations, which presumably retain a higher fraction of LRRK2 in a GTP-bound ‘on’-state, thereby promoting increased kinase activity and subsequent neurodegeneration (28, 29). Importantly, the PD-protective R1398H variant, which is in strong LD with the CD-protective N551K variant, is located in the Roc domain (Fig. 4A). To determine the effects of LRRK2 variants on LRRK2 GTPase activity, we compared the ratio of GDP/GTP-bound LRRK2 in vitro across the variants (Fig. 4C). We found that the GTPase activity was increased in both LRRK2 R1398H and N551K+R1398H-transfected HEK293T cells, but not in G2019S, N2081D, or N551K mutants (Fig. 4C).
Figure 4. Effect of LRRK2 mutations on protein kinase activity and GTPase activity.
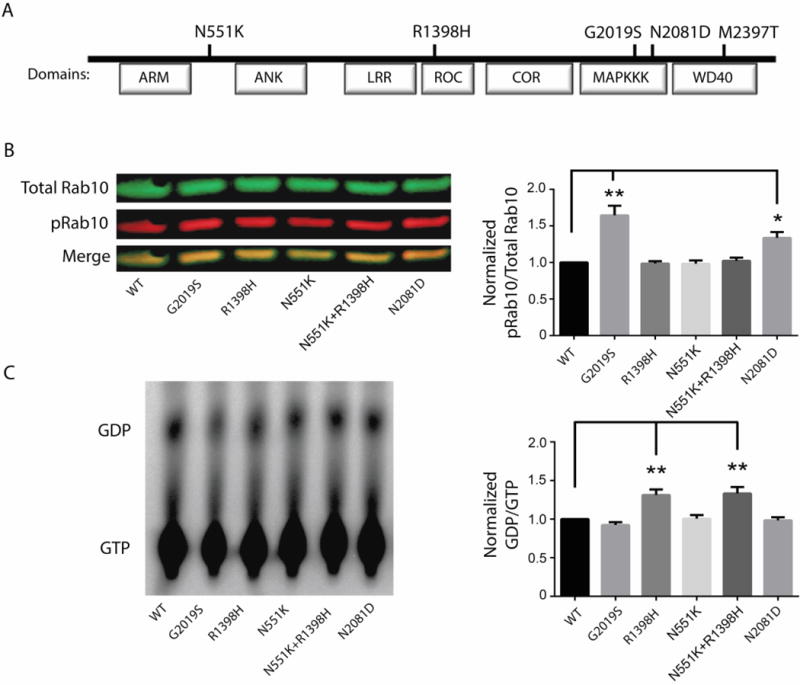
(A)Schematic representation of LRRK2 domain structure and the respective locations of the N551K, R1398H, and N2081D amino acid substitutions relative to the previously reported PD-associated G2019S mutation and CD-associated M2397T mutation. (B) Representative immunoblot (left panel) and quantification (right panel) of Rab10 phosphorylation by wild-type (WT) and LRRK2 variants in patient macrophages in vitro. (C) GTPase activity of WT and LRRK2 variants. Representative GTP hydrolysis assay (left) and the fraction of hydrolyzed GTP (GDP) over bound GTP (right panel). All values represent the mean of 3 independent experiments ± standard error, and significance was calculated by ANOVA. *P≤0.05, **P≤0.01.ARM, armadillo; ANK, ankyrin repeat region; LRR, leucine-rich repeat; Roc, Ras in complex protein; COR, C terminal of Roc; MAPKKK, MAP kinase kinase kinase, WD40, WD40 protein-protein interaction domain.
Role of LRRK2 mutations in cytoskeletal and autophagic function in macrophages from CD patients
To further investigate the properties of the LRRK2 mutations (Fig. 4A), we characterized human monocyte-derived M1 macrophages collected from CD patients who carried LRRK2 N2081D (n=4), N551K+R1398H (all samples selected for their 551K carrier status also carried 1398H; n=5) or neither mutation (n=4) in response to cellular serum-nutrient starvation (Fig. 5). No differences were detected in total LRRK2 expression by mutation status. As LRRK2 has been reported to influence acetylation of α-tubulin, thus regulating cellular protein trafficking via the microtubule cytoskeleton, we determined the effect of the LRRK2 mutations on α-tubulin protein acetylation (Fig. 5A). Lower acetylation of α-tubulin was detected in macrophages from N2081D carriers under normal and PBS-stressed conditions, suggesting impaired resting acetylation activity and a lack of response to cellular stress. In contrast, the highest basal acetylation of α-tubulin was detected in macrophages of non-carriers and carriers of the protective 551K+1398H mutations, which proportionally decreased following cellular stress induced by nutrient starvation. As α-tubulin acetylation is associated with autophagy (30), one of the major pathophysiological processes involved in CD (and in PD) development, we next investigated the effect of the mutations on autophagy markers, LC3-II, an autophagosome-bound form of the microtubule-associated protein 1 light chain 3β (LC3B), and sequestosome-SQSTM1/p62 (p62), a ubiquitin-associated protein facilitating cargo recognition. Following nutrient starvation, we observed a smaller reduction in p62 expression in N2081D macrophages compared to N551K+R1398H macrophages, whereas all cells displayed a similar LC3-II ratio (stress/control) regardless of LRRK2 genotype (Fig. 5A). Despite little change in LC3-II, which is sometimes insensitive to autophagy alterations, a low response of p62 expression to stress suggested an impairment of cargo clearance. Finally, using a lysosome permeable fluorescent pH indicator (lysosensor), we compared lysosomal acidity, a key factor in autophagy, in response to stress, between the LRRK2 N2081D and N551K mutant macrophages (Fig. 5B). We found that the relative change in mean fluorescence intensity following starvation, although varying among individuals, was decreased (alkaline) in risk N2081D carriers and increased (acidic) in carriers of the protective 551K+R1398H variant (Fig. 5B). These data suggest that N2081D and N551K+R1398H mutations in CD patient macrophages have opposing effects on LRRK2 protein function that, in turn, can alter the autophagy-lysosome response to cellular stress.
Figure 5. Effects of CD-associated LRRK2 mutations on human monocyte-derived macrophages.
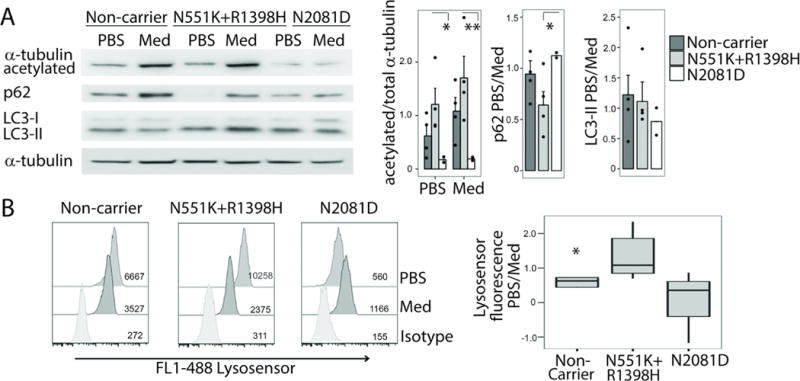
(A) Representative immunoblot showing expression of acetylated α-tubulin, p62, and LC3B (forms I-II) under control (culture medium, Med) or starvation (saline, PBS) conditions in macrophages from patients with different LRRK2 genotypes (left panel). Bar graphs depicting normalized protein expression ratio of acetylatedα-tubulin to total α-tubulin and the ratio during autophagy-inducing starvation. Ratio of protein expression during control (Med) and starvation (PBS) for p62 and LC3-II are also shown. Studies were performed in macrophages from non-carriers (n=4), and carriers of the N551K (n=4) or N2081D variants (n=2). Three independent technical replicates were performed for each sample.(B) Representative flow cytometry data presented as histograms illustrating lysosensor fluorescence after starvation (saline PBS, top), culture medium control (Med, middle) or isotype antibody control (bottom). Flow cytometry was performed on macrophages from non-carriers (n=4), and N551K (n=5) or N2081D variant carriers (n=4) (left panel). The mean Lysosensor fluorescence ratio for PBS versus culture medium control are shown. All values represent mean ± standard error, and significance was calculated by mixed model ANOVA with a random effect of a biological sample (panel A) or order-constrained ANOVA (57) (panel B). *P≤0.05, **P≤0.01.
Additive effects and phenotypic impact of LRRK2 variants
In contrast to the dominant effect of the G2019S mutation in PD risk, we observed an additive effect of N2081D mutations on CD risk, as testing for dominant and recessive disease models did not show any increase in association statistical significance (Table S4). To assess the strength of the combined effect across the LRRK2 variants, we calculated additive burden scores (defined as the log sum of the number of risk-conferring alleles carried by each individual, weighted by the CD odds ratio, which is highly correlated with PD odds ratio as shown in Fig. 2) based upon their genotypes. The additive effects of the LRRK2 risk alleles strongly correlated with both CD and PD risk (Fig. S6), indicating an overall similar genetic architecture throughout the LRRK2 locus underlying both diseases. There was no evidence of interaction effects between any of the nominally associated variants.
Moreover, because of a recent study implicating essential roles for both NOD2 and LRRK2 in proper lysosomal sorting in Paneth cells (31), a group of secretory cells in the ileum with a vital role in maintaining the function of the epithelial barrier, we next examined the effect of LRRK2 N2081D risk alleles on CD disease location. Whereas 80.5% of CD patients homozygous for the wildtype LRRK2 allele had ileal involvement, heterozygous and homozygous carriers of the N2018D variant demonstrated ileal involvement in 86.1% and 90.9% of individuals, respectively (P=0.01, chi-square test, Table 3). Also, carrying the N2081D allele was significantly associated with a younger age of onset (26.5 years for non-carriers, 24.6 years for heterozygous carriers, and 20.8 years for homozygous carriers; P=0.002, linear regression). Neither LRRK2 N551K nor R1398H showed any meaningful correlation with age of onset or ileal involvement in CD (Table 3).
Table 3.
Subphenotypic values by LRRK2 N2081D and R1398H genotype in pooled Ashkenazi Jewish and non-Jewish CD cohorts
| N2081D genotype | Age of CD onset (SD) [N] | Disease location in ileum [N] |
|---|---|---|
| AA | 26.5 (14.0) [5601] | 80.5% [5311] |
| GA | 24.6 (13.1) [482] | 86.1% [453] |
| GG | 20.8 (9.0) [12] | 90.9% [11] |
| P=0.002 | P=0.01 | |
|
| ||
| R1398H genotype | ||
| GG | 26.3 (13.9) [5365] | 81.1%[5095] |
| GA | 26.4 (14.1) [701] | 80.7% [652] |
| AA | 27.2 (19.4) [29] | 71.4% [28] |
| ns | ns | |
SD, standard deviation. N, group sample size. ns, not significant. Similar results were found for the N551K variant (in strong linkage disequilibrium with R1398H, r2=0.81). P values were calculated using simple linear regression.
DISCUSSION
In this study, we performed exome sequencing followed by array-based exome chip genotyping in several independent cohorts of Ashkenazi Jewish CD cases and controls. Among protein-coding variants, in addition to the well-established NOD2 and IL23R associations, we observed genome-wide significant associations for chromosome 12q12 S6N in SLC2A13 and N2081D in LRRK2 (P<5×10−8), in high LD with each other (r2=0.91), and an independent protective CD-association signal at LRRK2 N551K. All previous GWAS association signals in or near LRRK2, including the common coding variant, M2397T (16), reported in one study to lower post-transcriptional LRRK2 protein (18), were significantly attenuated after conditioning on N2081D. Given the high LD between S6N in SLC2A13 and N2081D in LRRK2, we applied co-expression approaches to define the likely contributing gene. In our Bayesian network analysis of IBD intestinal tissue, we observed a highly connected subnetwork with LRRK2, but with no other genes within the chromosome 12q12 region including SLC2A13, demonstrating similar connectivity. SLC2A13 (solute carrier family 2 member 13) is a glucose transporter that is not expressed in the gut or the immune system and has not been previously linked to IBD, further suggesting that the observed 12q12 signal is driven by the LRRK2 gene. Intriguingly, LRRK2 was tightly linked with GPR65, where the IBD-associated risk allele, I231L, is associated with impaired lysosomal function (24) and HLA-DPA1, with variants in this locus linked to both IBD (25) and PD (26).
Notably, both LRRK2 N2081D and N551K variants were also associated with PD in both Ashkenazi Jewish and non-Jewish cohorts (Table 2). Whereas previous reports have documented that LRRK2 N2081D confers PD risk, and the N551K-R1398H-K1423K haplotype confers protection (12–15), we now demonstrate that these specific non-synonymous variants in LRRK2 genetically link CD to PD. Importantly, despite the same direction of the effect, the effect size for the risk variant N2081D was substantially higher for CD compared to PD (Table 2). Of interest, G2019S, the maximally-associated risk allele in PD (32, 33) occurring in the same domain as N2081D (Fig. 4A), although not in LD with it, showed suggestive association with CD in the Ashkenazi Jewish discovery cohort only. Further association analysis of independent common variants in >24,500 PD and CD cases and controls suggested additional extensive genetic pleiotropy between CD and PD within the extended LRRK2 locus with a consistent pattern of correlated effect sizes (Fig. 2) in both Ashkenazi Jewish and non-Jewish datasets. Intriguingly, a recent independent report has suggested that PD is associated with an increased risk of IBD (34). Taken together, these results point toward potential shared genetic and epidemiological links between these two diseases and can help to identify a subgroup of patients with CD who are at a higher risk for developing PD.
Numerous functional roles for LRRK2 have been reported, including vesicular trafficking and endocytosis, protein synthesis, immune response regulation, inflammation, and cytoskeleton homoeostasis, among others (35). In addition to their association with PD and CD risk, variations in the LRRK2 locus have been also independently linked to excessive inflammatory responses in patients with leprosy (36) and risk of particular types of cancer (37). In the gastrointestinal tract of CD patients, LRRK2 expression is restricted to lamina propria macrophages, dendritic cells and B lymphocytes and is induced by interferon-γ, which is consistent with its role in IBD (38). A recent study has found high expression of LRRK2 in Paneth cells in the ileum demonstrating that both NOD2 and LRRK2 are required for proper lysosomal sorting within Paneth cells (31). Our correlations of N2081D in LRRK2 to an earlier age of CD onset and an ileal location mirror previously reported NOD2 risk allele phenotypic correlations. Specifically, we showed that carriers of two copies of the risk allele N2081D had almost a 6-year earlier age of onset compared to non-carriers and predominantly ileal disease involvement, which may be consistent with the recent report of LRRK2’s effects in Paneth cells (39) that are exclusively located in the small intestine. These findings are of clinical importance as a large recent phenotype-genotype analysis of all IBD-associated loci identified only a handful of mutations, including in NOD2, that had considerable effects on age of onset and disease location in CD; in that study, the LRRK2 N2081D variant was not specifically tested (40). Defining altered Paneth cell function stratified according to various LRRK2 and NOD2 genotype combinations should be a focus of future studies.
The majority of PD-causing mutations fall within the kinase and RocCOR domains, resulting in increased kinase activity or GTP-binding, leading to neurodegeneration. Our findings showed that both kinase domain disease-associated mutations, G2019S (PD) and N2081D (CD) increased the phosphorylation of the LRRK2 substrate Rab10. Previous studies have reported that the G2019S mutation increases phosphorylation of several RAB-family members leading to an abnormal cytosol-membrane Rab protein distribution, which could result in the disruption of autophagy (27). Consistent with this report, our studies in human monocyte-derived macrophages from CD patients carrying the N2081D mutation demonstrated faulty stress responses directly related to autophagy, including impaired autophagic cargo clearance, lysosomal acidification as well as defective tubulin acetylation, defects also found in PD models (41).
Moreover, we also showed the link between the protective Roc domain R1398H mutation and an increase in GTPase activity (42). Importantly, although our statistical analysis prioritized the N551K mutation as significantly associated with a reduced risk of CD, in our biochemical analysis, N551K alone did not yield any detectable effect. Based on a high LD between N551K and R1398H mutations and the fact that all N551K human carriers that were analyzed also carried R1398H, we tested the combined effect of N551K+R1398H on GTPase activity and concluded that the actual physiological protective effect is driven by R1398H and not N551K.Notably, human macrophages from N551K+R1398H carriers also demonstrated an enhanced autophagy response to cellular stress.
However, we speculate that the precise nature of the lysosomal alterations likely differs between these two diseases. Autosomal recessive mutations in the GBA (glucosylceramidase beta) gene, which cause Gaucher’s disease and are highly associated with PD (with most cases involving dominant transmission) also are prevalent in Ashkenazi Jewish populations. In our study, we did not find GBA mutations to be associated with CD. This would suggest that PD and CD pathophysiologies differ in the cell-specific properties of the lysosomes in neurons or glia versus inflammatory or Paneth cells, respectively, or with respect to distinct hydrolytic targets, namely glycolipids versus bacterial peptidoglycans, respectively. Nevertheless, naturally occurring protective alleles, such as the R1398H variant in LRRK2, are of particular importance as they define a desired functional effect for therapeutic development. Just as the loss-of-function, protective R381Q variant in IL23R would predict that blocking the IL-23 pathway would be safe and effective, our present findings suggest that targeting LRRK2-mediated signaling may be beneficial in the treatment of both CD and PD.
Among the limitations to our study is the fact that our CD cohorts were not explicitly screened for PD and vice versa, potentially allowing for the inclusion of individuals with both diseases in one disease category (either CD or PD). However, both CD and PD are relatively rare in the general population (~0.2% and ~1%, respectively) and misclassification of such patients would be expected to have minimal impact on any analyses. Also, we studied the Ashkenazi Jewish population given its higher CD prevalence, but this focus limited our cohort size and thus the power to identify new, rarer contributing alleles. Because the exome-sequencing phase of our study involved only 50 individuals, there are certainly many rare Ashkenazi Jewish - specific variants that were not tested in the association phases, and some of these likely play a role in CD pathogenesis. Finally, our Bayesian network analysis, while offering a method to examine gene function in an unbiased manner apart from disease association, did so indirectly and with only gene-expression data from whole tissue used to construct our network.
Our study strongly implicates the contribution of LRRK2 in CD risk as shown through multiple complementary approaches, including genome-wide screening, Bayesian network analysis, genotype-phenotype correlations, and functional studies. The LRRK2 N2081 risk allele and the N551K/R1398H protective alleles, as well as numerous other variants within the LRRK2 locus, revealed pleiotropy between CD and PD risk, providing a potential biological basis for clinical co-occurrence. Our findings suggest that LRRK2 may be a useful target for developing drugs to treat CD.
MATERIALS AND METHODS
Study design
We first performed exome sequencing of 50 Ashkenazi Jewish individuals with CD (44 independent individuals and 3 full-sibling pairs) having sufficient power to detect new variants with MAF>0.015 in order to catalog variation in the Ashkenazi Jewish population that may confer risk for CD (43). Because little genetic variation in Ashkenazi Jewish datasets was available from prior public genome sequencing, we sought to extend the coverage of available commercial genotyping platforms by adding new variants detected in our exome sequencinganalyses. In particular, we favored polymorphic sites that were less likely to be tagged in a previous well-powered genome-wide association study of CD in the Ashkenazi Jewish population. From these results, we selected 4,277 putatively high-yield new mutations that were added to the base content of the Illumina HumanExome 1.0 array to create a semi-custom genotyping platform. With this weperformed discovery-phase genotyping and association analyses in 1,477 CD cases and 2,614 controls with full genetic Ashkenazi Jewish ancestry (11), providing sufficient power to detect associations with modest effect sizes. The top association signals were then replicated in an independent cohort of 589 CD cases and 1,019 controls, recruited throughout North America, Europe, and Israel. Disease diagnosis was confirmed using standard criteria as described elsewhere and full Ashkenazi Jewish ancestry was validated using principal components analysis (11, 44). Our second stage genetic association analysis included a total of 8,619 independent Ashkenazi Jewish and 16,401 independent non-Jewish participants comprising CD cases, PD cases, and healthy controls, genotyped in previous studies (45, 46). PD diagnoses were supported by standard UK Brain Bank criteria (47), with a modification to allow the inclusion of cases that had a family history of PD. We performed imputation of genotypes across diseases and within populations in order to allow direct comparison of genetic association at each site between CD and PD. We next conducted experimental validation studies for LRRK2 N2081D and N551K/R1398H mutations using HEK293 cell lines and whole blood from human subjects enrolled in our prior studies, who consented to be contacted for future research, and who were recalled based on their LRRK2 genotype status. Four N551K+R1398 carriers and five N2081D carriers were matched to five non-carriers, all with CD, for age, sex and disease severity. All experiments were performed in at least 3 biological replicates.
Discovery and replication of new variants associated with CD
We performed chi square-based association testing on all variants genotyped by the Exome chip. We tabulated all non-synonymous variants with P-values suggestive of CD association (P<2×10−5), a threshold we estimated using Bonferroni correction with the approximate number of polymorphic variants genotyped using our platform. This enabled strong and widespread correlations among exomic variants (i.e. “chip-wide significance”). We collected genotypes at these markers in independent case and control cohorts with full Ashkenazi Jewish ancestry. These replication data were combined with those generated by Exome chip genotyping for a meta-analysis using the METAL program with default parameters (49); coding variation with genome-wide significant P-values (P<5×10−8) are presented as positive association signals (Table 1).
Imputation-based comparative analysis of CD and PD
Additional non-Jewish CD and PD and Ashkenazi Jewish PD datasets were added to the Ashkenazi Jewish CD data (imputation cohorts, Table S3), and reference-free imputation using MACH was performed in order to facilitate direct comparisons across groups at specific variants (50). Both unconditioned and conditional analyses were conducted using logistic regression on pooled empiric (directly genotyped) and probabilistic (imputed) genotypes.
Network analysis
We constructed an adult IBD Bayesian network, using previously described methodology (20), from gene expression data generated on 203 intestinal biopsies that included ileum, ascending colon, descending colon and transverse colon, and inflamed and non-inflamed sigmoid and rectum, all collected at baseline from 54 anti-TNFα resistant CD patients enrolled in the Ustekinumab (anti-IL12/IL23) clinical trial (21) with the goal of projecting these genes onto the intestinal network and identifying co-expressed genes that act together. This type of probabilistic causal network structure has previously been demonstrated to represent biologically functional pathways across a broad range of diseases including obesity and diabetes (20, 51–53), asthma and COPD (54, 55), and Alzheimer’s disease (56). We next excluded genes previously associated with PD (23), including LRRK2, as well as genes within 1 Mb of LRRK2 to see whether either LRRK2 or other genes could be “recovered” by the network as being co-expressed with the IBD-associated genes. We then identified the largest connected sub-graph from the set of IBD-associated genes projected onto the network. To focus on pathways potentially relevant to CD pathogenesis, we removed from our analysis all genes more than two edge lengths away from any of these IBD-associated genes.
RAB10 In Vitro Kinase Assay
LRRK2 was incubated with Rab10 or inhibitor for 30 min incubation on ice in 30uL kinase buffer (20mM Tris pH 7.5, 1mM DTT, 15mM MnCl 2, 20mM β-glycerophosphate). Reactions were initiated by adding 50μM cold ATP. After 30 minutes at 37°C, reactions were stopped by addition of Laemmli buffer and boiling at 95°C for 10 minutes. Samples were resolved on 4-12% SDS-PAGE pre-cast gels (Invitrogen, Madison, WI, USA). Samples were then subjected to Western blot, using anti-Rab 10 (Cell Signaling, #4262) and anti-pT73 Rab10 (University of Dundee, UK). Licor imaging was used to detect phospho- and total Rab10 on the same membrane and Image Studio Lite was used for quantification.
GTP Hydrolysis Assay
GTPase activity of LRRK2 was measured in 30uL GTPase buffer (20mM Tris pH 7.5, 150mM NaCl, 1mM DTT, 5mM MgCl2, 1mM EDTA) at 30°C for 90 minutes, where the reaction rate is still in a linear phase as previously established, allowing for quantification by densitometry (29). Reactions were initiated with the addition of 50μM cold GTP and [α-32P]GTP (3000Ci/mmol; PerkinElmer Life Sciences, Waltham, MA). Reactions were terminated by adding 0.5M EDTA. 2uL of the reaction mixture were dotted onto Thin-Layer Chromatography (TLC) plates (EMD Millipore, Darmstadt, Germany) and GDP and GTP were separated by TLC using 0.5M KH2PO4 pH 3.5 for 60 minutes. The TLC plate was dried for 15 minutes and radioactive signal was captured using a phosphor-screen (GE Lifesciences, Pittsburgh, PA, USA) and a Typhoon scanner. ImageQuant densitometry was used to quantify the phosphor-signal.
Autophagy studies in human samples
M1-macrophages from CD patients were derived from whole peripheral blood monocytes according to the manufacturer’s instructions (Promocell, Heidelberg, Germany). Monocytes were polarized to mature M1-macrophages in the DXF M1-macrophage generation medium (M1-medium, resting condition, Promocell) for 12 days and then incubated in PBS and M1 medium for 45 minutes. Cells were then lysed and 10 micrograms of total protein were loaded onto 4-12% Bis-Tris Plus precast SDS-polyacrylamide gels, transferred onto a PVDF membrane and probed with primary rabbit anti-LRRK2 antibody (ab133474, abcam), mouse anti-acetylated alpha-tubulin (T7451, Sigma-Aldrich, St. Louis, MO), rabbit anti-alpha tubulin (ab4074, abcam), mouse anti-SQSTM1 (sc-28359, Santa Cruz Biotechnology), and rabbit anti-LC3B (NB100-2220, Novus Biologicals). The corresponding HRP-conjugated secondary antibody was applied for detection. Total alpha-tubulin was used as a loading control for normalization and protein densitometry was performed using ImageJ software. LRRK2 degradation was assessed as the ratio of degraded LRRK2 to total LRRK2 (full length + degraded) protein. Alpha-tubulin acetylation was assessed as the ratio of acetylated to total alpha-tubulin.
Next, M1 macrophages (1×10ˆ5 cells per experiment), in M1-medium and PBS, were pulsed with lysosensor green DND-189 (L-7535, Life Technologies) for 45 minutes (58). Antibodies for cell surface markers were added and cells incubated for 30 minutes at 4°C. After staining, the cells were washed and analyzed on a CANTOII (BD) multi-parameter flow cytometer and data were analyzed using FlowJo software (Tree Star). A fluorescence minus one (FMO) was used for the FITC lysosensor control samples. The fluorescent ratio was calculated between PBS and M1-medium and compared by the LRRK2 genotype.
Statistical analysis
Genotyping quality control was performed following guidelines produced by the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) consortium (48). This procedure included removing samples with low quality metrics (genotype call rate < 0.96 and/or p10GC < 0.4125) and removing markers with overall low probe intensity. A subset of SNPs was subsequently excluded according to clustering criteria based on fluorescent probe intensities and genotype frequencies, as well as visual inspection of markers with uncertain genotyping quality. All experimental values represent mean±standard error, and significance was calculated by ANOVA, mixed model ANOVA with a random effect of a biological sample or order-constrained ANOVA (57).
Supplementary Material
Table S4. All variants with Ashkenazi Jewish CD discovery P values < 2 × 10−5 (Excel file)
Table S6. All imputed variants with nominal CD or PD association (P < 0.05) within the LRRK2 locus (Excel file)
Figure S1. Schematic workflow of genetic analysis, by analytic stages
Figure S2. Variants identified through exome sequencing, by MAF and imputation quality
Figure S3. Principal components analysis
Figure S4. Q-Q plot of CD association results shows enrichment of true positive signals below 10−3
Figure S5. Single-point association with CD and PD in the Ashkenazi Jewish cohort, conditioned and unconditioned on the CD-associated coding LRRK2 risk variant, N2081D
Figure S6. Log odds ratio-weighted additive risk allele burden scores
Table S1. Power calculations
Table S2. Ashkenazi Jewish enriched exome variants genotyped as custom content
Table S3. Sample cohorts description
Table S5. LRRK2 phased haplotype association
Single Sentence Summary.
A coding Crohn’s disease (CD)-associated risk variant in the LRRK2 gene, N2081D and the coding CD-protective LRRK2 N551K variant, mediate similar effects in Crohn’s disease and Parkinson’s disease.
Acknowledgments
We thank Alain Diaz of the University of Miami for technical assistance. Funding We acknowledge financial support from NIH research grants GM007205, DK098927 (to K.Y.H.), DK62429, DK062422, DK092235, DK106593 (to J.H.C.), DK062413, DK046763-19, AI067068, HS021747 (to D.P.B.M.), DK062431 (to S.R.B), AG042188 (to G.A.), NS050487, NS060113 (to L.N.C.), MH089964, MH095458, MH084098 (to T.L.), AG618381, AG021654, AG038072 (to N.B.), NS071674 (to J.M.V.), NS37167, NS036711 (to T.F.), NS076843 (to R.H.M.), NS036960 (to H.P.), DK062420 (to R.H.D.), CA141743 (to R.H.D.), CA121852 (to I.Pe’er), and NS060809 (to Z.Y.); NSF research grants 08929882 and 0845677 (to I.Pe’er); Human Frontier Science Program (to S.C.); Lewis and Rachel Rudin Foundation (to H.O.); North Shore–LIJ Health System Foundation (to T.L.); Brain & Behaviour Foundation (to T.L.); US-Israel Binational Science Foundation (to T.L.); Nathan Shock Center of Excellence for the Biology of Aging (to N.B.); the Glenn Center for the Biology of Human Aging (to N.B.); New York Crohn’s Foundation (to R.J.D; I.Peter; Y.P.); Edwin and Caroline Levy and Joseph and Carol Reich (to S.B.); SUCCESS grant (to J.H.C.; I Peter); the Sanford J. Grossman Charitable Trust (to J.H.C.); the Cedars-Sinai F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute Research Funds, the European Union, the Crohn’s and Colitis Foundation of America (CCFA), the Joshua L. and Lisa Z. Greer Chair in IBD Genetics (to D.P.B.M.); The Leona M. and Harry B. Helmsley Charitable Trust (to D.P.B.M; I.Peter), the Parkinson’s Disease Foundation (to L.N.C.), Meyerhoff Inflammatory Bowel Disease Center and the Atran Foundation (to S.R.B), University of Pittsburgh Inflammatory Bowel Disease Genetic Research Chair (to R.H.D.), The Robert P. & Judith N. Goldberg Foundation, the Bumpus Foundation and the Harvard NeuroDiscovery Center (to T.F.), The Charles Wolfson Charitable Trust (to A.W.S.;A.P.L.; E.R.S.; N.P.).Genotyping services for selected Parkinson’s disease cohorts were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C.
Footnotes
Overline: Crohn’s Disease
Author Contributions
Primary analysis and manuscript-writing (K.Y.H.); project conception and design (I.Peter; J.H.C.; R.J.D.; D.P.B.M.); patient recruitment, sample acquisition, phenotype data collection for Crohn’s disease, Parkinson’s disease, and control datasets for a total of >24,000 study subjects (N.B.; S.R.B.; A.S.C.; R.H.D.; S.K.; D.P.B.M.; J.D.R.; A.S.; N.P.; A.L.; E.R.S.; M.S.S.; S.B.; R.H.M.; L.O.; H.P.; W.K.S.; J.M.V.; T.F., G.A.; L.N.C.; T.L.; P.R.L.); data processing, preparation, and analysis (K.Y.H.; N.P.; M.R.; K.G.; S.C.; H.O.; T.H.; D.L.; M.J.D.; I. Pe’er; E.E.S.); functional study design (J.H.; Z.Y.; Y.P.;Y.I.; I.U.B.); performing experiments (H.F.H.; A.S.;J.H.; X.B.; X.L.; D.R.; N.V.; N.Y.H; L.S.C.; E.C.); manuscript writing (K.Y.H.; I.Peter; J.H.C.; J.H.; S.C.; H.F.H.; E.E.S.).
Competing Interests
RJD has consulted for Amicus Therapeutics, Alexion Pharmaceuticals, Genzyme-Sanofi, Kiniksa Pharmaceuticals,
Mitsubishi-Tanabe, Synageva Pharmaceuticals,
Recordati Rare Diseases,
Sangamo Therapeutics, and has received royalties from Shire. Shire: Royalties
YI has consulted for Neurotrope, Inc and Amathus Therapeutics, Inc.
SC has consulted for MyHeritage
ASC has consulted for AbbVie Pharmaceuticals, Janssen Pharmaceuticals, Takeda Pharmaceuticals, Pfeizer Pharmaceuticals, Ferring Pharmaceuticals, Miraca Life Sciences
DM has consulted for Janssen Pharmaceutical, UCB, Merck, Cidara, Qu Biologics.
The other authors declare no competing interests.
Data Availability: Samples from the Ashkenazi Genome Consortium are available from member institutions through a Material Transfer Agreement.
References
- 1.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, Cho J, Daryani NE, Franke L, Fuyuno Y, Hart A, Juyal RC, Juyal G, Kim WH, Morris AP, Poustchi H, Newman WG, Midha V, Orchard TR, Vahedi H, Sood A, Sung JJ, Malekzadeh R, Westra HJ, Yamazaki K, Yang SK, C. International Multiple Sclerosis Genetics, I. B. D. G. C. International. Barrett JC, Franke A, Alizadeh BZ, Parkes M, B KT, Daly MJ, Kubo M, Anderson CA, Weersma RK, I. B. D. G. Consortium Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh S, Kroe-Barrett RR, Canada KA, Zhu X, Sepulveda E, Wu H, He Y, Raymond EL, Ahlberg J, Frego LE, Amodeo LM, Catron KM, Presky DH, Hanke JH. Selective targeting of the IL23 pathway: Generation and characterization of a novel high-affinity humanized anti-IL23A antibody. mAbs. 2015;7:778–791. doi: 10.1080/19420862.2015.1032491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR, van Lookeren Campagne M. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–462. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 6.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hebuterne X, Harel-Bellan A, Mograbi B, Darfeuille-Michaud A, Hofman P. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein CN, Rawsthorne P, Cheang M, Blanchard JF. A population-based case control study of potential risk factors for IBD. Am J Gastroenterol. 2006;101:993–1002. doi: 10.1111/j.1572-0241.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, McElree C, Roth MP, Shanahan F, Targan SR, Rotter JI. Familial empirical risks for inflammatory bowel disease: differences between Jews and non-Jews. Gut. 1993;34:517–524. doi: 10.1136/gut.34.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Kenny EE, Pe’er I, Karban A, Ozelius L, Mitchell AA, Ng SM, Erazo M, Ostrer H, Abraham C, Abreu MT, Atzmon G, Barzilai N, Brant SR, Bressman S, Burns ER, Chowers Y, Clark LN, Darvasi A, Doheny D, Duerr RH, Eliakim R, Giladi N, Gregersen PK, Hakonarson H, Jones MR, Marder K, McGovern DP, Mulle J, Orr-Urtreger A, Proctor DD, Pulver A, Rotter JI, Silverberg MS, Ullman T, Warren ST, Waterman M, Zhang W, Bergman A, Mayer L, Katz S, Desnick RJ, Cho JH, Peter I. A genome-wide scan of ashkenazi jewish Crohn’s disease suggests novel susceptibility Loci. PLoS Genet. 2012;8:e1002559. doi: 10.1371/journal.pgen.1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A, Brighina L, Van Broeckhoven C, Carr J, Chartier-Harlin MC, Dardiotis E, Dickson DW, Diehl NN, Elbaz A, Ferrarese C, Ferraris A, Fiske B, Gibson JM, Gibson R, Hadjigeorgiou GM, Hattori N, Ioannidis JP, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C, Kruger R, Kyratzi E, Lesage S, Lin CH, Lynch T, Maraganore DM, Mellick GD, Mutez E, Nilsson C, Opala G, Park SS, Puschmann A, Quattrone A, Sharma M, Silburn PA, Sohn YH, Stefanis L, Tadic V, Theuns J, Tomiyama H, Uitti RJ, Valente EM, van de Loo S, Vassilatis DK, Vilarino-Guell C, White LR, Wirdefeldt K, Wszolek ZK, Wu RM, Farrer MJ, C. Genetic Epidemiology Of Parkinson’s Disease Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. The Lancet Neurology. 2011;10:898–908. doi: 10.1016/S1474-4422(11)70175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorostidi A, Marti-Masso JF, Bergareche A, Rodriguez-Oroz MC, Lopez de Munain A, Ruiz-Martinez J. Genetic Mutation Analysis of Parkinson’s Disease Patients Using Multigene Next-Generation Sequencing Panels. Molecular diagnosis & therapy. 2016 doi: 10.1007/s40291-016-0216-1. [DOI] [PubMed] [Google Scholar]
- 14.Biskup S, West AB. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson’s disease. Biochim Biophys Acta. 2009;1792:625–633. doi: 10.1016/j.bbadis.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benitez BA, Davis AA, Jin SC, Ibanez L, Ortega-Cubero S, Pastor P, Choi J, Cooper B, Perlmutter JS, Cruchaga C. Resequencing analysis of five Mendelian genes and the top genes from genome-wide association studies in Parkinson’s Disease. Molecular neurodegeneration. 2016;11:29. doi: 10.1186/s13024-016-0097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D’Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, Guthery SL, Halfvarson J, Verspaget HW, Hugot JP, Karban A, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, Mowat C, Newman W, Panes J, Phillips A, Proctor DD, Regueiro M, Russell R, Rutgeerts P, Sanderson J, Sans M, Seibold F, Steinhart AH, Stokkers PC, Torkvist L, Kullak-Ublick G, Wilson D, Walters T, Targan SR, Brant SR, Rioux JD, D’Amato M, Weersma RK, Kugathasan S, Griffiths AM, Mansfield JC, Vermeire S, Duerr RH, Silverberg MS, Satsangi J, Schreiber S, Cho JH, Annese V, Hakonarson H, Daly MJ, Parkes M. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12:1063–1070. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, Leonardson A, Castellini LW, Wang S, Champy MF, Zhang B, Emilsson V, Doss S, Ghazalpour A, Horvath S, Drake TA, Lusis AJ, Schadt EE. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, Ghosh S, de Villiers WJ, Panaccione R, Greenberg G, Schreiber S, Lichtiger S, Feagan BG, C. S. Group Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367:1519–1528. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Lee CH, Peters LA, Mastropaolo LA, Thoeni C, Elkadri A, Schwerd T, Zhu J, Zhang B, Zhao Y, Hao K, Dinarzo A, Hoffman G, Kidd BA, Murchie R, Al Adham Z, Guo C, Kotlarz D, Cutz E, Walters TD, Shouval DS, Curran M, Dobrin R, Brodmerkel C, Snapper SB, Klein C, Brumell JH, Hu M, Nanan R, Snanter-Nanan B, Wong M, Le Deist F, Haddad E, Roifman CM, Deslandres C, Griffiths AM, Gaskin KJ, Uhlig HH, Schadt EE, Muise AM. Variants in TRIM22 That Affect NOD2 Signaling Are Associated With Very-Early-Onset Inflammatory Bowel Disease. Gastroenterology. 2016;150:1196–1207. doi: 10.1053/j.gastro.2016.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farlow J, Pankratz ND, Wojcieszek J, Foroud T. In: SourceGeneReviews® [Internet] A M, Pagon RA, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. University of Washington; Seattle, WA: 2004. May 25, [updated 2014 Feb 27] [Google Scholar]
- 24.Lassen KG, McKenzie CI, Mari M, Murano T, Begun J, Baxt LA, Goel G, Villablanca EJ, Kuo SY, Huang H, Macia L, Bhan AK, Batten M, Daly MJ, Reggiori F, Mackay CR, Xavier RJ. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity. 2016;44:1392–1405. doi: 10.1016/j.immuni.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goyette P, Boucher G, Mallon D, Ellinghaus E, Jostins L, Huang H, Ripke S, Gusareva ES, Annese V, Hauser SL, Oksenberg JR, Thomsen I, Leslie S, C. International Inflammatory Bowel Disease Genetics, Australia, I. New Zealand, I. B. D. G. C. Belgium, I. B. D. G. C. Italian Group for, N. I. B. D. G. Consortium, I. United Kingdom, C. Wellcome Trust Case Control, I. B. D. G. C. Quebec. Daly MJ, Van Steen K, Duerr RH, Barrett JC, McGovern DP, Schumm LP, Traherne JA, Carrington MN, Kosmoliaptsis V, Karlsen TH, Franke A, Rioux JD. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat Genet. 2015;47:172–179. doi: 10.1038/ng.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrari R, Wang Y, Vandrovcova J, Guelfi S, Witeolar A, Karch CM, Schork AJ, Fan CC, Brewer JB, F. T. D. G. C. International, C. International Parkinson’s Disease Genomics, P. International Genomics of Alzheimer’s. Momeni P, Schellenberg GD, Dillon WP, Sugrue LP, Hess CP, Yokoyama JS, Bonham LW, Rabinovici GD, Miller BL, Andreassen OA, Dale AM, Hardy J, Desikan RS. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. Journal of neurology, neurosurgery, and psychiatry. 2017;88:152–164. doi: 10.1136/jnnp-2016-314411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife. 2016;5 doi: 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong Y, Dawson VL, Dawson TM. LRRK2 GTPase dysfunction in the pathogenesis of Parkinson’s disease. Biochem Soc Trans. 2012;40:1074–1079. doi: 10.1042/BST20120093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackeh R, Lorin S, Ratier A, Mejdoubi-Charef N, Baillet A, Bruneel A, Hamai A, Codogno P, Pous C, Perdiz D. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate alpha-tubulin acetyltransferase-1 (alphaTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J Biol Chem. 2014;289:11816–11828. doi: 10.1074/jbc.M113.507400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Pan Y, Yan R, Zeng B, Wang H, Zhang X, Li W, Wei H, Liu Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. 2015;16:918–926. doi: 10.1038/ni.3233. [DOI] [PubMed] [Google Scholar]
- 32.Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–680. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 34.Lin JC, Lin CS, Hsu CW, Lin CL, Kao CH. Association Between Parkinson’s Disease and Inflammatory Bowel Disease: a Nationwide Taiwanese Retrospective Cohort Study. Inflamm Bowel Dis. 2016 doi: 10.1097/MIB.0000000000000735. [DOI] [PubMed] [Google Scholar]
- 35.Wallings R, Manzoni C, Bandopadhyay R. Cellular processes associated with LRRK2 function and dysfunction. The FEBS journal. 2015;282:2806–2826. doi: 10.1111/febs.13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fava VM, Manry J, Cobat A, Orlova M, Van Thuc N, Ba NN, Thai VH, Abel L, Alcais A, Schurr E, T. Canadian Lrrk2 in Inflammation A Missense LRRK2 Variant Is a Risk Factor for Excessive Inflammatory Responses in Leprosy. PLoS neglected tropical diseases. 2016;10:e0004412. doi: 10.1371/journal.pntd.0004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devine MJ, Plun-Favreau H, Wood NW. Parkinson’s disease and cancer: two wars, one front. Nat Rev Cancer. 2011;11:812–823. doi: 10.1038/nrc3150. [DOI] [PubMed] [Google Scholar]
- 38.Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu TC, Naito T, Liu Z, VanDussen KL, Haritunians T, Li D, Endo K, Kawai Y, Nagasaki M, Kinouchi Y, McGovern DPB, Shimosegawa T, Kakuta Y, Stappenbeck TS. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight. 2017 doi: 10.1172/jci.insight.91917. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, Brant SR, Cho JH, Daly MJ, Dubinsky M, Duerr RH, Ferguson LR, Franke A, Gearry RB, Goyette P, Hakonarson H, Halfvarson J, Hov JR, Huang H, Kennedy NA, Kupcinskas L, Lawrance IC, Lee JC, Satsangi J, Schreiber S, Theatre E, van der Meulen-de Jong AE, Weersma RK, Wilson DC, C. International Inflammatory Bowel Disease Genetics. Parkes M, Vermeire S, Rioux JD, Mansfield J, Silverberg MS, Radford-Smith G, McGovern DP, Barrett JC, Lees CW. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387:156–167. doi: 10.1016/S0140-6736(15)00465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esteves AR, Cardoso SM. LRRK2 at the Crossroad Between Autophagy and Microtubule Trafficking: Insights into Parkinson’s Disease. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2016 doi: 10.1177/1073858415616558. [DOI] [PubMed] [Google Scholar]
- 42.Nixon-Abell J, Berwick DC, Granno S, Spain VA, Blackstone C, Harvey K. Protective LRRK2 R1398H Variant Enhances GTPase and Wnt Signaling Activity. Frontiers in molecular neuroscience. 2016;9:18. doi: 10.3389/fnmol.2016.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Hui KY, Gusev A, Warner N, Ng SM, Ferguson J, Choi M, Burberry A, Abraham C, Mayer L, Desnick RJ, Cardinale CJ, Hakonarson H, Waterman M, Chowers Y, Karban A, Brant SR, Silverberg MS, Gregersen PK, Katz S, Lifton RP, Zhao H, Nunez G, Pe’er I, Peter I, Cho JH. Extended haplotype association study in Crohn’s disease identifies a novel, Ashkenazi Jewish-specific missense mutation in the NF-kappaB pathway gene, HEATR3. Genes Immun. 2013 doi: 10.1038/gene.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peter I, Mitchell AA, Ozelius L, Erazo M, Hu J, Doheny D, Abreu MT, Present DH, Ullman T, Benkov K, Korelitz BI, Mayer L, Desnick RJ. Evaluation of 22 genetic variants with Crohn’s Disease risk in the Ashkenazi Jewish population: a case-control study. BMC Med Genet. 2011;12:63. doi: 10.1186/1471-2350-12-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, C. International Parkinson’s Disease Genomics, G. I. Parkinson’s Study Group Parkinson’s Research: The Organized, and Me, GenePd, C. NeuroGenetics Research, G. Hussman Institute of Human, I. Ashkenazi Jewish Dataset, H. Cohorts for, E. Aging Research in Genetic, C. North American Brain Expression, C. United Kingdom Brain Expression, C. Greek Parkinson’s Disease, G. Alzheimer Genetic Analysis. Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vacic V, Ozelius LJ, Clark LN, Bar-Shira A, Gana-Weisz M, Gurevich T, Gusev A, Kedmi M, Kenny EE, Liu X, Mejia-Santana H, Mirelman A, Raymond D, Saunders-Pullman R, Desnick RJ, Atzmon G, Burns ER, Ostrer H, Hakonarson H, Bergman A, Barzilai N, Darvasi A, Peter I, Guha S, Lencz T, Giladi N, Marder K, Pe’er I, Bressman SB, Orr-Urtreger A. Genome-wide mapping of IBD segments in an Ashkenazi PD cohort identifies associated haplotypes. Hum Mol Genet. 2014;23:4693–4702. doi: 10.1093/hmg/ddu158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. Journal of neurology, neurosurgery, and psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD, Hansen M, Borecki IB, Cupples LA, Fornage M, Gudnason V, Harris TB, Kathiresan S, Kraaij R, Launer LJ, Levy D, Liu Y, Mosley T, Peloso GM, Psaty BM, Rich SS, Rivadeneira F, Siscovick DS, Smith AV, Uitterlinden A, van Duijn CM, Wilson JG, O’Donnell CJ, Rotter JI, Boerwinkle E. Best Practices and Joint Calling of the HumanExome BeadChip: The CHARGE Consortium. PLoS ONE. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, Mouy M, Steinthorsdottir V, Eiriksdottir GH, Bjornsdottir G, Reynisdottir I, Gudbjartsson D, Helgadottir A, Jonasdottir A, Jonasdottir A, Styrkarsdottir U, Gretarsdottir S, Magnusson KP, Stefansson H, Fossdal R, Kristjansson K, Gislason HG, Stefansson T, Leifsson BG, Thorsteinsdottir U, Lamb JR, Gulcher JR, Reitman ML, Kong A, Schadt EE, Stefansson K. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 52.Zhong H, Beaulaurier J, Lum PY, Molony C, Yang X, Macneil DJ, Weingarth DT, Zhang B, Greenawalt D, Dobrin R, Hao K, Woo S, Fabre-Suver C, Qian S, Tota MR, Keller MP, Kendziorski CM, Yandell BS, Castro V, Attie AD, Kaplan LM, Schadt EE. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am J Hum Genet. 2010;86:581–591. doi: 10.1016/j.ajhg.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bunyavanich S, Schadt EE, Himes BE, Lasky-Su J, Qiu W, Lazarus R, Ziniti JP, Cohain A, Linderman M, Torgerson DG, Eng CS, Pino-Yanes M, Padhukasahasram B, Yang JJ, Mathias RA, Beaty TH, Li X, Graves P, Romieu I, Navarro Bdel R, Salam MT, Vora H, Nicolae DL, Ober C, Martinez FD, Bleecker ER, Meyers DA, Gauderman WJ, Gilliland F, Burchard EG, Barnes KC, Williams LK, London SJ, Zhang B, Raby BA, Weiss ST. Integrated genome-wide association, coexpression network, and expression single nucleotide polymorphism analysis identifies novel pathway in allergic rhinitis. BMC Med Genomics. 2014;7:48. doi: 10.1186/1755-8794-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoo S, Takikawa S, Geraghty P, Argmann C, Campbell J, Lin L, Huang T, Tu Z, Feronjy R, Spira A, Schadt EE, Powell CA, Zhu J. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015;11:e1004898. doi: 10.1371/journal.pgen.1004898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuiper R, Klugkist I, Hoijtink H. A Fortran 90 program for confirmatory analysis of variance. J Stat Softw. 2010;34:1–31. [Google Scholar]
- 58.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431–441. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, Hillman-Jackson J, Kuhn RM, Pedersen JS, Pohl A, Raney BJ, Rosenbloom KR, Siepel A, Smith KE, Sugnet CW, Sultan-Qurraie A, Thomas DJ, Trumbower H, Weber RJ, Weirauch M, Zweig AS, Haussler D, Kent WJ. The UCSC Genome Browser Database: update 2006. Nucleic acids research. 2006;34:D590–598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chuang LS, Villaverde N, Hui KY, Mortha A, Rahman A, Levine AP, Haritunians T, Ng SM, Zhang W, Hsu NY, Facey JA, Luong T, Fernandez-Hernandez H, Li D, Rivas M, Schiff ER, Gusev A, Schumm LP, Bowen BM, Sharma Y, Ning K, Remark R, Gnjatic S, Legnani P, George J, Sands BE, Stempak JM, Datta LW, Lipka S, Katz S, Cheifetz AS, Barzilai N, Pontikos N, Abraham C, Dubinsky MJ, Targan S, Taylor K, Rotter JI, Scherl EJ, Desnick RJ, Abreu MT, Zhao H, Atzmon G, Pe’er I, Kugathasan S, Hakonarson H, McCauley JL, Lencz T, Darvasi A, Plagnol V, Silverberg MS, Muise AM, Brant SR, Daly MJ, Segal AW, Duerr RH, Merad M, McGovern DP, Peter I, Cho JH. A Frameshift in CSF2RB Predominant Among Ashkenazi Jews Increases Risk for Crohn’s Disease and Reduces Monocyte Signaling via GMCSF. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.C. Genomes Project. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carmi S, Hui KY, Kochav E, Liu X, Xue J, Grady F, Guha S, Upadhyay K, Ben-Avraham D, Mukherjee S, Bowen BM, Thomas T, Vijai J, Cruts M, Froyen G, Lambrechts D, Plaisance S, Van Broeckhoven C, Van Damme P, Van Marck H, Barzilai N, Darvasi A, Offit K, Bressman S, Ozelius LJ, Peter I, Cho JH, Ostrer H, Atzmon G, Clark LN, Lencz T, Pe’er I. Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nature communications. 2014;5:4835. doi: 10.1038/ncomms5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guha S, Rosenfeld JA, Malhotra AK, Lee AT, Gregersen PK, Kane JM, Pe’er I, Darvasi A, Lencz T. Implications for health and disease in the genetic signature of the Ashkenazi Jewish population. Genome biology. 2012;13:R2. doi: 10.1186/gb-2012-13-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. All variants with Ashkenazi Jewish CD discovery P values < 2 × 10−5 (Excel file)
Table S6. All imputed variants with nominal CD or PD association (P < 0.05) within the LRRK2 locus (Excel file)
Figure S1. Schematic workflow of genetic analysis, by analytic stages
Figure S2. Variants identified through exome sequencing, by MAF and imputation quality
Figure S3. Principal components analysis
Figure S4. Q-Q plot of CD association results shows enrichment of true positive signals below 10−3
Figure S5. Single-point association with CD and PD in the Ashkenazi Jewish cohort, conditioned and unconditioned on the CD-associated coding LRRK2 risk variant, N2081D
Figure S6. Log odds ratio-weighted additive risk allele burden scores
Table S1. Power calculations
Table S2. Ashkenazi Jewish enriched exome variants genotyped as custom content
Table S3. Sample cohorts description
Table S5. LRRK2 phased haplotype association