

1. Introduction
1.1 Background and Scope of the Review
With over 4,700 halogenated natural products at last count[1] (approximately half with C-sp3–halogen bonds[2]), with the increasing importance of halogens in drug discovery,[3] and with the great potential of halogen-bearing stereogenic centers in asymmetric synthesis,[4] the development of methods for the stereocontrolled introduction of carbon–halogen bonds is a vibrant area of research. From long-known stereospecific anti-deoxyhalogenations of chiral secondary alcohols to modern methods for asymmetric alkene halogenation, a vast array of methods and tactics for the stereoselective synthesis of halogenated molecules has been amassed. In this review, we do not aim for a comprehensive treatment of methods for stereocontrolled halogenation; rather, we have chosen to focus largely on the strategies and methods utilized in the stereocontrolled synthesis of halogenated natural products, with a few digressions that we feel are justified for pedagogical reasons, or because some particularly new and promising advances have not yet seen application in natural product synthesis. To keep the review to a reasonable scope, the following very interesting topics will not be discussed: (1) the stereocontrolled synthesis of halogenated alkenes, and (2) the stereoconrolled introduction of (poly)halomethyl groups. Still, it is our hope that this review will provide a broad overview of the state-of-the-art in the field of stereocontrolled halogenation, and that the information accumulated and critically reviewed herein will be of significant utility to chemists working on halogenation methods, natural product synthesis, drug discovery, and more.
There is an incredible breadth of structural types among halogenated natural products. Often, unusual synthesis strategies have been adopted in order to deal with complex structural targets that are further complicated by the presence of the halogens; in other words, strategies that might be perfectly amenable to a non-halogenated congener would find the presence of halogens to be a liability to key transformations. As a result, strategies that are “halophobic”—in that the halogens are installed in reliable late-stage transformations (for example, alcohol deoxyhalogenation)—differ largely from strategies that are “halophilic” and embrace the halogens as opportunities to orchestrate key bond constructions. Indeed, in certain instances, the presence of the halogen can provide occasions to innovate.
Stereochemical control is critical in addressing the efficient and selective synthesis of halogenated natural products wherein the halogens define an element of stereogenicity. To effect stereocontrolled halogenation, chemists have relied on stereospecific reactions for absolute stereocontrol (SN2 displacements, for example) and relative stereocontrol (alkene halogenation and halofunctionalization), stereoselective reactions under substrate control, and stereoselective reactions under reagent and catalyst control, among others. In this review, we try to organize our discussion according to the underlying mechanism of halogenation, which unfortunately does not readily permit a logical progression with respect to the mechanisms of stereocontrol.
We applaud all of those who have made the incredible advances discussed below. Of course, we must apologize at the outset to any workers in this area whose work we did not discuss, either because of space limitations (this review is not meant to be comprehensive) or because of oversight on our part. We are also compelled to apologize for not including citations to all of the incredible isolation and structural elucidation work that provided the incredible structures that inspired the synthetic chemistry that we discuss at length.
1.2 Halogenation in Natural Product Biosynthesis
The known mechanisms of biosynthetic halogenation have been extensively reviewed elsewhere;[5] however, it is helpful to contextualize the purely chemical strategies for stereocontrolled halogenation that are the focus of this review with a brief overview of nature’s approaches to halogenation. In cases where possible, the advances discussed in the body of this review will be related to the ideas described in this section.
Most known instances of biosynthetic halogenation concern the chemistry of the halenium cation or its equivalent, generated by oxidation of halide anion via a handful of different enzymatic processes. The most important classes of enzymes that accomplish these transformations are the haloperoxidases,[6] which generate Cl+ and Br+ equivalents, likely as metal-coordinated O–X ligands, with heme iron and vanadium as the key metals. Necessarily, these halogenation reactions operate on electron-rich (nucleophilic) substrates that react with the generated electrophile in the same manner as chemists are accustomed to seeing in chemical synthesis: electrophilic aromatic substitution (halogenation), haloetherification and its close relatives, and halenium-induced π-cyclizations. Three examples of natural products thought to arise via the action of haloperoxidases are shown in Figure 1. Additionally, some natural products appear to be the products of alkene halogenation or interhalogenation. Owing to the high relative abundance of chloride and the reasonable prevalence of bromide coupled with its ease of oxidation, most natural products thought to arise from haloperoxidase-based biosyntheses contain either chlorine or bromine residues (or both). The poor abundance of iodide results in a relative dearth of naturally occurring organoiodides from haloperoxidases (the mammalian thyroid hormones are interesting examples) and the extremely difficult oxidation of fluoride prevents their incorporation in this way.
Figure 1.
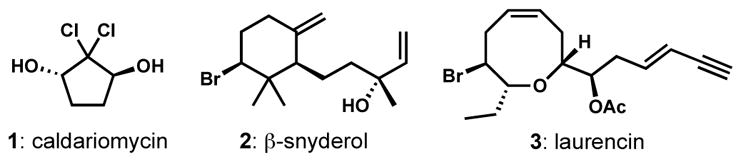
Representative natural products that arise from biosynthetic electrophilic halogenation by haloperoxidase enzymes
A second general mechanism for biosynthetic halogenation involves radical C–H functionalization manifolds. Those reactions of this type that have been well characterized are typically catalyzed by α-ketoglutarate-dependent non-heme iron enzymes that operate similarly to the structurally related monooxygenase enzymes but with an active site that has evolved to bind a halide; a rebound mechanism accounts for halogenation in face of possible hydroxylation.[7] There has been significant work in this area of late because of the remarkable transformations that these enzymes perform, including the selective monochlorination of threonine by the enzyme SyrB2 (see 4, Figure 2) and the highly selective di- or trichlorination of one of the diastereotopic methyl groups of leucine by the combination of BarB1 and BarB2, which provides precursors to natural products such as barbamide (5). Very recently, the chlorine residues in members of the hapalindole and fischerindole family of alkaloids (see 6) have been shown to arise from late-stage chlorination by the α-ketoglutarate-dependent non-heme iron enzyme WelO5,[8] which differs from SyrB2 and BarB2 in that the substrate is not covalently bound in the form of an aminoacylated peptidyl carrier protein. In other words, WelO5 site-specifically and stereoselectively chlorinates soluble small molecule precursors to the chlorinated alkaloids.
Figure 2.
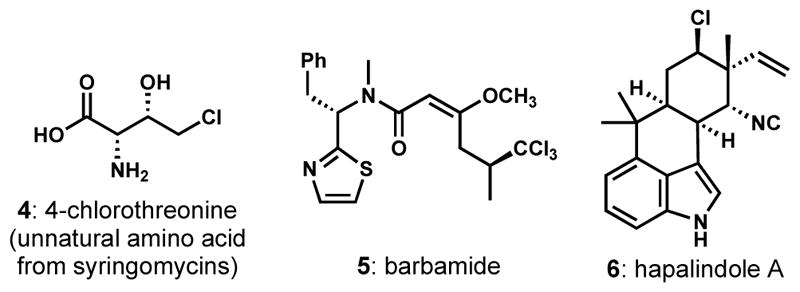
Representative products produced by α-ketoglutarate-dependent non-heme iron halogenases
Interestingly, experimental observations suggest that the chlorination events that form the chlorosulfolipids, such as danicalipin A (7, Figure 3), are radical in nature.[9,10] It seems likely that a small family of chlorinases installs these chlorides one at a time to ultimately yield these very highly halogenated and unusual natural products. The enzymatic halogenation process is not at all understood.
Figure 3.
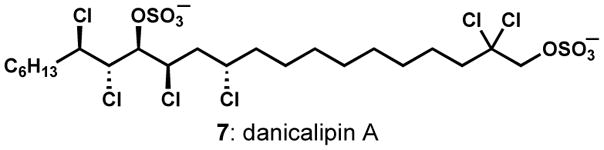
Danicalipin A, a representative chlorosulfolipid
Finally, there are some natural products understood to arise from biosynthetic nucleophilic halogenation, but they are rare in comparison with those that are made via electrophilic and radical halogenation processes. Of course, synthetic chemists have relied heavily on just this type of reaction to introduce halogen atoms, but the availability of a plethora of activating groups for alcohols and of organic solvents, in which the halide nucleophile is much less hindered by solvation, makes such reactions quite practical.
It is noteworthy that practitioners of chemical synthesis have made substantial use of each of these general reaction types: electrophilic, radical, and nucleophilic halogenations are pervasive in the literature. However, while radical intermediates have been used to introduce halogens in syntheses of halogenated natural products, no examples of direct C–H halogenation—as the counterpart to the enzymatic reactions discussed above—are yet known. Of course, chemists have also developed numerous halogenation reactions that do not have their counterpart in biosynthesis.
It should be mentioned that in many cases, the identity of the substrate of biological halogenation is not known with certainty; therefore, when chemists engage in biogenetically-inspired synthesis of halogenated natural products, it stands to reason that the laboratory substrates will often differ from the natural ones. In several cases in this review, when strong circumstantial evidence or close analogy to well-understood systems is available, we might use language reflecting confidence in a biosynthetic mechanism that might not have been rigorously validated experimentally. This approach is taken for conciseness and clarity of the discussion of the synthesis work.
2. Stereoselective Halogenation Methods and Applications in Synthesis
2.1 Nucleophilic Displacement
2.1.1 SN2 Displacement of Activated Alcohols
The SN2 displacement of activated alcohols with halide ions has been utilized myriad times for the straightforward installation of halogen-bearing stereocenters. As a result, the discussion will be limited to one particular context that demonstrates the potential hazards of relying upon such a transformation in sterically hindered contexts. While unencumbered secondary alcohols often undergo such deoxyhalogenation reactions with high efficiency, this approach suffers in more demanding situations because of possible halide equilibration and elimination, and predicting the reactivity of sterically encumbered substrates can be difficult. During the syntheses of (±)-prepinnaterpene (8, Scheme 1) and (±)-oppositol (9), Masamune and co-workers attempted the SN2 substitution of the neo-pentylic secondary alcohol 10, activated as its mesylate, with tetra-n-butylammonium bromide.[11] They obtained a diastereomeric mixture favoring equatorial bromide 11 (overall retention of configuration) over desired axial bromide 12, which was characterized as “labile”, possibly owing to facile elimination to the elimination product 13, which was also observed. At a lower temperature (80 °C, reaction not shown), the axial bromide 12 was the major product, suggesting that the more stable equatorial bromide 11 was formed through double inversion. To obtain the desired configuration under the halide equilibration conditions, the bromide substituent was relocated from an equatorial position to an axial position via epimerization of the cis-fused bicyclic skeleton to a trans-hydrindane scaffold via a few synthetic manipulations. The resulting axial bromide 14 was then converted to the more stable equatorial bromide 15 via substitution with tetra-n-butylammonium bromide. The formation of a substantial amount of elimination product 16 further diminished the efficiency of this strategy. Because of the preference for the more stable stereoisomer under these thermodynamically controlled conditions, the configuration of the reacting carbon center in the starting material is inconsequential. Kim and co-workers generated analogous equatorial bromide 18 following Masamune’s conditions starting from equatorial mesylate 17.[12] Whereas Masamune installed the bromide in a cis-hydrindane substrate to give the undesired diastereomer as a major product, which had to be inverted later in a trans-hydrindane substrate, Kim was able to obtain the desired bromide directly in a trans-hydrindane substrate. Incomplete double inversion and/or the substantial formation of elimination products diminished the efficiency of these processes, and serve as a cautionary tale in the displacement of sterically congested leaving groups.
Scheme 1.

Thermodynamically controlled halogenation via SN2 halide equilibration
2.1.2 Halogenative SN2′ Displacements
Halogen exchange reactions can also take place in a net allylic substitution fashion. In synthetic studies toward (±)-violacene (22, Scheme 2), Williard and co-workers attempted syn-dichlorination of allylic bromide 21 with phenyliodonium dichloride, which was known to effect syn-dichlorination of a cholesterol derivative.[13] However, the unexpected loss of bromine and the migration of the double bond resulted in the formation of a related natural product (±)-epi-plocamene (23) instead.[14] While the phenyliodonium dichloride reagent can effect alkene dichlorination via polar or radical mechanisms, depending upon the reaction conditions and substrate, it is not clear how the net allylic displacement occurred in this instance. The indirect introduction of the syn-dichloride of 22 via epoxide and chlorohydrin intermediates also failed because of the difficulty associated with the displacement of hindered tertiary alcohols.
Scheme 2.
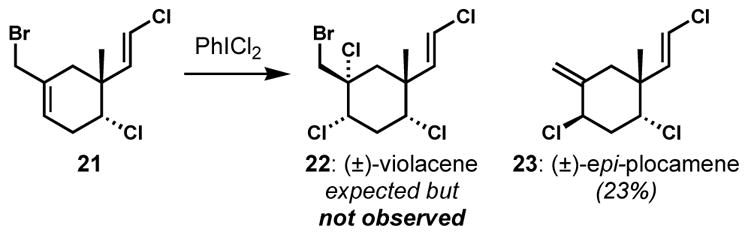
Allylic halogen displacement during attempted syn-dichlorination
The stereospecific anti-SN2′ displacement approach has been most widely employed for the late stage installation of bromoallenes from propargylic alcohol precursors. Although methods are now available for the synthesis of chiral secondary propargylic alcohols by enantioselective reduction of the corresponding propargylic ketones or by asymmetric acetylide additions to aldehydes, the stereoselective preparation of such precursors used to be a challenging problem. In addition, competing SN2 displacement pathways in the halogenation event have to be suppressed.[15] Selective bromoallene formation was first successfully accomplished by Overman and co-workers in their synthesis of (±)-kumausallene (27) (Scheme 3).[16] The propargylic alcohol 25 was synthesized with moderate diastereoselectivity by the titanium tetrachloride-promoted addition of a titanium trimethylsilylacetylide reagent to the aldehyde 24 under Felkin–Anh stereocontrol. The major diastereomer was isolated and activated as the corresponding sulfonate ester after removal of the trimethylsilyl group. It was critical to employ a bulky sulfonate group for the activation of the propargylic alcohol in order to suppress the undesired SN2 displacement pathway. Whereas bromination of the mesylate of the terminal alkyne derived from 25 with LiCuBr2 provided the desired bromoallene 26 (ca. 50%) along with a substantial amount of the propargylic bromide (ca. 25–30%), the use of the corresponding trisylate attenuated the formation of propargylic bromide (ca. 10–15%) and improved the yield of 26 (73%, >15:1 dr).
Scheme 3.

The preparation of a bromoallene via SN2′ displacement of an activated propargylic alcohol
The stereoselective preparation of propargylic alcohol substrates is not always straightforward. When Crimmins and co-workers attempted the direct conversion of the aldehyde 28 to the propagylic alcohol 31 for the synthesis of (−)-isolaurallene (33), the carbonyl addition of ethynylmagnesium bromide was unselective (Scheme 4).[17] Precedent suggests that the use of a titanium acetylide reagent would favor formation of the undesired diastereomer. Thus, the requisite intermediate 31 was prepared in a diastereomerically pure form via a longer sequence including Wittig olefination, Sharpless asymmetric epoxidation, and elimination of a chloroepoxide intermediate 30. Then, 31 was transformed to the desired bromoallene 32 following Overman’s procedure (8:1 dr, 9% SN2).
Scheme 4.

Difficulty in stereoselective formation of propargylic alcohol (SAE = Sharpless Asymmetric Epoxidation)
When only the undesired diastereomer of propargylic alcohol can be obtained with high stereoselectively, the desired diastereomer can be accessed through the inversion of the alcohol-bearing stereocenter via the Mitsunobu reaction (Scheme 5). Whereas Pagenkopf and co-workers could prepare the undesired anti-diastereomer of propargylic alcohol 34 by the substrate-controlled addition of an alkynyltitanium reagent to the aldehyde, attempts to produce the desired syn-diastereomer was unsuccessful even under reagent-controlled, asymmetric alkynylation conditions.[18] Eventually, the syn-configuration was generated via the Mitsunobu reaction of 34. Through these synthetic studies, the structure of (−)-aplysiallene was revised as 36. Alternatively and more efficiently, a direct installation of sulfonate with inversion of configuration was demonstrated by Kim and co-workers for the synthesis of microcladallene B (39).[19] After the diastereoselective reduction of a ynone intermediate 37 under Felkin–Anh control, the invertive trisylation was accomplished under the Mitsunobu conditions.[19,20] Kim and co-workers also revised the structure of (+)-itomanallene A through total synthesis employing the SN2′ approach.[21]
Scheme 5.
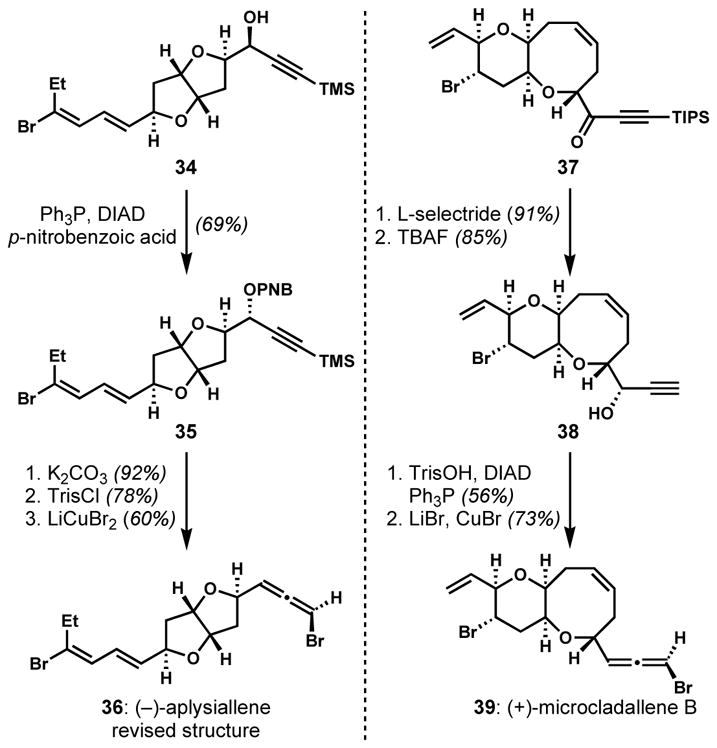
Correction of propargylic alcohol configuration via the Mitsunobu reaction
The versatility of the SN2′ approach was greatly improved by the application of chiral alkynylating reagents to overcome the well-precedented challenges in substrate-controlled alkynylation of aldehydes. Boukouvalas and co-workers employed enantioselective alkynylations[22] to produce both diastereomers (42 and 44, Scheme 6) of the propargylic alcohol precursor with high stereoselectivity under reagent control.[23] After converting 42 and 44 to their corresponding bromoallene epimers 43 and 45 following Overman’s protocol, the relative and absolute configurations of (−)-panacene were unambiguously assigned as 45.
Scheme 6.
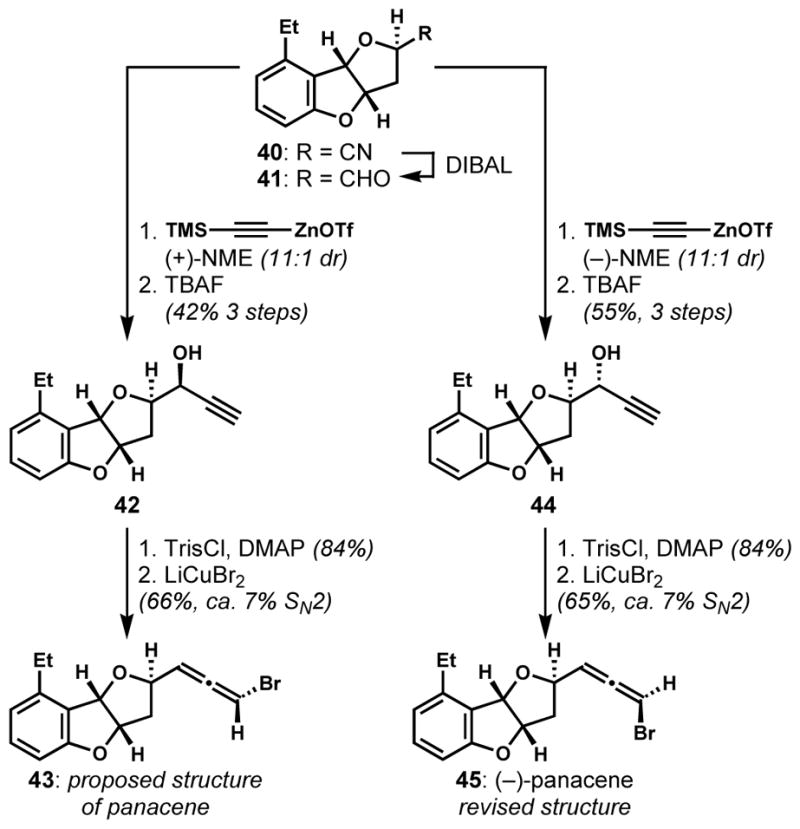
Stereodivergent synthesis via a reagent-controlled alkynylation
2.1.3 Nucleophilic Halogenolysis of Strained Rings
Halogen-bearing stereogenic centers can be introduced in a highly stereoselective manner via SN2 opening of epoxides with halide ions.[24] However, in this case, regioselectivity needs to be addressed because an epoxide has two reactive, electrophilic carbon centers. Two major strategies exist for regiocontrol with sterically undifferentiated 1,2-disubstituted epoxides: (1) inductive deactivation of one terminus by the proximity of electron-withdrawing groups, and (2) activation of one terminus of the epoxide, for example with vinyl epoxides. While direction of the halide nucleophile to one terminus of the epoxide cannot be ruled out in all cases, regiocontrol in the vast majority of ring-opening epoxide halogenolyses can be accounted for by electronic effects.
By modifying Sharpless’s conditions for nucleophilic ring openings of 2,3-epoxyalcohols,[25] Murai and co-workers developed general solutions for the site-selective openings of 3,4- or 2,3-epoxy alcohols and derivatives with halide nucleophiles.[26] Whereas a combination of a diethylammonium halide salt and titanium tetraisopropoxide is effective for the distal selective ring openings of unprotected epoxy alcohol substrates, diethylaluminum halide has been the reagent of choice for the openings of protected epoxy alcohol substrates. These conditions were employed as key steps for the synthesis of halogenated tetrahydropyran natural products, (−)-dactylyne (50) and (−)-isodactylyne (51) (Scheme 7).[26d] The 3,4-cis-epoxy alcohol intermediate 46 was treated with diethylammonium bromide in the presence of titanium tetraisopropoxide to give the bromohydrin 47 with high site-selectivity. At a later stage, a 2,3-trans-epoxy mesylate 48, which was prepared via Sharpless asymmetric epoxidation, was converted to the chlorohydrin 49 with perfect regioselectivity upon treatment with diethylaluminum chloride. The electron withdrawing mesylate group provides a substantial electronic bias via the selective deactivation of the proximal epoxide carbon center. Similar methods employing halotitanium triisopropoxide have also been developed by Raifeld and co-workers[27] and were recently employed by Carreira and coworkers on epoxyalcohol 52 en route to the synthesis of the reported structure of mytilipin C (54) (Scheme 8).[28]
Scheme 7.
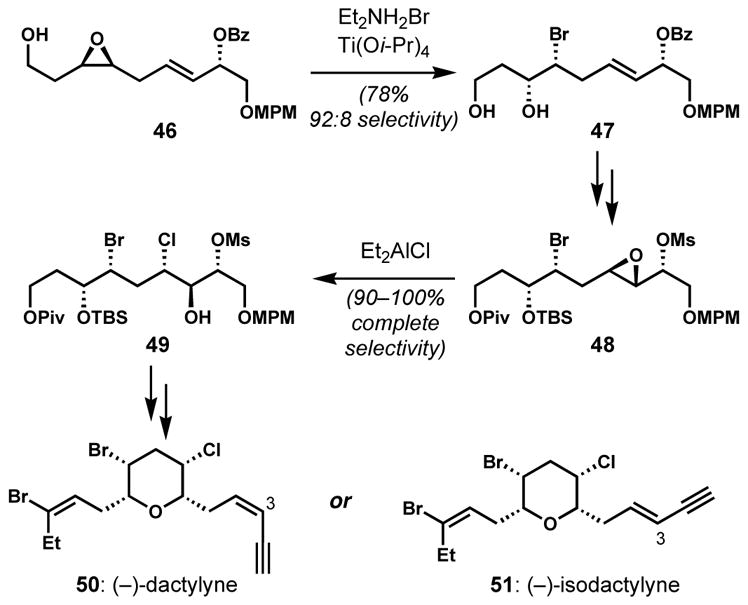
Stereoselective halogenations via regioselective epoxide halogenolyses en route to dactylyne and isodactylyne
Scheme 8.
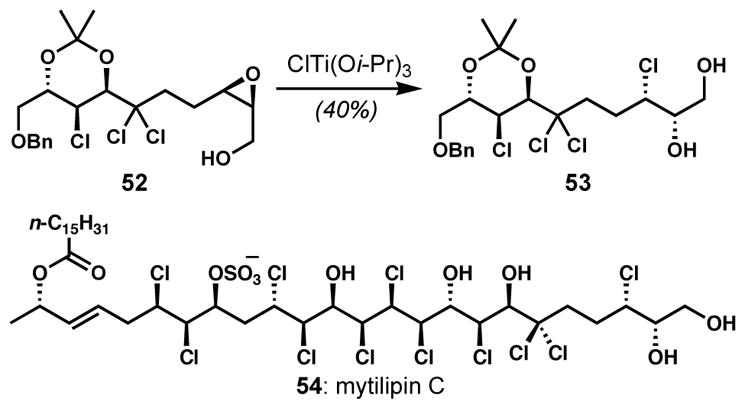
Application of site-selective epoxide opening to the synthesis of the chlorosulfolipid mytilipin C
The site-selective epoxide opening strategy was employed by Overman and co-workers for the syntheses of (−)-laurenyne (57)[29] and (+)-isolaurepinnacin (60)[30] (Scheme 9). In both cases, the cis-epoxides 55 and 58 were prepared via Sharpless asymmetric epoxidation. Then, the corresponding halohydrins 56 and 59 were produced using Sharpless’s and Murai’s conditions, respectively. Although the absolute stereochemical outcome of halohydrin formation relies on enantioselective epoxidation methods, regiocontrolled halogenolysis is critical to efficiency of the overall strategy.
Scheme 9.
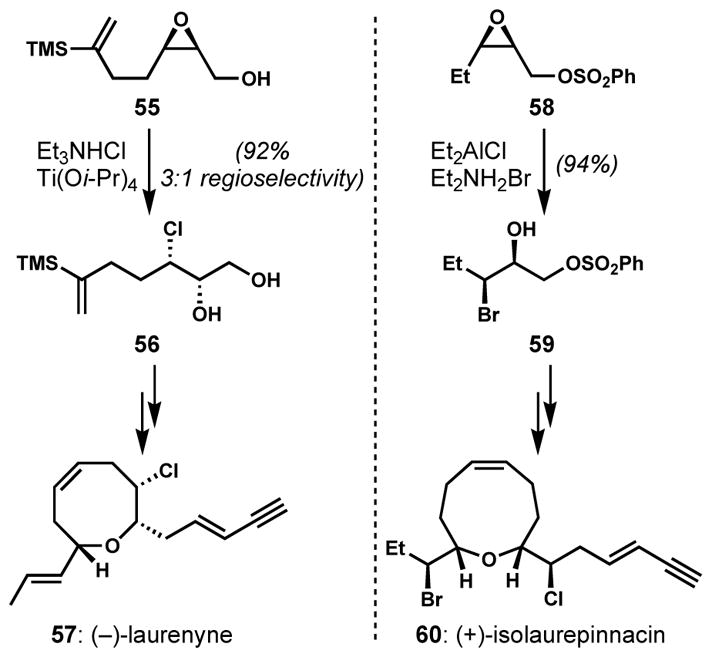
Applications of regioselective and stereospecific epoxide ring-opening to halogenated natural product synthesis
Unfortunately, a complementary method does not yet exist for the opening of epoxy alcohols with alternative regioselectivity. During the synthesis of the reported structure of mytilipin C (54, Scheme 8) by Carreira and co-workers, the desired chlorohydrin 63 (Scheme 10) could not be directly accessed from the secondary epoxy alcohol 61 via Murai’s method, which would give the undesired constitutional isomer.[26a] Thus, 63 was prepared via a longer sequence involving temporary oxidation of 61 to the corresponding epoxy ketone 62, the subsequent opening of the activated α-terminus with zirconium tetrachloride, and the immediate reduction of the unstable ketone.
Scheme 10.
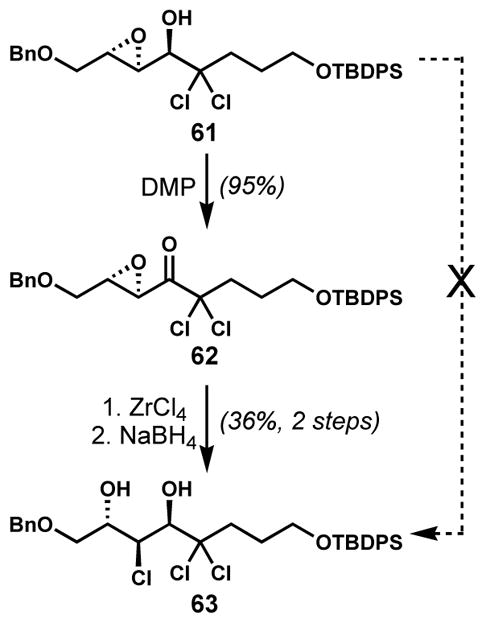
Access to opposite regioselectivity of epoxide chlorinolysis via an intermediate keto-epoxide
Vinyl epoxide halogenolysis is predictably regiocontrolled, with the allylic terminus activated for ring-opening. Examples from recent chlorosulfolipid syntheses demonstrate both the reliable regiocontrol and the potential complications when working with polychlorinated substrates. First reported by the Carreira group in their synthesis of mytilipin A, unexpected anchimeric assistance of one of the distal chlorides in 64 (Scheme 11) led to the undesired C5-configuration in 66 by overall halogenolysis with retention of configuration, and ultimately the formation of a stereoisomer of the natural product.[31] Seizing on this unusual reactivity, this group changed the configuration of their starting epoxide (see 68); the net stereoretentive epoxide chlorinolysis afforded the desired C5 configuration in 65 and permitted completion of the synthesis of target 69. This reactivity was also seen by the Vanderwal group with epoxide 70 en route to danicalipin A (7, Figure 3); the undesired reactivity was prevented with the use of an excess of soluble chloride and a change of Lewis acid.[32] Carreira and co-workers recently performed a careful mechanistic study, implicating the likely intermediacy of the four-membered ring cyclic chloronium ion (67) rather than the five-membered ring isomer.[33]
Scheme 11.

Unexpected anchimeric assistance by distal chloride residues in vinyl epoxide chlorinolyses en route to chlorosulfolipid natural products
Openings of other types of reactive three-membered rings have also been utilized for the stereoselective introduction of halogen atoms. Fukuyama and co-workers installed the chloride-bearing stereocenter of (−)-hapalindole G (76) via opening of activated cyclopropane intermediate 74, which was prepared from diazomalonate 73 through copper(II) bis(salicylidene-tert-butylamine)-catalyzed intramolecular cyclopropanation (Scheme 12).[34] Upon heating in the presence of lithium chloride and camphorsulfonic acid, 74 underwent chlorinative ring opening followed by decarboxylation to give the hindered, neo-pentylic chloride 75 in a site-selective and stereospecific manner.
Scheme 12.
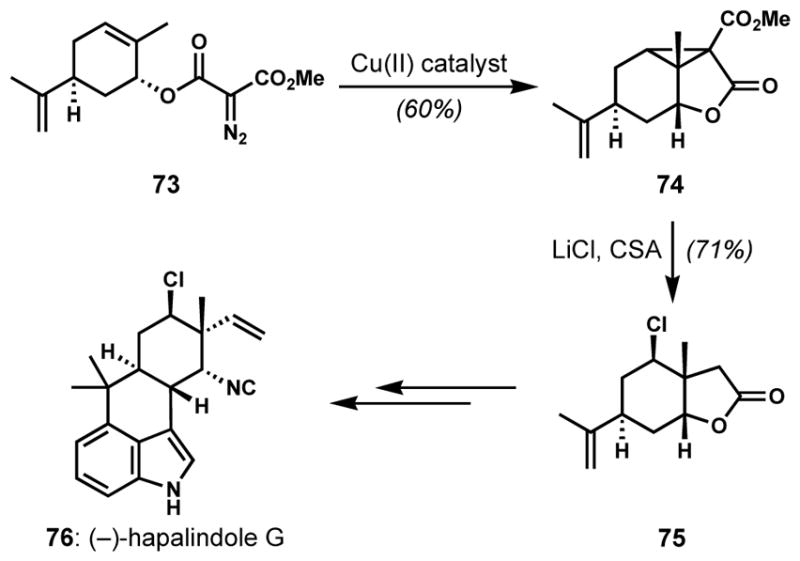
Stereoselective chlorination via site-selective opening of a cyclopropane intermediate
During the synthesis of (±)-virantmycin (80) by Raphael and co-workers, a stereoretentive chlorination of a secondary alcohol 77 delivered 78 upon treatment with thionyl chloride (Scheme 13).[35] This unexpected result was rationalized by the involvement of an aziridine intermediate and the following in situ opening with chloride ion, which would result in double inversion. This hypothesis was confirmed by Shirahama and co-workers through the preparation of the putative aziridine intermediate 79 under Mitsunobu conditions.[36] Subsequent ester hydrolysis and chlorinative opening of the aziridine ring with tetraethylammonium chloride and trifluoroacetic acid afforded the unnatural enantiomer (+)-virantmycin (80).
Scheme 13.
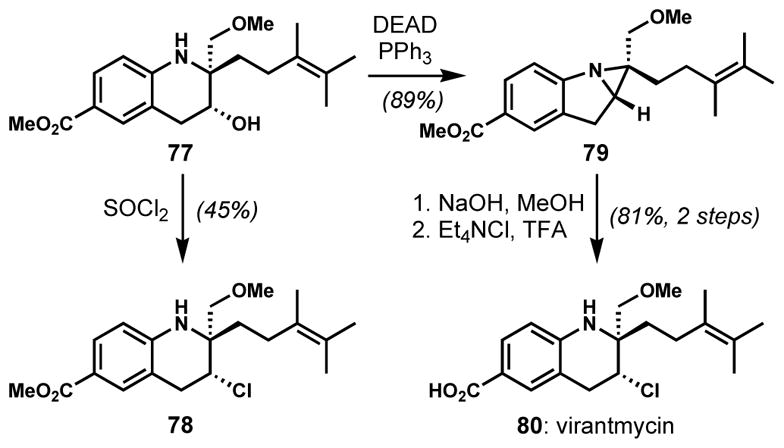
Stereoretentive chlorination via an aziridine intermediate. Although Raphael and co-workers made (±)-virantmycin and Shirahama and co-workers made unnatural (+)-virantmycin, the natural configuration is shown.
Hanessian and co-workers investigated site-selective openings of aziridines for the synthesis of chlorodysinosin A (84, Scheme 14), which contains an unprecedented (2S,3R)-3-chloroleucine residue.[37] Under the optimized conditions, both N-t-butylsulfonyl aziridines 81 and 82 underwent highly regioselective ring opening in the presence of an excess amount of cerium(III) chloride heptahydrate to deliver (2S,3R)-N-t-butylsulfonyl-3-chloroleucinol (83); deprotection of the t-butyldimethylsilyl group of 82 occurred under the reaction conditions. Although it might be expected that the electronic bias provided by the hydroxyl/silyloxy groups might be enough to encourage high levels of regiocontrol, experiments with smaller sulfonamide protecting/activating groups led these workers to attribute the high regioselectivity to the steric encumbrance between t-butylsulfonyl and isopropyl groups. For example, when the smaller trifluoromethylsulfonyl group was employed, a 1:1 mixture of constitutional isomers was obtained.
Scheme 14.
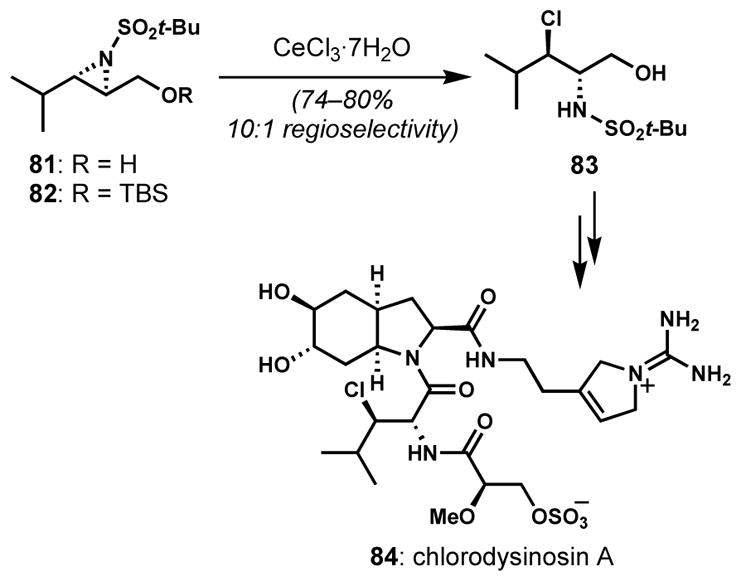
Stereocontrolled chlorination via regioselective opening of an activated aziridine
A three-membered oxonium species has been proposed as an in situ generated, highly reactive intermediate that can determine the stereoselectivity of the subsequent halogenative opening. In synthetic studies toward (±)-laurencial (90) by Iwata and co-workers, two epimeric tertiary alcohols 85 and 88 were accessed (Scheme 15).[38] Both compounds were treated with thionyl chloride in the presence of a catalytic amount of zinc chloride in dioxane.[39] These conditions are known to favor retention of configuration, and axial chloride 86 was obtained from the axial alcohol 85. However, the equatorial alcohol 88 also afforded 86 as the major product along with the expected equatorial chloride 89 with ca. 2:1 selectivity. This observed stereoconvergence was rationalized by the intermediacy of a three-membered oxonium species formed via participation of the neighboring ether oxygen. Thus, the invertive pathway was dominant in the case of 88. The epimeric chlorides 86 and 89 were converted to (±)-3-epilaurencial (87) and (±)-laurencial (90), respectively.
Scheme 15.
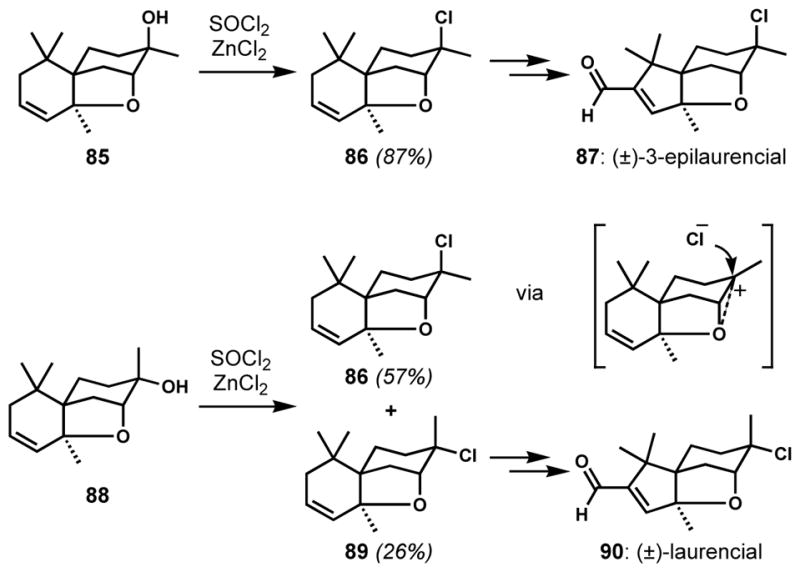
Stereoconvergent chlorination via a three-membered oxonium species
Chlorinolysis need not be restricted to three-membered ring substrates. A key step in the synthesis of (−)-N-methylwelwitindolinone B isothiocyanate (93, Scheme 16) by Garg and co-workers involved ether chlorinolysis in a particularly complex setting.[40] Installation of the neopentylic secondary alkyl chloride was accomplished by ring-opening of the ether bridge in the bridged oxabicyclic substructure of 91; not surprisingly, chloride introduction via SN2 displacement of related activated alcohols was unsuccessful; such a proposition was shown to be extremely challenging by Bhat and Rawal.[41] The successful regioselective chlorinative ring-opening provided exclusively the desired constitutional isomer of oxabicyclic intermediate 92. The Lewis acidic conditions shown were previously reported by Shea and co-workers in their synthetic studies toward the synthesis of 93.[42] Among a variety of Brønsted and Lewis acids, boron trichloride was uniquely effective for these systems.[43]
Scheme 16.
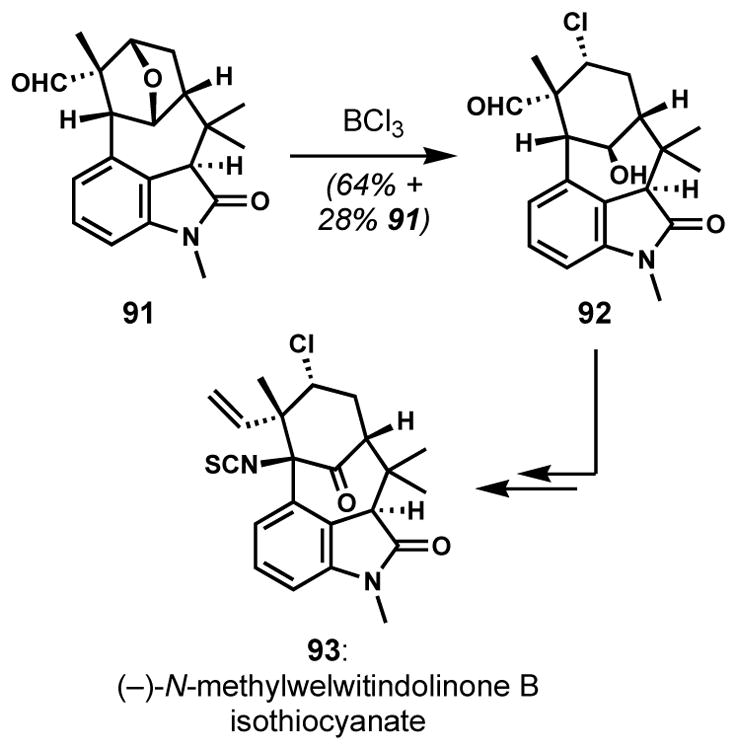
Preparation of hindered secondary chloride via opening of a bicyclic ether
2.2 Stereoselective Alkene Halogenation
There are a relatively small number of natural products containing vicinal dihalide arrangements. The two most visible families of natural products that contain vicinal dihalides are the chlorosulfolipids (see 7, 54, and 69 above, and malhamensilipin A, 94, Figure 4) and the polyhalogenated monoterpenoids from red algae (see 22 above, and 95 and 96). Vicinal dichlorides in cyclic contexts can also be found in the clionastatins and dichlorolissoclimide and congeners. Largely owing to great recent interest in acyclic polyhalogenated compounds as targets for chemical synthesis, there have been many recent methodological and strategic advances in the stereocontrolled introduction of vicinal dihalides.
Figure 4.
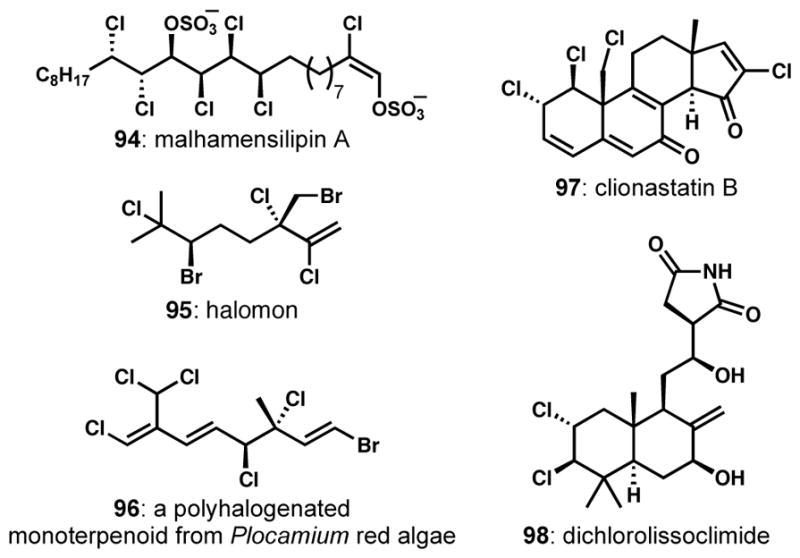
Representative natural products bearing vicinal dihalide motifs
2.2.1 Diastereoselective Dichlorination
The chlorosulfolipids are a family of polychlorinated lipids that bear sulfate esters in the midst of their chlorine-rich regions (see malhamensilipin A, 94). Inspired by its potential value to syntheses of these targets, Vanderwal and co-workers developed a diastereoselective vicinal dichlorination of chiral secondary Z-allylic alcohol derivatives (Scheme 17).[44] A wide variety of substituent groups on the alcohol were examined to modulate the steric and electronic environment and provide high diastereoselectivity. While the reaction was relatively insensitive to steric bulk, electron-deficient substituents such as trifluoro- and trichloroacetates were beneficial for high diastereoselectivity and clean reactivity without the unwanted participation of the carbonyl group, which was a problem for less electron-deficient acyl groups. Under the optimized conditions, the syn,syn-hydroxydichloride stereotriad, which is commonly found in chlorosulfolipids, was obtained by the anti-dichlorination of a range of structurally diverse Z-allylic trichloroacetates with Mioskowski’s reagent (tetraethylammonium trichloride) with high diastereoselectivities. The trichloroacetyl group could be cleanly removed without interference of the chloride residues such as epoxide formation from the resulting chlorohydrins. This reaction was applied to an anti-chlorohydrin substrate, generating product 101 with four contiguous stereogenic centers found in mytilipin A (69). The mechanisms of these reactions with the trichloride reagent are poorly understood and likely to be complex. As a result, anything more than pure speculation on the mechanism of stereocontrol, which likely does involve A1,3-strain minimization, cannot be made without further investigations.
Scheme 17.
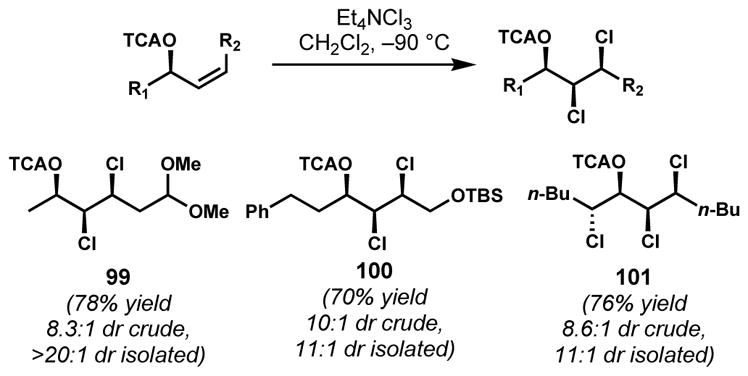
syn,syn-Selective dichlorination of chiral secondary Z-allylic trichloroacetates (TCA = trichloroacetyl)
The “left-hand” stereotriad of malhamensilipin A (94) has the syn,anti-configuration, which could not be accessed from a chiral Z-allylic alcohol derivative via Vanderwal’s diastereoselective dichlorination method. Because of the attractiveness of syn-diol precursors, which could be prepared enantioselectively via Sharpless asymmetric dihydroxylation, a range of derivatives was evaluated for diastereoselective dichlorination.[45] Unexpectedly, highly syn,anti-selective dichlorination was achieved with α-nosylate 102 (Scheme 18); fortuitously, the sulfate in the product 103 was exactly that required for a subsequent epoxide formation. Although remarkable for its influence on the stereochemical outcome, the role of the remote nosyl group remains unclear. This study led to the enantioselective synthesis of (+)-malhamensilipin A.
Scheme 18.
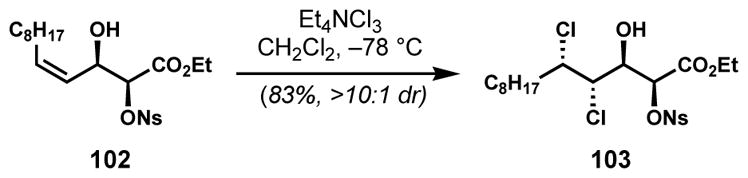
Diastereoselective dichlorination of Z-allylic alcohol with a remote nosyl ester
Diastereoselective dichlorination of Z-allylic alcohol derivative 104 was employed for the synthesis of reported structure of mytilipin C (54) by Carreira and co-workers (Scheme 19).[28] Whereas poor diastereoselectivities (ca. 2:1 dr) were observed in Vanderwal’s studies of simple Z-allylic t-butyldimethylsilyl ethers,[44] the dichlorination of complex substrate 104 under analogous conditions afforded the desired syn,syn-105 in good diastereoselectivity.
Scheme 19.
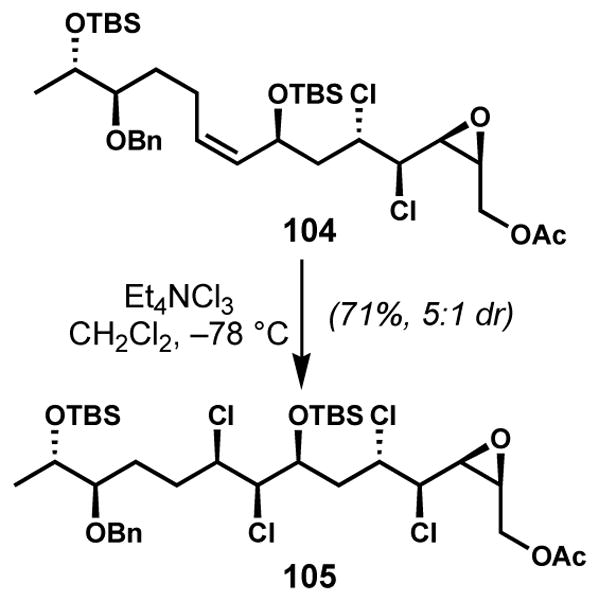
syn,syn-Selective dichlorination of a chiral secondary Z-allylic t-butyldimethylsilyl ether
Highly diastereoselective dichlorination of chiral secondary Z-allylic chlorides has been used in several instances for the synthesis of the chlorosulfolipids after first being described by Carreira and co-workers for the synthesis of (±)-mytilipin A (69).[31] The dichlorination of Z-allylic chloride 106 with Mioskowski’s reagent proceeded in high diastereoselectivity (Scheme 20). The utility of this transformation was further demonstrated by Vanderwal and co-workers for the enantioselective syntheses of (−)-mytilipin A (69)[46] and (+)-malhamensilipin A (94).[45] In the latter synthesis, Z-allylic chloride 108 was dichlorinated selectively to give 109. Recently, this method was employed with a more complex substrate during the synthesis of the reported structure of mytilipin C (59).[28]
Scheme 20.

syn,syn-Selective dichlorination of chiral secondary Z-allylic chlorides
At a late stage of their synthesis of (+)-mytilipin A (69), Yoshimitsu and co-workers investigated the diastereoselective installation of the “left-hand” anti-dichloride moiety which required diastereoselective dichlorination of a secondary E-allylic alcohol and derivatives (110/111, Scheme 21).[47] For this study, the Markó–Maguire reagent system was employed to generate an uncharacterized active dichlorinating reagent, possibly benzyltriethylammonium trichloride.[48] While stereocontrolled dichlorination of secondary Z-allylic alcohol derivatives might be explained via minimization of A1,3-strain, it is not straightforward to rationalize the observed diastereoselectivity of dichlorination of secondary E-allylic alcohol derivatives because of less severe allylic strain. Whereas the dichlorination of trichloroacetate derivative 110 afforded predominantly the undesired diastereomer 113 along with elimination products, the unprotected secondary E-allylic alcohol substrate 110 provided the desired diastereomer 114 with moderate selectivity. It is difficult to rationalize the reversal of stereoselectivity between substrates 110 and 111. The authors suggest that the formation of a substantial amount of 115, the product of a syn-dichlorination reaction, from allylic alcohol 111 might be explained by anchimeric assistance of a distal chloride under these conditions.
Scheme 21.
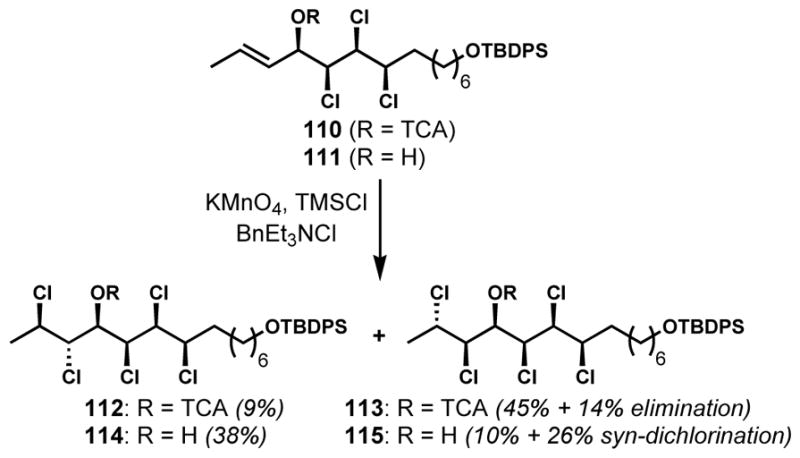
Moderately diastereoselective dichlorination of a chiral secondary E-allylic alcohol and a derivative
Substrate (protecting group)-dependent diastereoselectivity reversal in alkene dichlorination was also observed by Vanderwal and co-workers in their synthetic studies toward the tricyclic core of the clionastatin B (97, Figure 4).[49] Whereas the pseudodiaxial dichloride 117 (Scheme 22) was obtained as the major product from 116 via the expected diaxial addition pathway, dichlorination of the corresponding free alcohol 118 favored the desired pseudodiequatorial diastereomer 119 for the reasons that are currently unclear. Spectroscopic data were inconsistent with a change in ground state conformation between the protected and the unprotected substrates.
Scheme 22.
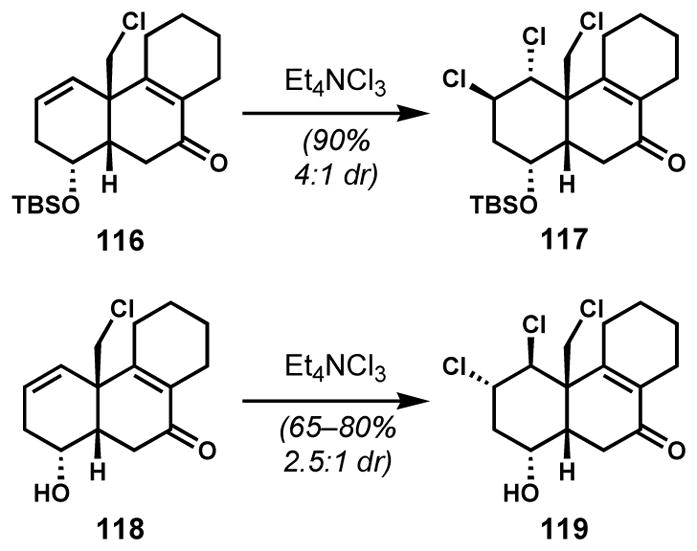
Alcohol protecting group dependent reversal of dichlorination diastereoselectivity
Diastereoselective dichlorination of trisubstituted allylic alcohol derivatives was also reported by Vanderwal and co-workers for the enantioselective syntheses of acyclic, polyhalogenated monoterpenoids from red algae of the genus Plocamium (Scheme 23).[50] With a general and divergent strategy, several natural products and unnatural analogs were synthesized efficiently from glyceraldehyde acetonide (120). One of the major challenges was the stereoselective construction of the vicinal secondary and tertiary dichlorides which are also each allylic. Because both syn- and anti-dichlorides are present in this family of natural products, the diastereoselective dichlorination was conducted with both E- and Z-alkene substrates 121 and 122. Although the direct dichlorination of the free alcohol was highly diastereoselective, the competing oxidation of the allylic alcohol to the corresponding unsaturated aldehyde was problematic. This side reaction was successfully avoided by the temporary protection of the alcohol as a trifluoroacetate. The subsequent in situ dichlorination and deacylation cleanly afforded the desired dichlorides 123 (anti) and 124 (syn) with high diastereoselectivities. Although the observed selectivities could be rationalized by the A1,3 strain minimization model, further studies are needed to formulate more detailed mechanistic picture. Short syntheses of a number of natural products, including 125–129, were facilitated by these diastereoselective alkene halogenation reactions.
Scheme 23.
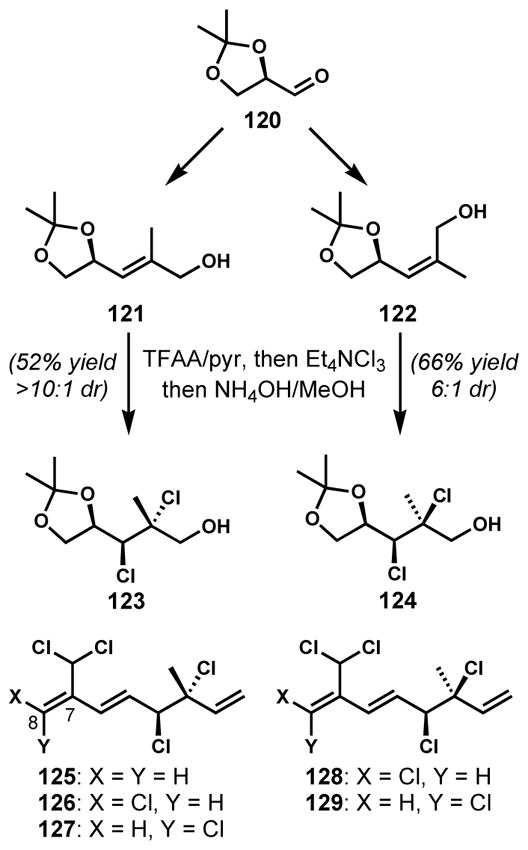
Diastereoselective dichlorination of trisubstituted E- and Z-allylic alcohol derivatives for the synthesis of polyhalogenated monoterpenoids
Halomon (95) is one of many polyhalogenated monoterpenoids from red algae that bear remote halogen-bearing stereogenic centers. Emulating its proposed biosynthesis, Hirama and co-workers completed a three-stage synthesis of (±)-halomon from myrcene (130).[51] After double bromochlorination of myrcene to give a 1:1 mixture of racemic 131 (Scheme 24) and its diastereomer, selective dehydrobromination gave 132. Selective bromochlorination of the non-chlorinated alkene provided (±)-halomon (95) and its diastereomer in about a 3:1 ratio. Even this moderate degree of diastereoselectivity is surprising; however, the reaction was relatively inefficient and it was not clear what became of the mass balance of this reaction that clearly proceeds via unstable intermediates. This synthesis, while incredibly direct, underscores the need for new methods for stereocontrolled alkene halogenation.
Scheme 24.
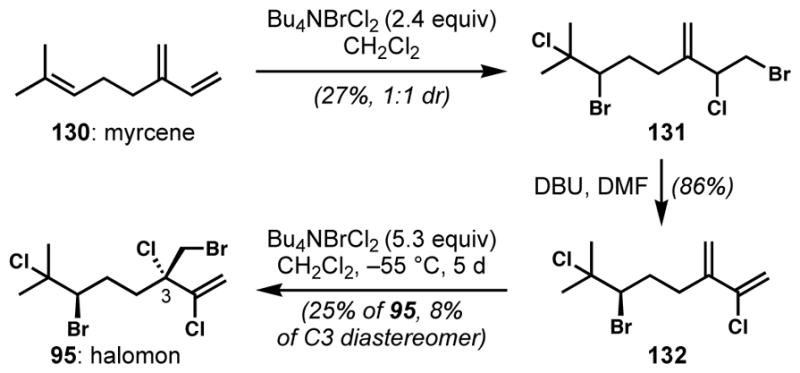
Moderate remote stereoinduction in the synthesis of halomon
In a fascinating synthesis from Shea and co-workers, bicyclic alkene 133, with its bridgehead alkene having arisen from a Type II intramolecular Diels–Alder reaction, is dichlorinated under the conditions shown to afford cis-dichloride 134 en route to 136, an unnamed cyclic monoterpenoid from a Plocamium red alga (Scheme 25).[52] This reaction is critical to the synthesis because it delivers the 1,3-cis-relationship of the two ring halogens, and is interesting because of the 1,2-cis-selectivity in the introduction of the two chlorides. Subsequent steps including lactone reduction with concomitant elimination of the silyl chloride to afford the exo-alkene in 135 and installation of the vinyl chloride to ultimately deliver the natural product. All of the examples shown up to this point have made use of conditions that are expected to proceed via stereospecific anti-addition to the alkenes, unless anchimeric assistance is operative. While the authors do not comment on the stereochemical outcome, it is known that some reagents—including phenyliodonium dichloride and sulfuryl chloride—can effect alkene halogenation via radical mechanisms under certain circumstances. Such a pathway might reasonably account for the interesting but ultimately inconsequential syn-dichlorination observed in this synthesis.
Scheme 25.
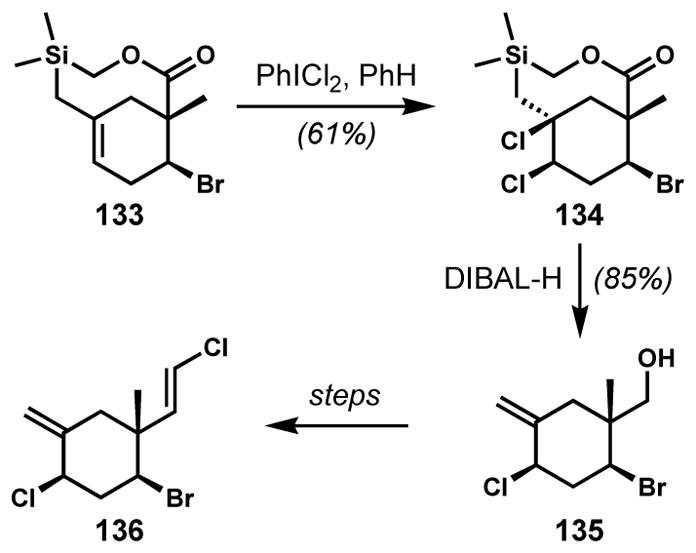
An unusual stereocontrolled syn-dichlorination
2.2.2 Stereospecific Deoxydichlorination
Although some progress has been made toward enantioselective dichlorination of alkenes (see below), a truly general solution has remained elusive. Therefore, chiral 1,2-dichloride moieties have been constructed indirectly via stereospecific deoxydichlorination of epoxides. Yoshimitsu and co-workers investigated the generality of this nucleophilic, Appel-type multiple chlorination process to access enantioenriched polychlorinated hydrocarbon motifs from chiral internal epoxides (Scheme 26).[53] Although various combinations of an organophosphine and a chlorinating reagent had been employed prior to this study, the substrate scope was limited to terminal epoxides and cyclic meso-epoxides. After surveying various known reagent combinations, the PPh3/NCS system was found to be optimal in terms of both reactivity and selectivity.[54a] Under these conditions, syn- or anti-dichlorides 138 and 141 could be prepared from trans- or cis-epoxides 137 and 140, respectively, along with alkenyl chloride side products, which were formed via E2 elimination of the reactive phosphonium intermediate, especially in sterically hindered environments. Side product formation was significant for aromatic and alkenyl epoxides. The application of a reactive Ph2PCl/NCS system suppressed side product formation to some degree. Notably, even diepoxides such as 143 could be transformed into the corresponding tetrachlorides (144) under optimized conditions. A catalytic variant has been reported.[54b] When combined with the Sharpless asymmetric epoxidation of allylic alcohols, this method provides enantioenriched polychlorides via a sequence that requires protection of the alcohol to avoid deoxychlorination of that group.
Scheme 26.
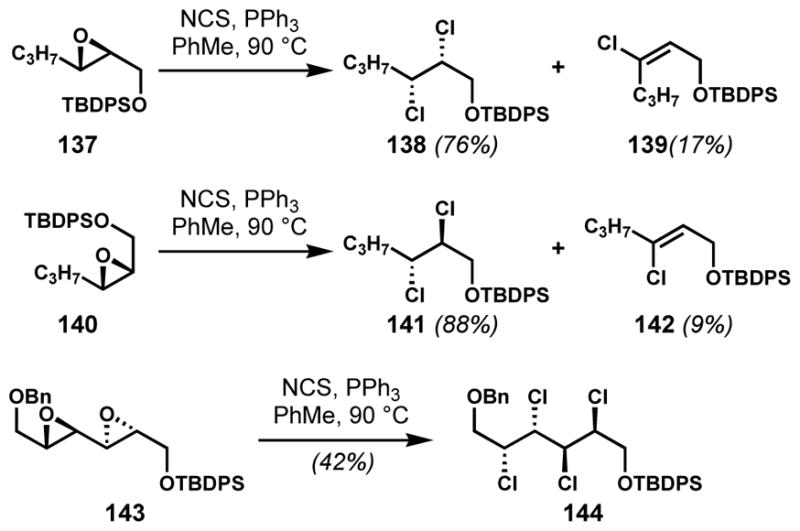
Stereospecific deoxydichlorination of chiral epoxides
Yoshimitsu and co-workers applied the epoxidation-deoxydichlorination (double chloride displacement) strategy to epoxides 145 and 147 in the enantioselective total syntheses of (+)-mytilipin A (69)[47] and (+)-danicalipin A (7),[55] respectively (Scheme 27). In both cases, the desired syn- or anti-1,2-dichlorides were obtained in high yields as single diastereomers.
Scheme 27.
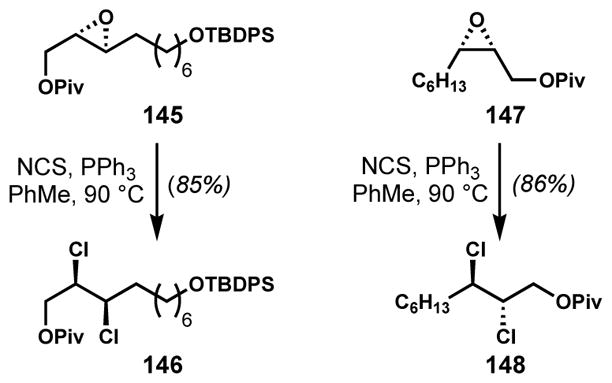
Application of the deoxydichlorination method to the preparation of enantioenriched syn- and anti-1,2-dichloride intermediates for the syntheses of chlorosulfolipids
The epoxide deoxydichlorination strategy found its utility again at a later stage of their mytilipin A synthesis when the chlorination of secondary homoallylic alcohol 149 was surprisingly problematic owing to competing elimination (Scheme 28).[47] A two-step sequence involving epoxide formation from the chlorohydrin followed by deoxydichlorination was more efficient for the production of trichloride 151 than the direct alcohol deoxychlorination, thus demonstrating the synthetic utility of this method.
Scheme 28.
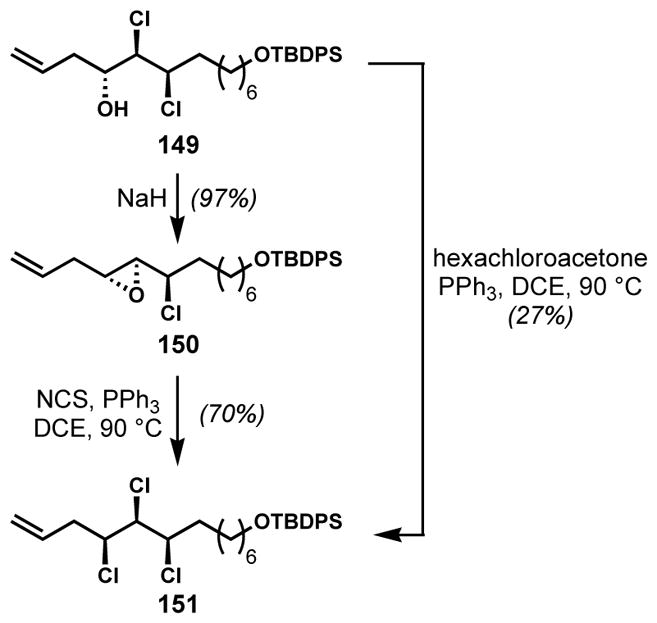
Two-step chlorination via deoxydichlorination of an epoxide intermediate
The frequently encountered vicinal bromochloride motif in halogenated natural products suggests a biosynthetic interhalogenation sequence that is likely initiated by bromonium ion formation followed by opening with a chloride ion.[56] Although a method for enantioselective bromochlorination of allylic alcohols has recently been invented (see below), a general enantioselective bromochlorination of unfunctionalized alkenes remains unknown. However, bromonium species can be stereoselectively generated from oxygenated precursors. In the synthesis of (±)-halomon (95) by Mioskowski and coworkers,[57] treatment of bromohydrin 152 with Viehe’s salt (153) provided the rearranged 1,2-bromochloride of (±)-halomon (95) directly as a mixture of two diastereomers along with allylic bromide side product 154 (Scheme 29). The intermediacy of a bromonium species generated via neighboring group participation of the bromine was proposed. The net process was not stereoselective simply because the starting material was not generated diastereoselectively; however, the complete 1,2-shift of the bromine in the presence of a chlorinating reagent was observed for the first time, and there is every reason to believe that this reaction would be stereospecific, permitting an enantio- or diastereomerically enriched bromohydrin intermediate to be converted to the corresponding vicinal bromochloride with effective transfer of stereochemical information.
Scheme 29.
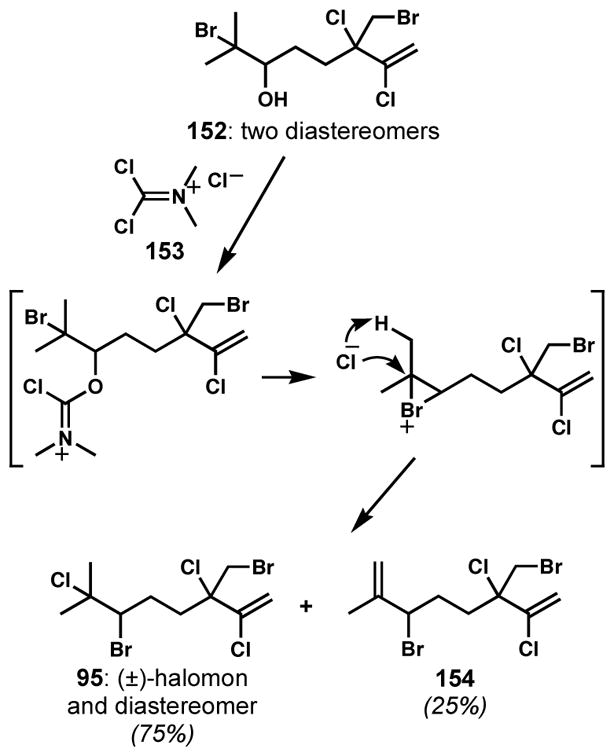
Generation of a bromonium intermediate from a bromohydrin for the synthesis of 1,2-bromochloride
This approach was applied to the enantioselective synthesis of a C2 symmetric oxasqualenoid, (+)-intricatetraol (160) by Morimoto and co-workers (Scheme 30).[58] The vicinal bromochloride moiety was constructed via stereospecific transformations of a stereoisomerically pure epoxide 155. Epoxide opening with dilithium tetrabromonickelate afforded the secondary bromide 156 with inversion of configuration at the less hindered carbon atom. The isomeric bromohydrin 157 was also prepared by treatment of 155 with magnesium bromide diethyl etherate. These bromohydrins were transformed into the fully functionalized fragment 158 via a sequence of chlorination and deprotection. The ratio of 158 and the dehydrated side product 159 was identical in both cases, strongly suggesting that the chlorinations proceed via a common bromonium intermediate, and indicating that this variant of the Mioskowski protocol was stereospecific.
Scheme 30.

Stereospecific stepwise deoxybromochlorination of an enantioenriched epoxide for the synthesis of (+)-intricatetraol
Virtually all enantioselective syntheses described above rely on asymmetric alkene oxidation processes, mainly Sharpless asymmetric epoxidation and dihydroxylation, which speaks to the far-reaching impact of these reactions. However, some promising progress has been made for enantioselective dihalogenation of alkenes, which will be discussed in the following section.
2.2.3 Enantioselective Alkene Halogenation
The catalytic enantioselective addition of two halogen atoms to an alkene in a single operation is inherently challenging. Such a catalytic system will require effective control of both alkene facial selectivity of the initial halonium ion formation (or its functional equivalent, such as a π complex of the alkene to a molecular halogen or interhalogen molecule) and regioselectivity of the subsequent halide attack, while navigating the possible reversibility of the initial reactive intermediate formation (Figure 5). Reversibility in halonium ion formation inevitably permits the possible interchange of the two enantiomeric halonium ion intermediates, and opposite regiochemistry of ring opening provides enantiomeric products in the case of dihalogenation. Of course, a selective interhalogenation is even more complicated, because regioisomeric products can also be formed.
Figure 5.
Challenges in enantioselective dihalogenation of alkenes
There have been only limited examples of enantioselective dihalogenation of alkenes. Henry and coworkers demonstrated the possibility of enantioselective dibromination via transition metal catalysis that has not seen application in natural product synthesis;[59] this method was based on their previous discovery of a related asymmetric hydroxychlorination of alkenes.[60] A catalytic enantioselective dichlorination of allylic alcohols was reported by Nicolaou and co-workers using chiral Lewis base catalysis.[61] Although apparently motivated by the chlorosulfolipids, this method has not yet been used in natural product synthesis, likely owing to the modest performance with relevant substrates. In its present form, it is reasonably selective only with cinnamyl alcohol substrates; optimization of the results with aliphatic allylic alcohols would permit direct application to chlorosulfolipid synthesis. [Removed old Schemes 31 and 33 and condensed two paragraphs into one]
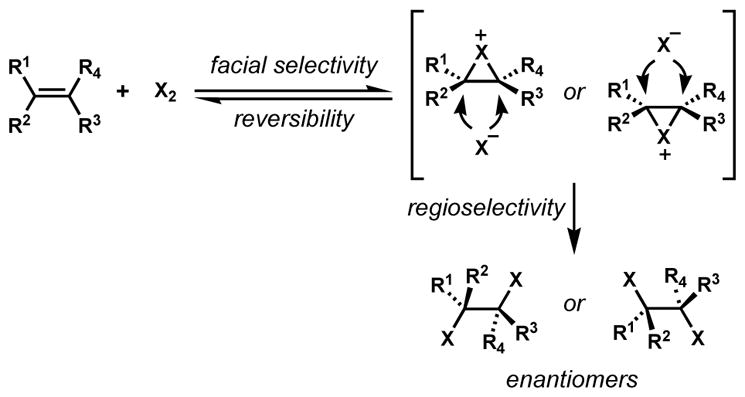
An isolated example of highly enantioselective dichlorination of an alkene was reported by Snyder and co-workers for the synthesis of napyradiomycin A1 (164) (Scheme 31).[62] The cyclic alkene substrate 161 was premixed with 4 equiv of chiral ligand 162 and 4 equiv of borane to form a chiral complex prior to dichlorination with elemental chlorine to afford 163 in enantioenriched form. This type of complexation was originally investigated for enantioselective Diels–Alder reactions.[63] Under these conditions, the enantioselectivity was remarkable, and this example stood as an important achievement in enantioselective halogenation, in spite of the superstoichiometric quantities of reagents needed. Stereocontrol was substantially diminished in the presence of less than 2 equiv of 162, thus supporting the authors’ proposed transition structure that involves two ligands and results in the shielding of one face of the reactive alkene and also prevents the chlorination of the aromatic ring.
Scheme 31.
Enantioselective dichlorination of an alkene using a superstoichiometric chiral controller
Burns and co-workers recently developed a catalytic enantioselective dibromination of E-cinnamyl alcohols with chiral Lewis acid catalysis.[64] This initial impressive result was followed by [removed old Scheme 34 and associated text] that group’s report of a new catalytic system involving N-bromosuccinimide, chlorotitanium triisopropoxide, and a chiral tridentate Schiff base catalyst 165 for enantioselective bromochlorination of allylic alcohols (Scheme 32).[65] Remarkably, the regioselectivity of this interhalogenation was also controlled by the catalyst, which could almost completely override the intrinsic electronic preference of Markovnikov addition. It was proposed that the chloride is transferred from the titanium metal to the proximal reactive carbon via an organized titanium complex. The coordination of the hydroxyl group to the titanium metal center allows for the selective bromochlorination of the neighboring alkene. These conditions were generally applicable to a wide range of structurally diverse allylic alcohols. The utility of this method was demonstrated by a short enantioselective synthesis of (+)-bromochloromyrcene (168), which had previously been synthesized as a racemate in nine steps.[57b] The potential of analogous catalytic systems for the enantioselective dichlorination (t-butyl hypochlorite instead of N-bromosuccinimide) and dibromination (bromotitanium triisopropoxide instead of chlorotitanium triisopropoxide) was also shown. Because of its breadth of scope with respect to the different halogens that can be installed and the ready availability of the reagents, catalyst and allylic alcohol substrates, this method and its variants are sure to see much future application in complex molecule synthesis.
Scheme 34.
Late stage bromocycloetherification for the syntheses of (+)-thyrsiferol and (+)-venustatriol
Scheme 32.

Catalytic enantioselective bromochlorination of allylic alcohols
2.2.4 Looking Forward: New Approaches
As a general rule, alkene halogenation processes are stereospecific anti-additions, as a result of the invertive halonium ring-opening event (or the analogous trans-addition to an alkene-halogen π complex). Two different exceptions, at least with respect to overall outcome, were noted above: (1) a net syn-addition when anchimeric assistance is operative, and (2) a net stereoselective (not stereospecific) syn-addition when a radical mechanism is likely operative. The ramification of the dependence upon anti-addition processes is that synthetic chemists did not have access to the complementary approach of syn-stereospecific additions; in the context of stereocontrolled synthesis of polyhalogenated molecules, the flexibility in synthesis design might be doubled with access to this type of reactivity.
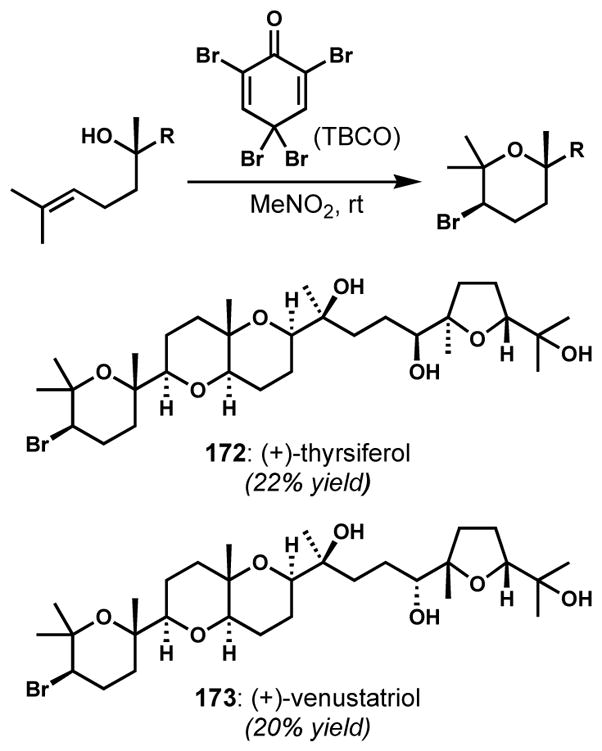
In response to this gap in synthesis technology, and likely as a stepping stone to a mechanistically distinct enantioselective alkene halogenation protocol, Denmark and co-workers recently unveiled a selenium(IV)-catalyzed stereospecific syn-dichlorination of alkenes (Scheme 33).[66] This reaction, which starts from diphenyl diselenide as the precatalyst and presumably proceeds via anti-chloroselenenylation and invertive chlorodeselenenylation (both steps that were known in isolation from previous work), shows broad scope in 1,2-disubstituted alkene syn-dichlorination, tolerating a broad range of functionality. Although it has not yet seen use in natural product synthesis, this new reactivity could easily be built into new approaches to the chlorosulfolipids that are complementary to those already completed.
Scheme 33.
Selenium(IV)-catalyzed syn-dichlorination of alkenes ([PyF]+[BF4]– = N-fluoropyridinium tetrafluoroborate)
2.3 Stereoselective Halocyclization
2.3.1 Diastereoselective Halocyclization
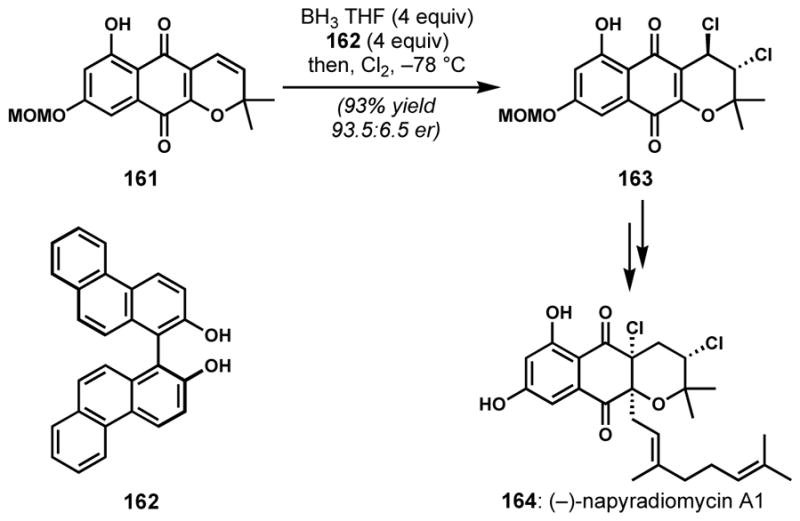
Halofunctionalization of an alkene with a pendant heteroatom nucleophile is a useful way to construct halogenated heterocycles. Moreover, this reactivity is common in the biosynthesis of natural products, especially among the terpenoids and acetogenins, wherein haloperoxidase enzymes are generally accepted to produce the vicinal haloether motif. Among various electrophilic reagents, the most widely employed is 2,4,4,6-tetrabromo-2,5-cyclohexadienone (TBCO), which was first described by Kato and co-workers for the bromocycloetherification of dehydrolinalool.[67] In spite of issues of stereoselectivity and regioselectivity, the bromocycloetherification approach was employed at the last step of the synthesis of complex natural products such as tetracyclic triterpenoid polyethers, (+)-thyrsiferol (172) and (+)-venustatriol (173) by Shirahama and co-workers (Scheme 34).[68] However, the competing formation of a diastereomeric mixture of the tetrahydrofuran side products diminished the efficiency of this process.
Corey and co-workers also constructed the bromoether subunit of (+)-venustatriol (173) via a similar strategy with TBCO.[69] The poor stereo- and regioselectivities were again problematic; cyclization of 174 yielded, in addition to the desired pyran 175, a small amount of the C3-epimer 176 and a mixture of the isomeric tetrahydrofurans 177 as major side products (Scheme 35). To increase throughput of the desired bromotetrahydropyran 175, the undesired isomers were converted back to the reactant 174 upon treatment with zinc and acetic acid.
Scheme 35.
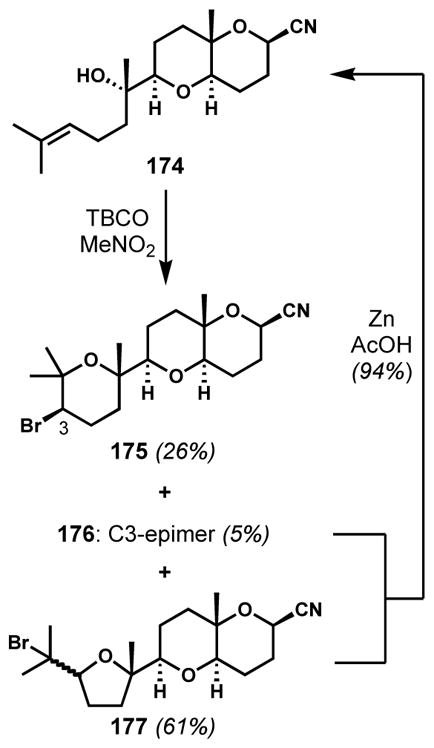
Improved efficiency of bromocycloetherification by recycling the undesired isomers
To avoid a low-yielding last step, Forsyth and co-workers aimed to employ bromocycloetherification at an earlier stage of the syntheses of thyrsiferyl 23-acetate and its derivatives.[70] Initial attempts with (S)-linalool (178) provided an inseparable mixture of tetrahydrofurans 179 and tetrahydropyrans 180 in poor selectivities (Scheme 36). The small steric difference between the methyl and the vinyl substituents is responsible for the low diastereoselectivity. Although the diastereoselectivity could be improved by introducing a bulkier substituent (not shown), the proportion of constitutional isomers (tetrahydrofurans) formed also increased. Because of the low efficiency of the direct bromocycloetherification, an indirect multi-step alternative was developed by employing mercury(II)-mediated cyclization (intramolecular oxymercuration). Upon sequential treatment of the racemic cyanohydrin 181 with mercuric trifluoroacetate and a saturated aqueous solution of potassium bromide and sodium bicarbonate, the desired organomercuric tetrahydropyran 182 was obtained as the sole product. Subsequent bromodemercuration with Br2 under photochemical conditions afforded racemic 183 with inversion of configuration. Unfortunately, at that time, it was difficult to prepare the enantioenriched (S)-cyanohydrin, which would have facilitated the enantioselective syntheses of the natural product targets.
Scheme 36.

More selective, indirect bromocycloetherification via mercury(II)-mediated cyclization (intramolecular oxymercuration)
When two alkene carbons are electronically equivalent, the formation of tetrahydropyrans is often disfavored because of 1,3-diaxial-like repulsions in the transition state. With such substrates, Martín and co-workers were able to selectively access halogenated tetrahydrofuran scaffolds for the synthesis of trans-(+)-deacetylkumausyne (189) (Scheme 37).[71] However, acyclic precursor 184 with the correct configurations at the oxygen-bearing carbon stereocenters suffered poor facial selectivity upon treatment with TBCO in dichloromethane despite the exclusive formation of tetrahydrofuran isomers. On the other hand, the desired syn-tetrahydrofuran could be obtained with epimeric precursor 187 with the inverted configuration at the silyloxy-bearing carbon, which provided 188 in 5:1 dr. The configuration of the secondary alcohol was later inverted via an additional sequence of oxidation and reduction en route to the natural product.
Scheme 37.
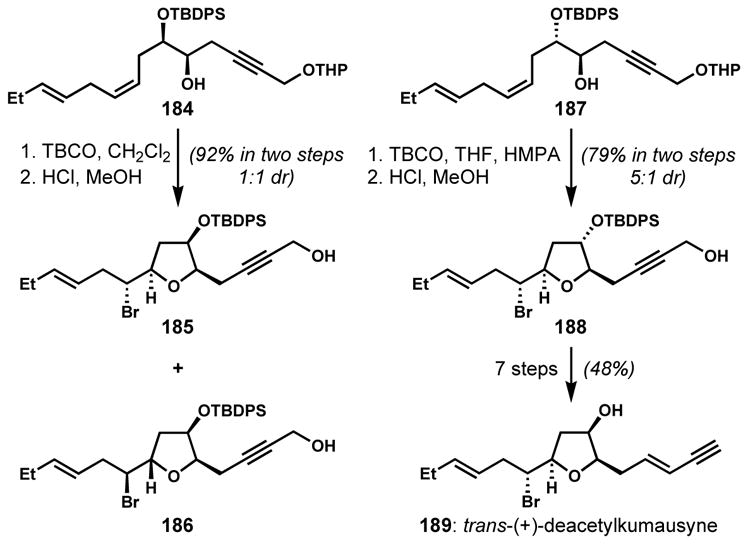
Regiocontrol in favor of tetrahydrofuran formation
It is possible to affect the cyclization regioselectivity by altering the electronic nature of the substrates. In particular, tetrahydropyran formation can be favored by having an electron-withdrawing group between the alkene and the alcohol. This type of electronically controlled bromocycloetherification (tetrahydropyran formation) was generally applicable as a key step to the syntheses of several aplysiapyranoids by Jung and co-workers.[72] In most instances, the relative stereochemistry of the bromotetrahydropyran product could be rationalized in a straightforward way by considering the most stable chair-like transition structure. For the enantioselective synthesis of an anti,anti-dihalo ether of aplysiapyranoid D (196), the cyclization precursor 191 was prepared by the titanium-mediated, regioselective chlorinative opening of a chiral 2,3-epoxy alcohol 190, which was prepared via Sharpless asymmetric epoxidation (Scheme 38).[72a] Upon treatment of 191 with TBCO, the electronegative chlorine substituent facilitates Markovnikov addition by inductively destabilizing the partial positive charge at the secondary carbon despite the competing directing effect of the destabilizing 1,3-diaxial-like repulsion. The modest directing role of the chlorine substituent was supported by the control experiment with the deschloro analogue, which provided a ca. 1:1 mixture of tetrahydropyran and tetrahydrofuran isomers under similar conditions. From the two reversibly generated bromonium ions, the more stable structure with fewer 1,3-diaxial interactions led to the desired all-equatorial tetrahydropyran 193 along with the tetrahydrofuran isomer 192. Only trace amounts of the other isomers 194 and 195 from the less stable bromonium species were observed. The desired isomer 193 was isolated in pure form after desilylation, then transformed to aplysiapyranoid D (196) in two more steps.
Scheme 38.
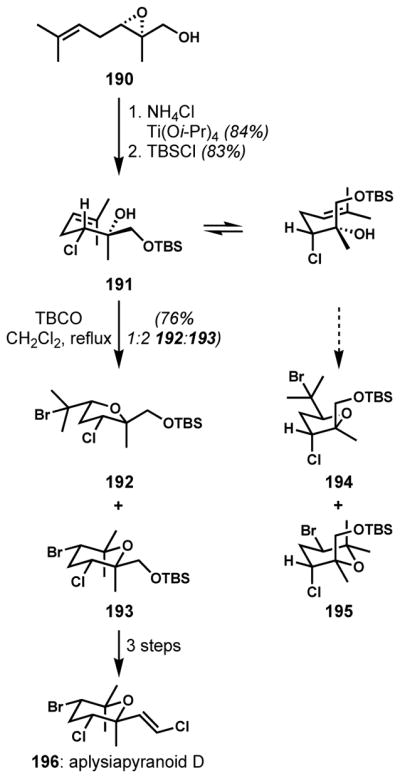
Improved regio- and stereoselectivity of bromocycloetherification with a chlorine substituent in the enantioselective synthesis of aplysiapyranoid D
For the anti,syn-dihalo ether characteristic of aplysiapyranoid A (198), a transition structure with an axial bromide substituent needs to be favored (Scheme 39).[72b] To overcome the inherent preference exerted by the bromine-bearing (homoallylic) stereocenter, the dominant diastereocontrol must arise from the alcohol-bearing (bishomoallylic) stereocenter, which requires a substituent that is smaller than the methyl group, such as an alkene. Thus, the bromocycloetherification of 197 with TBCO gave aplysiapyranoid A (198) as the major product along with two other isomers, demonstrating that the size of the substituents could predictably control the diastereoselectivity. Again, the presence of a halogen is necessary for the preferential formation of tetrahydropyrans because the quantity of tetrahydrofuran isomers increases without the bromine substituent. A related natural product, aplysiapyranoid C (202) could also be synthesized in a similar way (Scheme 40).[72c]
Scheme 39.
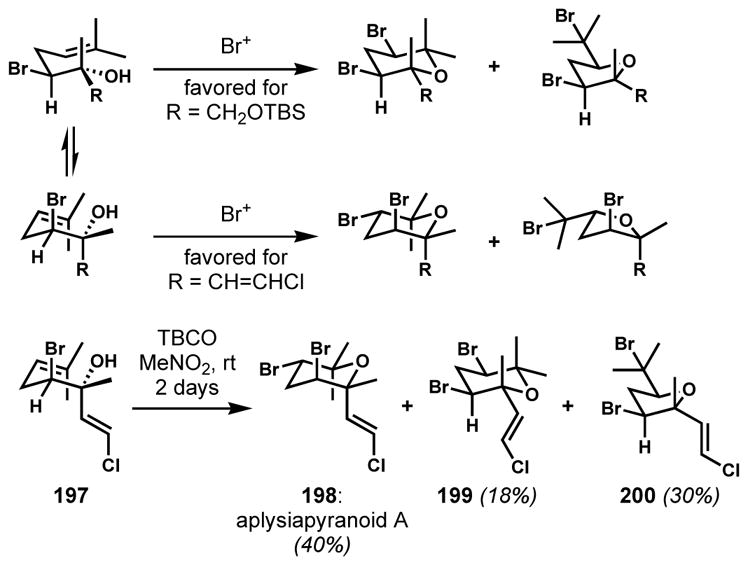
Diastereocontrol of bromocycloetherification by altering the size of substituents for the enantioselective synthesis of aplysiapyranoid A
Scheme 40.
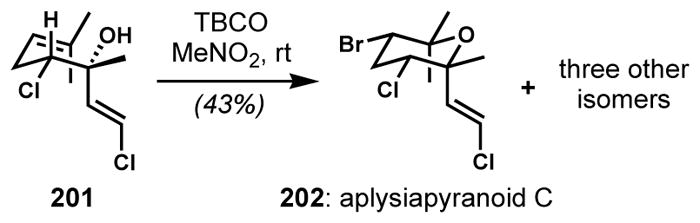
Enantioselective synthesis of aplysiapyranoid C via diastereoselective bromocycloetherification
The bromocycloetherification strategy can also be applied to the diastereoselective synthesis of brominated oxepanes. Morimoto and co-workers employed a highly diastereoselective 7-endo-trig bromocycloetherification initiated with N-bromosuccinimide in the highly polar, non-nucleophilic solvent 1,1,1,3,3,3-hexafluoro-2-propanol for the enantioselective syntheses of the bromotriterpene polyethers, (+)-aurilol (205) and (+)-enshuol (not shown) (Scheme 41).[73]
Scheme 41.

Diastereoselective synthesis of brominated oxepanes via 7-endo-trig bromocycloetherification
Reports of the direct construction of 8-membered rings via 8-endo bromocycloetherification are rare.[74] This process was inefficient even using a crude preparation of bromoperoxidase enzyme(s) from Laurencia, which is thought to be responsible for such bond constructions in nature; however, that datum does not necessarily indicate that such transformations are poorly effective in the organism (Scheme 42).[75]
Scheme 42.
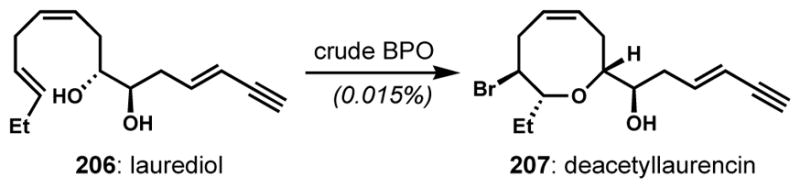
Enzymatic 8-endo bromocycloetherification
Snyder and co-workers developed a clever alternative solution for the synthesis of 8- and 9-membered Laurencia-type bromoethers through a ring-expanding bromocycloetherification strategy that takes advantage of a more favorable 5-membered ring-forming cyclization.[76] This idea was formulated on the basis of their observation of a pinacol-type rearrangement of an in situ generated bicyclic oxonium species to give tetrahydrofuranyl ketone (see 208 → 209, Scheme 43).[76a] Prior to Snyder’s work, the idea of ring expansion through oxonium formation (tandem bromonium-induced transannular oxonium ion formation-fragmentation) had been proposed for the formation of the 12-membered ring of obtusallenes and demonstrated with model substrates by Braddock and co-workers (not shown).[77]
Scheme 43.
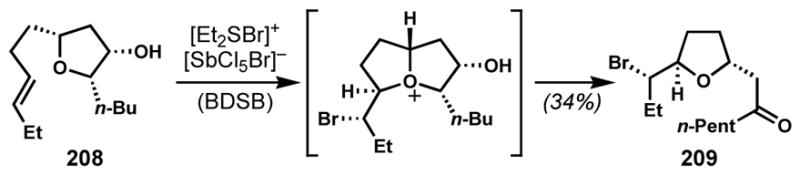
Pinacol-type rearrangement of an in situ generated bicyclic oxonium species (BDSB = bromodiethylsulfonium bromopentachloroantimonate)
Snyder and co-workers developed a readily prepared, stable, and highly reactive brominating reagent, bromodiethylsulfonium bromopentachloroantimonate (BDSB), that generates non-Lewis basic byproducts for efficient bromocyclizations.[78] The antimonate counterion sequesters the bromide as shown by X-ray crystallographic analysis. BDSB induces regio- and stereoselective transformations of tetrahydrofurans and tetrahydropyrans possessing pendant alkene side chains via the transient formation of bicyclic oxonium intermediates and subsequent opening at the ring junction to give 8- and 9-membered cyclic bromoethers that resemble the cores of the Laurencia C15 acetogenins (Scheme 44). Notably, 6-endo bromocyclization was not observed in the cases of the substrates with a homoallylic side-chain such as 212 and 214. Moreover, the products were obtained as single diastereomers. Although both faces of the pendant alkene are accessible to produce two diastereomeric bromonium species, only one (less hindered) oxonium intermediate was apparently selectively transformed into the product, presumably owing to facile bromonium transfer between alkenes and reversible oxonium formation (Figure 6).[79] To avoid steric hindrance, the exo-orientation of the bromo side chain of the oxonium intermediate is preferred. In this chemistry, as in many other bromocyclizations (see below), BDSB proved superior to other commonly used bromonium sources. Via analogous processes, 9-exo, and 9-endo products were also obtained.
Scheme 44.
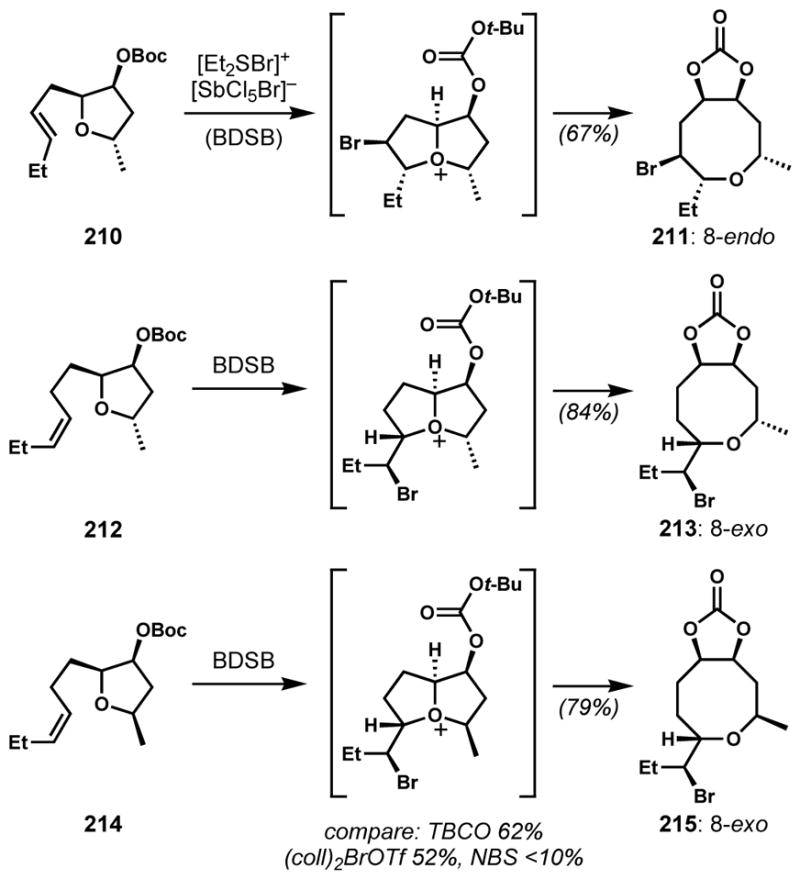
Ring-expanding bromocycloetherification via a bicyclic oxonium intermediate
Figure 6.
Proposed model for diastereoselection
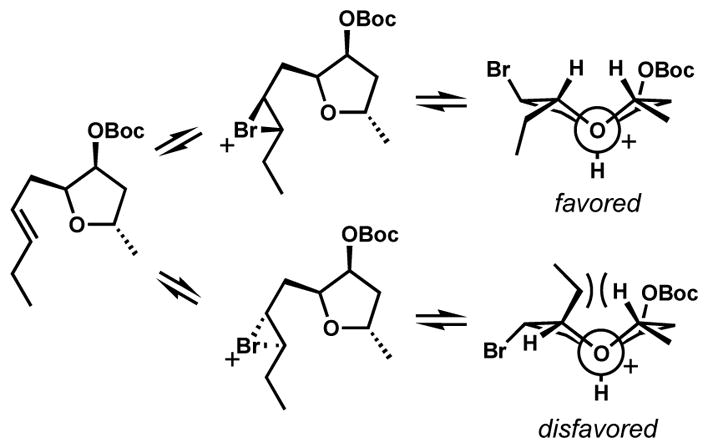
This diastereoselective ring-expanding bromocycloetherification strategy was applied to the formal synthesis of the lauroxocane family natural products.[80] By strategically positioning a pendant alkene and an internal nucleophilic trap around the tetrahydrofuran core, eight-membered rings with a diverse array of functionality could be produced diastereoselectively upon treatment with BDSB. An elaborated 8-endo bromoether 217 was prepared for the synthesis of laurefucin (219) via ring-expanding bromocycloetherification of 216 (Scheme 45). The bromonium ion formation was chemoselective even in the presence of excess reagent. Thus, the internal alkene reacts selectively over the terminal alkene, which is critical for the success of this strategy. The mild reaction conditions allowed the effective formation of the oxocane moiety as a single diastereomer without interference from the mesylate group.
Scheme 45.
Formal synthesis of (±)-laurefucin via a diastereoselective ring-expanding bromocycloetherification
Upon treatment of 217 with diisobutylaluminium hydride, the carbonate was reduced to the alkoxide, which displaced the mesylate in situ to form the bridging tetrahydrofuran in 218. The conversion of 218 to (±)-laurefucin (219) had been previously accomplished in additional two steps by Kim and coworkers.[81]
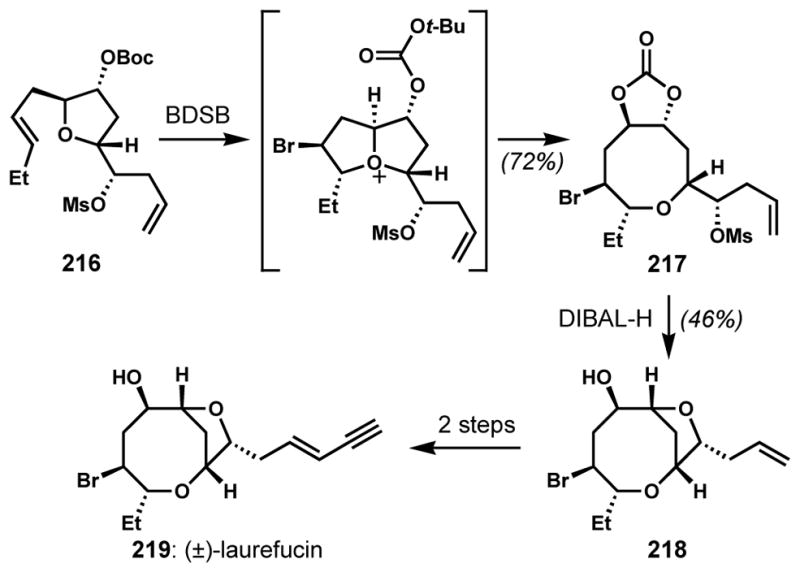
By altering the position of the nucleophilic trap, different scaffolds such as 8-exo bromoether 221 could be produced, again as a single diastereomer (Scheme 46). Unfortunately, attempts to convert the carbonate and its diol derivatives to the alkene, which can be found in several natural products, were unsuccessful. The installation of the desired alkene could be achieved via clever inclusion of an alkylsilane trap in tetrahydrofuran 222 to give the known synthetic oxocane intermediate 223, which had been previously prepared by Kim and co-workers for the synthesis of (±)-E- and Z-pinnatifidenyne (224).[82]
Scheme 46.
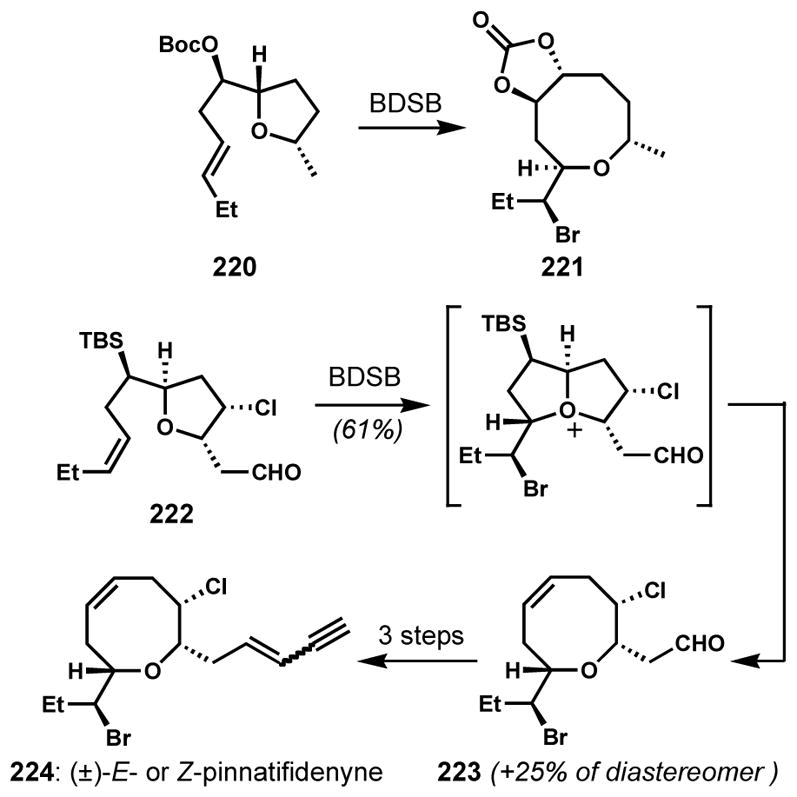
Formal syntheses of (±)-E- and Z-pinnatifidenyne via a diastereoselective ring-expanding bromocycloetherification
The intermediacy of a bicyclic oxonium species via the unexpected participation of an ether oxygen during bromocyclization of an oxocene intermediate has also been observed, resulting in the formation of bromotetrahydrofurans. Murai and co-workers performed an enzymatic bromocycloetherification of (+)-Z-prelaureatin (225) by bromoperoxidase (BPO) or lactoperoxidase (LPO) in the presence of sodium bromide and hydrogen peroxide to give (+)-Z-laureatin (226) and (+)-Z-isolaureatin (227) in low yields along with other products (Scheme 47).[83] However, when similar bromocycloetherifications of analogous substrate 228 were attempted with TBCO or N-bromosuccinimide for the chemical synthesis of 226/227 by Suzuki and co-workers, a rearranged bromotetrahydrofuran with a pendant ketone (231) was obtained instead of the expected bicyclic bromoethers 229 and/or 230. Presumably, this outcome arises from a sequence of transannular bromocyclization of the ether oxygen followed by pinacol-type rearrangement of the resulting oxonium ion.[84]
Scheme 47.

Bromonium-induced ring-contraction via a bicyclic oxonium intermediate
The selective participation of an ether oxygen over a hydroxyl group was also observed by Howell and co-workers in synthetic studies toward Z-laureatin (226).[85] Beginning from oxetane 232, 8-exo-bromocycloetherification was attempted to construct bicyclic core 233 (Scheme 48). However, upon treatment of 232 with N-bromosuccinimide, unexpected bromocyclization of the ether oxygen triggered a rearrangement to give a bromotetrahydrofuran containing a pendant epoxide (234) as a single diastereomer, instead of 233. This type of epoxyfuranyl scaffold can also be found in a related natural product, laureoxolane (236).[86] Another brominative rearrangement was observed while attempting a derivatization of 234. Instead of the SN2 bromination of the secondary alcohol on the tetrahydrofuran ring, the activated alcohol was displaced by the epoxide to form an oxonium species, which was opened by bromide ion to give a trans-fused bicycle 235, which has a fused bis-tetrahydrofuran motif that is structurally related to kumausallene (27) and aplysiallene (36).
Scheme 48.
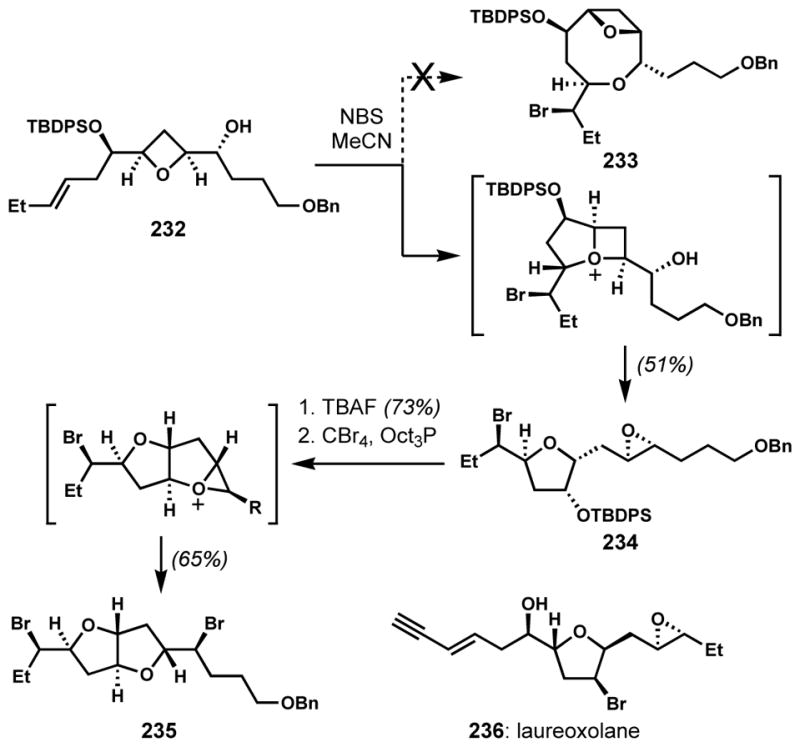
Brominative rearrangements by the participation of ether oxygens over hydroxyl groups
This type of epoxide ether participation can be deliberately orchestrated into highly productive cascade reactions. The reaction of an epoxide with an electrophile-activated alkene has been used for the preparation of bicyclic structures via the initial formation of a bromonium[87] or phenylselenium[88] species. Jamison and co-workers utilized a bromonium-initiated epoxide-opening cascade to construct the tricyclic subunit for the enantioselective synthesis of ent-dioxepandehydrothyrsiferol (239) (Scheme 49).[89] Highly regioselective Markovnikov openings directed by the methyl substituents were promoted in hexafluoro-2-propanol, a highly polar, non-nucleophilic solvent, which can stabilize partial positive charge. Upon treatment of triepoxide 237 with N-bromosuccinimide, the cascade of completely regiocontrolled and stereospecific epoxide openings afforded 238 in high efficiency (90% yield per epoxide). However, the product was obtained as a 1:1 mixture of diastereomers because of the unselective initial bromonium ion formation.
Scheme 49.
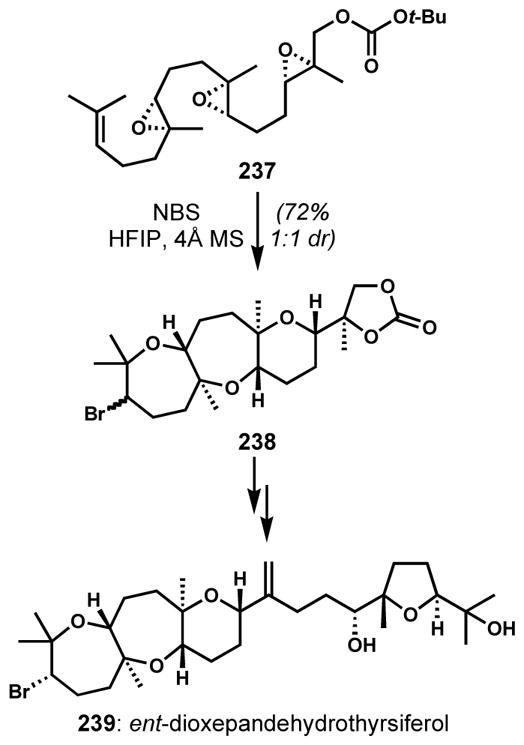
Synthesis of ent-dioxepandehydrothyrsiferol via bromonium-initiated epoxide-opening cascade
Halocyclization can sometimes lead to subsequent rearrangement, especially when strained intermediates are involved. Romo and co-workers developed a stereoselective tandem chlorination/ring contraction strategy for the synthesis of densely functionalized spirocyclic chlorocyclopentanes related to the core of the oroidin alkaloids, palau’amine (242) and axinellamines A and B (243 and 244) (Scheme 50).[90] The formation of a C=O bond presumably facilitates the halonium-induced rearrangement-cyclization sequence. Upon treatment of allylic alcohol substrate 240 with N-chlorosuccinimide, chlorination took place on the convex face. The subsequent epoxide opening by the neighboring electron lone pair was followed by a ring contraction via a suprafacial 1,2-alkyl migration to provide the spirocyclic hydantoin 241.
Scheme 50.

Construction of densely functionalized spirocyclic chlorocyclopentanes via tandem chloroepoxidation and ring contraction
A more direct halocyclization strategy was employed by Baran and co-workers for the racemic synthesis of spirocycle 247, which is a common synthetic intermediate for a variety of pyrrole-imidazole marine alkaloids (Scheme 51).[91] During optimization studies, they found that residual trifluoromethanesulfonamide from the previous step was beneficial for the clean and high yielding chloroguanidylation cyclization of allylic guanidine 245. The vicinal stereogenic centers were efficiently created by employing a combination of t-butyl hypochlorite and trifluoromethanesulfonamide to provide the desired chlorospirocycle 246 on gram scale and as a single diastereomer. This product was directly transformed into 247 via subsequent oxidation and deprotection. This highly efficient and scalable route led to the racemic synthesis of axinellamine A and B (243 and 244). During the course of this study, it was proposed that the active chlorinating agent was an N-chloroguanidine species. On the basis of this hypothesis, a series of chloroguanidines were developed as a new class of practical and reactive chlorinating reagents. A representative compound is chlorobis(methoxycarbonyl)guanidine (248, CBMG, Palau’chlor).[91b,c]
Scheme 51.
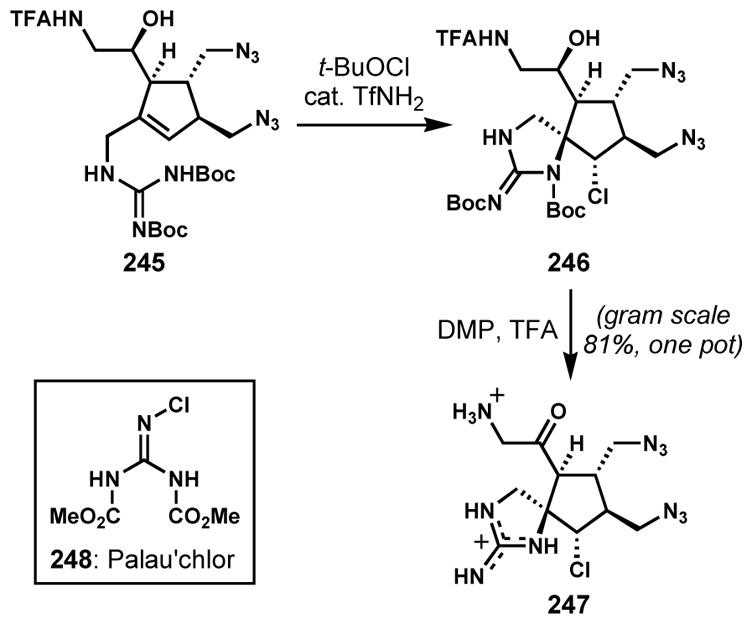
Stereoselective chloroguanidylation for the preparation of a key chlorospirocycle intermediate for the syntheses of axinellamine A and B
2.3.2 Halogenative Semi-Pinacol Rearrangements
In the correct setting, skeletal rearrangements can be induced by the transient formation of halonium intermediates. Semi-pinacol rearrangements instigated in this way have been used multiple times in the context of natural product synthesis;[92] however, there appears to be only a single example in which the activating halogen atom is retained in the structure of the natural product target. Wood and coworkers completed a synthesis of welwitindolinone A isonitrile (251, Scheme 52) wherein a chlorinative semi-pinacol rearrangement of tertiary allylic alcohol 249 furnished the α-chloro quaternary stereogenic center in 250, one of several challenging motifs present in this structurally unusual natural product.[93] The stereospecific transfer of the migrating group anti to the transient chloronium ion necessitated that the chloronium ion was formed on the α-face of the cyclohexene or, in the case that chloronium ion formation is reversible, that migration occurs preferentially in the α-chloronium ion context. Although the underlying mechanism of stereochemical control was not discussed by the authors, the reaction itself was high yielding and perfectly stereoselective, with the outcome consistent with chloronium formation/reaction from the face opposite to the bulky silyl ether.
Scheme 52.

Chlorinative semi-pinacol rearrangement for the generation of the vicinal quaternary/secondary chloride stereogenic centers
2.3.3 Diastereoselective 1,4-Bromocycloetherification of Enynes
1,4-Halocyclization has found application in halogenated natural product synthesis for the installation of bromoallene moieties via intramolecular 1,4-bromocycloetherification of enynes. In contrast to the SN2′ bromination of propargylic sulfonates, this approach enables the stereoselective simultaneous assembly of a cyclic ether with an appended bromoallene, which is a common motif among marine acetogenin natural products. The stereochemical course of this process has been elucidated by Braddock and coworkers.[94] Whereas E-enyne 252 underwent intramolecular 1,4-bromocycloetherification to give bromoallene 253 as a major product, diastereomeric bromoallene 255 was obtained from Z-enyne 254, demonstrating that these reactions preferentially proceed via syn-addition pathways (Scheme 53).
Scheme 53.
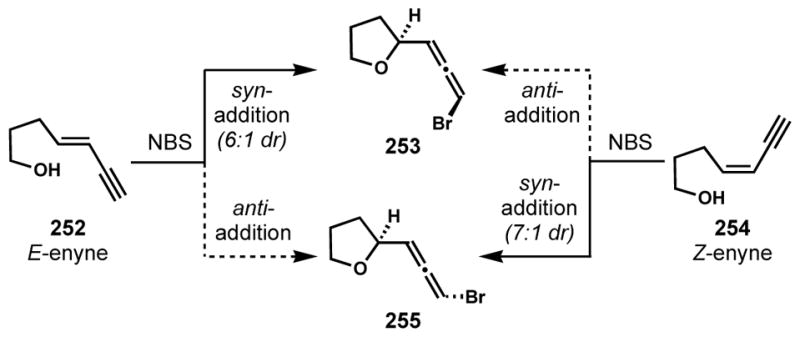
The syn-addition pathway in intramolecular 1,4-bromoetherification of achiral enynes
In the cases of more complex substrates, the diastereoselectivity can be attenuated by the influence of preexisting stereocenters. Murai and co-workers first investigated the 1,4-bromocycloetherification of (+)E- and Z-prelaureatins (256 and 225, Scheme 54).[83] In both cases, the bromocyclizations with TBCO were poorly selective, providing a mixture of (+)-laurallene (257) and its diastereomer 258 that is epimeric at the bromine-bearing allene carbon. The same endgame was employed by Crimmins and coworkers.[95] Low diastereoselectivity was also observed when Evans and co-workers conducted 1,4-bromocycloetherification of a cis-2,5-disubstituted tetrahydrofuran with an enyne side chain (259) for the enantioselective synthesis of (−)-kumausallene (27).[96] Bromocyclization of 259 with freshly prepared TBCO afforded bromoallene 260 as a 2.5:1 mixture of diastereomers favoring the unnatural epimer, which could be recycled through reductive fragmentation with samarium(II) iodide.
Scheme 54.

Attenuated diastereoselectivity of 1,4-bromocycloetherification of complex chiral enynes
Solvent-dependent diastereoselectivity was observed in the enyne 1,4-bromoetherication of 261 in the synthesis of (±)-panacene (45) by Canesi and co-workers (Scheme 55).[97] However, this reaction could only be driven to the highly selective formation of unnatural epipanacene (262) via anti-addition using non-polar solvents. The use of N-bromosuccinimide resulted in the bromination of the aromatic ring. To obtain the desired stereoisomer by taking advantage of inherent stereochemical preferences, bromine was installed on the terminal alkyne (263) prior to the cyclization. The following one-pot sequence of diastereoselective oxymercuration with mercury(II) acetate and stereoretentive demercuration with 1,2-ethanedithiol afforded panacene with the desired configuration of 45.
Scheme 55.
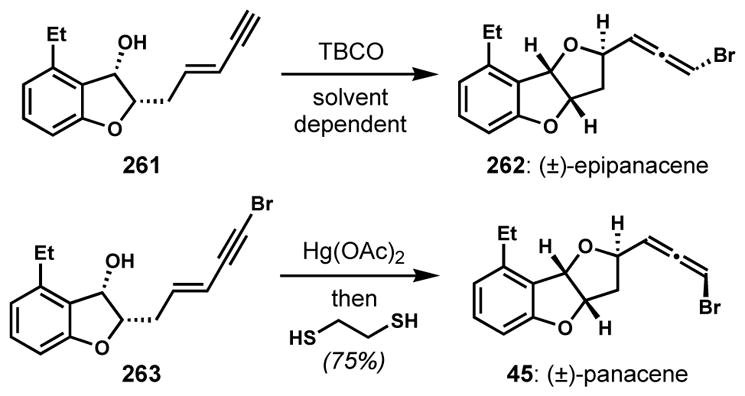
Solvent-dependent diastereoselectivity of enyne bromoetherification and a complementary approach via anti-mercurocyclization
Tang and co-workers evaluated the effect of Lewis basic additives (catalysts) on the diastereoselectivity of 1,4-bromocycloetherification of enynes with the goal of favoring syn-addition en route to another synthesis of (−)-kumausallene (27) (Scheme 56).[98] On the basis of their previous reports on the complementary syn-addition in the related 1,4-bromolactonization of conjugated enynes,[99] in which the degree of syn-selectivity was dependent on the structure of catalyst, it was postulated that catalysts might be able to alter the stereochemical course of the 1,4-bromocycloetherification of conjugated enynes. Whereas a complex mixture was obtained from the reactions of 264 with N-bromosuccinimide or freshly prepared TBCO under Evans’s conditions, the bromocycloetherification was promoted by Lewis basic additives such as N,N-dimethylformamide (DMF) or hexamethylphosphoramide. However, the desired bromoallene 265 was not produced as the major product. The structure of the C5 side chain had a significant influence on the stereoselecitvity of the bromocycloetherification of the C2 side chain. When the 1,4-bromocycloetherification was carried out with the aliphatic secondary bromide in place (see 266, the proposed precursor in biosynthesis), (−)-kumausallene (27) was cleanly produced via syn-addition using N-bromosuccinimide and DMF. A complex mixture was obtained without inclusion of DMF.
Scheme 56.
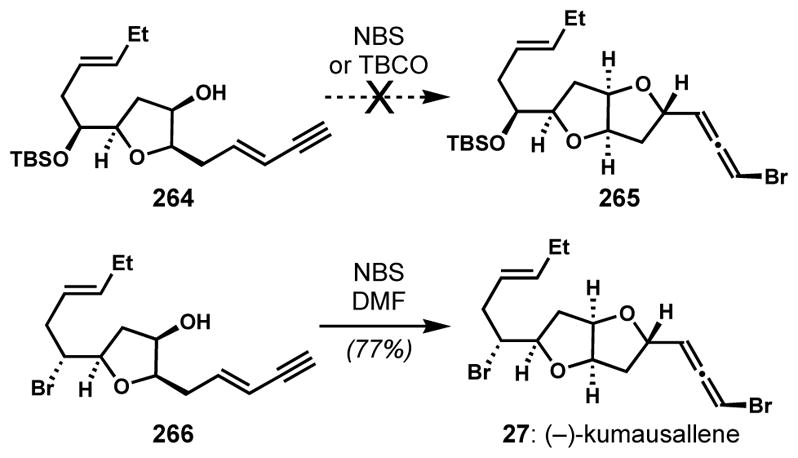
Lewis-base-catalyzed syn-1,4-bromocycloetherification
2.3.4 Enantioselective Halocyclization
The success of enantioselective halofunctionalizations of alkenes requires not only the selective generation of an enantioenriched halonium intermediate but also the prevention of the well-known competing racemization pathway via halonium transfer between alkene substrates, which has been identified as the major challenge by Brown and co-workers[100] and further investigated by Denmark and co-workers.[101] Moreover, the more pronounced carbocationic character in the case of chloronium species often leads to undesired side reactions, such as elimination to form alkenes and skeletal rearrangements. Despite these challenges, many creative solutions for enantioselective halocyclizations—wherein the presence of a tethered nucleophile improves the chances for capture of the fleeting intermediates—have been developed during the past decade and they have been recently reviewed a number of times by several authors.[102]
Although these methods have not been used for the direct incorporation of halogen-bearing chiral centers for natural product synthesis, useful synthetic intermediates can be accessed by an enantioselective halocyclization and the substitution of the resulting halide. For example, Yeung and coworkers employed their 1,3-diol desymmetrization via catalytic enantioselective bromoetherification with N-bromosuccinimide and chiral sulfide Lewis base catalyst 268 to generate enantioenriched tetrahydrofuran 269 (Scheme 57).[103] The brominated form of catalyst 268 can be considered as a chiral variant of Snyder’s BDSB. The protonation of succinimide by methanesulfonic acid is proposed to facilitate the formation of the active brominating species and the turnover of the catalyst.[104] The corresponding iodide of the bromotetrahydrofuran product 269 is a known precursor to the antifungal drugs posaconazole (270, Noxafil®) and Sch 51048 (271).
Scheme 57.
Desymmetrizing enantioselective bromoetherification with a chiral cyclic sulfide catalyst
2.4. Stereospecific Halonium-induced Polyene Cyclization
2.4.1 Difficulties Associated with Halonium-based Polyene Cyclizations
Polyene cyclizations are appreciated as particularly efficient tools for the rapid construction of many polycyclic natural products, predominantly in the realm of terpenoids. This area has been reviewed[105] and we will focus only the specifics of halonium-ion-induced cyclizations and their closely relevant analogues.
Bromonium-initiated polyene cyclizations appear to be widespread in the biosynthetic pathways of brominated marine natural products.[56] However, attempts to mimic such reactivity by chemical synthesis have proven challenging. Brominative polyene cyclizations with common electrophilic brominating reagents such as bromine, N-bromosuccinimide, or TBCO often suffer from low efficiency, most often because of unwanted side reactions via intermolecular participation of an external Lewis base, including the counterion from the electrophilic brominating reagent. In particular, owing to slow cation π-cyclization, electron-poor alkene substrates suffer from competing eliminations of proton or additions of nucleophile, each of which produces side products that are often difficult to separate from the desired products. The addition of a protic acid is often necessary to facilitate the second cyclization by regeneration of the cationic intermediate via protonation of eliminated compounds. For example, van Tamelen and Hessler showed that treatment of methyl farnesoate (272) with N-bromosuccinimide in aqueous tetrahydrofuran afforded the corresponding bromohydrin 273 as the major product along with only a small quantity of the desired cyclization product 274 (Scheme 58).[106] The addition of an external nucleophile, water in this case, was faster than the cyclization with a poorly nucleophilic pendant alkene. Even in the absence of competing nucleophiles, the basic counterion of the brominating reagent promoted elimination from cationic intermediates prior to the desired cyclization. Following their initial report on TBCO as a brominative cyclization reagent,[107] Kato and co-workers attempted a biomimetic synthesis of α- and β-snyderols (276 and 2) via the bromonium-induced cyclization of nerolidol (275).[108] The yields of the natural products were low, and it is unclear to what extent the cyclizations were diastereoselective with respect to the tertiary alcohol stereogenic center. Much of the mass balance of the reaction consisted of bromoetherification products of the central alkene (not shown), demonstrating that brominative carbocyclizations are generally unable to compete with heterocyclizations.
Scheme 58.

Competing side reactions of bromonium-initiated polyene cyclization
The problematic bromide counterion has been sequestered by precipitation as its silver salt or by complexation with a Lewis acid with limited success. Hoye and Kurth investigated the brominative polyene cyclization of geranyl derivatives with bromine in combination with a Lewis acid (not shown).[109] Under these conditions, proton-mediated cyclization remained a significant competing side reaction. The polyene cyclization of homogeranic acid (277) with bromine and silver tetrafluoroborate afforded the desired trans-fused bicyclic bromolactone 278 along with a mixture of the trans- and cis-fused norbromolactones 279 (Scheme 59). The almost exclusive formation of the less stable trans-isomer strongly supports a concerted cyclization mechanism. The bromocyclization is accompanied by the generation of acid, which is responsible for the competing acid-promoted cyclization to from 279. Although this side reaction could be suppressed in the presence of an excess of bromine, the yield was only marginally improved (3 equiv of Br2, 15% yield), because these conditions led to the formation of other side products.
Scheme 59.
Proton-mediated cyclization as a side reaction
As an alternative strategy, a multi-step sequence involving mercury(II)-mediated electrophilic polycyclization was developed by Hoye and Kurth (Scheme 60).[110] The subsequent stereospecific bromodemercuration of the bicyclic intermediate can be effected with either retention or inversion of configuration. The combination of these two well-known processes results in a net bromocyclization. Thus, homogeranic acid (277) was treated sequentially with mercury(II) trifluoroacetate followed by a saturated aqueous potassium bromide solution to produce an organomercuric bromide 280, which could be readily bromodemercurated in a solution of molecular bromine in pyridine to give a mixture of epimeric bromides (not shown). The β-bromo epimer 278 was selectively obtained upon addition of lithium bromide and oxygen to suppress the radical pathway, which is responsible for the generation of the other epimer. The α-bromo epimer of 278 was obtained selectively via radical chain reactions under photochemical conditions (not shown), which permits bromodemercuration with inversion of configuration resulting from preferential axial attack. This indirect bromocyclization strategy (with stereoretentive bromodemercuration) was employed for the synthesis of 3-bromo-8-epicaparrapi oxide (283). Unfortunately, the direct conversion of nerolidol (275) to 283 was unsuccessful, probably because of the ionization of allylic alcohol.
Scheme 60.
Indirect multi-step sequence involving mercury(II)-mediated polyene cyclization
The direct brominative polyene cyclization approach was marginally successful in the synthesis of aplysistatin (289) by Prestwich and co-workers (Scheme 61). Upon treatment of 284 with TBCO, the desired product 285 was obtained as a minor component of a 19:81 diastereomeric mixture in poor yield.[111] On the other hand, the groups of Hoye[112] and of White[113] both employed the mercury(II)-mediated indirect approach for the synthesis of 289. The direct cyclization 287 with TBCO was again not effective. Following Hoye’s multistep procedure,[110] White and co-workers converted 287 to the net bromocyclization products in a low but improved yield. The major diastereomer 288 had the desired configuration and was readily converted to 289. A similar mercury(II)-based approach was also employed for the synthesis of palisadin A by Tanaka and co-workers.[114]
Scheme 61.
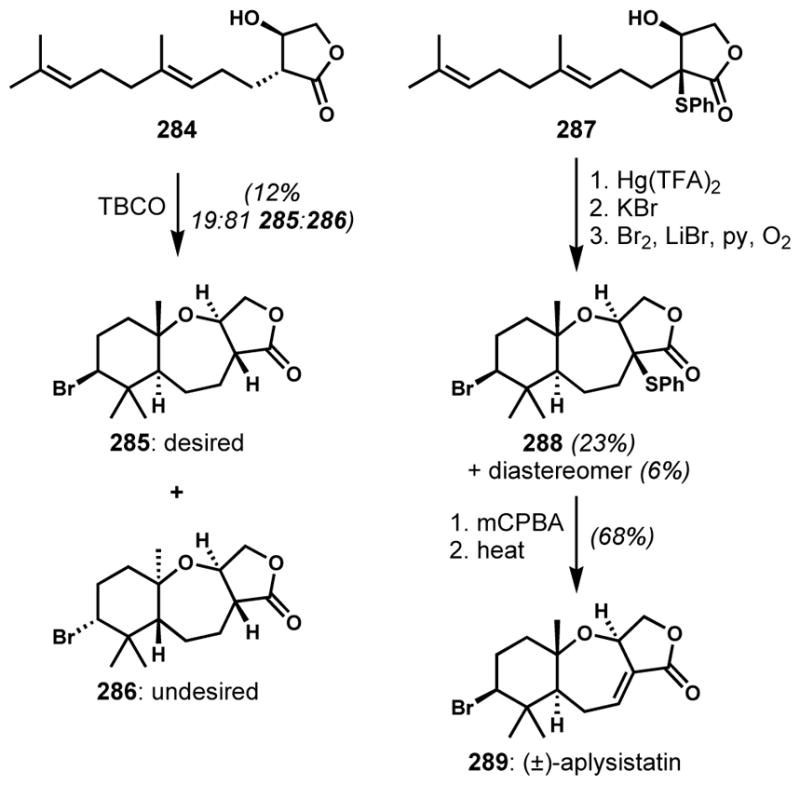
Direct and indirect brominative polyene cyclization approaches for the synthesis of aplysistatin
The multi-step approach was applied to extended polyene systems during the synthesis of isoaplysin-20 acetate (291) by Nishizawa and co-workers (Scheme 62).[115] Remarkably, the polyene cyclization of a tetraene substrate, E,E,E-geranylgeranyl acetate (290), proceeded via the all chair conformation even in the presence of four consecutive 1,3-diaxial interactions to give isoaplysin-20 acetate (291). Although the mercury(II)-mediated indirect method allowed the syntheses of several brominated polycyclic natural products, many drawbacks including generally low yields, the lengthy sequence, and the use of highly toxic mercury reagents diminishes the practicality.
Scheme 62.
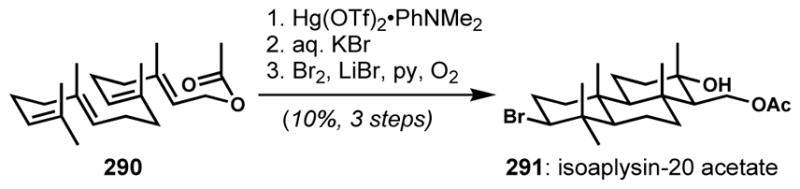
Mercury(II)-mediated brominative cyclization of a tetraene substrate
2.4.2 Direct Halonium-induced Polyene Cyclization
To this point, the problems associated with bromonium-induced polyene cyclization were addressed by presenting inefficient and unsuccessful examples. In addition, mercury(II)-promoted reactions were shown as an overall more effective, if not as desirable, alternative strategy. From here, efficient direct halonium-induced polyene cyclizations will be illustrated.
The major side reactions in halonium-induced polyene cyclizations are caused by the accompanying Lewis basic counterion. Thus, Faulkner and co-workers employed combinations of bromine with Lewis acids to produce bromonium ions without the generation of Lewis basic counterions (Scheme 63).[56a] In the presence of bromine and silver tetrafluoroborate, the polyene cyclization of geranyl acetate (292) proceeded in 20% yield. The formation of an alcohol (293) under anhydrous conditions was rationalized by the hydrolysis of a cyclic oxonium intermediate. The stereochemistry was consistent with a concerted cyclization mechanism. Similarly, geranyl acetone (294) was cyclized to bicyclic ether 295, which was converted in a few straightforward bioinspired steps to 10-bromo-α-chamigrene (296). In contrast to Hoye’s example (Scheme 59), acid is not generated under these conditions.
Scheme 63.
Sequestering a Lewis basic counterion by the use of silver tetrafluoroborate
As was seen in the synthesis of halomon (95, Scheme 29) and intricatetraol (160, Scheme 30), a bromonium ion can be transiently formed stereoselectively from a bromohydrin, providing an alternative to the direct reaction of an alkene with a halenium source that is often inefficient. Murai and coworkers employed such a reaction in the synthesis of (±)-aplysin-20 (not shown).[116] The use of this indirect method in combination with asymmetric alkene oxidation methods allows the formation of enantioenriched bromonium intermediates. Couladouros and co-workers prepared an enantioenriched epoxide 297, which was converted to the bromohydrin 298 via the subsequent regio- and stereospecific opening with lithium bromide under mildly acidic conditions (Scheme 64).[117] Activation of the hydroxyl group with tin tetrachloride triggered stereospecific polyene cyclization to give the brominated bicyclic lactone 278, which served as a synthetic precursor to several natural products including palisadins A (299) and B (300), and aplysistatin (289). This approach was further examined by Braddock and co-workers.[118]
Scheme 64.

Generation of an enantioenriched bromonium intermediate from a bromohydrin
The success of direct halonium-induced cyclizations depends critically upon the use of highly reactive halogenating reagents that generate only non-Lewis-basic byproducts. Snyder’s highly electrophilic reagent, BDSB, fits the criteria and efficiently promotes the bromonium-induced polyene cyclizations of a wide range of electronically diverse substrates; the reactivity of this family of reagents is an important advance in this area.[78] In general, BDSB-promoted reactions are faster and cleaner than cyclizations performed under other conditions. Surprisingly, electron-poor substrates such as geranyl cyanide (301) and trifluoroacetate (303) were effectively cyclized under these conditions (Scheme 65).[78a,b] Treatment of 301 with TBCO resulted in the exclusive dibromination of the more electron rich alkene[119] and no other reagents were able to promote the brominative cyclization of 303. Moreover, BDSD is chemoselective because it is compatible with electron-rich aromatic compounds such as 305. The relative configurations of the products arise from stereospecific cyclizations via highly organized, chair-like transition structures, as expected. In most cases, the polycyclizations proceeded well without the need of any extra protic acid additive because protonated dialkyl sulfide is generated in situ during the course of the reaction. This acidic byproduct can promote the later cyclizations when needed, and is likely responsible for high yield. However, significant challenges remain to render these polycyclizations catalytic and enantioselective. Because the acidic byproduct can also initiate the polyene cyclization to give non-halogenated compounds, it is important to consume most of the starting material at an early stage of the reaction, which is a difficult proposition under catalytic conditions. Under these effective brominating conditions, cis-decalin frameworks can also be generated, which had never been accomplished by brominative polyene cyclization (not shown).[78b]
Scheme 65.
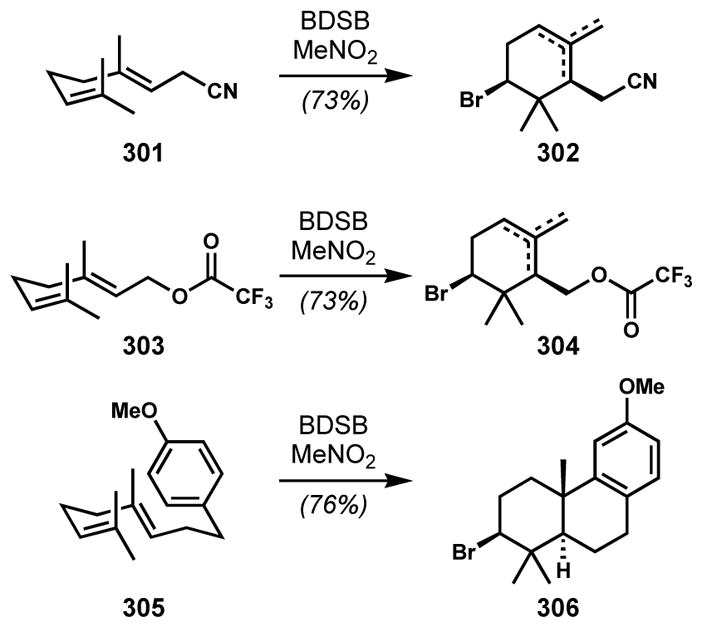
BDSB-promoted, highly effective and chemoselective polyene cyclizations
From the BDSB-promoted polyene cyclizations of the E,Z-farnesol-derived substrates 307 and 309, the substituent at C9 was installed at an axial position, which can be rationalized by a chair-like transition structure (Scheme 66). On the other hand, the reactions of E,E-farnesol-derived substrates 311 and 313 also afforded some of the bicyclic products 308 and 310 with an axial C9 substituent along with the expected products 312 and 314, even though a chair-like transition structure predicts high preference for the latter products with the equatorial orientation of the C9 substituent. These results suggest a deviation from an all-chair conformation.[120] These studies led to several total or formal syntheses of natural products including peyssonol A (315, revised structure), peyssonoic acid A, aplysin-20, loliolide, K-76, and stemodin.
Scheme 66.

Presumed deviation from the all-chair conformation in the transition structure leads to epimeric products
BDSB has also been applied to bromonium-induced transannular cyclization of a macrocyclic intermediate (316) in work toward the synthesis of bromophycolides A and D (318) by Krauss and coworkers (Scheme 67).[121] Although three very similar alkenes are present in the substrate, the preferred conformation of 316 allowed the initiation of bromocyclization only at the central alkene. Thus, upon treatment with BDSB, the desired products 317 were obtained as a mixture of alkene constitutional isomers in 19% yield. The cyclization was slow because of geometrical constraints in the bromonium intermediate, allowing competing deprotonation to take place to give allylic bromide side products. BDSB was superior to other brominating reagents because it does not generate any basic byproducts and the reaction can be carried out at low temperature. Unfortunately, bromohydrin formation—the last key step of the synthesis—could not be achieved, presumably because only one face of the alkene is exposed, and the other side cannot be accessed after the formation of a bromonium species.
Scheme 67.
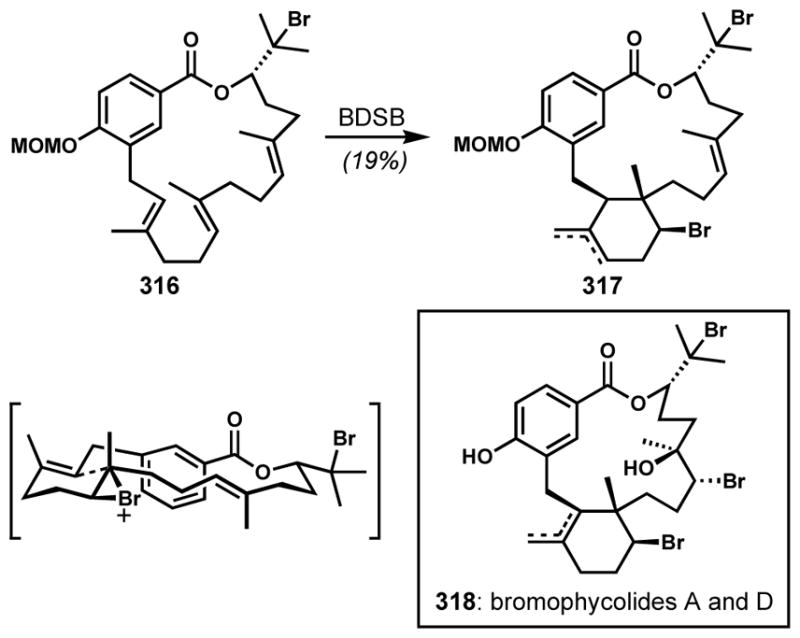
BDSB-promoted transannular polyene cyclization in a synthesis approach to the bromophycolides
A highly reactive brominating species can also be generated in situ from N-bromosuccinimide via Lewis base activation.[122] Chan, McErlean and co-workers employed a 3,3′-substituted 1,1′-bi-2-naphthol-derived phosphoramidite catalyst 320 for the synthesis of (+)-luzofuran (321) (Scheme 68).[123] These conditions are compatible with alcoholic substrates such as 319, which are not tolerated by BDSB. The 2,4,5-trichlorophenyltriazole substituent improves the reagent’s stability toward oxidation and its catalytic activity, which presumably involves intermediate 323. The authors concluded from control experiments that the diastereoselectivity of the reaction was primarily due to substrate control, with little influence from the chiral catalyst. The importance of the N-heterocyclic substituent in catalyst 320 was demonstrated by the lack of reactivity with simpler phosphoramidite 324. Important prior studies by the Ishihara group using phosphoramidite[124] and phosphite[125] activators for halocyclization have to date not been used in natural product synthesis.
Scheme 68.

Generation of a highly reactive brominating species with a Lewis base catalyst
Effective conditions for chloronium- or iodonium-induced polyene cyclizations are rare. Snyder and coworkers developed other halogenating reagents that are analogous to BDSB.[78b] The corresponding iodinating reagent, IDSI, exists as a chlorine-linked dimer as shown by X-ray crystallographic analysis (Scheme 69). Although IDSI is not as stable as BDSB, it affords similar rates, yields, and chemoselectivity as BDSB. The iodinated polycyclic compounds such as 326 were obtained as single diastereomers. In contrast to the conditions developed by the Ishihara laboratory, these reactions did not stall after the first cyclization and proceeded to the final products without protic acid additives. Similarly to BDSB, the generation of an acidic byproduct during the course of the reaction is responsible for this advantage. These conditions were also effective for the iodonium-induced polyene cyclization of electron-deficient substrates. Although these iodinated polycycles are not found in nature, they can be easily transformed into a range of other functional groups. In particular, a versatile alkene such as that in 327 was readily prepared via elimination of hydrogen iodide with 1,8-diazabicyclo[5.4.0]undec-7-ene; such eliminations are less facile from the more-stable bromides. A related process was used to synthesize the natural product loliolide (not shown).[78b] Of course, given the dearth of natural products containing iodides, to date iodonium-induced polyene cyclizations have not yet been responsible for the stereocontrolled incorporation of halogen-bearing stereogenic centers that are retained in the natural product targets.
Scheme 69.
IDSI-promoted polyene cyclization
Unfortunately, the corresponding chlorinating reagent, CDSC (chlorodiethylsulfonium hexachloroantimonate) is not as effective as BDSB or IDSI. Chloronium-induced polyene cyclization remains a challenge owing to the high reactivity of these intermediates and the potential for multiple competing pathways. Also, generally poor diastereoselectivities in these chloronium-induced cyclizations demonstrate the substantial challenge associated with the pronounced carbocation character of the initial chlorinated species. Polycyclic chlorides have been accessed via atom transfer radical cyclizations, which will be discussed briefly below.[126]
2.4.3 Enantioselective Halonium-induced Polyene Cyclization
As described in the previous sections, direct enantioselective halonium-induced transformation is a challenging process because of the potential for racemization of the halonium intermediate. Owing to the very difficult nature of this problem, we start with one important example that has not yet been applied to natural product synthesis. Following up on their earlier development of a phosphine-catalyzed, N-halosuccinimide-induced polyene cyclizations to give racemic products,[124,125] Ishihara and co-workers achieved a direct, enantioselective iodonium-induced polyene cyclization with N-iodosuccinimide in the presence of a chiral Lewis base promoter (Scheme 70).[124a] As various Lewis basic phosphorus compounds were identified as effective activators for N-bromosuccinimide and N-iodosuccinimide, a 1,1′-bi-2-naphthol-derived chiral phosphoramidite 329 with 3,3′-triisopropylsilyl substituents was employed for the enantioselective iodocyclization. Although a stoichiometric amount of 329 was necessary, and treatment of the crude mixture with strong acid was needed to fully convert partially cyclized intermediates, the desired iodinated polycyclic compounds 330 and 334 were obtained with excellent enantioselectivities. To accomplish efficient transfer of chiral information, it was critical to use a non-polar solvent to favor the formation of a tight ion pair involving the phosphonium salt. Although analogous reactions with N-bromosuccinimide or N-chlorosuccinimide were unsuccessful, the more naturally abundant chlorinated and/or brominated polycyclic compounds (331 and 332) could be accessed via a transhalogenation sequence involving reductive lithiation and bromination or chlorination with BrCF2CF2Br or ClCF2CF2Cl, respectively. Of course, with the ability to reductively deiodinate products of type 330 and 334, this method provides an excellent enantioselective approach to polycyclic terpenoids that are devoid of heteroatom substitution in the A rings.
Scheme 70.
Direct, enantioselective iodonium-induced polyene cyclization with N-iodosuccinimide and a chiral Lewis base promoter
Alternatively, an indirect procedure can be devised by taking advantage of chiral transition metal-mediated enantioselective polyene cyclization and stereospecific halogenative demetallation. Snyder and co-workers developed a two-step enantioselective sequence of mercury(II)-mediated polyene cyclization using a chiral bisoxazoline ligand 335 followed by stereoretentive halodemercuration analogous to Hoye’s procedure[110] to afford the corresponding polycyclic chloride 336, bromide 337, and iodide 326 (Scheme 71).[127] This strategy was applicable to a five-step enantioselective total synthesis of 4-isocymobarbatol (340).
Scheme 71.
Two-step enantioselective synthesis of polycyclic halides via asymmetric mercury(II)-mediated polyene cyclization
Snyder and co-workers attempted a direct, enantioselective halonium-induced polyene cyclization by employing chiral derivatives of CDSC, BDSB, and IDSI as halogenating reagents.[78b] Unfortunately, no promising data were obtained from their brief studies of polyene cyclization with C2-symmetric chiral halosulfonium ions (not shown). Finally, Gagné and co-workers have reported a fascinating asymmetric platinum-catalyzed formal fluorocyclization (not shown) that, while not directly relevant to natural product synthesis, might well be a lead toward a new general strategy to access A-ring-halogenated terpenoids.[128] Owing to the large number of chlorinated and brominated polycarbocyclic terpenoid natural products, and because of the sheer intellectual challenge posed, significant continued efforts to develop efficient catalytic and enantioselective methods for halonium-induced polyene cyclizations are anticipated.
2.5 Enantioselective α-Halogenation of Carbonyl Compounds
Without question, carbonyl α-halogenation is one of the most developed strategies to incorporate halogen-bearing stereogenic centers into organic molecules with control over absolute configuration. Auxiliary-based methods offered the first means of stereocontrol in the α-halogenation of carboxylic acid derivatives, as described by Evans and co-workers.[129] Lectka and co-workers achieved the highly enantioselective preparation of α-chloro or bromoesters from acid chlorides with a Lewis basic Cinchona alkaloid catalyst.[130] Methods for catalytic asymmetric α-halogenation of β-ketoesters and phosphonates involving chiral Lewis acids are also well developed;[131] however, the utility of these compounds for natural product synthesis is not obvious: natural products with halogen-bearing stereogenic centers between electron-withdrawing groups are uncommon, and the transformation of these compounds into higher-value-added compounds with retention of the halogen and without erosion of stereochemistry is not trivial. The enantioselective chlorination of enoxysilanes with a chiral chlorinating reagent also affords simple α-chloroketones.[132] Finally, over the past decade, the revolution in organocatalyzed asymmetric α-functionalization of aldehydes and ketones[133] has provided a number of useful protocols for access to highly enantioenriched α-haloaldehydes and α-haloketones.[134]
The versatility of α-haloaldehydes and ketones as synthetic building blocks and precursors to heterocyclic compounds has been recognized for a long time.[135] Still, to this day, the number of applications of asymmetric carbonyl α-halogenation chemistry to the stereocontrolled synthesis of halogenated natural products is incredibly small. Rather, these products tend to be used as building blocks wherein the halogen-bearing stereogenic center serves as a control element for further stereocontrolled reactions, or stereospecific displacement of the halide provides a solution to other stereochemical problems, or both. However, the connection between the stereogenicity of α-haloaldehydes and ketones and natural products bearing halogenated stereogenic centers is likely to be made more and more often now that simple organocatalytic methods to make these motifs are so readily available.
Umezawa, Matsuda, and co-workers made use of the enantioselective aldehyde α-chlorination developed by the Jørgensen group for their stereocontrolled synthesis of the chlorosulfolipid danicalipin A (7).[136] In this catalyst-controlled diastereoselective reaction, aldehyde 341 was treated with N-chlorosuccinimide in the presence of catalyst 342 and bromoacetic acid; the α-chloroaldehyde was produced as a single diastereomer (Scheme 72). The rather sensitive chloroaldehyde intermediate was directly converted to the corresponding unsaturated ester 344 by a Wittig reaction in the second stage of a one-pot process. From a strategic perspective, application of catalyst-controlled stereoselective chlorination permitted introduction of the C11 chloride without having to rely on potentially difficult remote stereocontrol. This work appears to be the only example of the use of asymmetric carbonyl α-halogenation in the stereocontrolled synthesis of halogenated natural products to date.[137]
Scheme 72.
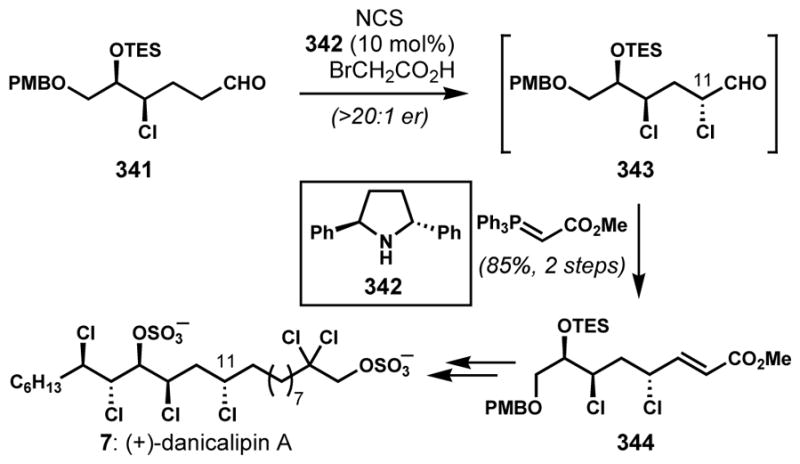
Organocatalytic asymmetric aldehyde α-chlorination in the synthesis of (+)-danicalipin A
The organocatalyzed asymmetric α-chlorinations developed by the MacMillan and the Jørgensen groups[138] were extensively utilized by Britton and co-workers for enantioselective syntheses of several natural products that do not contain halogen atoms (Figure 7).[4,139] In these syntheses, all of the stereocenters were constructed via asymmetric induction from these single chlorine-bearing stereogenic centers; diastereocontrolled nucleophilic additions to the α-haloaldehydes were the key steps in each case. Because it does not fit with the topic of the review, we simply offer a small subset of the natural products made by the Britton group using the sequence of asymmetric aldehyde α-chlorination/diastereocontrolled carbonyl addition/intramolecular chloride displacement. The bonds that were made by displacement of the chloride-bearing stereogenic center are indicated. This group has clearly demonstrated the versatility of chloride-bearing stereogenic centers as they play the roles both of temporary chiral auxiliary and precursor to key C–O or C–N bonds.
Figure 7.
Natural products made via the intermediacy of enantioenriched α-chloroaldehydes (indicated bonds were made by chloride displacements)
A fascinating example of substrate-controlled ketone α-bromination was reported by Stoltz and coworkers in their synthesis of elatol (351).[140] The final sequence in this synthesis is noteworthy for the stereocontrolled introduction of the unusual cis-bromohydrin by (di)bromination of 349 and an unusual hydride reduction of 350 that also provides the exocyclic alkene of the natural product (Scheme 73).
Scheme 73.
Generation of the unusual cis-bromohydrin of elatol by ketone α-bromination/reduction
2.6 Conversions of Alkenyl Halides to Halide-Bearing Stereogenic Centers
Geometrically-defined haloalkenes can serve as versatile starting materials for stereoselective formation of halogen-bearing stereogenic centers via a variety of stereospecific alkene functionalization reactions. The reaction types that have been used in natural product synthesis include alkene reductions, cycloadditions, and one example of a cyclization onto a chlorinated enoxysilane. It is likely that significant advances will continue to be made in this area.
2.6.1 Haloalkene Hydrogenations
Hydrogenation of haloalkenes is one simple and straightforward approach toward the installation of stereogenic centers featuring secondary halides. During the synthesis of (+)-isolaureatin (227), Kim and co-workers employed diimide to reduce cyclic bromoalkene intermediate 354 to install the secondary bromide on the bicyclic core (Scheme 74).[141] This strategy was particularly logical because the bromine atom is located on the more sterically congested concave face of the bridged bicyclic system. Bromination via SN2 displacement of an activated hydroxyl group or electrophilic alkene hydrobromination would require delivery of bromine from the concave side. Whereas deoxybromination of secondary alcohol 352 afforded the secondary bromide 353 in low yields with retention of configuration, desired bromide 355 was obtained via diimide reduction of alkenyl bromide 354 from the less hindered convex face.
Scheme 74.
Diastereocontrolled reduction of an alkenyl bromide to introduce a bromide-bearing stereogenic center
The alkenyl halide hydrogenation approach was a key strategic step in the synthesis of (−)-acutumine (357, Scheme 75) by Herzon and co-workers.[142] The secondary chloride was installed in the final step of the synthesis via homogeneous hydrogenation of a chloroalkene precursor, dehydroacutumine (356), using a rhodium catalyst under a high pressure of hydrogen gas. The natural product was obtained as a single diastereomer, likely owing to the directing effect of the neighboring amino or hydroxyl groups through the coordination to the catalyst. However, the hydrogenation had to be stopped at low conversion because dechlorination of the product became problematic at higher conversion. The use of various heterogeneous catalysts resulted in the exclusive formation of overreduced product dechloroacutumine (358). Recently, Herzon and co-workers developed new conditions for hydrogenation of haloalkenes (F, Cl, Br, I) with a cobalt catalyst and demonstrated that problematic dehalogenation can be suppressed under these conditions that operate via a hydrogen atom transfer mechanism.[143] This method is likely to become important for substrate-controlled alkenyl halide reductions in natural product synthesis.
Scheme 75.
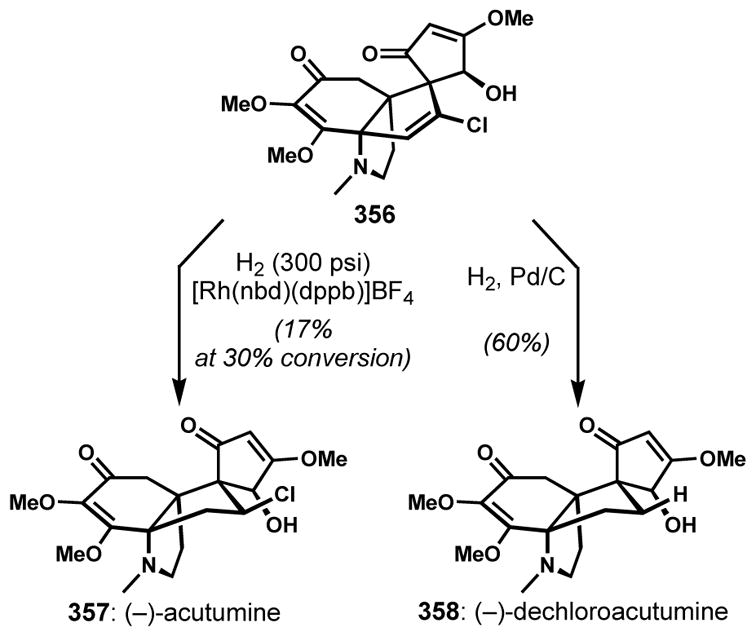
Homogeneous alkenyl chloride hydrogenation as the final step in a synthesis of acutumine
2.6.2 Diels–Alder Cycloadditions of Halogenated Dienes or Dienophiles
Halogen-bearing stereogenic centers in cyclic compounds can also be constructed via the cycloaddition reactions of alkenyl halides. Since the intramolecular Diels–Alder reaction of a β-chlorodienophile was reported by Corey and co-workers for the synthesis of gibberellic acid (which does not retain the chloride),[144] this approach has been utilized for the synthesis of halogenated natural products. Williard and co-workers first investigated the Diels–Alder reactions of β-halogenated dienophiles for the synthesis of cyclic polyhalogenated natural products from Plocamium red alga.[14] For example, the core of epi-plocamene (23) was formed stereoselectively via cycloaddition of butadiene (359) and Z-β-chloromethacrolein (360) (Scheme 76). The influence of the β-halogen substituent on the regio- or stereoselectivity was minimal. Unfortunately, this method could not be generalized because cycloadditions of substituted dienes proceeded with poor regio- and stereoselectivity to afford inseparable mixtures of products.
Scheme 76.
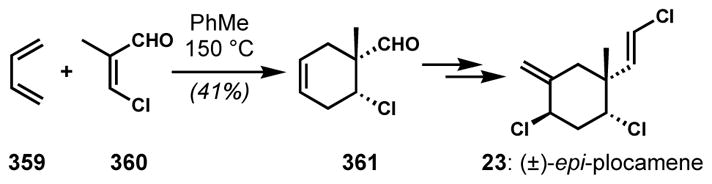
Diels–Alder cycloaddition using a chlorinated dienophile sets up a key stereochemical relationship in epi-plocamene
The regiochemical problem associated with this approach could be controlled by rendering the cycloaddition intramolecular using a temporary silane tether between diene and dienophile. From unsymmetrically substituted acyclic silane 362, Shea and co-workers were able to synthesize a bridgehead allylic silane 133 (Scheme 77), which was subsequently dichlorinated and elaborated over three additional steps to a Plocamium monoterpenoid (see Scheme 25).[52]
Scheme 77.
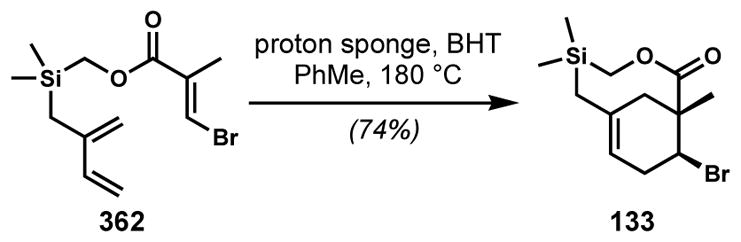
Temporary tether strategy to control regiochemistry of β-bromomethacryclic ester Diels–Alder cycloaddition
The intramolecular Diels–Alder cycloaddition approach was also employed for the installation of the chlorine-bearing stereocenter of (±)-virantmycin (80) by Corey and co-workers (Scheme 78).[145] Treatment of 363 with base resulted in the formation of an o-azaxylylene intermediate,[146] which underwent an intramolecular [4+2] cycloaddition to give 364, thereby installing the correct relationship between the two stereocenters in the natural product.
Scheme 78.
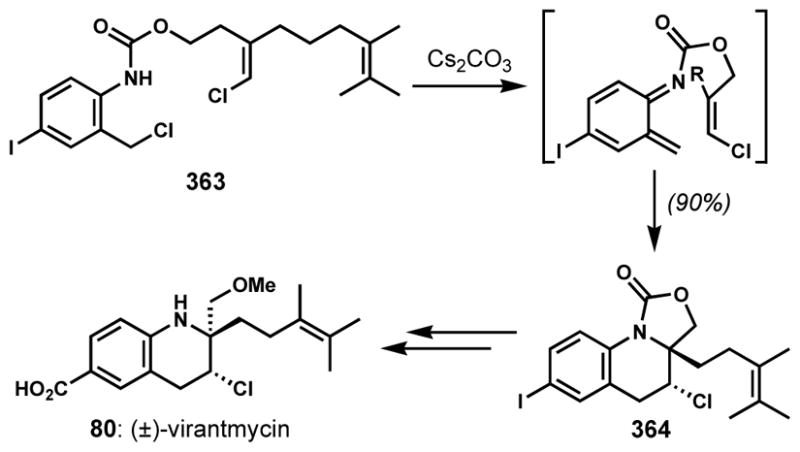
Intramolecular cycloaddition to install the α-chloro tertiary carbinolamine motif in virantmycin
Several members of the hapalindole family bear secondary chlorides with adjacent quaternary stereogenic centers, a motif seen above in several of the welwitindolinone alkaloids (see 93, Scheme 16, and 251, Scheme 52). Chandra and Johnston synthesized hapalindoles K (368) and A (6) via a Diels–Alder cycloaddition of two highly substituted reaction partners, trisubstituted diene 365 andβ-chloro-α-methyl enone 366 (Scheme 79).[147] With the appropriate choice of Lewis acid, this demanding cycloaddition proceeded in good yield to provide the endo-product (with respect to the ketone function), although the important stereochemical relationship that was forged was that between the chloride-bearing and quaternary stereogenic centers, because the silyloxy group was later replaced in the context of a substrate-controlled Ritter reaction.
Scheme 79.
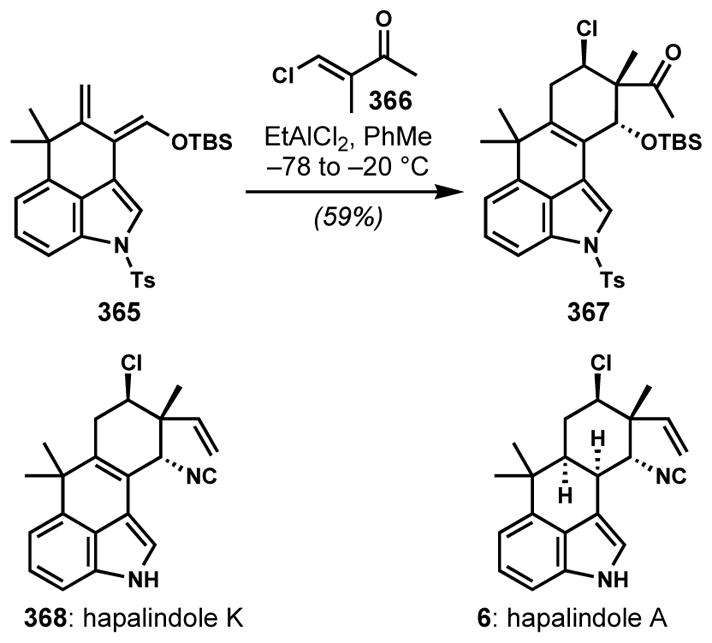
Challenging cycloaddition to install a key vicinal stereochemical arrangement in hapalindoles A and K
2.6.3 Other Cycloadditions or Cyclizations onto Haloalkenes
As seen in the previous section, halogen-bearing carbon stereocenters can be created via C–C bond-forming reactions of haloalkenes. A clever utilization of such reactivity was reported by Carreira and co-workers for the construction of one of the two contiguous tertiary chlorides in gomerone C (372, Scheme 80).[148] The Conia-ene reaction of a chlorinated enoxysilane 369 in the presence of Echavarren’s catalyst (370)[149] allowed for the simultaneous formation of the bridgehead tertiary chloride and the bicyclic scaffold. The subsequent hydrochlorination of the resulting electron-poor, exocyclic alkene required forcing reaction conditions with an excess of tin tetrachloride.[150] The addition of chloride from the less hindered exo-face afforded the desired axial chloride of the natural product. This two-step sequence is a clever solution to the problem posed by the vicinal tertiary chloride stereogenic centers that simultaneously addresses the bridging ring system.
Scheme 80.
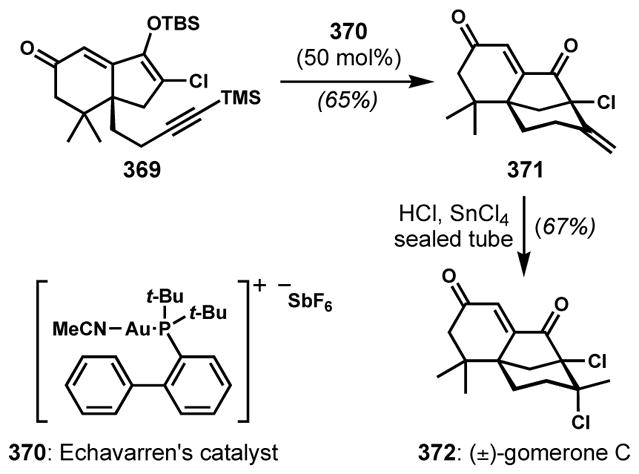
Synthesis of gomerone C via gold-catalyzed Conia-ene-like cyclization of a chlorinated enoxysilane
The trans-chlorocyclopropane moiety of callipeltoside A (379) has been synthesized by multiple research groups using several different approaches. Stereoselective and stereospecific cyclopropanations of E-chloroalkenes is a direct way to generate the vicinal stereogenic centers (Scheme 81). Evans and coworkers developed a highly diastereoselective cyclopropanation of D-mannitol-derived chiral E-chloroalkene 373.[151] Although this chloroalkene was poorly reactive under standard Simmons–Smith conditions, Shi’s highly active reagent system with trifluoroacetic acid was successfully employed to give 374 as a single diastereomer in high yield.[152] The groups of Paterson[153] and Panek[154] applied the enantioselective variant of the Simmons–Smith cyclopronanation developed by Charette and coworkers.[155] In the presence of the (S,S)-dioxaborolane ligand 376, the trans-chlorocyclopropylmethanol (377) was produced from chloroallylic alcohol (375) with high enantioselectivity.
Scheme 81.
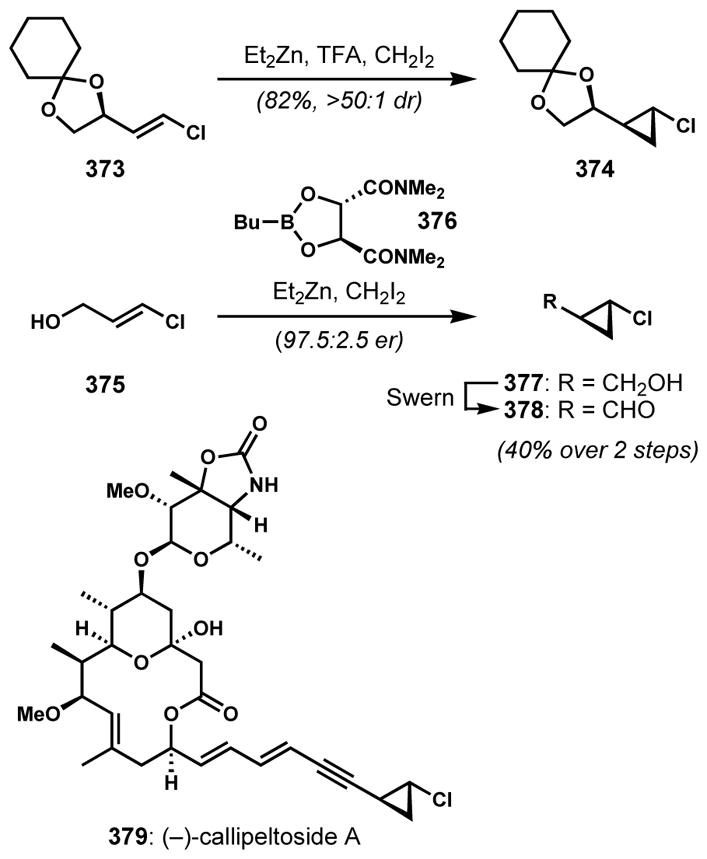
Synthesis of the enantiomerically enriched chlorocyclopropanes for callipeltoside A
A Claisen rearrangement of a chlorinated allylic ether chloroalkenes was used in the context of the halomon synthesis by Mioskowski and co-workers; however, this reaction did not control the formation of the new chlorine-bearing stereogenic center.[57a] Sigmatropic rearrangements involving chlorinated alkene substrates appear to be promising but underdeveloped strategies for the stereocontrolled synthesis of certain halogenated natural products.
2.6.4 Nucleophilic Haloallylation of Carbonyl Compounds
The use of allylmetal reagents terminally substituted with halogens is a powerful method for the stereocontrolled introduction of halogen-bearing stereogenic centers, yet it has seen relatively little use in the synthesis of halogenated natural products. The most compelling showcase of this strategy is in the synthesis of the spirastrellolide family (see spirastrellolide A methyl ester, 380, Scheme 82). In completed syntheses by the groups of Paterson[156] and of Fürstner,[157] the Oehlschlager chloroallylboration[158]—a variant of the Brown asymmetric allylboration—is used to efficiently introduce the C28 and C29 stereogenic centers, including the key chloride. Simple aldehyde 381 was treated with the chiral chloroallylborane 382 to afford building block 383 with near perfect diastereoselectivity and high enantioselectivity; the acetal was removed under workup conditions.[156a] That this reaction was able to support the synthesis of such a complex target indicates its robustness. A related bromoallylalumination reaction, wherein the bromide was eventually converted to a chloride via an intermediate epoxide, was utilized in chlorosulfolipid syntheses by Vanderwal and co-workers.[46]
Scheme 82.
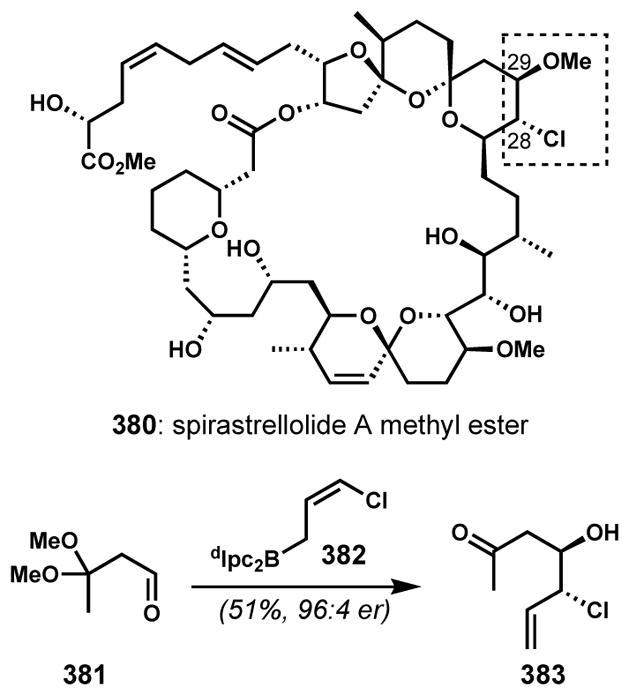
Asymmetric chloroallylboration en route to the spirastrellolides
2.7 Radical Processes
2.7.1 Radical Halodecarboxylation
Substrate-controlled, diastereoselective radical halogenation can be a useful strategy, particularly with conformationally rigid cyclic substrates. For example, the chlorine atom of callipeltoside A (379) was installed on the cyclopropane ring in a highly diastereoselective manner by such a reaction. Trost and co-workers developed a two-step, one-pot procedure for Barton decarboxylation/chlorination (the Barton version of a Hunsdiecker reaction)[159] for the highly stereoselective conversion of a chiral acyl chloride 384 to the corresponding trans-cyclopropyl chloride 385 (Scheme 32).[160] Presumably, the menthol auxiliary present on the molecule—needed for the stereocontrolled formation of the cyclopropane—plays little role in the stereochemical outcome of chlorination, and the major bias likely arises from preferential chlorine incorporation trans to the ester.
The Barton chlorodecarboxylation chemistry was just as effective in a much more complex setting. In the course of the enantioselective synthesis of the cyclopentane core of the axinellamines (243 and 244) by Carreira and co-workers,[161] diastereoselective chlorination of densely functionalized cyclopentane 386 was achieved using the Barton process to give the sterically hindered secondary chloride 387 (Scheme 84). Interestingly, after formation of the Barton ester in deoxygenated CCl4, spontaneous decarboxylation/chlorination occurred without the need for any added initiator.
Scheme 84.
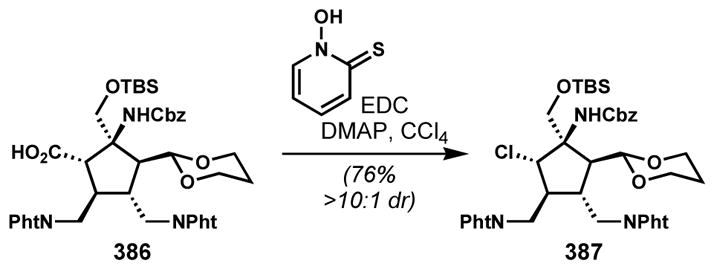
Barton chlorodecarboxylation in studies toward the axinellamines
2.7.2 Radical Dehalogenation and Cyclization Processes of gem-Dihalides
Other types of radical reactions such as dehalogenation and cyclization are also effective for the stereoselective synthesis of halogenated organic compounds. These sterically-controlled approaches were strategically employed by Lee and co-workers for the stereoselective installations of secondary halides in dactomelynes (392 and 393) (Scheme 85)[162] Because the axial chlorine substituent needs to be located on the more congested side, radical dehalogenantion of a gem-dichloride precursor such as 388 is perfectly suited for the delivery of the hydrogen from the less hindered side. While a range of tin hydride reagents promoted poorly selective dechlorination to give a mixture of diasteromers, the use of tris(trimethylsilyl)silane in combination with triethylborane successfully provided the desired product 389 in high yield and diastereoselectivity.[163] The equatorial bromine substituent was introduced via a radical cyclization in which the steric interaction around the bromine is minimized. Thus, desired bromopyran 391 was produced exclusively from 390 under tributyltin hydride-mediated radical cyclization conditions.
Scheme 85.

Syntheses of dactomelynes featuring radical dechlorination and cyclization steps
3. Conclusions and New Directions
Clearly, nature has provided a wealth of fascinating and complex halogenated compounds to inspire synthetic chemists, who have risen to the occasion with many outstanding accomplishments. Furthermore, nature’s methods for halogenation have motivated some great advances in synthesis. Still, from our survey of methods and strategies that have been used to introduce halogen-bearing stereogenic centers in the context of natural product synthesis, it has become apparent that there is room for many future accomplishments. Reaction types that would both be of tremendous general utility and that would represent significant advances in basic synthetic chemistry include, but are certainly not limited to: (1) general enantioselective alkene halogenation reactions, (2) asymmetric haloalkene reductions, (3) direct diastereoselective or enantioselective C–H halogenation, and (4) general enantioselective halonium-induced cationic cyclizations. Examples of reaction types that are known but so far have not, to our knowledge, been used in the synthesis of natural products include radical C–H halogenation and alkene hydrohalogenation via radical pathways. Surely, there will be many more stunning accomplishments in the synthesis of halogenated natural products.
Scheme 83.
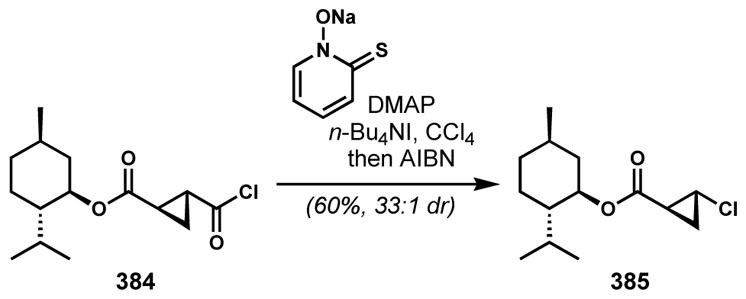
Stereocontrolled Hunsdiecker-type chlorination using a Barton ester for the synthesis of the chlorocyclopropane of callipeltoside A
Biographies

Won-jin Chung was born in Wonju, South Korea in 1979. He obtained a B.S. degree in Chemistry from Korea Advanced Institute of Science and Technology (KAIST) in 2002 and his Ph.D. degree from University of Illinois at Urbana–Champaign under the direction of Professor Scott E. Denmark in 2008. After completion of Korean military service in 2011, he conducted postdoctoral studies with Professor Christopher D. Vanderwal at the University of California, Irvine. In 2014, he joined Gwangju Institute of Science and Technology (GIST) as an assistant professor.

Christopher Vanderwal received a B.Sc. degree in Biochemistry (1995) and an M.Sc. degree in Chemistry (1998) from the University of Ottawa. He then moved to the Scripps Research Institute for doctoral studies in the group of Professor Erik Sorensen. After obtaining his Ph.D. in 2003, Chris joined the group of Professor Eric Jacobsen at Harvard University as a Jane Coffin Childs postdoctoral associate. In 2005, Chris began his independent academic career at the University of California, Irvine, where he is currently Professor of Chemistry.
References
- 1.a) Gribble GW. Environ Chem. 2015 doi: 10.1071/EN15002. in press. [DOI] [Google Scholar]; b) Gribble GW, Knölker H-J, editors. The Alkaloids: Chemistry and Biology. Vol. 71. chap. 1. Academic Press; Amsterdam, The Netherlands: 2012. Occurrence of Halogenated Alkaloids. [DOI] [PubMed] [Google Scholar]; c) Naturally Occurring Organohalogen Compounds–A Comprehensive Update. Gribble GW. Prog Chem Org Nat Prod. 2010;91:1–613. doi: 10.1007/978-3-031-26629-4_1. [DOI] [PubMed] [Google Scholar]; d) Gribble GW. Chemosphere. 2003;52:289–297. doi: 10.1016/S0045-6535(03)00207-8. [DOI] [PubMed] [Google Scholar]
- 2.Gribble GW. personal communication.
- 3.a) Gribble GW. Mar Drugs. 2015;13:4044–4136. doi: 10.3390/md13074044. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu Z, Yang Z, Liu Y, Lu Y, Chen K, Zhu W. J Chem Inf Model. 2014;54:69–78. doi: 10.1021/ci400539q. [DOI] [PubMed] [Google Scholar]; c) Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. J Med Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]; d) Lu Y, Liu Y, Xu Z, Li H, Liu H, Zhu W. Expert Opin Drug Discov. 2012;7:375–383. doi: 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]; e) Hernandes MZ, Cavalcanti SMT, Moreira DRM, de Azevedo WF, Jr, Leite ACL. Curr Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]; f) Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu W. J Med Chem. 2009;52:2854–2862. doi: 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]; g) Harris CM, Kannan R, Kopecka H, Harris TM. J Am Chem Soc. 1985;107:6652–6658. [Google Scholar]
- 4.Halperin SD, Britton R. Org Biomol Chem. 2013;11:1702–1705. doi: 10.1039/c3ob27462d. [DOI] [PubMed] [Google Scholar]
- 5.a) Neumann CS, Fujimori DG, Walsh CT. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]; b) Fujimori DG, Walsh CT. Curr Opin Chem Biol. 2007;11:553–560. doi: 10.1016/j.cbpa.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]; d) van Pée KH, Unversucht S. Chemosphere. 2003;52:299–312. doi: 10.1016/S0045-6535(03)00204-2. [DOI] [PubMed] [Google Scholar]
- 6.a) Leblanc C, Vilter H, Fournier JB, Delage L, Potin P, Rebuffet E, Michel G, Solari PL, Feiters MC, Czjzek M. Coord Chem Rev. 2015 doi: 10.1016/j.ccr.2015.02.013. in press. [DOI] [Google Scholar]; b) Butler A, Sandy M. Nature. 2009;460:848–854. doi: 10.1038/nature08303. [DOI] [PubMed] [Google Scholar]; c) Winter JM, Moore BS. J Biol Chem. 2009;284:18577–18581. doi: 10.1074/jbc.R109.001602. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Butler A, Carter-Franklin JN. Nat Prod Rep. 2004;21:180–188. doi: 10.1039/b302337k. [DOI] [PubMed] [Google Scholar]; e) Butler A. Coord Chem Rev. 1999;187:17–35. [Google Scholar]; f) Butler A. Curr Opin Chem Biol. 1998;2:279–285. doi: 10.1016/s1367-5931(98)80070-7. [DOI] [PubMed] [Google Scholar]; g) van Pée KH. Annu Rev Microbiol. 1996;50:375–399. doi: 10.1146/annurev.micro.50.1.375. [DOI] [PubMed] [Google Scholar]; h) Butler A, Walker JV. Chem Rev. 1993;93:1937–1944. [Google Scholar]
- 7.a) Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]; b) Galonić DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]; c) Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci USA. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 8.Hillwig ML, Liu X. Nat Chem Biol. 2014;10:921–923. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 9.a) Mercer EI, Davies CL. Phytochemistry. 1974;13:1607–1610. [Google Scholar]; b) Mercer EI, Davies CL. Phytochemistry. 1975;14:1545–1548. [Google Scholar]; c) Mercer EI, Davies CL. Phytochemistry. 1979;18:457–462. [Google Scholar]; d) Mooney CL, Mahoney EM, Pousada M, Haines TH. Biochemistry. 1972;11:4839–4844. doi: 10.1021/bi00775a030. [DOI] [PubMed] [Google Scholar]; e) Mooney CL, Haines TH. Biochemistry. 1973;12:4469–4472. doi: 10.1021/bi00746a026. [DOI] [PubMed] [Google Scholar]
- 10.Bedke DK, Vanderwal CD. Nat Prod Rep. 2011;28:15–25. doi: 10.1039/c0np00044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuzawa A, Sato H, Masamune T. Tetrahedron Lett. 1987;28:4303–4306. [Google Scholar]
- 12.Kim D, Kim IH. Tetrahedron Lett. 1997;38:415–416. [Google Scholar]
- 13.Barton DHR, Miller E. J Am Chem Soc. 1950;72:370–374. [Google Scholar]
- 14.Williard PG, De Laszlo SE. J Org Chem. 1985;50:3738–3749. [Google Scholar]
- 15.Feldman KS, Mechem CC, Nader L. J Am Chem Soc. 1982;104:4011–4012. [Google Scholar]
- 16.Grese TA, Hutchinson KD, Overman LE. J Org Chem. 1993;58:2468–2477. [Google Scholar]
- 17.a) Crimmins MT, Emmitte KA. J Am Chem Soc. 2001;123:1533–1534. doi: 10.1021/ja005892h. [DOI] [PubMed] [Google Scholar]; b) Crimmins MT, Emmitte KA, Choy AL. Tetrahedron. 2002;58:1817–1834. [Google Scholar]
- 18.Wang J, Pagenkopf BL. Org Lett. 2007;9:3703–3706. doi: 10.1021/ol701797e. [DOI] [PubMed] [Google Scholar]
- 19.Park J, Kim B, Kim H, Kim S, Kim D. Angew Chem Int Ed. 2007;46:4726–4728. doi: 10.1002/anie.200700854. [DOI] [PubMed] [Google Scholar]
- 20.a) Anderson NG, Lust DA, Colapret KA, Simpson JH, Malley MF, Gougoutas JZ. J Org Chem. 1996;61:7955–7958. doi: 10.1021/jo9609539. [DOI] [PubMed] [Google Scholar]; b) Galynker I, Still WC. Tetrahedron Lett. 1982;23:4461–4464. [Google Scholar]
- 21.Jeong W, Kim MJ, Kim H, Kim S, Kim D, Shin KJ. Angew Chem Int Ed. 2010;49:752–756. doi: 10.1002/anie.200905826. [DOI] [PubMed] [Google Scholar]
- 22.a) Frantz DE, Fässler R, Carreira EM. J Am Chem Soc. 2000;122:1806–1807. [Google Scholar]; b) Moore D, Pu L. Org Lett. 2002;4:1855–1857. doi: 10.1021/ol025825n. [DOI] [PubMed] [Google Scholar]
- 23.Boukouvalas J, Pouliot M, Robichaud J, MacNeil S, Snieckus V. Org Lett. 2006;8:3597–3599. doi: 10.1021/ol061385e. [DOI] [PubMed] [Google Scholar]
- 24.Bonini C, Righi G. Synthesis. 1994:225–238. [Google Scholar]
- 25.Caron M, Sharpless KB. J Org Chem. 1985;50:1557–1560. [Google Scholar]
- 26.a) Gao L-x, Murai A. Chem Lett. 1989;18:357–358. [Google Scholar]; b) Gao L-x, Murai A. Chem Lett. 1991;20:1503–1504. [Google Scholar]; c) Gao L-x, Saitoh H, Feng F, Murai A. Chem Lett. 1991;20:1787–1790. [Google Scholar]; d) Gao L-x, Murai A. Tetrahedron Lett. 1992;33:4349–4352. [Google Scholar]
- 27.Raifeld YE, Nikitenko AA, Arshava BM. Tetrahedron: Asymmetry. 1991;2:1083–1084. [Google Scholar]
- 28.Nilewski C, Deprez NR, Fessard TC, Li DB, Geisser RW, Carreira EM. Angew Chem Int Ed. 2011;50:7940–7943. doi: 10.1002/anie.201102521. [DOI] [PubMed] [Google Scholar]
- 29.Overman LE, Thompson AS. J Am Chem Soc. 1988;110:2248–2256. [Google Scholar]
- 30.Berger D, Overman LE, Renhowe PA. J Am Chem Soc. 1993;115:9305–9306. [Google Scholar]
- 31.Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]
- 32.Bedke DK, Shibuya GM, Pereira A, Gerwick WH, Haines TH, Vanderwal CD. J Am Chem Soc. 2009;131:7570–7572. doi: 10.1021/ja902138w. [DOI] [PubMed] [Google Scholar]
- 33.Shemet A, Sarlah D, Carreira EM. Org Lett. 2015;17:1878–1881. doi: 10.1021/acs.orglett.5b00558. [DOI] [PubMed] [Google Scholar]
- 34.Fukuyama T, Chen X. J Am Chem Soc. 1994;116:3125–3126. [Google Scholar]
- 35.a) Hill ML, Raphael RA. Tetrahedron Lett. 1986;27:1293–1296. [Google Scholar]; b) Hill ML, Raphael RA. Tetrahedron. 1990;46:4587–4594. [Google Scholar]
- 36.Morimoto Y, Shirahama H. Tetrahedron. 1996;52:10631–10652. [Google Scholar]
- 37.Hanessian S, Del Valle JR, Xue Y, Blomberg N. J Am Chem Soc. 2006;128:10491–10495. doi: 10.1021/ja0625834. [DOI] [PubMed] [Google Scholar]
- 38.Miyashita K, Tanaka A, Shintaku H, Iwata C. Tetrahedron. 1998;54:1395–1406. [Google Scholar]
- 39.Squires TG, Schmidt WW, McCandlish CS., Jr J Org Chem. 1975;40:134–136. [Google Scholar]
- 40.Weires NA, Styduhar ED, Baker EL, Garg NK. J Am Chem Soc. 2014;136:14710–14713. doi: 10.1021/ja5087672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhat V, Rawal VH. Chem Commun. 2011;47:9705–9707. doi: 10.1039/c1cc13498a. [DOI] [PubMed] [Google Scholar]
- 42.Cleary L, Pitzen J, Brailsford JA, Shea KJ. Org Lett. 2014;16:4460–4463. doi: 10.1021/ol5020043. [DOI] [PubMed] [Google Scholar]
- 43.a) Borthwick AD, Curry DJ, Poynton A, Whalley WB, Hooper JW. J Chem Soc, Perkin Trans. 1980;1:2435–2444. [Google Scholar]; b) Koreeda M, Gopalaswamy R. J Am Chem Soc. 1995;117:10595–10596. [Google Scholar]
- 44.Shibuya GM, Kanady JS, Vanderwal CD. J Am Chem Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]
- 45.Bedke DK, Shibuya GM, Pereira AR, Gerwick WH, Vanderwal CD. J Am Chem Soc. 2010;132:2542–2543. doi: 10.1021/ja910809c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.a) Chung W-j, Carlson JS, Bedke DK, Vanderwal CD. Angew Chem Int Ed. 2013;52:10052–10055. doi: 10.1002/anie.201304565. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chung W-j, Carlson JS, Vanderwal CD. J Org Chem. 2014;79:2226–2241. doi: 10.1021/jo5000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshimitsu T, Fukumoto N, Nakatani R, Kojima N, Tanaka T. J Org Chem. 2010;75:5425–5437. doi: 10.1021/jo100534d. [DOI] [PubMed] [Google Scholar]
- 48.Markó IE, Richardson PR, Bailey M, Maguire AR, Coughlan N. Tetrahedron Lett. 1997;38:2339–2342. [Google Scholar]
- 49.Tartakoff SS, Vanderwal CD. Org Lett. 2014;16:1458–1461. doi: 10.1021/ol500265v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogel CV, Pietraszkiewicz H, Sabry OM, Gerwick WH, Valeriote FA, Vanderwal CD. Angew Chem Int Ed. 2014;53:12205–12209. doi: 10.1002/anie.201407726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sotokawa T, Noda T, Pi S, Hirama M. Angew Chem Int Ed. 2000;39:3430–3432. doi: 10.1002/1521-3773(20001002)39:19<3430::aid-anie3430>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 52.Whitney JM, Parnes JS, Shea KJ. J Org Chem. 1997;62:8962–8963. [Google Scholar]
- 53.Yoshimitsu T, Fukumoto N, Tanaka T. J Org Chem. 2009;74:696–702. doi: 10.1021/jo802093d. [DOI] [PubMed] [Google Scholar]
- 54.Iranpoor N, Firouzabadi H, Azadi R, Ebrahimzadeh F. Can J Chem. 2006;84:69–75.; for a catalytic variant with (COCl)2, see: Denton RM, Tang X, Przeslak A. Org Lett. 2010;12:4678–4681. doi: 10.1021/ol102010h.
- 55.Yoshimitsu T, Nakatani R, Kobayashi A, Tanaka T. Org Lett. 2011;13:908–911. doi: 10.1021/ol1029518. [DOI] [PubMed] [Google Scholar]
- 56.a) Faulkner DJ. Pure Appl Chem. 1976;48:25–28. [Google Scholar]; b) Faulkner DJ. Tetrahedron. 1977;33:1421–1443. [Google Scholar]
- 57.Schlama T, Baati R, Gouverneur V, Valleix A, Falck JR, Mioskowski C. Angew Chem Int Ed. 1998;37:2085–2087. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2085::AID-ANIE2085>3.0.CO;2-J.; See also Jung ME, Parker MH. J Org Chem. 1997;62:7094–7095. doi: 10.1021/jo971371+.
- 58.Morimoto Y, Okita T, Takaishi M, Tanaka T. Angew Chem Int Ed. 2007;46:1132–1135. doi: 10.1002/anie.200603806. [DOI] [PubMed] [Google Scholar]
- 59.El-Qisairi AK, Qaseer HA, Katsigras G, Lorenzi P, Trivedi U, Tracz S, Hartman A, Miller JA, Henry PM. Org Lett. 2003;5:439–441. doi: 10.1021/ol0273093. [DOI] [PubMed] [Google Scholar]
- 60.El-Qisairi A, Hamed O, Henry PM. J Org Chem. 1998;63:2790–2791. doi: 10.1021/jo005627e. [DOI] [PubMed] [Google Scholar]
- 61.Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. J Am Chem Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Snyder SA, Tang Z-Y, Gupta R. J Am Chem Soc. 2009;131:5744–5745. doi: 10.1021/ja9014716. [DOI] [PubMed] [Google Scholar]
- 63.Kelly TR, Whiting A, Chandrakumar NS. J Am Chem Soc. 1986;108:3510–3512. [Google Scholar]
- 64.Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]
- 65.Hu DX, Seidl FJ, Bucher C, Burns NZ. J Am Chem Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]
- 66.Cresswell AJ, Eey ST-C, Denmark SE. Nat Chem. 2015;7:146–152. doi: 10.1038/nchem.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kato T, Ichinose I, Hosogai T, Kitahara Y. Chem Lett. 1976;5:1187–1190.; See also: Ting PC, Bartlett PA. J Am Chem Soc. 1984;106:2668–2671.
- 68.a) Hashimoto M, Kan T, Yanagiya M, Shirahama H, Matsumoto T. Tetrahedron Lett. 1987;28:5665–5668. [Google Scholar]; b) Hashimoto M, Kan T, Nozaki K, Yanagiya M, Shirahama H, Matsumoto T. Tetrahedron Lett. 1988;29:1143–1144. [Google Scholar]; c) Hashimoto M, Kan T, Nozaki K, Yanagiya M, Shirahama H, Matsumoto T. J Org Chem. 1990;55:5088–5107. [Google Scholar]
- 69.Corey EJ, Ha DC. Tetrahedron Lett. 1988;29:3171–3174. doi: 10.1016/0040-4039(88)85122-0.; for a similar study with NBS, see: Broka CA, Lin YT. J Org Chem. 1988;53:5876–5885.
- 70.a) González IC, Forsyth CJ. Org Lett. 1999;1:319–322. doi: 10.1021/ol990648k. [DOI] [PubMed] [Google Scholar]; b) González IC, Forsyth CJ. J Am Chem Soc. 2000;122:9099–9108. [Google Scholar]
- 71.a) Tonn CE, Palazón JM, Ruiz-Pérez C, Rodrlguez ML, Martín VS. Tetrahedron Lett. 1988;29:3149–3152. [Google Scholar]; b) Martín T, Soler MA, Betancort JM, Martín VS. J Org Chem. 1997;62:1570–1571. [Google Scholar]
- 72.a) Jung ME, Lew W. J Org Chem. 1991;56:1347–1349. [Google Scholar]; b) Jung ME, D’Amico DC, Lew W. Tetrahedron Lett. 1993;34:923–926. [Google Scholar]; c) Jung ME, Fahr BT, D’Amico DC. J Org Chem. 1998;63:2982–2987. [Google Scholar]
- 73.a) Morimoto Y, Nishikawa Y, Takaishi M. J Am Chem Soc. 2005;127:5806–5807. doi: 10.1021/ja050123p. [DOI] [PubMed] [Google Scholar]; b) Morimoto Y, Yata H, Nishikawa Y. Angew Chem Int Ed. 2007;46:6481–6484. doi: 10.1002/anie.200701737. [DOI] [PubMed] [Google Scholar]
- 74.a) Rousseau G, Homsi F. Chem Soc Rev. 1997;26:453–461. [Google Scholar]; b) Mendès C, Renard S, Rofoo M, Roux MC, Rousseau G. Eur J Org Chem. 2003:463–471. [Google Scholar]
- 75.Fukuzawa A, Aye M, Takasugi Y, Nakamura M, Tamura M, Murai A. Chem Lett. 1994;23:2307–2310. [Google Scholar]
- 76.Snyder SA, Treitler DS, Brucks AP, Sattler W. J Am Chem Soc. 2011;133:15898–15901. doi: 10.1021/ja2069449.; for a related ring-expanding iodocyclization, see: Fujioka H, Kitagawa H, Nagatomi Y, Kita Y. J Org Chem. 1996;61:7309–7315. doi: 10.1021/jo960714l.
- 77.Braddock DC. Org Lett. 2006;8:6055–6058. doi: 10.1021/ol062520q.Braddock DC, Millan DS, Pérez-Fuertes Y, Pouwer RH, Sheppard RN, Solanki S, White AJP. J Org Chem. 2009;74:1835–1841. doi: 10.1021/jo8026577.; for a similar strategy with selenenium-induced ring expansion, see: Kim B, Lee M, Kim MJ, Lee H, Kim S, Kim D, Koh M, Park SB, Shin KJ. J Am Chem Soc. 2008;130:16807–16811. doi: 10.1021/ja806304s.
- 78.a) Snyder SA, Treitler DS. Angew Chem Int Ed. 2009;48:7899–7903. doi: 10.1002/anie.200903834. [DOI] [PubMed] [Google Scholar]; b) Snyder SA, Treitler DS, Brucks AP. J Am Chem Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]; c) Snyder SA, Treitler DS. Org Synth. 2011;88:54–69. [Google Scholar]
- 79.a) Bennet AJ, Brown RS, McClung RED, Klobukowski M, Aarts GHM, Santarsiero BD, Bellucci G, Bianchini R. J Am Chem Soc. 1991;113:8532–8534. [Google Scholar]; b) Rodebaugh R, Fraser-Reid B. Tetrahedron. 1996;52:7663–7678. [Google Scholar]
- 80.Snyder SA, Brucks AP, Treitler DS, Moga I. J Am Chem Soc. 2012;134:17714–17721. doi: 10.1021/ja3076988. [DOI] [PubMed] [Google Scholar]
- 81.Baek S, Jo H, Kim H, Kim H, Kim S, Kim D. Org Lett. 2005;7:75–77. doi: 10.1021/ol047877d. [DOI] [PubMed] [Google Scholar]
- 82.Kim H, Choi WJ, Jung J, Kim S, Kim D. J Am Chem Soc. 2003;125:10238–10240. doi: 10.1021/ja035538u. [DOI] [PubMed] [Google Scholar]
- 83.Ishihara J, Shimada Y, Kanoh N, Takasugi Y, Fukuzawa A, Murai A. Tetrahedron. 1997;53:8371–8382. [Google Scholar]
- 84.Sugimoto M, Suzuki T, Hagiwara H, Hoshi T. Tetrahedron Lett. 2007;48:1109–1112. [Google Scholar]
- 85.Keshipeddy S, Martínez I, Castillo BF, II, Morton MD, Howell AR. J Org Chem. 2012;77:7883–7890. doi: 10.1021/jo301048z. [DOI] [PubMed] [Google Scholar]
- 86.Fukuzawa A, Aye M, Takaya Y, Fukui H, Masamune T, Murai A. Tetrahedron Lett. 1989;30:3665–3668. [Google Scholar]
- 87.Bravo F, McDonald FE, Neiwert WA, Hardcastle KI. Org Lett. 2004;6:4487–4489. doi: 10.1021/ol048212e. [DOI] [PubMed] [Google Scholar]
- 88.Zakarian A, Batch A, Holton RA. J Am Chem Soc. 2003;125:7822–7824. doi: 10.1021/ja029225v. [DOI] [PubMed] [Google Scholar]
- 89.Tanuwidjaja J, Ng S-S, Jamison TF. J Am Chem Soc. 2009;131:12084–12085. doi: 10.1021/ja9052366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.a) Dilley AS, Romo D. Org Lett. 2001;3:1535–1538. doi: 10.1021/ol015864j. [DOI] [PubMed] [Google Scholar]; b) Dransfield PJ, Wang S, Dilley A, Romo D. Org Lett. 2005;7:1679–1682. doi: 10.1021/ol0473602. [DOI] [PubMed] [Google Scholar]; c) Dransfield PJ, Dilley AS, Wang S, Romo D. Tetrahedron. 2006;62:5223–5247. [Google Scholar]; d) Wang S, Dilley AS, Poullennec KG, Romo D. Tetrahedron. 2006;62:7155–7161. [Google Scholar]
- 91.a) Su S, Rodriguez RA, Baran PS. J Am Chem Soc. 2011;133:13922–13925. doi: 10.1021/ja206191g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rodriguez RA, Pan CM, Yabe Y, Kawamata Y, Eastgate MD, Baran PS. J Am Chem Soc. 2014;136:6908–6911. doi: 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rodriguez RA, Steed DB, Kawamata Y, Su S, Smith PA, Steed TC, Romesberg FE, Baran PS. J Am Chem Soc. 2014;136:15403–15413. doi: 10.1021/ja508632y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Song Z-L, Fan C-A, Tu Y-Q. Chem Rev. 2011;111:7523–7556. doi: 10.1021/cr200055g. [DOI] [PubMed] [Google Scholar]
- 93.a) Reisman SE, Ready JM, Hasuoka A, Smith CJ, Wood JL. J Am Chem Soc. 2006;128:1448–1449. doi: 10.1021/ja057640s. [DOI] [PubMed] [Google Scholar]; b) Reisman SE, Ready JM, Weiss MM, Hasuoka A, Hirata M, Tamaki K, Ovaska TV, Smith CJ, Wood JL. J Am Chem Soc. 2008;130:2087–2100. doi: 10.1021/ja076663z. [DOI] [PubMed] [Google Scholar]
- 94.Braddock DC, Bhuva R, Pérez-Fuertes Y, Pouwer R, Roberts CA, Ruggiero A, Stokes ESE, White AJP. Chem Commun. 2008:1419–1421. doi: 10.1039/b800054a. [DOI] [PubMed] [Google Scholar]
- 95.Crimmins MT, Tabet EA. J Am Chem Soc. 2000;122:5473–5476. [Google Scholar]
- 96.Evans PA, Murthy VS, Roseman JD, Rheingold AL. Angew Chem Int Ed. 1999;38:3175–3177. [PubMed] [Google Scholar]
- 97.Sabot C, Bérard D, Canesi S. Org Lett. 2008;10:4629–4632. doi: 10.1021/ol801921d. [DOI] [PubMed] [Google Scholar]
- 98.Werness JB, Tang W. Org Lett. 2011;13:3664–3666. doi: 10.1021/ol201477u. [DOI] [PubMed] [Google Scholar]
- 99.a) Zhang W, Xu H, Xu H, Tang W. J Am Chem Soc. 2009;131:3832–3833. doi: 10.1021/ja8099008. [DOI] [PubMed] [Google Scholar]; b) Zhang W, Zheng S, Liu N, Werness JB, Guzei IA, Tang W. J Am Chem Soc. 2010;132:3664–3665. doi: 10.1021/ja100173w. [DOI] [PubMed] [Google Scholar]
- 100.a) Brown RS, Nagorski RW, Bennet AJ, McClung RED, Aarts GHM, Klobukowski M, McDonald R, Santarsiero BD. J Am Chem Soc. 1994;116:2448–2456. [Google Scholar]; b) Neverov AA, Brown RS. J Org Chem. 1996;61:962–968. [Google Scholar]; c) Brown RS. Acc Chem Res. 1997;30:131–137. [Google Scholar]
- 101.Denmark SE, Burk MT, Hoover AJ. J Am Chem Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]
- 102.a) Castellanos A, Fletcher SP. Chem Eur J. 2011;17:5766–5776. doi: 10.1002/chem.201100105. [DOI] [PubMed] [Google Scholar]; b) Snyder SA, Treitler DS, Brucks AP. Aldrichimica Acta. 2011;44:27–40. [Google Scholar]; c) Tan CK, Zhou L, Yeung YY. Synlett. 2011:1335–1339. [Google Scholar]; d) Hennecke U. Chem Asian J. 2012;7:456–465. doi: 10.1002/asia.201100856. [DOI] [PubMed] [Google Scholar]; e) Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chemler SR, Bovino MT. ACS Catal. 2013;3:1076–1091. doi: 10.1021/cs4001249. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Tan CK, Yeung YY. Chem Commun. 2013;49:7985–7996. doi: 10.1039/c3cc43950j. [DOI] [PubMed] [Google Scholar]; h) Tan CK, Yu WZ, Yeung YY. Chirality. 2014;26:328–343. doi: 10.1002/chir.22272. [DOI] [PubMed] [Google Scholar]; i) Zheng S, Schienebeck CM, Zhang W, Wang HY, Tang W. Asian J Org Chem. 2014;3:366–376. [Google Scholar]; j) Cheng YA, Yu WZ, Yeung YY. Org Biomol Chem. 2014;12:2333–2343. doi: 10.1039/c3ob42335b. [DOI] [PubMed] [Google Scholar]
- 103.Ke Z, Tan CK, Chen F, Yeung Y-Y. J Am Chem Soc. 2014;136:5627–5630. doi: 10.1021/ja5029155. [DOI] [PubMed] [Google Scholar]
- 104.Denmark SE, Kornfilt DJP, Vogler T. J Am Chem Soc. 2011;133:15308–15311. doi: 10.1021/ja2064395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yoder RA, Johnston JN. Chem Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Tamelen EE, Hessler EJ. Chem Commun (London) 1966:411–413. [Google Scholar]
- 107.a) Kato T, Ichinose I, Kumazawa S, Kitahara Y. Bioorg Chem. 1975;4:188–193. [Google Scholar]; b) Kitahara Y, Kato T, Ichinose I. Chem Lett. 1976;5:283–286. [Google Scholar]
- 108.a) Kato T, Ichinose I, Kamoshida A, Kitahara Y. J Chem Soc, Chem Commun. 1976:518–519. [Google Scholar]; b) Kato T, Ishii K, Ichinose I, Nakai Y, Kumagai T. J Chem Soc, Chem Commun. 1980:1106–1108. [Google Scholar]
- 109.Hoye TR, Kurth MJ. J Org Chem. 1978;43:3693–3697. [Google Scholar]
- 110.Hoye TR, Kurth MJ. J Org Chem. 1979;44:3461–3467. [Google Scholar]
- 111.Shieh H-M, Prestwich GD. Tetrahedron Lett. 1982;23:4643–4646. [Google Scholar]
- 112.a) Hoye TR, Kurth MJ. J Am Chem Soc. 1979;101:5065–5067. [Google Scholar]; b) Hoye TR, Caruso AJ, Dellaria JF, Jr, Kurth MJ. J Am Chem Soc. 1982;104:6704–6709. [Google Scholar]
- 113.White JD, Nishiguchi T, Skeean RW. J Am Chem Soc. 1982;104:3923–3928. [Google Scholar]
- 114.Tanaka A, Suzuki M, Yamashita K. Agric Biol Chem. 1986;50:1069–1071. [Google Scholar]
- 115.Nishizawa M, Takenaka H, Hirotsu K, Higuchi T, Hayashi Y. J Am Chem Soc. 1984;106:4290–4291. [Google Scholar]
- 116.Murai A, Abiko A, Masamune T. Tetrahedron Lett. 1984;25:4955–4958. [Google Scholar]
- 117.Couladouros EA, Vidali VP. Chem Eur J. 2004;10:3822–3835. doi: 10.1002/chem.200400407. [DOI] [PubMed] [Google Scholar]
- 118.Braddock DC, Marklew JS, Thomas AJF. Chem Commun. 2011;47:9051–9053. doi: 10.1039/c1cc13619d. [DOI] [PubMed] [Google Scholar]
- 119.Kato T, Ichinose I. J Chem Soc, Perkin Trans 1. 1980:1051–1056. [Google Scholar]
- 120.Shenvi RA, Corey EJ. Org Lett. 2010;12:3548–3551. doi: 10.1021/ol101410g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lin H, Pochapsky SS, Krauss IJ. Org Lett. 2011;13:1222–1225. doi: 10.1021/ol200099n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.a) Denmark SE, Burk MT. Org Lett. 2012;14:256–259. doi: 10.1021/ol203033k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Denmark SE, Burk MT. Proc Natl Acad Sci USA. 2010;107:20655–20660. doi: 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Recsei C, Chan B, McErlean CSP. J Org Chem. 2014;79:880–887. doi: 10.1021/jo402790x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- 125.Sawamura Y, Nakatsuji H, Sakakura A, Ishihara K. Chem Sci. 2013;4:4181–4186. [Google Scholar]
- 126.a) Yang D, Yan YL, Zheng BF, Gao Q, Zhu NY. Org Lett. 2006;8:5757–5760. doi: 10.1021/ol0623264. [DOI] [PubMed] [Google Scholar]; b) Helliwell M, Fengas D, Knight CK, Parker J, Quayle P, Raftery J, Richards SN. Tetrahedron Lett. 2005;46:7129–7134. [Google Scholar]
- 127.Snyder SA, Treitler DS, Schall A. Tetrahedron. 2010;66:4796–4804. [Google Scholar]
- 128.Cochrane NA, Nguyen H, Gagné MR. J Am Chem Soc. 2013;135:628–631. doi: 10.1021/ja3116795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Evans DA, Ellman JA, Dorow RL. Tetrahedron Lett. 1987;28:1123–1126. [Google Scholar]
- 130.Wack H, Taggi AE, Hafez AM, Drury WJ, III, Lectka T. J Am Chem Soc. 2001;123:1531–1532. doi: 10.1021/ja005791j. [DOI] [PubMed] [Google Scholar]
- 131.For lead references, see: Oestreich M. Angew Chem Int Ed. 2005;44:2324–2327. doi: 10.1002/anie.200500478.France S, Weatherwax A, Lectka T. Eur J Org Chem. 2005:475–479.
- 132.a) Zhang Y, Shibatomi K, Yamamoto H. J Am Chem Soc. 2004;126:15038–15039. doi: 10.1021/ja0454485. [DOI] [PubMed] [Google Scholar]; b) Hajra S, Bhowmick M, Maji B, Sinha D. J Org Chem. 2007;72:4872–4876. doi: 10.1021/jo070614n. [DOI] [PubMed] [Google Scholar]
- 133.For selected reviews, see: List B. Chem Commun. 2006:819–824. doi: 10.1039/b514296m.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016.Melchiorre P, Marigo M, Carlone A, Bartoli G. Angew Chem Int Ed. 2008;47:6138–6171. doi: 10.1002/anie.200705523.Kotsuki H, Ikishima H, Kotsuki AO. Heterocycles. 2008;75:757–797.Marigo M, Jørgensen KA. Chem Commun. 2006:2001–2011. doi: 10.1039/b517090g.Guillena G, Ramón DJ. Tetrahedron: Asymmetry. 2006;17:1465–1492.
- 134.Ueda M, Kano T, Maruoka K. Org Biomol Chem. 2009;7:2005–2012. doi: 10.1039/b901449g. [DOI] [PubMed] [Google Scholar]
- 135.De Kimpe N, Verhé R. In: The Chemistry of α-Haloketones, α-Haloaldehydes and α-Haloimines. Patai S, Rappoport Z, editors. Wiley; Chichester: 1988. [Google Scholar]
- 136.Umezawa T, Shibata M, Kaneko K, Okino T, Matsuda F. Org Lett. 2011;13:904–907. doi: 10.1021/ol102882a. [DOI] [PubMed] [Google Scholar]
- 137.For an example in which the α-chloroaldehyde is not formed stereoselectively, but is retained in the natural product, see: Shymanska NV, An IH, Pierce JG. Angew Chem Int Ed. 2014;53:5401–5404. doi: 10.1002/anie.201402310.
- 138.a) Brochu MP, Brown SP, MacMillan DWC. J Am Chem Soc. 2004;126:4108–4109. doi: 10.1021/ja049562z. [DOI] [PubMed] [Google Scholar]; b) Halland N, Braunton A, Bachmann S, Marigo M, Jørgensen KA. J Am Chem Soc. 2004;126:4790–4791. doi: 10.1021/ja049231m. [DOI] [PubMed] [Google Scholar]
- 139.For select references, see: Kang B, Britton R. Org Lett. 2007;9:5083–5086. doi: 10.1021/ol702273n.Draper J, Britton R. Org Lett. 2010;12:4034–4037. doi: 10.1021/ol101631e.Halperin S, Kang B, Britton R. Synthesis. 2011;12:1946–1953.Britton R, Kang B. Nat Prod Rep. 2013;30:227–236. doi: 10.1039/c2np20108a.Shand V, Draper JA, Moore J, Britton R. Org Lett. 2013;15:1914–1917. doi: 10.1021/ol400566j.Holmes MT, Britton R. Chem Eur J. 2013;19:12649–12652. doi: 10.1002/chem.201302352.Bergeron-Brlek M, Meanwell M, Britton R. Nat Commun. 2015;6:6903. doi: 10.1038/ncomms7903.
- 140.White DE, Stewart IC, Grubbs RH, Stoltz BM. J Am Chem Soc. 2008;130:810–811. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kim H, Lee H, Lee D, Kim S, Kim D. J Am Chem Soc. 2007;129:2269–2274. doi: 10.1021/ja068346i. [DOI] [PubMed] [Google Scholar]
- 142.a) King SM, Calandra NA, Herzon SB. Angew Chem Int Ed. 2013;52:3642–3645. doi: 10.1002/anie.201210076. [DOI] [PubMed] [Google Scholar]; b) Calandra NA, King SM, Herzon SB. J Org Chem. 2013;78:10031–10057. doi: 10.1021/jo401889b. [DOI] [PubMed] [Google Scholar]
- 143.King SM, Ma X, Herzon SB. J Am Chem Soc. 2014;136:6884–6887. doi: 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]
- 144.Corey EJ, Danheiser RL, Chandrasekaran S, Siret P, Keck GE, Gras JL. J Am Chem Soc. 1978;100:8031–8034. [Google Scholar]
- 145.Steinhagen H, Corey EJ. Org Lett. 1999;1:823–824. doi: 10.1021/ol9908575. [DOI] [PubMed] [Google Scholar]
- 146.Steinhagen H, Corey EJ. Angew Chem Int Ed. 1999;38:1928–1931. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1928::AID-ANIE1928>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 147.Chandra A, Johnston JN. Angew Chem Int Ed. 2011;50:7641–7644. doi: 10.1002/anie.201100957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huwyler N, Carreira EM. Angew Chem Int Ed. 2012;51:13066–13069. doi: 10.1002/anie.201207203. [DOI] [PubMed] [Google Scholar]
- 149.a) Barabé F, Bétournay G, Bellavance G, Barriault L. Org Lett. 2009;11:4236–4238. doi: 10.1021/ol901722q. [DOI] [PubMed] [Google Scholar]; b) Nieto-Oberhuber C, López S, Echavarren AM. J Am Chem Soc. 2005;127:6178–6179. doi: 10.1021/ja042257t. [DOI] [PubMed] [Google Scholar]
- 150.Yaday VK, Babu KG. Eur J Org Chem. 2005:452–456.Larock RC, Leong WW. In: Addition of H-X Reagents to Alkenes and Alkynes—Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 4. Pergamon Press; Oxford: 1991. pp. 269–327., and references therein.
- 151.a) Evans DA, Burch JD. Org Lett. 2001;3:503–505. doi: 10.1021/ol0155182. [DOI] [PubMed] [Google Scholar]; b) Evans DA, Burch JD, Hu E, Jaeschke G. Tetrahedron. 2008;64:4671–4699. doi: 10.1016/j.tet.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang Z, Lorenz JC, Shi Y. Tetrahedron Lett. 1998;39:8621–8624. [Google Scholar]
- 153.a) Paterson I, Davies RDM, Marquez R. Angew Chem Int Ed. 2001;40:603–607. doi: 10.1002/1521-3773(20010202)40:3<603::AID-ANIE603>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]; b) Paterson I, Davies RDM, Heimann AC, Marquez R, Meyer A. Org Lett. 2003;5:4477–4480. doi: 10.1021/ol0357853. [DOI] [PubMed] [Google Scholar]
- 154.Huang H, Panek JS. Org Lett. 2004;6:4383–4385. doi: 10.1021/ol0480325. [DOI] [PubMed] [Google Scholar]
- 155.a) Charette AB, Juteau H. J Am Chem Soc. 1994;116:2651–2652. [Google Scholar]; b) Charette AB, Prescott S, Brochu C. J Org Chem. 1995;60:1081–1083. [Google Scholar]
- 156.(a) Paterson I, Maltas P, Anderson EA. Pure Appl Chem. 2013;85:1133–1147. [Google Scholar]; (b) Paterson I, Anderson EA, Dalby SM, Lim JH, Maltas P, Loiseleur O, Genovino J, Moessner C. Org Biomol Chem. 2012;10:5861–5872. doi: 10.1039/c2ob25100k. [DOI] [PubMed] [Google Scholar]
- 157.Arlt A, Benson S, Schulthoff S, Gabor B, Fürstner A. Chem Eur J. 2013;19:3596–3608. doi: 10.1002/chem.201203965. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A, Fenster MDB, Fasching B, Godbout C, Radkowski K. Angew Chem Int Ed. 2006;45:5510–5515. doi: 10.1002/anie.200601655. [DOI] [PubMed] [Google Scholar]
- 158.Hu S, Jayaraman S, Oehlschlager AC. J Org Chem. 1998;63:8843–8849. [Google Scholar]
- 159.a) Barton DHR, Crich D, Motherwell WB. Tetrahedron Lett. 1983;24:4979–4982. [Google Scholar]; b) Barton DHR, Crich D, Motherwell WB. Tetrahedron. 1985;41:3901–3924. [Google Scholar]
- 160.a) Trost BM, Dirat O, Gunzner JL. Angew Chem Int Ed. 2002;41:841–843. doi: 10.1002/1521-3773(20020301)41:5<841::aid-anie841>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]; b) Trost BM, Gunzner JL, Dirat O, Rhee YH. J Am Chem Soc. 2002;124:10396–10415. doi: 10.1021/ja0205232. [DOI] [PubMed] [Google Scholar]
- 161.Starr JT, Koch G, Carreira EM. J Am Chem Soc. 2000;122:8793–8794. [Google Scholar]
- 162.Lee E, Park CM, Yun JS. J Am Chem Soc. 1995;117:8017–8018. [Google Scholar]
- 163.Giese B, Kopping B, Chatgilialoglu C. Tetrahedron Lett. 1989;30:681–684. [Google Scholar]