

Abstract
Helicobacter pylori infects half of the world's population, and strains that encode the cag type IV secretion system for injection of the oncoprotein CagA into host gastric epithelial cells are associated with elevated levels of cancer. CagA translocation into host cells is dependent on interactions between the H. pylori adhesin protein HopQ and human CEACAMs. Here, we present high‐resolution structures of several HopQ‐CEACAM complexes and CEACAMs in their monomeric and dimeric forms establishing that HopQ uses a coupled folding and binding mechanism to engage the canonical CEACAM dimerization interface for CEACAM recognition. By combining mutagenesis with biophysical and functional analyses, we show that the modes of CEACAM recognition by HopQ and CEACAMs themselves are starkly different. Our data describe precise molecular mechanisms by which microbes exploit host CEACAMs for infection and enable future development of novel oncoprotein translocation inhibitors and H. pylori‐specific antimicrobial agents.
Keywords: CagA, CEACAM, Helicobacter pylori, HopQ, X‐ray crystallography
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Introduction
Helicobacter pylori is a gastric pathogen that infects more than half of the human population worldwide and is a major cause of gastroduodenal diseases, such as chronic active gastritis and peptic ulcers, as well as stomach cancers including mucosa‐associated lymphoid tissue (MALT) lymphoma and gastric adenocarcinoma (Peek & Blaser, 2002; Suerbaum & Michetti, 2002). Certain H. pylori strains harbor the 37‐kilobase cytotoxin‐associated gene‐pathogenicity island (cag‐PAI) that encodes numerous Cag proteins that collectively form a membrane‐spanning type IV secretion system (T4SS), as well as the effector protein CagA. Upon contact with gastric epithelial cells, CagA is translocated through the T4SS into host cells (Odenbreit et al, 2000) prompting a pro‐inflammatory response (Fischer et al, 2001; Gorrell et al, 2013). Once inside the host cell, CagA is phosphorylated and subsequently interacts with numerous host cell proteins (Higashi et al, 2002; Tsutsumi et al, 2003; Selbach et al, 2009) to impair tight junctions and cause loss of epithelial cell polarity (Amieva et al, 2003; Saadat et al, 2007), disrupt kinase signaling, and modulate the apoptotic program (Buti et al, 2011). CagA is considered a bacterial oncoprotein (Hatakeyama, 2004) as its presence is correlated with the risk of developing cancer in humans, and it induces gastrointestinal and hematopoietic neoplasms in mice (Ohnishi et al, 2008).
Several host–pathogen interactions are functional contributors to H. pylori adherence to human gastric epithelial cells and CagA translocation. The α5β1‐integrin heterodimer is a target for proteins encoded by the cag‐PAI, including CagA (Kwok et al, 2007; Jimenez‐Soto et al, 2009) and CagL (Bonsor et al, 2015b). A family of H. pylori adhesin proteins, the Helicobacter outer proteins (Hops), bind to both glycans and proteins on the host cell surface. Of these, BabA and SabA bind to the human blood group antigens, Lewis b (Leb) and sialylated Lewis X (sLeX), respectively, to facilitate the bacterium's persistence at the gastric mucosa (Ilver et al, 1998; Mahdavi et al, 2002; Linden et al, 2004). Numerous X‐ray crystal structures of BabA alone and in complex with Leb carbohydrates (Hage et al, 2015; Moonens et al, 2016) provide a comprehensive structural basis for glycan recognition by H. pylori. Another Hop, HopQ, has been shown to exploit human carcinoembryonic antigen‐related cell adhesion molecule (CEACAM) proteins for both host cell adherence and CagA translocation (Javaheri et al, 2016; Koniger et al, 2016). Although a high‐resolution structure of HopQ has been reported (Javaheri et al, 2016), it remains unclear how HopQ binds to CEACAMs.
The CEACAM family forms a group of extracellular receptors involved in cell adhesion, differentiation, proliferation, signaling, tumor suppression, and survival (Benchimol et al, 1989; Singer et al, 2005, 2010; Gray‐Owen & Blumberg, 2006; Tchoupa et al, 2014). The 12 human CEACAMs are each comprised of: (i) a single N‐terminal variable immunoglobulin (IgV) domain that can homo‐ and/or heterodimerize with themselves or other CEACAMs (Bonsor et al, 2015a); (ii) a variable number of constant subtype 2 (IgC2) domains, ranging from zero to six, separating the IgV domain and the plasma membrane anchor, which is either a transmembrane helix or a glycophosphatidylinositol linker; and (iii) for some CEACAMs, an intracellular signaling motif. Beyond their role as cell adhesion and signaling molecules, CEACAMs, in particular CEACAM1, CEACAM5, and CEACAM6, are also targets of diverse pathogenic bacteria, including Neisseria gonorrhoeae and Escherichia coli (Tchoupa et al, 2014). Each of these bacteria has convergently evolved distinct cell surface adhesins to bind CEACAMs and exploit the human mucosa, on which the above‐mentioned CEACAMs are expressed on the apical side, as an attachment site for colonization, multiplication, and spread (Virji, 2009); or for endocytosis into epithelial cells and transcytosis through intact epithelial layers (Virji et al, 1996; Gray‐Owen et al, 1997a,b). More recently, CEACAMs have been shown to be exploited by the H. pylori adhesin protein HopQ for translocation of the oncoprotein CagA (Javaheri et al, 2016; Koniger et al, 2016).
Certain bacterial adhesins also bind to CEACAM3, which does not interact with itself or other CEACAM family members. CEACAM3 is expressed on granulocytes and contains a cytoplasmic ITAM motif. Host–pathogen interactions involving CEACAM3 promote phagocytosis and clearance of bacteria such as N. gonorrhoeae (McCaw et al, 2003; Schmitter et al, 2007), a process distinct from the transcytosis across the epithelia supported by other CEACAMs. Through this mechanism, CEACAM3 has been proposed to function as an innate immune receptor for human‐restricted pathogens that arose by an evolutionary response in the host to recognize and eliminate CEACAM‐coopting bacteria (Pils et al, 2008).
Here, we describe the molecular basis by which H. pylori HopQ engages human CEACAMs, thereby allowing CagA translocation. We determined X‐ray crystal structures of HopQ in complex with both CEACAM1 and CEACAM3 N‐terminal dimerization domains, the first high‐resolution structures of host–pathogen interactions involving CEACAMs. We find that HopQ recognizes CEACAMs through a coupled folding and binding mechanism and binds at the same site that CEACAMs use to homo‐ and heterodimerize. Despite this common interface, the molecular mechanisms of HopQ‐CEACAM interaction and CEACAM dimerization are entirely distinct.
Results
HopQ exploits the CEACAM dimer interface for binding
We crystallized and determined the 2.7 Å resolution structure of the complex formed by the recombinant extracellular domain of the H. pylori P12 strain HopQ and the N‐terminal IgV domain of human CEACAM1 (Fig 1A and Table EV1). In the crystal, the complex is comprised of one HopQ molecule bound to one CEACAM1 molecule. CEACAMs, including CEACAM1, form homo‐ and heterodimers with themselves and other CEACAMs via interactions of their N‐terminal IgV domains (Bonsor et al, 2015a). From the HopQ‐CEACAM1 crystal structure, it is evident that HopQ binds CEACAM1 by exploiting the dimer surface of the latter, thereby prohibiting CEACAM1 dimerization and consistent with a 1:1 binding stoichiometry. The overlap of the CEACAM1 interfaces used for HopQ binding and for dimerization is extensive (Fig 1A) and includes 26 common residues, including Phe29, Gln44, Ile91, and Val96. In order to confirm that HopQ‐CEACAM1 forms 1:1 complexes in solution and not just in a crystalline environment, we performed native mass spectrometry on the complex and observed a dominant species at 58.8 kDa, equivalent to one HopQ extracellular domain (46.8 kDa) and one CEACAM IgV domain (12.0 kDa; Fig 1B).
Figure 1. Crystal structure of the HopQ‐CEACAM1 complex.
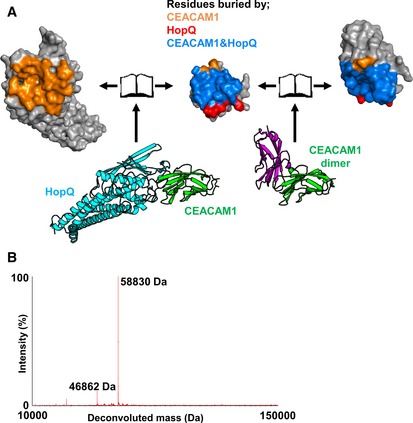
- Bottom left—Crystal structure of HopQ‐CEAMCAM1 (cyan and green, respectively). Bottom right—The CEACAM1 homodimer (pdb code 2gk2 is orientated to overlay with CEACAM1 (green) in the HopQ‐CEACAM1 structure. The other half of CEACAM1 dimer is shown as magenta. Both complexes are rendered as a molecular surface representation in an open‐book conformation with HopQ (top left), CEACAM1 of both complexes (center), and CEACAM1 of the dimer (top right). Surfaces are colored according to residues buried by just CEACAM1 (orange), just HopQ (red), and by both HopQ and CEACAM1 (blue).
- Deconvolution of raw native mass spectrometry data through maximum entropy displays a predominant population of 58,830 Da (mass of the HopQ‐CEACAM1 complex) and a minor population of 46,862 Da (uncomplexed HopQ).
Upon HopQ‐CEACAM1 complex formation, a total of 2,020 Å2 of surface area is buried, which is 400 Å2 more than in the CEACAM1 homodimer (Fedarovich et al, 2006). Four loops in HopQ make intermolecular contacts with CEACAM1: the α3‐β1 loop (residues 102–111); the β2‐α4 loop (residues 134–161); the α5‐α6 loop (residues 240–244); and the α7‐α8 loop (residues 363–385; Fig 2A). Of these, the β2‐α4 loop contributes approximately 60 percent of the HopQ buried surface area in the complex. The β‐hairpin motif of the β2‐α4 loop (residues 135–138) extends the CEACAM1 GFCC′C″ β‐sheet (i.e., the β sheet composed of the G, F, C, C′, and C″ β strands of the CEACAM1 Ig‐fold domain) by main‐chain hydrogen bonding to residues 95–97 of CEACAM1 (Fig 2B), similar to β‐zipper motifs. This places the small α‐helix and random coil of the β2‐α4 loop that follow the β‐hairpin across the face of GFCC′C″ sheet, forming extensive van der Waals and hydrogen bonding interactions across all of the CEACAM1 β‐strands (Fig 2C). These interactions include several residues that are known to be important for CEACAM1 dimerization including Phe29CEACAM1, Val39CEACAM1, Gln44CEACAM1, Gln89CEACAM1, and Leu95CEACAM1 (Fedarovich et al, 2006). The majority of hydrogen bonds formed between CEACAM1 and the HopQ β2‐α4 loop are from main‐chain atoms with only six of the 14 hydrogen bonds from side‐chain atoms. Only the most apical of the eleven residues in the HopQ α7‐α8 loop contact CEACAM1. Asn371HopQ forms van der Waals contacts with Leu95CEACAM1, and Thr370HopQ forms a hydrogen bond with the main‐chain carbonyl of Asp94CEACAM1 (Fig 2D). The remaining two HopQ loops, α3‐β1 and α5‐α6, form minimal interactions with CEACAM1, including van der Waals contacts between Ile102HopQ‐Phe29CEACAM1, Tyr106HopQ‐Thr56CEACAM1, Pro110HopQ‐Pro59CEACAM1/Gly41CEACAM1, and Gly111HopQ‐Gly41CEACAM1/Asn42CEACAM1 and a single hydrogen bond between the main‐chain amine of Ala243HopQ and the main‐chain carbonyl of Ser93CEACAM1 (Fig 2D).
Figure 2. Four loops of HopQ make interactions with CEACAM1.
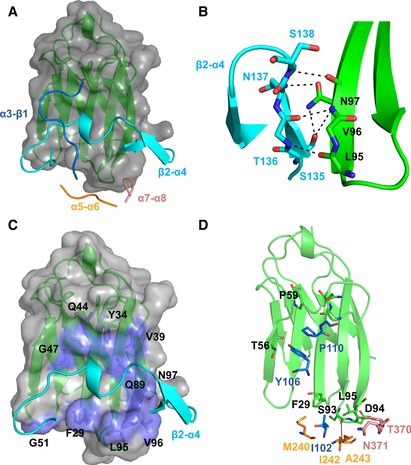
- Cartoon representation of CEACAM1 (green) with a molecular surface (gray) bound by the four loops of HopQ; α3‐β1 (blue); β2‐α4 (cyan); α5‐α6 (yellow); and α7‐α8 (pink).
- Residues 135–138 of the β2‐α4 loop (cyan) form a hydrogen bond network (black lines) to residues 95–97 of CEACAM1 (green).
- Residues 145–156 of the β2‐α4 loop (cyan) make extensive van der Waals contacts (lilac) across all five β‐strands of CEACAM1 (green).
- The α3‐β1, α5‐α6, and α7‐α8 loops (blue, yellow, pink, respectively) form several van der Waal contacts and hydrogen bonds (black lines) above and below the β2‐α4 loop to CEACAM1 (green).
Glycosylation contributes minimally to HopQ‐CEACAM interactions
Helicobacter pylori utilizes several adhesin proteins to adhere to host cell surfaces. These adhesins are members of the Helicobacter outer membrane protein (Hop) and Hop‐related (Hor) gene families, from which HopQ, BabA, and SabA cluster phylogenetically. BabA and SabA are both glycan recognition bacterial adhesins—BabA binds Leb antigens, while SabA binds sLeX antigens. HopQ shares sequence identities of 35 and 23% with BabA and SabA, respectively, and the extracellular domains of all three proteins are structurally related, comprising 4 + 3 α helical bundles sprouting from coiled‐coil stems formed by their N‐ and C‐termini that link them to their transmembrane domains. An insertion domain that varies in size and sequence for HopQ, BabA, and SabA is found between α helices 4 and 5. While the insertion domain comprises the glycan‐binding site for both BabA and SabA (Pang et al, 2014; Hage et al, 2015; Moonens et al, 2016), it is quite distant from the CEACAM‐binding site for HopQ (Fig 3A and B).
Figure 3. Characterization of glycans, reduction, and pH on the HopQ‐CEACAM1 interaction.
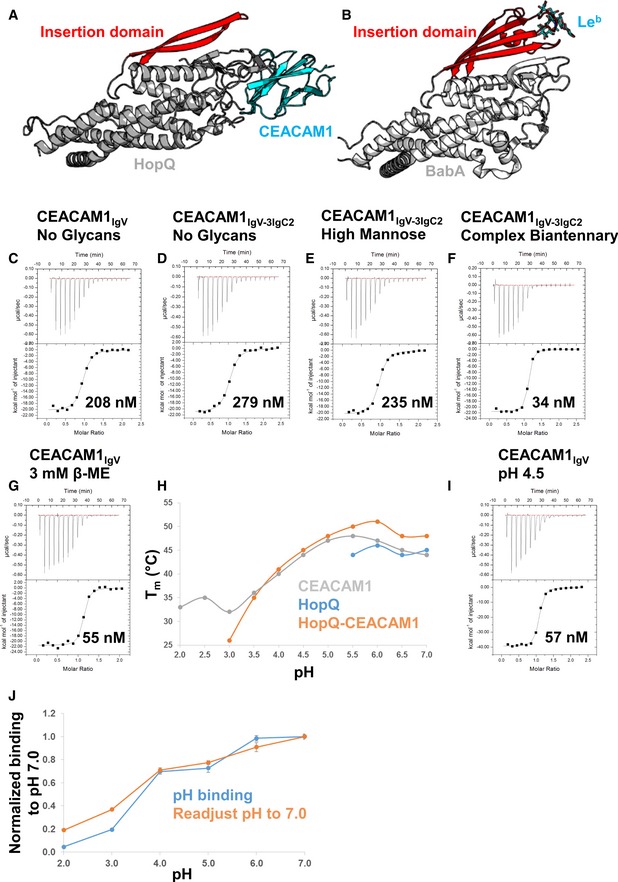
-
ACartoon representation of the HopQ‐CEACAM1 complex (gray and cyan, respectively) with the insertion domain of HopQ highlighted in red showing no contacts are made to CEACAM1.
-
BCartoon representation of BabA (pdb code 5f9d, gray). The insertion domain of BabA (red) is in complex with the Leb heptasaccharide antigen (cyan sticks).
-
C–GIsothermal titration calorimetry profiles of HopQ binding to (C) CEACAM1IgV no glycans, (D) CEACAM1IgV‐3IgC2 no glycans, (E) CEACAM1IgV‐3IgC2 high‐mannose glycans, (F) CEACAM1IgV‐3IgC2 complex biantennary glycans, and (G) CEACAM1IgV with 3 mM β‐mercaptoethanol.
-
HMelting temperatures (T m) of CEACAM1 (gray), HopQ (blue), and the HopQ‐CEACAM1 complex (orange) were measured by thermal shift assay at various pHs between pH 2.0 and 7.0.
-
IITC profile of HopQ‐CEACAM1IgV at pH 4.5.
-
JAcid sensitivity of HopQ binding to CEACAM1 as measured by ELISA at several pHs between 2.0 and 7.0 for 90 min is plotted as the blue line. The orange line shows the recovery of HopQ after 60 min of exposure to the various pHs by readjustment of the pH to 7.0. Two independent experiments were performed on four replicates (n = 8). Data were plotted as an average with error bars as standard errors of the mean.
Despite the abbreviated nature of the HopQ insertion domain, HopQ could potentially bind glycans and the lack of contacts between the insertion domain and CEACAM1 in our crystal structure could have resulted from our use of an aglycosylated CEACAM1 IgV domain expressed in E. coli. The N‐terminal IgV domain of CEACAM1 is predicted to contain three N‐linked glycosylation sites, Asn70CEACAM1, Asn77CEACAM1, and Asn81CEACAM1, of which Asn70CEACAM1 and Asn77CEACAM1 are found in the DE and EF loops on the back face of CEACAM1 pointing away from the HopQ insertion domain. Asn81CEACAM1 is also found in the EF loop but is facing toward the insertion domain; however, it is positioned more than 30 Å from the insertion domain. While previous studies identified that glycans were not necessary for HopQ binding (Javaheri et al, 2016; Koniger et al, 2016), only the N‐terminal IgV domain was tested. CEACAM1, though, also contains three IgC2 domains that can be glycosylated and could potentially contact the insertion domain. To assess any potential role for glycans in CEACAM1 recognition by HopQ, we expressed the entire ectodomain of CEACAM1 (CEACAM1IgV‐3IgC2; inclusive of one IgV and three IgC2 domains) in HEK293 cells in the presence of kifunensine, a mannosidase I inhibitor, to produce CEACAM1 decorated with high‐mannose glycans, half of which we treated with EndoA, an endoglycosidase that specifically hydrolyzes high‐mannose glycans, leaving only a single N‐linked N‐acetylglucosamine. We also expressed CEACAM1IgV‐3IgC2 in HEK293 cells in the absence of glycosidase inhibitors in order to produce the full‐length CEACAM1 ectodomain decorated predominately with complex biantennary‐type glycans. Using isothermal titration calorimetry (ITC), we measured no differences in binding affinity or thermodynamic parameters for high‐mannose and deglycosylated CEACAM1IgV‐3IgC2 binding to HopQ relative to CEACAM1IgV (Fig 3C–E), indicating that no significant roles in molecular recognition can be ascribed to either CEACAM high‐mannose‐type glycans or the HopQ insertion domain. However, we observed a 10‐fold increase in affinity between HopQ and CEACAM1IgV‐3IgC2 displaying complex‐type glycans caused by both a favorable increase in binding enthalpy and a smaller entropic penalty relative to deglycosylated CEACAM1IgV‐3IgC2 and its high‐mannose‐type glycoform (Fig 3F). If complex glycans were binding HopQ, we would expect a larger entropic penalty due to restriction of the CEACAM, suggesting that these glycans may help pre‐order CEACAM1 into a HopQ‐receptive conformation.
HopQ‐CEACAM and BabA‐Leb interactions are affected in opposite ways by pH and redox state
HopQ, BabA, and SabA contain 3, 4, and 2 disulfides, respectively. All three disulfides in HopQ are conserved in BabA, while they do not align with those in SabA. In all three proteins however, the disulfides are observed in loops, constricting conformational sampling of these loops. BabA has been shown to be redox sensitive—the presence of reducing agent substantially weakens its affinity for Leb and the redox‐active compound N‐acetylcysteine inhibits Leb binding, as well as BabA‐mediated adherence and inflammation by H. pylori (Moonens et al, 2016). By conducting ITC binding experiments in the presence of a large molar excess of the reducing agent β‐mercaptoethanol, we observed that the reduction in all disulfides in HopQ corresponded to a modest increase in affinity for CEACAM1IgV (Fig 3G). This is in contrast to BabA‐Leb interactions, for which reduction in BabA disulfide bonds prevents Leb ligand binding (Moonens et al, 2016).
Helicobacter pylori has evolved the ability to survive the pH gradient that exists in the stomach from pH 2.0 in the lumen to pH 7.0 found deep in the gastric mucosal layer close to the epithelial surface. The role of pH in BabA recognition of Leb antigens has been extensively studied (Bugaytsova et al, 2017)—BabA binds Leb antigens maximally at pH 5.0–6.0 and is observed to have a pH50 of ~ 2.4–3.8 (depending on the H. pylori strain); furthermore, when reconditioned to a higher pH after exposure to low pH, BabA recovers 75–100% of its binding capacity to Leb antigens. Accordingly, we sought to determine the pH responsiveness of HopQ to CEACAM1 and whether HopQ exhibits pH reconditioning. First, we determined the melting temperature (T m) of CEACAM1IgV and HopQ individually by differential scanning fluorimetry (DSF) at various pH values between 2.0 and 7.0. At low pH, CEACAM1IgV has an estimated T m of 31°C which increases with pH to a maximum of 47°C between pH 5.0 and 6.0 before decreasing to 43°C at pH 7.0 (Fig 3H). We found that HopQ was less stable than CEACAM1IgV over the same pH range. We observed fluorescence changes occurring only above pH 5.5, with a T m of approximately 45°C (Fig 3H). Formation of the HopQ‐CEACAM1IgV complex led to an increase in T m of 3–5°C at high pH, but also produced fluorescence changes down to pH 3.0, demonstrating that the HopQ‐CEACAM1IgV complex is substantially more stable over a broader pH range than is HopQ alone (Fig 3H). We also performed an ITC binding analysis at pH 4.5 for the CEACAM1IgV‐HopQ interaction and observed a threefold increase in affinity (K D ~ 60 nM; Fig 3I). Because ITC experiments at lower pH are not feasible due to the acid‐sensitive cell of this instrument, we measured binding of HopQ to a CEACAM1IgV‐coated plate at pHs between 2.0 and 7.0 in an ELISA. We observed that HopQ binds CEACAM1 above pH 3.0, with a pH50 of 3.5, with minimal binding at pH 2.0 (Fig 3J). Using the same ELISA setup, after incubation of the plate with HopQ at the various pHs, we readjusted the pH to pH 7.0 in order to determine whether CEACAM1 binding by HopQ was recoverable. We found that binding in the reconditioned medium nearly identically mirrors the binding curve of the pH‐dependent interaction indicating that, unlike the BabA‐Leb interaction, HopQ binding to CEACAM is not reversible when acidity is decreased (Fig 3J).
HopQ recognizes CEACAMs through a coupled folding and binding mechanism
In the structure of H. pylori strain G27 HopQ in the absence of CEACAM reported previously (Javaheri et al, 2016), several loops lack sufficient electron density required to model their conformations. Notably, these include the β2‐α4 and α7‐α8 loops, both of which are ordered and form intermolecular contacts in our HopQ‐CEACAM1 complex (Fig 4A and B). The amino acid sequences between the HopQ proteins from the H. pylori G27 and P12 strains differ by only a single residue in the β2‐α4 and three residues in the α7‐α8 loops, with Tyr144HopQ‐G27 to Ser144HopQ‐P12, and Ala364HopQ‐G27 to Met364HopQ‐P12, Ala370HopQ‐G27 to Thr370HopQ‐P12, and Asn376HopQ‐G27 to Ser376HopQ‐P12 polymorphisms, respectively (Appendix Fig S1). Only the HopQ residue at position 370 contacts CEACAM1 (see above), forming a hydrogen bond to Asp94CEACAM1.
Figure 4. Structural changes of HopQ and CEACAM1 upon complexation.
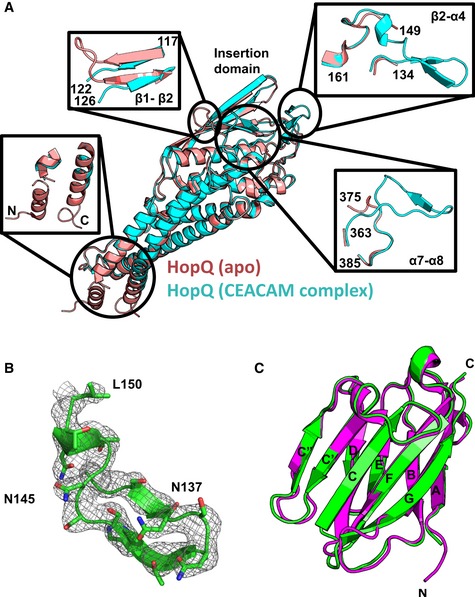
- Apo‐HopQ (pdb code 5lp2, pink) superimposes with HopQ of the HopQ‐CEACAM1 complex (cyan) with a RMSD of 0.60 Å. Order‐disordering events occur upon complexation including, disordering of the coiled‐coils (bottom left) and the β1‐β2 loop (top left), and ordering of the β2‐α4 (top right) and the α7‐α8 (bottom right) loops.
- Omit map contoured at 1.2 σ around residues 133–150 of the β2‐α4 loop.
- CEACAM1 (pdb code 2gk2, magenta) superimposes with CEACAM1 of the HopQ‐CEACAM1 complex (green) with a RMSD of 0.66 Å.
Conversely, several other structural features that are ordered in the unbound HopQ structure appear disordered in our CEACAM1‐bound structure of HopQ, including (i) the β1‐β2 loop in the middle of the two‐stranded β‐hairpin structure of the insertion domain and (ii) the coiled‐coil domain formed by the N‐ and C‐terminal α‐helices, α1 and α8, the latter of which links the HopQ extracellular domain to its β‐barrel transmembrane domain (Fig 4A). Neither the β1‐β2 loop nor the α1 and α8 coiled coil contacts CEACAM1. Beyond these above‐mentioned structural differences in the CEACAM‐bound and unbound HopQ structures, there are no gross structural changes in HopQ, which is reflected by a low root mean square deviation (RMSD) for Cα atoms of 0.60 Å.
The IgV domain of CEACAM1 in the HopQ‐CEACAM1 structure exhibits the classic IgV fold, consisting of two β‐sheets, ABED and GFCC′C″, the latter of which forms the dimerization interface observed in all CEACAMs. All residues of the CEACAM1 molecule, except for the N‐terminal residue, are resolved in the HopQ‐CEACAM1 structure. The RMSD of CEACAM1 Cα atoms in the complex relative to that of CEACAM1 (Fedarovich et al, 2006) is low (0.66 Å) with only the BC, C′C″, and GF loops exhibiting observable deviations in the HopQ‐bound structure (Fig 4C). Because HopQ loops that form a substantial part of the HopQ‐CEACAM interface adopt a single conformation in the complex, HopQ recognizes CEACAMs through a coupled folding and binding mechanism. At the same time, several structural elements outside of the HopQ‐CEACAM interface become disordered upon binding, with unknown, if any, functional consequences.
CEACAM1 dimerization and HopQ‐CEACAM1 interaction exhibit distinct energetic landscapes
To gain a more detailed understanding of the distribution of binding energy within the interfaces of the CEACAM dimer and HopQ‐CEACAM complexes, we undertook a comprehensive alanine scanning mutagenesis analysis in which we generated a total of 13 mutants in the IgV domain of CEACAM1 and the extracellular domain of HopQ and measured their effects on CEACAM1 dimerization and on HopQ‐CEACAM1 binding, by analytical ultracentrifugation (AUC) and ITC, respectively. This analysis allowed us to quantitatively map and compare the energetic landscapes of both complexes.
Protein–protein interfaces commonly exhibit a distribution of hot spot residues, in which the binding energy of the interaction is highly concentrated, and energetically silent residues. The CEACAM1 homodimer is no exception in this regard, as shown in Figs 5A and EV1, and Table EV2. We observed changes in binding free energy (ΔΔG) of up to 3 kcal mol−1 for several alanine mutations, including CEACAM1F29A, CEACAM1Q44A, and CEACAM1V96A, and as large as 4 kcal mol−1 for the CEACAM1N97A mutation, which effectively converts CEACAM1 into a monomer. Conversely, the CEACAM1Q89A and CEACAM1L95A mutations had little to no effect on CEACAM1 dimerization.
Figure 5. Binding analysis of CEACAM1 homodimerization and complexation with HopQ reveal different binding energies.
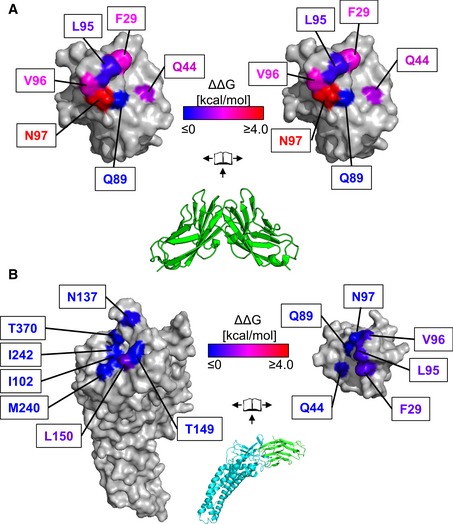
- Center—Crystal structure of CEACAM1 homodimer (pdb code 2gk2, green). The dimer is rendered as a molecular surface representation in an open‐book conformation (left and right) with the surfaces colored according to changes in binding energies as measured by AUC.
- Center—Crystal structure of HopQ (cyan) bound to CEACAM1 (green). The complex is rendered as a molecular surface representation in an open‐book conformation with the surfaces of HopQ (left) and CEACAM1 (right) colored according to changes in binding energies as determined by ITC. Whereas mutations in CEACAM1 cause changes in affinity which span at least a 1,000‐fold range in homodimerization, only small differences are observed in complexation with HopQ.
Figure EV1. Alanine scan of the CEACAM1 dimerization interface.
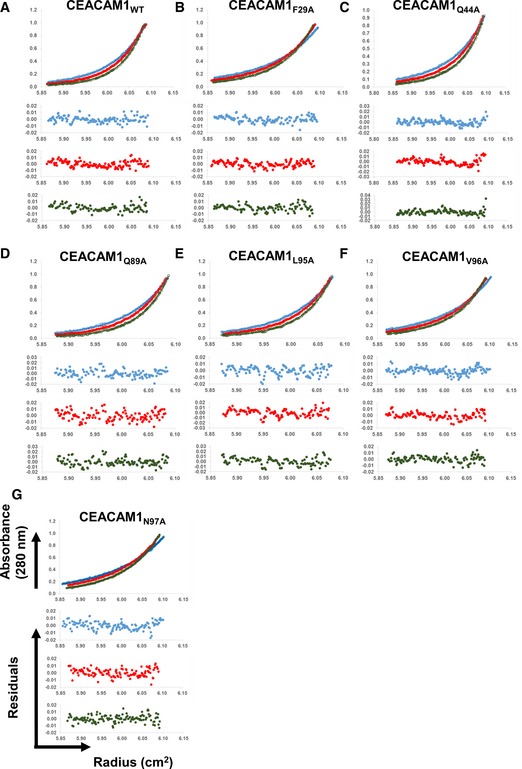
-
A–GSedimentation equilibrium analyses (Upper) and residuals of the fits for each curve (Lower) for (A) CEACAM1WT, (B) CEACAM1F29A, (C) CEACAM1Q44A, (D) CEACAM1Q89A, (E) CEACAM1L95AA, (F) CEACAM1V96A, and (G) CEACAM1N97A.
We next tested all of these single‐site alanine mutants in CEACAM1 for binding to HopQ. Contrary to CEACAM1 dimerization, we found that none of them had any significant impact on binding affinity, although the thermodynamics of binding did vary with the CEACAM1Q89A, CEACAM1L95A, and CEACAM1N97A exhibiting approximately 8 kcal mol−1 less favorable binding enthalpies, which were offset by favorable gains in entropy (Figs 5B—right and EV2, and Table EV2). The variant with the largest effect on the interaction, CEACAM1F29A, bound HopQ only threefold more weakly than did wild‐type CEACAM1.
Figure EV2. Mutational analysis of the HopQ‐CEACAM1 binding interface.
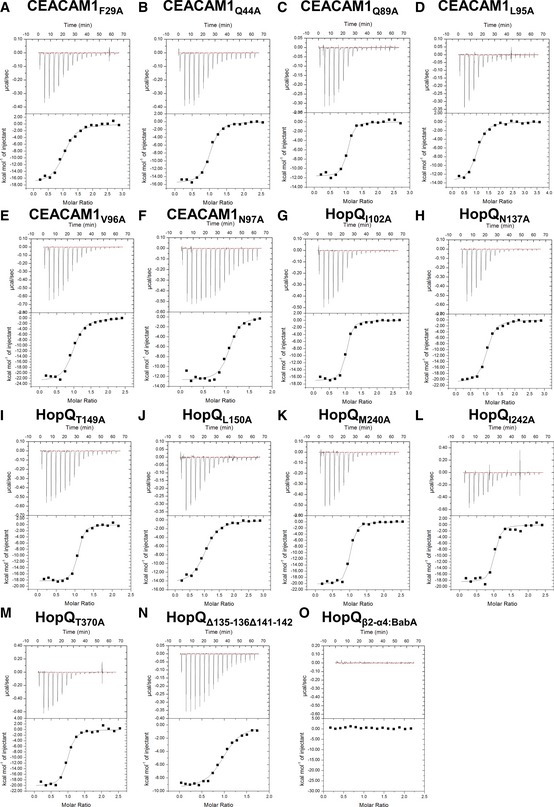
-
A–OITC binding curves of (A) CEACAM1F29A, (B) CEACAM1Q44A, (C) CEACAM1Q89A, (D) CEACAM1L95AA, (E) CEACAM1V96A, and (F) CEACAM1N97A titrated into HopQWT. ITC binding curves of (G) HopQI102A. (H) HopQN137A, (I) HopQT149A, (J) HopQL150A, (K) HopQM240A, (L) HopQI242A, (M) HopQT370A, (N) HopQΔ135–136Δ141–142, and (O) HopQβ2‐α4:BabA being titrated with CEACAM1WT.
In order to interrogate the HopQ side of the HopQ‐CEACAM1 complex interface, we made single‐site alanine mutations in HopQ, none of which had a significant effect on HopQ‐CEACAM‐binding affinities, mirroring the properties of the CEACAM1 alanine mutants (Figs 5B—left and EV2, and Table EV2). Of the seven variants that we were able to express and purify, most exhibited similar affinities to the WT interaction, with only changes in the thermodynamics observed, while a few resulted in only modest resulted affinity changes that, although quantifiable, are unlikely to be physiologically meaningful. One such variant, HopQL150A, resulted in a modest (~ 4.5‐fold) decrease in affinity (920 nM, Fig EV2J). Seeking a modification of the HopQ‐CEACAM1 molecular interface that could dramatically decrease the affinity, we tested HopQΔ135–136Δ141–142, which completely removes the β‐hairpin that folds in the β2‐α4 loop. However, these multiple sequence changes again resulted in only a modest (~ 4.5‐fold) reduction in affinity relative to wild‐type HopQ (1,000 nM, Fig EV2N). Thermodynamically, this interaction remains exothermic, although the enthalpy of binding is twofold less favorable and is only slightly entropically unfavorable, consistent, in accordance with the comparison of unbound and CEACAM‐bound HopQ structures described above, with the folding of the β2‐α4 loop upon CEACAM1 binding (Fig EV2N). The CEACAM1 Q89A, L95A, and N97A mutations resulted in similar thermodynamic signatures as that of the HopQΔ135–136Δ141–142 mutant. These CEACAM1 residues interact with the HopQ β‐hairpin motif in the β2‐α4 loop (residues 135–138), and thus, either by mutation of these CEACAM1 residues or deletion of the HopQ β‐hairpin, we observe a loss of enthalpy and reduction in entropy due to folding of this structural element upon binding.
A sequence alignment of HopQ and BabA reveals two key differences: (i) The BabA insertion domain, which is its recognition domain of glycans, is much larger than that of HopQ; and (ii) the β2‐α4 loop in BabA is much shorter than in HopQ and little sequence identity exists between the two (Appendix Fig S2). Several BabA structures have been solved both in the apo form and in complex with Leb antigens (Hage et al, 2015; Moonens et al, 2016) and in all of these structures the β2‐α4 loop is folded. Several intrinsically disordered prediction programs such as Globplot (Linding et al, 2003), IUPred (Dosztanyi et al, 2005), RONN (Yang et al, 2005), and PONDR (Peng et al, 2006), all predict the HopQ β2‐α4 loop to be disordered. To test whether this loop in HopQ contributes the majority of the binding energy to the HopQ‐CEACAM1 interaction, we constructed a HopQ‐BabA chimeric protein, HopQβ2‐α4:BabA, in which the β2‐α4 loop of HopQ (residues 133–158) was replaced with that of BabA (residues 136–148). We verified that the HopQβ2‐α4:BabA protein was folded by size‐exclusion chromatography and thermal shift assay analyses, but observed that it exhibited no binding to CEACAM1 (Fig EV2O), demonstrating that the β2‐α4 loop is specifically recognized by CEACAM1 and contributes substantial binding energy to the HopQ‐CEACAM1 interaction.
Molecular determinants of HopQ‐CEACAM interaction control CagA translocation
To determine whether the molecular determinants of binding described above also dictate function, we tested our CEACAM1 and HopQ mutants in a functional assay to quantify translocation of the H. pylori oncoprotein CagA into human cells. We and others have shown that CagA translocation is dependent on HopQ‐CEACAM interactions (Javaheri et al, 2016; Koniger et al, 2016). First, we transiently transfected HEK293 cells with wild‐type or mutant ceacam1 genes and tested CEACAM1 surface production by FACS analysis (Fig 6A). Next, we infected these cells with H. pylori strain P12, producing a TEM‐1‐CagA fusion protein, or its isogenic ΔhopQ, ΔcagI, or ΔcagT translocation‐deficient mutants, and measured TEM‐1 β‐lactamase activity as a proxy for CagA translocation in an established reporter assay system (Schindele et al, 2016; Fig 6B). In agreement with the highly distributed energetic landscape for the HopQ‐CEACAM1 interaction, no single‐site alanine mutants of ceacam1 significantly affected CagA translocation relative to wild‐type ceacam1 (Fig 6B).
Figure 6. Effects of targeted mutations in HopQ or CEACAM1 for CagA translocation.
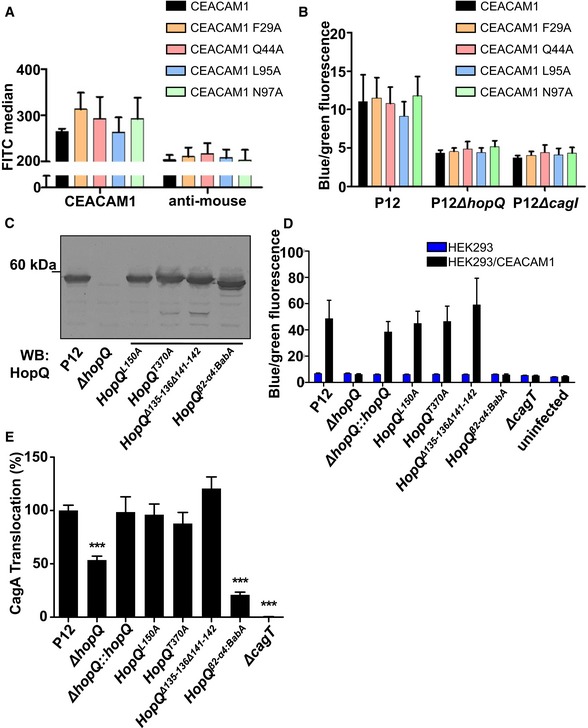
- Flow cytometry showing the surface localization of full‐length CEACAM1 or derivatives with defined amino acid changes in the interaction domains.
- HEK293 cells transiently transfected with CEACAM1 or CEACAM1 mutant derivatives, infected with P12 wt or mutant Helicobacter pylori strains producing TEM‐1‐CagA. Cells were analyzed using the TEM β‐lactamase assay.
- Immunoblot showing the production of wt and mutant HopQ proteins of strain P12.
- HEK293 or HEK293/CEACAM1‐producing cells infected with H. pylori P12 wt or defined hopQ mutant strains producing TEM‐1‐CagA.
- Gastric epithelial cells (AGS) infected with H. pylori P12 wt or mutant strains expressing mutant versions of HopQ.
Second, in order to investigate the role of HopQ mutants in CagA translocation, we complemented H. pylori P12[TEM‐1‐CagA]ΔhopQ with wild‐type or mutant hopQ genes and confirmed the stable production of recombinant mutant HopQ proteins by immunoblotting (Fig 6C). We then infected HEK293 cells, or HEK293 cells transiently transfected with wild‐type ceacam1, and quantified CagA translocation by β‐lactamase activity. Again, we found that no single‐site alanine substitution in hopQ had an effect on CagA translocation relative to wild type. The P12ΔhopQ::hopQ β2‐α4:BabA complemented strain, however, was completely defective in CagA translocation, despite the fact that it produced similar amounts of HopQ protein as found for other mutants (Fig 6C and D). We observed similar results when we infected gastric AGS cells with P12 producing the different HopQ variants; in these cells, the hopQ mutant is still capable of translocating CagA, albeit at a reduced efficiency (Fig 6E). Together, these data indicate that the molecular determinants that dictate distinct energetic landscapes in CEACAM dimerization and HopQ‐CEACAM interaction translate to the biological function of the HopQ‐CEACAM interaction.
Molecular bases of CEACAM self‐ and HopQ recognition extend to other CEACAMs
Our mutational analysis of CEACAM1 indicates that weakening CEACAM1 dimerization has little to no effect on HopQ binding by CEACAM1. In order to determine whether this held true for other CEACAMs known to bind HopQ and across an even broader range of CEACAM dimerization constants, we constructed a CEACAM IgV domain mutant that resulted in dimerization of an otherwise monomeric CEACAM, CEACAM3, which is found predominantly on phagocytes (Schmitter et al, 2004). Indeed, when we analyzed wild‐type CEACAM3 by AUC, we found it to be monomeric in solution (Fig EV3A and Table EV2). A Leu‐to‐Gln mutant at position 44 of CEACAM3, CEACAM3L44Q, resulted in homodimerization with a dimerization constant of approximately 4 μM (Fig EV3B and Table EV2). We solved the X‐ray crystal structures of both wild‐type CEACAM3 and CEACAM3L44Q to resolutions of 2.10 and 2.43 Å, respectively (Table EV1). In both of these structures, CEACAM3 packs identically in the respective crystals, even though our AUC data show CEACAM3 to be monomeric in solution (Fig 7A). The structures superimpose with a RMSD of 0.33 Å. These structures reveal that residue Gln44 in the CEACAM3L44Q mutant drives dimerization by forming two hydrogen bonds across the dimerization interface to the backbone of Asn97, one for each pair of residues, that cannot be made by Leu44 in wild‐type CEACAM3 (Fig 7A).
Figure EV3. Dimerization of CEACAM3 has no effect on HopQ binding.
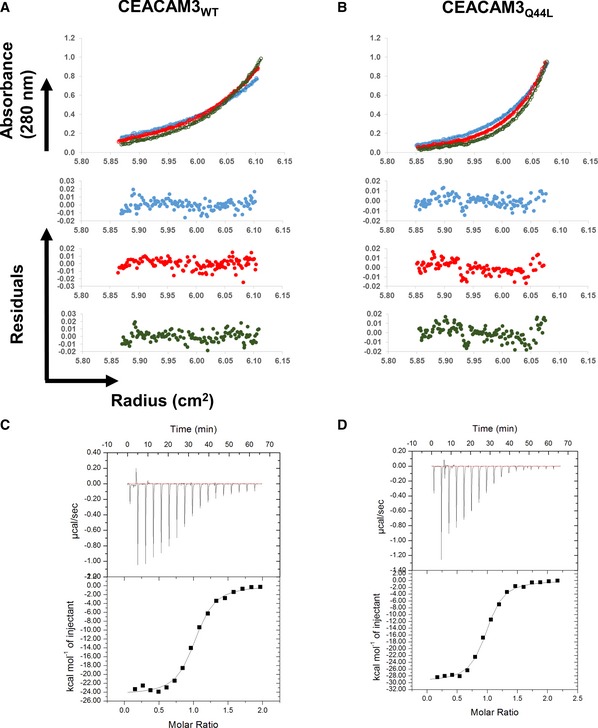
-
A–DSedimentation equilibrium analyses (Upper) and residuals of the fits for each curve (Lower) for (A) CEACAM3WT and (B) CEACAM3Q44L. ITC binding curves of (C) CEACAM3WT and (D) CEACAM3Q44L titrated into HopQWT.
Figure 7. Induced dimerization of CEACAM3 has no effect on binding to HopQ.
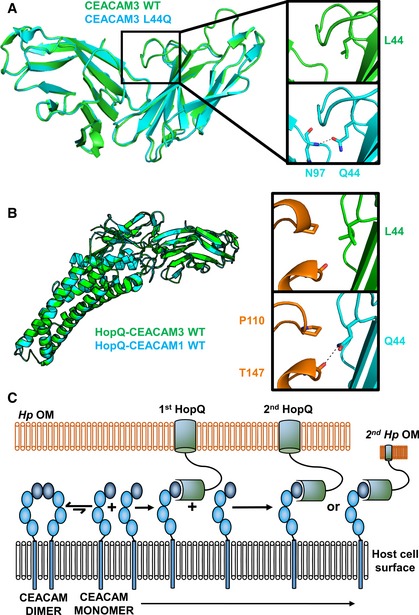
- CEACAM3 WT (green) at position 44 is a leucine. By AUC, it is monomeric; however, it adopts a dimeric conformation in the crystal structure. CEACAM3 Leu44Gln mutation (cyan) creates two symmetrical hydrogen bonds across the dimer interface to the backbone of Asn97. This causes dimerization of CEACAM3. No structural changes are observed between CEACAM3 and mutant with a RMSD of 0.33 Å.
- Superposition of the HopQ‐CEACAM1 complex (cyan) with the HopQ‐CEACAM3 WT complex reveals little structural changes between the complexes with a RMSD of 0.30 Å. In WT CEACAM3 (green), Leu44 forms van der Waal interactions with Pro110 of HopQ (orange). In CEACAM3, Leu44Gln (cyan) forms van der Waal interactions with Pro110 and a hydrogen bond to the main chain of Thr147 (orange).
- Model of HopQ recruitment of CEACAM. CEACAM1 (blue) is predominately dimeric. HopQ (green) binding of CEACAM monomers causes redistribution of dimers to monomers which can bind further HopQ on the same bacterium or recruit other bacteria to the host cell.
Wild‐type CEACAM3 binds HopQ with an affinity approximately twofold weaker than does CEACAM1 (Fig EV3 and Table EV2). Despite the dramatic effect that the CEACAM3L44Q mutant has on CEACAM dimerization (i.e., making CEACAM3 a dimer instead of a monomer), it binds with similar affinity to HopQ as does wild‐type CEACAM3. In order to determine the structural basis for this discrepancy in driving CEACAM dimerization versus HopQ‐CEACAM interaction, we determined the X‐ray crystal structures of HopQ in complex with both wild‐type CEACAM3 and the CEACAM3L44Q mutant (Fig 7B—left and Table EV1). These structures reveal that although Leu44 and Gln44 reside in the interface with HopQ in the respective structures, the distinct intermolecular contacts made by the mutated residue are not of energetic significance (Fig 7B—right). These data indicate that our findings for CEACAM1 dimerization and HopQ‐CEACAM1 interaction can be extended not only to different CEACAMs but also to mutations that either weaken or strengthen CEACAM dimerization.
Discussion
CEACAMs are used by numerous pathogenic bacteria in order to adhere to host cells. The first structures of CEACAMs in complex with a bacterial adhesin reported here clearly show that HopQ binds to the dimerization interface of CEACAMs. This is consistent with all known bacterial adhesins that engage CEACAMs: The Opa proteins from N. gonorrhoeae are membrane proteins that use two hypervariable loops to interact with the CEACAM dimerization interface (Billker et al, 2000); trimeric UspA1 of Moraxella catarrhalis contains a 600‐Å‐long coiled coil, of which a small region close to the OM bends upon binding to the dimerization interface of CEACAM1 (Conners et al, 2008); and the Dr adhesins of E. coli, which recognize CEACAM5, also bind to the dimerization interface (Korotkova et al, 2008). Although the structures of these adhesin proteins are diverse, they all recognize the dimerization interface (CC′C″FG face) of CEACAMs and not the ABDE face. The evolution of adhesion proteins to recognize the dimerization interface may be due to the presence of conserved glycosylation sites on the ABDE face, which could shield and prevent binding of the adhesins and other proteins.
Prior to our studies, the role of glycosylation in HopQ binding had not been fully explored. HopQ, like SabA and BabA, contains an insertion domain, the role of which in BabA and SabA is to bind Leb and sLeX antigens, respectively. It was suggested previously that the HopQ insertion domain was important for binding CEACAM1, as its deletion reduces the affinity of HopQ to CEACAM1 and a peptide derived from the HopQ insertion domain inhibits the induction of the elongation phenotype of gastric cells when treated with H. pylori (Javaheri et al, 2016). Our structures, however, clearly show that both CEACAM1 and CEACAM3 do not interact with the insertion domain. Additionally, as the ABDE face is pointing away from the insertion domain and is 30 Å away, it is unlikely that the insertion domain recognizes glycans on CEACAMs as suggested by both our work and that of others (Javaheri et al, 2016). However, the importance of the insertion domains of SabA and BabA raises the possibility that a putative function of the HopQ insertion domain is to bind a yet‐unknown co‐receptor that further facilitates H. pylori attachment to host cells.
Numerous molecules expressed by H. pylori are important for adherence, including both surface‐displayed adhesin proteins (e.g., glycan‐binding BabA and SabA, CEACAM‐binding HopQ) and proteins that comprise the pilus of the T4SS through which CagA translocates (e.g., β1‐integrin‐binding CagL; Barden et al, 2014; Bonsor et al, 2015b; Bonig et al, 2016; Frick‐Cheng et al, 2016). Our data indicate that HopQ and BabA binding to their respective host cell surface ligands are affected in distinct ways by the chemical environment, including pH. BabA is pH‐responsive such that it is released from Leb glycans in an acidic environment and is able to rebind these ligands upon encountering them again at neutral pH, a property that has been proposed to enable tight mucosal bacterial adherence while providing an effective escape from gastric epithelial cells shed into the acidic gastric lumen (Bugaytsova et al, 2017). We show here that HopQ‐CEACAM interactions do not exhibit the reversible pH responsiveness that BabA does. Additionally, we previously showed that CagL undergoes a dramatic conformational change in response to pH that affects access to its helical RGD motif required for β1‐integrin binding (Bonsor et al, 2015b), providing it a similar pH responsiveness to BabA‐Leb interactions. Collectively, these data suggest a hierarchy of host–pathogen adherence events in which certain steps are reversible, such as bacterial attachment to host cells via Bab‐Leb interactions and even T4SS anchoring via CagL‐β1 integrin interactions, prior to a committed step of HopQ‐CEACAM interaction that triggers CagA translocation. Although we have comprehensively described the molecular basis of HopQ‐CEACAM interactions, the mechanism(s) by which these interactions trigger or permit CagA translocation remains unclear at this time. It is unlikely to be a signal transduction phenomenon, however, as several CEACAMs with distinct cytoplasmic signaling domains permit CagA translocation, and HopQ has no known signaling properties. It may be primarily a result of the relative high affinities of HopQ‐CEACAM interactions, which are in the mid‐nM range, that provide sufficient “dwell time” for CagA to be translocated.
Our structures show that CEACAM binds to a HopQ molecular surface composed of several loops of the 3 + 4 helix bundle and that two of these loops fold upon binding. A limited alanine scan of both sides of the binding interface resulted in no clearly defined energetic hot spot, with only a modest threefold to fivefold reduction in binding observed, suggesting that binding energy is dispersed over many residues. We could, however, prevent binding by swapping the β2‐α4 loop with that of BabA, as measured by ITC analysis and CagA translocation assays. Our data further show that HopQ specifically contacts the dimerization surface of CEACAMs but in a manner that is biophysically distinct from CEACAM homodimerization. Indeed, our alanine scan of the CEACAM1 dimerization does not cause a dramatic effect on HopQ binding; it does, however, alter the dimerization of CEACAM1 over a 1,000‐fold range. Furthermore, we can induce dimerization of CEACAM3 through the Leu44Gln mutation without affecting HopQ binding.
Indeed, recognition of CEACAM monomers by HopQ, as opposed to dimers, may in fact be advantageous for H. pylori as it would further induce monomerization of CEACAM dimers due to depletion of the CEACAM monomer pool. This would shift the equilibrium of binding between HopQ and CEACAMs, leading to the binding of additional HopQ molecules on the same H. pylori, more firmly anchoring the bacterium to the host cell, and/or allowing adherence to the same host cell by additional H. pylori bacteria (Fig 7C).
The HopQ‐CEACAM interaction is essential for both host cell adhesion of H. pylori and translocation of CagA, making it an attractive target for therapeutic development since inhibitors of this interaction could act as anticancer agents at multiple levels (i.e., as anti‐adhesives and anti‐oncoprotein translocators). For instance, such inhibitors could be developed by engineering CEACAMs to bind with higher affinity and specificity to HopQ than do wild‐type cell surface‐expressed CEACAMs. Furthermore, if they were conjugated to antimicrobial agents, such affinity‐matured CEACAM variants could function as narrow‐spectrum antibiotics to kill H. pylori while mitigating damage to commensal bacteria in the gut microbiota.
Materials and Methods
Cloning and mutagenesis of plasmids for bacterial and eukaryotic cell manipulation and expression
Plasmids for expression of full‐length CEACAM1 in HEK293 cells and bacterial expression vector of the N‐terminal domain of CEACAM1 have been described elsewhere (Koniger et al, 2016). The N‐terminal domain of CEACAM3 (tagless) was synthesized as a Gene String (Thermo Fisher) and cloned into an NcoI/XhoI cut pET21d vector. The extracellular domain of HopQ (residues 38–462, Hp strain P12) was cloned into a BamHI/XhoI‐modified pRSFDuet vector that contains an N‐terminal hexahistidine tag for bacterial expression. The ectodomains of CEACAM1 were cloned into the Xmal/NotI pSG160 vector with an N‐terminal hexahistidine tag following the secretion peptide for expression of glycosylated CEACAM1 in HEK cells. Mutagenesis, loop swaps, and loop deletions were carried out by whole‐plasmid mutagenesis (QuikChange Agilent).
Protein expression and purification
The N‐terminal domain of CEACAM1, CEACAM3, and mutants was expressed and purified as described before (Koniger et al, 2016) with minor modifications. Briefly, BL21(DE3) pLysS cells were grown in 1 l of Miller's Luria Broth to an OD600 nm of ~ 0.6 at 37°C before induction with 1 mM IPTG. Cells were grown for a further 4 h at 37°C before harvesting. The cell pellet was lysed by sonication in 20 mM Tris–HCl, 500 mM NaCl, pH 7.5 (Buffer A) with 1% v/v Triton X‐100. The inclusion bodies (IBs) were isolated by centrifugation, suspended in Buffer A with 1% v/v Triton X‐100 and lysed by sonication. The IBs were isolated, suspended in 10 mM Tris, 1.0 M NaCl, pH 8.0, and lysed by sonication. Isolated IBs were washed with Buffer A, pelleted, and dissolved in ~ 5 ml 30 mM Tris, 150 mM NaCl, 8.0 M urea, pH 8.3 for 1 h at room temperature. Undissolved material was spun down, and the supernatant was slowly injected (0.1 ml min−1) into 50 ml of 50 mM CHES‐HCl, 500 mM l‐Arginine, pH 9.2 with stirring at 4°C. The protein was allowed to refold overnight before extensive dialysis against 5 mM Tris, pH 8.0. The protein was concentrated by MonoQ (GE Healthcare). CEACAM1, CEACAM3, and mutants were further purified by size‐exclusion chromatography using a Superdex 200 column (GE Healthcare) equilibrated with 20 mM Tris, 150 mM NaCl, pH 7.5.
HopQ and mutants were expressed in BL21(DE3) pLysS cells and grown in Miller's Luria Broth to an OD600 nm of ~ 0.6 at 37°C before induction with 1 mM IPTG and growth for 18 h at 18°C. Harvested cells were suspended in 20 mM Tris–HCl, 500 mM NaCl, pH 7.5, and lysed by sonication. Cell debris was removed by centrifugation, and the clarified supernatant was passed over 2 ml of HisPur Ni‐NTA resin (Thermo Fisher). The Ni‐resin was washed with 25 ml of Buffer A followed by 25 ml of Buffer A with 50 mM imidazole. HopQ was eluted with 10 ml of Buffer A with 400 mM imidazole and further purified by size‐exclusion chromatography using a Superdex 200 column (GE Healthcare) equilibrated with 20 mM Tris, 150 mM NaCl, pH 7.5.
The ectodomains of CEACAM1 were expressed in HEK cells grown in FreeStyle 293 with GlutaMAX and gentamicin (Thermo Fisher) through transfections with the plasmid described above using polyethyleneimine. To produce both glycosylated and non‐glycosylated forms of the ectodomains, HEK cells were treated to kifunensine (GlycoSyn, final concentration 1 μg ml−1) 30 min post‐transfection to produce high‐mannose glycoproteins. Cells were grown at 37°C with 8% CO2. Glycosylated protein was harvested from the supernatant 5 days post‐transfection. Non‐glycosylated protein was produced by adding EndoA, an endoglycosidase which cleaves high mannose leaving just GlcNAc attached the asparagine, to the cells (final concentration of 1 μg ml−1) 4 days post‐transfections. Non‐glycosylated protein was harvest from the supernatant 24 h later. Complex‐type glycosylated CEACAM1 ectodomain was produced by not treating the HEK cells with kifunensine or EndoA. Proteins were purified using a 5‐ml HisTrap Excel column and further purified on Superdex 200 column (GE Healthcare) equilibrated with 20 mM Tris, 150 mM NaCl, pH 7.5.
Isothermal titration calorimetry
Proteins were extensive dialyzed against 30 mM Tris, 150 mM NaCl, pH 7.5, or 30 mM potassium phosphate, 150 mM NaCl, pH 4.5. ITC experiments were performed using an iTC200 instrument (GE Healthcare). A typical experiment was carried out with the syringe loaded with CEACAM1 (200 μM) and the cell loaded with HopQ (20 μM). Titrations were performed at 25°C with 16 injections of 2.42‐μl aliquots, with 230‐s intervals between injections. All data were analyzed using Origin 7.0 software.
Thermal shift assays
Proteins were diluted with 20 mM Tris, 150 mM NaCl, pH 7.5 to a final concentration of 10 μM (CEACAM1) or 20 μM (HopQ, HopQβ2‐α4:BabA, HopQ‐CEACAM1, and HopQβ2‐α4:BabA‐CEACAM1). One millilitre of protein samples was mixed with 1 μl of 5,000× SYPRO Orange. 12.5 μl of each protein–SYPRO Orange was aliquoted to 2 × 12 wells of a MicroAmp Fast 96‐well reaction plate and mixed with 12.5 μl of 200 mM buffer. Twelve buffers were selected to cover pH 2.0–7.5 in 0.5 pH unit intervals; sodium phosphate 2.0 and 2.5; sodium citrate 3.0–6.0; Bis–Tris, pH 6.5; Bis–Tris propane, pH 7.0; and HEPES pH 7.5. The plate was sealed with iCycler Optical Tape, and melting curves were measured from 25 to 95°C on a StepOnePlus Real‐Time PCR System. Data were analyzed according to established methods (Niesen et al, 2007).
pH effect on the HopQ‐CEACAM1 interaction by ELISA
CEACAM1IgV (1.5 μg ml−1) in PBS was used to coat a 96‐well immunoplate (Nunc MaxiSorp) overnight at 4°C. Wells were blocked with PBS containing 5% w/v non‐fat dry milk powder (RPI) and 0.5% w/v BSA (Thermo Fisher Scientific) for 1 h at room temperature. 90 μl of 50 mM buffer system (sodium phosphate pH 2.0; sodium citrate pH 3.0, 4.0, 5.0 and 6.0; Bis–Tris propane pH 7.0) containing 0.5% w/v BSA and 0.05% v/v Tween‐20 were added to wells, followed by 10 μl of HopQ (100 μg ml−1) and mixed. Plates were incubated at room temperature for 1 h before the addition of either 100 μl of 200 mM of buffer system containing 0.5% w/v BSA and 0.05% v/v Tween‐20 or 200 mM Bis–Tris propane, pH 7.0, 0.5% w/v BSA, and 0.05% v/v Tween‐20 to readjust the pH to 7.0. Plates were incubated for a further 30 min before removal and washing three times with PBS containing 0.5% w/v BSA and 0.05% v/v Tween‐20 (PBSTB). Plates were incubated with 1:5,000 dilution of anti‐His HRP conjugate (Merck Millipore) in PBSTB with 5% w/v non‐fat milk powder for 1 h at RT before a further three washes with PBSTB. For detection, 1‐Step Turbo TMB‐ELISA Substrate Solution was used (Thermo Fisher Scientific). Enzymatic reactions were typical stopped 15–30 min later with 1 M sulfuric acid. Absorbances were measured at 450 nm on a SpectaMaxM2 plate reader (Molecular Devices).
Analytical ultracentrifugation measurements
Measurements were performed essentially as described in Bonsor et al (2015a). Briefly, sedimentation equilibrium measurements of CEACAM1, CEACAM3, and mutants were performed using an Optima XL‐I analytical ultracentrifuge fitted with a four‐hole An‐60Ti Rotor (Beckman Coulter) at 20°C. Proteins were extensively dialyzed against buffer containing 50 mM Tris, pH 7.5, 50 mM NaCl. Protein partial specific volume and solvent density values from the protein amino acid sequences and buffer composition, respectively, were calculated using SedenTerp (http://bitcwiki.sr.unh.edu). Samples were prepared at three concentrations (approximately 12, 18, and 30 μM) and loaded into cells equipped with six‐hole charcoal‐filled epon centerpieces (1.2 cm pathlength) with sapphire windows. Centrifugation was carried out at 63,222, 77,495, and 93,220 g, and scans were acquired at 280 nm with a step size of 0.001 and five averages per step. The data were globally analyzed to both single species and monomer–dimer self‐association models using the program HeteroAnalysis.
Crystallization and structure determination of CEACAMs and HopQ‐CEACAM complexes
The HopQ‐CEACAM1 complex was prepared by mixing a twofold excess of purified CEACAM1 over HopQ. The complex was incubated at room temperature for 30 min before concentration using an Amicon Ultra‐15 with a 50,000 NMWL membrane allowing the excess CEACAM1 and unbound HopQ to pass through. The retentate was diluted and concentrated three times using 20 mM Tris, 150 mM NaCl, pH 7.5. The complex was subjected to size‐exclusion chromatography using a Superdex 200 column (GE Healthcare) equilibrated with 20 mM Tris, 150 mM NaCl, pH 7.5. Complex fractions were pooled, concentrated to 7.8 mg ml−1, and screened against the Wizards I/II/III/IV (Rigaku) screens using a Crystal Gryphon Protein Crystallography System (Art Robbins Instruments). Crystals grew in condition G11 of the Wizards III/IV screen (20% w/v PEG 6000, 0.1 M HEPES‐NaOH, pH 7.0, 0.2 M NaCl). Extensive attempts to reproduce the crystals failed, and therefore, crystals were harvested directly from the initial screen drop. Crystals were washed and cyroprotected in mother liquor containing 20% w/v glycerol. Data were collected on beamline 12‐2 at the Stanford Synchrotron Radiation Lightsource (SSRL). Data were processed with XDS (Kabsch, 2010) and indexed and converted to structure factors with Aimless. The complex was solved by molecular replacement using MOLREP and the structures of HopQ (PDB entry 5lp2) and CEACAM1 (PDB entry 2gk2) as search models. The complex was refined with REFMAC and rebuilt in Coot (Winn et al, 2011). The final model and structure factors have been deposited to the RCSB PDB with the entry code of 6AW2.
The HopQ‐CEACAM3 WT and L44Q complexes were prepared as for HopQ‐CEACAM1 and screened against the Wizards I/II/III/IV screens (Rigaku) at 7.7 mg ml−1. Crystals grew in condition G4 of the Wizards I/II screen (20% w/v PEG 8000, 0.1 M MES‐NaOH, pH 6.0, 0.2 M calcium acetate). Crystals were optimized and were found to grown in 20% w/v PEG 3000, 0.1 M Bis–Tris–HCl, pH 6.5, 0.2 M calcium acetate, and 1% v/v glycerol. Crystals were washed and cyroprotected with mother liquor containing 20% v/v glycerol. Data were collected on beamline ID23‐D at the Advance Photon Source (HopQ‐CEACAM3 WT) 9‐2 at SSRL (HopQ‐CEACAM3 L44Q). Data were processed with XDS and indexed and converted to structure factors with Aimless. The complex was solved by molecular replacement using MOLREP and the structure of the HopQ‐CEACAM1 complex as the search model. The complex was refined with REFMAC and rebuilt in Coot. The final models and structure factors were deposited to the RCSB PDB with the entry code of 6AVZ and 6AW3.
CEACAM3 WT and L44Q mutant were dialyzed against 20 mM Tris, 150 mM NaCl, pH 7.5. WT CEACAM3 was screened against the JCSG+ screen (QIAgen) at 5.6 mg ml−1. Crystals grew in condition H2 (1.0 M ammonium sulfate, 0.1 M Bis–Tris–HCl, pH 5.5, 1% w/v PEG 3350). Crystals were not further optimized and were washed and cyroprotected with mother liquor containing 30% v/v glycerol. Data were collected on beamline 12‐2 at SSRL, processed with XDS, and indexed and converted to structure factors with Aimless. CEACAM1 (PDB entry 2gk2) was used as a search model in MOLREP, refined with REFMAC, and rebuilt in Coot. The final model and structure factors have been deposited to the RCSB PDB with the entry code of 6AW1. CEACAM3 L44Q crystallized in a slightly optimized condition (0.8 M ammonium sulfate, 0.1 M Bis–Tris–HCl, pH 5.5, 1% w/v PEG 3350). Crystals were washed and cyroprotected with mother liquor containing 20% v/v glycerol. A dataset was collected in‐house using a Rigaku rotating anode Micromax‐007 beam. Images were processed with iMosflm and intensities were converted to structure factors using Aimless. CEACAM3 WT was used a search model in MOLREP, refined using REFMAC, and rebuilt in Coot. The final model and structure factors have been deposited to the RCSB PDB with the entry code of 6AW0.
Native mass spectrometry
The HopQ‐CEACAM1 complex was buffer exchanged against 50 mM ammonium acetate pH 6.8. Native MS was acquired on a SynaptG2 with a nanoESI source (Waters). Samples were sprayed with gold‐coated glass capillaries. Acquisition conditions were as follows: capillary voltage 1.3 kV, sample cone voltage 30 V, extraction cone 3 V, source temperature 40°C, trap CE 10 V, and no transfer CE. Data were analyzed with the Masslynx software (Waters).
Helicobacter pylori complementation assays
The effects of specific amino acid exchanges in HopQ on functionality of the Cag type IV secretion system were determined with H. pylori P12 ΔhopQ mutants that were complemented with mutated hopQ expressed from the indigenous plasmid pHel12 (see below). For qualitative assessment of CagA translocation using tyrosine phosphorylation assays, hopQ expression plasmids were transformed into an isogenic ΔhopQ deletion mutant described previously (Koniger et al, 2016). For quantitative determination of CagA translocation into target cells, a marker‐free derivative of the previously described H. pylori P12 strain producing a TEM‐1 β‐lactamase‐CagA fusion (Schindele et al, 2016) was used. This strain was first transformed with a hopQ deletion plasmid (pFS10), generated by cloning 1‐kb hopQ upstream and downstream fragments obtained with primers VK57/VK58 or VK59/VK60, respectively (Koniger et al, 2016), together with an rpsL‐erm cassette (Rohrer et al, 2012) into pUC18. The obtained P12ΔhopQ [TEM1‐CagA] strain was then used for complementation with the respective hopQ expression plasmids. As negative controls, strains P12 or P12 [TEM‐1‐CagA] were transformed with either a cagT or a cagI deletion plasmid (Fischer et al, 2001) to disrupt the Cag type IV secretion apparatus.
The hopQ expression plasmids used for complementation were designed to integrate into the indigenous plasmid of strain P12 (pHel12). To achieve this, a pSMART‐hcKan vector harboring a fragment of pHel12 (Fischer et al, 2010) was amplified by inverse PCR with primers SR53 (5′‐GATCGGATCC CCTACAGGTC TCACACATCA‐3′) and SR54 (5′‐GATCGGATCC CCAAACTTTC CCTATGGGCA‐3′), thus generating flanking regions for integration into the intergenic region between hpp12_p11 and hpp12_p01 (nucleotides 9192–10072 and 10131‐[10225/1]‐946, respectively). Between these flanking regions, we inserted a hopQ expression module, consisting of (i) a cat GC chloramphenicol resistance cassette amplified from the shuttle vector pHel2 (Heuermann & Haas, 1998) with primers WS193 (5′‐GGGGTACCCG ATCGGCCATA TTGTGTTGAA AC‐3′) and LH2 (5′‐GATCGGATCC ATCCACGTTG AAAATCTC‐3′), (ii) the alpA promoter (Odenbreit et al, 1999) including the multiple cloning site from plasmid pIB6 (Bugaytsova et al, 2017), and (iii) the hopQ gene amplified with primers CE68 (5′‐GATCGTCGAC ATGAAAAAAA CGAAAAAAAC‐3′) and CE69 (5′‐GATCAGATCT TTTAATACGC GAACACATAA‐3′), resulting in plasmid pWS658. Site‐specific mutations were introduced into pWS658 by whole‐plasmid mutagenesis.
Expression of hopQ constructs by H. pylori P12ΔhopQ or P12[TEM‐1‐CagA]ΔhopQ was confirmed by immunoblotting using the polyclonal HopQ antiserum AK298 (Koniger et al, 2016).
For testing the influence of HopQ‐CEACAM interactions on type IV secretion efficiency, HEK293 cells were transiently transfected with pRc/CMV‐CEACAM1 plasmids harboring full‐length CEACAM1 or mutated derivatives, using lipofectamine 2000 transfection, as described previously (Koniger et al, 2016). Expression of CEACAM constructs was verified by flow cytometry using the anti‐CEACAM1 antibody GM‐8G5 (Genovac). Transfected cells or untransfected control cells, or alternatively AGS gastric epithelial cells, were infected with H. pylori under standard conditions and examined by phosphotyrosine or TEM‐1 β‐lactamase assays (either in a plate reader or by flow cytometry), as described previously (Schindele et al, 2016).
Data availability
The X‐ray crystal structures of HopQ‐CEACAM1, HopQ‐CEACAM3, HopQ‐CEACAM3 L44Q, CEACAM3 WT, and CEACAM3 L44Q from this publication have been deposited to the Protein Databank (http://www.rcsb.org) with the accession codes of 6AW2, 6AVZ, 6AW3, 6AW1, and 6AW0, respectively.
Author contributions
DAB and EJS conceived the project and wrote the paper; WF, DB, PLW, and RH edited the manuscript; DAB conducted X‐ray crystallography, mutagenesis, and ITC experiments; DD preformed the MS experiments; JW and DB performed the AUC experiments; QZ and BS performed the functional assays; EW, RB, BD, and WF provided reagents; WF, DB, PLW, RH, and EJS supervised research.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
This work was supported in part by an Alexander von Humboldt Foundation Experienced Researcher Fellowship to EJS, grants from the DFG (HA2697/16‐2 and SFB914 Project B05) to RH, and NIH grant S10 RR15899‐01 to DB. We wish to thank the support staff of Beamlines 12‐2 and 9‐2 at the Stanford Synchrotron Radiation Lightsource and ID23‐D at Advanced Photon Source for their aid in data collection.
The EMBO Journal (2018) 37: e98664
References
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S (2003) Disruption of the epithelial apical‐junctional complex by Helicobacter pylori CagA. Science 300: 1430–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barden S, Schomburg B, Conradi J, Backert S, Sewald N, Niemann HH (2014) Structure of a three‐dimensional domain‐swapped dimer of the Helicobacter pylori type IV secretion system pilus protein CagL. Acta Crystallogr D Biol Crystallogr 70: 1391–1400 [DOI] [PubMed] [Google Scholar]
- Benchimol S, Fuks A, Jothy S, Beauchemin N, Shirota K, Stanners CP (1989) Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell 57: 327–334 [DOI] [PubMed] [Google Scholar]
- Billker O, Popp A, Gray‐Owen SD, Meyer TF (2000) The structural basis of CEACAM‐receptor targeting by neisserial Opa proteins. Trends Microbiol 8: 258–260; discussion 260‐1 [DOI] [PubMed] [Google Scholar]
- Bonig T, Olbermann P, Bats SH, Fischer W, Josenhans C (2016) Systematic site‐directed mutagenesis of the Helicobacter pylori CagL protein of the Cag type IV secretion system identifies novel functional domains. Sci Rep 6: 38101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsor DA, Gunther S, Beadenkopf R, Beckett D, Sundberg EJ (2015a) Diverse oligomeric states of CEACAM IgV domains. Proc Natl Acad Sci USA 112: 13561–13566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsor DA, Pham KT, Beadenkopf R, Diederichs K, Haas R, Beckett D, Fischer W, Sundberg EJ (2015b) Integrin engagement by the helical RGD motif of the Helicobacter pylori CagL protein is regulated by pH‐induced displacement of a neighboring helix. J Biol Chem 290: 12929–12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaytsova JA, Bjornham O, Chernov YA, Gideonsson P, Henriksson S, Mendez M, Sjostrom R, Mahdavi J, Shevtsova A, Ilver D, Moonens K, Quintana‐Hayashi MP, Moskalenko R, Aisenbrey C, Bylund G, Schmidt A, Aberg A, Brannstrom K, Koniger V, Vikstrom S et al (2017) Helicobacter pylori adapts to chronic infection and gastric disease via pH‐responsive BabA‐mediated adherence. Cell Host Microbe 21: 376–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL (2011) Helicobacter pylori cytotoxin‐associated gene A (CagA) subverts the apoptosis‐stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci USA 108: 9238–9243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conners R, Hill DJ, Borodina E, Agnew C, Daniell SJ, Burton NM, Sessions RB, Clarke AR, Catto LE, Lammie D, Wess T, Brady RL, Virji M (2008) The Moraxella adhesin UspA1 binds to its human CEACAM1 receptor by a deformable trimeric coiled‐coil. EMBO J 27: 1779–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21: 3433–3434 [DOI] [PubMed] [Google Scholar]
- Fedarovich A, Tomberg J, Nicholas RA, Davies C (2006) Structure of the N‐terminal domain of human CEACAM1: binding target of the opacity proteins during invasion of Neisseria meningitidis and N. gonorrhoeae . Acta Crystallogr D Biol Crystallogr 62: 971–979 [DOI] [PubMed] [Google Scholar]
- Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R (2001) Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin‐8. Mol Microbiol 42: 1337–1348 [DOI] [PubMed] [Google Scholar]
- Fischer W, Windhager L, Rohrer S, Zeiller M, Karnholz A, Hoffmann R, Zimmer R, Haas R (2010) Strain‐specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res 38: 6089–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick‐Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD, Cover TL (2016) Molecular and structural analysis of the Helicobacter pylori cag Type IV secretion system core complex. MBio 7: e02001–e02015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrell RJ, Guan J, Xin Y, Tafreshi MA, Hutton ML, McGuckin MA, Ferrero RL, Kwok T (2013) A novel NOD1‐ and CagA‐independent pathway of interleukin‐8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol 15: 554–570 [DOI] [PubMed] [Google Scholar]
- Gray‐Owen SD, Dehio C, Haude A, Grunert F, Meyer TF (1997a) CD66 carcinoembryonic antigens mediate interactions between Opa‐expressing Neisseria gonorrhoeae and human polymorphonuclear phagocytes. EMBO J 16: 3435–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray‐Owen SD, Lorenzen DR, Haude A, Meyer TF, Dehio C (1997b) Differential Opa specificities for CD66 receptors influence tissue interactions and cellular response to Neisseria gonorrhoeae . Mol Microbiol 26: 971–980 [DOI] [PubMed] [Google Scholar]
- Gray‐Owen SD, Blumberg RS (2006) CEACAM1: contact‐dependent control of immunity. Nat Rev Immunol 6: 433–446 [DOI] [PubMed] [Google Scholar]
- Hage N, Howard T, Phillips C, Brassington C, Overman R, Debreczeni J, Gellert P, Stolnik S, Winkler GS, Falcone FH (2015) Structural basis of Lewis(b) antigen binding by the Helicobacter pylori adhesin BabA. Sci Adv 1: e1500315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M (2004) Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 4: 688–694 [DOI] [PubMed] [Google Scholar]
- Heuermann D, Haas R (1998) A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol Gen Genet 257: 519–528 [DOI] [PubMed] [Google Scholar]
- Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M (2002) SHP‐2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295: 683–686 [DOI] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T (1998) Helicobacter pylori adhesin binding fucosylated histo‐blood group antigens revealed by retagging. Science 279: 373–377 [DOI] [PubMed] [Google Scholar]
- Javaheri A, Kruse T, Moonens K, Mejias‐Luque R, Debraekeleer A, Asche CI, Tegtmeyer N, Kalali B, Bach NC, Sieber SA, Hill DJ, Koniger V, Hauck CR, Moskalenko R, Haas R, Busch DH, Klaile E, Slevogt H, Schmidt A, Backert S et al (2016) Helicobacter pylori adhesin HopQ engages in a virulence‐enhancing interaction with human CEACAMs. Nat Microbiol 2: 16189 [DOI] [PubMed] [Google Scholar]
- Jimenez‐Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, Terradot L, Fischer W, Haas R (2009) Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD‐independent manner. PLoS Pathog 5: e1000684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koniger V, Holsten L, Harrison U, Busch B, Loell E, Zhao Q, Bonsor DA, Roth A, Kengmo‐Tchoupa A, Smith SI, Mueller S, Sundberg EJ, Zimmermann W, Fischer W, Hauck CR, Haas R (2016) Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat Microbiol 2: 16188 [DOI] [PubMed] [Google Scholar]
- Korotkova N, Yang Y, Le Trong I, Cota E, Demeler B, Marchant J, Thomas WE, Stenkamp RE, Moseley SL, Matthews S (2008) Binding of Dr adhesins of Escherichia coli to carcinoembryonic antigen triggers receptor dissociation. Mol Microbiol 67: 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S (2007) Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449: 862–866 [DOI] [PubMed] [Google Scholar]
- Linden S, Mahdavi J, Hedenbro J, Boren T, Carlstedt I (2004) Effects of pH on Helicobacter pylori binding to human gastric mucins: identification of binding to non‐MUC5AC mucins. Biochem J 384: 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R, Russell RB, Neduva V, Gibson TJ (2003) GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res 31: 3701–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB et al (2002) Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297: 573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaw SE, Schneider J, Liao EH, Zimmermann W, Gray‐Owen SD (2003) Immunoreceptor tyrosine‐based activation motif phosphorylation during engulfment of Neisseria gonorrhoeae by the neutrophil‐restricted CEACAM3 (CD66d) receptor. Mol Microbiol 49: 623–637 [DOI] [PubMed] [Google Scholar]
- Moonens K, Gideonsson P, Subedi S, Bugaytsova J, Romao E, Mendez M, Norden J, Fallah M, Rakhimova L, Shevtsova A, Lahmann M, Castaldo G, Brannstrom K, Coppens F, Lo AW, Ny T, Solnick JV, Vandenbussche G, Oscarson S, Hammarstrom L et al (2016) Structural insights into polymorphic ABO glycan binding by Helicobacter pylori . Cell Host Microbe 19: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2: 2212–2221 [DOI] [PubMed] [Google Scholar]
- Odenbreit S, Till M, Hofreuter D, Faller G, Haas R (1999) Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol Microbiol 31: 1537–1548 [DOI] [PubMed] [Google Scholar]
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287: 1497–1500 [DOI] [PubMed] [Google Scholar]
- Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, Yamada G, Azuma T, Hatakeyama M (2008) Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA 105: 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang SS, Nguyen ST, Perry AJ, Day CJ, Panjikar S, Tiralongo J, Whisstock JC, Kwok T (2014) The three‐dimensional structure of the extracellular adhesion domain of the sialic acid‐binding adhesin SabA from Helicobacter pylori . J Biol Chem 289: 6332–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek RM Jr, Blaser MJ (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2: 28–37 [DOI] [PubMed] [Google Scholar]
- Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z (2006) Length‐dependent prediction of protein intrinsic disorder. BMC Bioinformatics 7: 208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pils S, Gerrard DT, Meyer A, Hauck CR (2008) CEACAM3: an innate immune receptor directed against human‐restricted bacterial pathogens. Int J Med Microbiol 298: 553–560 [DOI] [PubMed] [Google Scholar]
- Rohrer S, Holsten L, Weiss E, Benghezal M, Fischer W, Haas R (2012) Multiple pathways of plasmid DNA transfer in Helicobacter pylori . PLoS One 7: e45623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadat I, Higashi H, Obuse C, Umeda M, Murata‐Kamiya N, Saito Y, Lu H, Ohnishi N, Azuma T, Suzuki A, Ohno S, Hatakeyama M (2007) Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447: 330–333 [DOI] [PubMed] [Google Scholar]
- Schindele F, Weiss E, Haas R, Fischer W (2016) Quantitative analysis of CagA type IV secretion by Helicobacter pylori reveals substrate recognition and translocation requirements. Mol Microbiol 100: 188–203 [DOI] [PubMed] [Google Scholar]
- Schmitter T, Agerer F, Peterson L, Munzner P, Hauck CR (2004) Granulocyte CEACAM3 is a phagocytic receptor of the innate immune system that mediates recognition and elimination of human‐specific pathogens. J Exp Med 199: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitter T, Pils S, Weibel S, Agerer F, Peterson L, Buntru A, Kopp K, Hauck CR (2007) Opa proteins of pathogenic neisseriae initiate Src kinase‐dependent or lipid raft‐mediated uptake via distinct human carcinoembryonic antigen‐related cell adhesion molecule isoforms. Infect Immun 75: 4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Paul FE, Brandt S, Guye P, Daumke O, Backert S, Dehio C, Mann M (2009) Host cell interactome of tyrosine‐phosphorylated bacterial proteins. Cell Host Microbe 5: 397–403 [DOI] [PubMed] [Google Scholar]
- Singer BB, Klaile E, Scheffrahn I, Muller MM, Kammerer R, Reutter W, Obrink B, Lucka L (2005) CEACAM1 (CD66a) mediates delay of spontaneous and Fas ligand‐induced apoptosis in granulocytes. Eur J Immunol 35: 1949–1959 [DOI] [PubMed] [Google Scholar]
- Singer BB, Scheffrahn I, Kammerer R, Suttorp N, Ergun S, Slevogt H (2010) Deregulation of the CEACAM expression pattern causes undifferentiated cell growth in human lung adenocarcinoma cells. PLoS One 5: e8747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S, Michetti P (2002) Helicobacter pylori infection. N Engl J Med 347: 1175–1186 [DOI] [PubMed] [Google Scholar]
- Tchoupa AK, Schuhmacher T, Hauck CR (2014) Signaling by epithelial members of the CEACAM family – mucosal docking sites for pathogenic bacteria. Cell Commun Signal 12: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M (2003) Attenuation of Helicobacter pylori CagA × SHP‐2 signaling by interaction between CagA and C‐terminal Src kinase. J Biol Chem 278: 3664–3670 [DOI] [PubMed] [Google Scholar]
- Virji M, Makepeace K, Ferguson DJ, Watt SM (1996) Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic neisseriae. Mol Microbiol 22: 941–950 [DOI] [PubMed] [Google Scholar]
- Virji M (2009) Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat Rev Microbiol 7: 274–286 [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZR, Thomson R, McNeil P, Esnouf RM (2005) RONN: the bio‐basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21: 3369–3376 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Data Availability Statement
The X‐ray crystal structures of HopQ‐CEACAM1, HopQ‐CEACAM3, HopQ‐CEACAM3 L44Q, CEACAM3 WT, and CEACAM3 L44Q from this publication have been deposited to the Protein Databank (http://www.rcsb.org) with the accession codes of 6AW2, 6AVZ, 6AW3, 6AW1, and 6AW0, respectively.