

Abstract
The human gastric pathogen Helicobacter pylori is a major causative agent of gastritis, peptic ulcer disease, and gastric cancer. As part of its adhesive lifestyle, the bacterium targets members of the carcinoembryonic antigen‐related cell adhesion molecule (CEACAM) family by the conserved outer membrane adhesin HopQ. The HopQ–CEACAM1 interaction is associated with inflammatory responses and enables the intracellular delivery and phosphorylation of the CagA oncoprotein via a yet unknown mechanism. Here, we generated crystal structures of HopQ isotypes I and II bound to the N‐terminal domain of human CEACAM1 (C1ND) and elucidated the structural basis of H. pylori specificity toward human CEACAM receptors. Both HopQ alleles target the β‐strands G, F, and C of C1ND, which form the trans dimerization interface in homo‐ and heterophilic CEACAM interactions. Using SAXS, we show that the HopQ ectodomain is sufficient to induce C1ND monomerization and thus providing H. pylori a route to influence CEACAM‐mediated cell adherence and signaling events.
Keywords: bacterial adhesion, CagA delivery, CEACAM1, Helicobacter pylori, HopQ
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Introduction
Helicobacter pylori chronically colonizes the stomach of about half the world population. Genomic analyses underscore a strong association and co‐evolution with human populations predating ancient migrations (Atherton & Blaser, 2009). H. pylori's extensive host adaptation allows it to avoid or temporarily withstand the noxious gastric low pH environment (Moore et al, 2011; Bugaytsova et al, 2017) and to evade innate and adaptive immune responses (Salama et al, 2013), resulting in a lifelong infection of the gastric mucosa. Infection results in usually asymptomatic gastritis, but prolonged exposure to more virulent strains leads to severe clinical symptoms such as gastric and duodenal ulcers, and malignant transformation resulting in gastric lymphomas and adenocarcinomas (Peek & Blaser, 2002; Polk & Peek, 2010). Strong virulence‐associated traits include the secretion and delivery of the CagA oncoprotein by a type IV secretion system (T4SS) encoded by the cytotoxin‐associated genes (cag) pathogenicity island (Backert et al, 2011), secretion of the VacA vacuolating toxin (Palframan et al, 2012), and the presence of outer membrane adhesins such as BabA, SabA, and HopQ (Ilver et al, 1998; Gerhard et al, 1999; Mahdavi et al, 2002; Posselt et al, 2013; Javaheri et al, 2016).
HopQ was recently shown to engage into a virulence‐enhancing interaction with epithelial carcinoembryonic antigen (CEA)‐related cell adhesion molecules (CEACAMs; Javaheri et al, 2016; Koniger et al, 2016). In humans, the CEACAM family comprises 12 heavily glycosylated cell surface anchored proteins that share a common architecture with an amino‐terminal variable immunoglobulin‐like (IgV‐like) domain followed by a variable number of membrane‐proximal constant immunoglobulin‐like domains (Gray‐Owen & Blumberg, 2006; Appendix Fig S1). The CEACAM family proteins mediate intercellular adhesion and outside‐in signaling events implicated in cellular processes associated with cell differentiation, proliferation, apoptosis, motility, and immunological responses (Gray‐Owen & Blumberg, 2006; Kuespert et al, 2006). CEACAMs have a wide but tissue‐specific distribution, with for example CEACAM1 being expressed in leukocytes, endothelial and epithelial cells, CEACAM3 and CEACAM8 in granulocytes, CEACAM5 and CEACAM7 in epithelial cells, and CEACAM6 in epithelia and granulocytes (Hammarstrom, 1999). CEACAMs mediate homo‐ and heterophilic interactions on a single‐cell surface (cis interactions) or across cell junctions (trans interactions). CEACAMs are also exploited by pathogenic and non‐pathogenic bacteria such as Neisseria gonorrhoeae, N. meningitides, Moraxella catarrhalis, Haemophilus influenzae, and Escherichia coli during mucosal colonization (Leusch et al, 1991; Virji et al, 1996, 2000; Hill & Virji, 2003; Berger et al, 2004; Barnich et al, 2007). Bacterial proteins interacting with CEACAMs have been described to inhibit T lymphocyte activation and proliferation, B‐cell antibody production, and innate inflammatory responses by epithelia, such as infection‐induced exfoliation (Muenzner et al, 2005, 2010; Slevogt et al, 2008; Sadarangani et al, 2011; Javaheri et al, 2016). In H. pylori, the HOP (Helicobacter outer membrane proteins) family adhesin HopQ was identified to enable a tight and specific nanomolar interaction with the N‐terminal IgV‐like domain of CEACAM1, CEACAM3, CEACAM5, and CEACAM6, but not with CEACAM8 (Javaheri et al, 2016). Two allelic variants of HopQ exist, type I and type II, sharing about 70% amino acid sequence homology (Cao & Cover, 2002). The type I HopQ is more frequently found in cag(+)/s1‐vacA type strains and is associated with higher inflammation and gastric atrophy rates (Ohno et al, 2009). In healthy, non‐infected gastric tissue, no significant levels of CEACAM1, 5, and 6 are detected on the epithelial cells. However, the levels of these CEACAM receptors were up‐regulated during gastritis and in gastric tumors (Javaheri et al, 2016). H. pylori binding induced tyrosine‐phosphorylation of the cytoplasmic CEACAM1‐L immunoreceptor tyrosine‐based inhibitory motif (ITIM) domain, triggering downstream signaling cascades (Slevogt et al, 2008; Javaheri et al, 2016). The HopQ–CEACAM1 interaction was also found to facilitate the translocation of CagA into host cells and enhances the release of pro‐inflammatory mediators such as interleukin‐8, unlike other known HOP adhesins (Belogolova et al, 2013; Javaheri et al, 2016; Koniger et al, 2016). However, the mode of interaction with human CEACAMs and the mechanism by which HopQ facilitates CagA delivery and phosphorylation remain poorly understood.
In this study, we present crystal structures of the two major allelic variants of HopQ, type I and type II, in complex with the N‐terminal domain of CEACAM1. These structures enabled us to rationalize the observed binding specificity of HopQ toward various CEACAMs. A combination of X‐ray crystallography and small‐angle X‐ray scattering (SAXS) demonstrated the HopQ adhesin domain is able and sufficient to dissociate CEACAM1‐N dimers into monomers, revealing a potential molecular route to modulate host immune responses.
Results
Type I HopQ binds CEACAM1 via an induced fit in disulfide‐clasped interaction loops
To gain structural insight in the specificity of HopQ toward human CEACAMs, we elucidated the X‐ray structure of the HopQ type I adhesin domain (HopQAD‐I, comprising residues 22–425 of the mature protein) bound to the N‐terminal domain of human CEACAM1 (C1ND; residues 1–108) to a resolution of 2.8 Å (Appendix Table S1; Figs 1A and EV1A). Since C1ND is extensively glycosylated (with N‐glycosylation sites at residues 70, 77, and 81), it was recombinantly produced in E. coli in its non‐glycosylated form to enhance homogeneity and crystallization potential. We previously showed that the HopQ–CEACAM1 interaction is driven by protein–protein contacts and that the HopQ dissociation constant (K D) for non‐glycosylated C1ND is similar to that for material produced in HEK293 cells (Javaheri et al, 2016). The HopQ structure features an α‐helical core domain arranged in a 3 + 4 helix bundle topology (Fig 1A), a fold that is conserved among all hitherto crystallized adhesins of the Hop family, like BabA and SabA (Pang et al, 2013; Hage et al, 2015; Javaheri et al, 2016; Moonens et al, 2016). The HOP family also shares a pattern of paired cysteines that stabilize extended, partially flexible disulfide‐clasped loops that protrude from the protein surface (Moonens et al, 2016). In HopQ, two of the three conserved cysteine pairs (cysteine‐clasped loop 1 and 2, hereafter CL1 and 2) cluster together at the tip of the core domain and form the main interaction site with the C1ND domain (Fig 1A). The CEACAM 1 Ig variable (Igv) domain (C1ND) consists of a β‐sandwich fold with 9 anti‐parallel β‐sheets organized in two opposing β‐sheets (ABED and GFCC'C’’; Fig 1A). HopQ binds the GFCC'C” interaction surface of C1ND through hydrophobic and H‐bond contacts with HopQ loop CL1 (connecting Cys 103 to Cys 132), the loop connecting CL1 to Helix 4 (CL1‐H4; residues 133–159) and CL2 (connecting Cys 238 to Cys 270; Figs 1A and EV1B). While Hop adhesins like SabA, BabA, and LabA are lectins (Ilver et al, 1998; Mahdavi et al, 2002; Rossez et al, 2014), HopQ was found to show a glycan‐independent interaction with CEACAMs (Javaheri et al, 2016). In agreement with this, the three N‐glycosylation sites in C1ND are positioned away from the HopQ interaction surface (Fig EV1D), and the full 1,024 Å2 interaction surface is made up of protein–protein contacts. Comparison of the bound and unbound HopQAD structures shows a strong conformational stability in the α‐helical core region, but reveals a conformational rearrangement and a disorder–order transition in CL1 and CL1‐H4 loops upon C1ND binding (Fig EV1B and C). In the unbound HopQAD structure, residues 135–148 of the CL1‐H4 loop are too flexible to be observed in the electron density map. Upon complex formation, these residues become ordered, forming a 3‐residue helix (residues 147–149) that inserts into the concave surface formed by the GFCC'C’’ β‐sheet of C1ND and creating an ordered loop (residues 134–145) that wraps around strand G at the edge of the IgV β‐plate (Figs 1A and B, and EV1B and D). In the CL1‐H4 helix, Thr149 contributes 3 H‐bonds with C1ND residues Gln44, Tyr34, and Ser32; and in the loop region, Ser135‐Thr136‐Asn137‐Ser138 contribute five H‐bonds with the C1ND strand G (Asn97 and main chain of L95; Figs 1B and EV2A). In CL1, the loop connecting strands S1 and S2 reaches over CL1‐H4 and forms an additional H‐bond through Tyr106 and Thr56 in C1ND strand C” (Fig EV2A). The conserved hydrophobic residues Phe29, Ile91, and Leu95 in C1ND are bound by a hydrophobic platform created by Ile102 (in loop connecting H3 and CL1), Ile240, and Ile242 in CL2, and L150 in CL1‐H4 (Figs 1B and EV2A). This gives HopQAD the appearance of an outstretched hand in which CL2 forms the palm that supports the hydrophobic tip of C1ND, and where CL1 and CL1‐H4 loops form the fingers and thumb that grab onto C1ND GFCC'C’’ β‐sheet (Figs 1A, and EV1B and D). The induced folding of the CL1‐H4 loop during HopQ–C1ND complex formation is further stabilized by the restructuring of an H‐bond network of Asn113 and Gln115 that interact with S3 in apo HopQ, and reorient to bind His142 and Tyr144 in the CL1‐H4 loop during C1ND complex formation (Fig EV1C). Strands S3 and S4 form a two‐stranded β‐hairpin domain that is inserted between helices 4 and 5, in an equivalent position to a 4 stranded β‐sheet that forms the blood group antigen binding site in BabA (Hage et al, 2015; Moonens et al, 2016). S3‐S4 removal was previously found to reduce HopQ‐binding activity approximately 10‐fold (Javaheri et al, 2016). The HopQ–C1ND X‐ray structure revealed that S3‐S4 is not directly implicated in the C1ND interaction, but shows that S3‐S4 interaction with S1‐S2 helps the ordering of the CL1 and CL1‐H4 loops into their binding conformation (Figs 1A and EV1C).
Figure 1. Structure of HopQAD ‐I bound to the N‐terminal domain of CEACAM1.
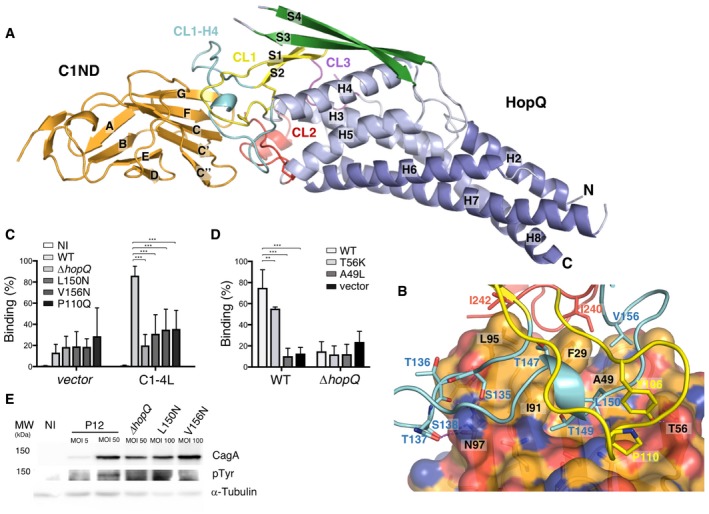
- Ribbon representation of the N‐terminal domain of CEACAM1 (C1ND; orange) bound to type I HopQ adhesin domain (HopQAD‐I). The adhesin binds the GFCC'C’’ sheet of its immunoglobulin‐like fold. HopQAD‐I shows the conserved 3 + 4‐helix bundle topology (α‐helices colored in light blue and blue) with three Cys‐bound loops CL1 (Cys102–Cys131), CL2 (Cys237–Cys269) and CL3 (Cys361–Cys384) colored in yellow, red, and purple, respectively. In the complex, the flexible CL1‐H4 loop (colored cyan) becomes structured upon C1ND binding. The β‐strands of the HopQAD‐I insertion domain are colored in green.
- Close‐up of the interaction surface between C1ND (ribbon and transparent surface representation) with HopQAD‐I (ribbon representation). Coloring scheme as in (A); carbon, nitrogen, and oxygen atoms of C1ND are, respectively, in orange, blue, and red. Interacting residues of HopQAD‐I and C1ND are depicted as stick model and named (residues in C1ND are labeled in black).
- Binding of H. pylori P12 wild‐type and HopQ mutants to MKN28 cells transfected with full‐length CEACAM1 (CC1‐4L) or empty vector. n = 3. Error bars = SD. Two‐way ANOVA with Bonferroni post‐test. ***P < 0.001.
- Binding of H. pylori P12 wild‐type and ∆hopQ to CHO cells transfected with CC1‐4L, CC1‐4L mutants or empty vector. n = 4. Error bars = SD. Two‐way ANOVA with Bonferroni post‐test. **P < 0.01. ***P < 0.001.
- Western blots of phospho‐Tyr, CagA, and loading control alpha‐tubulin in MKN28 CEACAM1‐4L cells infected at indicated multiplicity of infection (MOI) for 6 h with the wild‐type P12 or P12 ∆hopQ complemented with indicated HopQ point mutants. CagA translocation is indicated by the appearance of a phosphorylated product running just below 150 kDa.
Source data are available online for this figure.
Figure EV1. The CL1‐H4 loop of HopQAD ‐I becomes structured upon C1ND binding.
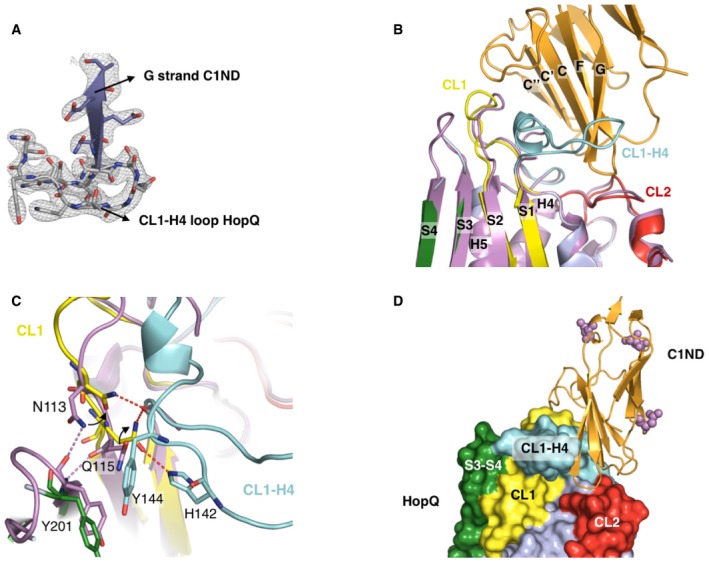
- Electron density map contoured at 1.2 σ around the interaction interface of the HopQAD‐I‐C1ND complex. The G strand of C1ND and the CL1‐H4 loop of HopQAD‐I are depicted.
- Comparison of the apo (purple) and C1ND (light blue; C1ND in orange) bound structures of HopQAD‐I. The CL1‐H4 loop is partially unstructured in the apo structure with no observable electron density.
- Overview on the structural rearrangements occurring around the CL1‐H4 loop between apo and C1ND bound HopQAD‐I. Upon C1ND binding, the CL2‐H4 loop becomes ordered and Asn113 and Gln115 reorganize to interact with the CL2‐H4 loop. Hydrogen bonds depicted by dashed lines.
- Overview on the HopQAD‐I‐C1ND complex with potential C1ND N‐glycosylation sites depicted by purple spheres.
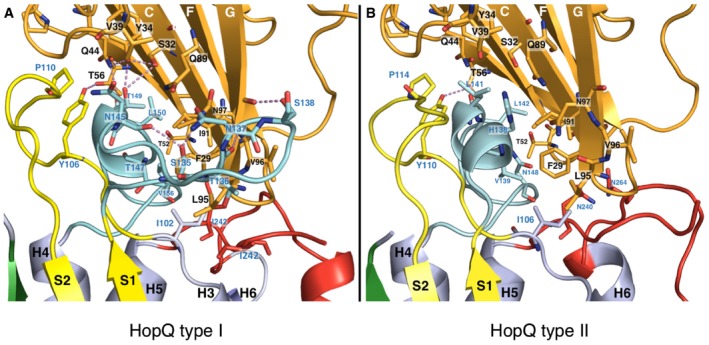
Figure EV2. Comparison of C1ND interacting with either HopQAD ‐I or HopQAD ‐ II .
-
A, BClose‐up on the formed interactions between HopQAD‐I and C1ND (A) and HopQAD‐II and C1ND (B). Interacting residues are shown in stick model and indicated (blue for HopQ residues, black for C1ND residues) with oxygen and nitrogen atoms, respectively, colored in red and blue. C1ND is colored in orange, CL1 in yellow, CL2 in red, and CL1‐H4 in cyan.
To confirm that the binding interface between HopQ and CEACAM1 as observed by X‐ray crystallography corresponds to the CEACAM‐binding paratope on the bacterial cell surface, we generated a number of H. pylori strains bearing directed point mutations in hopQ predicted to disrupt the HopQAD‐C1ND contact and performed binding assays. In the HopQ CL1, the conserved Pro110 was mutated to Gln to create a steric clash with C1ND Gln44, or in the CL1‐H4 loop of HopQ, Leu150 or Val156 was mutated to Asn to insert a sterically bulky polar group in the hydrophobic binding interface with C1ND. MKN28 control and MKN28‐CC14L cells were infected with the different mutant strains, and bacterial binding to the epithelial cells was assessed by flow cytometry. While wild‐type H. pylori P12 efficiently bound to CEACAM1‐4L‐expressing MKN28 cells, exchange of single amino acid residues in HopQ–CEACAM1 interface (Leu150Asn, Val156Asn, Pro110Gln) led to a significant reduction in binding, comparable to the background binding levels of a hopQ deletion mutant (Fig 1C). In the absence of CEACAM1, we barely observed bacterial binding independent of the hopQ status (Fig 1C). A hallmark of H. pylori infection is the translocation and phosphorylation of the CagA oncoprotein into host cells, a process shown to depend on HopQ‐mediated adherence (Javaheri et al, 2016; Koniger et al, 2016). Unlike for wild‐type P12, we could not detect CagA translocation when infecting MKN28 CC1‐4L cells with the HopQ point mutants, suggesting functional knockdown of the HopQ–CEACAM interaction and associated virulence pathways (Fig 1E). We note that compared to wild‐type P12, the tested HopQ mutant showed somewhat reduced HopQ expression levels (Appendix Fig S2) so that HopQ loss of function in the biological assays may in part have resulted from reduced binding avidity. Nevertheless, purified HopQAD‐I mutant Leu150Arg lacked any detectable binding to C1ND in solution when analyzed by ITC, showing the functional importance of Leu 150 for the HopQ–CEACAM1 interaction (Fig 2, Appendix Fig S2). Next, MKN28 cells were transfected with CEACAM1 in which we mutated Thr56 to Lys or Ala49 to Leu to create a possible steric clash in the HopQ‐binding interface. While MKN28 cells expressing the CEACAM1 Thr56Lys mutant showed a ~20% reduction in bacterial binding compared with WT CEACAM1, bacterial binding to cells expressing the Ala49Leu mutant dropped to background levels seen for cells transfected with empty vector or cells exposed to bacteria lacking hopQ (Fig 1D), thus confirming the functional importance of Ala49 for the interaction with the CL1‐H4 loop.
Figure 2. Comparison of C1ND bound structures of HopQAD ‐I and HopQAD ‐ II .
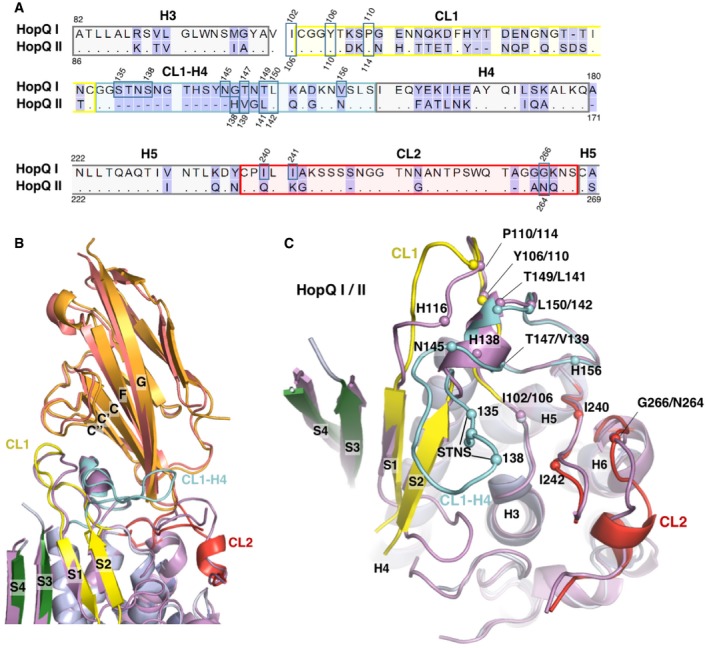
- Sequence alignment of HopQAD‐I and HopQAD‐II. Secondary structure elements, CL1, CL2, and the CL1‐H4 loop are indicated and boxed.
- Overlay of the C1ND bound structures of HopQAD‐I (purple; C1ND in orange) and HopQAD‐II (blue; C1ND in deep salmon). HopQAD‐II CL1 is colored in yellow, the CL2 in red, CL1‐H4 in cyan, and β‐strands of the insertion domain in green.
- Top view of the overlay of HopQAD‐I and HopQAD‐II (C1ND removed for clarity). The CL1‐H4 loop is missing in HopQAD‐II, and minor additional structural rearrangements between HopQAD‐I and HopQAD‐II are observed in the CL2 and H3‐S1 loop. Corresponding CEACAM‐interacting residues of HopQAD‐I and HopQAD‐II are shown as a sphere and labeled; color scheme is as in (B).
Allelic variation in HopQ–C1ND interaction
Phylogenetic analysis of clinical isolates shows that H. pylori genomes can hold one or both of two distinct families of hopQ alleles, with adhesin domains that share an average 70% identity on the amino acid level (Fig 2A; Cao & Cover, 2002). Type I HopQ is most prevalent in Eastern type strains with cagA‐positive and S1 vacA genotypes, while HopQ type II is found more commonly in Western strains with cagA‐negative, vacA S2 genotype. To gain insight into the possible differences in CEACAM binding of type I and II HopQ, we determined the X‐ray structure of type II HopQAD bound to C1ND (type I and II HopQ adhesin domains are referred to as HopQAD‐I and HopQAD‐II hereafter). The overlay of both complexes reveals HopQAD‐II targets the same binding site on the C1ND surface as the type I allele (Fig 2B). Although C1ND undergoes a small 5° rotation in relation to HopQAD‐I or HopQAD‐II, no significant conformational changes are observed in C1ND (with average root‐mean‐squared deviation (RMSD) for equivalent Cα atoms of 0.427). On the contrary, comparison of type I and type II HopQ reveals significant changes in the binding interface region (Figs 2B and C, and EV2B). In HopQAD‐II, the CL1‐H4 loop is shortened by 11 residues (Fig 2). The type II CL1‐H4 loop retains and extends the short α‐helical element by 2 residues, but this directly connects to the S2 strand rather than forming the loop region that grabs around the edge of the C1ND β‐plate, as seen in HopQAD‐I. In the α‐helical element, Thr147 and Thr149 are replaced with Val and Leu, respectively, so that a total of 7 H‐bond interactions are lost in the type II CL2‐H4–C1ND interaction (Figs 2C and EV2B). In CL1, the H‐bond interaction of Tyr106/110 (HopQ I/II) with C1ND Thr57 is conserved, as is the presence of Pro in position 110/114, while in CL2, the hydrophobic platform formed by Ile240 and Ile242 in HopQAD‐I is replaced in HopQAD‐II by a H‐bond interaction of Asn264 with the backbone carbonyl of C1ND Ser93 (Figs 2C and EV2B). A total of seven hydrogen bonds are lost in the HopQ type II‐C1ND complex compared to the HopQ type I‐C1ND complex. Unexpectedly, however, the affinity of HopQ type II for C1ND is around sixfold higher when compared to the affinity of HopQ type I for C1ND (K D values of 417 and 69 nM for type I and type II, respectively, determined by isothermal titration calorimetry; Appendix Fig S2). In agreement with the net loss of hydrogen bonds in the type II complex, the thermodynamic signature of the interaction shows a reduced enthalpic contribution compared to that seen in the type I complex (−4.4 and −16.7 kcal/mol resp.). However, this is compensated by a strongly favorable entropic binding contribution of 18.1 cal/mol/deg, unlike the HopQ type I interaction, which is associated with a negative entropic contribution of −26.8 cal/mol/deg, likely due to the disorder–order transition in the CL1‐H4 loop (Appendix Fig S2). These thermodynamic data suggest that in HopQ type II, the shortened CL1‐H4 loop is likely pre‐structured prior to C1ND binding.
HopQ specificity toward human CEACAMs
Humans and higher primates represent the only host for H. pylori, and the bacterium has had a long history of co‐evolution with humans (Atherton & Blaser, 2009). We previously found that type I HopQ bound human, but not mouse CEACAM1, as well as human CEACAM3, 5, and 6, but not human CEACAM8 (Javaheri et al, 2016). The structures of the type I and II HopQ bound to human C1ND now allow a closer inspection of the molecular determinants of the HopQ‐binding tropism. Multiple sequence alignment and superimposition of the structures of human CEACAM1, 3, 6, and 8 (2QSQ, 4Y8A, 4Y88; Korotkova et al, 2008; Bonsor et al, 2015) show the high degree of sequence and structural conservation in the N‐domain (rmsd values ranging from 0.49 to 0.58 Å; only backbone atoms; Fig 3A and B). Within the HopQ‐binding interface, Ser32, Gly41, Gly51, Ile91, and Val96 are conserved in CEACAM1, 3, 5, and 6, while Ala49 is replaced by the more bulky hydrophobic residue Val in CEACAM3, 5, 6, and 8 (Fig 3A). Mutation of Ala49 to Val in C1ND increased the binding affinity for HopQAD‐I, from a dissociation constant of 416.7 ± 48.4 to 252.5 ± 29.6 nM (Fig EV3C). In CEACAM 6, Phe29 is replaced by the similarly sized hydrophobic residue Ile, and in CEACAM3 and 6, a hydrogen bond with HopQ CL1‐H4 is lost by substitution of Gln44 to Leu (Fig 3A). In CEACAM 8, which does not support C1ND binding (Javaheri et al, 2016), each of these seven interface residues is mutated, introducing bulkier side chains in the HopQ–C1ND interface. This results in steric hindrance between CEACAM8 and HopQ, reflected by a significantly higher calculated MolProbity clash score between HopQ I and CEACAM8 in the binding interface (73.6; calculated across HopQ residues 102–159 and 237–273, and CEACAM residues 26–60 and 90–100) when compared to the clash scores of the HopQ–C1ND structure (9.94) or the modeled HopQ–CEACAM3 (26.5) and HopQ–CEACAM6 (18.1) complexes. Although targeting the same CEACAM epitope, the structure of the HopQ type I and II paratope and the thermodynamic signature of the CEACAM‐binding interaction is very different (Fig EV2, Appendix Fig S2A and B). Both proteins showed similar binding levels to MKN45 cells, a gastric adenocarcinoma cell line expressing CEACAM1, 5, and 6 (Javaheri et al, 2016), but did not bind MKN28 cells, which lack surface‐exposed CEACAMs (Fig 3C). HopQAD‐I and HopQAD‐II both retained similar binding levels to MKN45 at pH 7 or pH 2, showing that, unlike the BabA adhesin, HopQ‐binding is resistant to low pH conditions (Fig 3D; Bugaytsova et al, 2017).
Figure 3. Specificity of HopQ toward human CEACAMs.

- Superimposition of HopQAD‐I‐C1ND (blue; CL1 in yellow, CL2 in red, and CL2‐H4 in cyan, β‐strands of the insertion domain in green) and HopQAD‐II‐C1ND (gray) with the structures of CEACAM3‐N (PDB:2QSQ; salmon), CEACAM6‐N (PDB:4Y8A; purple), and CEACAM8‐N (PDB:4Y88; green).
- Binding of purified HopQAD‐I or HopQAD‐II to MKN45 cells naturally expressing CEACAM 1, 3, and 6.
- Binding of purified HopQAD‐I or HopQAD‐II to MKN45 at pH 7 or pH 2.
- Binding of H. pylori strains P12 and TX30 to CHO cells transfected with human CEACAM1, 3, 5, 6, 8, or a vector control. Strains P12 and TX30 hold and express the type I and II HopQ alleles, respectively. The binding percentage for P12 and TX30 was normalized to that of P12 ∆HopQ (not shown) to present binding as a fold over the control P12 ∆HopQ. n = 3. Two‐way ANOVA with Bonferroni post‐test **P < 0.01. ***P < 0.001. Error bars = SD.
- Binding of purified HopQAD‐I and HopQAD‐II protein to CHO cells transfected with human CEACAM1, 3, 5, 6, 8 or a vector control.
- Binding of different H. pylori strains to CHO cells transfected with human CEACAM1, 6, or vector control. H. pylori strains P12, G27, J99, and 2808 hold and express the type I HopQ allele, while strains 60190 and TX30 express the type II allele. Fold binding is defined as in panel (C). n = 2, except 60190 where n = 1. Error bars = SD.
Figure EV3. Species specificity of HopQ type I.
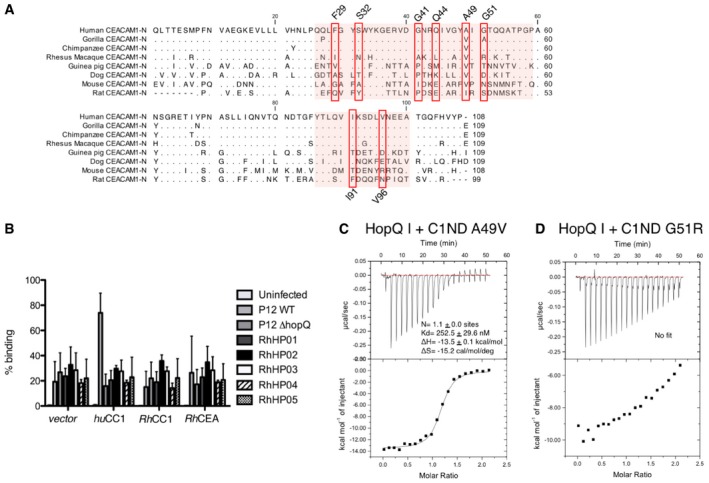
-
ASequence alignment of the N‐terminal domain of human CEACAM1 (P13688), chimpanzee CEACAM1 (H2QGG8), gorilla CEACAM1 (G3S247), rhesus macaque CEACAM1 (F7BJE0), guinea pig CEACAM1 (H0WD59), mouse CEACAM1 (P31809), dog CEACAM1 (Q004B2), rat CEACAM1 (P16573). The GFCC'C’’ sheet of CEACAM1‐N that interacts with HopQ is indicated by a red background. Residues of CEACAM1 shown to interact with HopQ are indicated by red boxes.
-
BBinding of H. pylori strain P12 wild‐type and P12 ∆HopQ, as well as several isolates from rhesus macaque to CHO cells transfected with human CEACAM1, rhesus macaque CEACAM1, CEA, or a vector control (n = 6. error bars = SD).
-
C, DIsothermal titration calorimetry (ITC) injection heats (upper) and normalized binding isotherm (lower) of C1ND A49V (C) or G51R (D) titrated with HopQAD‐I. Binding affinity and thermodynamic profile are shown below. Black line shows nonlinear regression fit of raw data.
To determine whether the allelic structural changes in the HopQ‐binding paratope result in different receptor specificities, transfected CHO cells expressing either CEACAM1, 3, 5, 6, or 8 were probed by flow cytometry with labeled H. pylori strains P12 or TX30, carrying type I and II HopQ, respectively (Fig 3E, Appendix Fig S3A). These data show that both strains bind CEACAM1, 3, and 5 with similar affinities, but that unlike TX30, P12 shows additional binding to CEACAM6. However, when probed with purified HopQAD‐I or HopQAD‐II robust binding was seen for CEACAM1, 3, or 5 expressing CHO cells, but no binding was apparent for CEACAM6 expressing cells (Fig 3F). Nevertheless, a loss of binding in P12 ΔhopQ showed that the adherence of the P12 strain to CEACAM6 expressing cells was dependent on HopQ, suggesting that the HopQ CEACAM6 interaction requires full‐length HopQ in its native context of the H. pylori outer membrane (Fig 3C and E). Binding assays with additional H. pylori strains carrying the type I (strains P12, G27, J99, and 2808) or the type II hopQ allele (strains TX30 and 60190) showed all strains bind CEACAM1‐transfected CHO cells, while the type I HopQ carrying strains P12, G27, and 2808, but not J99 also bound CEACAM6‐expressing CHO cells (Fig 3G). Thus, HopQ type I and II alleles differ in their ability to bind CEACAM6, although unlike CEACAM1 binding, the CEACAM6 binding capacity of the type I alleles was found to be polymorphic among H. pylori isolates, with some strains lacking any detectable CEACAM6 binding. Analyzed HopQ type I alleles showed an average 85% pairwise sequence identity in the adhesin domain. It is unclear at present which regions in non‐binding type I polymorphs or the type II alleles discriminate against CEACAM6 binding. Based on sequence conservation in the HopQ‐binding epitope (Fig 3A), it is also unclear why CEACAM6 would not form a general HopQ receptor as is the case for CEACAM1, 3, and 5.
We next compared the sequence conservation in the CEACAM1 N‐domain in rat, mouse, dog, guinea pig as well as the rhesus macaque, which is a frequently used primate infection model for H. pylori disease, and our close relatives chimpanzee and gorilla (Fig EV3). Except for the binding enabling mutation Ala49Val and Gly51Ala in gorilla, the interface residues in chimpanzee and gorilla CEACAM1 N‐domain are fully conserved. In the rhesus monkey, Phe29, Ser32, Gly41, Gln44, and Ala49 are replaced with Ile, Asn, Ala, Leu, and Val, respectively. In addition, RhC1ND holds the Gly51Arg mutation, here shown to result in a dramatic reduction in HopQ‐binding affinity (Fig EV3D). To test whether HopQ derived from human clinical isolates could bind rhesus CEACAM1, we performed flow cytometric binding studies of H. pylori‐mediated adherence to MKN28 cells expressing human CEACAM1, or rhesus CEACAM 1 or 5. The human isolate P12 showed robust binding to human CEACAM1 only, an interaction that was lost in its isogenic hopQ knockout mutant (Fig EV3B). Also in rhesus‐adapted strains, that is primary isolates from H. pylori‐infected macaques, no binding to rhesus CEACAM1 or 5 was seen (Fig EV3B). Strikingly, these strains also lacked binding to MKN28 cells expressing human CEACAM1, despite their hopQ‐positive genotype.
HopQ induces CEACAM1 monomerization
CEACAM1 is involved in homo‐ and heterophilic cis‐ (among CEACAMs on a single cell) and trans‐(across cells) dimerization interactions that are important for propagating signals from the cell exterior to the cytosol of the cell (Gray‐Owen & Blumberg, 2006; Klaile et al, 2009; Müller et al, 2009). While same‐cell, cis oligomerization involves the C1ND surface formed by β‐sheet ABED, cross‐cell trans dimerization of C1ND has been demonstrated to be mediated through the non‐glycosylated GFCC'C’’ sheet of the CEACAM1 N‐terminal domain (Watt et al, 2001; Klaile et al, 2009; Fig 4A), the same surface here seen to form the HopQ‐binding epitope. Superimposition of the crystallographic C1ND trans or cis dimer (PDB identifiers 4WHD and 2GK2, respectively; Fedarovich et al, 2006) with the HopQ–C1ND complex shows HopQ binding is incompatible with CEACAM1 trans dimerization (Fig 4B). To verify the oligomeric state of our C1ND preparations, we performed SAXS measurements on soluble C1ND. Comparison of the experimental scattering data with the theoretical scattering curves for monomeric, or cis or trans dimeric C1ND reveal the CEACAM1 N‐domain is present as a trans dimer at the high micromolar protein concentrations used in our study (Fig 4C). In agreement with the crystallographic HopQAD‐C1ND complexes, solution scattering curves of the two proteins show them present in a 1:1 stoichiometric complex as seen in the crystals (Fig 4D). Together these data reveal C1ND is present as a trans dimer in solution, and that in the presence of HopQ, the C1ND dimer is lost in favor of the HopQ–C1ND complex. These findings suggest that C1ND trans dimers are in fast equilibrium with the monomeric form or that HopQ can disrupt the C1ND trans dimer during its binding process. To evaluate whether HopQ binding induces dimer to monomer conversion of full‐length CEACAM1 exposed on the host cell surface, we monitored CEACAM1 cross‐linking by the bifunctional amine cross‐linker dithiobis (succinimidyl propionate) in non‐infected MKN28 CEACAM1‐4L cells, as well as cells infected with H. pylori strain P12 or P12 ΔHopQ (Fig 4E and F). These data show that infection with P12, but not P12 ΔHopQ results in a reduction of cross‐linking efficiency in CEACAM1. Thus, in vitro and in vivo data show that HopQ‐dependent adherence induces dimer to monomer conversion of host CEACAMs.
Figure 4. HopQ type I is able to monomerize CEACAM1‐N in solution.

- Ribbon and schematic representation of the crystallographic C1ND cis and trans dimers (PDB codes 2GK2 and 4WHD, respectively). In trans, C1ND dimers the monomers contact each other through their GFCC'C’’ sheets, whereas in cis dimers, contacts are made through the ABED face.
- Superimposition of the crystallographic trans and cis C1ND dimers with the HopQAD‐I‐C1ND structure. Steric clashes would prevent the trans dimer from interacting with HopQAD‐I.
- SAXS profile of E. coli produced C1ND (data points indicated by red dots) superimposed with the theoretical scattering curves of the C1ND monomer, cis C1ND dimer, and trans C1ND dimer (green, blue and black, resp.).
- Experimental SAXS scattering curve of the HopQAD‐I‐C1ND complex (4 mg/ml) superimposed with the theoretical calculated scattering curve of the HopQAD‐I‐C1ND structure (red curve) by CRYSOL (Svergun et al, 1995). The inset shows a typical DAMMIF ab initio shape determination superimposed with the HopQAD‐I‐C1ND complex by SUPCOMB (Kozin & Svergun, 2001).
- Western analysis of CEACAM1 on CHO cells transfected with CEACAM1 and treated with dithiobis(succinimidyl propionate) (DSP)‐cross‐linker after infection with P12, P12 ΔHopQ or buffer control (uninfected). Total CC1 represents CEACAM1 run under reducing conditions, and cross‐linked CC1 corresponds to a CEACAM1 dimer after cross‐linking with DSP.
- Quantitative measure of the CEACAM1 cross‐linking efficiencies in DSP‐treated cells upon infection P12, P12 ΔHopQ, or buffer control (Uninfected). Data are shown as relative cross‐linking efficiencies as percent CEACAM1 cross‐linking compared to uninfected cells (n = 3; error bars = SD).
Source data are available online for this figure.
CEACAM1 trans dimerization influences outside—in signaling likely by enhancing cis oligomerization on the cell surface and thereby clustering of the receptors’ cytoplasmically localized ITIMs of CEACAM1‐L (Gray‐Owen & Blumberg, 2006; Klaile et al, 2009; Müller et al, 2009). To determine whether ITIM clustering is required for the HopQ‐dependent translocation and phosphorylation of CagA (CagAPY), the formation of CagAPY was monitored in MKN28 cells transfected with full‐length CEACAM1 (CC1‐4L) or a splice form that lacks the cytoplasmic ITIM sequences (CC1‐4S; Gray‐Owen & Blumberg, 2006; Klaile et al, 2009; Müller et al, 2009). To first investigate whether the 4L or 4S CEACAM1 receptors influence the level of HopQ‐mediated H. pylori binding to the MKN28 cells, we measured their interaction under live‐cell conditions. The LigandTracer system was used to monitor the interaction of live and motile H. pylori G27 wild‐type (wt) or its isogenic ΔhopQ mutant with MKN28 wt cells, which are CEACAM‐negative, as well as MKN28 cells stably expressing either the CC1‐4S or CC1‐4L receptor, respectively (Fig 5A and B). The experiments with the MKN28‐CEACAM1‐4S and ‐4L cells revealed equally strong binding of HopQ carrying wt bacteria as judged by fluorescence signal intensity in the binding curve over time, and almost no interaction with the ΔhopQ mutant (Fig 5A and B). This demonstrates directly that expression of HopQ is sufficient and necessary for H. pylori to bind to the corresponding CEACAM receptor. The curves for the MKN28‐CEACAM1‐4S and ‐4L cells are highly similar for HopQ‐expressing H. pylori wt, demonstrating it binds comparably well to both CEACAM isoforms, while the HopQ‐deficient bacteria cannot (Fig 5A and B). As a control, the CEACAM‐negative MKN28 wt cells exhibited significantly reduced signals and a similar flat binding curve for both the wt and the ΔhopQ mutant H. pylori G27 (Fig EV4). Next, the level of CagA translocation and phosphorylation of two H. pylori clinical isolates was monitored on MKN28 cells expressing either the CC1‐4S or CC1‐4L (Fig 5C). Anti‐phosphotyrosine immunoblots show that the translocation and phosphorylation of CagA require expression of CEACAM1 in MKN28 and reveal that CC1‐4S and CC1‐4L result in similar levels of CagAPY, demonstrating that CagA translocation and phosphorylation do not require the presence of the ITIM domain or ITIM‐dependent signaling pathways (Fig 5C).
Figure 5. The cytoplasmic ITIM containing domain of CEACAM1 is not essential for CagA translocation and phosphorylation.

-
A, BAdhesion of H. pylori strain G27 wild‐type or its isogenic ΔhopQ mutant to MKN28 cells stably expressing either CEACAM1‐4 with (A; CC1‐4L) or without (B; CC1‐4S) cytoplasmic ITIM sequences, assayed by the LigandTracer Green System (n = 3; error bars = SD).
-
CAnti‐phosphotyrosine immunoblots of AGS cells (positive control) and MKN28 cells expressing either empty vector, CC1‐4S, or CC1‐4L that were uninfected (mock) or infected with H. pylori strains Ka88 or HPAG1. Anti‐CagA staining of the Ka88 or HPAG1‐infected MKN28‐C1‐4S cells is shown as control and reference for CagA position relative to the MW marker.
Source data are available online for this figure.
Figure EV4. Helicobacter pylori G27 interacts with MKN28 cells expressing CEACAM5.
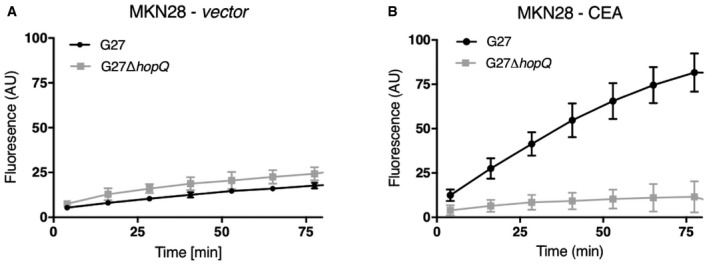
-
A, BAdhesion of H. pylori strain G27 wild‐type or its isogenic ΔhopQ mutant to MKN28 cells transfected with either a control vector (A) or CEACAM5 (B) by the LigandTracer Green System. (n = 3; error bars = SD).
Discussion
Several mucosa‐associated bacterial pathogens interact with CEACAM family proteins found on mucosal surfaces of the human body. The Neisseria gonorrhoeae and N. meningitidis Opacity (Opa) proteins (Virji et al, 1996), Haemophilus influenza outer membrane protein P1 (Tchoupa et al, 2015), Moraxella catarrhalis ubiquitous surface protein A (UspA; Hill & Virji, 2003), the E. coli type 1 and Afa/Dr pilus adhesins (Leusch et al, 1991; Berger et al, 2004; Barnich et al, 2007), and the H. pylori HopQ proteins (Javaheri et al, 2016; Koniger et al, 2016) are structurally different, non‐homologous adhesins that all target CEACAM receptors on the apical side of mucosal cells (Tchoupa et al, 2014). Here, we report on the structure of the H. pylori adhesins HopQ type I and II in complex with the N‐terminal domain of human CEACAM1 (C1ND), a first example of a H. pylori outer membrane adhesin bound to a protein receptor in a glycan‐independent manner. Although type I and II HopQ only share an average 70% amino acid identity in the adhesin domain, they bind the same epitope on the CC'C”FG sheet of human CEACAMS, albeit with a drastically different paratope and thermodynamic binding signature. Whereas the HopQ type I–C1ND interaction is enthalpically driven and entropically disfavored, the HopQ type II–C1ND binding is a largely hydrophobic, entropically driven interaction. The CC'C”FG sheet is the non‐glycosylated face of C1ND and forms the trans dimerization interface in homo‐ and heterophilic CEACAM interactions. Strikingly, available structures or mutational analyses show that this same interface is also the binding epitope of the E. coli Dr adhesins (Korotkova et al, 2008), as well as Neisseria Opa proteins, M. catarrhalis UspA and H. influenza P1 (Villullas et al, 2007). Thus, it appears that these diverse adhesins and pathogens have converged on a common binding interface in their human CEACAM receptors.
The interaction of pathogens with host CEACAMs and the diversification of some of these human CEACAMs are thought to reflect a bilateral struggle for survival and eons of co‐evolution between bacteria and host. CEACAM1 forms an attractive target receptor for pathogenic bacteria as it is expressed on most epithelial cells, thereby enabling the pathogens to gain a foothold in the host. Also, CEACAM1 expression on leukocytes allows the pathogen to interfere with host immune responses by inducing ITIM‐dependent signaling (Gray‐Owen & Blumberg, 2006). The human host on the other hand can react either by genetic adaptation of the HopQ‐binding epitopes in the targeted receptors, but also by the expression of decoy receptors, which can take the form of soluble decoys that compete with bacterial binding of cell‐associated receptors, or can be expressed as decoy receptors on pathogen‐clearing immune cells. For example, Neisseria binds to CEACAM1 but also to GPI‐anchored CEACAM5 and CEACAM6 which can be released from the cell surface (Kolla et al, 2009; Klaile et al, 2013). Although the N‐domain has some specific amino acid substitutions in the different CEACAMs (Fig 3A), it is sufficiently conserved that most bacterial CEACAM1‐binding adhesins will bind multiple CEACAMs. Neisseria also interacts with granulocyte‐specific human CEACAM3 that contains an N‐terminal domain closely related to human CEACAM1 but it carries an immunoreceptor tyrosine‐based activation motif (ITAM) in its cytoplasmic domain, thereby promoting uptake and destruction of the pathogens by granulocytes (Schmitter et al, 2004). ITAM is the antagonistic equivalent of ITIM, in that it relays activating signals whereas the latter has an inhibitory role. Here, CEACAM3, which has arisen after CEACAM1 in mammals (Kammerer & Zimmermann, 2010), can be interpreted as an evolutionary host response to pathogens binding CEACAM1, 5, and 6, facilitating their removal. A second example is canine CEACAM28 that bears high sequence identity to canine CEACAM1 but acts as a decoy receptor. Canine CEACAM28 is expressed by cells of the immune system and has, like human CEACAM3, a functional cytoplasmic ITAM‐containing domain that is important for leukocyte‐mediated immune responses (Kammerer et al, 2007). H. pylori HopQ type I was found not only to interact with human CEACAM1 but as well with CEACAM3, CEACAM5, and CEACAM6 (Javaheri et al, 2016). Indeed, when superimposing the previously deposited structures of CEACAM3 and CEACAM6 on the HopQ I‐C1ND co‐complex structure, it becomes clear that changes between the different CEACAMs do not interfere with HopQ binding thus allowing CEACAM 3, 5, and 6 to act as decoy receptors. Interestingly, type II HopQ as well as individual polymorphs of the type I allele were found to discriminate against CEACAM6 binding, while retaining high‐affinity binding of human CEACAM1. CEACAM6 can be found as cell‐associated receptor on human epithelia in lung and the gastrointestinal tract, including esophagus and stomach (Uhlén et al, 2015). It is also found in spleen and lymph nodes, however, and at least in lung, it was shown to be abundantly secreted as soluble receptor (Kolla et al, 2009; Klaile et al, 2013). It is tempting to speculate that type II HopQ and some type I HopQ may be discriminating against CEACAM6 binding to avoid deleterious binding interactions. The HopQ‐binding epitopes in CEACAM1 and 6 are highly similar such that it is unclear at present how these non‐binding HopQ polymorphs can discriminate against CEACAM6 binding.
The selective pressure of host–pathogen co‐evolution may also form a driving force behind the sequence diversification of CEACAM1 (especially the CC'C”FG face, Fig EV3A) across different species (Voges et al, 2010). HopQ type I was demonstrated to be specific for human CEACAM1 (Javaheri et al, 2016), but non‐binding to mouse, canine, or bovine CEACAM1 orthologues. These findings could explain the highly efficient T4SS‐dependent translocation and phosphorylation of CagA in human MKN28 cells expressing CEACAM1 isoforms, while defects for CagA delivery have been detected for various non‐human cell lines (Backert et al, 2011). In addition, Voges et al could show that the Opa and UspA adhesins from pathogenic Neisseriae and Moraxella catarrhalis, respectively, displayed a high selectivity for human CEACAM1, reflecting humans as the preferred host niche for these pathogens. Changes in host CEACAMs that abolish the host–pathogen interaction are countered by changes in bacterial adhesins to restore binding in an ever‐continuing hide‐and‐seek. When aligning C1NDs of different species (Fig EV3A), only the C1ND of the closest to humans relatives, the great apes (e.g. gorilla, chimpanzee), reveals a sequence conservation in C1ND that would accommodate binding toward HopQ. In chimpanzee, C1ND only one amino acid is substituted in the binding site (Ala49Val) and when introducing this mutation an increase in affinity is observed (Fig EV3C), demonstrating chimpanzee CEACAM1 can interact with HopQ type I. However, when comparing with more distant species, the correlation in the CC'C”FG sheet on the amino acid level is lost, as is binding by HopQ. For example, in murine CEACAM1, the majority of residues on the CC'C”FG sheet are different compared to human CEACAM1 (Fig EV3). Both Ig‐like structures superimpose with an RMSD of 0.68 Å, revealing a high structural similarity between both. Like in CEACAM8, more bulky side chains are introduced in murine CEACAM1 as reflected by a significant higher MolProbity clash score (55.8), explaining why murine CEACAM1 is unable to interact with HopQ I.
In humans, 12 splice variants of CEACAM1 occur that vary in the number of Ig‐like extracellular domains, the presence of a membrane anchor, and the size of the cytoplasmic domain (Gray‐Owen & Blumberg, 2006). The four major expressed isoforms CEACAM1‐4L, CEACAM1‐3L, CEACAM1‐4S, and CEACAM1‐3S are composed out of a string of four or three glycosylated Ig‐like domains, a single‐pass transmembrane domain and followed by either a long (71 amino acids) or short (10 amino acids) cytoplasmic domain, respectively. These isoforms are co‐expressed but only CEACM1‐4L contains two functional ITIM motifs that, upon phosphorylation by Src family tyrosine kinases, can interact with cytoplasmic effectors such as protein tyrosine phosphatases SHP‐1 and SHP‐2, which are important for downstream inhibitory or stimulatory CEACAM1‐L related signaling events. Klaile et al (2009) could show the monomeric CEACAM1‐4 ectodomain is highly flexible with extended, kinked, and back‐folded shapes. Using a combination of chemical cross‐linking, molecular tomography and surface plasmon resonance the authors demonstrated the CEACAM1 ectodomain occurs as a complex equilibrium between monomers, dimers, trimers, and oligomers. Two types of dimers were distinguished that can participate simultaneously in homophilic binding: “C‐dimers” that are mediated only by the CC'C”FG sheet of C1ND and responsible for anti‐parallel intermembrane adhesion (Watt et al, 2001; Klaile et al, 2009); and “A‐dimers” that are formed by the close association of all Ig‐like domains in a parallel manner. Their data highlighted the important role of C1ND in the functional activity of CEACAM1 and proposed a model for adhesion‐induced signaling. First, C‐dimers mediate trans‐homophilic interactions during intercellular adhesion, which induces the formation of cis interactions on the cell surface by means of parallel A‐dimers and oligomers. This induced cis interactions bring the cytoplasmic domains together and alter the interactions with intracellular signaling molecules (Klaile et al, 2009). The monomer/dimer equilibrium of the cytoplasmic domain of CEACAM1‐L was found important for the association of effectors such as SHP‐1/SHP‐2, which preferably interact with dimeric over monomeric CEACAM1‐L, whereas tyrosine kinase c‐Src (Müller et al, 2009) does not exhibit preference toward any oligomeric state of CEACAM1‐L. Interestingly, the eukaryotic cell can self‐regulate the formation of dimeric CEACAM1‐L by the expression of a CEACAM1‐S isoform with a shortened cytoplasmic domain that lacks the ITIM domains. CEACAM1‐S is unable to interact with SHP‐1/SHP‐2 and c‐Src and will form CEACAM1‐L/CEACAM1‐S heterodimers that resemble functional CEACAM1‐L monomers in that they interact with lessened affinity with SHP‐1/SHP‐2 (Müller et al, 2009). Together, this points toward the importance of the CEACAM1 oligomeric state in the production of downstream signaling events. Using a combination of X‐ray crystallography and SAXS, we could demonstrate HopQ interferes with the formation of a CEACAM1‐N homodimer by interacting with the CC'C”FG face of C1ND and thereby preventing the formation of the above‐described C‐dimer that is responsible for mediating both trans and cis interactions. The K D for the formation of A‐ and C‐dimers based on the observed oligomeric species in molecular tomography was calculated to be in the 40–260 μM range (Klaile et al, 2009). Size exclusion chromatography and analytical ultracentrifugation demonstrated the N‐terminal domain of human carcinoembryonic antigen (CEA; 85% sequence identity to human CEACAM1‐N) forms a homodimer with a reported K d of 800 nM. These values are well above the here determined HopQ–C1ND interaction affinities and confirm HopQ is able to dissociate CEACAM trans dimerization, which will result in reduced CEACAM1 ITIM clustering and altered signal transduction.
The targeting and disruption of the CEACAM1 trans dimerization site by H. pylori HopQ enables this pathogen to interfere with CEACAM1‐mediated signaling by altering the equilibrium between oligomeric states (Fig 6). The HopQ–CEACAM binding was shown to facilitate the T4SS‐dependent intracellular injection and phosphorylation of the oncoprotein CagA by a yet unknown mechanism (Javaheri et al, 2016; Koniger et al, 2016). However, CagA delivery and activation are intact in CEACAM splice variants lacking the cytoplasmic ITIM domains, suggesting this step is independent of altered signaling cascades in the host cell. Whether CagA delivery is the primary virulence‐enhancing activity of HopQ or whether additional inhibitory or stimulatory events are triggered by altering CEACAM1‐dependent signaling through the HopQ–CEACAM interaction is presently unclear. Interestingly, CEACAM1 engagement by Neisseria adhesins also targets the trans dimerization site and led to expression of CD105 on the host cell surface, which prevented bacteria‐induced exfoliation and detachment of epithelial cells (Muenzner et al, 2005, 2010). In the human stomach, epithelial cells are continuously being shed, thereby exposing H. pylori cells to the acidic pH in the stomach. A similar mechanism of CEACAM1‐induced protection against defoliation would protect H. pylori from being exposed to the harsh environment of the stomach lumen, which merits further exploration.
Figure 6. Proposed model for Helicobacter pylori HopQ's interference with CEACAM1 cell–cell‐mediated adherence and downstream signaling events.
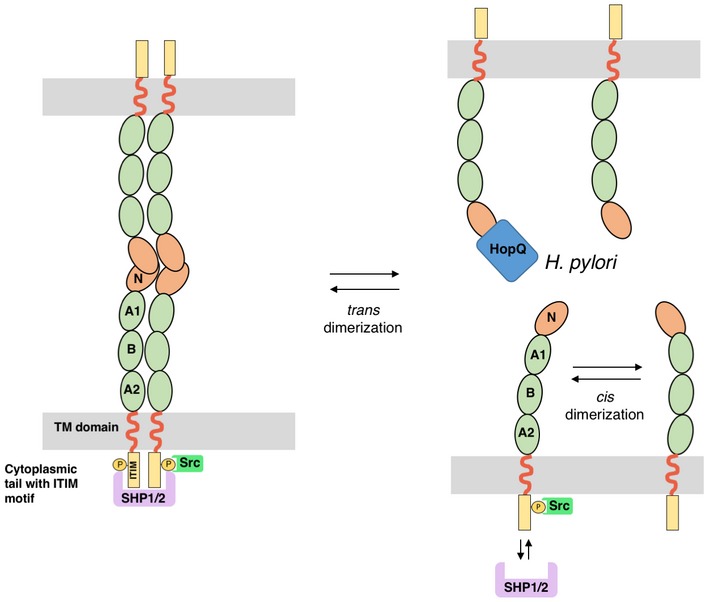
Intercellular CEACAM1 interactions are mediated by the formation of trans CEACAM1 dimers through the N‐terminal domain of CEACAM1 (CEACAM1‐N). On the cell surface, cis CEACAM1 dimers and oligomers can be formed by lateral interactions through Ig‐like domains and cis contacts in the N‐domain. This CEACAM clustering is required for the recruitment of cytoplasmic protein tyrosine phosphatases SHP1/2 to the cytoplasmic ITIM domains. HopQ is able and sufficient to monomerize dimeric CEACAM1‐N. The loss of trans CEACAM dimerization is known to destabilize cis dimer formation, thereby altering downstream signaling events by interfering with SHP1/2 but not Src recruitment to the cytoplasmic ITIMs (Klaile et al, 2009; Müller et al, 2009).
Materials and Methods
Production of wild‐type and mutant CEACAM1‐N
Cloning and purification of the N‐terminal domain of human CEACAM1 (C1ND) in Escherichia coli were described earlier (Javaheri et al, 2016). In brief, the amino acid sequence of CEACAM1 (residues 35–142, Uniprot ID: P13688) was codon optimized for expression in E. coli, synthesized by GeneArt de novo gene synthesis (Life Technologies) and cloned with a C‐terminal His6 tag in the pDEST™14 vector using Gateway technology (Invitrogen). The resulting construct was transformed in E. coli C43(DE3) cells and grown in LB supplemented with 100 μg/ml ampicillin at 37°C while shaking. At an optical density at 600 nm (OD600) of 1 C1ND expression was induced with 1 mM isopropyl‐β‐D‐thiogalactopyranoside overnight at 30°C. Cells were collected by centrifugation at 6,238 g for 15 min at 4°C and resuspended in 50 mM Tris–HCl pH 7.4, 500 mM NaCl (4 ml/g wet cells) supplemented with 5 μM leupeptin and 1 mM 4‐(2‐aminoethyl)benzenesulfonyl fluoride (AEBSF), 100 μg/ml lysozyme, and 20 μg/ml DNase I. Subsequently, cells were lysed by a single passage in a Constant System Cell Cracker at 20 kpsi at 4°C and debris was removed by centrifugation at 48,400 g for 40 min. The cytoplasmic extract was filtrated through a 0.45‐μm pore filter and loaded on a 5 ml pre‐packed Ni‐NTA column (GE Healthcare) equilibrated with buffer A (50 mM Tris–HCl pH 7.4, 500 mM NaCl, and 20 mM imidazole). The column was then washed with 40 bed volumes of buffer A, and bound proteins were eluted with a linear gradient of 0–75% buffer B (50 mM Tris–HCl pH 7.4, 500 mM NaCl, and 500 mM imidazole). Fractions containing C1ND, as determined by SDS–PAGE, were pooled and concentrated in a 10‐kDa molecular‐weight cutoff spin concentrator to a final volume of 5 ml. To remove minor protein contaminants, the concentrated samples were injected onto the Hi‐Prep 16/60 Sephacryl S‐100 HR column (GE Healthcare) pre‐equilibrated with a buffer containing 50 mM Tris–HCl pH 8.0, 150 mM NaCl. Fractions containing the C1ND complex were pooled and concentrated using a 10‐kDa molecular‐weight cutoff spin concentrator.
Cloning of HopQ constructs
HopQ type I constructs were designed as we described earlier (Javaheri et al, 2016). In brief, the HopQ type I adhesin domain fragment (HopQAD‐I) is derived from H. pylori strain G27 hopQ gene (accession no. CP001173; Region 1228696..1230621) and spans the residues 43–446 (residues 22–425 of the mature protein), thus removing the N‐terminal β‐strand and signal peptide, as well as the C‐terminal β‐domain expected to represent the transmembrane domain. DNA coding sequences corresponding to the hopQ type I fragments were PCR‐amplified from H. pylori G27 genomic DNA using primers (forward: GTTTAACTTTAAGAAGGAGATATACAAATGAACGCCGACAAGGTGCAAAAAC; reverse: TCAAGCTTATTAATGATGATGATGATGGTGGGCGCCGTTATTCGTGGTTG) containing 30‐bp overlap to the flanking target vector sequences of pPRkana‐1, a derivative of pPR‐IBA 1 (IBA) with the ampicillin resistance cassette replaced by the kanamycin resistance cassette, under a T7 promotor. In parallel, the vector was PCR‐amplified using primers (forward: CACCATCATCATCATCATTAATAAGCTTGATCCGGCTGCTAAC; reverse: GTTTAACTTTAAGAAGGAGATATACAAATG) using the same overlapping sequences in reversed orientation. The forward primer also carried the sequence for a 6× His‐tag. The amplicons were seamlessly cloned using Gibson Assembly (New England Biolabs).
The HopQ type II fragment was constructed as described for the HopQ type I fragment with minor deviations. The HopQ type II adhesin domain fragment (HopQAD‐II) is derived from the H. pylori strain Tx30a hopQ gene (locus accession no. AY147201) and spans the residues 37–454 (residues 16–433 of the mature protein). The DNA coding sequence was codon optimized using the JCat algorithm (Grote et al, 2005; organism: E. coli K12, no additional options selected). The optimized gene was synthesized by GeneArt de novo gene synthesis (Life Technologies) and inserted into the vector pPRkana‐1 as described above.
Design and construction of HopQ and C1ND mutants
The various HopQ and C1ND mutants were designed and constructed by site‐directed mutagenesis according to the QuikChange protocol (Stratagene).
Generation of H. pylori expressing HopQ mutants by natural transformation
P12 ∆HopQ bacteria were recovered from WC dent plates and resuspended in BHI medium, then mixed with 2 μg of the plasmid pSB1001 containing the mutated HopQ gene. The mixture was plated on WC dent plates with no selection antibiotic and the bacteria allowed to recover for 24 h, before plating on WC Dent with added kanamycin to select for bacteria expressing the mutant HopQ.
Production and purification of wild‐type and mutant HopQAD‐I
The pPRkana‐1 construct carrying the gene for the expression of HopQAD‐I was transformed in E. coli BL21(DE3) cells and grown at 37°C in terrific broth (TB) medium supplemented with 100 mg/l kanamycin–sulfate (Duchefa) while shaking, until an OD of 1 was reached. At that time, the temperature was lowered to 25°C and expression of HopQAD‐I was induced overnight with 1 mM isopropyl‐β‐D‐thiogalactopyranoside. Cells were the next day collected by centrifugation at 6,000 g for 15 min at 4°C using an JLA‐8.1000 rotor in an Avanti J‐26XP centrifuge (Beckman Coulter). Subsequently, cells were resuspended in 4 ml cold buffer A (500 mM NaCl, 100 mM Tris–HCl, 25 mM imidazole, pH 7.4) per gram of wet cells, supplemented with 5 μM leupeptin, 1 mM AEBSF, 100 μg/ml lysozyme, and 20 μg/ml DNase I. The cells were lysed by a single passage in a Constant System Cell Cracker at 20 kpsi at 4°C, and debris was removed by centrifugation at 48,400 g for 40 min and remaining particles were removed by filtration through a 0.2‐μm filter.
Next, the cell lysate was loaded onto a 5 ml pre‐packed Ni‐NTA HisTrap FF crude column (GE Healthcare) pre‐equilibrated with buffer A, washed with 10 column volumes (CVs) of buffer A, and the bound protein eluted with a 5 CV linear gradient to 75% buffer B (500 mM NaCl, 100 mM Tris–HCl, 500 mM imidazole, pH 7.4). Eluted peak fractions were collected and analyzed by SDS–PAGE and HopQ containing fractions were pooled and concentrated to a final concentration of 8–10 mg/ml using a 10‐kDa molecular‐weight cutoff spin concentrator. Subsequently, 5 ml of the concentrated protein was loaded onto a Hi‐Prep 26/60 Sephacryl S‐100 HR column (GE Healthcare) pre‐equilibrated with a buffer containing 20 mM Tris–HCl, 150 mM NaCl, pH 8.
Production, purification, and refolding of HopQAD‐II from inclusion bodies
The pPRkana‐1 construct carrying the gene for the expression of HopQAD‐II was transformed in E. coli BL21(DE3) cells and grown at 37°C in terrific broth (TB) medium supplemented with 100 mg/l kanamycin–sulfate (Duchefa) while shaking, until an OD of 1 was reached. At that time, expression of HopQAD‐II was induced with 1 mM isopropyl‐β‐D‐thiogalactopyranoside at 37°C overnight. Cells were the next day collected by centrifugation at 5,000 rpm for 15 min at 4°C using an JLA‐8.1000 rotor in an Avanti J‐26XP centrifuge (Beckman Coulter). Subsequently, cells were resuspended in a cold buffer containing 500 mM NaCl, 100 mM Tris–HCl pH 7.4 (4 ml/g of wet cells), supplemented with 5 μM leupeptin, 1 mM AEBSF, 100 μg/ml lysozyme and 20 μg/ml DNase I. The cells were lysed by a single passage in a Constant System Cell Cracker at 20 kpsi at 4°C. Next, inclusion bodies were collected by a centrifugation step at 48,400 g for 40 min and resuspended in buffer A [500 mM NaCl, 100 mM Tris–HCl, 25 mM imidazole, pH 7.4, 6 M guanidinium HCl, 5 mM dithiothreitol (DTT)]. The solution was stirred for 30 min at 4°C and clarified by centrifugation at 39,200 g for 30 min at 4°C. Before loading the DTT containing supernatant, the HisTrap FF column (GE Healthcare) was prepared for reducing conditions according to the instructions of the manufacturer and equilibrated with buffer A. Subsequently, the supernatant (containing the unfolded HopQAD‐II) was loaded, washed with ten column volumes (CVs) of buffer A, and the bound protein eluted with a 5 CV linear gradient to 100% buffer B (buffer A + 500 mM imidazole). Eluted peak fractions were collected, pooled, and concentrated to a final concentration of at least 10 mg/ml using a 10‐kDa molecular‐weight cutoff spin concentrator. HopQAD‐II was refolded by slow drop‐wise dilution in 20‐fold excess of cold refolding buffer (330 mM arginine phosphate, 5 mM DTT, pH 7.4) at 4°C while stirring, then incubated for 45 min at 4°C, and filtered through a 0.2‐μm filter. The filtrate was reapplied to a HisTrap FF column that was prepared for reducing conditions, but this time equilibrated in buffer A without guanidinium HCl. The loaded refolded HopQ II was subsequently washed by buffer A without guanidinium HCl and DTT, with a subsequent residence time of 45 min that allows the controlled oxidization of disulfide bonds. Afterward, the protein was eluted by applying a step gradient to 75% of elution buffer (500 mM NaCl, 100 mM Tris–HCl, 500 mM imidazole, pH 7.4) and concentrated using a 10‐kDa molecular‐weight cutoff spin concentrator, and 5 ml was loaded onto a Hi‐Prep 26/60 Sephacryl S‐100 HR column (GE Healthcare) that was equilibrated with a buffer a buffer containing 150 mM NaCl, 20 mM Tris pH 7.4.
Crystallization and structure determination of HopQAD‐I‐C1ND and HopQAD‐II‐C1ND
HopQAD‐I‐C1ND and HopQAD‐II‐C1ND were concentrated to 20 mg/ml and crystallized by sitting drop vapor diffusion at 20°C in, respectively, condition B3 of the Morpheus HT‐96 screen [Molecular Dimensions; 0.09 M halogens mix (0.3 M sodium fluoride; 0.3 M sodium bromide; 0.3 M sodium iodide), 0.1 M Buffer System 1 (1.0 M imidazole; MES monohydrate (acid); pH 6.5], 50% v/v Precipitant Mix 3 (40% v/v glycerol; 20% w/v PEG 4000) and condition A4 of the JB Screen Classic HTS II screen (Jena Bioscience; 1 M ammonium sulfate, 100 mM Tris; pH 8.5). Crystals were loop‐mounted and flash‐cooled in liquid nitrogen. Data were collected at 100 K at beamlines Proxima1 (SOLEIL, Gif‐sur‐Yvette, France) and IO2 (Diamond Light Source, Didcot, UK) and were indexed, processed, and scaled using the XDS package (Kabsch, 2010). Phases were obtained by molecular replacement using the HopQAD‐I structure (PDB:5LP2; Javaheri et al, 2016) and the program phaser (McCoy et al, 2007). The models were refined by iterative cycles of manual rebuilding in the graphics program COOT (Emsley & Cowtan, 2004) and maximum‐likelihood refinement using Refmac5 (Murshudov et al, 1997). Appendix Table S1 summarizes the crystal parameters, data processing and structure refinement statistics.
Isothermal titration calorimetry
ITC measurements were performed on a MicroCal iTC200 calorimeter (Malvern). C1ND (25 μM) was loaded into the cell of the calorimeter and 250 μM HopQ type I or II (wild‐type or mutant) was loaded into the syringe. All measurements were performed at 25°C, with a stirring speed of 600 rpm, in 20 mM HEPES buffer (pH 7.4), 150 mM NaCl, 5% (vol/vol) glycerol, and 0.05% (vol/vol) Tween 20. Binding data were analyzed using the MicroCal LLC ITC200 software.
Small‐angle X‐ray scattering
Data were collected in‐house on an BioSAXS‐2000 instrument (Rigaku). Different concentrations of the HopQAD‐I‐C1ND complex (4, 2 and 1 mg/ml) and C1ND (6, 3, 1.5, 0.8, 0.4 mg/ml) were prepared in a buffer containing 20 mM Tris pH 8.0, 150 mM NaCl and loaded into the capillary by the automatic sample changer. Three images with an exposure time per image of 10 min were collected, and the resulting scattering curves were assessed on radiation damage (Appendix Fig S4). Further analysis of the resulting scattering curve was performed using the ATSAS package (Franke et al, 2017). Buffer subtraction and data merging/averaging were performed using PRIMUS (Konarev et al, 2003). The indirect transform program GNOM (Svergun, 1992) was used to calculate the particle distance distribution function p(r). Ab initio shape reconstruction for the HopQAD‐I‐C1ND complex was performed using DAMMIF (Franke & Svergun, 2009).
Fluorescence labeling of H. pylori
FITC labeling of H. pylori was performed as described by Aspholm et al (2006). Wild‐type (wt) strain G27 and its isogenic ΔhopQ mutant were grown on horse serum GC agar plates supplemented with nystatin (1 μg/ml), vancomycin (10 μg/ml), and trimethoprim (5 μg/ml) and if necessary with 4 μg/chloramphenicol per ml. Growth was performed for 2 days at 37°C in anaerobic chambers containing a CampyGen gas mix (Oxoid, Wesel, Germany) at 37°C. H. pylori was harvested and resuspended in phosphate‐buffered saline (PBS, pH 7.4) using sterile cotton swabs (Carl Roth, Karlsruhe, Germany). After the labeling process, the bacteria were resuspended in BHI medium (Oxoid, Wesel, Germany) with 20% glycerol (Carl Roth, Karlsruhe, Germany) and stored at −20°C. To stain cells with CFSE for flow cytometry‐based adhesion assays, H. pylori cells were grown for 1–3 days on WC dent plates, with additional selection antibiotics as necessary, recovered, and resuspended in BHI medium. Ten micromoles of carboxyfluorescein succinimidyl ester (CFSE, eBioscience) was added to 2 × 108 bacteria, with the mixture left shaking for 30 min in a microaerobic incubator. Labeled bacteria were then used in adhesion assays.
Real‐time adhesion of H. pylori to MKN28 cells by the LigandTracer Green System
Human MKN28 cells used in this study were kindly provided by Motomo Kuroki (Fukuoka University/Japan) and are phenotypically different from the T4SS‐susceptible MKN28 cell line (JCRB cell bank, 0253) described previously (Schneider et al, 2011; Tegtmeyer et al, 2017). The MKN28 cell lines permanently expressing hCEACAM1‐4L, hCEACAM1‐4S, hCEACAM5, Mac‐CEACAM1, and Mac‐CEACAM5, respectively, were generated by stably transfecting cells with 3 μg plasmids utilizing the Lipofectamine 2000 approach according to the manufacturer's protocol (Invitrogen). Stable transfected cells were selected in culture medium containing 1 mg/ml of geneticin sulfate (G418 sulfate, PAA). The surface expression of hCEACAM1 and 5 in individual clones growing in log phase was determined by staining with monoclonal antibody 6G5j (B.B. Singer, University Duisburg‐Essen) and subsequent flow cytometric analyses (FACSCalibur, BD, Appendix Fig S3B).
The LigandTracer Green instrument (Ridgeview Instruments AB, Uppsala, Sweden) was used to quantify the interaction of H. pylori cells to cultured MKN28 cells in real time and under live‐cell conditions. The device was placed in a Hera Cell 150i CO2 incubator (Thermofisher, Waltham, MA, USA) to maintain experimental conditions of 37°C and 5% CO2. MKN28 wt cells, which are CEACAM‐negative, and MKN28 cells stably expressing the human CEACAM1 (CC1) 4S or 4L receptor were used for the experiments. The cells were grown in DMEM (Invitrogen, Karlsruhe, Germany) and 10% FCS (Invitrogen). The CEACAM‐expressing cells were additionally grown with 500 μg/ml G418 (Sigma‐Aldrich, Germany). To obtain monolayer‐grown cells, 2 × 106 freshly split MKN28 cells were resuspended in 3.5 ml of fresh medium and applied to the positive read‐out domain in a Nunclone™ Surface Petri dish (Nunc A/S, Roskilde, Denmark). The opposite non‐treated side of the dish was used as the reference. The plate was incubated at 37°C under 5% CO2 atmosphere in a BBD 6620 CO2 incubator (Thermofisher) until the confluence of monolayer reached 90%, which was confirmed by light microscopy and counting. Before the measurement experiments, the cells were washed three times with DMEM and 10% FCS. Three milliliters of fresh media was added, and the plate was placed into the LigandTracer Green system (Ridgeview Instruments AB, Uppsala, Sweden). The assay was performed with the “general assay” template which is included into the LigandTracer Control software (Ridgeview Instruments AB). After a 30‐min long baseline measurement, 100 μl of FITC‐labeled H. pylori G27 wt or G27ΔhopQ of OD600 = 1.0 was applied to the medium in the dish. The fluorescence intensity of bacterial cells binding to MKN28 wt cells or MKN28‐CEACAM cells were measured in the rotating dish for 150 min, registered once every minute by the fluorescence detector and compared to the reference signal of the opposite part of the dish.
Adhesion of H. pylori to MKN28 or CHO cells as measured by flow cytometry
Adhesion assays were performed as described by Hytönen et al (2006) and Javaheri et al (2016). MKN28 or CHO cells transfected with CEACAM variants, or vector controls were grown and maintained as described above. CFSE‐labeled H. pylori were washed 2× with PBS and added to 2.5 × 105 CHO or MKN28 cells at MOI 10 in DMEM supplemented with 10% BHI medium and left to shake for 30 min in a microaerobic incubator. After washing 3× with PBS, cells were fixed in 1% paraformaldehyde for 10 min and adhesion was measured by flow cytometry (using a Beckman Coulter Cyan ADP), where the eukaryotic cells are gated and then the percentage of cells emitting green fluorescence (signifying binding of CFSE‐stained bacteria) is calculated.
CagA translocation and phosphorylation assays
After H. pylori infection of the MKN28 cells for 6 h at multiplicity of infection (MOI) of 5, 50, or 100, total cell proteins were run on 6% SDS polyacrylamide gels and blotted onto PVDF membranes (Immobilon‐P, Merck Millipore) as described (Mueller et al, 2012). To investigate translocation and phosphorylation of CagA, the blots were subsequently probed with mouse monoclonal α‐phosphotyrosine antibody PY‐99 (Santa Cruz), stripped with Restore Western Blot Stripping buffer (Thermo Scientific), and reprobed with rabbit polyclonal α‐CagA antibody (Austral Biologicals). After blocking the membranes in TBS‐T (140 mM NaCl; 20 mM Tris–HCl, pH 7.4; 0.1% Tween‐20) with 3% BSA or 5% skim milk for 1 h at room temperature, or overnight at 4°C, they were incubated with the antibodies, overnight at 4°C, or for 2 h at room temperature (Mueller et al, 2012). Protein detection was performed using horseradish peroxidase‐conjugated α‐mouse or α‐rabbit polyvalent goat immunoglobulin secondary antibodies (Promega/Thermo Fisher Scientific) and the ECL Plus chemiluminescence Western Blot Detection Reagents (GE Healthcare Life Sciences) or Clarity Western ECL blotting substrates (Bio‐Rad). In Fig 4, H. pylori strains Ka88 and HPAG1 were chosen because they express CagA that runs at a molecular mass well above that of a tyrosine‐phosphorylated host cell protein at ~125 kDa present in MKN28. For the complementation assays using selected HopQ mutants, we used strain P12 because of the availability of the HopQ knockout (Fig 1).
Amino acid sequence alignment
Amino acid sequence alignments were performed using CLC main Workbench (CLC bio).
Data availability
Structure factors and coordinates for the X‐ray structures of the HopQAD‐I‐C1ND and HopQAD‐II‐C1ND complexes are deposited in the PDB with accession numbers 6GBG and 6GBH, respectively. Sequences for rhesus macaque CEACAM1 and CEACAM5 were deposited in GenBank under accession numbers MG874670 and MG874671, respectively.
Author contributions
KM performed the structural studies and acquired protein interaction data; MN, YH and NT performed LigandTracer and CagA translocation experiments; TK, YH, MR, RK, RM‐L, and BBS performed CEACAM mutagenesis and specificity studies. KM, RK, RM‐L, BBS, SB, MG, and HR conceived the experiments and analyzed the data. KM and HR wrote the manuscript with contributions of all authors. All authors read and approved the final manuscript.
Conflict of interest
M.G., B.B.S., S.B., H.R., K.M., and T.K. are named as inventors on a patent application related to HopQ. T.K. is an employee and Shareholder of ImevaX GmbH. The other authors declare no competing financial interests.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Birgit Maranca‐Hüwel for excellent technical support. K.M. and H.R. acknowledge use of the Soleil synchrotron, Gif‐sur‐Yvette, France, under proposal 20,131,370, Diamond Light Source under proposal MX12718, and support by VIB and the Flanders Science Foundation (FWO) through the Odysseus programme, a postdoctoral fellowship to KM and Hercules Funds UABR/09/005. The work of S.B. and N.T. is supported by the German Science Foundation (project A04 in CRC‐1181 and project TE776/3‐1), and Y.H. and M.G. acknowledge support through DFG project GE 2042/5‐1 and project SI‐1558/3‐1 to B.B.S.
The EMBO Journal (2018) 37: e98665
References
- Aspholm M, Kalia A, Ruhl S, Schedin S, Arnqvist A, Lindén S, Sjöström R, Gerhard M, Semino‐Mora C, Dubois A, Unemo M, Danielsson D, Teneberg S, Lee WK, Berg DE, Borén T (2006) Helicobacter pylori adhesion to carbohydrates. Methods Enzymol 417: 293–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JC, Blaser MJ (2009) Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest 119: 2475–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S, Clyne M, Tegtmeyer N (2011) Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori . Cell Commun Signal 9: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel JF, Darfeuille‐Michaud A (2007) CEACAM6 acts as a receptor for adherent‐invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest 117: 1566–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogolova E, Bauer B, Pompaiah M, Asakura H, Brinkman V, Ertl C, Bartfeld S, Nechitaylo TY, Haas R, Machuy N, Salama N, Churin Y, Meyer TF (2013) Helicobacter pylori outer membrane protein HopQ identified as a novel T4SS‐associated virulence factor. Cell Microbiol 15: 1896–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger CN, Billker O, Meyer TF, Servin AL, Kansau I (2004) Differential recognition of members of the carcinoembryonic antigen family by Afa/Dr adhesins of diffusely adhering Escherichia coli (Afa/Dr DAEC). Mol Microbiol 52: 963–983 [DOI] [PubMed] [Google Scholar]
- Bonsor DA, Günther S, Beadenkopf R, Beckett D, Sundberg EJ (2015) Diverse oligomeric states of CEACAM IgV domains. Proc Natl Acad Sci USA 112: 13561–13566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaytsova JA, Björnham O, Chernov YA, Gideonsson P, Henriksson S, Mendez M, Sjöström R, Mahdavi J, Shevtsova A, Ilver D, Moonens K, Quintana‐Hayashi MP, Moskalenko R, Aisenbrey C, Bylund G, Schmidt A, Åberg A, Brännström K, Königer V, Vikström S et al (2017) Helicobacter pylori adapts to chronic infection and gastric disease via pH‐responsive BabA‐mediated adherence. Cell Host Microbe 21: 376–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Cover TL (2002) Two different families of hopQ alleles in Helicobacter pylori . J Clin Microbiol 40: 4504–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fedarovich A, Tomberg J, Nicholas RA, Davies C (2006) Structure of the N‐terminal domain of human CEACAM1: binding target of the opacity proteins during invasion of Neisseria meningitidis and N. gonorrhoeae . Acta Crystallogr D Biol Crystallogr 62: 971–979 [DOI] [PubMed] [Google Scholar]
- Franke D, Svergun DI (2009) DAMMIF, a program for rapid ab‐initio shape determination in small‐angle scattering. J Appl Crystallogr 42: 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI (2017) ATSAS 2.8: a comprehensive data analysis suite for small‐angle scattering from macromolecular solutions. J Appl Crystallogr 50: 1212–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C (1999) Clinical relevance of the Helicobacter pylori gene for blood‐group antigen‐binding adhesin. Proc Natl Acad Sci USA 96: 12778–12783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray‐Owen SD, Blumberg RS (2006) CEACAM1: contact‐dependent control of immunity. Nat Rev Immunol 6: 433–446 [DOI] [PubMed] [Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33: W526–W531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage N, Howard T, Phillips C, Brassington C, Overman R, Debreczeni J, Gellert P, Stolnik S, Winkler GS, Falcone FH (2015) Structural basis of Lewis(b) antigen binding by the Helicobacter pylori adhesin BabA. Sci Adv 1: e1500315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom S (1999) The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 9: 67–81 [DOI] [PubMed] [Google Scholar]
- Hill DJ, Virji M (2003) A novel cell‐binding mechanism of Moraxella catarrhalis ubiquitous surface protein UspA: specific targeting of the N‐domain of carcinoembryonic antigen‐related cell adhesion molecules by UspA1. Mol Microbiol 48: 117–129 [DOI] [PubMed] [Google Scholar]
- Hytönen J, Haataja S, Finne J (2006) Use of flow cytometry for the adhesion analysis of Streptococcus pyogenes mutant strains to epithelial cells: investigation of the possible role of surface pullulanase and cysteine protease, and the transcriptional regulator Rgg. BMC Microbiol 6: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T (1998) Helicobacter pylori adhesin binding fucosylated histo‐blood group antigens revealed by retagging. Science 279: 373–377 [DOI] [PubMed] [Google Scholar]
- Javaheri A, Kruse T, Moonens K, Mejias‐Luque R, Debraekeleer A, Asche CI, Tegtmeyer N, Kalali B, Bach NC, Sieber SA, Hill DJ, Koniger V, Hauck CR, Moskalenko R, Haas R, Busch DH, Klaile E, Slevogt H, Schmidt A, Backert S et al (2016) Helicobacter pylori adhesin HopQ engages in a virulence‐enhancing interaction with human CEACAMs. Nat Microbiol 2: 16189 [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer R, Popp T, Härtle S, Singer BB, Zimmermann W (2007) Species‐specific evolution of immune receptor tyrosine based activation motif‐containing CEACAM1‐related immune receptors in the dog. BMC Evol Biol 7: 196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer R, Zimmermann W (2010) Coevolution of activating and inhibitory receptors within mammalian carcinoembryonic antigen families. BMC Biol 8: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaile E, Vorontsova O, Sigmundsson K, Müller MM, Singer BB, Ofverstedt LG, Svensson S, Skoglund U, Obrink B (2009) The CEACAM1 N‐terminal Ig domain mediates cis‐ and trans‐binding and is essential for allosteric rearrangements of CEACAM1 microclusters. J Cell Biol 187: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaile E, Klassert TE, Scheffrahn I, Müller MM, Heinrich A, Heyl KA, Dienemann H, Grünewald C, Bals R, Singer BB, Slevogt H (2013) Carcinoembryonic antigen (CEA)‐related cell adhesion molecules are co‐expressed in the human lung and their expression can be modulated in bronchial epithelial cells by non‐typable Haemophilus influenzae, Moraxella catarrhalis, TLR3, and type I and II interferons. Respir Res 14: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolla V, Gonzales LW, Bailey NA, Wang P, Angampalli S, Godinez MH, Madesh M, Ballard PL (2009) Carcinoembryonic cell adhesion molecule 6 in human lung: regulated expression of a multifunctional type II cell protein. Am J Physiol Lung Cell Mol Physiol 296: L1019–L1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS : a Windows PC‐based system for small‐angle scattering data analysis. J Appl Crystallogr 36: 1277–1282 [Google Scholar]
- Koniger V, Holsten L, Harrison U, Busch B, Loell E, Zhao Q, Bonsor DA, Roth A, Kengmo‐Tchoupa A, Smith SI, Mueller S, Sundberg EJ, Zimmermann W, Fischer W, Hauck CR, Haas R (2016) Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat Microbiol 2: 16188 [DOI] [PubMed] [Google Scholar]
- Korotkova N, Yang Y, Le Trong I, Cota E, Demeler B, Marchant J, Thomas WE, Stenkamp RE, Moseley SL, Matthews S (2008) Binding of Dr adhesins of Escherichia coli to carcinoembryonic antigen triggers receptor dissociation. Mol Microbiol 67: 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin M, Svergun D (2001) Automated matching of high‐ and low‐resolution structural models. J Appl Crystallogr 34: 33–41 [Google Scholar]
- Kuespert K, Pils S, Hauck CR (2006) CEACAMs: their role in physiology and pathophysiology. Curr Opin Cell Biol 18: 565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leusch HG, Drzeniek Z, Markos‐Pusztai Z, Wagener C (1991) Binding of Escherichia coli and Salmonella strains to members of the carcinoembryonic antigen family: differential binding inhibition by aromatic alpha‐glycosides of mannose. Infect Immun 59: 2051–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB et al (2002) Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297: 573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonens K, Gideonsson P, Subedi S, Bugaytsova J, Romao E, Mendez M, Norden J, Fallah M, Rakhimova L, Shevtsova A, Lahmann M, Castaldo G, Brannstrom K, Coppens F, Lo AW, Ny T, Solnick JV, Vandenbussche G, Oscarson S, Hammarstrom L et al (2016) Structural insights into polymorphic ABO glycan binding by Helicobacter pylori . Cell Host Microbe 19: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ME, Borén T, Solnick JV (2011) Life at the margins: modulation of attachment proteins in Helicobacter pylori . Gut Microbes 2: 42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S (2012) c‐Src and c‐Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest 122: 1553–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzner P, Rohde M, Kneitz S, Hauck CR (2005) CEACAM engagement by human pathogens enhances cell adhesion and counteracts bacteria‐induced detachment of epithelial cells. J Cell Biol 170: 825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzner P, Bachmann V, Zimmermann W, Hentschel J, Hauck CR (2010) Human‐restricted bacterial pathogens block shedding of epithelial cells by stimulating integrin activation. Science 329: 1197–1201 [DOI] [PubMed] [Google Scholar]
- Müller MM, Klaile E, Vorontsova O, Singer BB, Obrink B (2009) Homophilic adhesion and CEACAM1‐S regulate dimerization of CEACAM1‐L and recruitment of SHP‐2 and c‐Src. J Cell Biol 187: 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Ohno T, Sugimoto M, Nagashima A, Ogiwara H, Vilaichone RK, Mahachai V, Graham DY, Yamaoka Y (2009) Relationship between Helicobacter pylori hopQ genotype and clinical outcome in Asian and Western populations. J Gastroenterol Hepatol 24: 462–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palframan SL, Kwok T, Gabriel K (2012) Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front Cell Infect Microbiol 2: 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang SS, Nguyen ST, Perry AJ, Day CJ, Panjikar S, Tiralongo J, Whisstock JC, Kwok T (2013) The three‐dimensional structure of the extracellular adhesion domain of the sialic acid‐binding adhesin SabA from Helicobacter pylori . J Biol Chem 289: 6332–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek RM, Blaser MJ (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2: 28–37 [DOI] [PubMed] [Google Scholar]
- Polk DB, Peek RM (2010) Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10: 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posselt G, Backert S, Wessler S (2013) The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi‐step process in pathogenesis. Cell Commun Signal 11: 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossez Y, Gosset P, Boneca IG, Magalhaes A, Ecobichon C, Reis CA, Cieniewski‐Bernard C, Joncquel Chevalier Curt M, Leonard R, Maes E, Sperandio B, Slomianny C, Sansonetti PJ, Michalski JC, Robbe‐Masselot C (2014) The lacdiNAc‐specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J Infect Dis 210: 1286–1295 [DOI] [PubMed] [Google Scholar]
- Sadarangani M, Pollard AJ, Gray‐Owen SD (2011) Opa proteins and CEACAMs: pathways of immune engagement for pathogenic Neisseria . FEMS Microbiol Rev 35: 498–514 [DOI] [PubMed] [Google Scholar]
- Salama NR, Hartung ML, Müller A (2013) Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori . Nat Rev Microbiol 11: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitter T, Agerer F, Peterson L, Munzner P, Hauck CR (2004) Granulocyte CEACAM3 is a phagocytic receptor of the innate immune system that mediates recognition and elimination of human‐specific pathogens. J Exp Med 199: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Carra G, Sahin U, Hoy B, Rieder G, Wessler S (2011) Complex cellular responses of Helicobacter pylori‐colonized gastric adenocarcinoma cells. Infect Immun 79: 2362–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevogt H, Zabel S, Opitz B, Hocke A, Eitel J, N'guessan PD, Lucka L, Riesbeck K, Zimmermann W, Zweigner J, Temmesfeld‐Wollbrueck B, Suttorp N, Singer BB (2008) CEACAM1 inhibits Toll‐like receptor 2‐triggered antibacterial responses of human pulmonary epithelial cells. Nat Immunol 9: 1270–1278. [DOI] [PubMed] [Google Scholar]
- Svergun DI (1992) Determination of the regularization parameter in indirect‐transform methods using perceptual criteria. J Appl Crystallogr 25: 495–503 [Google Scholar]
- Svergun DI, Barberato C, Koch MHJ (1995) CRYSOL ‐ a program to evaluate X‐ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28: 768–773 [Google Scholar]
- Tchoupa AK, Schuhmacher T, Hauck CR (2014) Signaling by epithelial members of the CEACAM family ‐ mucosal docking sites for pathogenic bacteria. Cell Commun Signal 12: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchoupa AK, Lichtenegger S, Reidl J, Hauck CR (2015) Outer membrane protein P1 is the CEACAM‐binding adhesin of Haemophilus influenzae . Mol Microbiol 98: 440–455 [DOI] [PubMed] [Google Scholar]
- Tegtmeyer N, Wessler S, Necchi V, Rohde M, Harrer A, Rau TT, Asche CI, Boehm M, Loessner H, Figueiredo C, Naumann M, Palmisano R, Solcia E, Ricci V, Backert S (2017) Helicobacter pylori employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host Microbe 22: 552–560 [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Villullas S, Hill DJ, Sessions RB, Rea J, Virji M (2007) Mutational analysis of human CEACAM1: the potential of receptor polymorphism in increasing host susceptibility to bacterial infection. Cell Microbiol 9: 329–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virji M, Watt SM, Barker S, Makepeace K, Doyonnas R (1996) The N‐domain of the human CD66a adhesion molecule is a target for Opa proteins of Neisseria meningitidis and Neisseria gonorrhoeae . Mol Microbiol 22: 929–939 [DOI] [PubMed] [Google Scholar]
- Virji M, Evans D, Griffith J, Hill D, Serino L, Hadfield A, Watt SM (2000) Carcinoembryonic antigens are targeted by diverse strains of typable and non‐typable Haemophilus influenzae . Mol Microbiol 36: 784–795 [DOI] [PubMed] [Google Scholar]
- Voges M, Bachmann V, Kammerer R, Gophna U, Hauck CR (2010) CEACAM1 recognition by bacterial pathogens is species‐specific. BMC Microbiol 10: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt SM, Teixeira AM, Zhou GQ, Doyonnas R, Zhang Y, Grunert F, Blumberg RS, Kuroki M, Skubitz KM, Bates PA (2001) Homophilic adhesion of human CEACAM1 involves N‐terminal domain interactions: structural analysis of the binding site. Blood 98: 1469–1479 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
Structure factors and coordinates for the X‐ray structures of the HopQAD‐I‐C1ND and HopQAD‐II‐C1ND complexes are deposited in the PDB with accession numbers 6GBG and 6GBH, respectively. Sequences for rhesus macaque CEACAM1 and CEACAM5 were deposited in GenBank under accession numbers MG874670 and MG874671, respectively.