

Abstract
PURPOSE OF REVIEW
To review the evidence for genetic modifier effects in the neurodegenerative diseases Huntington’s Disease (HD), Frontotemporal Lobar Degeneration (FTLD), Alzheimer’s Disease (AD), and Parkinson’s Disease (PD).
RECENT FINDINGS
Increasingly, we understand human disease genetics less through the lens of single-locus/single-trait effects, and more through that of polygenic contributions to disease risk. In addition, specific examples of genetic modifier effects of the chromosome 7 gene TMEM106B on various target genes including those causal for Mendelian classes of FTLD – GRN and c9orf72 – have emerged from both genetic cohort studies and mechanistic examinations of biological pathways.
SUMMARY
Here, we summarize the literature reporting genetic modifier effects in HD, FTLD, AD, and PD. We further contextualize reported genetic modifier effects in these diseases in terms of insight they may lend to the concept of a polygenic landscape for the major neurodegenerative diseases.
Keywords: Genetic modifier, neurodegeneration, FTLD, Alzheimer’s Disease, Parkinson’s Disease, TMEM106B, APOE
INTRODUCTION
Neurodegeneration – the progressive loss of neurons with ensuing effects on cognition, motor function, and other brain activities – affects millions worldwide, with numbers suffering from neurodegenerative diseases expected to increase at an alarming rate as the population ages [1,2]. Currently, there are no disease modifying therapies for the major neurodegenerative diseases: Huntington’s disease (HD), Frontotemporal lobar degeneration (FTLD), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Amyotrophic lateral sclerosis (ALS). However, many genetic studies – ranging from traditional family linkage studies, to genomewide association studies (GWAS), to investigations of genetic effects on endophenotypes within each disease – have uncovered a wealth of loci, and in some cases, specific genetic variants, that confer varying levels of predisposition to specific diseases or specific manifestations of these diseases. Indeed, ~200 different loci have been linked to FTLD, AD, PD, ALS, and related neurodegenerative disorders by GWAS alone [3,4].
Concurrent with our discovery of ever-expanding numbers of genetic loci associated with the various neurodegenerative diseases is an evolving understanding of the landscape of human disease genetics. Specifically, the one gene-one trait model that dominated much of early human disease genetic investigations is giving way to a polygenic model, whereby multiple genes may interact to influence various traits in additive, synergistic, or even opposing ways [5,6]. Additionally, epistasis, the phenomenon whereby the interactions of genes are non-linear (i.e. not additive), has received considerable attention [7], although specific examples of epistatic effects are surprisingly rare in the human disease literature [8,9].
Thus, it is timely to consider the role of genetic modifiers in the neurodegenerative diseases. Genetic modifiers – defined as genes that alter the expression of other “target” genes – have traditionally been studied in the context of genetic modifier loci that affect the penetrance, severity, or other clinically-important features of diseases caused by rare mutations in target genes. These diseases are inherited in Mendelian fashion, and include examples such as cystic fibrosis [10]. We review here the evidence for traditional genetic modifiers in HD, a Mendelian neurodegenerative disease, as well as Mendelian subgroups of FTLD, ALS, PD, and AD. In broader terms, however, genetic modifiers serve as examples of the phenomenon of polygenic contributions to trait determination, be they by linear or epistatic effects. We thus also review the evidence for polygenic contributions to neurodegenerative disease phenomenology outside of the strictly Mendelian forms of disease. Finally, as our intent is to highlight the ways in which insight derived from genetic studies might inform therapeutic strategy, we focus particularly on areas where genetic modifier loci – both identified and to-be-identified – might be reasonable targets for therapeutic intervention.
HUNTINGTON’S DISEASE
HD is a rare, progressive neurodegenerative condition characterized by dementia and behavioral abnormalities [11]. Unlike the other diseases in this review, HD consists only of autosomal dominant cases defined by mutations in a single gene, HTT, encoding the protein huntingtin [12,13]. Specifically, HD is caused by a CAG repeat expansion in HTT exon 1, resulting in the translation of a polyglutamine tract of varying sizes, with age at disease onset inversely correlated with the length of the expansion [14,15].
Despite the evidence that the size of the CAG repeat expansion affects age at HD onset, considerable variability in presentation exists that is not explained by repeat size; for example, HD individuals carrying 44 CAG repeats may demonstrate disease onset ranging from 31 to 66 years of age [15]. To investigate the role of other genetic modifiers in HD, a recent GWAS was performed to identify loci associated with age at disease onset, finding genetic variants at two loci – on chromosome 15 (chr15) and chromosome 8 (chr8), with what appear to be three independent effects – that associate with this endophenotype [16]. The genes associated with these loci that mediate these effects on age at onset are yet unknown, but likely candidates include nearby genes MTMR10 and FAN1 and pseudogene HERC2P10 at the chr15 locus and the genes RRM2B and UBR5 at the chr8 locus [16]. While results are promising, they await replication and further investigation into biological mechanism.
FRONTOTEMPORAL LOBAR DEGENERATION
FTLD is the second most common form of dementia in individuals under the age of 65. A progressive brain disorder with degeneration of the frontal and/or temporal lobes, FTLD affects a patient’s behavior and language [17,18]. A subset of FTLD patients additionally experience motor neuron degeneration, with ensuing symptoms that resemble those of Amyotrophic Lateral Sclerosis (ALS) [17] – these patients are described as having FTLD-MND.
Autosomal dominant mutations in the genes encoding human progranulin (GRN) and the microtubule-associated protein tau (MAPT), as well as hexanucleotide expansions in c9orf72, have been shown to cause FTLD [19–23]. While MAPT mutations cause a form of FTLD characterized neuropathologically by inclusions containing the tau protein (FTLD-tau), GRN mutations and c9orf72 expansions cause FTLD characterized neuropathologically by inclusions containing the HIV TAR DNA-binding Protein of 43 kD (TDP43), termed FTLD-TDP [24]. Additionally, while MAPT mutations are very rare, GRN mutations and c9orf72 mutations are not, together affecting over half of all familial cases of FTLD [17,18,25–27]. Pathogenic mutations in GRN, MAPT, and c9orf72 are all highly-penetrant causes of FTLD [28].
In contrast with these rare-variant/strong-effect FTLD genetic loci, all of which were found by family linkage studies, common variants in the gene encoding Transmembrane Protein 106B (TMEM106B) have been shown by GWAS to confer slightly increased risk of FTLD, with an odds ratio of ~1.6 for the risk-associated haplotype at the TMEM106B locus [29]. While the original GWAS focused on neuropathologically-confirmed cases of FTLD-TDP, and included a significant group of GRN mutation carriers in whom the TMEM106B locus risk association appeared to be particularly strong, subsequent studies have replicated the finding that common variants at this locus associate with risk for FTLD in additional clinical cohorts as well [30,31].
Additional rare genetic causes of FTLD have been reported, as reviewed previously [32]. However, here we focus on the more commonly-found genes associated with FTLD – namely, GRN and c9orf72 – and the effects of common variation in TMEM106B on clinical presentation in FTLD individuals who harbor GRN mutations, c9orf72 expansions, or no Mendelian mutations.
Modifier Effects in GRN Mutation-associated FTLD-TDP
Since mutations in GRN were first identified as a cause for FTLD [20,21,33], two major themes have emerged. First, all autosomal dominant FTLD-causing mutations in GRN appear to be haploinsufficiency mutations, suggesting that a scarcity of progranulin leads to neurodegeneration [33,34]. Second, among GRN mutation carriers, clinical presentation varies greatly, even within the same family [20,21,35], suggesting the presence of genetic or environmental modifiers of phenotype.
Progranulin is a secreted growth factor that may enhance neuronal survival [36]; both progranulin, and daughter granulin peptides derived from progranulin, have also been reported to function in wound healing and inflammation [37]. More recently, Sortilin-1, encoded by SORT1, has been reported as the neuronal receptor for progranulin, conferring on neurons the ability to internalize progranulin [38], although multiple groups have also described sortilin-independent effects of progranulin [39–41]. As progranulin and sortilin-1 may function as a ligand-receptor pair, there is strong scientific rationale for SORT1 as a genetic modifier of progranulin-mediated effects. Indeed, the rs646776 SNP near SORT1, previously linked to SORT1 expression levels as an expression quantitative trait locus (eQTL), has also been reported to associate with plasma progranulin levels [42].
Mechanistic data linking TMEM106B to progranulin exist as well. In particular, we and others have shown that manipulation of TMEM106B expression levels in cell culture results in changes in progranulin protein measures [43–46]. Moreover, TMEM106B deletion from GRN null animals ameliorates abnormal lysosomal phenotypes and rescues retinal degeneration seen in GRN null animals, possibly through a mechanism involving TMEM106B’s interaction with components of the vacuolar ATPase complex (and particularly V-ATPase AP1) responsible for lysosomal acidification [47]. From a human genetics standpoint, TMEM106B common variants associated with risk for FTLD-TDP in the general population also associate with earlier age at FTLD onset for GRN mutation carriers [43].
Modifier Effects in C9ORF72 Mutation-associated FTLD-TDP
TMEM106B has also been mechanistically linked to c9orf72. Specifically, we have shown that aberrant lysosomal phenotypes (vacuolar morphology, defect in acidification) induced by over-expression of TMEM106B are rescued with concomitant knockdown of c9orf72 [48]. Moreover, genotypes at the sentinel single nucleotide polymorphism associated with FTLD-TDP by GWAS, rs1990622, associate significantly with age at onset and age at death for FTLD-TDP patients carrying expansions in c9orf72 in our study of 89 neuropathologically-confirmed FTLD-TDP cases from 31 sites around the world [49].
Intriguingly, however, the direction of association differs for TMEM106B effects on GRN mutation carriers vs. c9orf72 mutation carriers. That is, whereas the rs1990622 G allele associated with decreased risk of FTLD-TDP by GWAS is found in GRN mutation carriers with a later age at disease onset, this same rs1990622 G allele is found in c9orf72 expansion carriers with an earlier age at disease onset and death. In theoretical genetic terms, this constitutes an example of sign epistasis, a situation whereby the same genetic variation that is beneficial on one genetic background may be deleterious in another genetic background.
Further complication of the intriguing relationship between TMEM106B and c9orf72 comes from the observation that, unlike GRN carriers, who manifest almost exclusively with FTLD-TDP, c9orf72 expansion carriers may manifest with FTLD-TDP or with ALS/MND (or with a combination of the two) [34]. Indeed, in a study of 325 c9orf72 expansion carriers, homozygous carriers of the TMEM106B rs1990622 G allele were significantly under-represented among FTLD patients, but not among MND patients. While the authors interpret this result to suggest that the rs1990622 GG genotype is highly protective, decreasing the penetrance of c9orf72 expansions, reports that of >36,000 control samples screened for c9orf72 expansions, only 40 (0.1%) asymptomatic individuals were found to harbor expansions suggest that there might be a more complicated interplay between TMEM106B genotype, c9orf72 expansions, and ultimate clinical manifestation [50].
Additional genetic modifier effects for TMEM106B
TMEM106B may exert modifier effects in groups beyond individuals carrying GRN mutations or c9orf72 expansions. Specifically, we first showed that TMEM106B genotypes correlate with cognitive phenotype in ALS, with carriers of the rs1990622 G allele more likely to show preserved cognition and lesser TDP-43 pathology in five brain regions [51]. Further support for a role for TMEM106B in modifying phenotypes beyond subsets of individuals with FTLD due to known Mendelian mutations comes from recent data demonstrating that the rs1990622 G allele (or proxy markers linked to this allele) may show a protective effect with respect to (1) hippocampal sclerosis of the aging [52], (2) general cognition among elderly individuals in the Religious Orders Study and Rush Memory and Aging Project [53], and (3) a frontal cortex brain expression profile representative of “aging” [54]. As the latter two results come from genomewide screens for modifiers of cognitive aging, support for a role for TMEM106B in these processes is substantial.
The evidence suggesting that TMEM106B genotypes may act as genetic modifiers of cognitive aging is furthermore supported by mechanistic work from multiple groups defining a role for TMEM106B in lysosomal function [44–46,48,55,56]. As genotypes at rs1990622 and linked SNPs act as TMEM106B eQTLs [3,29,57], likely through a mechanism involving differential recruitment of the chromatin organizing protein CTCF [3], the data, taken together, suggest that levels of TMEM106B expression impact lysosomal function, with ensuing effects on cellular health and brain aging.
ALZHEIMER’S DISEASE
AD is the most common form of dementia in the elderly, affecting ~50% of individuals 85 years or older [58]. AD is a progressive brain disorder associated with decline in memory and other cognitive domains [59–61]. Mutations in the genes encoding amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) have long been known to cause autosomal dominant, early-onset forms of AD [62–66]. However, unlike FTLD, familial forms of AD are rare, encompassing less than 5% of total cases [67,68].
In addition to rare, Mendelian causes of AD, common variation in the Apolipoprotein E gene (APOE) has been extensively studied as a genetic risk factor for development of AD [69]. Three major ApoE isoforms exist in the general population, termed the ApoE ε2, ε3, and ε4 isoforms, with corresponding ε2, ε3, and ε4 alleles [69]. The ε4 allele has been well established as a strong genetic risk factor for development of AD [70–72], with a reported odds ratio of 14.9 for a Caucasian population carrying the ε4/ε4 genotype [73]. As the ε4 allele is not uncommon (~14% frequency) [73], APOE is an important contributor to AD pathogenesis in terms of both effect size and numbers affected. In addition to contributing to risk for development of AD, the APOE ε4 allele associates with earlier age at onset for AD as a whole, as well as PSEN1-mutation and PSEN2-muation associated AD [73–77]. The ε2 allele has also been reported to exert effects on AD risk; specifically, ε2 allele carriers may be protected against late-onset AD [78]. Additionally a large-scale genome wide survival analysis reported that the rs1057233 G allele, within a previously reported CELF1 AD risk locus, associates with a later age of disease onset for AD [79].
Apolipoprotein E is believed to function in the clearance of beta-amyloid (Aβ42), the major protein that accumulates in the senile plaques that, along with tau-filled neurofibrillary tangles, characterize AD neuropathologically [80]. As such, it is perhaps unsurprising that APOE genotypes have also been linked to other processes involving Aβ42. For example, the APOE ε4 allele has been associated with CSF levels of Aβ42 [81,82]. Moreover, in individuals who at baseline were without evidence of Aβ42 deposition by imaging, or dementia clinically, the APOE ε4 allele associated with subsequent brain accumulation of Aβ42 [83]. In addition, in PD, a distinct neurodegenerative disease in which Aβ42 deposition is also frequently observed [84–87], the ε4 allele of APOE has also been linked to cognitive decline and dementia [88–90].
PARKINSON’S DISEASE
PD is the second most common neurodegenerative disorder after AD, affecting 2–3% of the population older than 65 [91]. PD is characterized by neuronal loss in the substantia nigra, the development of inclusions including aggregates of alpha-synuclein (encoded by the gene SNCA), and development of many motor, as well as non-motor, symptoms [92–96]. The discovery of SNCA mutations, as well as SNCA duplications and triplications, as causes of familial PD, established alpha-synuclein as a central player in PD pathogenesis [92,97]. However, these mutations are rare, limited, in some cases, to a few families. In contrast, mutations in the leucine-rich repeat kinase 2 (LRRK2), also causal for PD, are more common, affecting approximately 5% of all PD [98,99], and higher proportions in PD patients from specific ancestral backgrounds, such as Ashkenazi Jews (~18% for LRRK2+ PD) [100], and North African Arabs (37–41% for LRRK2+ PD) [101,102]. Surpassing even LRRK2 mutations in frequency are mutations in the gene encoding β-glucocerebrosidase (GBA), found in 7% of PD patients [103]. Long understood to be the autosomal recessive cause of the childhood-onset lysosomal storage disorder Gaucher’s disease, GBA mutations were linked to increased risk for PD in 2009 [103]. Specifically, the presence of one GBA mutation is associated with an odds ratio of ~5 for development of PD [103]. Moreover, GBA mutations have been reported to modify the clinical presentation in PD, with carriers of GBA mutations as well as the GBA E326K polymorphism at increased risk for GBA-related cognitive deficits [104,105].
Unlike the situation with GRN mutations or c9orf72 expansions in FTLD, or APP, PSEN1, or PSEN2 mutations in AD, all of which are highly penetrant, neither LRRK2 nor GBA mutations are highly penetrant in PD. In the case of LRRK2, age-related penetrance for the most common LRRK2 Gly2019Ser mutation can range from ~30% to 70% [106]. In the case of GBA, age-related penetrance can range from ~7% to 30% [107].
Thus, given the high prevalence of LRRK2 and GBA mutations, as well as their variable penetrance, the question of what additional genetic loci may modify the effects of LRRK2 or GBA is an important one to answer in the field. No clear genetic modifier loci are known at this time. However, such genetic modifier loci, if they can be found, might be targets for manipulation to significantly delay PD onset (or avoid it entirely) in the sizeable number of individuals with LRRK2 or GBA mutations.
CONCLUSION
We live in a data-rich age. Reflecting this, hundreds of genetic loci – be they in Mendelian “causal” genes, common risk variants, or loci representative of other types of effects – have been linked to the various adult-onset neurodegenerative diseases [3,4]. An understanding of their interplay and biological function is needed, however, to translate any of these discoveries into potential therapy for patients suffering from these diseases.
The insight that genetic loci may act together, in complex ways, has been valuable in the creation of models to derive meaning from the wealth of newly-available genetic/genomic data. Equally valuable, however, may be “sanity check” real-world examples derived from the preponderance of the evidence. For example, the recent advent of an “omnigenic” model [108] – in effect, the extreme example of a polygenic model – posits that complex traits may have a preponderance of heritability explained by effects of genes outside of core driver pathways because, essentially, most or all genes expressed in the relevant tissue types are connected to genes on driver pathways, leading to their “discovery” as risk factors for disease. If this is true of neurodegenerative disease genetics, implications for the common practice of finding genetic risk factors in order to identify targetable pathways are sobering.
Fortunately, the evidence as reviewed here suggests a less “omnigenic” landscape for at least FTLD and AD (the diseases in which we have the most data for genetic modifier effects) as we currently understand them (Figure 1). That is, genetic modifier loci, even the ones found by GWAS (i.e. TMEM106B), appear to interact with target genes in biologically specific pathways that are disease-relevant (e.g. lysosomal pathways for TMEM106B/GRN/c9orf72, receptor-ligand interactions for SORT1/GRN, APP processing for APOE/PSEN1/PSEN2). Thus, we continue to hope that an understanding of polygenic effects on human neurodegenerative diseases will lead to insight that can benefit the many millions worldwide suffering from these diseases.
Figure 1.
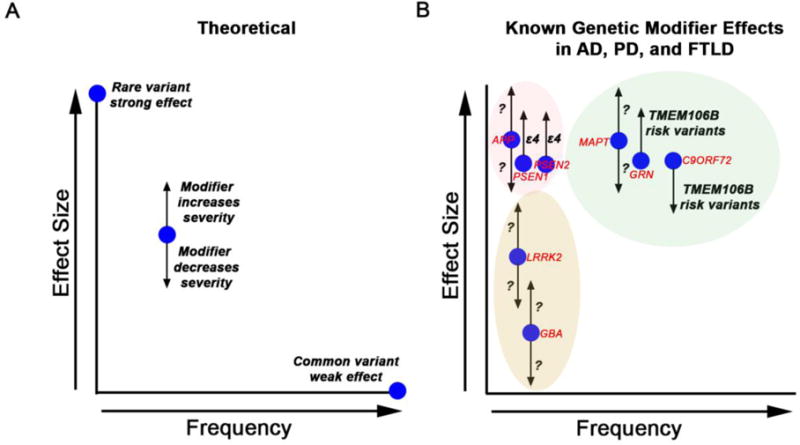
A. Representation of types of genetic variants and effects on trait, with variant frequency on the X-axis and effect size on the Y-axis. Genetic modifier effects are represented by arrows emanating from target gene (blue dot) loci.
B. Known genetic modifier effects in AD, PD, and FTLD. Target gene names are shown in red, and modifier loci names are shown in black, with direction of effect indicated by arrow. AD loci are highlighted by the pink oval, FTLD loci by the green oval, and PD loci by the tan oval. Arrows are not drawn to scale, and some genetic modifier loci are unknown (question marks).
Table 1.
Directional effect of modifier genes in Mendelian disease-causing mutations and all-comer populations
| Target Gene | Modifier Gene | Directional Effect | References |
|---|---|---|---|
| Huntington’s Disease | |||
| HTT | loci on chromosome 8 and 15 | chr8 locus associated with age at onset chr15 locus may harbor two loci with independent, opposing effects | Lee et al. 2015 |
| Frontotemporal Lobar Degeneration | |||
| GRN | TMEM106B | rs1990622 G allele associated with older age at onset | Cruchaga et al., 2011 Finch et al., 2011 |
| SORT1 | rs646776 C allele associated with decreased GRN plasma expression | Carrasquillo et al., 2010 Hu et al., 2010 |
|
| C9ORF72 | TMEM106B | rs1990622 G allele associated with younger age at onset and death | Gallagher et al., 2014 Blitterswijk et al., 2014 |
| MAPT | ? | - | - |
| - | TMEM106B | rs1990622 G allele may show a protective effect on “cognitive aging” | Katsumata et al., 2017 White et al., 2017 Rhinn et al., 2017 |
| Amyotrophic Lateral Sclerosis | |||
| - | TMEM106B | rs1990622 G allele associated with less cognitive impairment | Vass et al., 2011 |
| Alzheimer’s Disease | |||
| APP | APOE | - | - |
| PSEN1 | APOE | ε4 allele associated with younger age at onset and death | Pastor et al., 2003 |
| PSEN2 | APOE | ε4 allele associated with younger age at onset and death | Wijsman et al., 2005 |
| - | APOE | ε4 allele associated with younger age at onset and death | Corder et al., 1993 Rebeck et al., 1993 Farrer et al., 1997 |
| - | CELF1 | rs1057233 G allele associated with older age at onset | Huang et al., 2017 |
| Parkinson’s Disease | |||
| LRRK2 | ? | - | - |
| GBA | ? | - | - |
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Both authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
CITATIONS
- 1.Vos T, Barber RM, Bell B, Bertozzi-Villa A, Biryukov S, Bolliger I, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800. doi: 10.1016/S0140-6736(15)60692-4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertram L. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–57. doi: 10.1172/JCI24761.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL, et al. A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am J Hum Genet. 2017 doi: 10.1016/j.ajhg.2017.09.004. TMEM106B genetic modifier effect in FTLD due to c9orf72 expansion, demonstrated in 31-site international FTLD cohort. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plomin R, Haworth CMA, Davis OSP. Common disorders are quantitative traits. Nat Rev Genet. 2010;10:872. doi: 10.1038/nrg2670.. [DOI] [PubMed] [Google Scholar]
- 6.Riordan JD, Nadeau JH. From Peas to Disease: Modifier Genes, Network Resilience, and the Genetics of Health. Am J Hum Genet. 2017;101:177–91. doi: 10.1016/j.ajhg.2017.06.004.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sackton TB, Hartl DL. Genotypic Context and Epistasis in Individuals and Populations. Cell. 2016;166:279–87. doi: 10.1016/j.cell.2016.06.047.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva RF, Mendonça SCM, Carvalho LM, Reis AM, Gordo I, Trindade S, et al. Pervasive Sign Epistasis between Conjugative Plasmids and Drug-Resistance Chromosomal Mutations. PLoS Genet. 2011;7:e1002181. doi: 10.1371/journal.pgen.1002181.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schenk MF, Szendro IG, Salverda MLM, Krug J, de Visser JAGM. Patterns of Epistasis between Beneficial Mutations in an Antibiotic Resistance Gene. Mol Biol Evol. 2013;30:1779–87. doi: 10.1093/molbev/mst096.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. 2010;1214:57–69. doi: 10.1111/j.1749-6632.2010.05879.x.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–16. doi: 10.1038/nrneurol.2014.24.. [DOI] [PubMed] [Google Scholar]
- 12.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-E.. [DOI] [PubMed] [Google Scholar]
- 13.Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, et al. A Worldwide Study of the Huntington’s Disease Mutation: The Sensitivity and Specificity of Measuring CAG Repeats. N Engl J Med. 1994;330:1401–6. doi: 10.1056/NEJM199405193302001.. [DOI] [PubMed] [Google Scholar]
- 14.Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet. 1993;4:393–7. doi: 10.1038/ng0893-393.. [DOI] [PubMed] [Google Scholar]
- 15.Lee J-M, Ramos EM, Lee J-H, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–5. doi: 10.1212/WNL.0b013e318249f683.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J-M, Wheeler VC, Chao MJ, Vonsattel JPG, Pinto RM, Lucente D, et al. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell. 2015;162:516–26. doi: 10.1016/j.cell.2015.07.003.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386:1672–82. doi: 10.1016/S0140-6736(15)00461-4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seelaar H, Rohrer JD, Pijnenburg YAL, Fox NC, van Swieten JC. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry. 2011;82:476–86. doi: 10.1136/jnnp.2010.212225.. [DOI] [PubMed] [Google Scholar]
- 19.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508.. [DOI] [PubMed] [Google Scholar]
- 20.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. doi: 10.1038/nature05016.. [DOI] [PubMed] [Google Scholar]
- 21.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017.. [DOI] [PubMed] [Google Scholar]
- 22.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72:257–68. doi: 10.1016/j.neuron.2011.09.010.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baborie A, Griffiths TD, Jaros E, McKeith IG, Burn DJ, Richardson A, et al. Pathological correlates of frontotemporal lobar degeneration in the elderly. Acta Neuropathol. 2011;121:365–71. doi: 10.1007/s00401-010-0765-z.. [DOI] [PubMed] [Google Scholar]
- 25.Mackenzie IRA, Baborie A, Pickering-Brown S, Plessis D Du, Jaros E, Perry RH, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: Classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–49. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, et al. Pathological Heterogeneity of Frontotemporal Lobar Degeneration with Ubiquitin-Positive Inclusions Delineated by Ubiquitin Immunohistochemistry and Novel Monoclonal Antibodies. Am J Pathol. 2006;169:1343–52. doi: 10.2353/ajpath.2006.060438.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AMT, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: Comparison with patients with MAPT and no known mutations. Brain. 2008;131:721–31. doi: 10.1093/brain/awm331.. [DOI] [PubMed] [Google Scholar]
- 28.Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7:1–19. doi: 10.3389/fnagi.2015.00171.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang L-S, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–9. doi: 10.1038/ng.536.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, DeJesus-Hernandez M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–74. doi: 10.1212/WNL.0b013e31820a0e3b.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134:808–15. doi: 10.1093/brain/awr007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 2016;138:32–53. doi: 10.1111/jnc.13622.. [DOI] [PubMed] [Google Scholar]
- 33.Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241.. [DOI] [PubMed] [Google Scholar]
- 34.Chen-Plotkin AS, Martinez-Lage M, Sleiman PM a, Hu W, Greene R, Wood EM, et al. Genetic and Clinical Features of Progranulin-Associated Frontotemporal Lobar Degeneration. Arch Neurol. 2011(68):488. doi: 10.1001/archneurol.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging. 2009;30:739–51. doi: 10.1016/j.neurobiolaging.2007.08.022.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, Van Swieten J, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He Z, Ong CHP, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–9. doi: 10.1038/nm816.. [DOI] [PubMed] [Google Scholar]
- 38.Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, et al. Sortilin-Mediated Endocytosis Determines Levels of the Frontotemporal Dementia Protein, Progranulin. Neuron. 2010;68:654–67. doi: 10.1016/j.neuron.2010.09.034.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Muynck L, Herdewyn S, Beel S, Scheveneels W, Van Den Bosch L, Robberecht W, et al. The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiol Aging. 2013;34:2541–7. doi: 10.1016/j.neurobiolaging.2013.04.022.. [DOI] [PubMed] [Google Scholar]
- 40.Zhou X, Sun L, de Oliveira FB, Qi X, Brown WJ, Smolka MB, et al. Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J Cell Biol. 2015;210:991–1002. doi: 10.1083/jcb.201502029.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, et al. Progranulin regulates neuronal outgrowth independent of Sortilin. Mol Neurodegener. 2012;7:33. doi: 10.1186/1750-1326-7-33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Carrasquillo MM, Nicholson AM, Finch N, Gibbs JR, Baker M, Rutherford NJ, et al. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet. 2010;87:890–7. doi: 10.1016/j.ajhg.2010.11.002. Genomewide analysis identifying SNP near SORT1 (coding for sortilin-1, reported to be the neuronal receptor for progranulin) as a predictor of plasma progranulin levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Cruchaga C, Graff C, Chiang H, Wang J, Hinrichs AL, Spiegel N, et al. Association of TMEM106B Gene Polymorphism With Age at Onset in Granulin Mutation Carriers and Plasma Granulin Protein Levels. Arch Neurol. 2011;68:581–6. doi: 10.1001/archneurol.2010.350. TMEM106B genetic modifier effect in FTLD due to GRN mutations, demonstrated in 4 large families. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, et al. TMEM106B, the Risk Gene for Frontotemporal Dementia, Is Regulated by the microRNA-132/212 Cluster and Affects Progranulin Pathways. J Neurosci. 2012;32:11213–27. doi: 10.1523/JNEUROSCI.0521-12.2012.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem. 2012;287:19355–65. doi: 10.1074/jbc.M112.365098.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet. 2013;22:685–95. doi: 10.1093/hmg/dds475.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TKT, et al. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron. 2017;95:281–296.e6. doi: 10.1016/j.neuron.2017.06.026. Provides in vivo evidence of a specific interaction between TMEM106B and GRN, as lysosomal defects in GRN-deficient mice are rescued by deletion of TMEM106B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Busch JI, Unger TL, Jain N, Skrinak RT, Charan RA, Chen-Plotkin AS. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet. 2016;25:2681–97. doi: 10.1093/hmg/ddw127. Provides mechanistic evidence of a specific interaction between TMEM106B and c9orf72, as aberrant lysosomal phenotypes induced by TMEM106B over-expression are rescued by knockdown of c9orf72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127:407–18. doi: 10.1007/s00401-013-1239-x.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chiò A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep. 2017;7:2116. doi: 10.1038/s41598-017-02364-1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121:373–80. doi: 10.1007/s00401-010-0782-y.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katsumata Y, Nelson PT, Ellingson SR, Fardo DW. Gene-based association study of genes linked to hippocampal sclerosis of aging neuropathology: GRN, TMEM106B, ABCC9, and KCNMB2. Neurobiol Aging. 2017;53:193.e17–193.e25. doi: 10.1016/j.neurobiolaging.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White CC, Yang HS, Yu L, Chibnik LB, Dawe RJ, Yang J, et al. Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. 2017;14:1–23. doi: 10.1371/journal.pmed.1002287.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rhinn H, Abeliovich A. Differential Aging Analysis in Human Cerebral Cortex Identifies Variants in TMEM106B and GRN that Regulate Aging Phenotypes. Cell Syst. 2017;4:404–415.e5. doi: 10.1016/j.cels.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 55.Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM. Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci. 2014;61:226–40. doi: 10.1016/j.mcn.2014.07.006.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwenk JM, Lindberg J, Sundberg M, Uhlén M, Nilsson P. Determination of Binding Specificities in Highly Multiplexed Bead-based Assays for Antibody Proteomics. Mol Cell Proteomics. 2007;6:125–32. doi: 10.1074/mcp.T600035-MCP200.. [DOI] [PubMed] [Google Scholar]
- 57.Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology. 2015;84:927–34. doi: 10.1212/WNL.0000000000001313.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evans DA. Prevalence of Alzheimer’s Disease in a Community Population of Older Persons. JAMA. 1989;262:2551. doi: 10.1001/jama.1989.03430180093036.. [DOI] [PubMed] [Google Scholar]
- 59.Wisniewski T, Golabek A, Matsubara E, Ghiso J, Frangione B. Apolipoprotein E: binding to soluble Alzheimer’s beta-amyloid. Biochem Biophys Res Commun. 1993;192:359–65. doi: 10.1006/bbrc.1993.1423.. [DOI] [PubMed] [Google Scholar]
- 60.Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer’s disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–93. doi: 10.1146/annurev.genet.32.1.461.. [DOI] [PubMed] [Google Scholar]
- 61.Dickson DW. Neuropathology of Alzheimer’s disease and other dementias. Clin Geriatr Med. 2001;17:209–28. doi: 10.1016/S0749-0690(05)70066-5.. [DOI] [PubMed] [Google Scholar]
- 62.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0.. [DOI] [PubMed] [Google Scholar]
- 63.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0.. [DOI] [PubMed] [Google Scholar]
- 64.Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J Neurol. 2006;253:139–58. doi: 10.1007/s00415-005-0019-5.. [DOI] [PubMed] [Google Scholar]
- 65.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–8. doi: 10.1038/376775a0.. [DOI] [PubMed] [Google Scholar]
- 66.Levy-Lahad E, Wasco W, Poorkaj P, Romano D, Oshima J, Pettingell W, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science (80-) 1995;269:973–7. doi: 10.1126/science.7638622.. [DOI] [PubMed] [Google Scholar]
- 67.Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: A retrospective cohort study. Lancet Neurol. 2011;10:213–20. doi: 10.1016/S1474-4422(10)70323-9.. [DOI] [PubMed] [Google Scholar]
- 68.Barber RC. The Genetics of Alzheimer’s Disease. Scientifica (Cairo) 2012;2012:1–14. doi: 10.6064/2012/246210.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18. doi: 10.1038/nrneurol.2012.263.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934.. [DOI] [PubMed] [Google Scholar]
- 71.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poirier J, Bertrand P, Poirier J, Kogan S, Gauthier S, Poirier J, et al. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-Q.. [DOI] [PubMed] [Google Scholar]
- 73.Farrer LA. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. JAMA. 1997;278:1349. doi: 10.1001/jama.1997.03550160069041.. [DOI] [PubMed] [Google Scholar]
- 74.Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, García G, et al. Apolipoprotein Eε4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54:163–9. doi: 10.1002/ana.10636.. [DOI] [PubMed] [Google Scholar]
- 75.Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, et al. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am J Med Genet Part B Neuropsychiatr Genet. 2005;132B:14–20. doi: 10.1002/ajmg.b.30087.. [DOI] [PubMed] [Google Scholar]
- 76.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4. doi: 10.1038/ng0694-180.. [DOI] [PubMed] [Google Scholar]
- 77.William Rebeck G, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron. 1993;11:575–80. doi: 10.1016/0896-6273(93)90070-8.. [DOI] [PubMed] [Google Scholar]
- 78.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4. doi: 10.1038/ng0694-180.. [DOI] [PubMed] [Google Scholar]
- 79.Huang K, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052–61. doi: 10.1038/nn.4587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE Isoforms Differentially Regulate Brain Amyloid- Peptide Clearance. Sci Transl Med. 2011;3:89ra57–89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–56. doi: 10.1007/s00401-017-1685-y.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Shaw LM, Trojanowski JQ, et al. Effect of APOE on biomarkers of amyloid load and neuronal pathology in AD. Ann Neurol. 2009;67:NA–NA. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lim YY, Mormino EC, Initiative ADN. APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology. 2017;89:1028–34. doi: 10.1212/WNL.0000000000004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Montine TJ, Shi M, Quinn JF, Peskind ER, Craft S, Ginghina C, et al. CSF Aβ 42 and tau in Parkinson’s disease with cognitive impairment. Mov Disord. 2010;25:2682–5. doi: 10.1002/mds.23287.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petrou M, Bohnen NI, Muller MLTM, Koeppe R a, Albin RL, Frey K a. Abeta -amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology. 2012;79:1161–7. doi: 10.1212/WNL.0b013e3182698d4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Irwin DJ, Lee VM, Trojanowski JQ. Amyloid beta-peptide and the dementia of Parkinson’s disease. Nat Rev Neurosci. 2013;14:626–636. doi: 10.1038/nrn3549.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72:587–98. doi: 10.1002/ana.23659.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siderowf A, Xie SX, Hurtig H, Weintraub D, Duda J, Chen-Plotkin A, et al. CSF amyloid β 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–61. doi: 10.1212/WNL.0b013e3181f39a78.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE ε4 Increases Risk for Dementia in Pure Synucleinopathies. JAMA Neurol. 2013;70:223. doi: 10.1001/jamaneurol.2013.600.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tropea TF, Chen-Plotkin AS. Unlocking the mystery of biomarkers: A brief introduction, challenges and opportunities in Parkinson Disease. Parkinsonism Relat Disord. 2017 doi: 10.1016/j.parkreldis.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–35. doi: 10.1016/S1474-4422(06)70471-9.. [DOI] [PubMed] [Google Scholar]
- 92.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science (80-) 1997;276:2045–7. doi: 10.1126/science.276.5321.2045.. [DOI] [PubMed] [Google Scholar]
- 93.FEARNLEY JM, LEES AJ. Ageing and Parkinson’s Disease: Substantia Nigra Regional Selectivity. Brain. 1991;114:2283–301. doi: 10.1093/brain/114.5.2283.. [DOI] [PubMed] [Google Scholar]
- 94.Aarsland D, Andersen K, Larsen JP, Lolk A. Prevalence and Characteristics of Dementia in Parkinson Disease. Arch Neurol. 2003;60:387. doi: 10.1001/archneur.60.3.387.. [DOI] [PubMed] [Google Scholar]
- 95.Buter TC, van den Hout A, Matthews FE, Larsen JP, Brayne C, Aarsland D. Dementia and survival in Parkinson disease: A 12-year population study. Neurology. 2008;70:1017–22. doi: 10.1212/01.wnl.0000306632.43729.24.. [DOI] [PubMed] [Google Scholar]
- 96.Hely MA, Reid WGJ, Adena MA, Halliday GM, Morris JGL. The Sydney Multicenter Study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov Disord. 2008;23:837–44. doi: 10.1002/mds.21956.. [DOI] [PubMed] [Google Scholar]
- 97.Singleton AB. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science (80-) 2003;302:841–841. doi: 10.1126/science.1090278.. [DOI] [PubMed] [Google Scholar]
- 98.Li J-Q, Tan L, Yu J-T. The role of the LRRK2 gene in Parkinsonism. Mol Neurodegener. 2014;9:47. doi: 10.1186/1750-1326-9-47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bardien S, Lesage S, Brice A, Carr J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord. 2011;17:501–8. doi: 10.1016/j.parkreldis.2010.11.008.. [DOI] [PubMed] [Google Scholar]
- 100.Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 G2019S as a Cause of Parkinson’s Disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–5. doi: 10.1056/NEJMc055509.. [DOI] [PubMed] [Google Scholar]
- 101.Lesage S, Belarbi S, Troiano A, Condroyer C, Hecham N, Pollak P, et al. Is the common LRRK2 G2019S mutation related to dyskinesias in North African Parkinson disease? Neurology. 2008;71:1550–2. doi: 10.1212/01.wnl.0000338460.89796.06.. [DOI] [PubMed] [Google Scholar]
- 102.Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger A-L, Janin S, et al. LRRK2 G2019S as a Cause of Parkinson’s Disease in North African Arabs. N Engl J Med. 2006;354:422–3. doi: 10.1056/NEJMc055540.. [DOI] [PubMed] [Google Scholar]
- 103.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N Engl J Med. 2009;361:1651–61. doi: 10.1056/NEJMoa0901281.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016;98108:1–8. doi: 10.1001/jamaneurol.2016.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord. 2016;31:95–102. doi: 10.1002/mds.26359.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–90. doi: 10.1016/S1474-4422(08)70117-0.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78:417–20. doi: 10.1212/WNL.0b013e318245f476.. [DOI] [PubMed] [Google Scholar]
- 108.Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169:1177–86. doi: 10.1016/j.cell.2017.05.038.. [DOI] [PMC free article] [PubMed] [Google Scholar]