

Abstract
The recent introductions of low-cost, long-read, and read-cloud sequencing technologies coupled with intense efforts to develop efficient algorithms have made affordable, high-quality de novo sequence assembly a realistic proposition. The result is an explosion of new, ultracontiguous genome assemblies. To compare these genomes, we need robust methods for genome annotation. We describe the fully open source Comparative Annotation Toolkit (CAT), which provides a flexible way to simultaneously annotate entire clades and identify orthology relationships. We show that CAT can be used to improve annotations on the rat genome, annotate the great apes, annotate a diverse set of mammals, and annotate personal, diploid human genomes. We demonstrate the resulting discovery of novel genes, isoforms, and structural variants—even in genomes as well studied as rat and the great apes—and how these annotations improve cross-species RNA expression experiments.
Short-read sequencing prices continue to drop and new technologies are being combined to produce assemblies of quality comparable to those previously created through intensive manual curation (Chaisson et al. 2015; Gordon et al. 2016; Putnam et al. 2016; Weisenfeld et al. 2017; Jain et al. 2018; Kronenberg et al. 2018). These advances have allowed researchers to perform clade genomics, producing assemblies for multiple members of a species or clade (Jarvis et al. 2014; Lilue et al. 2018; Thybert et al. 2018), and are required for the ambitious goals of projects such as Genome 10K (Genome 10K Community of Scientists 2009), which aim to produce thousands of assemblies of diverse organisms. In addition, efforts are growing to produce de novo assemblies of individual humans to evaluate the human health implications of structural variation and variation within regions not currently accessible with reference-assisted approaches (Steinberg et al. 2014; Pendleton et al. 2015; Schneider et al. 2017).
These advances in genome assembly require subsequent advances in genome comparison. Central to this comparison is annotation. The challenge of finding functional elements in genome assemblies has been considered for at least the past 20 years (Haussler et al. 1996). This problem is traditionally approached by ab initio prediction (using statistical models of sequence composition) (Stanke et al. 2004) and sequence alignment of known mRNAs or proteins (Aken et al. 2016). The former has limited accuracy, whereas the latter is limited by the existence of useful sequence information. Annotation pipelines such as MAKER (Cantarel et al. 2008), RefSeq (Pruitt et al. 2006), and AUGUSTUS (Stanke et al. 2006) make use of both approaches (for a review of genome annotation methods, see Hoff and Stanke 2015).
A huge amount of effort has gone into the annotation of model organisms, in particular human and mouse. For the past five years, the GENCODE Consortium (Harrow et al. 2012) has used a wide range of sequencing and phylogenetic information to manually build and curate comprehensive annotation sets, with more than 43,281 and 60,297 open-reading frames in mouse and human, respectively. The GENCODE databases give a glimpse into the diversity of alternative isoforms and noncoding transcripts present in vertebrate genomes. Similarly, efforts in other model organisms, such as zebrafish (Westerfield et al. 1998), C. elegans (Stein et al. 2001), A. thaliana (Swarbreck et al. 2008), and many others, have produced high-quality annotation sets for their respective assemblies.
As we enter a third era of genome assembly, consideration should be given to scaling annotation. Here, we present a method and toolkit to make use of multiple genome alignments produced by Progressive Cactus (Paten et al. 2011) and existing high-quality annotation sets to simultaneously project well-curated annotations onto lesser studied genomes. In contrast to most earlier alignment methods (Blanchette et al. 2004; Miller et al. 2007; Earl et al. 2014), Progressive Cactus alignments are not reference based, include duplications, and are thus suitable for the annotation of many-to-many orthology relationships. We show how the output of this projected annotation set can be cleaned up and filtered through special application of AUGUSTUS (Stanke et al. 2008) and how novel information can be introduced by combining the projected annotation set with predictions produced by Comparative Augustus (König et al. 2016). These predictions can be further supplemented and validated by incorporating long-range RNA-sequencing (RNA-seq) data, such as those generated by the Iso-Seq protocol (Gordon et al. 2015). We provide a fully featured annotation pipeline, the Comparative Annotation Toolkit (CAT), that can perform this annotation process reproducibly on any combination of a local computer, a compute cluster, or on the cloud. We show that this pipeline can be applied to a wide range of genetic distances, from distant members of the same clade to individualized assemblies of the same species.
Results
Comparative Annotation Toolkit
CAT provides a software toolkit designed to perform end-to-end annotation; Figure 1 gives an overview. The only required inputs are a hierarchical alignment format (HAL) (Hickey et al. 2013) multiple genome alignment as produced by Progressive Cactus and a GFF3 format annotation file for the previously annotated genome(s). CAT can take as optional input a set of aligned RNA-seq or Iso-Seq BAM format files, as well as protein FASTA files, which are used to construct hints for AUGUSTUS.
Figure 1.

CAT pipeline schematic. The CAT pipeline takes as input a HAL alignment file, an existing annotation set, and aligned RNA-seq reads. CAT uses the Cactus alignment to project annotations to other genomes using transMap (Stanke et al. 2008). These transcript projections are then filtered and paralog resolved. Optionally, AUGUSTUS can be run in as many as four parameterizations. All transcripts are classified for extrinsic support and structure, and a “chooser” algorithm picks the best representative for each input transcript, incorporating ab initio transcripts when they provide novel supported information. The final consensus gene set, as well as associated feature tracks, are used to create an assembly hub ready to be loaded by the UCSC Genome Browser (for more detail, see Supplemental Fig. S1; Supplemental Methods).
TransMap (Zhu et al. 2007; Stanke et al. 2008) is used to project existing annotations between genomes using the Progressive Cactus alignment. TransMap projections are filtered based on a user-tunable flag for minimum coverage, and then the single highest scoring alignment is chosen. If this results in transcripts for a given gene mapping to multiple loci, these are resolved to one locus based on the highest average score of a locus, rescuing lower scoring alignments.
Based on input parameters, CAT will run AUGUSTUS in up to four distinct parameterizations, two of which rely on transMap projections (AugustusTMR) and two that perform ab initio predictions (AugustusCGP and AugustusPB) using extrinsic information to guide prediction. AugustusCGP performs simultaneous comparative prediction (König et al. 2016) on all aligned genomes, whereas AugustusPB uses long-read RNA-seq to discover novel isoforms. The output of these modes of AUGUSTUS are evaluated alongside the original transMap projections using a combination of classifiers as well as the output from homGeneMapping (Stanke et al. 2004), which uses the Cactus alignments to project features such as annotations and RNA-seq support between the input genomes. AugustusCGP and AugustusPB transcript predictions are assigned to transMap genes based on genomic and exonic overlap. If they overlap projections that were filtered out in the paralog resolution process, then they are flagged as putatively paralogous; if they do not overlap any transMap projections, they are flagged as putatively novel. A consensus-finding algorithm combines these gene sets.
The consensus-finding algorithm combines all sources of transcript evidence into an annotation set. On a per-gene basis, it evaluates the transMap transcripts for passing user-tunable flags for RNA-seq and annotation support. It then considers the inclusion of ab initio transcripts based on their assignment to this locus and their contribution of novel splice junctions supported by RNA-seq or Iso-Seq. Finally, it evaluates ab initio transcripts not assigned to a gene as novel loci if they are supported by RNA-seq or Iso-Seq as defined by user-tunable flags. For a more detailed description of CAT, see Supplemental Methods.
Annotation of great apes
The previous generation of great ape assemblies (panTro4, ponAbe2, and gorGor4), as well as the new SMRT Pacific Biosciences (PacBio) great ape assemblies (Gordon et al. 2016; Kronenberg et al. 2018), were annotated by CAT by using GRCh38 and GENCODE V27 as the reference. On average, CAT identified 141,477 more transcripts and 25,090 more genes in the new SMRT assemblies of the great apes compared to the Ensembl V91 annotation of the previous generation of great ape assemblies. Relative to the existing human annotation, the CAT annotations represent an average of 95.0% of GENCODE gene models and 94.3% of GENCODE isoforms in the SMRT great ape assemblies. This increase in isoform representation is mostly due to the large number of isoforms annotated by GENCODE and reproduced in these genomes, whereas the increase in gene content is due to the mapping over of noncoding genes poorly represented in the Ensembl annotation. Comparing the CAT annotations of SMRT great apes and older assemblies, we see an average increase of 610 genes (1.9%) and 3743 isoforms (1.0%) (Supplemental Fig. S2) in the SMRT assemblies; given this relatively small increase, most of the observed increase in genes and isoforms in the CAT annotations relative to the Ensembl annotations are therefore a result of the CAT annotation process rather than the updated assemblies.
In contrast to the overall increases in genes and isoforms, CAT identifies on average 3553 fewer protein-coding genes than Ensembl. However, this brings the total number of coding genes more closely in line with the GENCODE annotation of human, as Ensembl has an average of 2081 more protein-coding genes in great apes than GENCODE has for human (Supplemental Fig. S2).
To evaluate these annotations in a non-species-biased fashion, consensus isoform sequences created from Iso-Seq reads for each species were compared to their respective species annotations. As a baseline comparison, equivalent human data were compared to the high-quality human GENCODE V27 annotation. The CAT annotation of both the SMRT and older great ape assemblies (which used the raw Iso-Seq reads during the annotation process) and the Ensembl annotation of the older assemblies were compared. We calculated the rate of isoform concordance, that is the fraction of consensus Iso-Seq sequences that match either exactly or fuzzily an annotated isoform (Fig. 2A; Methods). Fuzzy matching allows for the intron boundaries to shift slightly in a isoform. For the SMRT chimpanzee (74.0%/82.1% exact/fuzzy matching) and orangutan (71.4%/80.4%) genome assemblies the isoform concordance rates were comparable to the rate for human (74.6%/82.1%). The gorilla GSMRT3.2 assembly showed lower concordance (67.6%/76.9%), likely due to the higher indel error rate in that assembly (Supplemental Fig. S3). In contrast, the isoform concordance rate for the older assemblies was lower (on average 60.0%/69.6%), mostly reflecting exons in gaps and mis-joins, and was lower still for the existing Ensembl annotations (on average 47.9%/57.6%).
Figure 2.

Primate annotation. (A) Validating CAT annotations using Iso-Seq data. As a baseline comparison, Iso-Seq data from human iPSCs were compared to the GENCODE V27 annotation. Iso-Seq data from chimpanzee, gorilla, and orangutan iPSC lines were compared to respective species-specific annotations. The Iso-Seq data were clustered with isoform-level clustering (ICE) and collapsed using ToFU (Gordon et al. 2015). CAT annotation of PacBio great apes showed similar isoform concordance to human and improvement over the older assemblies. (B) Kallisto (Bray et al. 2016) was used to quantify liver Illumina RNA-seq from each species on both the gene and transcript level on the existing and new great ape assemblies. Solid bars are transcripts or genes with transcripts per million (TPM) >0.1, whereas shaded hatched bars are the remainder of the annotation sets. CAT annotation of great apes shows nearly the same number of expressed genes and isoforms as the GENCODE reference on human with the exception of orangutan. (C) The number of novel isoforms and paralogous genes with Iso-Seq support discovered by analysis of AugustusPB and AugustusCGP predictions for each species. (D) Kallisto protein-coding gene-level expression for chimpanzee iPSC RNA-seq is compared to human across all of the chimpanzee annotation and assembly combinations as well as when mapped directly to human. In all cases, the x-axis is the TPM of human iPSC data mapped to human. The highest correlation (Pearson r = 0.96) is seen when comparing Clint annotated with CAT to GRCh38 annotated with GENCODE V27.
To assess the utility of CAT annotations for short-read analysis of RNA expression, species-specific induced pluripontent stem cell (iPSC) Illumina RNA-seq data were quantified (Fig. 2B). Comparing the annotations of the older assemblies, CAT identified an average of 9518 more genes and 54,107 more transcripts with measurable expression compared to Ensembl.
We might expect the per-gene abundance estimates of the majority of genes in matched cell types to agree between species, particularly for closely related species. It is reasonable to therefore prefer a priori an annotation of the great apes that produces expression estimates that agree with expression estimates from the matched human data using the GENCODE annotation. Doing these comparisons, we find better correlations on average using the CAT annotation of the older assemblies (average Pearson r = 0.63) (Fig. 2D; Supplemental Fig. S4) than the Ensembl annotations of the older assemblies (average Pearson r = 0.44). However, we find by far the highest correlation when CAT annotates the SMRT primate assemblies (average Pearson r = 0.90). This reflects the increased representation in the updated assemblies of transcript sequence, especially 3′ UTRs that are important for quantifying poly(A) primed RNA-seq (Kronenberg et al. 2018). Notably, we find that the correlations between the CAT annotations of the SMRT assemblies and the matched human data are higher than when mapping the species-specific data back to the human GENCODE annotations and comparing to the human data (Fig. 2D; Supplemental Fig. S4), demonstrating the benefit of having species-specific annotations within closely related species that have clear cross-species orthology relationships. Analysis at the isoform level showed the same patterns (Supplemental Fig. S5), albeit with slightly weaker correlations.
Predictions performed by AugustusCGP and AugustusPB were incorporated into the gene sets based on the presence of splice junctions supported by RNA-seq or Iso-Seq and not present in the transMap/AugustusTMR-derived annotations (Fig. 2C). An average of 1677 novel isoforms and 64 novel loci were found across the assemblies with at least one Iso-Seq read supporting the prediction.
CAT provides new metrics for diagnosing assembly quality. In the process of annotating the great ape genomes, we noticed that assemblies that had undergone Quiver and Pilon (Walker et al. 2014) correction still exhibited a systematic bias toward coding deletions. These were identified to be related to heterozygosity in the input data set, and a variant calling–based correction method (Kronenberg et al. 2018) was developed to resolve these issues, dramatically lowering the coding indel rate and reducing systematic bias (Supplemental Fig. S3). CAT can also measure gene assembly contiguity by reporting the number of genes whose transcripts end up split across multiple contigs, or on disjoint intervals in the same contig. Comparison of split gene metrics between the old and new primate assemblies shows 504 fewer split genes in chimpanzee, 560 fewer in gorilla, and 1858 fewer in orangutan (Supplemental Fig. S6).
Annotation of personal human diploid assemblies
High-quality de novo assembly of a human genome is increasingly feasible; both Pacific Biosciences (Chin et al. 2016; Huddleston et al. 2017; Korlach et al. 2017) (Falcon) and 10x Genomics (Weisenfeld et al. 2017) (Supernova) provide tools to construct phased, diploid assemblies. Annotating diploid assemblies provides a window into haplotype-specific structural variation that may affect gene expression. To evaluate the ability of CAT to provide this analysis, Progressive Cactus alignments were generated between hg38 and the two haploid cell line assemblies, CHM1 (GCA 001297185.1) and CHM13 (GCA 000983455.2), as well as the 10x Genomics diploid assemblies of four individuals (NA12878, NA24385, HG00512, and NA19240).
An average of 98.5% of genes present in GENCODE V27 were identified in CHM1/CHM13, and an average of 97.3% of genes were identified in the 10x Genomics Supernova assemblies (Fig. 3A). After filtering, an average of 552 genes in the PacBio assemblies and 461 genes in the 10x assemblies had frame-shifting indels (Fig. 3B). Compared to ExAC, which found between 75 and 125 putative truncating events per individual (Karczewski et al. 2017), this result suggests indel errors in the assemblies are producing false positives. All assemblies exhibit systematic overrepresentation of deletions, including the PacBio assemblies despite coming from haploid cell lines (Fig. 3B). Split gene analysis found the CHM1 assembly to be the most gene contiguous, with only 39 genes split across multiple contigs, and the PacBio assemblies overall more contiguous (Fig. 3C). Gene contiguity is measured by looking at genes with multiple alignments post-paralog resolution that start and end nearby in transcript coordinates.
Figure 3.

Pseudodiploid human annotation metrics. (A) The number and fraction of genes comparatively annotated from GENCODE V27 in each assembly. GENCODE biotypes are simplified into protein coding, lncRNA, ncRNA, pseudogene, and other. Other includes processed transcripts, nonsense-mediated decay, and immune-related genes. (B) Frame-shifting insertions, deletions, and multiple of three indels that do not shift frame are reported for each assembly. Consistent with the great ape genomes, there is a systematic overrepresentation of coding deletions in Falcon assemblies, despite these assemblies coming from haploid cell lines. 10x Genomics Supernova assemblies also exhibit similar properties. (C) Split gene analysis reports how often paralog-resolved transcript projections end up on different contigs, which can measure assembly gene-level contiguity. PacBio assemblies, especially CHM1, are the most contiguous.
Manual analysis of genes with different transMap coverage in CHM13 relative to CHM1 led to the discovery of the example region in Figure 4A. This deletion removes most of the exons of TRIB3, a pseudokinase associated with type 2 diabetes (Supplemental Fig. S7; Shi et al. 2009). Similar analysis in the diploid assembly of NA12878 led to the discovery of a tandem duplication involving an exon of TAOK3 in one haplotype (Fig. 4B).
Figure 4.

Pseudodiploid human annotation examples. (A) UCSC Assembly Hub (Nguyen et al. 2014) showing TRIB3 deletion in CHM1. Analysis of genes found in one genome and not the other led to the discovery of a novel structural variant specific to CHM1, which disables the gene TRIB3. Spanning reads were found in both PacBio and Illumina whole-genome sequencing that validate the deletion. (B) An example insertion near an exon of TAOK3 seen in one haplotype of NA12878. It was not possible to determine if this insertion affects transcription of this gene.
Reannotating the rat genome
We tested CAT's ability to reannotate the rat genome using information from the mouse genome. These genomes differ by approximately 0.18 substitutions/site, much more, for example, than the 0.04 substitutions/site separating the human and orangutan genomes (Karolchik et al. 2003).
CAT was run on a Cactus alignment between mouse (mm10) and rat (rn6) using rabbit (oryCun2), Egyptian jerboa (jacJac1), and human (hg38) as outgroups. To provide hints to AUGUSTUS, RNA-seq data were obtained from the NCBI Sequence Read Archive (SRA) (Supplemental Table S2; Cortez et al. 2014; Fushan et al. 2015; Liu et al. 2016). For comparison we used existing Ensembl and RefSeq rat annotations and ran the MAKER2 pipeline (Holt and Yandell 2011) to generate an annotation set. MAKER2 was provided both a Trinity (Haas et al. 2013) de novo assembly of the input RNA-seq data provided to CAT (MAKER2 does not process raw RNA-seq) as well as the mouse protein sequences from GENCODE VM11, together providing a comparable input set to what CAT had.
CAT comparatively annotated 78.1% of genes and 91.9% of protein-coding genes present in GENCODE VM11 on rn6 (Supplemental Fig. S8), representing an increase of 14,675 genes and 74,308 transcripts over Ensembl V90, 5104 genes and 32,157 transcripts over RefSeq, and 14,541 genes and 81,022 transcripts over MAKER2. A total of 13,651 loci were identified with no overlap to any other annotation set (Supplemental Fig. S9).
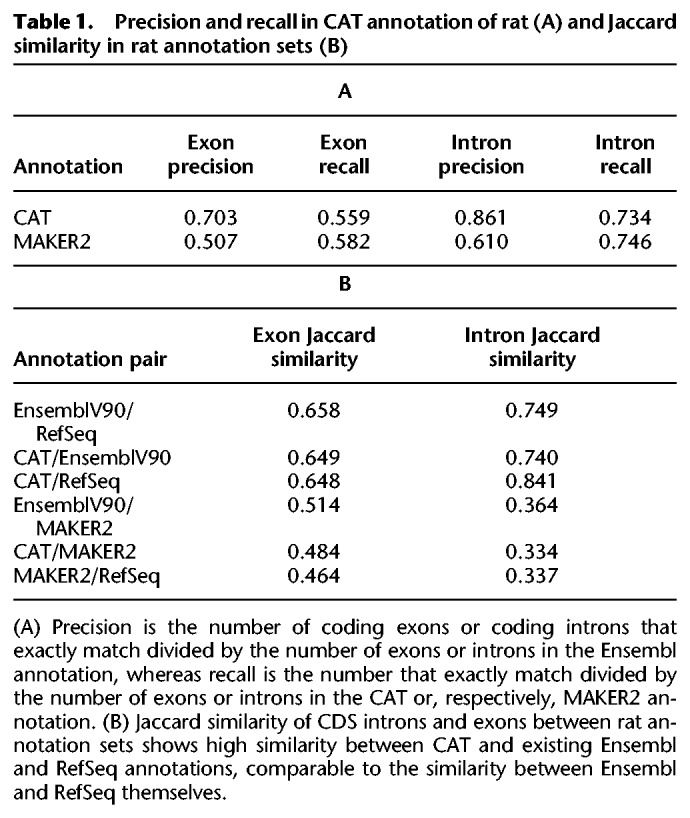
We compared CDS exon and CDS intron predictions between annotation sets (Table 1A; Supplemental Fig. S10). We measured precision and recall of coding intron and exon intervals based on comparing the CAT annotation to EnsemblV90, in which precision is the proportion of CAT exons/introns that exactly match Ensembl, and recall is the proportion of CAT exons/introns that exactly match Ensembl over the number of exons/introns CAT annotated. Ensembl, RefSeq, and CAT CDS exon annotations were all comparably similar (between 0.648 and 0.659 Jaccard similarity); for CDS introns, CAT and RefSeq displayed the highest Jaccard similarity (0.841). In all comparisons, MAKER2 was the outlier (Table 1B) with the lowest similarity to the other sets.
Table 1.
Precision and recall in CAT annotation of rat (A) and Jaccard similarity in rat annotation sets (B)
The input RNA-seq data set was used for isoform quantification against the CAT, MAKER2, Ensembl, and RefSeq transcrip- tomes (Fig. 5A). CAT identified 1881 protein-coding genes and 1011 lncRNAs with measurable expression not present in either Ensembl or RefSeq. CAT also identified 27,712 expressed coding splice junctions not in the union of RefSeq and Ensembl, for a total of 21,267 novel expressed isoforms. Of the 13,651 loci, 5526 unique to CAT had measurable expression.
Figure 5.
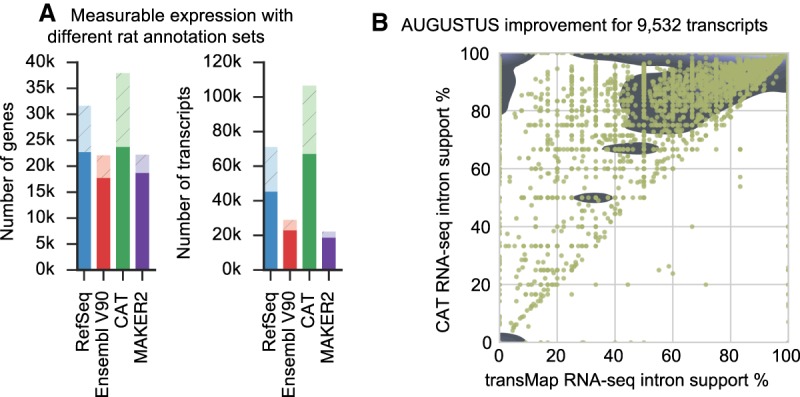
Validation of CAT annotation using rat. (A) Each transcript set was used to construct a Kallisto (Bray et al. 2016) index, and then all the input RNA-seq for annotation were quantified. Solid bars are genes or transcripts with nonzero expression (TPM>0.1) estimates, and light hatched bars are the remainder of the annotation set. CAT provides an annotation set with slightly more detectable genes than other annotation methods and far more detectable isoforms. (B) AugustusTMR provides a mechanism to clean up transcript projections and shift splice sites, fixing alignment errors as well as real evolutionary changes. Most of the 9532 AugustusTMR transcripts chosen in consensus finding show an improvement in RNA-seq support, which is one of the features used in consensus finding.
AugustusTMR, which uses transMap and RNA-seq, provides CAT with a way to improve transcript predictions projected between species. Comparing the 9532 multiexon protein-coding transcripts in which the AugustusTMR prediction differed from the input transMap projection, we see considerable overall improvement in resulting RNA-seq support of predicted splice boundaries in the AugustusTMR transcripts (Fig. 5B).
Annotation of a diverse set of mammals
Finally, to test CAT's ability to annotate across a substantial and diverse range of genomes, 13 mammalian genomes were comparatively annotated using the mouse (mm10) GENCODE VM15 as the reference transcript set (Fig. 6A). Species-specific RNA-seq was used for every genome (Supplemental Table S2). To assess the completeness of these annotation sets, 4104 benchmarking universal single-copy orthologs (BUSCO) were used (Simão et al. 2015), which by design should be nearly uniformly present in each of these genomes. On average, only 108 BUSCO genes (2.63%) were not annotated by CAT in each genome (Supplemental Table S1).
Figure 6.

Thirteen-way annotation. (A) The phylogenetic guide tree for 14-mammal alignment. See Methods for the exact Newick format tree. (B) The gene annotation sets for each of the 13 mammalian genomes were quantified against the mixed input RNA-seq sets obtained from SRA. Genes or transcripts with TPM>0.1 are solid colors, whereas genes or transcripts with no measurable expression are shaded. An average of 2.8 isoforms per gene per genome had quantifiable expression, suggesting that CAT can infer isoform information across long branch lengths.
To estimate the usefulness of these annotation sets, the input RNA-seq data sets were used to quantify expression of the annotation sets (Fig. 6B). The main factor in measurable expression is the variety of the input RNA-seq data sets, as exemplified by the ability to measure expression of 88.9% of genes annotated in the sheep genome.
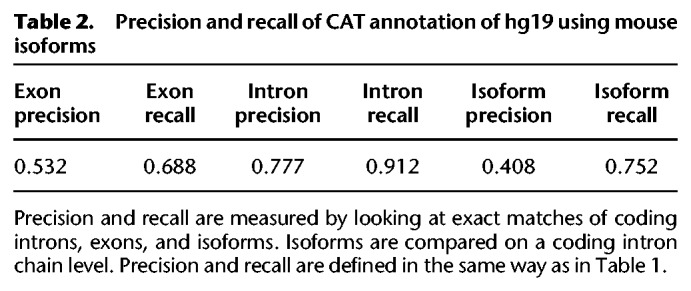
To assess the CAT translation of annotations over large phylogenetic distances, as well as provide a baseline validation of CAT performance, the annotation of human hg19 (GRCh37) produced in the representative mammalian genome annotation was compared to the current human GENCODE annotation set for that assembly (GENCODE V27lift37). Of the 19,233 ICE isoforms detected when running ToFU (Gordon et al. 2015) against hg19, 12,911 (67.2%) fuzzy matched a CAT isoform compared to 15,920 (82.8%) of the human GENCODE annotations. Precision and recall analysis shows results similar to the rat annotation, with better matches in introns; 91.2% of CAT introns and 75.2% of CAT protein coding isoforms match GENCODE (Table 2).
Table 2.
Precision and recall of CAT annotation of hg19 using mouse isoforms
Discussion
Gene annotation is a longstanding and critical task in genome informatics that must now be scaled to handle the rapidly increasing number of available genomes. At the time of writing, there were 570 vertebrate genomes available from NCBI, but only 100 (17.5%) and 237 (41.6%) had Ensembl and RefSeq annotations, respectively.
We introduce CAT to help meet this need, building around a number of key innovations. First, CAT utilizes the reference- free, duplication-aware multiple genome alignments we have developed. This allows CAT to annotate multiple genomes symmetrically and simultaneously, breaking from the traditional pattern of annotating each new genome individually, as is currently the practice for the RefSeq, Ensembl, and MAKER2 gene-building pipelines. Not only does this solve a key scalability issue by annotating multiple genomes simultaneously and consistently, CAT is able to produce orthology mappings, naming each equivalence class of orthologs based upon an initial reference annotation, and add to this sets of newly discovered genes. This can provide valuable evolutionary insights. For example, the analysis of the rat genome shows that many of the alternative isoforms and projected transcription start sites identified by GENCODE in mouse genes are supported by expression analysis in rat (Supplemental Fig. S11).
A second key innovation made by CAT is its leveraging of existing reference annotations. A huge amount of effort has been placed into the annotation of key species, such as human and mouse, using a myriad of technologies and extensive, labor-intensive manual curation. It is very unlikely that this effort will be replicated across a significant fraction of other genomes, so instead we propose the “project and augment” strategy used by CAT to annotate related genomes. Here, we show that this strategy is very clearly able to improve the annotation of great ape genomes, using the human GENCODE set as the reference, and we make the case that we can even improve the annotation of a genome as well studied as the rat.
To circumvent the reference bias of existing annotations and to discover new genes and isoforms, CAT is able to integrate multiple forms of extrinsic information, using multiple, novel parameterizations of the AUGUSTUS algorithms. This includes use of new long-read RNA data, in particular Iso-Seq data, and shortly will integrate Nanopore-based long-read data (Byrne et al. 2017). Using this expression data not only allowed us to confirm expression of a substantial fraction of isoforms, but allowed us to discover thousands of novel isoforms and dozens of novel genes in the great ape genomes.
With the advent of more affordable de novo genome assembly, there is renewed interest in the generation of de novo human genomes, and in general, the creation of multiple de novo genomes for a species. This has the advantage of providing fully independent reconstruction and is particularly appropriate for sequences that are highly divergent from the reference, e.g., structural variations. However, such assemblies do not negate the need for genome comparison. Cactus can be parameterized to rapidly create sensitive whole-genome alignments of human genomes, and here we have demonstrated that CAT can be used to build upon this to produce a high-quality diploid gene annotation and ortholog mapping.
CAT works best when provided RNA-seq data, but for many species this may not be possible. From our experience, a reasonable amount (on the order of 50 million reads) of RNA-seq from tissues like brain and liver is fairly informative. Using poly(A)-selected libraries is recommended, because it greatly reduces false positive predictions in AugustusCGP. Iso-Seq data allowed for the discovery of thousands of novel isoforms in the great apes but may be too expensive for many projects. In clade genomics projects, we would suggest generating RNA-seq for a few of the species and then relying on the coordinate mapping that AugustusCGP and homGeneMapping provide to evaluate support in other members of the clade.
A key barrier to the use of bioinformatics tools is their ease of use; we have focused on providing cloud agnostic distributions of the CAT software so that, despite its complexity, it can be run within a uniform computational environment by external groups.
CAT is not without limitations. In the future it would be good to use the genome alignments to not only project transcripts, but to use the evolutionary conservation signatures to predict the potential likelihood of projected annotations being coded (Lin et al. 2011). CAT also does not yet provide means to detect new processed, unprocessed, and unitary pseudogene predictions other than via projection of existing annotations. CAT's current implementation also does not attempt to put weights on the features used for constructing a consensus gene set. Instead, it simply scores transcripts based on the sum of all features evaluated. In the future, deep learning methods could be added to CAT to construct feature weights and improve consensus finding, better mimicking the labor-intensive efforts of manual annotators who currently weigh such evidence.
An earlier version of CAT was used to annotate the PacBio-based assembly of the gorilla genome (Gordon et al. 2016) as well as produce the current Ensembl annotations for 16 laboratory mouse strains as part of the Mouse Genomes Project (Lilue et al. 2018) (http://www.sanger.ac.uk/science/data/mouse-genomes-project). In addition, CAT has been proposed for the Vertebrate Genomes Project (VGP), which aims to be a pilot project to assemble and annotate one member of every order of vertebrate species. CAT also will be used on the 200 Mammals Project, which aims to add about 140 new mammalian genome assemblies to the existing set (https://karlssonlab.org/2017/08/03/the-200-mammals-project/). These projects will provide a new understanding of gene evolution.
Methods
CAT produces as output a series of diagnostic plots, an annotation set for each target genome, and a UCSC comparative assembly hub (Nguyen et al. 2014). Both the pipeline and associated documentation can be found at https://github.com/ComparativeGenomicsToolkit/Comparative-Annotation-Toolkit. CAT is constructed using the Luigi workflow manager (https://github.com/spotify/luigi), with Toil (Vivian et al. 2017) used for computationally intensive steps that work best when submitted to a compute cluster.
RNA-seq
CAT annotation is improved when species-specific RNA-seq data are provided. These data are used as hints for AugustusTMR and AugustusCGP. In AugustusTMR, RNA-seq helps fill in missing information in the alignment and resolve evolutionary changes. In AugustusCGP, RNA-seq additionally helps prevent false positives inherent in ab initio gene finding. For these reasons, RNA-seq was obtained from SRA for all species annotated in this paper. All RNA-seq were aligned to their respective genomes with STAR (Dobin et al. 2013), and the resulting BAM files were passed to CAT to construct the extrinsic hints database. See Supplemental Table S2 for accessions and tissue types of RNA-seq data used for annotation. In addition, for the PacBio great ape annotation, RNA-seq data were generated using iPSC lines for human, chimpanzee, gorilla, and orangutan derived from cells from the same individuals as the assemblies (Kronenberg et al. 2018). For all expression analyses, Kallisto (Bray et al. 2016) was used.
Annotation set similarity analysis
Jaccard similarity analysis was performed with BEDTools (Quinlan and Hall 2010). The rat locus overlap analysis was performed with the Kent tool clusterGenes, which requires exonic overlap on the same strand.
Iso-Seq
Iso-Seq full-length nonchimeric reads (FLNC) were also generated from the great ape iPSC lines and aligned with GMAP (Wu and Watanabe 2005). To perform isoform-level validation in the primates, the Iso-Seq data used as input to CAT were also clustered with isoform-level clustering (ICE) and then collapsed into isoforms using ToFU (Gordon et al. 2015). Ensembl provided a pre-release of their new V91 annotations for panTro4 and gorGor4, but did not yet run their updated pipeline on ponAbe2.
ICE validation
The output transcripts from ICE were compared to various annotation sets in both an exact and fuzzy matching scheme. In the exact scheme, the genomic order and positions of all of the introns (an intron chain) of a transcript are compared to any ICE isoforms which overlap it. In the fuzzy matching scheme, each annotated intron chain is allowed to move up to 8 bases in either direction and still be called a match.
BUSCO
The mammalian BUSCO (Simão et al. 2015) analysis was performed using the mammalia odb9 set of 4104 genes. BUSCO was run against the complete protein-coding sequence repertoire produced by CAT in that species in the “protein” mode.
Progressive Cactus
All Cactus alignments, except the 14-way mammal alignment, were generated using Progressive Cactus (https://github.com/glennhickey/ProgressiveCactus) commit 91d6344. For the mouse-rat alignment, the guide tree was
(((Lesser_Egyptian_jerboa:0.1,(Mouse:0.084509,Rat:0.091589)mouse_rat:0.107923)rodent :0.148738,Rabbit:0.21569)glires:0.015313,Human:0.143908).
For the primate alignments, the guide tree was
(((((((Susie_Gorilla:0.008964,(Human:0.00655,Clint_Chimp:0.00684)human_chimp:0.00122) gorilla_chimp_human:0.009693, Susie_Orangutan: 0.01894)great_ape:0.003471,Gibbon:0.02227) great_ape_gibbon:0.01204,Rhesus:0.004991)old_ world_monkey:0.02183,Squirrel_monkey:0.01035)monkey:0.05209,Bushbaby:0.1194)primate_anc: 0.013494,Mouse:0.084509).
An identical tree (with different assembly names) was used for the alignment of current reference great apes.
For the diploid human alignments, the two haploid cell lines (PacBio) or all human haplotypes (10x) were placed under the same node with a very short branch length, with chimpanzee as outgroup. The guide trees were
((hg38:0.001,chm1:0.001,chm13:0.001)human:0.01,chimp:0.01)
and
((hg38:0.001,HG00512-H1:.001,HG00512-H2:.001,NA12878-H1:.001,NA12878-H2:.001,NA19240-H1:.001, NA19240-H2:.001,NA24385-H1:.001, NA24385-H2:.001)human:0.01,chimp:0.01),
representing a star phylogeny of the three human assemblies. For the 14-way mammal alignment, the Progressive Cactus commit used was e3c6055 and the guide tree was
((((oryCun2:0.21,((Pahari_EiJ:0.03,mm10:0.025107)1:0.02,rn6:0.013)1:0.252)1:0.01,((((hg19: 0.00642915,panTro4:0.00638042)1:0.00217637,gorGor3:0.00882142)1:0.00935116,ponAbe2: 0.0185056)1:0.00440069,rheMac3:0.007)1:0.1)1:0.02,((oviAri3:0.019,bosTau8:0.0506) 1:0.17, (canFam3:0.11,felCat8:0.08)1:0.06)1:0.02)1:0.02,loxAfr3:0.15).
Slightly out-of-date versions of some assemblies (hg19 and rheMac3) were used because a collaborator had data on those assemblies that they wished to use the alignment to analyze. The rodent and primate subtrees were first aligned separately (the rodent subtree originally included additional mouse strains) (Lilue et al. 2018; Thybert et al. 2018). The two subtrees were then “stitched” together into a single alignment by aligning together their roots along with several Laurasiatheria genomes. This was done to save alignment time by reusing existing alignments.
CAT
CAT was run on the UCSC Genome Browser compute cluster for all annotation efforts in this publication. CAT commit f89a814 was used. For a detailed description of how CAT works, see both the Supplemental Text as well as the README.md on GitHub (https://github.com/ComparativeGenomicsToolkit/Comparative-Annotation-Toolkit).
Pipeline runtime
CAT is relatively efficient, taking on the order of thousands of core hours to run. The largest considerations for runtime are running the various parameterizations of AUGUSTUS as well as generating the required Cactus alignment. AugustusCGP may run significantly faster on alignments with many genomes by reducing the chunk size from the default, but at the cost of lower quality predictions. AugustusTMR runtime scales linearly with the number of protein-coding transcripts in the input annotation set, but scales nonlinearly with the number of extrinsic hints provided, particularly if the hints are contradictory.
All of the analyses in this paper were run on the UCSC cluster, which uses the cluster management tool Parasol and has 1024 cores with 8 GB of RAM per core. CAT was optimized for this and should not need more memory per core in any case except the AugustusCGP step when the number of aligned genomes exceeds approximately 10. This can be adjusted by reducing the alignment chunk size that AugustusCGP is given to work with. For example, for the 14-way mammalian analysis, the flags --maf-chunksize 1000000 --maf-overlap 200000 were set, which kept memory usage under 8 GB.
Cactus alignments take on the order of 120 CPU days (2880 core h) per internal node on the guide tree, assuming a binary tree. This number can fluctuate by a factor of 2–4 depending on how similar the two genomes being aligned at that node are. Cactus alignments are a mix of high CPU low memory steps with a few high memory steps, with some jobs requiring ∼240 GB of RAM.
Running CAT on the PacBio primate genomes took a total of 7030 core hours, with 3437 of those dedicated to running AugustusTMR, 1191 dedicated to running AugustusPB, and 2190 dedicated to running AugustusCGP. Running CAT on the 14-way mammalian alignment took a total of 24,122 core h, with 14,045 of those dedicated to running AugustusTMR and 8225 dedicated to running AugustusCGP.
Software availability
CAT is available on GitHub (https://github.com/ComparativeGenomicsToolkit/Comparative-Annotation-Toolkit). The exact commit used for these analyses is also in Supplemental Materials.
Competing interest statement
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc., and I.T.F. is an employee of 10x Genomics.
Supplementary Material
Acknowledgments
We thank Brian Raney, Hiram Clawson, and the rest of the UCSC Genome Browser team for help creating browser tracks. We also thank James Kent for allowing UCSC Genome Browser compute resources to be used for this project. Finally, we thank Fergal Martin and Paul Flicek for revising the paper and providing a pre-release of the Ensembl V91 annotations on chimpanzee and gorilla, as well as the whole GENCODE Consortium for their support and advice. This work was supported, in part, by US National Institutes of Health (NIH) grants U24HG009081 and U41HG007635 to E.E.E., HG007990 to D.H. and B.P., and HG007234 to B.P. E.E.E. and D.H. are investigators of the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.233460.117.
References
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, et al. 2016. The Ensembl gene annotation system. Database 2016: baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. [DOI] [PubMed] [Google Scholar]
- Byrne A, Beaudin A, Olsen H, Jain M, Cole C, Palmer T, DuBois R, Forsberg E, Akeson M, Vollmers C. 2017. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun 8: 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Alvarado AS, Yandell M. 2008. Maker: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18: 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Wilson RK, Eichler EE. 2015. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16: 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, Grützner F, Kaessmann H. 2014. Origins and functional evolution of Y chromosomes across mammals. Nature 508: 488–493. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. Star: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl D, Nguyen N, Hickey G, Harris RS, Fitzgerald S, Beal K, Seledtsov I, Molodtsov V, Raney BJ, Clawson H, et al. 2014. Alignathon: a competitive assessment of whole-genome alignment methods. Genome Res 24: 2077–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushan AA, Turanov AA, Lee SG, Kim EB, Lobanov AV, Yim SH, Buffenstein R, Lee SR, Chang KT, Rhee H, et al. 2015. Gene expression defines natural changes in mammalian lifespan. Aging Cell 14: 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genome 10K Community of Scientists. 2009. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered 100: 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SP, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, Kang D, Underwood J, Grigoriev IV, Figueroa M, et al. 2015. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One 10: e0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Huddleston J, Chaisson MJ, Hill CM, Kronenberg ZN, Munson KM, Malig M, Raja A, Fiddes I, Hillier LW, et al. 2016. Long-read sequence assembly of the gorilla genome. Science 352: aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8: 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler D, Reese MG, Eeckman FH. 1996. A generalized hidden Markov model for the recognition of human genes in DNA. In Proceedings of the international conference on intelligent systems for molecular biology, pp. 134–142, St. Louis, MO. [PubMed] [Google Scholar]
- Hickey G, Paten B, Earl D, Zerbino D, Haussler D. 2013. HAL: a hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics 29: 1341–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff K, Stanke M. 2015. Current methods for automated annotation of protein-coding genes. Curr Opin Insect Sci 7: 8–14. [DOI] [PubMed] [Google Scholar]
- Holt C, Yandell M. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12: 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Chaisson MJ, Steinberg KM, Warren W, Hoekzema K, Gordon DS, Graves-Lindsay TA, Munson M, Kronenberg ZN, Vives L, et al. 2017. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res 27: 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, Ho SY, Faircloth BC, Nabholz B, Howard JT, et al. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346: 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha KE, Cummings BB, et al. 2017. The ExAC browser: displaying reference data information from over 60,000 exomes. Nucleic Acids Res 45: D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu Y, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res 31: 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König S, Romoth L, Gerischer L, Stanke M. 2016. Simultaneous gene finding in multiple genomes. Bioinformatics 32: 3388–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J, Gedman G, Kingan SB, Chin CS, Howard JT, Audet JN, Cantin L, Jarvis ED. 2017. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience 6: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, Underwood JG, Nelson BJ, Chaisson MJP, Dougherty ML, et al. 2018. High-resolution comparative analysis of great ape genomes. Science 360: eaar6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilue J, Doran AG, Fiddes IT, Abrudan M, Armstrong J, Bennett R, Chow W, Collins J, Czechanski A, Danecek P, et al. 2018. Multiple laboratory mouse reference genomes define strain specific haplotypes and novel functional loci. bioRxiv 10.1101/235838. [DOI] [PMC free article] [PubMed]
- Lin MF, Jungreis I, Kellis M. 2011. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27: i275–i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Pan L, Zhang M, Bo L, Li C, Liu Q, Wang L, Jin F. 2016. Identification of distinct genes associated with seawater aspiration-induced acute lung injury by gene expression profile analysis. Mol Med Rep 14: 3168–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Rosenbloom K, Hardison RC, Hou M, Taylor J, Raney B, Burhans R, King DC, Baertsch R, Blankenberg D, et al. 2007. 28-way vertebrate alignment and conservation track in the UCSC Genome Browser. Genome Res 17: 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N, Hickey G, Raney BJ, Armstrong J, Clawson H, Zweig A, Karolchik D, Kent WJ, Haussler D, Paten B, et al. 2014. Comparative assembly hubs: web-accessible browsers for comparative genomics. Bioinformatics 30: 3293–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, Earl D, Nguyen N, Diekhans M, Zerbino D, Haussler D. 2011. Cactus: algorithms for genome multiple sequence alignment. Genome Res 21: 1512–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, Stütz AM, Stedman W, Anantharaman T, Hastie A, et al. 2015. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods 12: 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. 2006. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35(suppl 1): D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, O'Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. 2016. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res 26: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Liu J, Guo Q, Ma X, Shen L, Xu S, Gao H, Yuan X, Zhang J. 2009. Association of TRB3 gene Q84R polymorphism with type 2 diabetes mellitus in Chinese population. Endocrine 35: 414–419. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. Augustus: a web server for gene finding in eukaryotes. Nucleic Acids Res 32(suppl 2): W309–W312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S. 2006. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D. 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644. [DOI] [PubMed] [Google Scholar]
- Stein L, Sternberg P, Durbin R, Thierry-Mieg J, Spieth J. 2001. WormBase: network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res 29: 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Schneider VA, Graves-Lindsay TA, Fulton RS, Agarwala R, Huddleston J, Shiryev SA, Morgulis A, Surti U, Warren WC, et al. 2014. Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res 24: 2066–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, Li D, Meyer T, Muller R, Ploetz L, et al. 2008. The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36(suppl 1): D1009–D1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thybert D, Roller M, Navarro FC, Fiddes I, Streeter I, Feig C, Martin-Galvez D, Kolmogorov M, Janousěk V, Akanni W, et al. 2018. Repeat associated mechanisms of genome evolution and function revealed by the Mus caroli and Mus pahari genomes. Genome Res 10.1101/gr.234096.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, et al. 2017. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35: 314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. 2017. Direct determination of diploid genome sequences. Genome Res 27: 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M, Doerry E, Kirkpatrick AE, Douglas SA. 1998. Zebrafish informatics and the ZFIN database. Methods Cell Biol 60: 339–355. [DOI] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. 2005. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21: 1859–1875. [DOI] [PubMed] [Google Scholar]
- Zhu J, Sanborn JZ, Diekhans M, Lowe CB, Pringle TH, Haussler D. 2007. Comparative genomics search for losses of long-established genes on the human lineage. PLoS Comput Biol 3: e247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.