

Abstract
Variation in RNA splicing (i.e., alternative splicing) plays an important role in many diseases. Variants near 5′ and 3′ splice sites often affect splicing, but the effects of these variants on splicing and disease have not been fully characterized beyond the two “essential” splice nucleotides flanking each exon. Here we provide quantitative measurements of tolerance to mutational disruptions by position and reference allele–alternative allele combinations. We show that certain reference alleles are particularly sensitive to mutations, regardless of the alternative alleles into which they are mutated. Using public RNA-seq data, we demonstrate that individuals carrying such variants have significantly lower levels of the correctly spliced transcript, compared to individuals without them, and confirm that these specific substitutions are highly enriched for known Mendelian mutations. Our results propose a more refined definition of the “splice region” and offer a new way to prioritize and provide functional interpretation of variants identified in diagnostic sequencing and association studies.
RNA splicing and alternative splicing are fundamental regulatory processes connecting transcription and translation. Splicing defects have been shown to make major contributions to the allelic architecture of numerous diseases including spinal muscular atrophy, retinitis pigmentosa, Parkinson's disease, familial dysautonomia, schizophrenia, autism spectrum disorder and various types of cancers (Lorson et al. 1999; Radisky et al. 2005; Ibrahim et al. 2006; Law et al. 2007; Samaranch et al. 2010; Poulikakos et al. 2011; Tanackovic et al. 2011; Voineagu et al. 2011; Ferrarese et al. 2014). For example, Ferrarese et al. (2014) showed that alternative splicing between exon 5 and exon 7 of ANXA7 gene enhances EGFR signaling and contributes to cancer progression in glioblastomas. Law et al. (2007) found higher expression levels of ERBB4 splice variants containing metalloprotease cleavable extracellular domains and PI3K binding sites in schizophrenia, and these splice variant isoforms were also highly associated with several risk SNP. Alternative splicing is particularly widespread in the central nervous system and generates isoform diversity important to neuronal development and normal functioning (Raj and Blencowe 2015).
Many previous studies focused on how mutations affect splicing. Large-scale RNA-seq studies and smaller-scale minigene-based approaches identified hundreds of eQTLs and sQTLs (Erkelenz et al. 2014; The GTEx Consortium 2015; Rivas et al. 2015; Soukarieh et al. 2016), confirming a widespread influence of mutations on splicing variation. Based on these results as well as an understanding of cis- and trans-acting elements that affect splicing (such as branch site, polypyrimidine tract, and splicing enhancer and silencer motifs), computational algorithms have been developed to predict the effect of mutations on both general and tissue-specific splicing patterns (Barash et al. 2010; Di Giacomo et al. 2013; Erkelenz et al. 2014; Rosenberg et al. 2015; Xiong et al. 2015). One important goal of studying mutations affecting splicing is to aid in the functional interpretation of the numerous single-nucleotide variants (SNVs) identified in disease-mapping studies (from common alleles implicated by GWAS to rare and de novo mutations found in both Mendelian and common diseases). For instance, The GTEx Consortium (2015) demonstrated significant enrichment of sQTLs in ENCODE functional domains; Xiong et al. (2015) sifted through variants in disease candidate genes and prioritized mutations using the predicted likelihood that they will disrupt normal splicing, but did not find significant enrichment of splice-disrupting variants in GWAS hits. Using a different method to identify sQTLs, Li et al. (2016) showed that the enrichment of sQTLs among GWAS SNPs is comparable or even larger in some cases than that of eQTLs.
Of the variants confirmed or predicted to affect splicing, many are outside the two ultraconserved positions at both the 5′ (donor, typically GT) and 3′ (acceptor, typically AG) splice sites (ss). These so-called “essential” or “canonical” splice bases are customarily included as loss-of-function annotations, while others nearby are most often ignored in such annotations. Establishment of the consensus sequence motif several base pairs around the canonical splice sites early on (Shapiro and Senapathy 1987; Stephens and Schneider 1992) provided early evidence suggesting that additional near-splice-site bases are important. More recently, Rivas et al. (2015) quantified the proportion of variants disrupting splicing at ±25 bp from the splice sites based on RNA-sequencing results, clearly demonstrating signal beyond the essential sites.
The recent availability of the ExAC data set (Lek et al. 2016), a deep-coverage exome sequencing data set with 60,706 individuals, permits a closer look at the near-splice-site region since standard exome capture generally provides deep coverage 20–40 nucleotides (nt) to both sides of captured coding exons. Of particular importance, to assess the degree of mutational tolerance in different genes, Lek et al. (2016) developed an expectation-maximization approach to quantify the lack of protein-truncating variants compared to expectation in each gene (the probability of being loss-of-function intolerant, or pLI). As a result, genes with pLI value ≥0.9 (n = 3230, 17.7%) are particularly intolerant of disruptive mutations. Following this line of thought, we utilized the relative incidence of variation in splice regions of intolerant and tolerant genes to characterize the deleteriousness of mutations at each individual near-splice sites (see Methods).
Although different studies have focused on mutations in this region, none of them directly quantified the level of deleteriousness of mutations by both positions and reference/alternative allele types. Here we offer a systematic analysis of near-splice-site mutations, identifying which sites are intolerant of mutation and confirming their impact through RNA-seq data and analysis of known Mendelian mutations, thereby developing a refined definition of “splice region” suitable for use in disease genetics. Beyond standard intolerance and previous examination of the splice region in Rivas et al. (2015), we provide here reference nucleotide-specific intolerance information accounting for the known reference biases of these positions that was not available in Rivas et al. (2015). It is worth noting that here we use human polymorphism rates and disease mutation occurrences to examine strong selective pressure against mutations.
Results
We first sought to define which near-splice-site positions are important for normal splicing. The range of near-splice sites we focused on in all of our analyses is ±10 bp around splice junctions, and nomenclature is shown in Figure 1A. The essential/canonical splice sites have well-known patterns GT (5′ splice site, D + 1 and D + 2) and AG (3′ splice site, A − 2 and A − 1) and are included for comparisons where applicable.
Figure 1.

Quantification of intolerance to mutations split by positions and reference alleles. (A) Nomenclature used throughout this study; (B) χ2 statistics at the 5′ end; (C) χ2 statistics at the 3′ end; (D) odds ratios at the 5′ end; (E) odds ratios at the 3′ end.
To determine potential functional significance, we used χ2 statistics and odds ratios to measure whether mutation rates at our sites of interest are significantly lower in loss-of-function intolerant genes (see Methods; for an example, see Supplemental Fig. S1). Consistent with implications from earlier RNA-seq studies, the distribution of the χ2 statistics over ±10 bp around splice junctions shows that, in addition to the four “essential splice” nucleotides, positions D + 3, D + 4, D + 5, D + 6, D − 1, and A + 1 are very significantly intolerant of mutations. χ2 statistics of exonic regions are, as expected, on average higher than intronic regions due to background genic mutation; even considering this background, A + 1 and D − 1 (the initial and terminal coding nucleotides in each exon) are unusually constrained (Supplemental Table S1).
Both χ2 statistics and odds ratios for the four reference alleles at each near-splice-site position demonstrate that a specific reference allele is more intolerant of mutations than others at the same positions. Figure 1, B and C, shows that reference base T at D + 6, G at D + 5, A at D + 4 and D + 3, G at D − 1, and G at A + 1 are significantly less tolerant of mutational alteration than are the other three reference bases at those same positions. This difference is clearly evident in the odds ratio of the variation rate between tolerant and intolerant genes (Fig. 1D,E), indicating that the statistical excess is not simply a function of sample size. Although, for G at A + 1, commonness of this allele does seem to contribute to the high χ2 statistics especially when compared to its odds ratio. Also the specificity of the G allele at this position is not as clear cut, which is also evident in Figure 2. In the following analyses, we name these reference bases that are particularly sensitive to mutations as “constrained reference bases.”
Figure 2.
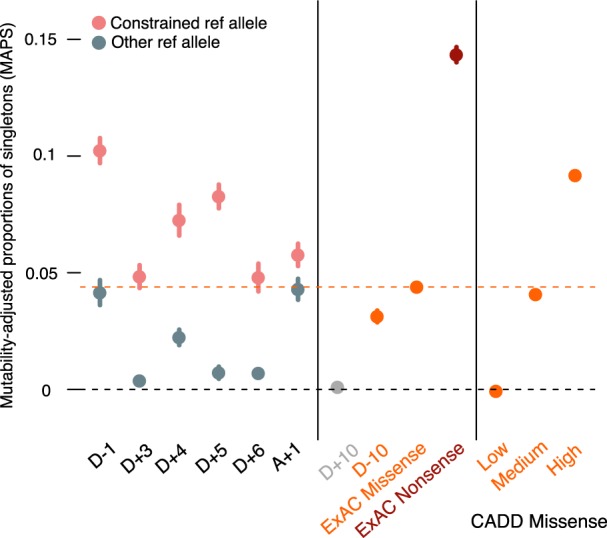
Singleton ratios adjusted by mutability at near-splice sites (left panel) compared to ExAC missense, nonsense (middle panel), and missense split by CADD (right panel) as references. “D + 10” and “D − 10” represent singleton ratios from an arbitrary intronic site and an arbitrary exonic site, respectively, and therefore serve as negative controls.
Negative selection not only reduces the rate at which we find variants but reduces the site frequency of observed sites when compared with neutral sites. To confirm the inference that selection is acting against specific nucleotide substitutions in the splice region, we first looked at the mutability-adjusted proportion of singletons (MAPS) as proposed in Lek et al. (2016) at constrained reference bases versus other three reference bases at the same positions (Fig. 2). If mutations changing the constrained reference bases are indeed more deleterious, it is expected that there is a higher singleton ratio among these mutations because deleterious variants tend to be rarer. As we are comparing different reference bases, MAPS rather than the direct proportion of singletons is needed in order to account for systematic mutability differences given local nucleotide context. Consistent with the prediction from the χ2 analysis, MAPS is significantly higher at constrained reference bases than at other reference bases at the same positions. Compared to the average MAPS of functional classes reported in Lek et al. (2016), the constrained reference bases fall mostly between “missense variant” (0.0439) and “stop gained” (0.143), confirming their functional relevance. As a negative control, we also calculated MAPS for D + 10 (a nonsignificant intronic site) as a comparator for the D + 3 to D + 6 sites and D − 10 (an exonic site, which includes a mixture of missense and synonymous mutations) as a comparator for the D − 1 and A + 1 sites.
After demonstrating the importance of individual constrained reference bases, we were also interested in whether combinations of such bases enhance or modify the effect. As the constrained reference bases are also the most common reference alleles at their respective positions (frequencies of respective constrained reference bases at D + 6, D + 5, D + 4, D + 3, D − 1, and A + 1 are 0.49, 0.77, 0.7, 0.62, 0.8, and 0.49, respectively), we hypothesized that they together form a consistent pattern. Since all of the constrained reference bases except for A + 1 are on the 5′ side of the intron, we tested whether mutations disrupting the pattern at the 5′ side tend to co-occur. More specifically, we used the Breslow-Day test on a three-way contingency table (Breslow and Day 1980) to study whether the presence of one constrained reference base-disrupting mutation (“Independent Pos”) modifies the odds ratios of such mutations occurring nearby (“Combination Pos”). An example three-way contingency table as well as the results are in Supplemental Table S2. For each of the independent positions, we tested for both its effect on one specific nearby position (only one position listed as the combination position) and on any of the nearby positions (more than one position listed as the combination position). Although mutations generally act independently (i.e., the presence of a mutation disrupting the constrained reference base does not affect chances of nearby similar mutations; Breslow-Day test P-value >0.05), we do observe a general property that some mutations (particularly those disrupting the D + 5 T, D + 3 A, or D − 1 G alleles) significantly reduce the impact of additional mutations. This may suggest that individual disruptions to a consensus motif are sufficient to disturb splicing—but could also reflect that some of the observed disruptions in constrained genes in reference databases might be relatively benign owing to the availability of a suitable backup splice site, and thus not additionally punished by the presence of a second mutation.
One of the first steps of the splicing process is the recognition of 5′ss by U1 snRNP, which is one of the five snRNPs making up the spliceosome. Later on, U1 will be replaced by U6, which brings the 5′ss and the branch point closer together to prepare for the first catalytic reaction. Therefore, we speculate that any mutation disrupting the G (GT) AAGT motif (i.e., disrupting the constrained reference bases) on the 5′ side would disrupt U1 and U6 snRNP binding, which in turn leads to abnormal splicing.
Given the clear evidence that natural selection does not tolerate substitutions in these “near-splice” regions in genes generally sensitive to heterozygous mutation, we then sought to characterize the functional effects on splicing and genetic impact on disease risk contributed by these variants. The availability of GTEx and GEUVADIS data provides an excellent opportunity to examine the effect of mutations at near-splice sites on splicing (Lappalainen et al. 2013; The GTEx Consortium 2015). Specifically, we tested if heterozygous carriers of variants changing the constrained reference bases resulted in significantly less correct splice junction reads compared to the homozygous reference allele carriers, with correct splice junction reads defined as the number of split-mapping reads consistent with canonical transcript structure normalized to sequencing depth and standardized to population level. Wilcoxon test shows that this is indeed the case for D + 6, D + 5, D + 4, and D − 1 (Table 1). As a negative control, the same tests for unconstrained reference bases at the same locations do not show a statistically significant result. These results strongly support the idea that mutations changing the constrained reference bases disrupt splicing and are less tolerated. To better understand this effect, we also calculated the correlations between MAPS of constraint reference alleles and effect sizes of splicing disruption (Pearson's correlation coefficient = 0.165 for GTEx and −0.592 for GEUVADIS). Although correlation in GTEx is weak, there is a strong negative correlation between MAPS and effect sizes of splicing disruption in GEUVADIS (constrained reference bases with higher MAPS also tend to have stronger negative effect on normal splicing). Beyond splicing disruption, no clear patterns of alternative splicing subtypes can be determined.
Table 1.
Comparison of the number of correct splice junction reads between mutation carriers and homozygous reference individuals in GTEx and GEUVADIS studies
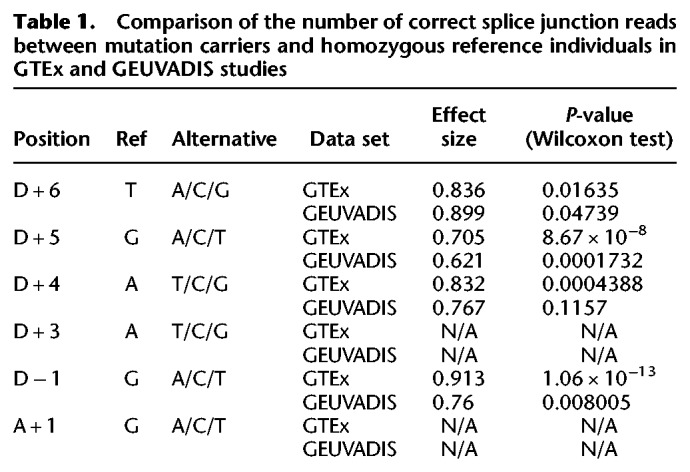
Although it would be very helpful to include the four essential nucleotides for comparison in Table 1, we did not have sufficient data to carry out the tests, because they rely on having a sufficient number of common polymorphic sites in GTEx and GEUVADIS to explore the effect of mutation and on having alternatives to the reference as negative controls for the assertion that mutation of a particular conserved reference site was functionally relevant.
After confirming their impact on splicing, we next explored the impact these specific substitutions have on known, largely Mendelian, diseases. In the ClinVar data set, mutations are assigned clinical significance categories based on different levels of evidence. Taking clinical significance categories “likely pathogenic,” “pathogenic,” “risk factor,” and “association” as the deleterious group (39.3% of all variants within 10 bp to the splice sites) and categories “benign” and “likely benign” as the benign group, we found that mutations changing the constrained reference bases are significantly enriched in the deleterious group but not the benign group (Table 2). This result further supports the idea that mutations changing the constrained reference alleles are more likely to be disease-related.
Table 2.
Enrichment analysis of constrained reference base mutations in the ClinVar data set

In addition to Mendelian and rare diseases, mutations disrupting the constrained reference alleles are also pathogenic in common diseases. For instance, a loss-of-function variant in ABCA7 gene (rs200538373, D + 5G > C) confers risk to Alzheimer's disease (Steinberg et al. 2015). A group of variants with the majority fitting pattern D − 1G > H, collectively named as “last-base exonic mutations” (LBEMs) by the authors, were found to be the most frequent mutation type disrupting normal splicing in several cancer types and enriched in tumor suppressor genes such as TP53 (Jung et al. 2015).
These findings can be used to better annotate potential strong-acting mutations in established disease genes. As an example, we looked at a curated list of genes having more than three de novo protein-truncating variants each from the published studies of de novo variation in autism spectrum disorder, developmental delay (DD), and intellectual disability (ID), which have recently been jointly analyzed (Kosmicki et al. 2017). Ten genes also harbor at least one de novo near-splice-site variant at the key positions identified above, indicating that these near-splice-site variants may have a strong role in disease pathology (Supplemental Table S3). Additionally, five genes (NCKAP1, NIN, EFTUD2, HNRNPK, and KDM6B) that have two protein-truncating variants each harbor a near-splice-site variant at key positions, pushing these genes into the more convincing disease-related range.
To estimate the overall addition to disease mutation annotations by taking into account the constrained reference bases, we compared the number of credible deleterious mutations occurring at the essential splice sites versus the constrained reference bases at near-splice sites in ClinVar and the de novo variant data sets. Among ClinVar variants that meet the clinical significance category criteria mentioned above, 1284 and 973 mutations disrupt the 5′ and 3′ essential splice sites, respectively. In comparison, 806 mutations disrupt the six constrained references bases (D + 3 A allele: 61; D + 4 A allele: 27; D + 5 G allele: 181; D + 6 T allele: 19; D − 1 G allele: 372; A + 1 G allele: 146). In the de novo variant data sets including both autism and DD/ID individuals, 59 and 39 de novo variants occur at the 5′ and 3′ essential splice sites and a total of 35 mutations occur at the constrained reference bases in genes with pLI ≥ 0.9. Overall, ∼36% more mutations in the splice region acquire new functional annotations if we take specifically intolerant splice junction mutations into account.
Discussion
Variants outside splice sites are known to affect splicing, but a detailed evaluation of which mutations at nonessential sites near splice junctions affect splicing, and to what degree these contribute to disease, is still lacking. Our study provides a quantitative measure of the deleteriousness of mutations in near-splice sites and shows that tolerance to mutations is reference allele specific. To validate this inference from the initial analysis, we showed that a mutability-adjusted proportion of singletons (a site-frequency–based metric correlated with negative selective pressure) at constrained reference bases are significantly higher compared to other reference bases at the same positions, and then we further used RNA-sequencing data from GEUVADIS and GTEx studies to show that mutations changing constrained reference bases resulted in significantly fewer correctly spliced reads. Importantly, we additionally confirmed that mutations changing the constrained reference bases are significantly enriched in ClinVar variants and make a meaningful addition to the allelic architecture of rare disease. In summary, by providing a detailed analysis of selective pressure and impact on splicing, we propose a refinement to the “splice region” definition suitable for use in Mendelian and complex disease exome analysis. As the specific pairings of positions and reference alleles have high impact on normal splicing if disrupted, they can therefore be used to prioritize and provide functional interpretations for mutations identified in association-type studies.
In contrast, genome interpretation at present often annotates but quite frequently ignores the vastly larger, and largely benign, category of any variant in the “splice region” within 10 or 20 bp of a splice junction—the annotation suggested here will enable a stronger consequence be attached to a much smaller number of variants. Specifically, surveying ExAC, we find that an average participant sampled contains 2918.6 variants in the splice region (within 10 bp of the splice junction)—only 106 of which are in the refined set of nucleotides and reference alleles listed here, and only 20.4 (0.7% of all near-splice-site mutations) are in genes with pLI ≥ 0.9. Restricting to only rare variants with MAF < 0.01, this number further drops to 1.28 mutations per individual. In clinical exome evaluation, therefore, rather than a general but uncertain consideration of variants “near splice junctions,” the refined set of sites and reference nucleotides identified in this study are demonstrated (Fig. 2; Table 2) as a category to confer risk comparable to damaging missense variants and should be considered similarly in evaluating plausible causal mutations.
Instead of using the constrained reference alleles criteria independently, one interesting question is whether it is possible to incorporate these positions into existing scores that measure mutation/gene deleteriousness, for example the pLI. We note that since the number of sites involved is not so large compared to potential nonsense plus essential splice nucleotides, the additional information would not make a very substantial difference. Indeed as the ExAC and other resources increase in size, particular splicing intolerance metrics should become an interesting possibility.
One limitation of our study is that we only considered variation at canonical transcripts. Although we used canonical transcripts to capture overall deleteriousness of mutations at near-splice sites, to fully understand splicing in the context of specific diseases, it would be best to look at tissue-specific transcripts and isoforms.
Methods
We defined “near-splice-site regions” as ±10 bp around the 5′ and 3′ splice sites. For all the subsequent analysis, only canonical transcripts were considered (GENCODE v19). See Figure 1A for additional nomenclature used throughout.
We used variation from the ExAC data set (version 0.3; http://exac.broadinstitute.org) mapped to human reference genome (hg19) to scan for evidence of selection against variation in the near-splice-site regions. First, we tallied the number of A, T, C, and G's in the reference genome at each near-splice-site position across all canonical exons along with the number of variants observed in ExAC, correcting the numbers by coverage following previous methods (Lek et al. 2016) in order to account for variants missed due to lower sequencing coverage. Briefly, the reference allele count at each base pair is multiplied by a factor determined by the median of coverage at that position. If median coverage >50, factor = 1; if 1 ≤ median coverage ≤ 50, factor = 0.089 + 0.217 × log(median coverage); if median coverage < 1, factor = 0.089. The corrected number of reference bases as well as number of mutations by reference and alternative allele are shown in Supplemental Table S1.
The probability of a gene being loss-of-function intolerant (pLI) was developed in Lek et al. (2016) by comparing observed with expected rates of truncating mutations and identified that in 15%–20% of genes, such mutations are under strong selection consistent with that seen in severe Mendelian haploinsufficiencies (Lek et al. 2016; Cassa et al. 2017). Analyses that followed comparing loss-of-function intolerant (pLI ≥ 0.9, n = 3230) genes with others (Genovese et al. 2016; Lek et al. 2016; Kosmicki et al. 2017) established that indeed heterozygous truncating mutations (nonsense, frameshift, and essential splice site mutations) in these genes often have significant medical consequences. We therefore theorized that any splice-region variants (beyond the essential splice sites) disrupting normal gene function should also be significantly depleted in loss-of-function intolerant genes. Thus we asked whether rates of mutations near splice junctions were significantly different between the same two groups of genes (LoF-tolerant and LoF-intolerant). We created a contingency table according to gene group (pLI < 0.9 versus pLI ≥ 0.9) and mutation count (number of bases with mutations versus number of bases lacking mutation, corrected for coverage) for each reference allele-alternative allele combination and calculated the Pearson's χ2 statistic (Agresti 2007). We noticed that at some positions, the χ2 statistics are consistently high for mutations from a specific reference allele regardless of the alternative allele to which they mutate; we also created a Pearson's χ2 statistic for each reference base at all positions (Supplemental Table S1). An example of the per-reference-base contingency table (D + 6 T allele) is in Supplemental Figure S1.
One caveat with comparing Pearson's χ2 statistics of different reference allele-alternative allele combinations directly is that this statistic increases as counts in contingency tables increase (reflecting more power to detect distortions when sample size is larger). In other words, more common reference alleles tend to have higher statistics. Although the level of significance that we observe is well above what can be explained by commonness of alleles alone, we report odds ratios to better quantify the differences in mutation rates in the two groups of genes.
Since nucleotide context is such a major determinant of mutation rate, we used the mutability-adjusted proportion of singletons (MAPS) as calculated in Lek et al. (2016). Briefly, the singleton ratio at each mutational site is adjusted by the mean singleton ratio of all mutations surrounded by the same 3-nt sequence context. We compared MAPS of mutations changing reference alleles with particularly high χ2 statistics and odds ratios (named as constrained reference bases) and other reference alleles in the splice region with that of ExAC missense and nonsense mutations, as well as ExAC missense mutations split by CADD categories, taken from Lek et al. (2016) directly.
RNA-seq analysis was carried out using GTEx and GEUVADIS data sets, with quality control and mapping steps completed as part of the respective project. For GTEx, the pilot phase and tissue types adipose tissues, artery tibial, heart left ventricle, lung, skeletal muscle, nerve tibial, sun-exposed skin, thyroid, and blood were used. Theses tissues were used because they have the highest numbers of samples in GTEx. To test whether mutations identified as deleterious from mutational data alone have an effect on splicing, the median of correct splice junction reads was compared between individuals carrying a mutation disrupting one of the constrained reference bases and individuals that are homozygous (for the constrained reference bases) by Wilcoxon rank-sum test. Correct splice junction reads were defined as the number of split-mapping reads consistent with structures of canonical transcripts normalized by sequencing depth and standardized to the population level. Mutations from different exons and genes are combined by their relative positions and disrupted reference alleles, and for each individual carrying a mutation, a homozygous wild-type individual is sampled from the population. Identical analyses at variation at unconstrained reference nucleotides (thus matched for DNA variation content compared to reference) were performed, and no deviation from 50–50 allele balance among heterozygote carriers was observed, thereby establishing no impact of DNA variation on read mapping efficiency. A “N/A” result was reported when there was not enough data to perform the test. Effect sizes (odds ratios of an individual being heterozygous as opposed to homozygous as correct splice junction reads increase) were also reported.
We tried to categorize the kind of alternative splicing events associated with the constrained reference base-disrupting mutations into one of exon skipping, exon elongations/reading into intron, or a mixture of the above, but no clear pattern was shown as to whether a particular event type is preferred. The results of this exploration and methods can be found in the supplemental text of Rivas et al. (2015). Briefly, a Gaussian mixture model was used to classify alternative splicing event types based on expression values of the nearest exon and intron of the mutation. In general, it is difficult to classify the type of event with great certainty at the current sequencing depth of GEUVADIS and GTEx.
The ClinVar data set was downloaded from https://www.ncbi.nlm.nih.gov/clinvar/docs/maintenance_use/ (accessed February 2016). Excesses of disease-causing variants were calculated using variants with clinical significance categories “likely pathogenic,” “pathogenic,” “risk factor,” and “association.” We tested for the enrichment of mutations changing constrained reference alleles in ClinVar variants with a binomial test comparing proportions of mutations with constrained reference alleles with that in the general population (ExAC, including both pLI ≥ 0.9 and pLI < 0.9 genes). Since the same nucleotides at the same positions were compared, frequency differences of the four reference alleles at each position were implicitly accounted for. A one-tailed test was used because the alternative hypothesis is that the true proportions of such mutations in ClinVar are higher than those naturally occurring in the general population. We also tested for enrichment in “benign/likely benign” categories as a negative control.
In all our analyses, reference versus alternative alleles were used instead of major versus minor alleles. Given the fact that the reference allele and major allele are the same for the vast majority of variants (e.g., 99.78% in ExAC), switching between using reference alleles and major alleles does not significantly change our results and does not change at all the primary analyses of ClinVar and ultra-rare/de novo mutations in neurodevelopmental disease.
In all the data sets used, reads were mapped to human reference genome GRCh37/hg19. Although GRCh38 is the most updated assembly with improvements in the mitochondrial genome and the centromeres, our results do not depend on these regions. Additionally, the number of nucleotides corrected in GRCh38 is relatively small and there is no reason to believe that they are enriched in the near-splice-site region of canonical transcripts. Therefore, we feel that using GRCh38 will not significantly change our results.
Supplementary Material
Acknowledgments
We would like to thank the ATGU community for their helpful discussions and insightful comments. K.J.K. is supported by a National Institute of General Medical Sciences Fellowship (NIGMS, NIH; F32GM115208). This work was supported by the Simons Foundation (SFARI 342292) and by the Leona M. and Harry B. Helmsley Charitable Trust (#2015PG-IBD001).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.231902.117.
References
- Agresti A. 2007. An introduction to categorical data analysis. Statistics. John Wiley, Hoboken, NJ. [Google Scholar]
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. 2010. Deciphering the splicing code. Nature 465: 53–59. [DOI] [PubMed] [Google Scholar]
- Breslow NE, Day NE. 1980. Classical methods of analysis of grouped data. In Statistical methods in cancer research. Volume I - The analysis of case-control studies, pp. 122–159. International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- Cassa CA, Weghorn D, Balick DJ, Jordan DM, Nusinow D, Samocha KE, O'Donnell-Luria A, MacArthur DG, Daly MJ, Beier DR, et al. 2017. Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat Genet 49: 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giacomo D, Gaildrat P, Abuli A, Abdat J, Frébourg T, Tosi M, Martins A. 2013. Functional analysis of a large set of BRCA2 exon 7 variants highlights the predictive value of hexamer scores in detecting alterations of exonic splicing regulatory elements. Hum Mutat 34: 1547–1557. [DOI] [PubMed] [Google Scholar]
- Erkelenz S, Theiss S, Otte M, Widera M, Peter JO, Schaal H. 2014. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res 42: 10681–10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarese R, Harsh GR IV, Yadav AK, Bug E, Maticzka D, Reichardt W, Dombrowski SM, Miller TE, Masilamani AP, Dai FP, et al. 2014. Lineage-specific splicing of a brain-enriched alternative exon promotes glioblastoma progression. J Clin Invest 124: 2861–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, Moran JL, Purcell SM, Sklar P, Sullivan PF, et al. 2016. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 19: 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2015. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim EC, Hims MM, Shomron N, Burge CB, Slaugenhaupt SA, Reed R. 2006. Weak definition of IKBKAP exon 20 leads to aberrant splicing in familial dysautonomia. Hum Mutat 28: 41–53. [DOI] [PubMed] [Google Scholar]
- Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, Lee E. 2015. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 47: 1242–1248. [DOI] [PubMed] [Google Scholar]
- Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K, et al. 2017. Refining the role of de novo protein truncating variants in neurodevelopmental disorders using population reference samples. Nat Genet 49: 504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ’t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. 2013. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501: 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Kleinman JE, Weinberger DR, Weickert CS. 2007. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum Mol Genet 16: 129–141. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Van De Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. 2016. RNA splicing is a primary link between genetic variation and disease. Science 352: 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth BA. 1999. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci 96: 6207–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. 2011. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480: 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. 2005. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436: 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Blencowe BJ. 2015. Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 87: 14–27. [DOI] [PubMed] [Google Scholar]
- Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, Deluca DS, Fromer M, et al. 2015. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 348: 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Patwardhan RP, Shendure J, Seelig G. 2015. Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163: 698–711. [DOI] [PubMed] [Google Scholar]
- Samaranch L, Lorenzo-Betacor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, et al. 2010. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133: 1128–1142. [DOI] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. 1987. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15: 7155–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukarieh O, Gaildrat P, Hamieh M, Drouet A, Baert-Desurmont S, Frébourg T, Tosi M, Martins A. 2016. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet 12: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, et al. 2015. Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 47: 445–447. [DOI] [PubMed] [Google Scholar]
- Stephens RM, Schneider TD. 1992. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J Mol Biol 228: 1124–1136. [DOI] [PubMed] [Google Scholar]
- Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL, Rivolta C. 2011. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet 88: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor R, Blencowe BJ, Geschwind D. 2011. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. 2015. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.