

Abstract
For more than 25 years, MDM2 and its homolog MDMX (also known as MDM4) have been shown to exert oncogenic activity. These two proteins are best understood as negative regulators of the p53 tumor suppressor, although they may have additional p53-independent roles. Understanding the dysregulation of MDM2 and MDMX in human cancers and how they function either together or separately in tumorigenesis may improve methods of diagnosis and for assessing prognosis. Targeting the proteins themselves, or their regulators, may be a promising therapeutic approach to treating some forms of cancer.
Keywords: cancer, MDM2, MDMX, p53, tumorigenesis
INTRODUCTION
Elucidating the molecular changes that have a role in both transformation and tumor progression remains an essential aspect of cancer diagnosis, the assessment of prognosis, and treatment. In this review, we discuss findings related to the roles that MDM2 and MDMX have in tumorigenesis, their expression in human cancers, their uses as biomarkers, and the therapeutic agents that target these proteins. We note that the major roles of MDM2 and MDMX are in regulating p53, the most extensively studied human tumor suppressor. Largely through its function as a sequence-specific transcriptional activator, p53 regulates a plethora of genes whose products mediate a variety of cellular pathways, including different forms of cell-cycle arrest, cell death, metabolic processes, DNA repair, and others (1, 2). The TP53 gene is mutated in approximately one-half of all sporadic human cancers, although mutation frequency varies greatly with tumor type (3). Cancer-prone Li-Fraumeni families bear germ-line mutations in one of their p53 alleles, further demonstrating the critical role of p53 in tumor suppression (4). Extensive evidence for the role of p53 also comes from mouse studies, and p53-null mice acquire tumors (lymphomas and sarcomas) with 100% frequency, from which they succumb by about 6 months of age (5). Importantly, many human tumors express wild-type p53 (overall 50%, but the frequency varies with tumor type), and some of these overexpress MDM2 or MDMX, or both, as a result of gene amplification or other mechanisms. Due to the diversity of human tumors that harbor wild-type p53, targeting these two important regulators of p53 has considerable therapeutic potential. Alternatively, harnessing the potential of MDM2 to repress mutant forms of p53 is another worthy goal. We start this review with a brief outline of the history of key discoveries related to MDM2 and MDMX.
HIGHLIGHTS IN THE HISTORY OF MDM2 AND MDMX
Originally cloned by Donna George and colleagues (6) from a double-minute amplicon present within a spontaneously transformed murine cell line, Mdm2 was then shown to function as an oncogene (7). The first hint about the molecular mechanism of the function of Mdm2 came when it was shown that it can bind to, and inhibit transactivation by, p53 (8, 9). Mdm2 (whose human homolog is occasionally referred to as HDM2) was then shown to be a transcriptional target of p53, thereby forming a negative-feedback loop—i.e., an autoregulatory cycle (10, 11). What cemented the central importance of MDM2 as a p53 regulator were studies demonstrating that the very early embryonic lethality of Mdm2-null mice was fully rescued in a p53-null background (12, 13). These findings, now confirmed and extended in multiple mouse-model studies discussed below, reveal the critical importance of keeping p53 in check, not only in the developing embryo but also in somatic tissues in mice. Furthermore, it was demonstrated that MDM2 not only inhibits p53 transactivation of its target genes but also mediates degradation of p53 (14, 15) by serving as an E3 ubiquitin ligase (16). Relatedly, it was discovered that phosphorylation of key sites in the p53 N terminus weakens the p53–MDM2 association, and such sites are phosphorylated in cells in response to DNA damage (17). The discovery that MDM2 targets p53 for proteasome-mediated degradation provided a plausible explanation as to why and how basal p53 protein levels are normally kept low until cells are subjected to numerous stresses, including DNA damage, hypoxia, and ribosomal stresses, each of which disables the interaction between MDM2 and p53 (18). Relevantly, the discovery that the p19ARF protein product of the INK4a locus binds to and disables the E3 ligase activity of MDM2 in response to various oncogenic stresses provided the explanation for how hyperproliferative signals activate p53 (19). When the MDM2 homolog MDMX (also known as MDM4; human MDMX is also known as HDMX or HDM4) was discovered, this added another level of complexity to the already dense p53–MDM2 field (20–22). MdmX was then shown to be as important as Mdm2 in embryonic development because both Mdm2- and MdmX-knockout mice result in p53-dependent embryonic lethality that is rescued by p53 loss (23, 24).
MDM2 AND MDMX: PLAYERS AND PATHWAYS
Structural Features
Two decades of research on human MDM2 have revealed a complicated and highly regulated protein whose 491 residues comprise multiple functional domains, signal sequences, and sites of modification (Figure 1b). It is also expressed as different isoforms generated by alternative splicing (reviewed in 25 and described below). The domain architecture of MDM2 illuminates its functions. The N-terminal region is essential for interacting with, and inhibiting the ability of, p53 to be transcriptionally active. The linker region that follows the N-terminal domain spans a nuclear localization signal (NLS) at position 178 and a nuclear export signal (NES) at position 192. The central acidic domain has a crucial auxiliary role in the degradation of p53 (26, 27) and is followed by the zinc-finger domain, which is thought to aid in interactions with various proteins, including ribosomal and nucleolar proteins, as well as the tumor suppressor p14ARF (reviewed in 28). The MDM2 C terminus comprises a RING domain (Really Interesting New Gene; amino acids 436–482) that harbors a cryptic nucleolar localization signal (NoLS; residues 466–473) and residues at the extreme C terminus (amino acids 485–491) that play a part in interactions among RING domains (29, 30). The MDM2 RING domain is responsible for the E3 ligase activity of the protein, and this activity is, in turn, critical for MDM2 to restrain p53 during early embryogenesis. This is supported by the finding that point mutations in the MDM2 RING domain promote p53-dependent mortality at a level comparable to that of the deletion of the entire gene (31).
Figure 1.
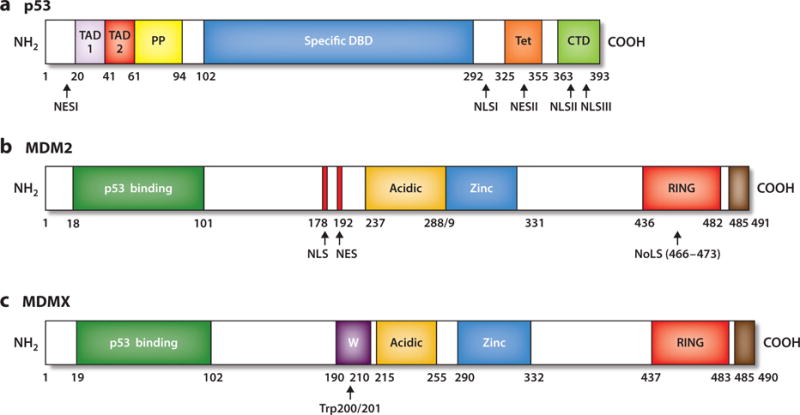
Landmarks of p53, MDM2, and MDMX proteins. (a) The N-terminal region of p53 (residues 1–101) contains two distinct transactivation domains, TAD 1 and TAD 2; a proline-rich region (PP) with five PXXP sequence motifs that are essential for its ability to induce apoptosis; and a nuclear export signal (NES) between residues 11 and 27. The central core domain (amino acids 102–292) spans the sequence-specific DNA-binding domain (DBD). The majority of missense mutations in the p53 gene are located within this domain with highly conserved subregions. The carboxyl terminus of p53 (residues 292–393) contains a flexible linker region (residues 292–324) that connects the core domain to the tetramerization domain (Tet, residues 325–355) and a basic regulatory domain (CTD, residues 363–393). The carboxyl terminus also encompasses both NES and nuclear localization signal (NLS) sequences. (b) MDM2 landmarks include the N-terminal p53-binding domain (residues 18–101); the centrally located acidic domain that binds ARF and also has a role in the ubiquitination of p53 (residues 237–288); and the zinc-finger motif, where ribosomal proteins interact (residues 289–330). The MDM2 carboxyl terminus contains a RING domain that displays E3 ubiquitin ligase activity (residues 436–482), and the carboxyl terminus tail (residues 485–491) regulates the RING motif via MDM2 homodimer formation as well as MDM2 and MDMX heterodimerization. MDM2 has NLS, nucleolar localization signal (NoLS), and NES sequences. (c) MDMX has structural domain similarity to MDM2, with an N-terminal p53 binding region (residues 19–102), central acidic (residues 215–255) and zinc-finger (residues 290–331) regions, and a carboxyl-terminally located RING domain (residues 437–483). MDMX lacks NLS, NES, or NoLS sequences but has a unique WWW motif (W, residues 190–210), which inhibits interaction with p53. In contrast to the MDM2 RING E3 ligase domain, the MDMX RING domain lacks E3 ligase activity. The carboxyl-terminal tail (residues 485–490) of MDMX is required for formation of MDM2–MDMX heterodimers.
To date, there are three identified regions of interaction between MDM2 and p53 (Figure 2). The initial documented interaction occurs between the MDM2 N-terminal domain and the p53 N-terminal transactivation domain 1, TAD 1 (amino acids 20–40). This interaction inhibits the binding of p53 to the transcriptional machinery (32). A crystal structure analysis of the MDM2 N terminus bound to a p53 peptide, encompassing amino acids 15–29, identified a specific hydrophobic pocket in MDM2, encompassing the amino acids phenylalanine 19, tryptophan (Trp) 23, and leucine 26 in p53 (33). Subsequent studies have demonstrated an interaction between the central acidic domain of MDM2 and the specific DNA-binding domain of p53 (34–37). This interaction is essential for efficient p53 ubiquitination. In addition, the MDM2 acidic region has been reported to promote a conformational change in p53 and to inhibit p53 interaction with DNA (38). A third interaction between the MDM2 N terminus and the p53 C terminus was described in 2010, demonstrating that p53–MDM2 interaction is decreased upon deletion of this p53 region and that modifications of the p53 C terminus can regulate the interaction between p53 and MDM2 (39).
Figure 2.
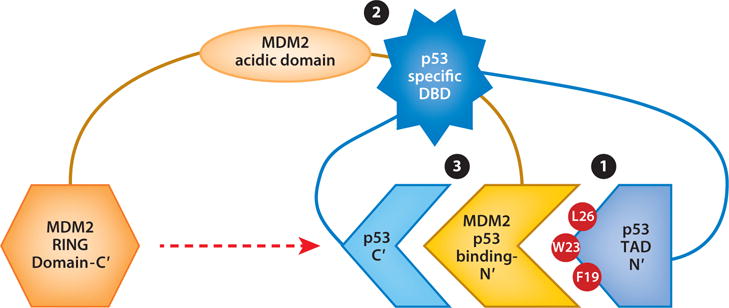
MDM2–p53 interactions involve three regions of each protein. (❶) The primary interaction regions are within the MDM2 N-terminal domain and the p53 N-terminal transactivation domain, TAD 1. (❷) The central regions of MDM2 (acidic domain) interact with the sequence-specific DNA-binding domain (DBD) of p53. (❸) The MDM2 N-terminal domain interacts with the p53 carboxyl-terminal domain, which harbors lysines whose ubiquitination requires the MDM2 RING domain (red dashed arrow). Abbreviations: C′, carboxyl terminus; F, phenylalanine; L, leucine; N′, N terminus; W, tryptophan.
The human MDMX protein consists of 490 amino acids (Figure 1c, although multiple isoforms have been described) and, when overexpressed, inhibits p53-activated transcription. This effect depends on the p53-binding domain of MDMX (40). MDMX does not contain NLS, NoLS, or NES sequences and localizes primarily to the cytoplasm. That said, upon association with different partners (MDM2 in particular), or in response to DNA damage, MDMX can localize to the nucleus. Intriguingly, the precise amino acids that are essential for interaction with both MDM2 and MDMX in the p53 N terminus are identical (41). Yet although MDM2 and MDMX share high homology in their p53-binding domains, structural analysis has revealed significant differences in their p53-binding pockets, with two residues moderately blocking the binding pocket of MDMX (42). MDMX does not exhibit E3 ligase activity in its RING domain and does not directly promote degradation of p53, but it interacts with MDM2 via its RING motif, and this interaction affects levels of both p53 and MDM2 (21, 22; reviewed in 43). Both MDM2 and MDMX have P-loop regions within their respective RING domains that allow them to bind specifically to ATP (adenosine triphosphate) (29, 44–47).
An MDMX autoinhibitory sequence motif has been described near residues Trp 200 and Trp 201, called the WWW element (48). The lack of this domain increases 32-fold the interaction with p53. The WWW motif binds to MDMX’s own N-terminal domain and prevents its interaction with p53. This element may rationalize the existence, as well as the oncogenic impact, of alternatively spliced MDMX proteins lacking this motif that are present in some aggressive tumors, and raises the possibility of an additional level of regulation.
Consequences of the MDM2–p53 Interaction
MDM2 has an essential role in regulating levels of p53 protein, both in unstressed cells and following genotoxic stress (49–51). The p53 protein is degraded by the 26S proteasome after undergoing ubiquitination at several C-terminally located lysine amino acids in a manner that requires the MDM2 E3 ubiquitin-ligase function. MDM2 also has a postubiquitination role in enabling direct p53 interaction with the proteasome (52). Finally, MDM2 is essential for regulating p53 function by mediating the export of p53 from the nucleus in a manner that involves monoubiquitination and sumoylation (53).
The MDM2 gene contains two promoters: P1 and P2. P1 is constitutively active in many cells at low levels. The p53-responsive P2 promoter contains two p53 binding sites and is stimulated in response to cellular stress in a p53-dependent manner (54, 55). It is through interaction with these sites that p53 mediates transcription of the MDM2 gene and thereby forms the p53 component of the p53–MDM2 negative-feedback loop. The autoregulatory circuit formed between p53 and MDM2 is critical for both keeping p53 in check in unstressed cells and restoring low levels of p53 after milder forms of stress. This relationship results in oscillation of the cellular levels of the two proteins, and this has been studied both in populations of cells (56) and at the single-cell level (57). This oscillation is altered in cells that express relatively high levels of MDM2 due to a single-nucleotide polymorphism (SNP) in the MDM2 promoter (discussed below) (58).
Intriguingly, it recently has been demonstrated in vivo that in the hematopoietic system, this feedback loop is important in regulating p53 activity, mainly in response to DNA damage, but it is not essential for homeostasis, development, or longevity (59). Specifically, in mice, point mutations in the two p53-binding sites of the Mdm2 promoter that were introduced into the endogenous Mdm2 locus resulted in increased response to DNA damage, although p53 degradation kinetics in various tissues remained similar to the wild-type control. This highlights the importance of understanding the distinct roles of MDM2 in different tissues.
Adding further complexity to the understanding of p53 regulation, a functional p53-response element has been identified in the MDMX promoter, potentially forming another negative-feedback loop (60). The p53-responsive promoter, named P2, produces a longer human MDMX transcript, HDMX-L, in which 18 residues are added at the N terminus. HDMX-L plays an essential part in MDM2-mediated p53 ubiquitination by reducing p53 levels to normal following stress activation (40). There is still work to be done to determine when, and under what circumstances, p53 regulates MDMX via its P2 promoter.
The MDM2–MDMX–p53 Axis
The best-understood, and likely the most important, role of MDM2 and MDMX in oncogenesis is via their interaction with p53. So far, two primary models have been proposed for this interaction (43). The first model proposes that MDM2 and MDMX independently regulate specific activities of p53, whereby MDM2 controls the cellular levels of p53, and MDMX solely modulates p53 transcriptional activity. Alternatively, the interaction between MDMX and MDM2 could regulate the activity of these two proteins, and they could collaboratively modulate p53 function. Although the evidence largely supports the second model—for example, the p53 degrading function of MDM2 has been shown to be enhanced by interaction with MDMX (reviewed in 61)—the situation is complex, especially as revealed by mouse models. MDMX has been shown to be as important as MDM2 in embryonic development because both Mdm2- and MdmX-knockout mice exhibit p53-dependent embryonic lethality that is rescued by p53 loss, although Mdm2-null mice die at the preimplantation stage and MdmX-null embryos succumb later, at embryonic day 7.5–9.5 (62; reviewed in 63). That embryonic lethality results from the loss of either Mdm2 or MdmX individually suggests that they are not functionally redundant. Yet p53 and MdmX double-null mice that overexpress Mdm2 are viable, suggesting that when expressed at high enough levels Mdm2 can compensate for the phenotype seen with the loss of MdmX (64). In aggregate, the literature on these two proteins provides strong evidence that they work together to restrain p53.
The RING-finger motifs of both MDM2 and MDMX are essential for their activity toward each other as well as toward p53. MDM2 and MDMX interact through their RING-finger motifs to form a complex (21, 22). Quantitative assays have indicated that there is 5–10 times more cellular MDM2 than MDMX in normal human fibroblasts and mammary epithelial cells, suggesting that most MDMX binds to MDM2 because the binding affinity for establishing a heterodimer is higher than for forming MDM2 homodimers (22, 65). It has been reported that heterodimerization of the RING domains of MDM2 and MDMX results in a complex that can mediate p53 polyubiquitination by MDM2, which alone mainly monoubiquitinates p53 (66). According to this study, previous observations that MDM2 alone can polyubiquitinate p53 were due to artificial activation caused by the use of GST (glutathione S-transferase)-tagged MDM2 used in in vitro assays. Further evaluation of this important issue will shed more light on the mechanism of mono-and polyubiquitination, and the potential intervention point it brings for the design of suitable inhibitors.
In several reports, the binding of MDM2 and MDMX through their RING domains has also been demonstrated to be crucial in adult tissues for efficient p53 degradation (67, 68). Additionally MDM2 has been proposed to have an indirect role in regulating stress-induced levels of p53, by downregulating MDMX protein levels; specifically, MDM2 can induce nuclear localization and E3-dependent degradation of MDMX following γ irradiation of cancer cell lines, and downregulation of MDMX contributes to p53 activation by irradiation (69–71).
Furthermore, the extreme C-terminal five residues of MDM2 have been reported to be crucial for E3 ligase function both in vivo and in vitro (29, 30). The formation of the Mdm2–MdmX heterodimer would appear to be crucial for embryonic development as shown in mouse models in which either deletion of, or a single missense mutation within, the MdmX RING domain, which prevents interaction with Mdm2, results in early embryonic lethality (72, 73). Despite the requirements for RING-domain interactions, the requirement of MDM2 E3 ligase activity for regulating p53 function during embryogenesis has recently been brought into question. Using a mouse model with an Mdm2 that lacked E3 ligase activity but retained its ability to bind MdmX (Y487A knockin), it was shown that in the absence of stress, the development of these mice was normal into adulthood (74). The discrepancy between this and earlier reports remains to be resolved.
Although MDM2 and MDMX show a high degree of homology, there are some distinct functions for the two RING-finger proteins. MDM2 inhibits the interactions between p53 and DNA via its acidic domain, but MDMX only slightly mitigates this interaction (38). MDMX can stimulate a conformational change in p53 and protect it from proteasomal degradation after DNA damage in an MDM2- and ubiquitin-independent manner, but MDM2 lacks this activity (75). Furthermore, the MDM2- and MDMX-binding pockets for p53 are not identical, despite the high similarity between them, which has been shown using structural analysis of these domains bound to a p53 N-terminal peptide (42). The WWW inhibitory sequence in MDMX but not in MDM2, mentioned above, suggests there is a new mechanism of regulation in which it competes with p53 TAD 1 for binding with the MDMX N-terminal domain. This highlights the potential oncogenic role of MDMX splice variants that lack this inhibitory sequence and are present in some aggressive tu-mors. In addition, functional deactivation occurring as a result of overexpression and amplification of either MDM2 or MDMX has been demonstrated in many human cancers that do not have a p53 mutation. Tumors overexpressing MDM2 (for example, liver tumors) are distinct from those that overexpress MDMX (for example, colon tumors), implying distinct functions for MDM2 and MDMX in cancer (reviewed in 76, 77). Despite these differences, and the fact that they cannot complement each other in mouse knockout models, there is strong evidence that they work together, and their codependence is what likely accounts for the requirement for both to restrain p53 during embryogenesis.
It should be noted that many tumors exhibit either a p53 mutation or MDM2 and MDMX amplification. For example, in a 2014 study analyzing samples of infiltrating ductal breast carcinoma, overexpression of MDM2 or MDMX was observed in more than half of cases, but was rarely present in the samples with p53 dysfunction (78). Interestingly, overexpression of MDMX has been detected in a wide spectrum of human cancers and can preclude tumor suppression mediated by p53 (79).
Paradoxically, a role for MDMX as a potential tumor suppressor has been reported and may either be p53 dependent or p53 independent, in different experimental settings. MDMX can suppress tumorigenesis in a p53-independent manner by promoting centrosome clustering and bipolar mitosis (80, 81). The p53 and MdmX double-null mice that overexpress MDM2 exhibit accelerated rates of tumor formation that are not detected in mice having both MDMX alleles, suggesting a context-dependent tumor-suppressive function for MDMX (64). In response to DNA damage, MDMX has been shown to translocate to the mitochondria and, thereby, enhance phosphorylation of p53 serine 46 to facilitate p53-mediated apoptosis in cisplatin-treated cells (82) in a manner that requires the activity of TAB1 (83). Whether these unanticipated roles of MDMX are relevant to human cancer remains to be determined.
MDM2 AND MDMX IN HUMAN CANCER
Deregulated Expression of MDM2 and MDMX in Cancer
Several aspects of the biology of MDM2 and MDMX are particularly relevant to human cancer. In this section, we highlight recent evidence from human cancers that largely supports the description of MDM2 and MDMX as oncogenes, and describe the alterations in expression in specific cancers.
Although MDM2 is rarely lost or mutated in human cancers, it is often amplified or overexpressed, or both (reviewed in 84). One review found the overall rate of MDM2 gene amplification to be 7%, with the highest amplification frequency observed in soft tissue tumors (reviewed in 85). Overexpression of MDM2 and MDMX has been shown to correlate with both tumor grade and prognosis. For example, one study reported that high levels of MDM2 expression correlated with poor survival in mesothelioma patients (86). Significantly, in a 2015 study, MDM2 has been shown to be an important prognostic and predictive marker for response to platin–pemetrexed therapy (antifolates and platin derivatives) in patients with malignant pleural mesothelioma (87).
The role of MDM2 in tumorigenesis is further supported by mouse models: For instance, in an early study, transgenic mice overexpressing Mdm2 exhibited spontaneous tumorigenesis, with a spectrum of tumors similar to that seen in p53-knockout mice (88). More recent models have shown that Mdm2 overexpression can lead to tumorigenesis via a loss of p53 activity, as well as by induction of p53-independent tumorigenesis pathways (reviewed in 89).
One line of research into the regulation of MDM2 transcriptional levels, and the consequent biological outcome, has focused on SNPs. Two opposing MDM2 SNPs, SNP 309 and SNP 285, have been identified and shown to affect cancer risk through differential Sp1 transcription- factor binding. In 2004, Bond and colleagues (90) demonstrated that a polymorphism in MDM2, an A to G change at nucleotide 309 in intron 1 of the second MDM2 promoter P2 (SNP 309T > G), affected the binding affinity of the Sp1 regulatory transcription factor. SNP 309G causes enhanced Sp1 binding to the MDM2 promoter, leading to increased MDM2 transcription. Mouse models bearing the tumor-associated SNP were generated; these had higher levels of Mdm2 expression, and exhibited a decrease in p53 function and an increase in tumorigenesis compared with mice bearing the noncancer associated SNP (SNP 309T) (91).
Demonstrating its tumorigenic potential, the 309G allele has been shown to be associated with early-onset cancer diagnosis in individuals with the hereditary Li-Fraumeni syndrome. The 309G allele has also been associated with early-onset diagnosis of sporadic cancers, such as soft tissue sarcoma, large B cell lymphoma, and colorectal cancer, as well as with the early diagnosis of estrogen receptor–positive breast cancer. However, the results from subsequent studies aimed at evaluating the association between SNP 309G and either cancer risk or early cancer diagnosis in other cancer types have been conflicting. Initially, there appeared to be an ethnic link between the 309G variant allele and increased cancer risk or early-onset diagnosis. Most of the studies in which the G-variant alleles showed an association with cancer risk or early-onset diagnosis had evaluated Asian or Ashkenazi Jewish populations. Assessing the role of SNP 309G in Caucasian populations provided mixed results. One group found no effect of this allele on cancer risk in Caucasian participants (92). This was proposed to be related to a second MDM2 polymorphism, SNP 285C, which was identified in a study of Norwegian patients with breast cancer (92). This polymorphism results from a G to C transversion, and is located 24 base pairs upstream of SNP 309 in the MDM2 promoter, near a predicted Sp1-binding site. The 285C and 309G polymorphisms could interact to form the 285C/309G haplotype. In vitro (in silico and surface plasmon resonance) analysis of different 285/309 haplotype combinations revealed that the enhanced transcription caused by the 309G allele was reduced in the presence of the 285C allele. In vivo analysis of the distribution of the two SNPs revealed that the presence of the 285C allele correlated with a decreased cancer risk for breast, ovarian, and endometrial cancer in patients who harbored at least one 309G allele (309GG or 309TG). Notably, Sp1 is not the only transcription factor that has a role in MDM2 expression in cancer. RXR γ-mediated MDM2 expression plays a part in cone-precursor-derived retinoblastoma (93).
To date, many meta-analyses (too numerous to cite here) have documented that there is an increased risk of several cancers, including hepatocellular carcinoma, leukemia, and colorectal, gastric, cervical, endometrial, breast, and other cancers. This was seen with the T309G variation, but was even more pronounced with the homozygous G/G genotype. Yet, as is often the case, there are exceptions: In one meta-analysis, which included studies of 5,151 cases and 1,003 controls, there was a decreased risk of prostate cancer in Caucasian patients harboring the 309G allele (94). In this same meta-analysis, no association with cancer risk was observed in an Asian ethnic group (94). And another study looking at the effect of SNPs 285 and 309 in a cohort including both Caucasians and African-Americans showed no association for either SNP with the risk of lung cancer or with survival (95). These disparate findings suggest that there are ethnic, and possibly tumor-type, variations in the SNP 309 and 285 phenotypes.
MDM2 levels are also regulated posttranscriptionally by microRNAs (miRNAs). In fact, multiple miRNAs that target MDM2 have been described, including miR-143/145, miR-605, miR-25, miR-32, miR-18b, and miR-661 (reviewed in 25). Some of these miRNAs are regulated by p53, and their modulation of MDM2 levels upregulates p53 levels and activity, effectively making this part of the negative-feedback loop. Due to their role in stabilizing p53, MDM2-regulating miRNAs function as tumor suppressors, and, indeed, a decrease in their expression levels has been observed in human cancers. Interestingly, the tumor-suppressor function of these miRNAs depends on p53 status: for example, one study found that in tumors with mutant p53, high expression levels of miR-661 correlated with a more aggressive cancer (96). In this study, miR-661 was also shown to modulate MDMX levels. In another study, published in 2010, miR-191 has been shown to play a part in the downregulation of MDM4 expression in tumors bearing MDM4-C but not MDM4-A splice variants (described below). The miR-191 target site is generated by SNP 34091, and the MDM4-A variant that lacks this SNP is associated with higher-grade carcinomas. It should be noted that the effect of miR-191 appears to be p53 independent (97). Other miRNAs that specifically target MDMX have been identified (98). The detection of these miRNAs can provide an invaluable diagnostic tool, as well as a therapeutic target.
Similarly to MDM2, MDMX is rarely mutated in cancer, but is frequently overexpressed. In fact, many tumors harboring MDM2 gene amplification, such as soft tissue sarcomas, hepatoblastomas, and urothelial cell carcinomas, have been shown to have MDMX gene deregulation as well (99–101). In addition, several MDMX-specific gene amplifications have been demonstrated: for example, in retinoblastoma, where the mechanism of p53 inactivation occurs via MDMX overexpression (102). There is a paucity of information on the upregulation of MDMX transcription. One study found that MDMX was overexpressed in approximately 50% of human colon tumors analyzed and was correlated with extracellular signal-regulated kinase phosphorylation, indicating regulation by mitogenic signaling that may promote p53 inactivation (103). As with MDM2, specific SNPs in MDMX (SNPs 1, 9, 7, and 12) may affect p53 tumor-suppression activity (104). In Ashkenazi Jewish and European cohorts, the presence of these SNPs has been associated with an increased risk of early-onset breast and ovarian cancers. It remains to be determined how these variants contribute to tumorigenesis. In 2012, it was reported that MDMX rs116197192G expression was elevated in retinoblastoma patients; however, the mechanism remains to be elucidated (105).
The role of MDMX overexpression in tumorigenesis has been explored in mouse models, with variable results. A conditional MdmX overexpression model demonstrated an increase in tumorigenesis, predominantly of sarcomas (106). However, a second model reported embryonic lethality for the homozygous mice, and there was no increase in tumorigenesis in hemizygous mice (107). Several explanations for the discrepancy are possible, including a difference in expression levels. Thus, the role of MDMX overexpression in tumorigenesis remains to be conclusively resolved.
In aggressive melanomas harboring wild-type p53 expression, a study found that upregulation of MDMX protein levels had significant clinical relevance (108). In this study, MDMX overexpression was reported in approximately 65% of stage I–IV human melanomas. MDMX deregulation has also been observed in acute lymphocytic leukemias (80%) and head and neck squamous cell carcinomas (50%) (109, 110). These studies highlight the importance of therapeutically targeting MDMX, at least in melanoma and other human cancers where p53 is rarely mutated.
Alternatively Spliced Isoforms of MDM2 and MDMX in Cancer
Although missense mutations in MDM2 and MDMX have been only rarely observed in human tumors, sequence changes due to alternative splicing have been reported, in both tumors and cell lines. Multiple isoforms of the resulting proteins are generated by use of an alternative promoter and alternative splicing, adding further complexity.
In general, MDM2 isoforms generated by alternative splicing lack the full N-terminal p53-binding domain and varying extents of the central acidic domain but, with only a few exceptions, retain the C-terminal RING domain (111). The loss of these p53-interacting regions suggests p53-independent function (or functions) for these MDM2 splice variants. An oncogenic role for these proteins has been proposed, and it is supported by the finding that human cancers, such as invasive breast cancer and soft tissue sarcoma, often express high levels of alternatively spliced MDM2 variants (reviewed in 112). In mice, the expression of Mdm2 isoforms lacking the p53-interacting domain has also been shown to predispose them to form tumors and thus further suggests a p53-independent function (113). Interestingly, although the MDM2-A variant has a protective function in the p53 wild-type background, it can contribute to tumor progression in a p53-null background (114). In one study from 2013, similar p53-independent transforming activity has been found in another MDM2 splice variant, MDM2-C (115).
As discussed above, genotoxic stress has a role in regulating isoform expression. Under these conditions, the alternative P2 promoter of MDM2 is utilized [and activated by p53, Sp1, and estrogen receptor (ER)], which leads to increased expression and possibly a greater variability in splicing. Emerging evidence has suggested that in cells harboring wild-type p53, these MDM2 isoforms may activate p53 by competing with full-length MDM2, but in cells lacking functional p53, these splice variants may take on pro-oncogenic functions (reviewed in 112). The putative role of MDM2 splice variants lacking p53-binding sites in tumorigenesis is supported by mouse models: When overexpressed, these splice variants can lead to increased tumor formation (113, 116). The aberrant expression of MDM2 splice variants has been identified in several human cancers, including colorectal cancer (117) and oral squamous cell carcinoma (118).
The existence of MDMX splice variants has also been documented, primarily in tumors and cancer cell lines; at least six variants have been reported, with differing functions. For example, the MDMX-B splice variant has been described in human glioma (119), and its expression levels are associated with cancer stage. In terms of mechanism, the S splice variant, which is characterized by the loss of exon 6 and which contains 26 unique residues, has been shown to be a more potent inhibitor of p53 function (120, 121). MDMX-S is overexpressed in some carcinomas and sarcomas (99, 122, 123). The overexpression of MDMX and MDMX-S has also been demonstrated in mantle cell lymphoma, in which it has been suggested that it suppresses p21 and therefore promotes cell-cycle progression (124).
Challenges remain in detecting the protein isoforms, not just mRNA (messenger RNA), which leaves many unanswered questions about the presence and level of expression of MDM2 and MDMX splice variants in tumors. Improving their detection may enhance the use of MDM2 and MDMX as biomarkers, and contribute to developing a better choice of chemotherapeutic agents, such as Nutlins (also referred to as imidazolines), which target the p53-dependent activity of MDM2 and would not be useful in targeting the shorter splice variants (125).
MDM2 and Regulation of Ribosomal Stress
The interaction of MDM2 with p53 is extensively regulated by posttranslation modifications and protein–protein interactions, which can modulate p53 function in response to a number of cellular stressors (Figure 3) (126). Among the myriad binding partners of MDM2 are a growing number of ribosomal proteins (RPs) (127). Cancer-derived mutations of MDM2 (mainly in the zinc-finger motif) have been reported to disrupt binding to RPL11 (128). In addition, heterozygous mutations in RPs, some of which have been shown to interact with MDM2, are associated with the human disorder Diamond–Blackfan syndrome, the symptoms of which include congenital anomalies, anemia, and a predisposition to developing leukemia and other malignancies. Several additional human ribosomopathies associated with increased cancer risk have been described, and these highlight the importance of better defining the mechanisms involved in impaired ribosome biogenesis in order to design suitable therapeutic approaches (reviewed in 129–132). Information about the link between MDM2 and such ribosomopathies, although still scarce, is intriguing.
Figure 3.
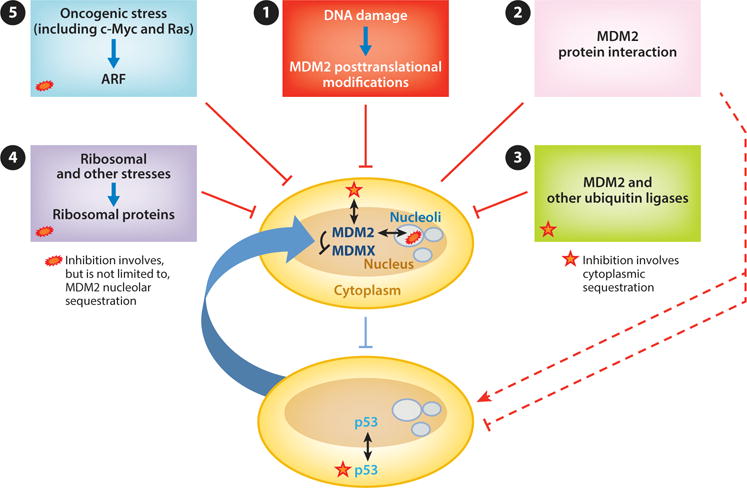
Regulation of the MDM2– and MDMX–p53 axis. In clockwise order from top: (❶) DNA damage signaling leads to posttranslational modifications of MDM2 that inhibit its interaction with and degradation of p53. Some forms of DNA damage also lead to MDM2 degradation of MDMX, thereby further activating p53. (❷) Numerous proteins interact with MDM2 and regulate its E3 ligase ability to activate or repress p53 (red dashed lines). (❸) MDM2 is degraded by as yet unidentified E3 ligases as well as by MDM2 itself in some settings. (❹) Ribosomal and other stresses cause the release of ribosomal proteins that bind and inhibit MDM2. (❺) Oncogenic stressors (including c-Myc and Ras) cause induction of ARF, which binds MDM2 and can either inhibit its E3 ligase activity or facilitate its translocation to the nucleolus.
Ribosomal stress—caused by the disruption of ribosome biosynthesis—and treatments that affect nucleolar function—such as serum starvation, experimental reduction of RPS6 levels, or drugs such as 5-fluorouracil or actinomycin D—are major sources of activation of p53 (reviewed in 127). Increasing numbers of RPs have been shown to play a part in regulating the p53–MDM2 pathway upon ribosomal stress (reviewed in 133). Several ribosomal proteins, including RPL5, RPL11, and RPL23, have been shown to interact with the MDM2 central acidic motif in a similar, yet distinct, manner. These interactions limit MDM2 E3 ubiquitin ligase activity and, hence, activate p53 function. Additional RPs, including RPL26 and RPS7, have also been reported to interact with MDM2 and to serve as substrates for its E3 ligase activity (134, 135). It is not fully understood why multiple RPs regulate MDM2 upon stress, but some work has suggested that the RP–MDM–p53 pathway may function as a more general response to various stress signals caused by different forms of DNA damage (127, 134, 136).
It has been suggested in one study that only the 60S RPs (RPL5 and RPL11) are crucial for p53 activation upon impaired ribosomal biogenesis (137). Highlighting the critical role of these RPs, it was reported that as a result of ribosomal stress, RPL5 and RPL11 exclusively accumulate in the nonribosomal fraction due to their mutual protection from proteasomal degradation and interaction with MDM2, while the other RPs are degraded by the proteasome (138). These observations, however, are not in full agreement with several previous studies showing there is a requirement for other RPs. In addition, it has been demonstrated that 5S rRNA (ribosomal RNA) is part of the RPL5–RPL11 complex (139). Intriguingly, 5S rRNA has a significant role in regulating MDMX stability (140). Further research needs to be done in order to fully comprehend the impact of RPs on the MDM2–MDMX–p53 circuit.
The role of MDM2 and ribosomal subunit interaction in oncogenesis is supported by mouse models. A disruption of the ribosome-interacting domain of MDM2 (C305F substitution) showed a reduction in response to ribosomal stress, although there was no change in the p53 DNA-damage response (141). Accelerated lymphomagenesis was observed in mice bearing this mutation in an Eμ-Myc background. Another recent mouse model described an interesting mode of action for the ribosomal protein S27-like (Rps27l), a physiological regulator of p53. In contrast to what has been reported for other RPs, and for Rps27l in cell lines (142), in vivo Rps27l stabilizes MDM2 and MDMX, and enhances p53 ubiquitination and degradation. Intriguingly, the study reported that in the same mouse model, Rps27l acts as a tumor suppressor by maintaining genomic integrity (143).
p53-Independent MDM2 and MDMX Tumorigenesis Pathways
Although the role of MDM2 overexpression in tumorigenesis via the inhibition of p53 has been extensively investigated, multiple lines of evidence also point to a p53-independent function of MDM2 in tumorigenesis and cancer progression. A clear hint of this comes from mouse models, where, in some cases, overexpression of Mdm2 leads to tumor development even in the absence of p53, and a different spectrum of tumor types and decreased tumor latency is observed. The finding that MDM2 splice variants that cannot bind p53 can still promote tumorigenesis further supports the p53-independent role for MDM2 (113, 116). Several other emerging p53-independent roles for MDM2 and MDMX in tumorigenesis and cancer progression are discussed below.
Although the role of p53 in cell-cycle arrest has been extensively studied, MDM2 also appears to have a p53-independent role in cell-cycle progression, as seen in the increased ploidy occurring in a targeted MDM2 overexpression model (144). Through ubiquitination and degradation of cell-cycle regulators, such as Rb, p21, and FoxO3A, MDM2 can directly impact the cell cycle (reviewed in 28). MDM2 has also been shown to have a p53-independent role in suppressing apoptosis: MDM2 enhances the expression of the antiapoptotic protein XIAP by enhancing its translation (145). MDMX has been demonstrated to bind with and inhibit members of the E2F and SMAD transcription-factor families (80, 146, 147).
MDM2 and MDMX have also been implicated in the induction of chromosomal instability in a p53-independent manner. Both proteins can interact with the DNA damage-response MRN complex (specifically Nsb1), as well as with ATM. Interaction with ATM slows the DNA-damage response, causing genomic instability, to an extent, when either MDM2 or MDMX is overexpressed (reviewed in 148). Interestingly, MDMX exerts this function in both a p53-and MDM2-independent manner (149). The molecular mechanism of this function, however, is unclear.
MDM2 may also play a part in the epithelial–mesenchymal transition (EMT), an essential step in the progression to metastatic cancer. MDM2 has been shown to ubiquitinate E-cadherin, targeting it for degradation (150). Because the loss of E-cadherin expression is associated with EMT and tumor grade, this finding may explain the observation that in some cancers, MDM2 amplification may correlate with poor prognosis (151). In tumor specimens from patients with metastatic breast cancer, a significant correlation has been observed between high levels of MDM2 and low levels of E-cadherin protein. This is supported by in vitro data, showing that overexpression of MDM2 in breast cancer cells can disrupt cell–cell contacts and enhance cell motility and invasive potential (150).
INTO THE CLINIC: MDM2 AND MDMX AS DIAGNOSTIC TOOLS AND THERAPEUTIC TARGETS
MDM2 and MDMX as Biomarkers
Variations in levels of MDM2 and MDMX expression (due to gene amplification and polymorphisms), and the expression of different isoforms, have been extensively described in tumors. Thus, the use of MDM2 and MDMX as both diagnostic and prognostic biomarkers has been proposed. Challenges remain in the method of detection and the identification of relevant alterations in MDM2 and MDMX: For example, MDM2 overexpression can correlate with either shorter or longer survival, depending on the tumor type (reviewed in 151). However, given the roles of these proteins in the p53 pathway, as well as their p53-independent oncogenic functions, MDM2 and MDMX may be useful biomarkers for detecting cancer earlier and ensuring more precise staging, for more accurately determining prognosis, and for improving treatment approaches.
Increased levels of MDM2 have been reported in a wide range of cancers, such as sarcomas, gliomas, melanomas, carcinomas, and hematologic malignancies (reviewed in 151). These are frequently, but not always, a result of gene amplification. In the case of MDMX as well, upregulation is most frequently due to gene amplification, and has been described in cancers such as melanoma, retinoblastoma, breast cancer, head and neck squamous cell carcinoma, glioma, and soft tissue sarcoma (reviewed in 152). In some of these tumor types, the prognostic value of alterations in MDM2 and MDMX expression has been assessed. Several research articles have assessed the use of MDM2 status in the diagnosis of liposarcomas (153–155). For example, in one study using fluorescence in situ hybridization to assess MDM2 gene amplification in well-differentiated versus dedifferentiated liposarcoma, a significant difference in amplification levels was reported, suggesting that MDM2 copy number could be used as a biomarker for tumor type (153). MDMX copy number may also have prognostic value: analyses of data from The Cancer Genome Atlas revealed lower survival rates in patients with uterine carcinoma who had amplified MDMX. A similar, although not statistically significant trend, has been observed in glioblastoma (154, 155).
The presence of specific MDM2 polymorphisms is also being investigated for use as a prognostic biomarker. There is an extensive literature elucidating the prognostic value of SNP 309 in different ethnic groups and across different cancers, as well as its value in predicting the response to treatment. For example, in a study of patients with diffuse large B cell lymphoma, those who were homozygous for the SNP 309T allele had longer overall and disease-free survival compared with those carrying the SNP 309G allele. Interestingly, SNP 309 had no effect on survival following rituximab treatment (156). In another article, the differential effects of both MDM2 and MDMX polymorphisms in hereditary pediatric retinoblastoma were assessed in relation to tumor development and survival (105). MDM2 SNP 309G (versus T) was seen significantly more frequently in controls, as was MDM4 rs4252668C (versus G). However, in patients with a constitutional RB1 mutation, longer survival correlated with the MDM2 SNP 309GG and GT genotypes.
Stress-induced isoforms of MDM2 and MDMX have been shown to correlate with high-grade disease in rhabdomyosarcoma (157), making them potentially useful as biomarkers. Relative expression of MDMX isoforms has also been reported as a prognostic marker. In osteosarcoma, a higher ratio of the S (short-form) to the FL (full-length) isoforms of MDMX correlated with less MDMX expression, and, more importantly, faster metastatic progression and overall shorter survival time. The authors postulated that this isoform ratio was associated with a perturbation of the p53 pathway (158).
A less-investigated approach to utilizing an overexpressed oncogene as a biomarker relies on detecting autoantibodies generated to it, a method that could allow for early tumor diagnosis. A study that assessed the prevalence of anti-MDM2 autoantibodies in samples from hepatocellular carcinoma tumors reported significantly higher levels compared with controls (159). Increases in MDM2 and p53 autoantibodies have also been reported in serum samples from patients with esophageal squamous cell carcinoma (160). The success of this approach in the early diagnosis of cancer remains to be determined.
MDM2 and MDMX status have also been assessed for use as predictors of responses to common therapeutic agents. For example, in tumor-derived cell lines, MDM2 copy number has been reported to be a predictor of increased sensitivity to alkylating agents and topoisomerase inhibitors, as has the MDM2 SNP 309GG genotype in a wild-type p53 background (161). MDM2 status has also been linked to radiosensitivity: The SNP 309GG genotype correlated with shorter disease-free survival times in oral squamous cell carcinoma patients undergoing postoperative radiotherapy (162). These patients were also shown to be less sensitive to radiotherapy. Conversely, in patients undergoing platinum-based chemotherapy for bladder cancer, the GG or GT SNP 309 genotype correlated with a higher survival rate (163). In gastric cancer, high expression of MDM2 correlated with shorter overall survival time without adjuvant treatment, but following treatment with fluorouracil–leucovorin–oxaliplatin, high expression of MDM2 correlated with higher overall survival (164). Increased MDM2 and MDMX levels in liposarcomas have been shown to correlate with enhanced sensitivity to Nutlin 3a, a well-studied molecule that disrupts the p53–MDM2 interaction in vivo (165). This underscores the importance of MDM2 and MDMX as biomarkers in human cancer and as predictors of therapeutic response.
Assays to determine MDM2 and MDMX amplification status, the SNP genotype, the expression level overall, and the expression of specific isoforms are under investigation in cancer patients. Given the important role these genes have in oncogenesis and cancer progression, identifying their specific expression patterns may be useful in enabling better diagnosis of tumor types, in more precisely tailoring treatment, and in determining overall prognosis.
MDM2 and MDMX as Therapeutic Targets
Deciphering the properties and functions of p53 and its two main inhibitors, MDM2 and MDMX, as well as their interactions, has proved essential to developing potential agents for cancer therapy. Roughly one-half of all cancers (with varying frequencies depending on tumor type) harbor mutant forms of p53. The approximately 50% of tumors that express wild-type p53 are potential targets for therapies aimed at reactivating p53 function. Some wild-type p53 tumors overexpress MDM2 or MDMX, or both, mainly by gene amplification or mutations (reviewed in 166). These tumors are potential targets for MDM2- and MDMX-based therapies, with the goal of restoring the tumor-suppression function of the p53 protein or enhancing its therapeutic response, or both. To achieve these goals, the most direct approach is to develop potent inhibitors against the p53-binding pockets of MDM2 and MDMX to prevent their interaction with p53. Other strategies that involve p53 itself have been described, including gene therapy, adenovirus-based therapy, p53 vaccines, and antisense and synthetic RNA approaches; these have been summarized recently and are not discussed in our review (167, 168). Currently, there are multiple small-molecule inhibitors of the MDM2–p53 interaction, and some advanced compounds are already in Phase I clinical trials. In this section we summarize progress in the field.
Targeting protein–protein interactions with small-molecule inhibitors is extremely complicated on many levels, but mainly due to the fact that their binding surfaces are usually too large or flat, or both, for effective interference by small molecules. That said, a success for this strategy has been reported and additional proof that it can be achieved has been demonstrated by the generation of efficient inhibitors for the interaction between p53 and MDM2, which is based on their structure and high-throughput screening methods (Table 1) (reviewed in 84, 169). What makes the p53–MDM2 interaction unique and targetable are the features of the deep pocket in MDM2 and the three residues in p53 that play a major part in this binding, which have been well defined by structural biology assays. In 2004, a group of small-molecule antagonists known as Nutlins, which target the MDM2 N-terminal p53-binding pocket and, hence, displace p53, were generated and shown to interfere with the interaction between p53 and MDM2, and to activate p53 function (170, 171). Indeed, the success of Nutlin 3a in disrupting the p53–MDM2 interaction has been something of a poster child for being able to efficiently disrupt interactions between two proteins. An improved derivative of Nutlin 3a, RG7112 (RO5045337, Hoffmann-La Roche), is currently in Phase I clinical trials and its use has been reported in patients (ClinicalTrials.gov numbers: NCT01677780, NCT01143740, NCT01164033, NCT00559533, and NCT00623870). The outcome of the first preliminary biomarker studies of RG7112 in a group of 20 patients has been published, and it proved the potency of RG7112 in activating p53 function (172). In patients with liposarcomas, a tumor type that frequently has amplified MDM2 and wild-type p53, RG7112 reached the tumor site, but the effect on p53 was inconclusive. Additional studies with more participants will be necessary in order to draw more solid conclusions about this drug. Some of the other compounds that target the MDM2 pocket include other small molecules, such as MI-219 (173), MI-63, MI-319 (174), the improved analog MI-773 that has entered Phase I clinical trials (Sanofi-Aventis) (175), benzodiazepinediones, AM-8553 (reported by Amgen) (176, 177), MK-8242 (reported by Merck in two Phase I trials; ClinicalTrials.gov numbers: NCT01451437 and NCT01463696), and AMG 232 (178, 179), which is currently in clinical trial. The use of some of these small-molecule inhibitors resulted in a partial decrease in the size of tumors in SJSA-1 xenograft models in mice and in human tumors (reviewed in 180). Although MI-219 and Nutlin 3a activate p53 in cancer cells that overexpress MDM2, they do not always have an effect on cancer cells expressing high levels of MDMX due to differences in MDM2 and MDMX p53-binding pockets (181–183).
Table 1.
Several approaches to generate antagonists to MDM2 and MDMX and to their interaction with p53 as well as their resultant products
| Strategy | Compound |
|---|---|
| Interruption of p53 interaction with MDM2 | Small molecules that bind the MDM2 N′ pocket: Nutlin 3a (RG7112), MI-219, MI-63, MI-319, MI-773, AM-8553, AMG 232, benzodiazepinediones, MK-8242 Small molecules that bind the p53 N-terminal domain: RITA |
| Interruption of p53 interaction with MDMX | Small molecules: WK 298, SJ-172550 |
| Interruption of p53 interactions with both MDM2 and MDMX | Peptidic compounds: SAH-p53s, PMI peptide, pDI peptide Small molecules: RO-5963, CGM097 |
| Inhibition of MDM2 ubiquitin ligase function | Small molecules: HLI98, MPD, MEL23, MEL24 |
| Inhibition of MDM2 E3 function | Small molecule: JNJ-26854165 |
| Inhibition of MDMX transcription | Small molecule: NSC 207895 |
A compound termed RITA (reactivation of p53 and induction of tumor cell apoptosis, also known as NSC 652287) binds the p53 N-terminal domain and disrupts the interaction between p53 and MDM2 without binding MDM2 (184). Treatment with RITA leads to an effect similar to that of molecules that bind the MDM2 pocket; however, RITA has also been shown to disrupt the interaction between MDM2 and iASPP. Yet another class of peptides that blocks MDM2 and, in some cases, MDMX function are the stapled peptides that compete for MDM2 and MDMX binding. Peptides such as SAH-p53s can bind MDM2 and inhibit its ability to bind p53 (185–192).
MDM2 E3 ligase activity has also been assessed as a therapeutic target. The rationale and importance of developing compounds that target this activity is supported by the fact that compounds that disrupt the interaction between p53 and MDM2 do not inhibit MDM2 E3 ligase function and, hence, do not halt the p53-independent activity of MDM2. MDM2 E3 ubiquitin ligase inhibitors, such as HLI98 (193), MPD (194), or MEL23 and MEL24 (195), have been shown to inhibit the ubiquitination of p53 and to stabilize the protein. Another small molecule, JNJ-26854165, inhibits both MDM2 E3 activity and proteasome binding, thus activating p53 function. This molecule from Forschungszentrum Karlsruhe and Janssen Pharmaceuticals entered a Phase I clinical study of use in cancer patients (ClinicalTrials.gov number: NCT00676910), but reportedly caused cardiotoxicity and exhibited off-target effects (196–198).
Recent research has highlighted the importance of generating inhibitors against MDMX for anticancer therapy. In melanoma, in which the majority of cases express wild-type p53, MDMX is abnormally upregulated (108, 199). Importantly, inhibition of the interaction between p53 and MDMX has been shown to restore the function of p53, suggesting that, at least in melanoma, p53 is the prime target of MDMX. The melanoma studies are of particular clinical relevance due to the fact that MDMX overexpression seems to be predominant in these tumors, with infrequent p53 mutations.
Several small molecules that target MDMX have been reported, including a small molecule called WK 298, which disrupts the interaction between p53 and MDMX by resembling the binding of a p53 peptide (200), and SJ-172550, which targets the MDMX N terminus–p53 interaction pocket (201). Careful analysis of SJ-172550 has revealed that this compound affects the conformation of MDMX instead of acting as a competitive inhibitor (202). The complex mechanism of this compound could prevent its further development for therapeutic purposes. In addition, some small molecules impede MDMX transcription—such as NSC 207895, a benzofuroxan derivative (203)—or indirectly destabilize MDMX through the inhibition of heat shock protein 90—such as 17-AAG, also known as tanespimycin (204).
Because many tumors overexpress MDMX, molecules that target solely MDM2 may be insufficient. Several compounds that target both MDM2 and MDMX, and therefore disrupt both p53–MDM2 binding and p53–MDMX binding, have been generated and are being tested. These include the small molecule RO-5963, which interferes with the p53–MDM2–MDMX interaction by promoting MDMX and MDM2 homo- and heterodimerization (205), and CGM097, developed by Novartis, which also sponsored a Phase I clinical trial for this molecule (ClinicalTrials.gov number: NCT01760525). Additional inhibitors of both MDM2 and MDMX include some synthetic peptidic compounds, such as SAH-p53-8, PMI peptide, and pDI peptide (185, 206–208). Notably, one study showed that high levels of SAH-p53-8 were effective in a melanoma mouse model expressing high levels of MDMX, in which applying SAH-p53-8 enhanced the effect of treatment with cisplatin and vemurafenib (a BRAF-V600E inhibitor) (108). This study is an example of the potential of using treatments that combine compounds or approaches. Using combination therapy, as opposed to more focused, conventional treatment, may reduce the problematic phenomenon of resistance and allow cancer treatment strategies to be tailored to each patient (209). (For a review of combination therapies with an emphasis on the inhibitor compounds, see 180.)
FUTURE DIRECTIONS
As the complexity of the roles that MDM2 and MDMX have in tumorigenesis becomes better appreciated, there is the potential for developing new and better diagnostic and therapeutic uses of these proteins in cancer. The MDM2 and MDMX research community can move forward in both refining previously utilized approaches and designing new strategies for targeting MDM2 and MDMX.
MDM2
The amplification of MDM2 in cancer has been widely appreciated. However, the part that mutations and polymorphisms, as well as posttranscriptional regulation, play in the expression of MDM2 (and MDMX) has not yet been fully elucidated. Although research looking at the effect of SNPs, such as SNP 309, has revealed their great potential as prognostic markers, further research into the role of this and other polymorphisms in different populations, their interactions with one another, and their involvement in the response to hormones such as estrogen will contribute to a more complete picture of MDM2 genetics. The emerging work on mutations in the MDM2 gene, and their effect on tumorigenesis, may lead to improved diagnostics.
Regulation at the posttranscriptional level has also been shown to have an important role in modulating MDM2 levels and functions. Utilizing MDM2 and MDMX for diagnostic purposes requires a clear understanding of the specific protein being expressed. Emerging data on MDM2 and MDMX isoforms makes it clear that such variants have distinct roles in oncogenesis. Better tools to assay isoform expression at the protein level will be essential for using MDM2 and MDMX as biomarkers. It will also be important to understand the stoichiometry of isoform expression in normal tissues and in tumors to design therapeutics that do not disrupt the balance of isoform expression. Finally, because some isoforms do not interact with p53, better tests for isoform expression can lead to better predictions of responses to therapeutic agents.
The regulation of MDM2 expression by miRNAs also provides an avenue of research into both better diagnostics and therapeutics. The correlation between specific miRNA expression and tumor aggressiveness may provide an important prognostic tool. And miRNA-based cancer therapies may offer an attractive alternative to small-molecule approaches to targeting MDM2 expression.
The MDM2–MDMX Heterodimer
In recent years, MDMX has emerged as an important component in the regulation of p53 and MDM2, and as a promising therapeutic target. Because the MDM2–MDMX heterodimer is likely to be physiologically relevant to the p53 ubiquitin ligase, it is essential to understand the balance of expression of these proteins and their roles in modulating p53 levels and oncogenesis. Indeed the MEL compounds have been shown to specifically target the MDM2–MDMX heterodimer (195). Promising research findings showing that disrupting the MDM2–MDMX interaction can lead to stabilization of p53 in the clinic indicates that this could provide an alternative to Nutlins and related molecules in the treatment of p53 wild-type cancers. In addition, targeting both MDM2 and MDMX may provide a more effective therapeutic strategy.
Interaction with Mutant p53
There is strong evidence that, at least in mice, Mdm2 may also play a part in modulating the activity of mutant p53 (210). This important finding mandates careful consideration when evaluating patients for MDM2-based therapeutics. Although targeting MDM2 in p53 wild-type tumors is an established treatment strategy, the heterogeneity of tumors raises the risk of the presence of p53-mutant tumor cells; therefore, this treatment may result in an undesired oncogenic effect. Moreover, different p53 mutations may have different activities. A suitable, tailored treatment needs to be considered in conjunction with genetic diagnosis. However, a promising line of attack for some tumors, such as serous ovarian cancer, for which the frequency of p53 mutation is extremely high (>95%), may be to find ways to stimulate the activity of MDM2.
Fully p53-Independent Functions
The rising appreciation of the p53-independent functions of MDM2 creates both new opportunities and new challenges. The ability to target MDM2 in p53-null tumors may provide a novel therapeutic strategy. However, in devising therapeutic approaches, it is essential to have a clearer understanding of all pathways affected by MDM2 to avoid unwanted effects, even in p53 wild-type tumors.
Developing New Detection Techniques
The existence of multiple isoforms of p53, MDM2, and MDMX adds additional levels of complexity. We still only poorly understand the role and expression of the different isoforms in different tumors, mainly due to the lack of good reagents, specifically isoform-specific antibodies. It will be essential to develop new reagents that uniquely detect a given expressed variant while avoiding cross-reactivity with other isoforms. Intriguingly, a promising new technique using nanobodies has recently been described (211). Nanobodies, which derive from the variable regions of Camelidae atypical immunoglobulins, are single-domain antibodies that function similarly to regular antibodies, but their small size (about one-tenth of an antibody), simple production in bacteria factories, high affinity for their respective epitopes, and great diversity make them attractive candidates for such reagents. We predict that nanobodies will be powerful general tools for diagnostic and therapeutic interventions involving p53, MDM2, and MDMX.
Understanding MDM2 and MDMX as Diagnostic and Prognostic Markers
Despite the wealth of knowledge that has already been accrued about MDM2 and MDMX, evaluating their respective or combined expression for use as a diagnostic tool will require a better understanding of the roles the two proteins have in different tissues, in different genetic backgrounds, as well as with their interaction partners. To ensure their safety, it will also be important to evaluate the impact of prolonged treatment with MDM2 or MDMX inhibitors, due to the potential toxicity that may occur when p53 is being activated and upregulated.
In summary, our increasing knowledge about the parts that MDM2 and MDMX play in both p53-dependent and -independent pathways may lead to exciting new approaches to treating cancer in the clinic. Whether acting as oncogenes or tumor suppressors, a better understanding of these proteins will set the stage for many new diagnostic and therapeutic advances.
Acknowledgments
The authors apologize for not being able to cite many important studies due to space limitations. We would like to thank Dr. Nicole Okoh for her help in initial stages of this manuscript and Dr. Yan Zhu (Columbia University) for critically reading the review and for insightful comments. Work on this review was supported by a grant from the National Institutes of Health (CA 58316).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Errata
An online log of corrections to Annual Review of Pathology: Mechanisms of Disease articles may be found at http://www.annualreviews.org/errata/pathol
LITERATURE CITED
- 1.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malkin D. Li-Fraumeni syndrome. Genes Cancer. 2011;2:475–84. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donehower LA. Insights into wild-type and mutant p53 functions provided by genetically engineered mice. Hum Mutat. 2014;35:715–27. doi: 10.1002/humu.22507. [DOI] [PubMed] [Google Scholar]
- 6.Cahilly-Snyder L, Yang-Feng T, Francke U, George DL. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat Cell Mol Genet. 1987;13:235–44. doi: 10.1007/BF01535205. [DOI] [PubMed] [Google Scholar]
- 7.Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991;10:1565–69. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 9.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 10.Perry ME, Piette J, Zawadzki JA, Harvey D, Levine AJ. The mdm-2 gene is induced in response to UV light in a p53-dependent manner. PNAS. 1993;90:11623–27. doi: 10.1073/pnas.90.24.11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–68. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 13.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 14.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–99. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 15.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 16.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 17.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 18.Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–50. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 19.Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998;12:2984–91. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- 20.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349–57. [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem. 1999;274:38189–96. doi: 10.1074/jbc.274.53.38189. [DOI] [PubMed] [Google Scholar]
- 22.Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447:5–9. doi: 10.1016/s0014-5793(99)00254-9. [DOI] [PubMed] [Google Scholar]
- 23.Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–38. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parant JM, Reinke V, Mims B, Lozano G. Organization, expression, and localization of the murine mdmx gene and pseudogene. Gene. 2001;270:277–83. doi: 10.1016/s0378-1119(01)00432-2. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Yu H, Hu W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sinica. 2014;46:180–89. doi: 10.1093/abbs/gmt147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai H, Wiederschain D, Yuan ZM. Critical contribution of the MDM2 acidic domain to p53 ubiquitination. Mol Cell Biol. 2003;23:4939–47. doi: 10.1128/MCB.23.14.4939-4947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meulmeester E, Frenk R, Stad R, de Graaf P, Marine JC, et al. Critical role for a central part of Mdm2 in the ubiquitylation of p53. Mol Cell Biol. 2003;23:4929–38. doi: 10.1128/MCB.23.14.4929-4938.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manfredi JJ. The Mdm2– p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24:1580–89. doi: 10.1101/gad.1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, et al. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007;26:90–101. doi: 10.1038/sj.emboj.7601465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26:102–12. doi: 10.1038/sj.emboj.7601469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clegg HV, Itahana K, Zhang Y. Unlocking the Mdm2-p53 loop: Ubiquitin is the key. Cell Cycle. 2008;7:287–92. doi: 10.4161/cc.7.3.5358. [DOI] [PubMed] [Google Scholar]
- 32.Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994;8:1235–46. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- 33.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–53. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 34.Ma J, Martin JD, Zhang H, Auger KR, Ho TF, et al. A second p53 binding site in the central domain of Mdm2 is essential for p53 ubiquitination. Biochemistry. 2006;45:9238–45. doi: 10.1021/bi060661u. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu H, Burch LR, Smith AJ, Dornan D, Wallace M, et al. The conformationally flexible S9–S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. J Biol Chem. 2002;277:28446–58. doi: 10.1074/jbc.M202296200. [DOI] [PubMed] [Google Scholar]
- 36.Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell. 2006;23:251–63. doi: 10.1016/j.molcel.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 37.Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez-Fernandez MR, Fersht AR. The central region of HDM2 provides a second binding site for p53. PNAS. 2006;103:1227–32. doi: 10.1073/pnas.0510343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cross B, Chen L, Cheng Q, Li B, Yuan ZM, Chen J. Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J Biol Chem. 2011;286:16018–29. doi: 10.1074/jbc.M111.228981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol. 2010;17:982–89. doi: 10.1038/nsmb.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips A, Teunisse A, Lam S, Lodder K, Darley M, Emaduddin M, et al. HDMX-L is expressed from a functional p53-responsive promoter in the first intron of the HDMX gene and participates in an autoregulatory feedback loop to control p53 activity. J Biol Chem. 2010;285:29111–27. doi: 10.1074/jbc.M110.129726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bottger V, Bottger A, Garcia-Echeverria C, Ramos YF, van der Eb AJ, et al. Comparative study of the p53–mdm2 and p53–MDMX interfaces. Oncogene. 1999;18:189–99. doi: 10.1038/sj.onc.1202281. [DOI] [PubMed] [Google Scholar]
- 42.Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–43. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 43.Shadfan M, Lopez-Pajares V, Yuan ZM. MDM2 and MDMX: alone and together in regulation of p53. Transl Cancer Res. 2012;1:88–89. [PMC free article] [PubMed] [Google Scholar]
- 44.Priest C, Prives C, Poyurovsky MV. Deconstructing nucleotide binding activity of the Mdm2 RING domain. Nucleic Acids Res. 2010;38:7587–98. doi: 10.1093/nar/gkq669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poyurovsky MV, Jacq X, Ma C, Karni-Schmidt O, Parker PJ, et al. Nucleotide binding by the Mdm2 RING domain facilitates Arf-independent Mdm2 nucleolar localization. Mol Cell. 2003;12:875–87. doi: 10.1016/s1097-2765(03)00400-3. [DOI] [PubMed] [Google Scholar]
- 46.Stevens C, Pettersson S, Wawrzynow B, Wallace M, Ball K, et al. ATP stimulates MDM2-mediated inhibition of the DNA-binding function of E2F1. FEBS J. 2008;275:4875–86. doi: 10.1111/j.1742-4658.2008.06627.x. [DOI] [PubMed] [Google Scholar]
- 47.Singh RK, Iyappan S, Scheffner M. Hetero-oligomerization with MdmX rescues the ubiquitin/Nedd8 ligase activity of RING finger mutants of Mdm2. J Biol Chem. 2007;282:10901–7. doi: 10.1074/jbc.M610879200. [DOI] [PubMed] [Google Scholar]
- 48.Bista M, Petrovich M, Fersht AR. MDMX contains an autoinhibitory sequence element. PNAS. 2013;110:17814–19. doi: 10.1073/pnas.1317398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000. [PubMed] [Google Scholar]
- 50.Jain AK, Barton MC. Making sense of ubiquitin ligases that regulate p53. Cancer Biol Ther. 2010;10:665–72. doi: 10.4161/cbt.10.7.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harbor Perspect Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulikov R, Letienne J, Kaur M, Grossman SR, Arts J, Blattner C. Mdm2 facilitates the association of p53 with the proteasome. PNAS. 2010;107:10038–43. doi: 10.1073/pnas.0911716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brooks CL, Gu W. p53 regulation by ubiquitin. FEBS Lett. 2011;585:2803–9. doi: 10.1016/j.febslet.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barak Y, Gottlieb E, Juven-Gershon T, Oren M. Regulation of mdm2 expression by p53: Alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994;8:1739–49. doi: 10.1101/gad.8.15.1739. [DOI] [PubMed] [Google Scholar]
- 55.Wu X, Bayle JH, Olson D, Levine AJ. The p53 –mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 56.Lev Bar-Or R, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. PNAS. 2000;97:11250–55. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, et al. Dynamics of the p53–Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 58.Hu W, Feng Z, Ma L, Wagner J, Rice JJ, et al. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007;67:2757–65. doi: 10.1158/0008-5472.CAN-06-2656. [DOI] [PubMed] [Google Scholar]
- 59.Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, et al. The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013;27:1857–67. doi: 10.1101/gad.227249.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 61.Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol Cancer Res. 2009;7:1–11. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, et al. Rescue of embryonic lethality in Mdm 4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 63.Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927–34. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- 64.Steinman HA, Hoover KM, Keeler ML, Sands AT, Jones SN. Rescue of Mdm4-deficient mice by Mdm2 reveals functional overlap of Mdm2 and Mdm4 in development. Oncogene. 2005;24:7935–40. doi: 10.1038/sj.onc.1208930. [DOI] [PubMed] [Google Scholar]
- 65.Wang YV, Wade M, Wong E, Li YC, Rodewald LW, Wahl GM. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. PNAS. 2007;104:12365–70. doi: 10.1073/pnas.0701497104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Wang J, Jiang X. MdmX protein is essential for Mdm2 protein-mediated p53 polyubiquitination. J Biol Chem. 2011;286:23725–34. doi: 10.1074/jbc.M110.213868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING domain-mediated interaction is a requirement for MDM2’s E3 ligase activity. Cancer Res. 2007;67:6026–30. doi: 10.1158/0008-5472.CAN-07-1313. [DOI] [PubMed] [Google Scholar]
- 68.Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15:841–44. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- 69.Li C, Chen L, Chen J. DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol. 2002;22:7562–71. doi: 10.1128/MCB.22.21.7562-7571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol. 2003;23:5113–21. doi: 10.1128/MCB.23.15.5113-5121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278:45946–53. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- 72.Huang L, Yan Z, Liao X, Li Y, Yang J, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. PNAS. 2011;108:12001–6. doi: 10.1073/pnas.1102309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. PNAS. 2011;108:11995–2000. doi: 10.1073/pnas.1102241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tollini LA, Jin A, Park J, Zhang Y. Regulation of p53 by Mdm2 E3 ligase function is dispensable in embryogenesis and development, but essential in response to DNA damage. Cancer Cell. 2014;26:235–47. doi: 10.1016/j.ccr.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Conza G, Mancini F, Buttarelli M, Pontecorvi A, Trimarchi F, Moretti F. MDM4 enhances p53 stability by promoting an active conformation of the protein upon DNA damage. Cell Cycle. 2012;11:749–60. doi: 10.4161/cc.11.4.19208. [DOI] [PubMed] [Google Scholar]
- 76.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 77.Perry ME. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harbor Perspect Biol. 2010;2:a000968. doi: 10.1101/cshperspect.a000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu Q, Li Y, Mu K, Li Z, Meng Q, et al. Amplification of Mdmx and overexpression of MDM2 contribute to mammary carcinogenesis by substituting for p53 mutations. Diagn Pathol. 2014;9:71. doi: 10.1186/1746-1596-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835–43. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matijasevic Z, Krzywicka-Racka A, Sluder G, Jones SN. MdmX regulates transformation and chromosomal stability in p53-deficient cells. Cell Cycle. 2008;7:2967–73. doi: 10.4161/cc.7.19.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX promotes bipolar mitosis to suppress transformation and tumorigenesis in p53-deficient cells and mice. Mol Cell Biol. 2008;28:1265–73. doi: 10.1128/MCB.01108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mancini F, Di Conza G, Pellegrino M, Rinaldo C, Prodosmo A, et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J. 2009;28:1926–39. doi: 10.1038/emboj.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu Y, Regunath K, Jacq X, Prives C. Cisplatin causes cell death via TAB1 regulation of p53/MDM2/MDMX circuitry. Genes Dev. 2013;27:1739–51. doi: 10.1101/gad.212258.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–59. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mairinger FD, Walter RF, Ting S, Vollbrecht C, Kollmeier J, et al. Mdm2 protein expression is strongly associated with survival in malignant pleural mesothelioma. Future Oncol. 2014;10:995–1005. doi: 10.2217/fon.13.261. [DOI] [PubMed] [Google Scholar]
- 87.Walter RF, Mairinger FD, Ting S, Vollbrecht C, Mairinger T, et al. MDM2 is an important prognostic and predictive factor for platin–pemetrexed therapy in malignant pleural mesotheliomas and deregulation of P14/ARF (encoded by CDKN2A) seems to contribute to an MDM2-driven inactivation of P53. Br J Cancer. 2015;112:883–90. doi: 10.1038/bjc.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. PNAS. 1998;95:15608–12. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Senturk E, Manfredi JJ. Mdm2 and tumorigenesis: evolving theories and unsolved mysteries. Genes Cancer. 2012;3:192–98. doi: 10.1177/1947601912457368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 91.Post SM, Quintas-Cardama A, Pant V, Iwakuma T, Hamir A, et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell. 2010;18:220–30. doi: 10.1016/j.ccr.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Knappskog S, Bjornslett M, Myklebust LM, Huijts PE, Vreeswijk MP, et al. The MDM2 promoter SNP285C/309G haplotype diminishes Sp1 transcription factor binding and reduces risk for breast and ovarian cancer in Caucasians. Cancer Cell. 2011;19:273–82. doi: 10.1016/j.ccr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 93.Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–31. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang J, Gao W, Song NH, Wang W, Zhang JX, et al. The risks, degree of malignancy and clinical progression of prostate cancer associated with the MDM2 T309G polymorphism: a meta-analysis. Asian J Androl. 2012;14:726–31. doi: 10.1038/aja.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ryan BM, Calhoun KM, Pine SR, Bowman ED, Robles AI, et al. MDM2 SNP285 does not antagonize the effect of SNP309 in lung cancer. Int J Cancer. 2012;131:2710–16. doi: 10.1002/ijc.27573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoffman Y, Bublik DR, Pilpel Y, Oren M. miR-661 downregulates both Mdm2 and Mdm4 to activate p53. Cell Death Differ. 2014;21:302–9. doi: 10.1038/cdd.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wynendaele J, Bohnke A, Leucci E, Nielsen SJ, Lambertz I, et al. An illegitimate microRNA target site within the 3– UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010;70:9641–49. doi: 10.1158/0008-5472.CAN-10-0527. [DOI] [PubMed] [Google Scholar]
- 98.Mandke P, Wyatt N, Fraser J, Bates B, Berberich SJ, Markey MP. MicroRNA-34a modulates MDM4 expression via a target site in the open reading frame. PLOS ONE. 2012;7:e42034. doi: 10.1371/journal.pone.0042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bartel F, Schulz J, Bohnke A, Blumke K, Kappler M, et al. Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int J Cancer. 2005;117:469–75. doi: 10.1002/ijc.21206. [DOI] [PubMed] [Google Scholar]
- 100.Veerakumarasivam A, Scott HE, Chin SF, Warren A, Wallard MJ, et al. High-resolution array-based comparative genomic hybridization of bladder cancers identifies Mouse Double Minute 4 (MDM4) as an amplification target exclusive of MDM2 and TP53. Clin Cancer Res. 2008;14:2527–34. doi: 10.1158/1078-0432.CCR-07-4129. [DOI] [PubMed] [Google Scholar]
- 101.Arai Y, Honda S, Haruta M, Kasai F, Fujiwara Y, et al. Genome-wide analysis of allelic imbalances reveals 4q deletions as a poor prognostic factor and MDM4 amplification at 1q32.1 in hepatoblastoma. Genes Chromosomes Cancer. 2010;49:596–609. doi: 10.1002/gcc.20770. [DOI] [PubMed] [Google Scholar]
- 102.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 103.Gilkes DM, Pan Y, Coppola D, Yeatman T, Reuther GW, Chen J. Regulation of MDMX expression by mitogenic signaling. Mol Cell Biol. 2008;28:1999–2010. doi: 10.1128/MCB.01633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Atwal GS, Kirchhoff T, Bond EE, Montagna M, Menin C, et al. Altered tumor formation and evolutionary selection of genetic variants in the human MDM4 oncogene. PNAS. 2009;106:10236–41. doi: 10.1073/pnas.0901298106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, Ferman SE, Lucena E, et al. Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer. 2012;59:39–43. doi: 10.1002/pbc.24014. [DOI] [PubMed] [Google Scholar]
- 106.Xiong S, Pant V, Suh YA, Van Pelt CS, Wang Y, et al. Spontaneous tumorigenesis in mice overexpressing the p53-negative regulator Mdm4. Cancer Res. 2010;70:7148–54. doi: 10.1158/0008-5472.CAN-10-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.De Clercq S, Gembarska A, Denecker G, Maetens M, Naessens M, et al. Widespread overexpression of epitope-tagged Mdm4 does not accelerate tumor formation in vivo. Mol Cell Biol. 2010;30:5394–405. doi: 10.1128/MCB.00330-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18:1239–47. doi: 10.1038/nm.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Han X, Garcia-Manero G, McDonnell TJ, Lozano G, Medeiros LJ, et al. HDM4 (HDMX) is widely expressed in adult pre-B acute lymphoblastic leukemia and is a potential therapeutic target. Mod Pathol. 2007;20:54–62. doi: 10.1038/modpathol.3800727. [DOI] [PubMed] [Google Scholar]
- 110.Valentin-Vega YA, Barboza JA, Chau GP, El-Naggar AK, Lozano G. High levels of the p53 inhibitor MDM4 in head and neck squamous carcinomas. Hum Pathol. 2007;38:1553–62. doi: 10.1016/j.humpath.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bartel F, Harris LC, Wurl P, Taubert H. MDM2 and its splice variant messenger RNAs: expression in tumors and down-regulation using antisense oligonucleotides. Mol Cancer Res. 2004;2:29–35. [PubMed] [Google Scholar]
- 112.Okoro DR, Rosso M, Bargonetti J. Splicing up mdm2 for cancer proteome diversity. Genes Cancer. 2012;3:311–19. doi: 10.1177/1947601912455323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fridman JS, Hernando E, Hemann MT, de Stanchina E, Cordon-Cardo C, Lowe SW. Tumor promotion by Mdm2 splice variants unable to bind p53. Cancer Res. 2003;63:5703–6. [PubMed] [Google Scholar]
- 114.Volk EL, Schuster K, Nemeth KM, Fan L, Harris LC. MDM2-A, a common Mdm2 splice variant, causes perinatal lethality, reduced longevity and enhanced senescence. Dis Model Mech. 2009;2:47–55. doi: 10.1242/dmm.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Okoro DR, Arva N, Gao C, Polotskaia A, Puente C, et al. Endogenous human MDM2-C is highly expressed in human cancers and functions as a p53-independent growth activator. PLOS ONE. 2013;8:e77643. doi: 10.1371/journal.pone.0077643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steinman HA, Burstein E, Lengner C, Gosselin J, Pihan G, et al. An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis. J Biol Chem. 2004;279:4877–86. doi: 10.1074/jbc.M305966200. [DOI] [PubMed] [Google Scholar]
- 117.Yu Z, Zhang B, Cui B, Wang Y, Han P, Wang X. Identification of spliced variants of the proto-oncogene hdm2 in colorectal cancer. Cancer. 2012;118:1110–18. doi: 10.1002/cncr.26330. [DOI] [PubMed] [Google Scholar]
- 118.Sam KK, Gan CP, Yee PS, Chong CE, Lim KP, et al. Novel MDM2 splice variants identified from oral squamous cell carcinoma. Oral Oncol. 2012;48:1128–35. doi: 10.1016/j.oraloncology.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 119.Wang X, Sheng P, Guo X, Wang J, Hou L, et al. Identification and expression of a novel MDM4 splice variant in human glioma. Brain Res. 2013;1537:260–66. doi: 10.1016/j.brainres.2013.07.054. [DOI] [PubMed] [Google Scholar]
- 120.de Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG. Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J Biol Chem. 2003;278:38315–24. doi: 10.1074/jbc.M213034200. [DOI] [PubMed] [Google Scholar]
- 121.Rallapalli R, Strachan G, Cho B, Mercer WE, Hall DJ. A novel MDMX transcript expressed in a variety of transformed cell lines encodes a truncated protein with potent p53 repressive activity. J Biol Chem. 1999;274:8299–308. doi: 10.1074/jbc.274.12.8299. [DOI] [PubMed] [Google Scholar]
- 122.Bartel F, Schulz J, Blumke K, Kappler M, Bache M, et al. [HDMX amplification and high levels of HDMX-S splice variant are correlated with a poor prognosis in soft tissue sarcomas] Verh Dtsch Ges Pathol. 2004;88:199–206. [PubMed] [Google Scholar]
- 123.Prodosmo A, Giglio S, Moretti S, Mancini F, Barbi F, et al. Analysis of human MDM4 variants in papillary thyroid carcinomas reveals new potential markers of cancer properties. J Mol Med. 2008;86:585–96. doi: 10.1007/s00109-008-0322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liang M, Han X, Vadhan-Raj S, Nguyen M, Zhang YH, et al. HDM4 is overexpressed in mantle cell lymphoma and its inhibition induces p21 expression and apoptosis. Mod Pathol. 2010;23:381–91. doi: 10.1038/modpathol.2009.170. [DOI] [PubMed] [Google Scholar]
- 125.Bozzi F, Conca E, Laurini E, Posocco P, Lo Sardo A, et al. In vitro and in silico studies of MDM2/MDMX isoforms predict Nutlin-3A sensitivity in well/de-differentiated liposarcomas. Lab In-vestig. 2013;93:1232–40. doi: 10.1038/labinvest.2013.107. [DOI] [PubMed] [Google Scholar]
- 126.Meek DW, Hupp TR. The regulation of MDM2 by multisite phosphorylation—opportunities for molecular-based intervention to target tumours? Semin Cancer Biol. 2010;20:19–28. doi: 10.1016/j.semcancer.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 127.Zhang Y, Lu H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell. 2009;16:369–77. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lipton JM, Ellis SR. Diamond Blackfan anemia 2008–2009 broadening the scope of ribosome biogenesis disorders. Curr Opin Pediatr. 2010;22:12–19. doi: 10.1097/MOP.0b013e328334573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Suzuki A, Kogo R, Kawahara K, Sasaki M, Nishio M, et al. A new PICTure of nucleolar stress. Cancer Sci. 2012;103:632–37. doi: 10.1111/j.1349-7006.2012.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Teng T, Thomas G, Mercer CA. Growth control and ribosomopathies. Curr Opin Genet Dev. 2013;23:63–71. doi: 10.1016/j.gde.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 132.Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, Lu H. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord. 2014;7:7–16. doi: 10.4137/CMBD.S16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2– p53 pathway revisited. J Biomed Res. 2013;27:254–71. doi: 10.7555/JBR.27.20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhu Y, Poyurovsky MV, Li Y, Biderman L, Stahl J, et al. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol Cell. 2009;35:316–26. doi: 10.1016/j.molcel.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32:180–89. doi: 10.1016/j.molcel.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miliani de Marval PL, Zhang Y. The RP–Mdm2–p53 pathway and tumorigenesis. Oncotarget. 2011;2:234–38. doi: 10.18632/oncotarget.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fumagalli S, Ivanenkov VV, Teng T, Thomas G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012;26:1028–40. doi: 10.1101/gad.189951.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bursac S, Brdovcak MC, Pfannkuchen M, Orsolic I, Golomb L, et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. PNAS. 2012;109:20467–72. doi: 10.1073/pnas.1218535109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2–p53 checkpoint. Cell Rep. 2013;4:87–98. doi: 10.1016/j.celrep.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li M, Gu W. A critical role for noncoding 5S rRNA in regulating Mdmx stability. Mol Cell. 2011;43:1023–32. doi: 10.1016/j.molcel.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Macias E, Jin A, Deisenroth C, Bhat K, Mao H, et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein–Mdm2 interaction. Cancer Cell. 2010;18:231–43. doi: 10.1016/j.ccr.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xiong X, Zhao Y, He H, Sun Y. Ribosomal protein S27-like and S27 interplay with p53–MDM2 axis as a target, a substrate and a regulator. Oncogene. 2011;30:1798–811. doi: 10.1038/onc.2010.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xiong X, Zhao Y, Tang F, Wei D, Thomas D, et al. Ribosomal protein S27-like is a physiological regulator of p53 that suppresses genomic instability and tumorigenesis. eLife. 2014;3:e02236. doi: 10.7554/eLife.02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lundgren K, Montes de Oca Luna R, McNeill YB, Emerick EP, Spencer B, et al. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997;11:714–25. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- 145.Gu L, Zhu N, Zhang H, Durden DL, Feng Y, Zhou M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell. 2009;15:363–75. doi: 10.1016/j.ccr.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kadakia M, Brown TL, McGorry MM, Berberich SJ. MdmX inhibits Smad transactivation. Oncogene. 2002;21:8776–85. doi: 10.1038/sj.onc.1205993. [DOI] [PubMed] [Google Scholar]
- 147.Wunderlich M, Ghosh M, Weghorst K, Berberich SJ. MdmX represses E2F1 transactivation. Cell Cycle. 2004;3:472–78. [PubMed] [Google Scholar]
- 148.Melo AN, Eischen CM. Protecting the genome from Mdm2 and Mdmx. Genes Cancer. 2012;3:283–90. doi: 10.1177/1947601912454139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carrillo AM, Bouska A, Arrate MP, Eischen CM. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene. 2014;34:846–56. doi: 10.1038/onc.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang JY, Zong CS, Xia W, Wei Y, Ali-Seyed M, et al. MDM2 promotes cell motility and inva-siveness by regulating E-cadherin degradation. Mol Cell Biol. 2006;26:7269–82. doi: 10.1128/MCB.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Onel K, Cordon-Cardo C. MDM2 and prognosis. Mol Cancer Res. 2004;2:1–8. [PubMed] [Google Scholar]
- 152.Markey MP. Regulation of MDM4. Front Biosci. 2011;16:1144–56. doi: 10.2741/3780. [DOI] [PubMed] [Google Scholar]
- 153.Ware PL, Snow AN, Gvalani M, Pettenati MJ, Qasem SA. MDM2 copy numbers in well-differentiated and dedifferentiated liposarcoma: characterizing progression to high-grade tumors. Am J Clin Pathol. 2014;141:334–41. doi: 10.1309/AJCPLYU89XHSNHQO. [DOI] [PubMed] [Google Scholar]
- 154.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBio Portal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hedstrom G, Thunberg U, Amini RM, Zainuddin N, Enblad G, Berglund M. The MDM2 polymorphism SNP309 is associated with clinical characteristics and outcome in diffuse large B-cell lymphoma. Eur J Haematol. 2014;93:500–8. doi: 10.1111/ejh.12388. [DOI] [PubMed] [Google Scholar]
- 157.Jacob AG, O’Brien D, Singh RK, Comiskey DF, Jr, Littleton RM, et al. Stress-induced isoforms of MDM2 and MDM4 correlate with high-grade disease and an altered splicing network in pediatric rhabdomyosarcoma. Neoplasia. 2013;15:1049–63. doi: 10.1593/neo.13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lenos K, Grawenda AM, Lodder K, Kuijjer ML, Teunisse AF, et al. Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res. 2012;72:4074–84. doi: 10.1158/0008-5472.CAN-12-0215. [DOI] [PubMed] [Google Scholar]
- 159.Liu M, Zheng SJ, Chen Y, Li N, Ren PF, et al. Autoantibody response to Murine Double Minute 2 protein in immunodiagnosis of hepatocellular carcinoma. J Immunol Res. 2014;2014:906532. doi: 10.1155/2014/906532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chai Y, Peng B, Dai L, Qian W, Zhang Y, Zhang JY. Autoantibodies response to MDM2 and p53 in the immunodiagnosis of esophageal squamous cell carcinoma. Scand J Immunol. 2014;80:362–68. doi: 10.1111/sji.12202. [DOI] [PubMed] [Google Scholar]
- 161.Liu W, He L, Ramirez J, Ratain MJ. Interactions between MDM2 and TP53 genetic alterations, and their impact on response to MDM2 inhibitors and other chemotherapeutic drugs in cancer cells. Clin Cancer Res. 2009;15:7602–7. doi: 10.1158/1078-0432.CCR-09-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tu HF, Chen HW, Kao SY, Lin SC, Liu CJ, Chang KW. MDM2 SNP 309 and p53 codon 72 polymorphisms are associated with the outcome of oral carcinoma patients receiving postoperative irradiation. Radiother Oncol. 2008;87:243–52. doi: 10.1016/j.radonc.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 163.Shinohara A, Sakano S, Hinoda Y, Nishijima J, Kawai Y, et al. Association of TP53 and MDM2 polymorphisms with survival in bladder cancer patients treated with chemoradiotherapy. Cancer Sci. 2009;100:2376–82. doi: 10.1111/j.1349-7006.2009.01331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ye Y, Li X, Yang J, Miao S, Wang S, et al. MDM2 is a useful prognostic biomarker for resectable gastric cancer. Cancer Sci. 2013;104:590–98. doi: 10.1111/cas.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ohnstad HO, Castro R, Sun J, Heintz KM, Vassilev LT, et al. Correlation of TP53 and MDM2 genotypes with response to therapy in sarcoma. Cancer. 2013;119:1013–22. doi: 10.1002/cncr.27837. [DOI] [PubMed] [Google Scholar]
- 166.Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39:1476–82. doi: 10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harbor Perspect Biol. 2010;2:a001222. doi: 10.1101/cshperspect.a001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–30. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 169.Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. 2013;19:34–41. doi: 10.1158/1078-0432.CCR-12-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vassilev LT. p53 Activation by small molecules: application in oncology. J Med Chem. 2005;48:4491–99. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 171.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–48. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 172.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, et al. Effect of the MDM2 antagonist RG7112 on the p53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–40. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 173.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2– p53 interaction. J Med Chem. 2006;49:3432–35. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 174.Mohammad RM, Wu J, Azmi AS, Aboukameel A, Sosin A, et al. An MDM2 antagonist (MI-319) restores p53 functions and increases the life span of orally treated follicular lymphoma bearing animals. Mol Cancer. 2009;8:115. doi: 10.1186/1476-4598-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zak K, Pecak A, Rys B, Wladyka B, Domling A, et al. Mdm2 and MdmX inhibitors for the treatment of cancer: a patent review (2011–present) Expert Opin Ther Pat. 2013;23:425–48. doi: 10.1517/13543776.2013.765405. [DOI] [PubMed] [Google Scholar]
- 176.Lucas BS, Fisher B, McGee LR, Olson SH, Medina JC, Cheung E. An expeditious synthesis of the MDM2–p53 inhibitor AM-8553. J Am Chem Soc. 2012;134:12855–60. doi: 10.1021/ja305123v. [DOI] [PubMed] [Google Scholar]
- 177.Rew Y, Sun D, Gonzalez-Lopez De Turiso F, Bartberger MD, et al. Structure-based design of novel inhibitors of the MDM2–p53 interaction. J Med Chem. 2012;55:4936–54. doi: 10.1021/jm300354j. [DOI] [PubMed] [Google Scholar]
- 178.Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, et al. The MDM2 inhibitor AMG 232 demonstrates robust anti-tumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther. 2015;14:649–58. doi: 10.1158/1535-7163.MCT-14-0710. [DOI] [PubMed] [Google Scholar]
- 179.Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2–p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–72. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 180.Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2–MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281–319. doi: 10.1007/978-94-017-9211-0_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030–35. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 182.Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res. 2006;66:3169–76. doi: 10.1158/0008-5472.CAN-05-3832. [DOI] [PubMed] [Google Scholar]
- 183.Wade M, Rodewald LW, Espinosa JM, Wahl GM. BH3 activation blocks Hdmx suppression of apoptosis and cooperates with Nutlin to induce cell death. Cell Cycle. 2008;7:1973–82. doi: 10.4161/cc.7.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, et al. Small molecule RITA binds to p53 blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–28. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 185.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, et al. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell. 2010;18:411–22. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc. 2007;129:2456–57. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chee SM, Wongsantichon J, Soo Tng Q, Robinson R, Joseph TL, et al. Structure of a stapled peptide antagonist bound to Nutlin-resistant Mdm2. PLOS ONE. 2014;9:e104914. doi: 10.1371/journal.pone.0104914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wei WJ, Wang YL, Li DS, Wang Y, Wang XF, et al. Association between the rs2910164 polymorphism in pre-miR-146a sequence and thyroid carcinogenesis. PLOS ONE. 2013;8:e56638. doi: 10.1371/journal.pone.0056638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. PNAS. 2013;110:E3445–54. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, et al. Stapled peptides with improved potency and specificity that activate p53. ACS Chem Biol. 2013;8:506–12. doi: 10.1021/cb3005148. [DOI] [PubMed] [Google Scholar]
- 191.Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, et al. Structure of the stapled p53 peptide bound to Mdm2. J Am Chem Soc. 2012;134:103–6. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- 192.Madden MM, Muppidi A, Li Z, Li X, Chen J, Lin Q. Synthesis of cell-permeable stapled peptide dual inhibitors of the p53–Mdm2/Mdmx interactions via photoinduced cycloaddition. Bioorganic Med Chem Lett. 2011;21:1472–75. doi: 10.1016/j.bmcl.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–59. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 194.Roxburgh P, Hock AK, Dickens MP, Mezna M, Fischer PM, Vousden KH. Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis. 2012;33:791–98. doi: 10.1093/carcin/bgs092. [DOI] [PubMed] [Google Scholar]
- 195.Herman AG, Hayano M, Poyurovsky MV, Shimada K, Skouta R, et al. Discovery of Mdm2–MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011;1:312–25. doi: 10.1158/2159-8290.CD-11-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chargari C, Leteur C, Angevin E, Bashir T, Schoentjes B, et al. Preclinical assessment of JNJ-26854165 (Serdemetan), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 2011;312:209–18. doi: 10.1016/j.canlet.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 197.Kojima K, Burks JK, Arts J, Andreeff M. The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther. 2010;9:2545–57. doi: 10.1158/1535-7163.MCT-10-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Smith MA, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of JNJ-26854165 (Serdemetan) by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;59:329–32. doi: 10.1002/pbc.23319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jochemsen AG. Reactivation of p53 as therapeutic intervention for malignant melanoma. Curr Opin Oncol. 2014;26:114–19. doi: 10.1097/CCO.0000000000000033. [DOI] [PubMed] [Google Scholar]
- 200.Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, et al. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53–MDMX/MDM2 antagonist drug discovery. Cell Cycle. 2010;9:1104–11. doi: 10.4161/cc.9.6.10956. [DOI] [PubMed] [Google Scholar]
- 201.Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010;285:10786–96. doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bista M, Smithson D, Pecak A, Salinas G, Pustelny K, et al. On the mechanism of action of SJ-172550 in inhibiting the interaction of MDM4 and p53. PLOS ONE. 2012;7:e37518. doi: 10.1371/journal.pone.0037518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther. 2011;10:69–79. doi: 10.1158/1535-7163.MCT-10-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vaseva AV, Yallowitz AR, Marchenko ND, Xu S, Moll UM. Blockade of Hsp90 by 17AAG antagonizes MDMX and synergizes with Nutlin to induce p53-mediated apoptosis in solid tumors. Cell Death Dis. 2011;2:e156. doi: 10.1038/cddis.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Graves B, Thompson T, Xia M, Janson C, Lukacs C, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. PNAS. 2012;109:11788–93. doi: 10.1073/pnas.1203789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hu B, Gilkes DM, Chen J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007;67:8810–17. doi: 10.1158/0008-5472.CAN-07-1140. [DOI] [PubMed] [Google Scholar]
- 207.Lehman JA, Hauck PM, Gendron JM, Batuello CN, Eitel JA, et al. Serdemetan antagonizes the Mdm2-HIF1α axis leading to decreased levels of glycolytic enzymes. PLOS ONE. 2013;8:e74741. doi: 10.1371/journal.pone.0074741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pazgier M, Liu M, Zou G, Yuan W, Li C, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. PNAS. 2009;106:4665–70. doi: 10.1073/pnas.0900947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–92. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 210.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods. 2014;11:1253–60. doi: 10.1038/nmeth.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]