

After the catastrophic 1964 Alaskan earthquake, marine stickleback colonized newly created ponds on seismically uplifted islands. Bassham and Catchen et al. show that, in replicate populations, as much as a quarter...
Keywords: contemporary evolution, ecological divergence, population genomics, Gasterosteus aculeatus, threespine stickleback
Abstract
Heterogeneous genetic divergence can accumulate across the genome when populations adapt to different habitats while still exchanging alleles. How long does diversification take and how much of the genome is affected? When divergence occurs in parallel from standing genetic variation, how often are the same haplotypes involved? We explore these questions using restriction site-associated DNA sequencing genotyping data and show that broad-scale genomic repatterning, fueled by copious standing variation, can emerge in just dozens of generations in replicate natural populations of threespine stickleback fish (Gasterosteus aculeatus). After the catastrophic 1964 Alaskan earthquake, marine stickleback colonized newly created ponds on seismically uplifted islands. We find that freshwater fish in these young ponds differ from their marine ancestors across the same genomic segments previously shown to have diverged in much older lake populations. Outside of these core divergent regions the genome shows no population structure across the ocean–freshwater divide, consistent with strong local selection acting in alternative environments on stickleback populations still connected by significant gene flow. Reinforcing this inference, a majority of divergent haplotypes that are at high frequency in ponds are detectable in the sea, even across great geographic distances. Building upon previous population genomics work in this model species, our data suggest that a long history of divergent selection and gene flow among stickleback populations in oceanic and freshwater habitats has maintained polymorphisms of alternatively adapted DNA sequences that facilitate parallel evolution.
WHEN populations of a species adapt to different environments, regions of the genome may chart divergent evolutionary courses, leading to heterogeneous patterns of genomic differentiation (Turner et al. 2005; Nosil et al. 2009; Ellegren et al. 2012; Reid et al. 2016; Wolf and Ellegren 2017). These genomic patterns can occur neutrally via genetic drift in isolated populations, but genetic isolation is not a prerequisite for such divergence (Charlesworth et al. 1997; Hey 2010; Feder et al. 2012; Akerman and Burger 2014; Cruickshank and Hahn 2014). Local adaptation combined with gene flow among the populations (Yeaman and Otto 2011; Yeaman and Whitlock 2011; Berg and Coop 2015; Lee and Coop 2017) can lead to differentiation at selected loci and homogenization of neutral regions of the genome (Feder and Nosil 2010; Nadeau et al. 2013; Poelstra et al. 2014).
Cases of rapid adaptation (Hendry and Kinnison 1999; Hendry et al. 2000; Pritchard et al. 2010; Burke et al. 2014; Kopp and Matuszewski 2014)—so-called contemporary evolution (Thompson 1998; Hendry and Kinnison 2001; Bell et al. 2004; Lotterhos and Schaal 2014; Lucek et al. 2014)—often invoke the use of standing genetic variation because the extended time needed to accumulate new beneficial mutations is presumed to limit the rate of adaptation (Kimura and Ota 1971; Orr 2005; Pritchard and Di Rienzo 2010; Radwan and Babik 2012; Savolainen et al. 2013). Although standing genetic variation permits accelerated change (Levin 1995; Barrett and Schluter 2008; Messer and Petrov 2013), the patterns of heterogeneous genomic differentiation that materialize during contemporary evolution still remain largely unexplored in natural systems (Hahn 2008; Schluter and Conte 2009; Cruickshank and Hahn 2014). It is still not known what proportion of the genome is affected during contemporary evolution by direct selection on standing adaptive variants (Berg and Coop 2015), and how much of the genome is influenced by linked selection such as genetic hitchhiking and genetic draft (Nordborg et al. 1996; Slatkin and Wiehe 1998; Charlesworth 2012; Cutter and Payseur 2013; Flaxman et al. 2013; Burri 2017). When a species exists across diverse habitats with distinct selective pressures, the extent to which independent populations adapting to similar habitats initially make use of the same haplotypes, and at what frequencies these adaptive haplotypes are distributed and maintained species-wide, is unclear.
Across the northern hemisphere, marine threespine stickleback (Gasterosteus aculeatus) have invaded and adapted to countless freshwater habitats over multiple timescales, making them a fruitful model for vertebrate evolution (Bell and Foster 1994; Cresko et al. 2007) and for population genomics of rapid adaptation (Lescak et al. 2015). Marine and freshwater stickleback differ considerably in many morphological (Walker and Bell 2000; Albert et al. 2008; Miller et al. 2014; Milligan-Myhre et al. 2016), as well as physiological and behavioral [(Kusakabe et al. 2017); reviewed in Kitano et al. (2012)], traits, and remarkably parallel phenotypic and genomic evolution has followed independent invasions by marine fish into freshwater habitats, including those on landscapes deglaciated only thousands of years ago (Bell 1984; Thompson et al. 1997; Cresko et al. 2004; Shapiro et al. 2004; Colosimo et al. 2005). At least some of this phenomenon might be due to the reuse of standing genetic variation (Hohenlohe et al. 2010; Feulner et al. 2013; Terekhanova et al. 2014; Marques et al. 2016), enabling the rapid and parallel adaptive evolution documented in this species (Schluter et al. 2004; Boughman et al. 2005; DeFaveri et al. 2011; Jones et al. 2012; Kaeuffer et al. 2012; Hirase et al. 2014). Schluter and Conte (2009) framed one possible scenario to explain these dynamics: a freshwater-adapted stickleback genotype, scattered into the marine population by gene flow and recombination, might be brought together and reintegrated during subsequent colonizations of freshwater habitats.
Here, we explore the nature of this “freshwater genome scattering and reassembly” during the earliest stages of freshwater adaptation by marine stickleback, using a remarkable naturally replicated experiment. Convincing genetic and geologic evidence indicates that stickleback colonized and adapted to new freshwater habitats within 50 years on marine islands in the Gulf of Alaska and Prince William Sound (Lescak et al. 2015). Submarine terraces that encircled the islands were suddenly thrust above sea level in 1964 by the Great Alaska Earthquake (Plafker 1969; Plafker and Rubin 1978; Gelmond et al. 2009), and the resulting changes in sediment deposition and erosion over the following years created new freshwater ponds (Gelmond et al. 2009; Lescak et al. 2015). Threespine stickleback fish that now inhabit many of these ponds have become as morphologically distinct in degree and form from their immediate marine ancestors as have much older freshwater populations in mainland Alaska and throughout the northern hemisphere (Bell and Foster 1994; Walker and Bell 2000; Kimmel et al. 2012). Previously, using 1000 randomly chosen genetic markers in these new populations, we showed that marine fish likely adapted to fresh water several independent times on different islands and even among ponds on the same island (Lescak et al. 2015).
The present study wields a data set of markers that is 30 times denser, in over 1200 fish from independently derived stickleback populations on three seismically uplifted marine islands (Danger, Middleton, and Montague), as well as from mainland Alaska and Oregon. We use this data set to dissect genome-wide patterns of haplotype frequency divergence across populations that range in age from just decades to thousands of years old. By exploring these replicate natural experiments in adaptation to alternative environments, we find that a much larger proportion of the genome is affected—likely through combined effects of direct and linked selection—than had been previously reported in stickleback. Loci with alternative haplotypes that dominate in either marine or freshwater populations are clustered in the genome, particularly across broad regions of divergence where marine fish are relatively depleted in haplotype diversity. We show how marine stickleback, even at great geographic distances, act as carriers of a common set of freshwater genotypes, facilitating repeated selection and reassembly of the same scattered haplotypes in independently colonized lakes and ponds.
Materials and Methods
Biological collections
Freshwater ponds and marine sites for stickleback collection (Supplemental Material, Figure S1) were chosen on the basis of maps and aerial imagery created before and after 1964. As described in Lescak et al. (2015), uplift island stickleback fish were trapped during the summers of 2005 (Montague Island), 2010 (Danger and Middleton Islands), and 2011 (Montague and Middleton Islands), and genomic DNA was isolated from fin tissue. All research was approved by the Institutional Animal Care and Use Committees (IACUCs) of the University of Alaska Anchorage and the University of Oregon. Fish were collected under Alaska Department of Fish and Game permits SF-2005-020, SF-2010-029, and SF-2011-153, as well as Oregon Department of Fish and Wildlife scientific taking permits OR2007_3495 and 13920.
Restriction site-associated DNA sequencing library preparation and sequence analysis
We used restriction site-associated DNA sequencing (RAD-seq) data sets, subsamples of which were previously used for population structure analyses in Lescak et al. (2015) and in Catchen et al. (2013a). As described in Lescak et al. (2015), RAD-seq libraries for 1057 fish were created using restriction endonuclease SbfI, and sequenced to 101 nucleotides (including a 6-nt inline barcode) on an Illumina HiSequation 2000 platform (Table S6). We used RAD-seq data from an additional 98 fish from Cushman Slough, Oregon, produced by similar methods, as previously described Catchen et al. (2013a). Individual fish were represented by an average of 1,265,744 ± 13,864 SE sequences each, of which 1,225,729 ± 13,744 SE sequences (97%) were aligned to the reference genome, and these produced an average of 43,174 ± 165.7 SE loci with an average depth of coverage of 27× using the software Stacks (Catchen et al. 2011, 2013b; Hohenlohe et al. 2012), version 1.46 (Table S7). Raw sequence data were demultiplexed according to barcode and filtered for quality using the process_radtags module (-c, -q, and -r parameters) in Stacks. Cleaned reads were aligned to the stickleback reference genome (version BROADS1, Ensembl release 86) using the mem module from the BWA aligner (Li 2013) with default settings. Stacks were assembled, single-nucleotide polymorphisms (SNPs) were called, and a population-level catalog was constructed by executing the pstacks (-m 3), cstacks (–aligned), and sstacks (–aligned) modules. Population-level corrections were made to the data by running rxstacks (–model_type bounded,–bound_high 0.1,–prune_haplo, and–conf_lim 0.25 parameters), and then rebuilding and matching to the catalog with cstacks (–aligned) and sstacks (–aligned). Final filtering was done with the populations module, as well as the calculation of population genetic statistics and associated kernel smoothing.
Our full data set comprises 48,307 RAD loci encompassing a total of 266,902 SNPs. We applied two general filtering steps for most analyses: one when considering populations individually and another when considering STRUCTURE-defined population clusters. In the former, for a locus to be included in downstream analyses, we required it to be present in ≥ 20 populations (-p 20 parameter) and in 75% of individuals of each population (-r 0.75), and we applied a minor allele frequency filter of 1% (–maf 0.01). This resulted in 29,911 RAD loci and 74,556 SNPs. In the latter analysis, a locus had to be present in nine populations and in 75% of individuals, with a minor allele frequency of 1%. This resulted in 30,285 RAD loci and 75,476 SNPs. The increase in total SNP number from that reported previously in Lescak et al. (2015), for the subset of sequencing data that overlap those reported here, is due primarily to the use of an improved alignment method (BWA vs. GSnap) and to the application of the rxstacks pipeline module, which makes individual model call corrections based on the alleles present in the population.
To compare the Middleton Island samples against those found previously in the Cook Inlet Basin, we downloaded sequences used in Hohenlohe et al. (2010) from the National Center for Biotechnology Information Sequence Read Archive using accession SRA010788.9. In addition, we added 54 more recently sequenced individuals from Rabbit Slough and 24 from High Ridge Lake, both also located in the Cook Inlet Basin. As these data stem from sequencing runs using different versions of Illumina technology, we retained 94 individuals that had read lengths of at least 49 bp and a minimum average depth of coverage of 10× (Table S8). These data were aligned to the stickleback reference genome and processed with Stacks, in the same manner as the Middleton Island data. We grouped the individuals into freshwater and marine clusters, and required loci to be present in both clusters and at a frequency of 75% among individuals in either separate cluster. Because of the smaller number of samples in this analysis, we applied a minor allele frequency filter of 6%. Table S8 summarizes the differences among the data sets and lists which were used in each analysis.
Statistical approach
We confirmed the population structure groupings defined in Lescak et al. (2015) using the revised data set of the current work. Filtered SNP data from all RAD loci across all uplift island populations were output into a file formatted for STRUCTURE (Pritchard et al. 2000). We randomly chose three subsets of 1000 SNPs each to complete the analysis, because of computational limitations. Because these three subsets yielded comparable results, data from only one subset are presented in Figure S1. STRUCTURE analyses were performed separately for each island. In total, 10,000 burn-in steps and 10,000 replicates were used, with 10 runs for each potential K (number of genotypic groups). The optimal K for each analysis was chosen using the ΔK method (Evanno et al. 2005).
The populations that were shown to be united into genetic groupings by STRUCTURE analysis here (Figure S1) and in Lescak et al. (2015) were treated together here as groups rather than as single populations. Also, the three geographically proximate Cook Inlet Basin freshwater populations sampled in Hohenlohe et al. (2010) are here treated as a group to increase sampling robustness for a regional comparison. Latitude and longitude, respectively, for these three populations are 61.6 and −149.75 (Bear Paw Lake), 61.72 and −150.12 (Boot Lake), and 61.93 and −150.97 (Mud Lake). See Figure S1 for information on island sampling locations and population groupings. In only one analysis, the FSTʹ comparison of sympatric ecotypes in Figure S11 were samples split by phenotype. In all other analyses, all samples from a given population or population group were considered together, regardless of phenotype.
Population genetic fixation and differentiation statistics were calculated using the populations program in Stacks (Catchen et al. 2011, 2013b; Hohenlohe et al. 2012), in which several common parametric and Analysis of MOlecular VAriance (AMOVA)-based statistical estimators have been implemented. For this paper we used FST and FSTʹ to estimate divergence due to changes in allele frequencies, and ΦST to integrate additional signal from sequence distances among haplotypes (Weir and Cockerham 1984; Holsinger and Weir 2009; Meirmans and Hedrick 2011).
FST is calculated as the comparison of pairwise nucleotide diversity within and among pairs of populations for each locus using SNPs at each locus. In contrast, calculations of haplotype diversity, FSTʹ and ΦST, are based on the haplotypic state of each ∼100-bp RAD locus in each individual instead of individual SNPs. The different observed combinations of fixed and variable nucleotides that comprise each locus are considered a set of haplotypes, and each individual is assigned a diploid haplotype using Stacks. While SNP-based statistical analyses are confined to biallelic nucleotide positions (for most populations of multicellular organisms), the haplotype-based statistics can have multiple haplotype alleles per locus. In addition, the distance measured between haplotypes provides information on the genealogical history of those alleles.
FSTʹ is a standardized version of FST that scales among-population divergence based upon within-population diversity by considering the maximum value of FST that can be observed. It is well documented that high levels of within-population polymorphism can sometimes cause loci to have a maximum value of FST far greater than 1.0 (Meirmans 2006). AMOVA estimators of FSTʹ and ΦST are implemented for population pairs, and they are calculated as ratios of among-population variance to total variance. For both FSTʹ and ΦST, haplotypes are compared at each locus in a two-dimensional, square matrix, with 2n rows and columns representing diploid haplotypes of n individuals; the matrix is further subdivided to group the haplotypes according to population. The cell values are 0 or 1 for FSTʹ (haplotypes either match or are different), or positive integers for ΦST (the sequence edit-distance between haplotypes). Sum-of-squares calculations are used to estimate within- and among-population variation. The value of FSTʹ is additionally scaled by the maximum divergence possible among populations for observed haplotypes at each locus [for full details see Bird et al. (2011)].
Genome-wide circle plots (also known as Large Hadron Collider Plots) were generated by a Python program, lhc_plot.py, which is distributed with the Stacks package. Population genetic statistics were kernel smoothed along the genome using default smoothing parameters in Stacks software.
Calculating linkage disequilibrium
All SNP outputs from Stacks were imported into Beagle (version 3.3.2) to phase SNPs across each chromosome of each population. We then transferred the resulting Beagle output into the phasedstacks program of Stacks to calculate Dʹ, the measure of linkage disequilibrium (LD) (Nei 1987; Weir 1996), and values of Dʹ were plotted as heat maps in Gnuplot 4.6 patchlevel 4 (http://gnuplot.info).
Calculating gene and haplotype diversity
We calculated two separate diversity measures for each RAD locus in each population. A RAD locus contains one or more haplotypes, which are the physically phased combination of SNPs present at that locus across the population. The first measure, gene diversity (Nei 1987; Weir 1996), can be thought of as the probability that two randomly chosen haplotypes at that locus are different. Given the number of samples n, and the ith haplotype p, it is calculated as:
The second measure, haplotype diversity (Nei 1987; Weir 1996), is similar to gene diversity, but it is scaled by the substitution distance, d, between the two randomly chosen haplotypes:
Haplotype diversity measures were kernel smoothed along the genome using default smoothing parameters in Stacks software. For haplotype diversity difference, the continuous smoothed values for the marine group were subtracted from those for the freshwater group, and these raw values were plotted on a nonlinear color scale.
Segmenting the genome via the hidden Markov Model
We used the general hidden Markov Model Library (GHMM) (Schliep et al. 2005), version 0.9-rc3), to segment the genome using haplotype-based FSTʹ. To make the FSTʹ measures discrete, we binned them into four categories; in each population we hierarchically clustered the FSTʹ measures in R (R Core Team 2013). Using the Python bindings for GHMM, we designed a two-state HMM representing diverged and not-diverged. The emission parameters for each of the two states were defined using the FSTʹ categories in regions of marked divergence between freshwater and oceanic populations on LG IV (diverged state) and in regions of nearly zero divergence on LG XV (not diverged state), respectively. For each pairwise comparison (e.g., MiFW vs. OC, MoSW vs. OC, and Cook Inlet vs. OC, where OC comprises the three island marine populations), the HMM was trained by executing the Baum–Welch algorithm of the GHMM library on the genomic FSTʹ values. After training, each LG was segmented into the diverged and not-diverged states by executing the Viterbi algorithm in the GHMM library. These processes were implemented in a custom Python program segment.py. Finally, we compared the set of states for each of the data sets (MiFW, MoSW, and CI vs. OC) to find the diverged regions in common using our shared_segments.py program. Plots of the HMM data were generated using Gnuplot 4.6 patchlevel 4.
Defining cases from haplotype data
Classification of cases was done using custom Python programs. For each locus in an analysis, we summed the haplotypes in order from the haplotype with the largest frequency in a population to the smallest. The subset of ordered haplotypes that occupied at least 60% of the haplotypes at that locus in that population were considered majority haplotypes. Loci were classified as case 1 when one or more majority haplotype/s occurred in a marine population, but the corresponding freshwater population had a single, alternative majority haplotype (i.e., one that is not a majority haplotype in the marine population). Loci were classified as case 2 when the marine population had one or more majority haplotype/s at a locus and the freshwater population had one alternative majority haplotype, and one or more haplotype/s also in the majority in the marine population. Loci were classified as case 3 when the marine population had one or more majority haplotype/s at a locus, but the corresponding freshwater population had two or more alternative majority haplotypes and no majority haplotypes in common with the marine population. Loci were classified as case 4 when the freshwater population had only a subset of majority haplotypes in the marine population. Circular genomic plots of cases were created with the lhcp_plot.py script while the linear plots of cases were created with a custom Python script.
Data availability
Sequence data are available in the NCBI Sequence Read Archive under accession SRP148115. Supplemental material available at Figshare: https://doi.org/10.25386/genetics.6272432. RAD consensus sequences for each locus with its aligned genomic position are available in FASTA format for populations (File_S01) and for population groups (File_S06). SNP and haplotype calls are available in VCF format for populations (SNP: File_S02; haplotype: File_S03) and for population groups (SNP: File_S07; haplotype: File_S08). Values of divergence (FST or FSTʹ and ΦST) at each locus are available for populations (SNP: File_S05; haplotype: File_S04), and for population clusters (SNP: File_S10; haplotype: File_S09). Figure S1 contains genetic groupings and a reference map of studied populations. Figure S2 depicts continuous genetic divergence across the genome for marine vs. each freshwater genetic group. Figure S3 shows the full HMM analysis across all LGs. Figure S4 contains gene and haplotype diversity measures. Figure S5 shows Cases along the genome in independent populations. Figure S6 depicts alternative freshwater haplotypes across LGI. Figure S7 contains per-chromosome allele frequency spectra. Figure S8 contains boxplots of alternative freshwater allele frequencies. Figure S9 contains per-marine fish counts of alternative freshwater alleles. Figure S10 is a comparison of genomic divergence patterns between this and an independent published analysis. Figure S11 shows LD patterns in freshwater fish. Figure S12 shows an aerial photograph of collection sites on Montague Island. Table S1 lists genome-wide genetic divergence values for freshwater vs. marine comparisons. Table S2 reports fractions of the diverged genome that overlap among populations. Table S3 quantifies categories of divergent loci (Cases) in different populations. Table S4 contains the mean, median, and SD of the frequencies of alternative freshwater alleles in marine populations. Table S5 contains the fraction of alternative freshwater alleles found on each chromosome for each marine fish. Tables S6–S8 report sequencing statistics. Table S9 contains a key for which data set was used in each figure/table.
Results
Parallel patterns of genomic divergence involving one-quarter of the genome assemble in decades on a background of no population structure
Despite their young age and their isolation by ocean expanses, we find that stickleback populations newly adapted to fresh water on seismically uplifted Middleton and Montague Islands (south of mainland Alaska) reiterate genomic patterns first described in fish from much older, postglacial lakes on the Alaskan mainland (Hohenlohe et al. 2010) (Figure 1). For each locus we calculated population genetic differentiation statistics across the genome (SNP-based FST and its normalized haplotype-based version, FSTʹ, both of which use allele identity, and haplotype-based ΦST which incorporates sequence distance; Table S1) using RAD-seq data in stickleback sampled from multiple populations. We then performed genome scans using continuous divergence values (Figure 1 and Figure 2).
Figure 1.

Nascent and old freshwater stickleback populations share parallel patterns of genomic divergence from marine fish. Smoothed, SNP-based FST is plotted along the genome in comparisons of mainland coastal marine vs. mainland postglacial Cook Inlet freshwater fish, and for island marine fish vs. freshwater populations that formed after the 1964 earthquake on geographically separated Middleton Island and Montague Island. Of the island populations studied, these freshwater groups [defined by population genetic structure analysis; Figure S1 and Lescak et al. (2015)] showed a pattern of regionalized high genomic divergence on a background of nearly zero divergence from marine fish. Is., island; MiFW, Middleton Island freshwater; MoSW, Montague Island freshwater.
Figure 2.

Patterns of genomic divergence are similar among young, independently colonized freshwater locales. Smoothed, haplotype-based FSTʹ is plotted along the genome for island freshwater population groups, including those least diverged from marine fish. Concentric rings are numbered from the center out, and labeled with 5-Mb intervals. Marine populations [Middleton Island (MiOC1) vs. Danger Island (DaOC)] from different islands ∼100 km apart show little differentiation (ring 1). While Middleton Island (MiFW, ring 2) and southwest Montague Island (MoSW, ring 3) all show parallel patterns of marine–freshwater divergence, the possibly younger populations on Montague (MoSE, ring 4) and Danger Island (DaFW, ring 5) differ strongly from marine on only a subset of the typically divergent linkage groups. In rings 2–5, marine comprises DaOC, MiOC1, and MiOC2.
A history of multiple founding events from the sea is reflected in the clear population structure that we documented previously (Lescak et al. 2015) among groups of freshwater ponds on three seismically uplifted islands (Figure S1). Because freshwater-adapted stickleback perish quickly in sea water [e.g., Kusakabe et al. (2017)] and because, despite intensive sampling by many research groups, morphological freshwater stickleback are not found in the sea, it is widely accepted that freshwater habitats isolated from each other by the ocean are colonized by marine stickleback rather than by direct migration of freshwater-adapted fish (Schluter and Conte 2009). We here consider a conservative minimum of three separate freshwater founding events, one on each island. However, within Middleton and Montague Islands, genetically distinct groups of freshwater populations could indicate there have been more than one independent founding event even within an island, such as the populations we sampled on the southeast and southwest sides of Montague Island (Lescak et al. 2015) (Figure S1).
We found that genomic patterns of divergence from marine genomes on Montague and Middleton Islands, despite their independent colonization, are broadly congruent, and resemble patterns typical of much older freshwater populations (Figure 1 and Figure 2). Importantly, as predicted if the increased divergence is due to parallel evolution after independent invasion, freshwater groups on different islands show little divergence from each other across the selected regions (Figure S2). A striking pattern is that, outside of foci of high divergence, baseline genetic divergence approaches zero between marine fish and freshwater fish in the new ponds. By contrast, the single freshwater population on Danger Island (DaFW) and a genetically distinct population (MoSE) on the east coast of Montague Island, exhibit only a subset of the FSTʹ signatures of selection common in freshwater-adapted fish (Figure 2 and Figure S1).
To quantify what proportion of the genome has diverged in allele frequencies over such a short time frame, and to what degree these regions of divergence are shared across independently evolved populations (Figure 1 and Figure 2), we needed to bound the divergent genomic regions that our FST-based selection scans identified. To infer from this continuous population genomic data which genomic regions fall into one of two categories – those regions affected only by neutral processes or those regions affected also by selection – we employed a Hidden Markov Model (HMM) in a similar vein to that previously applied in stickleback population genomics (Jones et al. 2012). We compared an older, mainland population to independently founded island populations that have a visually clear pattern of regionalized high genomic divergence on a background of nearly zero divergence from marine fish. The emission and transition parameters of the HMM for each population group were trained on patterns of fresh water vs. marine FSTʹ values for RAD loci across the genome. The trained model delineated two states: diverged or not diverged (Figure 3 and Figure S3). We found that a significant proportion of the genome falls within well-defined regions that diverge between marine and freshwater populations in all comparisons: 29% for Middleton (MiFW), 24% for southwest Montague (MoSW), and 18% for the mainland group (Table 1 and Table S2). The genomic blocks are largely nested when compared among all three freshwater groups; Figure 1 and Figure 2). The divergent regions of MiFW and MoSW overlap by > 70%, and these island populations overlap by between 51 and 59% with the much older mainland populations (Table 1 and Table S2). Clearly, the majority of the patterns of genomic divergence are shared across populations, young and old, and this shared, persistent divergence amounts to nearly 15% of the genome.
Figure 3.

Genomic blocks of freshwater–marine divergence largely overlap among freshwater populations. (A) A HMM was trained on FSTʹ patterns to discriminate between diverged and not-diverged regions. Shown here, for example, is the outcome of the model on three LGs that have large regions of divergence and one LG that has little divergence. From this, the proportion of the genome diverged in each freshwater group (dashed lines) and the divergence overlap (bold green line) among these populations were calculated. (B) The total proportion of the genome diverged from marine in the compared freshwater populations ranges nearly to 30%, the majority of which (totaling ≈15% of the genome) overlaps across freshwater populations. FW, freshwater; HMM, hidden Markov Model; MI, Middleton Island; MO, Montague Island; OC, marine; SW, southwestern.
Table 1. Regions of the stickleback genome that diverge because of adaptation highly overlap between freshwater populations, both young and old.
| MiFW (%) | MoSW (%) | Cook Inlet (%) | |
|---|---|---|---|
| MiFW | 29.06 | 22.01 | 16.27 |
| MoSW | 70.15 | 24.33 | 15.95 |
| Cook Inlet | 51.79 | 59.02 | 18.64 |
The total amount of the genome allocated to the freshwater vs. marine “diverged” state by a hidden Markov Model is listed, along the diagonal in bold, for each of the three island population groupings that showed a pattern of regionalized high genomic divergence on a background of nearly zero divergence from marine fish. Above the diagonal are the proportions of the genome that populations have in common. Below the diagonal is the degree of overlap of the divergent stretches of the genome for each population comparison. Despite the much older time since founding, the Cook Inlet freshwater group still shares > 50% of divergent regions with the much younger uplift populations. MiFW, Middleton Island freshwater; MoSW, Montague Island southwest.
Marine and freshwater populations differ in patterns of haplotype diversity and composition
FST statistics quantify relative divergence affected by differences in genetic frequencies among, as well as diversities within, populations (Charlesworth 1998; Holsinger and Weir 2009; Jakobsson et al. 2013; Lotterhos and Schaal 2014). RAD-seq also generates genome-wide DNA sequence data, allowing us to examine both the relative and absolute haplotype diversities within differentiated genomic regions in each population, as well as document the distribution and abundance of particular haplotypes across populations. We found that divergent genome segments correspond to regions where marine and freshwater populations differ in haplotype diversity. Marine genomes have lower absolute haplotype diversity across these regions, relative to nondivergent parts of the genome (Figure 4 and Figure S4). To distinguish whether parallel patterns of divergence stem wholly from this reduction in diversity in the marine populations, or also from parallel selection of alleles in fresh water, we explored patterns of haplotype sharing across freshwater populations.
Figure 4.

Divergent genomic regions differ also in haplotype diversity (Div) between freshwater and marine fish. (A) Concentric rings are numbered from the center out. Blocks of elevated FSTʹ in a comparison of Middleton freshwater (MiFW) vs. the combined island marine populations (ring 1) highly correspond with regions of relative increase in haplotype diversity in fresh water (ring 2). On ring 2, blue represents higher diversity in MiFW than in marine, and red is higher diversity in marine than MiFW. Smoothed haplotype diversity is plotted separately for freshwater (ring 3) and marine (ring 4) populations. While a fraction of haplotype diversity outliers exceed the value range shown, > 99.9% of the smoothed values were ≤ 1.0. See Materials and Methods for how diversities and difference (Diff) are calculated. (B–D) The freshwater and marine populations are compared across LG IV, an LG to which many morphological traits map that differ between marine and freshwater ecotypes (Albert et al. 2008): (B) divergence statistics, (C) gene diversity, (D) haplotype diversity, and (E) gene and haplotype diversity difference. Where values in (E) are greater than zero, fresh water has greater diversity than marine.
At individual RAD loci with high estimates of FSTʹ (≥ 0.2), both the marine and freshwater populations each often have a single but different majority haplotype (Table 2 and Table S3). We define majority haplotypes as the highest frequency haplotypes that—singly or in combination—account for ≥ 60% of genotypes at a given locus (Figure 5). Independent freshwater populations most often have the same majority haplotype across all shared loci at which there is a single, alternative majority freshwater haplotype. These we call “case 1 loci” (see Figure 5 for definitions of four qualitatively different cases). For instance, nearly 100% (427 of 428) of the case 1 loci shared by both MiFW and MoSW have the same majority haplotype. In populations from these two islands, loci with majority haplotypes alternative to those in the combined island marine populations (MiOC1, MiOC2, and DaOC) are clustered in the divergent regions delineated by the FSTʹ-based HMM (Figure 5). Outside of these divergent regions, loci with freshwater vs. marine alternative haplotypes occur, but the density of them differs by population group, with MoSW having the fewest, MiFW having more, and MoSE having the most (Figure 5 and Figure S5).
Table 2. Freshwater vs. marine divergent loci categorized by haplotype characteristics.
| Population | Total loci | Case 1 | %Rare | %Strict alt | Case 2 | %Rare | Case 3 | %Rare | Case 4 |
|---|---|---|---|---|---|---|---|---|---|
| MiFW | 3021 | 1294 | 84 | 75.73 | 1230 | 91.14 | 286 | 98.25 | 192 |
| MoSW | 2114 | 494 | 75.51 | 70.24 | 1408 | 90.63 | 87 | 93.1 | 108 |
| MoSE | 4370 | 1121 | 38.72 | 29.17 | 1703 | 73.69 | 261 | 89.27 | 1245 |
| DaFW | 3800 | 1063 | 29.44 | 19.47 | 1249 | 67.17 | 188 | 85.11 | 1265 |
Loci with FSTʹ ≥ 0.2 are tallied for each population group. Of these, the number that fall into each case (as defined in Figure 5) is given. Also listed, where applicable, is the proportion (% Rare) of these loci carrying a freshwater majority haplotype that occurs only rarely (i.e., < 10% allele frequency) in the sea. For case 1, the proportion (% Strict alt) of loci that satisfy these criteria is listed: they carry a single majority haplotype in fresh water that is rare in the sea, and they carry a different single majority haplotype in the sea. The island populations with a pattern of divergence that closely parallels older mainland populations have the largest proportion these of loci with radically alternative freshwater vs. marine haplotype frequencies. DaFW, Danger Island freshwater; MiFW, Middleton Island freshwater; MoSE, Montague Island southeast; MoSW, Montague Island southwest.
Figure 5.

Regions of parallel divergence contain loci with alternative freshwater vs. marine haplotypes. (A) To characterize the nature of loci with high FSTʹ in marine–freshwater comparisons, we first rank haplotypes at these loci from highest to lowest frequency per locus per population, define “majority” haplotypes as the highest frequency haplotypes that singly or in combination account for ≥ 60% of the population haplotypes, and then assign each locus to one of four categories: at case 1 loci, marine and freshwater populations have singular but alternative majority haplotypes; at case 2 loci, the freshwater population has one or more haplotypes that is/are majority only in fresh water, and these loci also have at least one majority haplotype that is also a majority haplotype in the sea; at case 3 loci, the freshwater population has more than one majority haplotype, none of which are majority in the sea; and at case 4 loci, the freshwater population has majority haplotypes that are a subset of the majority haplotypes in the sea. (B) Divergent loci are assigned to each of the four cases and plotted across the genome for two freshwater groups from Middleton Island (MiFW) and one from Montague Island (MoSW). Only loci with FSTʹ ≥ 0.2 are plotted, colored by case. While these cases are not strictly discreet, they broadly capture qualitatively different patterns of population genomic processes. Sampling effects could move some loci with borderline allele frequencies (i.e., those with frequencies near our parameter boundaries) between cases 1–3, but are less likely to move such loci to case 4 (for example, see Table S3).
Another important qualitative difference between southwest Montague and Middleton can be seen in the loci falling within the divergent regions. While MoSW does have alternative haplotypes, it retains more of the common marine haplotypes at these divergent loci, reflected in the observed lower FSTʹ of this group vs. marine (Figure 2 and Figure S3). We define these as case 2 loci (plotted in blue in Figure 5). Taken together, these distinctions are expected if MoSW has greater introgression between freshwater and marine fish, as was inferred by Lescak et al. (2015), who described morphological intermediates in this population. In other words, such loci could indicate a transitory state, one in which freshwater adaptive allele frequencies are intermediate because of recent gene flow from marine fish (and see Figure S1A). MoSE (which lacks most of the peaks of high FSTʹ relative to marine) has higher diffuse FSTʹ than do the other pond populations across genomic regions that do not usually diverge in fresh water (Figure 2). This pattern, perhaps due to small founding population size, is a consequence of the low overall haplotype diversity in MoSE (Table 3 and Figure S4). An interesting correlate of this pattern in MoSE is that FSTʹ relative to marine drops across regions that are highly divergent in other freshwater–marine comparisons, because these genomic segments have low diversity in both MoSE and marine. The scattered loci of higher FSTʹ found in MoSE predominantly result from demographic subsampling of majority marine haplotypes (such as from 7 to 21 Mb on LG VII; Figure 2, Figure 6, and Figure S4). Such loci that are haplotype-depleted, likely via neutral processes, form the rationale for defining the case 4 category of loci.
Table 3. Genome-wide averages of gene and haplotype diversity in the uplift island populations.
| Population | Gene diversity | Haplotype diversity |
|---|---|---|
| MiFW | 0.3106 | 0.4408 |
| MoSW | 0.3071 | 0.4367 |
| MoSE | 0.2136 | 0.2971 |
| DaFW | 0.2036 | 0.2844 |
Diversity is lower in the potentially younger pond populations on Danger Island and in southeast Montague Island. DaFW, Danger Island freshwater; MiFW, Middleton Island freshwater; MoSE, Montague Island southeast; MoSW, Montague Island southwest.
Figure 6.

Changes in haplotype frequency reveal ongoing processes in the youngest freshwater populations. The set of haplotypes at each divergent locus is represented as a stacked bar chart of haplotype frequencies. Only loci with FSTʹ ≥ 0.2 are plotted. Red haplotypes correspond to marine majority haplotypes and blue to fresh water majority haplotypes. (A) In the Danger Island freshwater pond (DaFW), an LG I region of elevated FSTʹ [relative to OC, where OC comprises three island marine populations, DaOC (Danger Island), MiOC1 (Middleton Island 1), and MiOC2 (Middleton Island 2)] encompassing a known inversion polymorphism (dashed box) shows a strong pattern of freshwater–marine alternative haplotypes (case 1 and case 3 loci) flanked by a region (solid box) with increased density of loci that are depleted in haplotype diversity relative to OC (i.e., loci classed as case 4). (B) In the southeast Montague ponds (MoSE), LG VII lacks the high FSTʹ typically seen across the middle of the chromosome in other freshwater vs. marine comparisons, and instead there is elevated FSTʹ at the chromosome ends. This inverse pattern is generated by the sparsity of loci with freshwater–marine alternative haplotypes (case 1 and case 3 loci) and an increase in the number of loci at which marine fish are more haplotype-rich (case 4). A clear red-to-blue shift of haplotypes is apparent between marine and Middle Island freshwater (MiFW) loci on LGVII. For comparison, the FSTʹ heat-map plots to the right are as in Figure 2. For additional help interpreting these stacked bar charts, see Figure S6. MoSW, Montague Island southwest.
Danger Island presents another qualitatively distinct scenario from the other freshwater populations in this study; only a fraction of the parallel pattern of freshwater–marine divergence—a single FSTʹ peak on LG I—is prominent in this island’s single freshwater pond (DaFW). This segment encompasses a known inversion whose orientation differs consistently between freshwater and marine genomes (Jones et al. 2012). Relative to the combined island marine populations, DaFW also has diffusely elevated FSTʹ due to genome-wide reduction in haplotype diversity (Figure 2, Figure S4, and Table 3), but FSTʹ crescendos at the LG I inversion. Unlike in the other freshwater groups in which this FSTʹ peak relative to marine typically has sharp bounds around the inversion borders, in DaFW, the elevated FSTʹ extends to the inversion’s flanking regions (Figure 2 and Figure 6). We find that two types of loci primarily occupy the inversion and closely linked regions. In the inversion itself, there is a concentration of loci that have increased in alternative freshwater haplotypes, and flanking this are loci depleted for common marine haplotypes (Figure 6 and Figure S6). Together, these could signal a recent or ongoing selective sweep in DaFW.
Freshwater majority haplotypes are readily detectable at thousands of loci in the marine population
At loci with FSTʹ ≥ 0.2, most haplotypes that are in the majority in fresh water can be found in marine populations, though they are typically rare (frequency ≤ 10%) (Figure 7, Figure S7, Table 2, Table S3, and Table S4). For example, 68% of alternative majority haplotypes identified in MiFW can be found in the marine comparators (131 marine fish from MiOC1, MiOC2, and DaOC) (Table S3). Freshwater allele frequencies in the sea were similar regardless of whether they were calculated for sets of freshwater majority haplotypes defined by freshwater populations that are geographically nearby or distant from a marine population (Figure S8). However, there is appreciable variation among marine chromosomes with regards to median and mean freshwater allele frequencies and allele frequency spectra (Figure S7, Figure S8, and Table S4), as might be expected given different patterns of divergence across the genome. Strikingly, marine populations that were sampled at great geographic distances from one another—at Middleton Island, at Danger Island (> 100 km from Middleton, mainly across open ocean) and even as far as the southern Oregon coast (> 2500 km away, along a coastal route)—carry many of the freshwater majority haplotypes identified on Middleton Island (Figure 8).
Figure 7.

Marine stickleback carry rare haplotypes that are found at high frequency in freshwater fish. Distributions of allele frequencies in marine fish of freshwater majority haplotypes from case 1 and case 3 loci (as defined in Figure 5) from the comparison between the combined island marine population [DaOC (Danger Island), MiOC1 (Middleton Island 1), and MiOC2 (Middleton Island 2)] and the MiFW (Middle Island freshwater) population. Allele frequency histograms are presented for 0.2 bins.
Figure 8.

Freshwater majority haplotypes are widely distributed in marine fish. Plotted for three geographically distant marine populations are all restriction site-associated DNA loci that fit two criteria: the locus falls into case 1 or case 3 (as defined in Figure 5) in a comparison between the marine population and a representative Middleton Island freshwater pond (Mi11), and at the same locus at least one marine fish carries a freshwater majority haplotype that is alternative to majority haplotype/s in the sea. Rings are numbered from the center out. Loci are colored by the case to which they belong. DaOC, Danger Island marine; MiOC1, Middleton Island marine 1.
The same minor freshwater allele frequencies in the sea can be generated if many marine fish each carry a few freshwater haplotypes, or if a few marine fish carry a majority or “jackpot” numbers of such haplotypes. To test whether freshwater haplotypes are primarily carried by only a fraction of marine individuals and to assess the patterns of freshwater haplotypes across the genomes of marine fish, we examined haplotype states in each marine individual at all case 1–3 loci, which carry alternative freshwater majority haplotypes. For example, most of the marine fish in the three island marine populations carried < 10% of these freshwater haplotypes, most often at heterozygous loci, but some fish had much larger numbers of them (Figure S9). Loci-bearing freshwater haplotypes are scattered diffusely among LGs in most individual marine fish, but in the fish that carry them at the highest proportions, these haplotypes often reside at loci that are densely concentrated on various subsets of the LGs that diverge the most in freshwater–oceanic comparisons. For example, one MiOC1 individual carried 51% of the case 1–3 freshwater majority haplotypes identified on LG VII, but carried < 6% of those identified on LG IV, and, conversely, another fish carried only 5% of the LG VII freshwater haplotypes but > 27% of those from LG IV (Table 4 and Table S5).
Table 4. Alternative freshwater majority haplotypes are carried in a variety of patterns by individual marine fish.
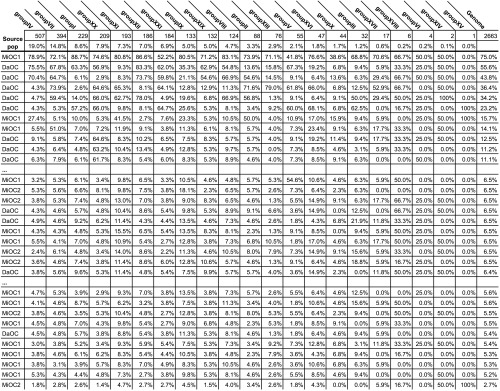 |
Out of the total number of possible freshwater majority haplotypes at case 1, 2, and 3 loci that were identified by comparing MiFW to the combined island marine populations, the proportion that each marine fish harbors is listed for each LG. The total number of possible freshwater haplotypes is listed first for each chromosome, which are ordered left to right from the highest to lowest number of these loci. Some chromosomes, such as LG XV, have relatively few loci with high freshwater–marine divergence, so there are few possible freshwater haplotypes on them. Others, such as LG IV and LG VII, have very large numbers of these loci. Individuals are ranked from those that carry the most to the fewest freshwater haplotypes, and of the 131 marine fish sampled, representative blocks of fish at the top, middle, and bottom of this range are shown. Da, Danger Island; Mi, Middleton Island; Mo, Montague Island.
Discussion
Selection affects a large proportion of the stickleback genome during divergence
By comparing patterns of freshwater–marine divergence in stickleback, we find that as much as 15% of the genome is affected in common by alternative adaptation to ponds and lakes vs. the sea in both mainland postglacial populations and young uplift island populations (Figure S3, Table 1, and Table S2). By comparison, Jones et al. (2012) sequenced 20 genomes in a geographically broad survey, using pairs of a single marine and a single freshwater stickleback fish from each sampling location. Use of a strict standard, of requiring a phylogenetic dichotomy of freshwater and marine alleles in this global sample of fish, revealed that < 0.5% of the genome was divergent. Our findings are strikingly distinct, and several factors may explain this 30-fold difference between the studies. It is possible that estimating divergence by smoothing across RAD loci could falsely gloss over some nondivergent regions between closely spaced divergent ones. However, differences in the methodologies employed between Jones et al. (2012) and the current study in terms of geographic breadth vs. population genomic depth of coverage make it more likely that the findings are compatible but provide different and complementary information. A specific subset of selected loci—those that are both likely ancient and house strictly alternative haplotypes—can be revealed in a broad geographic survey of few individuals, such as presented in Jones et al. (2012), whereas polygenic selection may involve more subtle changes in allele frequencies at multiple loci (Mather 1943; Burke et al. 2010; Pritchard and Di Rienzo 2010; Pritchard et al. 2010; Lotterhos and Schaal 2014; Laporte et al. 2016) that can only be detected with deeper population genomic sampling, such as we present here. Similarly, the immediate effects of linked selection can be revealed by the kind of deep population genomic sampling we performed (Cutter and Payseur 2013; Burri et al. 2015; Burri 2017; Wolf and Ellegren 2017), particularly in very young populations such as on these uplifted islands.
While we might expect to see broader genomic regions of divergence in these young freshwater ponds because of the effects of linkage (Table 1), others investigating stickleback populations that could be of similar age to those studied here reported much less total divergence across the genome. Terekhanova et al. (2014) studied marine–freshwater genomic divergence along the shores of the White Sea (Russia) in artificially seeded populations of known age and natural populations of inferred age, ranging from several decades to several centuries. The proportion of the genome estimated to be affected by marine–freshwater divergence in these Russian populations is far less than what we report for young populations on the uplift islands or even for much older Alaskan populations (Figure S10, Table 1, and Table S2). Small biological sample sizes from the White Sea populations, and subsequent analytical approaches that this undersampling necessitated (Terekhanova et al. 2014), could have led to a significant underestimate of the extent of linked selection there.
In contrast, because of the high level of biological sampling at the level of individuals and populations, we provide clear evidence that at least a quarter of the stickleback genome is initially affected by the action of strong divergent natural selection and that this effect is replicated independently across populations. A key, novel finding that emerges from such a sufficiently powered population genomic study is an unexpected change in haplotype diversities in each habitat. Regions of reduced haplotype diversity in the marine populations correspond to most of the genomic segments elevated in FSTʹ (Figure 4), a pattern that may have gone unrecognized in previous oceanic–freshwater comparisons because of the primary use of SNPs (Roesti et al. 2014; Marques et al. 2016) or sampling that does not allow accurate diversity estimation within populations (Jones et al. 2012; Terekhanova et al. 2014). For example, Terekhanova et al. (2014) report that nucleotide diversity in the ocean is higher than in fresh water within regions of divergence. This important difference from what we describe may be due to sampling or to uncertainties in using nucleotide diversity as a proxy for haplotype diversity. Regions of reduced absolute and relative haplotype diversity in the sea highlight an underappreciated likelihood of strong selection in both habitats, not just in fresh water. Greater purifying selection in the ocean across genomic regions subject to divergent selection could also contribute to the magnitude of the divergence we document.
That haplotype diversity is relatively higher in freshwater stickleback across the regions of divergence perhaps runs counter to an intuition that hard selective sweeps could reduce haplotype diversity in nascent freshwater populations relative to genetically diverse holarctic marine populations from which they emerge. One potential explanation for elevated diversity in these young populations is that freshwater-adapted haplotypes have not completely displaced marine haplotypes and are in a transitory state due to limited time since founding, or they are in a stable equilibrium due to lack of isolation from the sea. However, a high magnitude of FSTʹ divergence can also manifest through a relative increase in freshwater haplotype diversity via selection of an ancient allele embedded in a small number of syntenic genotypes, a process that would transiently increase the relative haplotype diversity in neighboring genomic regions in freshwater populations (Charlesworth et al. 1997; Berg and Coop 2015; Lee and Coop 2017). The majority of selected genomic regions could comprise such old alleles—for example a region highlighted in the recurrent evolution of armor loss (Cresko et al. 2004; Shapiro et al. 2004; Colosimo et al. 2005; Marchinko et al. 2014; O’Brown et al. 2015)—which drive the broadly parallel trait changes in freshwater fish. At the same time, the broader phenotypic diversity documented among freshwater populations than has been observed in the globally distributed marine population could result from diversity in their habitats (Walker and Bell 2000), but we suggest that it might also be attributed to linked genetic variants in which the selected ancient alleles are embedded.
Heterogeneous genomic divergence occurs in just decades despite gene flow
The heterogeneous divergence across the stickleback genome in these very young populations is unlikely to have occurred simply by neutral demographic processes or by the action of background selection (Charlesworth et al. 1993; Nordborg et al. 1996; Ellegren et al. 2012; Roesti et al. 2013; Burri et al. 2015; Burri 2017). Furthermore, a historically central [but not exclusive; see Defaveri and Merilä (2013) and Guo et al. (2015)] focus of stickleback phenotypic and population genomic studies has been on the action of natural selection in freshwater habitats as marine fish colonize them. The present study underscores the action of similarly strong divergent selection in marine habitats. However, the increased FST in each freshwater population as compared to the marine is not simply due to a reduction in diversity in the ocean in those regions, but also to an increase in frequency of largely the same haplotypes in independently derived freshwater populations. The reuse of ancient haplotypes can be seen in comparisons among the independently colonized islands, supporting the action of directional selection in fresh water. In fact, we observed as much as 99% of freshwater majority haplotypes that are shared between independent freshwater populations are also those for which marine and freshwater populations have strongly alternative haplotypes.
Although it has been predicted that a pool of freshwater-adapted haplotypes must be rare but present in and dispersed by the marine population [e.g., Colosimo et al. (2005) and Schluter and Conte (2009)], such haplotypes had so far not been quantified with a genome-wide approach. In a sample of only 131 phenotypically marine stickleback collected along the shores of the uplifted islands, we found a large proportion of freshwater majority haplotypes that had been identified at divergent loci in island pond populations, but they were rare and were most often carried heterozygously in marine stickleback. Nonetheless, the allele frequency spectra of freshwater majority haplotypes borne by the marine populations argue that continuous and substantial gene flow from myriad freshwater habitats maintains appreciable levels of these genotypes despite selection against them in the sea.
Evidence for the possibility of such gene flow exists on Middleton Island. Reduction in body size and bony lateral armor plates is typical of freshwater stickleback (Albert et al. 2008), and we documented previously that many uplift ponds on Middleton house only small fish with minimal armor (Lescak et al. 2015). However, several low-lying ponds still allow an influx of marine stickleback and harbor both low-armored and well-armored fish, as well as some phenotypic and genetic intermediates (Lescak et al. 2015). Even here, despite cohabitation and evidence of hybridization, the same divergent genome segments seen in older, more isolated freshwater populations (Hohenlohe et al. 2010; Roesti et al. 2014) manifest when sympatric marine and freshwater morphological ecotypes are compared against each other (seen in FSTʹ and in patterns of LD; Figure S11), and suggest that these habitats are zones of secondary contact where strong selection inhibits genomic homogenization. A testable hypothesis that emerges from our study is that such migration–selection balance under high gene flow would lead to significant genetic load (Akerman and Burger 2014) of maladaptive alleles in marine stickleback.
The “transporter hypothesis” and the nature of genomic changes in the first few generations after colonization
Migration–selection balance and the reuse of ancient haplotypes underlie the popularly cited transporter hypothesis to interpret parallel evolution in threespine stickleback (Schluter and Conte 2009), which predicts that the freshwater-adapted genome should enter new habitats piecemeal via marine colonizers over a period of time. Though capped at a maximum age by the 1964 earthquake, the island populations studied here probably span a range of younger ages. Montague’s southeast ponds (MoSE) are perched on a narrow peninsula < 2 m above sea level (Figure S12), and the pond on Danger Island is even lower. They are vulnerable to periodic inundation from the sea, which could cause osmotic stress, local extinction of stickleback adapting to fresh water, or an influx of marine stickleback leading to recolonization or genetic introgression. These populations show only a subset of the pattern of divergence seen in the other freshwater groups [e.g., (Hohenlohe et al. 2010) and this study]. For instance, an inversion-spanning region on LG I (Jones et al. 2012) alone stands out in DaFW. While other freshwater populations have a sharp peak of FST coinciding with this small inversion, in DaFW, the inversion nests within a broader window of divergence that includes reduced haplotype diversity, the clear signature of an ongoing selective sweep. The sparser pattern of genomic divergence in these precarious habitats may reflect initial random sampling of freshwater-adapted genotypes from the sea, and could also imply that these subsets of loci are sufficient for initial persistence of pioneers in fresh water. The LG I inversion, for example, includes the atp1a1 gene encoding a sodium–potassium ATPase that is important for osmoregulation in fishes (Jampol and Epstein 1970; McCairns and Bernatchez 2010; Jones et al. 2012).
Though fewer chromosomes show divergence from marine fish in MoSE, it nonetheless has elevated FSTʹ across the full length of the massive region of parallel divergence on LG IV. This is perhaps a testament to a low recombination rate across this part of the chromosome (Roesti et al. 2013), making it more likely for LG IV haplotypes alternative to majority marine haplotypes to enter new ponds as a unit. If true, this genomic architecture should speed the time that the parallel pattern of a diverged freshwater-adapted genome can be reassembled from what might be a relatively small number of large genomic segments.
Most of the alternative freshwater majority haplotypes we identified on Middleton (MiFW) and Montague (MoSW) were also present in marine stickleback, where in most individuals they occur in low numbers across the genome. However, a subset of the marine fish in our sample carried densely colocalized freshwater haplotypes on some LGs. These individuals could be early generation hybrids between freshwater and oceanic stickleback. Some of the freshwater haplotypes may also be in regions that resist recombination, such as those that lie in inversions or across large parts of LG IV and LG VII (Roesti et al. 2013). Discovery of these patterns of freshwater majority haplotypes in a relatively small sample of fish from the sea suggests that the rapid time of adaptation to new freshwater habitats could hinge on jackpot carriers, and that a freshwater-adapted genome might effectively be reassembled by migrants in new habitats one or more chromosomes at a time. Together, our findings provide a potential answer to the question of how enough low-frequency standing genetic variation can be reconstituted in new freshwater ponds even with few initial stickleback colonists: luck of the draw. A small number of migrants may in total carry enough chromosome-scale freshwater haplotypes to colonize newly opened habitats. Even if the initial colonists do not have an entire set of variants, they might have enough to stay in the game and subsequent migrants could quickly complete the winning hand.
General lessons about migration–selection balance and the genomic architecture of rapid adaptation
Together, our data and other recent studies in stickleback argue for a view of the stickleback species as a metapopulation, with strong divergent selection occurring in both marine and freshwater habitats despite bidirectional gene flow. This view contrasts with a more traditional, “raceme” or “bottle-brush” concept of stickleback, with a core of marine populations that harbor the majority of genetic diversity in this species, while freshwater populations are the bottle-brush “whiskers,” ephemeral in nature (so-called “evolutionary dead-ends”). In contrast, our data argue strongly that both population types serve as reservoirs for maintaining genetic diversity through a long-term dynamic of bidirectional migration–selection balance. Although the relative impact of any particular freshwater population may be slight, the aggregated hundreds of thousands (and likely millions) of freshwater populations along the coastlines of the Northern Hemisphere could have a significant impact on the pool of standing genetic variation in marine populations. This is particularly likely when, as has been documented, haplotypes that are identical by descent are selected repeatedly in independently derived freshwater populations.
The stickleback radiation may provide more general evolutionary genomic lessons because adaptation from standing genetic variation on such a short time frame may be common in the wild. This could be particularly true in spatially structured species that experience spatially or temporally variable selection and gene flow that maintains ancient diversity. We argue that this scenario is more likely to be the rule than the exception in natural populations of most species, such as has been documented in the study of Darwin’s finches (Han et al. 2017). If common, then, for many species standing genetic variation maintained and shaped by molecular evolutionary processes operating over thousands or millions of generations could permit population genomic changes and organismal adaptation to new environments over just decades. Fully understanding rapid adaptive dynamics may often require a deep molecular evolutionary perspective of the haplotypes’ changing frequencies.
The long-term association of the stickleback species with both oceanic and freshwater habitats leads to an intriguing hypothesis. Although each bout of adaptation might require only decades, the genomic regions underlying the change are a much older polymorphism maintained through variable selection in the metapopulation. If so, the molecular evolutionary history of locally adapted genomic regions could actually be quite deep. Alternatively adapted freshwater and marine haplotypes in genomic regions with lower recombination rates (Roesti et al. 2013) may be millions of years old (Colosimo et al. 2005), leading to increased absolute divergence by the accumulation of mutations. Aligning with this hypothesis, coalescent analysis of Sanger length RAD-based sequences sampled across the genome demonstrates that, in many of the genomic regions with the most significant relative divergence of allele frequencies, haplotypes also show more ancient evolutionary divergence that is millions of years old (Nelson and Cresko 2018). Therefore, a deep evolutionary history underlies the ability of stickleback to adapt so rapidly. Similar analyses of other species with populations experiencing divergent selection and gene flow could show whether this phenomenon of old genetic variants fueling rapid adaptation is unique to stickleback, or a common theme in nature.
Acknowledgments
We thank M. S. Christy, S. A. Hatch, B. Lohman, V. M. Padula, L. Smayda, and K. Walton for assistance with fieldwork logistics and fish collection; T. Wilson and M. Currey for technical help; P. Hohenlohe for discussions during early stages of the development of the project; members of the W.A.C., P. C. Phillips, and M. A. Streisfeld laboratories at the University of Oregon for discussions and critical comments; and Genetics associate editor J. Novembre and the anonymous reviewers for their valuable input. This research was supported primarily by National Science Foundation DEB 0949053 and IOS 102728 (to W.A.C.) and DEB 0919234 (to F.A.v.H.). Additional support came from NIH Grant 1R24GM079486-01A1 (to W.A.C.), NIH NRSA Ruth L. Kirschstein Fellowship F32GM095213-01 (to J.C.). E.A.L. was supported with funds from NIH Institutional Development Award (IDeA) P20GM103395.
Footnotes
Supplemental material available at Figshare: https://doi.org/10.25386/genetics.6272432.
Communicating editor: J. Novembre
Literature Cited
- Akerman A., Burger R., 2014. The consequences of gene flow for local adaptation and differentiation: a two-locus two-deme model. J. Math. Biol. 68: 1135–1198. 10.1007/s00285-013-0660-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert A. Y., Sawaya S., Vines T. H., Knecht A. K., Miller C. T., et al. , 2008. The genetics of adaptive shape shift in stickleback: pleiotropy and effect size. Evolution 62: 76–85. 10.1111/j.1558-5646.2007.00259.x [DOI] [PubMed] [Google Scholar]
- Barrett R. D., Schluter D., 2008. Adaptation from standing genetic variation. Trends Ecol. Evol. 23: 38–44. 10.1016/j.tree.2007.09.008 [DOI] [PubMed] [Google Scholar]
- Bell M. A., 1984. Evolutionary phenetics and genetics. The threespine stickleback, Gasterosteus aculeatus, and related species, pp. 431–528 in Evolutionary Genetics of Fishes, edited by Turner B. J. Springer-Verlag, Berlin: 10.1007/978-1-4684-4652-4_9 [DOI] [Google Scholar]
- Bell M. A., Foster S. A., 1994. The Evolutionary Biology of the Threespine Stickleback. Oxford University Press, Oxford. [Google Scholar]
- Bell M. A., Aguirre W. E., Buck N. J., 2004. Twelve years of contemporary armor evolution in a threespine stickleback population. Evolution 58: 814–824. 10.1111/j.0014-3820.2004.tb00414.x [DOI] [PubMed] [Google Scholar]
- Berg J. J., Coop G., 2015. A coalescent model for a sweep of a unique standing variant. Genetics 201: 707–725. 10.1534/genetics.115.178962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird C., Karl S., Mouse P., Toonen R., 2011. Detecting and measuring genetic differentiation, pp. 31–55 in Phylogeography and Population Genetics in Crustacea. CRC Press, Boca Raton, FL. [Google Scholar]
- Boughman J. W., Rundle H. D., Schluter D., 2005. Parallel evolution of sexual isolation in sticklebacks. Evolution 59: 361–373. 10.1111/j.0014-3820.2005.tb00995.x [DOI] [PubMed] [Google Scholar]
- Burke M. K., Dunham J. P., Shahrestani P., Thornton K. R., Rose M. R., et al. , 2010. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature 467: 587–590. 10.1038/nature09352 [DOI] [PubMed] [Google Scholar]
- Burke M. K., Liti G., Long A. D., 2014. Standing genetic variation drives repeatable experimental evolution in outcrossing populations of Saccharomyces cerevisiae. Mol. Biol. Evol. 31: 3228–3239. 10.1093/molbev/msu256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burri R., 2017. Linked selection, demography and the evolution of correlated genomic landscapes in birds and beyond. Mol. Ecol. 26: 3853–3856. 10.1111/mec.14167 [DOI] [PubMed] [Google Scholar]
- Burri R., Nater A., Kawakami T., Mugal C. F., Olason P. I., et al. , 2015. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 25: 1656–1665. 10.1101/gr.196485.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J., Bassham S., Wilson T., Currey M., O’Brien C., et al. , 2013a The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing. Mol. Ecol. 22: 2864–2883. 10.1111/mec.12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J., Hohenlohe P. A., Bassham S., Amores A., Cresko W. A., 2013b Stacks: an analysis tool set for population genomics. Mol. Ecol. 22: 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J. M., Amores A., Hohenlohe P., Cresko W., Postlethwait J. H., 2011. Stacks: building and genotyping Loci de novo from short-read sequences. G3 (Bethesda) 1: 171–182. 10.1534/g3.111.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., 1998. Measures of divergence between populations and the effect of forces that reduce variability. Mol. Biol. Evol. 15: 538–543. 10.1093/oxfordjournals.molbev.a025953 [DOI] [PubMed] [Google Scholar]
- Charlesworth B., 2012. The effects of deleterious mutations on evolution at linked sites. Genetics 190: 5–22. 10.1534/genetics.111.134288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Morgan M. T., Charlesworth D., 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Nordborg M., Charlesworth D., 1997. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet. Res. 70: 155–174. 10.1017/S0016672397002954 [DOI] [PubMed] [Google Scholar]
- Colosimo P. F., Hosemann K. E., Balabhadra S., Villarreal G., Jr., Dickson M., et al. , 2005. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307: 1928–1933. 10.1126/science.1107239 [DOI] [PubMed] [Google Scholar]
- Cresko W. A., Amores A., Wilson C., Murphy J., Currey M., et al. , 2004. Parallel genetic basis for repeated evolution of armor loss in Alaskan threespine stickleback populations. Proc. Natl. Acad. Sci. USA 101: 6050–6055. 10.1073/pnas.0308479101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresko W. A., McGuigan K. L., Phillips P. C., Postlethwait J. H., 2007. Studies of threespine stickleback developmental evolution: progress and promise. Genetica 129: 105–126. 10.1007/s10709-006-0036-z [DOI] [PubMed] [Google Scholar]
- Cruickshank T. E., Hahn M. W., 2014. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23: 3133–3157. 10.1111/mec.12796 [DOI] [PubMed] [Google Scholar]
- Cutter A. D., Payseur B. A., 2013. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat. Rev. Genet. 14: 262–274. 10.1038/nrg3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defaveri J., Merilä J., 2013. Evidence for adaptive phenotypic differentiation in Baltic Sea sticklebacks. J. Evol. Biol. 26: 1700–1715. 10.1111/jeb.12168 [DOI] [PubMed] [Google Scholar]
- DeFaveri J., Shikano T., Shimada Y., Goto A., Merila J., 2011. Global analysis of genes involved in freshwater adaptation in threespine sticklebacks (Gasterosteus aculeatus). Evolution 65: 1800–1807. 10.1111/j.1558-5646.2011.01247.x [DOI] [PubMed] [Google Scholar]
- Ellegren H., Smeds L., Burri R., Olason P. I., Backstrom N., et al. , 2012. The genomic landscape of species divergence in Ficedula flycatchers. Nature 491: 756–760. 10.1038/nature11584 [DOI] [PubMed] [Google Scholar]
- Evanno G., Regnaut S., Goudet J., 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14: 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Feder J. L., Nosil P., 2010. The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64: 1729–1747. 10.1111/j.1558-5646.2009.00943.x [DOI] [PubMed] [Google Scholar]
- Feder J. L., Egan S. P., Nosil P., 2012. The genomics of speciation-with-gene-flow. Trends Genet. 28: 342–350. 10.1016/j.tig.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Feulner P. G., Chain F. J., Panchal M., Eizaguirre C., Kalbe M., et al. , 2013. Genome-wide patterns of standing genetic variation in a marine population of three-spined sticklebacks. Mol. Ecol. 22: 635–649. 10.1111/j.1365-294X.2012.05680.x [DOI] [PubMed] [Google Scholar]
- Flaxman S. M., Feder J. L., Nosil P., 2013. Genetic hitchhiking and the dynamic buildup of genomic divergence during speciation with gene flow. Evolution 67: 2577–2591. 10.1111/evo.12055 [DOI] [PubMed] [Google Scholar]
- Gelmond O., von Hippel F. A., Christy M. S., 2009. Rapid ecological speciation in three-spined stickleback Gasterosteus aculeatus from Middleton Island, Alaska: the roles of selection and geographic isolation. J. Fish Biol. 75: 2037–2051. 10.1111/j.1095-8649.2009.02417.x [DOI] [PubMed] [Google Scholar]
- Guo B., DeFaveri J., Sotelo G., Nair A., Merila J., 2015. Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks. BMC Biol. 13: 19 10.1186/s12915-015-0130-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. W., 2008. Toward a selection theory of molecular evolution. Evolution 62: 255–265. 10.1111/j.1558-5646.2007.00308.x [DOI] [PubMed] [Google Scholar]
- Han F., Lamichhaney S., Grant B. R., Grant P. R., Andersson L., et al. , 2017. Gene flow, ancient polymorphism, and ecological adaptation shape the genomic landscape of divergence among Darwin’s finches. Genome Res. 27: 1004–1015. 10.1101/gr.212522.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry A. P., Kinnison M. T., 1999. Perspective: the pace of modern life: measuring rates of contemporary microevolution. Evolution 53: 1637–1653. 10.1111/j.1558-5646.1999.tb04550.x [DOI] [PubMed] [Google Scholar]
- Hendry A. P., Kinnison M. T., 2001. An introduction to microevolution: rate, pattern, process. Genetica 112–113: 1–8. 10.1023/A:1013368628607 [DOI] [PubMed] [Google Scholar]
- Hendry A. P., Wenburg J. K., Bentzen P., Volk E. C., Quinn T. P., 2000. Rapid evolution of reproductive isolation in the wild: evidence from introduced salmon. Science 290: 516–519. 10.1126/science.290.5491.516 [DOI] [PubMed] [Google Scholar]
- Hey J., 2010. Isolation with migration models for more than two populations. Mol. Biol. Evol. 27: 905–920. 10.1093/molbev/msp296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase S., Ozaki H., Iwasaki W., 2014. Parallel selection on gene copy number variations through evolution of three-spined stickleback genomes. BMC Genomics 15: 735 10.1186/1471-2164-15-735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe P. A., Bassham S., Etter P. D., Stiffler N., Johnson E. A., et al. , 2010. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 6: e1000862 10.1371/journal.pgen.1000862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe P. A., Catchen J., Cresko W. A., 2012. Population genomic analysis of model and nonmodel organisms using sequenced RAD tags. Methods Mol. Biol. 888: 235–260. 10.1007/978-1-61779-870-2_14 [DOI] [PubMed] [Google Scholar]
- Holsinger K. E., Weir B. S., 2009. Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat. Rev. Genet. 10: 639–650. 10.1038/nrg2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M., Edge M. D., Rosenberg N. A., 2013. The relationship between F(ST) and the frequency of the most frequent allele. Genetics 193: 515–528. 10.1534/genetics.112.144758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jampol L. M., Epstein F. H., 1970. Sodium- potassium-activated adenosine triphosphatase and osmotic regulation by fishes. Am. J. Physiol. 218: 607–611. 10.1152/ajplegacy.1970.218.2.607 [DOI] [PubMed] [Google Scholar]
- Jones F. C., Grabherr M. G., Chan Y. F., Russell P., Mauceli E., et al. , 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61. 10.1038/nature10944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeuffer R., Peichel C. L., Bolnick D. I., Hendry A. P., 2012. Parallel and nonparallel aspects of ecological, phenotypic, and genetic divergence across replicate population pairs of lake and stream stickleback. Evolution 66: 402–418. 10.1111/j.1558-5646.2011.01440.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Cresko W. A., Phillips P. C., Ullmann B., Currey M., et al. , 2012. Independent axes of genetic variation and parallel evolutionary divergence of opercle bone shape in threespine stickleback. Evolution 66: 419–434. 10.1111/j.1558-5646.2011.01441.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M., Ota T., 1971. Theoretical aspects of population genetics. Monogr. Popul. Biol. 4: 1–219. [PubMed] [Google Scholar]
- Kitano J., Ishikawa A., Kume M., Mori S., 2012. Physiological and genetic basis for variation in migratory behavior in the three-spined stickleback, Gasterosteus aculeatus. Ichthyol. Res. 59: 293–303. 10.1007/s10228-012-0289-8 [DOI] [Google Scholar]
- Kopp M., Matuszewski S., 2014. Rapid evolution of quantitative traits: theoretical perspectives. Evol. Appl. 7: 169–191. 10.1111/eva.12127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe M., Ishikawa A., Ravinet M., Yoshida K., Makino T., et al. , 2017. Genetic basis for variation in salinity tolerance between stickleback ecotypes. Mol. Ecol. 26: 304–319. 10.1111/mec.13875 [DOI] [PubMed] [Google Scholar]
- Laporte M., Pavey S. A., Rougeux C., Pierron F., Lauzent M., et al. , 2016. RAD sequencing reveals within-generation polygenic selection in response to anthropogenic organic and metal contamination in North Atlantic Eels. Mol. Ecol. 25: 219–237. 10.1111/mec.13466 [DOI] [PubMed] [Google Scholar]
- Lee K. M., Coop G., 2017. Distinguishing among modes of convergent adaptation using population genomic data. Genetics 207: 1591–1619. 10.1534/genetics.117.300417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescak E. A., Bassham S. L., Catchen J., Gelmond O., Sherbick M. L., et al. , 2015. Evolution of stickleback in 50 years on earthquake-uplifted islands. Proc. Natl. Acad. Sci. USA 112: E7204–E7212. 10.1073/pnas.1512020112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D., 1995. Metapopulations: an arena for local speciation. J. Evol. Biol. 8: 635–644. 10.1046/j.1420-9101.1995.8050635.x [DOI] [Google Scholar]
- Li, H., 2013 Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN]. [Google Scholar]
- Lotterhos K. E., Schaal S. M., 2014. Genome scans for the contemporary response to selection in quantitative traits. Mol. Ecol. 23: 4435–4437. 10.1111/mec.12853 [DOI] [PubMed] [Google Scholar]
- Lucek K., Lemoine M., Seehausen O., 2014. Contemporary ecotypic divergence during a recent range expansion was facilitated by adaptive introgression. J. Evol. Biol. 27: 2233–2248. 10.1111/jeb.12475 [DOI] [PubMed] [Google Scholar]
- Marchinko K. B., Matthews B., Arnegard M. E., Rogers S. M., Schluter D., 2014. Maintenance of a genetic polymorphism with disruptive natural selection in stickleback. Curr. Biol. 24: 1289–1292. 10.1016/j.cub.2014.04.026 [DOI] [PubMed] [Google Scholar]
- Marques D. A., Lucek K., Meier J. I., Mwaiko S., Wagner C. E., et al. , 2016. Genomics of rapid incipient speciation in sympatric threespine stickleback. PLoS Genet. 12: e1005887 10.1371/journal.pgen.1005887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather K., 1943. Polygenic inheritance and natural selection. Biol. Rev. Camb. Philos. Soc. 18: 32–64. 10.1111/j.1469-185X.1943.tb00287.x [DOI] [Google Scholar]
- McCairns R. J., Bernatchez L., 2010. Adaptive divergence between freshwater and marine sticklebacks: insights into the role of phenotypic plasticity from an integrated analysis of candidate gene expression. Evolution 64: 1029–1047. 10.1111/j.1558-5646.2009.00886.x [DOI] [PubMed] [Google Scholar]
- Meirmans P. G., 2006. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60: 2399–2402. 10.1111/j.0014-3820.2006.tb01874.x [DOI] [PubMed] [Google Scholar]
- Meirmans P. G., Hedrick P. W., 2011. Assessing population structure: F(ST) and related measures. Mol. Ecol. Resour. 11: 5–18. 10.1111/j.1755-0998.2010.02927.x [DOI] [PubMed] [Google Scholar]
- Messer P. W., Petrov D. A., 2013. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol. Evol. 28: 659–669. 10.1016/j.tree.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. T., Glazer A. M., Summers B. R., Blackman B. K., Norman A. R., et al. , 2014. Modular skeletal evolution in sticklebacks is controlled by additive and clustered quantitative trait Loci. Genetics 197: 405–420. 10.1534/genetics.114.162420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan-Myhre K., Small C. M., Mittge E. K., Agarwal M., Currey M., et al. , 2016. Innate immune responses to gut microbiota differ between oceanic and freshwater threespine stickleback populations. Dis. Model. Mech. 9: 187–198. 10.1242/dmm.021881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau N. J., Martin S. H., Kozak K. M., Salazar C., Dasmahapatra K. K., et al. , 2013. Genome-wide patterns of divergence and gene flow across a butterfly radiation. Mol. Ecol. 22: 814–826. 10.1111/j.1365-294X.2012.05730.x [DOI] [PubMed] [Google Scholar]
- Nei M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York. [Google Scholar]
- Nelson T. C., Cresko W. A., 2018. Ancient genomic variation underlies repeated ecological adaptation in young stickleback populations. Evolution Letters 2: 9–21. 10.1002/evl3.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M., Charlesworth B., Charlesworth D., 1996. The effect of recombination on background selection. Genet. Res. 67: 159–174. 10.1017/S0016672300033619 [DOI] [PubMed] [Google Scholar]
- Nosil P., Funk D. J., Ortiz-Barrientos D., 2009. Divergent selection and heterogeneous genomic divergence. Mol. Ecol. 18: 375–402. 10.1111/j.1365-294X.2008.03946.x [DOI] [PubMed] [Google Scholar]
- O’Brown N. M., Summers B. R., Jones F. C., Brady S. D., Kingsley D. M., 2015. A recurrent regulatory change underlying altered expression and Wnt response of the stickleback armor plates gene EDA. Elife 4: e05290 10.7554/eLife.05290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr H. A., 2005. The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 6: 119–127. 10.1038/nrg1523 [DOI] [PubMed] [Google Scholar]
- Plafker G., 1969. The Alaska Earthquake, March 27, 1964: Regional Effects – the Tectonics of the March 27, 1964 Alaska Earthquake 543-I. U.S. Government Printing Office, Washington, DC, pp. 1–73. [Google Scholar]
- Plafker, G., and M. Rubin, 1978 Uplift history and earthquake recurrence as deduced from marine terraces on Middleton Island, Alaska. Open-File Report 78, pp. 687–721, U.S. Geological Survey, Fairbanks, AK. [Google Scholar]
- Poelstra J. W., Vijay N., Bossu C. M., Lantz H., Ryll B., et al. , 2014. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 344: 1410–1414. 10.1126/science.1253226 [DOI] [PubMed] [Google Scholar]
- Pritchard J. K., Di Rienzo A., 2010. Adaptation - not by sweeps alone. Nat. Rev. Genet. 11: 665–667. 10.1038/nrg2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J. K., Stephens M., Donnelly P., 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J. K., Pickrell J. K., Coop G., 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr. Biol. 20: R208–R215. 10.1016/j.cub.2009.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team , 2013. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/ [Google Scholar]
- Radwan J., Babik W., 2012. The genomics of adaptation. Proc. Biol. Sci. 279: 5024–5028. 10.1098/rspb.2012.2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid N. M., Proestou D. A., Clark B. W., Warren W. C., Colbourne J. K., et al. , 2016. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science 354: 1305–1308. 10.1126/science.aah4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesti M., Moser D., Berner D., 2013. Recombination in the threespine stickleback genome--patterns and consequences. Mol. Ecol. 22: 3014–3027 (erratum: Mol. Ecol. 22: 5270); (erratum: Mol. Ecol. 22: 3652) 10.1111/mec.12322 [DOI] [PubMed] [Google Scholar]
- Roesti M., Gavrilets S., Hendry A. P., Salzburger W., Berner D., 2014. The genomic signature of parallel adaptation from shared genetic variation. Mol. Ecol. 23: 3944–3956. 10.1111/mec.12720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen O., Lascoux M., Merila J., 2013. Ecological genomics of local adaptation. Nat. Rev. Genet. 14: 807–820. 10.1038/nrg3522 [DOI] [PubMed] [Google Scholar]
- Schliep A., Costa I. G., Steinhoff C., Schönhuth A., 2005. Analyzing gene expression time-courses. IEEE/ACM Trans. Comput. Biol. Bioinform. 2: 179–193. 10.1109/TCBB.2005.31 [DOI] [PubMed] [Google Scholar]
- Schluter D., Conte G. L., 2009. Genetics and ecological speciation. Proc. Natl. Acad. Sci. USA 106: 9955–9962. 10.1073/pnas.0901264106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D., Clifford E. A., Nemethy M., McKinnon J. S., 2004. Parallel evolution and inheritance of quantitative traits. Am. Nat. 163: 809–822. 10.1086/383621 [DOI] [PubMed] [Google Scholar]
- Shapiro M. D., Marks M. E., Peichel C. L., Blackman B. K., Nereng K. S., et al. , 2004. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 428: 717–723. 10.1038/nature02415 [DOI] [PubMed] [Google Scholar]
- Slatkin M., Wiehe T., 1998. Genetic hitch-hiking in a subdivided population. Genet. Res. 71: 155–160. 10.1017/S001667239800319X [DOI] [PubMed] [Google Scholar]
- Terekhanova N. V., Logacheva M. D., Penin A. A., Neretina T. V., Barmintseva A. E., et al. , 2014. Fast evolution from precast bricks: genomics of young freshwater populations of threespine stickleback Gasterosteus aculeatus. PLoS Genet. 10: e1004696 10.1371/journal.pgen.1004696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. E., Taylor E. B., McPhail J. D., 1997. Parallel evolution of lake-stream pairs of threespine sticklebacks (Gasterosteus) inferred from mitochondrial DNA variation. Evolution 51: 1955–1965. 10.1111/j.1558-5646.1997.tb05117.x [DOI] [PubMed] [Google Scholar]
- Thompson J. N., 1998. Rapid evolution as an ecological process. Trends Ecol. Evol. 13: 329–332. 10.1016/S0169-5347(98)01378-0 [DOI] [PubMed] [Google Scholar]
- Turner T. L., Hahn M. W., Nuzhdin S. V., 2005. Genomic islands of speciation in Anopheles gambiae. PLoS Biol. 3: e285 10.1371/journal.pbio.0030285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J. A., Bell M. A., 2000. Net evolutionary trajectories of body shape evolution within a microgeographic radiation of threespine sticklebacks (Gasterosteus aculeatus). J. Zool. (Lond.) 252: 293–302. 10.1111/j.1469-7998.2000.tb00624.x [DOI] [Google Scholar]
- Weir B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Weir B. S., Cockerham C. C., 1984. Estimating F-statistics for the analysis of population-structure. Evolution 38: 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Wolf J. B., Ellegren H., 2017. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet. 18: 87–100. 10.1038/nrg.2016.133 [DOI] [PubMed] [Google Scholar]
- Yeaman S., Otto S. P., 2011. Establishment and maintenance of adaptive genetic divergence under migration, selection, and drift. Evolution 65: 2123–2129. 10.1111/j.1558-5646.2011.01277.x [DOI] [PubMed] [Google Scholar]
- Yeaman S., Whitlock M. C., 2011. The genetic architecture of adaptation under migration-selection balance. Evolution 65: 1897–1911. 10.1111/j.1558-5646.2011.01269.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequence data are available in the NCBI Sequence Read Archive under accession SRP148115. Supplemental material available at Figshare: https://doi.org/10.25386/genetics.6272432. RAD consensus sequences for each locus with its aligned genomic position are available in FASTA format for populations (File_S01) and for population groups (File_S06). SNP and haplotype calls are available in VCF format for populations (SNP: File_S02; haplotype: File_S03) and for population groups (SNP: File_S07; haplotype: File_S08). Values of divergence (FST or FSTʹ and ΦST) at each locus are available for populations (SNP: File_S05; haplotype: File_S04), and for population clusters (SNP: File_S10; haplotype: File_S09). Figure S1 contains genetic groupings and a reference map of studied populations. Figure S2 depicts continuous genetic divergence across the genome for marine vs. each freshwater genetic group. Figure S3 shows the full HMM analysis across all LGs. Figure S4 contains gene and haplotype diversity measures. Figure S5 shows Cases along the genome in independent populations. Figure S6 depicts alternative freshwater haplotypes across LGI. Figure S7 contains per-chromosome allele frequency spectra. Figure S8 contains boxplots of alternative freshwater allele frequencies. Figure S9 contains per-marine fish counts of alternative freshwater alleles. Figure S10 is a comparison of genomic divergence patterns between this and an independent published analysis. Figure S11 shows LD patterns in freshwater fish. Figure S12 shows an aerial photograph of collection sites on Montague Island. Table S1 lists genome-wide genetic divergence values for freshwater vs. marine comparisons. Table S2 reports fractions of the diverged genome that overlap among populations. Table S3 quantifies categories of divergent loci (Cases) in different populations. Table S4 contains the mean, median, and SD of the frequencies of alternative freshwater alleles in marine populations. Table S5 contains the fraction of alternative freshwater alleles found on each chromosome for each marine fish. Tables S6–S8 report sequencing statistics. Table S9 contains a key for which data set was used in each figure/table.