

Abstract
Background
A statistical pipeline was developed and used for determining candidate genes and candidate gene co-expression networks involved in two alcohol (i.e., ethanol) metabolism phenotypes, namely alcohol clearance and acetate area under the curve (AUC) in a recombinant inbred (HXB/BXH) rat panel. The approach was also used to provide an indication of how ethanol metabolism can impact the normal function of the identified networks.
Methods
RNA was extracted from alcohol-naïve liver tissue of 30 strains of HXB/BXH recombinant inbred rats. The reconstructed transcripts were quantitated and data was used to construct gene co-expression modules and networks. A separate group of rats, comprising the same 30 strains, were injected with ethanol (2 gm/kg) for measurement of blood ethanol and acetate levels. These data were used for QTL analysis of the rate of ethanol disappearance and circulating acetate levels. The analysis pipeline required calculation of the module eigengene values, the correction of these values with ethanol metabolism rates and acetate levels across the rat strains and the determination of the eigengene QTLs. For a module to be considered a candidate for determining phenotype, the module eigengene values had to have significant correlation with the strain phenotypic values and the module eigengene QTLs had to overlap the phenotypic QTLs.
Results
Of the 658 transcript co-expression modules generated from liver RNA sequencing data, a single module satisfied all criteria for being a candidate for determining the alcohol clearance trait. This module contained two alcohol dehydrogenase genes, including the gene whose product was previously shown to be responsible for the majority of alcohol elimination in the rat. This module was also the only module identified as a candidate for influencing circulating acetate levels. This module was also linked to the process of generation and utilization of retinoic acid as related to the autonomous immune response.
Conclusions
We propose that our analytical pipeline can successfully identify genetic regions and transcripts which predispose a particular phenotype and our analysis provides functional context for co-expression module components.
Keywords: Alcohol metabolism, RNA sequencing, Weighted gene co-expression network analysis, Quantitative trait locus mapping, HXB/BXH recombinant inbred rat panel, Liver
Introduction
Genome-wide association studies (GWASs) were originally designed to leverage the principle of linkage disequilibrium at the population level by scanning millions of variants in the genome across unrelated individuals to identify loci associated with (and presumably predisposing) complex traits (Visscher et al., 2012). Since its first applications (Klein et al., 2005), hundreds of GWASs have been implemented and a dedicated catalog of the published studies has been developed (Welter et al., 2014, GWAS Catalog, https://www.ebi.ac.uk/gwas/. Accessed 04 Dec. 2017). Often the quantitative trait loci (QTLs) identified in GWASs do not fully explain the heritability of the complex trait anticipated from epidemiologic studies (e.g. alcohol dependence) (Edenberg and Foroud, 2013), and the relationship between the identified loci and the biology underlying complex diseases may not be easily deciphered (Nicolae et al., 2010).
The advent of next-generation RNA sequencing (RNA-seq) technologies has provided researchers with new tools for gaining insight into the genetic basis of health and disease. Namely, researchers can now incorporate RNA expression levels in a “use all data” (Perez-Enciso et al., 2007) systems biology approach to extract meaningful genetic information about complex traits. Integrating transcriptome expression data with genotype information, i.e. genetical genomics, can provide insight into the mechanisms predisposing disease phenotypes (Schadt et al., 2003). A now standard approach for integrating information on RNA expression with genotypic information, to elucidate mechanisms by which DNA polymorphisms contribute to complex traits, is to identify the areas of the genome that are associated with a complex trait (QTLs) and that contribute to determining the levels of gene expression (expression quantitative trait loci; eQTLs). Moreover, the use of methods to generate information on networks arising from analysis of gene co-expression and the genetic loci driving such co-expression (module eigengene quantitative trait loci; meQTLs), can contribute additional knowledge to the underlying biology (Ghazalpour et al., 2006, Mackay et al., 2009). For example, co-expressed genes may not only be controlled by the same transcriptional regulatory program (Mackay et al., 2009), but also may be functionally linked (Stuart et al., 2003). The co-expressed gene products may be members of the same metabolic pathway or protein complex (Ge et al., 2001). Additionally, co-expression modules can be used to functionally annotate (“guilt by association”) novel or under annotated genes (Serin et al., 2016), including non-coding elements. Zhang and Horvath (2005) developed a statistical technique for quantifying gene co-expression networks and identifying co-expression modules from RNA expression data. This methodology, termed weighted gene co-expression analysis (WGCNA), has been employed in numerous studies (Fuller et al., 2007, Oldham et al., 2008, Konopka et al., 2009, DiLeo et al., 2011, Xue et al., 2014) to statistically describe the relationship amongst gene products.
Our previous work has integrated eQTL information and WGCNA with phenotypic QTL (pQTL) analysis in a hypothesis generating approach, to identify candidate modules predisposing complex traits (Tabakoff et al., 2009, Vanderlinden et al., 2013, Saba et al., 2015, Harrall et al., 2016). In our current study, we investigated whether this approach can be valuable in a “hypothesis testing” mode. We sought to provide a proof-of-concept that an unsupervised, statistically-based systems biology approach can identify predisposition co-expression module(s) in alcohol naïve animals that contain components known to influence alcohol (i.e., ethanol) metabolism. It is well-documented that the majority of alcohol (~ 95%) is eliminated via metabolism in the liver (Bosron et al., 1993, Ramchandani et al., 2001, Norberg et al., 2003). Hepatic oxidation of ethanol, in which alcohol dehydrogenase(s) (ADH) convert ethanol to acetaldehyde, and acetaldehyde is converted to acetate by aldehyde dehydrogenase (ALDH), is the major metabolic pathway for elimination of ingested ethanol (Norberg et al., 2003, Guindalini et al., 2005, Zakhari, 2006, Cederbaum, 2012). Prior studies seeking genetic explanations for differences in ethanol metabolism using QTL analysis in mice have produced results which were difficult to interpret, and did not relate to prior literature (Grisel et al., 2002). Therefore, we were attempting to validate our approach by verifying whether co-expression module(s) which were associated with alcohol clearance, derived through our analysis, reflected the extensive information in the literature on known pathways of alcohol clearance. Assuming that our analytical framework produced credible results, we further postulated that the module information that we generated would provide insight into the normal physiologic network that could be perturbed by ingestion of ethanol.
Materials and Methods
Unless otherwise noted, all analyses were performed using R (v. 3.3.2).
Animals
The HXB/BXH Recombinant Inbred (RI) rat panel used in this study was derived from the congenic Brown Norway strain with polydactyly-luxate syndrome (BN-Lx/Cub) and the spontaneous hypertensive rat strain (SHR/OlaIpcv) using gender reciprocal crossing and more than 80 generations of brother/sister mating after the F2 generation (Pravenec et al., 1989). While this panel was originally constructed to examine genetic control of cardiovascular phenotypes, many other complex traits have been found to vary across this panel and are thus amenable to genetic studies (Tabakoff et al., 2009, Vanderlinden et al., 2013, Saba et al., 2015, Kunes et al., 1994, Bielavska et al., 2002, Conti et al., 2004, Pravenec et al., 2004).
Male rats at the age of 90 days were used for our studies. These animals were bred and maintained at the Institute of Physiology of the Czech Academy of Sciences, Prague, Czech Republic. All experiments involving the administration of ethanol and blood sampling, as well as liver harvesting, were performed in accordance with the Animal Protection Law of the Czech Republic and were approved by the Ethics Committee of the Institute of Physiology, Czech Academy of Sciences, Prague.
Alcohol Clearance and Blood Acetate Level Measurements in the HXB/BXH Recombinant Inbred Rat Panel
Three male rats per strain across 30 strains of the HXB/BXH RI panel (90 rats total) were intraperitoneally injected with a 2 g/kg dose of ethanol (15% w/v). (See Supplementary Methods in Appendix S1 for a detailed description of ethanol dose choice rationale.) Blood draws from the tail vein were collected for quantifying alcohol and acetate concentration at the following time points post-alcohol administration: 20, 40, 60, 90, 120, 180, 240, 300, and 400 minutes. In addition, a 0 time point sample was gathered immediately prior to alcohol administration. For each sampling, approximately 100 μL of blood was collected. Following collection, two volumes of ice cold 0.6 N perchloric acid were added to each sample, and the resulting supernatant, after centrifugation at 13000 g and 4 °C for 10 minutes, was kept for analysis. Samples were stored at −80 °C and shipped in dry ice to the University of Colorado Anschutz Medical Campus for analysis.
Alcohol Clearance Quantitation in the HXB/BXH Recombinant Inbred Rat Panel
Blood alcohol levels were determined using a Varian 3800 gas chromatograph (Varian, Palo Alto, CA, USA) equipped with an Agilent Technologies DB-ALC1 column (Agilent Technologies, Santa Clara, CA, USA; part number 123-9134) and a Varian 8200 AutoSampler (Varian, Palo Alto, CA, USA). Prior to gas chromatographic analysis, the thawed blood samples were centrifuged at 13000 g and 4 °C for three minutes, and 10 μL of 100 mM 2-propanol internal standard was added to 80 μL aliquots of the supernatant. With each batch of samples assayed, standard curves were generated using 0, 5, 10, 20, 40, 60, 80, and 100 mM alcohol standards that were prepared in blood taken from control rats in a manner identical to the experimental samples. The alcohol concentration/time curves for each rat were fit to a one-compartment pharmacokinetic (PK) model with first-order absorption and first-order elimination by employing the nonlinear Levenberg-Marquardt fitting algorithm using the minpack.lm package (v. 1.2-1) (Elzhov et al., 2012) in R. The first-order clearance values represent the alcohol clearance phenotype (see Supplementary Methods in Appendix S1 for a detailed description of alcohol quantitation). We used intraperitoneal injection of ethanol to avoid the confounding of metabolism of ethanol by the high KM stomach ADH system (Vaglenova et al., 2003). (See Supplementary Methods in Appendix S1 for a detailed description of ethanol delivery method rationale.) The pharmacokinetics of some strains of the HXB/BXH RI panel resembled pseudo zero-order alcohol elimination kinetics (e.g., BXH10 in Fig. 1). We, however, used a uniform phenotype derived using first-order kinetic parameters to enable comparison of alcohol clearance across strains. Both statistical (Table S1) and visual comparison of zero-order (straight line) and first-order (exponential decay) kinetic models indicated little difference in fits between the two models for each animal.
Figure 1. Representative alcohol and acetate profiles in blood after 2 g/kg alcohol administration.

Concentrations in millimolar for individual animals are represented by circles at each time point for (a) blood alcohol concentrations and (b) blood acetate concentrations. The lines represent strain-specific one-compartment pharmacokinetic models with first-order absorption and elimination (a) generated from the mean of the parameter estimates from the individual rats and the lines connecting the strain mean concentrations of acetate at each time point (b).
Acetate Area under the Curve (AUC) Quantitation in the HXB/BXH Recombinant Inbred Rat Panel
Blood acetate levels were determined using the Sigma-Aldrich acetate colorimetric assay kit (Sigma-Aldrich, St. Louis, MO, USA; catalog number MAK086) using the manufacturer’s recommended protocol. To prepare the blood samples for analysis, 48 μL of 0.5 M potassium hydroxide was added to 60 μL aliquots of the blood samples. Samples were centrifuged at 13000 g and 4 °C for one minute, and 35.7 μL of the supernatant was combined with 14.3 μL of assay buffer. For the standard curves, blood from control rats was processed in an identical manner to the experimental samples, and standard curves for concentrations of 0.00, 0.25, 0.50, 1.00, 1.50, and 2.00 mM were constructed. Absorbance values were measured using the BioTek Synergy HT plate reader (BioTek, Winooski, VT, USA). Acetate AUC from 0 to 400 minutes was calculated from the acetate concentration-time curves for each individual rat by employing the linear trapezoidal rule using the PK package (v. 1.3-3) (Jaki and Wolfsegger, 2010) and these values were used as the acetate AUC phenotype (see Supplementary Methods in Appendix S1 for a detailed description of acetate quantitation).
Whole Liver RNA Sequencing for the HXB/BXH Recombinant Inbred Rat Panel
Liver tissue was stored in liquid nitrogen and shipped to the University of Colorado Anschutz Medical Campus for RNA extraction and cDNA library preparation. Total RNA extracted from livers obtained from 49 male rats (90 days old) was sequenced. Of the 49 liver samples, 44 were from the HXB/BXH RI panel (1-2 livers/strain) and five samples were from the progenitor strains (BN-Lx/Cub and SHR/OlaIpcv). The rats from these RI strains are genetically identical to the rats used for phenotyping but the rats used for RNA-Seq analysis were not exposed to alcohol. These animals were housed in identical environments as the rats that received ethanol.
Livers were processed in three batches and included seven technical replicates (56 libraries). Total RNA (>200 bases) was extracted and cleaned using the RNeasy Plus Universal Midi Kit and RNeasy Mini Kit, respectively (Qiagen, Valencia, CA, USA). Four μL of a 1:100 dilution of either ERCC Spike-In Mix 1 or Mix 2 (ThermoFisher Scientific, Wilmington, DE, USA) were added to each extracted RNA sample. Construction of sequencing libraries was done using the Illumina TruSeq Stranded RNA Sample Preparation kit (Illumina, San Diego, CA, USA) in accordance with the manufacturer’s protocol. Part of this process included ribosomal RNA depletion via the Ribo-Zero rRNA reduction chemistry. An Agilent Technologies Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) was utilized to assess sequencing library quality, and samples were sequenced (2×100 paired-end (PE) reads, three to four samples multiplexed per lane) on an Illumina HiSeq2500 (Illumina, San Diego, CA, USA) in High Output mode.
Quantitation of Whole Liver RNA for the HXB/BXH Recombinant Inbred Rat Panel
Raw reads were trimmed to remove adapter sequences as well as low quality bases using Trim Galore! (v. 0.4.0). Low quality bases were determined using the default parameters. The trimmed reads were initially aligned to ribosomal RNA (rRNA) from the RepeatMasker database (Smit et al., 1996) accessed through the UCSC Genome Browser (Kent et al., 2002b, UCSC Genome Browser. https://genome.ucsc.edu/. Accessed 15 April 2016) using TopHat (v. 2.0.14) (Trapnell et al., 2009). PE reads which did not map to rRNA were quantified into Ensembl gene-level abundance estimates (Ensembl Release 81) (Cunningham et al., 2015) using the RSEM (RNA-Seq by Expectation Maximization package) (v. 1.2.21) (Li and Dewey, 2011) and strain-specific Ensembl transcriptomes generated in our laboratories. (See Supplementary Methods in Appendix S1 for a detailed description of Trim Galore!, TopHat, and RSEM settings). Initially, strain-specific genomes for the RI strains were constructed from the Rat Genome Sequencing Consortium (RGSC) Rnor_6.0 version of the rat genome (Gibbs et al., 2004) by imputing single nucleotide polymorphism (SNP) information for each strain based on their STAR Consortium genotypes (Star Consortium et al., 2008) and DNA sequencing (DNA-Seq) data from male rats of the progenitor strains [86]. These data are publicly available on the PhenoGen website (Saba et al., 2015, PhenoGen. http://phenogen.ucdenver.edu. Accessed 04 Dec. 2017). Strain-specific transcriptomes were generated from these imputed genomes and the Ensembl database (Ensembl Release 81) (Cunningham et al., 2015).
To prepare the expression estimates for analysis, genes with an average RSEM-estimated read count of less than one across the 56 samples were considered undetectable above background and not used in the analysis. Quantitated samples were initially examined for quality using hierarchical clustering. The RUV (Removal of Unwanted Variance) algorithm (Risso et al., 2014) based on empirically-derived control genes was used to eliminate batch effects and other technical factors contributing to variance. Empirically-derived control genes were identified as the 5,000 least significant genes in a negative binomial generalized linear model (McCarthy et al., 2012) with RI strain as the covariate using the edgeR package (v. 3.14.0) (Robinson et al., 2010) in R. The RUVg function from the RUVSeq package (v. 1.6.2) (Risso et al., 2014) was used to derive three normalization factors. The number of normalization factors used was determined by the clustering of technical replicates. Normalized counts were used in subsequent analyses.
After normalization, samples were reduced to only include RI animals (not the progenitors) and only one technical replicate per biological sample, i.e., multiple RNA-Seq libraries from the same animal (the technical replicate with the highest normalized read count was retained to ensure the most powerful library for that animal). Because of the reduction in the number of libraries included and the normalization of read counts, genes were again filtered based on the criteria of an average normalized read count greater than one. The normalized expression data were transformed into regularized log (rlog) values using the DESeq2 package (v. 1.12.4) (Love et al., 2014). This function 1) transformed the data to a log2 scale and 2) stabilized the within gene variance to avoid the dependence of the variance on the mean.
Weighted Gene Co-Expression Network Analysis for the HXB/BXH Recombinant Inbred Rat Panel
The WGCNA package (v. 1.51) (Langfelder and Horvath, 2008) was used to build co-expression modules from the rlog-transformed RNA expression estimates for the HXB/BXH RI panel collected for this study. For HXB/BXH RI strains with multiple biological replicates, i.e., measurements on independent animals but of the same strain, mean strain values were used to calculate connectivity. Two settings of the network-building function for WGCNA were changed from their default settings: the minimum module size parameter (set to five instead of 30) and the deepSplit parameter (set to four instead of two). Both of these alterations promote the identification of smaller modules, which was desirable because it facilitates subsequent independent expert analysis of how genes contained within any identified candidate modules are interrelated for ascertaining biological insights. By allowing smaller modules, we are retaining genes and their respective co-expression modules that would have otherwise not been placed into a module at all. The absolute value of the Pearson correlation coefficient was used to determine the adjacency matrix, i.e. an unsigned network. Furthermore, the soft-thresholding index, β, was set to six to approximate a scale-free topology. The value for the soft-thresholding index was determined using the methods and critical values proposed in Zhang and Horvath (2005) (Fig. S1). A module eigengene (first principal component) was used to summarize the gene expression profiles within a module across strains for subsequent analyses (Langfelder and Horvath, 2008).
Molecular Markers for Genetic Mapping
The molecular marker set used for quantitative trait loci (QTL) analyses was derived from the existing publicly available strain-specific genomes created using SNPs genotyped by the STAR Consortium (2008). The SNP positions within the marker set that was publically released were based on the RN3.4 version of the rat genome. To convert the SNP positions to the RN6 version of the rat genome, we aligned the probes from the SNP arrays used to generate the public STAR Consortium marker set to the RGSC Rnor_6.0 rat genome assembly (Gibbs et al., 2004, Havlak et al., 2004) using the UCSC command line BLAST-like alignment tool (BLAT) (Kent, 2002a), and both the genome and the alignment tool were downloaded from the UCSC Genome Browser (Kent et al., 2002b, UCSC Genome Browser. https://genome.ucsc.edu/. Accessed 15 April 2016). We retained markers for QTL analysis in this study if the following criteria were met: 1) their probe sequence aligned perfectly and uniquely to the genome, 2) their genotypes differed between progenitor strains, 3) neither progenitor strain was heterozygous for the SNP, and 4) less than 5% of the HXB/BXH RI strains were missing or heterozygous for the SNP. In addition, markers with large estimated genetic distance compared to physical distance from adjacent markers (improbable recombination events, flanked by more than 10 cM on each side) and double recombinant markers were removed. Genetic distances were estimated using the R/qtl package (v. 1.40-8) (Broman et al., 2003). Prior to QTL analyses, the marker set was reduced to unique strain distribution patterns, i.e. multiple adjacent markers with the same genotype pattern across strains were represented by a single marker, in order to reduce the computational burden. The RN6 version of the STAR Consortium SNP map is available on the PhenoGen website.
Quantitative Trait Loci Analysis (QTL)
Marker regression was used to calculate module eigengene QTLs using strains of the HXB/BXH RI panel that had both genotype information from the STAR Consortium and RNA expression/eigengene estimates from the PhenoGen database. The pQTLs for alcohol clearance and acetate AUC in the HXB/BXH RI panel were also determined using marker regression with strain means. Empirical genome-wide p-values were calculated using 1000 permutations (Churchill and Doerge, 1994). For the two phenotypes, both significant (p < 0.05) and suggestive (p < 0.63) pQTLs were considered. The definitions of significant and suggestive p-values were taken from Lander and Kruglyak (1995) and have been adopted by others (The Complex Trait Consortium, 2003). The 95% Bayesian credible interval of each meQTL and pQTL was calculated using the methods detailed in Sen and Churchill (2001). All QTL analyses and graphics were generated using the R/qtl package (v. 1.40-8) (Broman et al., 2003).
Identification of Candidate Modules for Alcohol Clearance and Acetate Area under the Curve in the HXB/BXH Recombinant Inbred Rat Panel
The first step for identifying candidate co-expression modules for alcohol clearance and acetate AUC was to evaluate their association with each phenotype. Strain mean values of alcohol clearance and acetate AUC were used for correlation analysis. A Pearson correlation coefficient between the module eigengene and the phenotype across the strains of the HXB/BXH RI panel was estimated for each module and phenotype. Only modules significantly associated (nominal p-value < 0.01) with at least one of the two phenotypes were considered in subsequent steps.
For modules correlated with alcohol clearance and/or acetate AUC, additional criteria were imposed in order to be considered a candidate module for either phenotype. Candidate modules were required to have a genome-wide significant (p-value < 0.01) meQTL, and the module meQTL must fall within the 95% Bayesian credible interval of a significant or suggestive pQTL for the given phenotype.
Results
Alcohol Clearance and Acetate Area Under the Curve in the HXB/BXH Recombinant Inbred Rat Panel
Representative blood alcohol and acetate profiles (Fig. 1) demonstrate the diversity of the blood alcohol and acetate profiles across the HXB/BXH RI panel. Overall, 82 rats and 691 measurements were used for alcohol clearance calculations after quality control. (See Supplementary Results in Appendix S1 for detailed results from quality control imposed on alcohol and acetate measures). Average alcohol clearance varied approximately 10-fold among strains in the recombinant inbred (RI) rat panel (0.8 to 7.5 mL/min/kg; Fig. 2A). Furthermore, the panel exhibited high broad-sense heritability (81%) for this phenotype, estimated as the coefficient of determination from a one-way ANOVA. After quality control, 89 rats and 888 measurements were used for acetate AUC calculations. Peak circulating blood acetate levels varied from 0.20 to 2.74 mM (interquartile range: 0.84 to 1.79 mM) among strains, and acetate (AUC) varied from 82 to 617 mM*min and displayed a high broad-sense heritability (66%; Fig. 2B). Using strain means, alcohol clearance and acetate AUC were positively correlated (Pearson’s correlation coefficient = 0.43, 95% CI = 0.09 to 0.69, p-value = 0.016; Fig. 2C).
Figure 2. Distribution of alcohol clearance and acetate AUC across the HXB/BXH recombinant inbred rat panel.

The bars represent mean values of the biological replicates within the strain denoted on the x-axis for (a) first-order alcohol clearance (blue) and (b) acetate AUC (red). The error bars represent plus/minus standard error of the mean. If error bars are missing, only one biological replicate was available for the given strain. Alcohol clearance estimate and acetate AUCs were determined in each rat separately. The broad sense heritability of each phenotype was estimated as the R-squared value from a one-way ANOVA using strain as the predictor. Mean values for the two phenotypes were plotted against each other by strain to examine the (c) association between alcohol clearance and acetate AUC. Each point is labeled by its respective strain.
Whole Liver RNA Sequencing for the HXB/BXH Recombinant Inbred Rat Panel
RNA-Seq was performed on RNA extracted from livers of naïve (non-alcohol exposed) rats in three batches (56 libraries including technical replicates). Over three billion total paired-end (PE) reads were generated from these samples. This amounts to approximately 60 million PE reads per sample. After trimming and removal of reads that aligned to ribosomal RNA (rRNA), the average number of PE reads per sample was 59.5 million and 58.4 million, respectively. A detailed summary of the RNA-Seq results by sample is in Table S2.
Quantitation of Whole Liver RNA for the HXB/BXH Recombinant Inbred Rat Panel
We eliminated Ensembl genes with an average estimated RSEM count of less than one across the 56 rat liver RNA-Seq libraries. This resulted in a reduction from 32285 to 16093 Ensembl genes. Based on visual inspection of the Pearson correlation between samples using log2(RSEM counts + 1) transformed data, four samples were identified as outliers and removed (Fig. S2). The dendrograms of the samples (including information on technical replicates and batches) before and after implementation of the RUV algorithm provided evidence that unwanted variance, such as that introduced by batch effects, was markedly reduced. Removal of data from progenitor strains and technical replicates (used for normalization) left data from 41 HXB/BXH RI samples (1-2 rats/strain; 29 strains), and the further removal of genes with normalized counts less than this final number of samples left 15984 Ensembl gene identified in liver. These data were utilized in WGCNA.
Weighted Gene Co-Expression Network Analysis for the HXB/BXH Recombinant Inbred Rat Panel
For strains in which RNA-Seq data were obtained (29 HXB/BXH RI strains), the expression estimates were subjected to WGCNA to identify co-expression modules. A total of 658 modules were identified (median module size = 8 genes; Fig. S3) along with 205 genes that could not be assigned to a module. The module eigengenes captured much of the within-module variability in expression across strains (interquartile range: 59% to 67%).
Quantitative Trait Loci Analyses
A total of 20283 SNPs were originally contained in the STAR dataset (2008). After processing (see Supplementary Results in Appendix S1 for detailed results from processing), we identified 1529 unique strain distribution patterns, i.e. haplotype blocks, for the 32 HXB/BXH RI strains genotyped by the STAR Consortium. Of 32 HXB/BXH RI strains with genotype information, 29 strains had expression/eigengene estimates and were used to calculate meQTLs, and 30 strains had alcohol clearance and acetate AUC data and were used for pQTL analysis. The HXB21 RI strain had alcohol clearance and acetate AUC data and was therefore used in pQTL analysis, but the RNA-Seq data were removed as outliers and therefore were not used in meQTL analysis. The pQTL analysis using 1,000 permutations identified one significant (genome-wide p-value < 0.05) and two suggestive (genome-wide p-value < 0.63) pQTLs for alcohol clearance (Fig. 3A) and four suggestive pQTLs for acetate AUC (Fig. 3B).
Figure 3. Quantitative trait loci for alcohol clearance and acetate AUC in the HXB/BXH recombinant inbred panel.

Strain means were used in a marker regression to determine phenotypic QTL for (a) alcohol clearance and (b) acetate AUC. The red lines represent the logarithm of odds (LOD) score threshold for a significant QTL (genome-wide p-value = 0.05), and the blue lines represent the LOD threshold for a suggestive QTL (genome-wide p-value = 0.63). Significant and suggestive QTL are labeled with their location, 95% Bayesian credible interval, LOD score, and genome-wide p-value.
Identification of Candidate Modules for Alcohol Clearance and Acetate Area under the Curve in the HXB/BXH Recombinant Inbred Rat Panel
RNA expression data from alcohol-naïve rats were used to identify the transcriptional predisposing factors for alcohol clearance and circulating acetate levels after administration of ethanol. Module eigengenes were correlated with strain mean values of alcohol clearance and acetate AUC separately. Ten modules were significantly (nominal p-value < 0.01) correlated with alcohol clearance, and ten modules were significantly associated with acetate AUC; moreover, three modules were correlated with both phenotypes (Table S3). The same marker set used for pQTL analyses was used to identify the meQTL with the greatest logarithm of odds (LOD) score for each module that had a significant correlation with either alcohol clearance or acetate AUC. Of these modules, one module associated with alcohol clearance had a significant (genome-wide p-value < 0.01) module eigengene QTL. The examination of overlap between the 95% Bayesian credible intervals of the alcohol tolerance pQTL and the location of the peak LOD score for the module eigengene QTL (Fig. 4) demonstrated that only this candidate module (orange3) met all the criteria to be identified as a candidate module influencing alcohol clearance. This module was also identified as a candidate module for acetate AUC. The genes comprising the orange3 module are listed in Table 1, and the connectivity between genes of the candidate module is visualized in Fig. 5.
Figure 4. Chromosome 2 QTLs for alcohol clearance, acetate AUC, and the orange3 module eigengene.
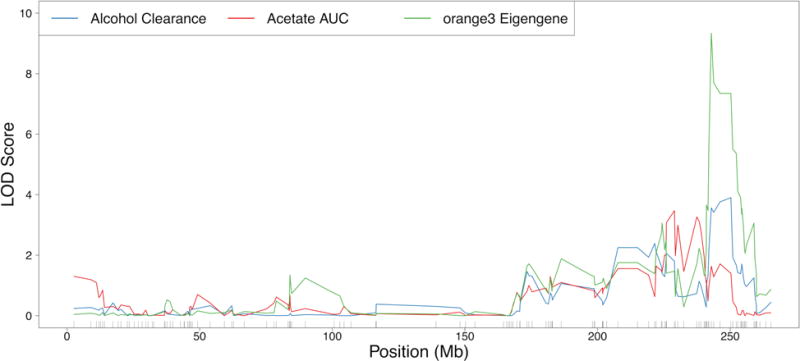
Strain means were used in a marker regression to determine phenotypic QTLs (pQTLs) for alcohol clearance (blue) and acetate AUC (red), and the orange3 module eigengenes were used in marker regression to elucidate module eigengene QTLs (meQTLs, shown in green). The maximum meQTL for the orange3 module was significant (genome-wide p-value < 0.01) and overlapped a significant (genome-wide p-value < 0.05) pQTL for alcohol clearance and a suggestive (genome-wide p-value < 0.63) pQTL for acetate AUC.
Table 1. Genes comprising the orange3 candidate module for both alcohol clearance and acetate AUC.
The candidate module was identified based on eigengene association (nominal p-value < 0.01) with the phenotypes across the HXB/BXH panel and a maximum module eigengene quantitative trait locus that was both significant (genome-wide p-value < 0.01) and overlapped significant (genome-wide p-value < 0.05) or suggestive (genome-wide p-value < 0.63) phenotypic quantitative trait loci for the phenotypes in the HXB/BXH RI rat panel after 2 g/kg alcohol administration. Genes are ordered by intra-modular connectivity.
| Associated Gene Name | Chromosome | Gene Starting Position (Mb) | Type | Description | Intra-modular Connectivity |
|---|---|---|---|---|---|
| Adh4 | 2 | 243.70 | Protein coding | Alcohol dehydrogenase 4 (class II) | 1.18 |
| Adh1 | 2 | 243.55 | Protein coding | Alcohol dehydrogenase 1 (class I) | 1.17 |
| Hs2st1 | 2 | 250.47 | Protein coding | Heparan sulfate 2-O-sulfotransferase 1 | 1.14 |
| Arl16 | 10 | 109.64 | Protein coding | ADP-ribosylation factor-like 16 | 1.13 |
| Gbp5 | 2 | 248.18 | Protein coding | Guanylate binding protein 5 | 1.11 |
| Camk2n1 | 5 | 156.88 | Protein coding | Calcium/calmodulin-dependent protein kinase II inhibitor 1 | 1.11 |
| LOC685067 | 2 | 248.22 | Protein coding | Similar to guanylate binding protein family, member 6 | 1.10 |
| Tmem79 | 2 | 187.70 | Protein coding | Transmembrane protein 79 | 1.08 |
| Zfp143 | 1 | 174.70 | Protein coding | Zinc finger protein 143 | 1.07 |
| Piwil2 | 15 | 52.04 | Protein coding | Piwi-like RNA-mediated gene silencing 2 | 1.05 |
Figure 5. Connectivity within the candidate co-expression module for both alcohol clearance and acetate AUC.
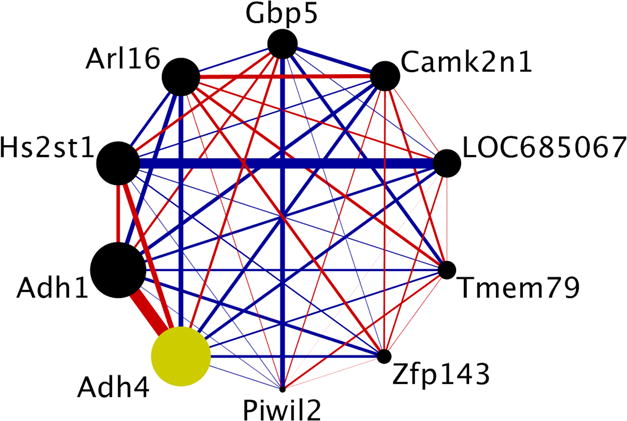
Each circle represents a gene from the co-expression module. The size of each circle is weighted based on its intra-modular connectivity (not to scale), and the thickness of each edge is weighted based on the magnitude of the connectivity between the two genes (not to scale). The edge colors indicate the direction of the connectivity (red = positive, blue = negative). The hub gene, defined here as the single gene with the largest intra-modular connectivity, is colored in yellow (Adh4 = alcohol dehydrogenase 4), and its expression is positively associated with both alcohol clearance and acetate AUC. The figure was generated using Cytoscape (v. 3.4.0) (Shannon et al., 2003).
The module eigengene for orange3 explained 56% of the genetic variance in alcohol clearance and 32% of the genetic variance in acetate AUC across the HXB/BXH RI rat panel, estimated as the coefficient of determination from a linear model using the module eigengene as a predictor of each phenotype. The hub gene, i.e. the gene with the highest intra-modular connectivity within the orange3 candidate module, was alcohol dehydrogenase 4 (Adh4).
Characterization of Alcohol Dehydrogenase Genes in the orange3 Candidate Module
Two alcohol dehydrogenase Ensembl genes, alcohol dehydrogenase 6 (Adh6) and Adh4, were initially identified in the orange3 candidate module. We further examined these genes at the transcript-level to verify their identities. The Adh6 gene expression estimate was found to be derived from pooled estimates of three Ensembl transcripts. Using the University of California – Santa Cruz (UCSC) Genome Browser (Kent et al., 2002b, UCSC Genome Browser. https://genome.ucsc.edu/. Accessed 15 April 2016), we found that the RefSeq database annotated two of these transcripts as separate genes: namely Adh6 and the class I alcohol dehydrogenase 1 (Adh1). The remaining transcript was unannotated in RefSeq, and closer inspection in Ensembl (Ensembl Release 88) (Aken et al., 2016) revealed that this transcript represented a fusion of Adh1 and Adh6. To disentangle the two Adh genes, we first examined the pile-up of the total RNA-Seq reads from the livers of the two progenitor strains. The vast majority of reads aligned to the RefSeq Adh1 gene, with very few reads aligning to Adh6, and there was no evidence for expression of the fusion gene (Fig. S4A). To verify that the variation in expression levels of the original gene-level estimate mimic variation in Adh1, pairwise Pearson’s correlation analysis was performed between the Ensembl gene-level expression estimate, Ensembl transcript-level expression estimates of the three transcripts, and the phenotypes, using strain mean values. Expression estimates of the transcripts were calculated in an identical manner as the gene-level estimate, i.e., rlog-transformed batch-corrected RNA expression values. The expression of the Ensembl transcript corresponding to the RefSeq Adh1 gene was most strongly correlated with the overall Ensembl Adh6 gene expression (Pearson’s r = 0.95) and closely matched its correlation with the phenotypes (Table S4). Moreover, the Ensembl database used for our initial annotation (Ensembl Release 81) (Cunningham et al., 2015) did not annotate any transcripts as Adh1, in spite of the fact that RNA from Adh1 is known to be present in rat liver (Hoog et al., 2001). Therefore, we concluded that the gene-level expression estimates originally annotated as Adh6 in fact most likely represented Adh1 and changed the annotation throughout this manuscript accordingly.
Likewise, Adh4 included pooled estimates from three Ensembl transcripts. Again, two of the Ensembl transcripts were annotated by RefSeq as separate genes, Adh4 and class III alcohol dehydrogenase 5 (Adh5), and the third represented a fusion of these genes that was unannotated in other databases. In this case, the expression of the Ensembl transcript corresponding to the RefSeq Adh4 gene was most closely correlated with the overall Ensembl Adh4 gene-level expression (Pearson’s r = 0.97) and most closely resembled its correlation with the phenotypes (Table S4). While the liver total RNA-Seq reads from the progenitor strains mapped to both the Adh4 and Adh5 RefSeq genes, indicating that both were expressed in the liver (Fig. S4B), the Ensembl transcript/RefSeq Adh4 gene demonstrated greater variation in expression across strains. Taken together, we surmised Adh4 was indeed the gene represented in the orange3 candidate module, and the original nomenclature was retained. Furthermore, BLAST analysis (Zhang et al., 2000) revealed that the Ensembl transcript/RefSeq Adh4 that we identified shared > 99% sequence similarity with the experimentally cloned and sequenced class II rat Adh4 gene (Hoog and Ostberg, 2011), thereby supporting the identity of the transcript that we sequenced as the class II alcohol dehydrogenase gene product in rat.
Further Review of Alcohol Dehydrogenase Genes, Aldehyde Dehydrogenase Genes, and Other Genes That Can Contribute to Alcohol Metabolism in the HXB/BXH RI Panel
All Ensembl annotated alcohol dehydrogenase genes and aldehyde dehydrogenase genes with expression estimates, as well as the genes encoding catalase (Cat) and Cytochrome P450 2E1 (Cyp2e1), were examined to ascertain their expression and determine the correlation of their expression estimates with alcohol clearance and acetate AUC in the HXB/BXH RI panel (Table S5). Using pairwise Pearson correlation analysis on strain mean values, significant (nominal p-value < 0.05) associations were only found between the following: expression of Adh1 and alcohol clearance (Pearson’s r = 0.76, p < 0.001), expression of Adh1 and acetate AUC (Pearson’s r = 0.57, p-value = 0.0012), expression of Adh4 and alcohol clearance (Pearson’s r = 0.64, p-value = 0.0002), and expression of Adh4 and acetate AUC (Pearson’s r = 0.52, p-value = 0.0037). However, we noted that the correlation between expression of Aldh1a1 (aldehyde dehydrogenase 1 family, member A1) and acetate AUC was marginally significant (Pearson’s r = 0.36, p-value = 0.052). If we accept the premise that expression levels of Aldh1a1 are contributing to the variation of acetate AUC in the panel, then Aldh1a1 transcript levels explain approximately 13% of the variance in acetate AUC.
Discussion
The systems genetics approach which utilizes quantitative genetics analysis to integrate physiological, behavioral, and transcriptomic information has been used to uncover a number of genetic factors for predisposition to cardiovascular, metabolic, and certain behavioral traits (Langfelder et al., 2012, Hasin et al., 2017). We have adopted this approach and integrated several filters (Fig. S5) to focus attention on the role of co-expression modules, and we have required conditions that have to be met to categorize a module as a candidate for influencing the quantitative character of a chosen trait. To diminish false positives, a co-expression module had to satisfy three conditions to be considered a candidate: 1) the module eigengene had to be significantly correlated with the quantitative phenotype measured across strains, 2) the module eigengene had to have a statistically significant QTL, and 3) the module eigengene QTL had to reside within the 95% Bayesian credible interval of a significant or suggestive physiologic/behavioral QTL. In the current work we chose to apply our approach to analysis of two phenotypes related to alcohol (ethanol) metabolism: 1) alcohol clearance rate and 2) the measure of circulating acetate levels over time after alcohol administration (i.e., the “area under the curve” for acetate). With regard to the alcohol clearance phenotype, our hypothesis was that if our approach was viable, the identified module would contain components, such as alcohol dehydrogenases, which are accepted determinants of the rate of alcohol metabolism in mammals. For the phenotype of acetate “area under the curve”, the approach was being used as hypothesis-generating rather than a hypothesis testing entity.
Candidate Module for Alcohol Clearance – orange3
Of the 658 co-expression modules built from the liver “total” RNA-Seq data across naïve rats of the HXB/BXH RI panel, only one module satisfied all criteria as a candidate module for alcohol clearance. The orange3 candidate module contained two alcohol dehydrogenase transcripts, Adh1 and Adh4, which produce the alcohol dehydrogenase enzymes class I Adh1 and class II Adh4, respectively. Adh1 is a high affinity, i.e., low Km, enzyme for alcohol (~ 1.4 mM) that is mainly expressed in liver, where it accounts for the majority of alcohol elimination in rats (Julia et al., 1987, Boleda et al., 1989). Adh4 in rat is analogous to the human variant (Julia et al., 1988) and is also expressed in the liver (Estonius et al., 1993, Svensson et al., 1999). While the Km value of rat Adh4 may be greater than that of its human counterpart (Svensson et al., 1999), similar to human ADH4, the rat enzyme most likely contributes to the metabolism of alcohol in the liver at higher concentrations of alcohol (Zakhari, 2006). Two other liver alcohol dehydrogenase genes in the rat have been reported in the literature – class III alcohol dehydrogenase 5 (Adh5) and class IV alcohol dehydrogenase 7 (Adh7) (Plapp et al., 2015). The Rnor_6.0 version of the rat genome lacked any Ensembl annotation for Adh5; however, RefSeq annotation indicates that one of the Ensembl transcripts quantitated in our RNA-Seq data may actually represent Adh5. Indeed, research has demonstrated that Adh5 is expressed in the rat liver (Julia et al., 1988), and the pile-up of liver total RNA-Seq reads from the progenitor strains indicates significant levels of Adh5 expression. Nevertheless, the gene product of Adh5 is believed to have no detectable ethanol metabolizing activity at concentrations reached in our studies (Julia et al., 1987, Julia et al., 1988, Plapp et al., 2015). An additional alcohol dehydrogenase gene, Adhfe1, existed in Ensembl annotation but is known for metabolizing 4-hydroxybutyrate in mammals rather than alcohol (Kardon et al., 2006). Finally, expression of both human and rat ADH7/Adh7 has been established as exclusive to the stomach (Pares et al., 1992). Our findings corroborated this view, as we found little Adh7 expression in the liver (Fig. S6). Overall, we were satisfied that our unsupervised statistically-based systems biology approach could clearly reproduce an accepted fact about the importance of Adh1 and Adh4 in the metabolism of alcohol when alcohol is present at levels attained in our studies.
The contradictory information regarding Ensembl and RefSeq annotations (see Results) with regard to Adh1 and Adh4, however, highlights both the need to carefully examine the results obtained from high throughput RNA-Seq analyses, and the intrinsic advantages of next-generation sequencing technologies like RNA-Seq over methods such as microarrays. Namely, RNA-Seq allows one to examine post-hoc where reads aligned to the genome, and accordingly make annotation adjustments as necessary. Indeed, updated Ensembl versions, for example Ensembl Release 88 (Aken et al., 2016) and newer, changed the Adh6 gene annotation in the Rnor_6.0 rat genome to Adh1 in agreement with the annotation used throughout this manuscript.
Characterization of Other Genes in the orange3 Module and Common Genetic Pathways
The added benefit of a systems biology approach is that it provides biologic context for the statistically-derived relationships between transcripts contained in a module. Gene products composing the orange3 candidate module are listed in Table 1. While products of Adh4 and Adh1 are well known for their ethanol metabolizing function, alcohol dehydrogenases also metabolize a wide variety of other substances, such as longer chain aliphatic alcohols (David et al., 1976, Ehrig et al., 1988), omega-hydroxy-fatty acids (Hoog and Ostberg, 2011, Boleda et al., 1993), hydroxysteroids (Hoog and Ostberg, 2011), and lipid peroxidation products (Boleda et al., 1993, Sellin et al., 1991, Hartley et al., 1995). Another substrate for ADH enzymes is all-trans-retinol which is the alcohol form of vitamin A (Kumar et al., 2012). The active metabolite of vitamin A is retinoic acid and the initial step in the conversion of the retinol to retinoic acid is catalyzed by Adh1 as well as members of the retinol dehydrogenase families (Kumar et al., 2012). The importance of Adh1 in this metabolic step is demonstrated by the knock-out of this enzyme, which results in accumulation of retinol in adult mice, and a greater retinol toxicity in the adult tissues (Kumar et al., 2012). The role of Adh1 in retinol metabolism provides one of the links to explain the association of Adh and the other gene products in the orange3 module. Retinoic acid plays a number of physiological roles through binding to cellular retinoic acid receptors (RARs) that control transcription (Shiota and Kanki, 2013). With regard to the components of the orange3 module, the induction of the retinoic-acid inducible gene I (RIG-I) is of interest (Matsumiya and Stafforini, 2010). RIG-I is a helicase which functions to destroy a number of RNA viruses that may enter the cell. The RIG-I pathway is tightly regulated to maximize antiviral immunity while minimizing immune-related pathology. The product of the orange3 module member ADP-ribosylation factor-like 16 (Arl16) is a protein that interacts with RIG-I and inhibits its activity. Arl16 is part of the extended ADP ribosylation factor (ARF) family of GTPases, and although the ADP-ribosylation factor-like (ARL) proteins have actions beyond those exhibited by the ARF GTPases, they also participate in the regulation of secretion, phagocytosis, endocytosis and signal transduction characteristic of the ARF GTPases (Burd et al., 2004). RIG-I activation also leads to the production of interferon (IFN), which in turn is the major inducer of transcription of guanylate-binding proteins (GBPs). The guanylate binding protein 5 (Gbp5) gene is a member of the orange3 module, and synthesis of its protein (Gbp5) is responsive to IFN-γ (Britzen-Laurent et al., 2010). Gbp5 is a member of the dynamin family of GTPases and recent studies have shown it to be a critical factor in the assembly of inflammasomes. Overexpression of Gbp5 enhances the expression of IFN and other pro-inflammatory factors (Feng et al., 2017), which generates a feed forward immune response and antiviral activity (Hotter et al., 2017). Since LOC685067 is an under-annotated gene described as “similar to guanylate binding protein family, member 6”, the fact that it shares membership with another guanylate binding protein in the orange3 module may add rationale to its description.
Hs2st1 (heparin sulfate 2-O-sulfotransferase) encodes a member of the heparin sulfate biosynthesis pathway (Xu et al., 2007). Heparin sulfate is part of a family of heparin/heparin sulfate glycosaminoglycans that organize at the cell surface to act as recognition and binding sites for chemokines (Schenauer et al., 2007, Monneau et al., 2016), transforming growth factors (Lyon et al., 1997), and viruses (Khanna et al., 2017, Kim et al., 2017). It should be noted that the quantity and location of sulfate groups on the heparin sulfate polysaccharide is a determining factor in the selectivity of the cell surface polyglycan for various ligands (Sasisekharan and Venkataraman, 2000, Kreuger et al., 2006). Acetylation of heparin sulfate is a further modification of the glycosaminoglycans and is important in determining the recognition of various chemokines (Kreuger et al., 2006). We postulate that the production of acetate from ethanol could influence this molecular modification. On the whole, the production and modification of heparin sulfate is an important component of both the capacity of pathogens, particularly viruses, to infect cells, and for the cell to mount an immune response to the pathogen. There also exists an under-investigated interaction between the heparin/heparin-sulfate glycosaminoglycans generated by the actions of the Hs2st1 protein and another member of the orange3 module. Heparin has been shown to inhibit phosphorylation and the generation of autonomous activity of the calcium/calmodulin-dependent protein kinase II (CaMKII) (Mishra-Gorur et al., 2002). The protein product of the module member calcium/calmodulin-dependent protein kinase II inhibitor (Camk2n1) is also an inhibitor of CaMKII. CaMKII is a multipurpose calcium/calmodulin signal transduction enzyme, best known for its role in generating cellular memory (specifically, long-term potentiation) in the hippocampus. In relation to the liver and the orange3 module, CaMKII has an important role in controlling tumor necrosis factor alpha (Tnf-α)-induced expression of CD44 (Mishra et al., 2005). CD44 is a transmembrane glycoprotein expressed in many cell types. A key event in the activation of monocytes and their transformation, cytokine release, and migration to sites of inflammation and tissue injury is the induction of CD44 expression (Mishra et al., 2005). Interestingly, ethanol is known to increase circulating levels of Tnf-α, and the Camk2n1 gene product may be a modifier of this response.
Zfp143 (zinc finger protein 143) encodes a zinc finger transcriptional regulator. Its human counterpart, ZNF143 (zinc finger protein 143), exhibits the interesting property of connecting promoter regions of DNA with distant regulatory elements through looping of chromatin (Bailey et al., 2015), and the protein product of ZNF143 has been hypothesized to influence differentiation and cell identity (Bailey et al., 2015). The Piwil2 (piwi like RNA-mediated gene silencing 2) gene product has previously been shown to be expressed in liver, and is considered to be involved in regeneration of liver after damage (Rizzo et al., 2014). The function of the product of the Tmem79 (transmembrane protein 79) gene is not known with regard to liver, but its inclusion in this module may provide some insights.
The module as a whole provides the impression that in a basal state (without having substantial amount of ethanol in the milieu) the co-expressed genes are functioning as components which contribute to cell interaction with pathogens (possibly viruses), cellular response to pathogens and immune system signals, and components of liver regeneration (if it sustains damage). The alcohol dehydrogenases included in this module may be the enzymes that generate the necessary ligands (e.g., retinoic acid or acetate) critical for the function of the other module components. The association of the alcohol dehydrogenase genes with the other module components may also indicate cross-cell type communication, with retinoic acid production in hepatocytes being utilized for function of other liver cell types (e.g., Kupffer cells and/or infiltrating macrophages). The relationship of the alcohol dehydrogenase-containing orange3 module with immune function may be through promoting what is called the autonomous immune response directed at viral infections. The chronic consumption of alcohol in excess of 50 g/day increases an individual’s vulnerability to the hepatitis C virus (HCV) (Taylor et al., 2016). McCartney et al. (2008) produced evidence that ethanol metabolism, rather than ethanol per se, promotes the replication of HCV and diminishes the antiviral action of interferon α. Our data may contribute to the interpretation of mechanisms by which ethanol metabolism promotes the development of viral hepatitis.
The orange3 module was additionally identified as a candidate module for influencing acetate AUC. This result indicates that the rate of alcohol clearance influences acetate AUC, i.e. systemic blood acetate levels are at least partially determined by the rate at which alcohol is metabolized. Such an observation logically follows what has been reported in the literature; that is, the majority of alcohol is cleared via oxidative metabolism (Ramchandani et al., 2001, Norberg et al., 2003) that generates acetate (Zakhari, 2006).
A somewhat surprising outcome was that, although the alcohol dehydrogenase-containing module contributed to the determining circulating levels of acetate, enzymes essential for conversion of acetaldehyde to acetate (aldehyde dehydrogenases) were not identified through the process of co-expression module analysis as being responsible for circulating levels of acetate. A cogent explanation of this fact would be, that under our experimental conditions, the aldehyde dehydrogenases that catalyze an irreversible production of acetate are not rate-limiting in the transition of ethanol to acetate. On the other hand, not all expressed genes can be assigned to co-expression modules, or even if assigned to a module, the module eigengene values for that module may not generate a statistically significant meQTL. With regard to Aldh1a1, we noted that it was included in an identified module but that the module’s eigengene values did not generate a meQTL which overlapped the pQTL for acetate AUC. On the other hand, an eQTL for the expression levels of Aldh1a1 per se was associated with a SNP located at chr20: 44.71 Mb (p-value = 0.006 via permutation on PhenoGen) and overlapped the pQTL for acetate AUC (chr20: 39.94 Mb, 95% Bayesian credible interval = 36.33-48.67 Mb, LOD score = 3.50). The expression levels of Aldh1a1 did, also, nominally correlate with the acetate AUC values although no correlation was evident with values for ethanol clearance. These results indicate to us that under some circumstances, transcript products may act outside of the context of the module to which they belong in order to carry out metabolic functions not normally part of the repertoire of the module.
The current conceptualization of the metabolism of acetaldehyde produced from ethanol is that it is rapidly metabolized by the protein product of Aldh2 (aldehyde dehydrogenase 2 family), which resides in the mitochondria (Klyosov et al., 1996). There is, however, ample evidence for the involvement of Aldh1a1 when higher levels of acetaldehyde are present. Thus, the contribution of Aldh1a1 to acetate AUC may be primarily evident under conditions of high ethanol clearance rates.
Conclusions
Overall, the unsupervised, statistically-based, systems biology approach that we instituted for analyzing factors influencing ethanol metabolism and resultant acetate levels produced some rewarding results. First, out of 658 modules, our approach identified one module related to the genomic locus determining the rate of ethanol clearance. This liver module contained two alcohol dehydrogenase transcripts that would be fully expected, from ample literature (Julia et al., 1987, Boleda et al., 1989), to be responsible for ethanol oxidation in the rat. The identification of a module with two alcohol dehydrogenases also substantiates the belief that alcohol dehydrogenase isoforms with different KM values for ethanol can contribute to metabolism depending on the blood levels achieved after a particular dose of ethanol (2 g/kg in our work). Our results also pointed to the functional context for inclusion of these alcohol dehydrogenases in a module which, in the rat, under normal conditions rarely, if at all, experiences alcohol concentrations of 40 mM or higher. The same can be said for most humans and this module’s involvement in generation and utilization of retinoic acid is another relevant component of our results.
We would suggest that the protocol we illustrate in Fig. S5, coupled with QTL or GWAS analysis of physiologic, pathologic and behavioral traits in animals, including humans, can bring credence to anticipated results and introduce unexpected but plausible systems genetic explanations of complex traits. Furthermore, since the RI rats utilized in this study represent a renewable genetic resource due to their inbred nature, and we have developed a rich database of their attributes for public use, studies addressing alternative research questions, e.g., the influence of genes and/or modules on different phenotypes or how they predispose response to various environmental factors, can easily be employed in this panel and perhaps extrapolated to humans for valuable insights. The evidence for the possible contribution of Aldh1a1 to acetate AUC, must, however, temper the absolute utility of our approach and indicates that careful inspection of all forms of gene expression data in relationship to a given phenotype is still necessary to reach optimum conclusions. Additionally, it should be noted that our study design included only male rats, and gender differences in alcohol metabolism have been reported (Rachamin et al., 1980).
Supplementary Material
Acknowledgments
The authors are grateful to Alena Musilova for assistance with blood collection, the expert technical assistance with blood alcohol and acetate levels and RNA-Seq provided by Yinni Yu and Seija Tillanen, for the expert computational assistance provided by Spencer Mahaffey, and for access to the computational resources provided by Joseph Lombardo and the National Supercomputing Center & Dedicated Research Network at the University of Nevada, Las Vegas.
Funding: This work was supported by the Czech Science Foundation (14-36804G) to MP. BT, PLH, LMS, LV, and RL derived support from National Institutes of Health (R24AA013162 and P30DA044223).
Footnotes
The authors declare that they have no competing interests.
Authors’ contributions:
BT and MP devised the study design. MP did the alcohol administration, blood draws, and tissue harvesting. BT and PLH supervised the biochemical analysis of alcohol and acetate concentrations. RL, LV, and LS participated in the statistical analyses including pharmacokinetics and systems genetics. RL and BT wrote the manuscript with input from PLH and LS. All authors read and approved the final manuscript.
Contributor Information
Ryan Lusk, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy & Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, CO 80045.
Laura M. Saba, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy & Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, CO 80045.
Lauren A. Vanderlinden, Department of Biostatistics and Informatics, Colorado School of Public Health, University of Colorado Anschutz Medical Campus, Aurora, CO 80045.
Vaclav Zidek, Department of Model Diseases, Institute of Physiology of the Czech Academy of Sciences, Czech Republic.
Jan Silhavy, Department of Model Diseases, Institute of Physiology of the Czech Academy of Sciences, Czech Republic.
Michal Pravenec, Department of Model Diseases, Institute of Physiology of the Czech Academy of Sciences, Czech Republic.
Paula L. Hoffman, Department of Pharmacology School of Medicine, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy & Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, CO 80045.
Boris Tabakoff, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy & Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, 12850 E. Montview Blvd., Aurora, CO 80045, Fax number: 303-724-4050, Telephone number: 303-724-3668.
References
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, Garcia Giron C, Hourlier T, Howe K, Kahari A, Kokocinski F, Martin FJ, Murphy DN, Nag R, Ruffier M, Schuster M, Tang YA, Vogel JH, White S, Zadissa A, Flicek P, Searle SM. The Ensembl gene annotation system. Database (Oxford) 2016;2016 doi: 10.1093/database/baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SD, Zhang X, Desai K, Aid M, Corradin O, Cowper-Sal Lari R, Akhtar-Zaidi B, Scacheri PC, Haibe-Kains B, Lupien M. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat Commun. 2015;2:6186. doi: 10.1038/ncomms7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielavska E, Kren V, Musilova A, Zidek V, Pravenec M. Genome scanning of the HXB/BXH sets of recombinant inbred strains of the rat for quantitative trait loci associated with conditioned taste aversion. Behav Genet. 2002;32:51–56. doi: 10.1023/a:1014407928865. [DOI] [PubMed] [Google Scholar]
- Boleda MD, Julia P, Moreno A, Pares X. Role of extrahepatic alcohol dehydrogenase in rat ethanol metabolism. Arch Biochem Biophys. 1989;274:74–81. doi: 10.1016/0003-9861(89)90416-5. [DOI] [PubMed] [Google Scholar]
- Boleda MD, Saubi N, Farres J, Pares X. Physiological substrates for rat alcohol dehydrogenase classes: aldehydes of lipid peroxidation, omega-hydroxyfatty acids, and retinoids. Arch Biochem Biophys. 1993;307:85–90. doi: 10.1006/abbi.1993.1564. [DOI] [PubMed] [Google Scholar]
- Bosron WF, Ehrig T, Li TK. Genetic factors in alcohol metabolism and alcoholism. Semin Liver Dis. 1993;13:126–135. doi: 10.1055/s-2007-1007344. [DOI] [PubMed] [Google Scholar]
- Britzen-Laurent N, Bauer M, Berton V, Fischer N, Syguda A, Reipschlager S, Naschberger E, Herrmann C, Sturzl M. Intracellular trafficking of guanylate-binding proteins is regulated by heterodimerization in a hierarchical manner. PLoS One. 2010;5:e14246. doi: 10.1371/journal.pone.0014246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Burd CG, Strochlic TI, Setty SR. Arf-like GTPases: not so Arf-like after all. Trends Cell Biol. 2004;14:687–694. doi: 10.1016/j.tcb.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–685. doi: 10.1016/j.cld.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti LH, Jirout M, Breen L, Vanella JJ, Schork NJ, Printz MP. Identification of quantitative trait Loci for anxiety and locomotion phenotypes in rat recombinant inbred strains. Behav Genet. 2004;34:93–103. doi: 10.1023/B:BEGE.0000009479.02183.1f. [DOI] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kahari AK, Keenan S, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Overduin B, Parker A, Patricio M, Perry E, Pignatelli M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Aken BL, Birney E, Harrow J, Kinsella R, Muffato M, Ruffier M, Searle SM, Spudich G, Trevanion SJ, Yates A, Zerbino DR, Flicek P. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David JR, Bocquet C, Arens MF, Fouillet P. Biological role of alcohol dehydrogenase in the tolerance of Drosophila melanogaster to aliphatic alochols: utilization of an ADH-null mutant. Biochem Genet. 1976;14:989–997. doi: 10.1007/BF00485131. [DOI] [PubMed] [Google Scholar]
- DiLeo MV, Strahan GD, den Bakker M, Hoekenga OA. Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS One. 2011;6:e26683. doi: 10.1371/journal.pone.0026683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Foroud T. Genetics and alcoholism. Nat Rev Gastroenterol Hepatol. 2013;10:487–494. doi: 10.1038/nrgastro.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrig T, Bohren KM, Wermuth B, von Wartburg JP. Degradation of aliphatic alcohols by human liver alcohol dehydrogenase: effect of ethanol and pharmacokinetic implications. Alcohol Clin Exp Res. 1988;12:789–794. doi: 10.1111/j.1530-0277.1988.tb01347.x. [DOI] [PubMed] [Google Scholar]
- Elzhov TV, Mullen KM, Spiess AN, Bolker B. R interface to the Levenberg-Marquardt nonlinear least-squares algorithm found in MINPACK, plus support for bounds. 2012 Retrieved from CRAN http://doi.org/10.2172/803290.
- Estonius M, Danielsson O, Karlsson C, Persson H, Jornvall H, Hoog JO. Distribution of alcohol and sorbitol dehydrogenases. Assessment of mRNA species in mammalian tissues. Eur J Biochem. 1993;215:497–503. doi: 10.1111/j.1432-1033.1993.tb18059.x. [DOI] [PubMed] [Google Scholar]
- Feng J, Cao Z, Wang L, Wan Y, Peng N, Wang Q, Chen X, Zhou Y, Zhu Y. Inducible GBP5 Mediates the Antiviral Response via Interferon-Related Pathways during Influenza A Virus Infection. J Innate Immun. 2017;9:419–435. doi: 10.1159/000460294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18:463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, Brozell A, Schadt EE, Drake TA, Lusis AJ, Horvath S. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2:e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan-Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera, Holt RA, Adams MD, Amanatides PG, Baden-Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C, Glodek A, Gu Z, Jennings D, Kraft CL, Nguyen T, Pfannkoch CM, Sitter C, Sutton GG, Venter JC, Woodage T, Smith D, Lee HM, Gustafson E, Cahill P, Kana A, Doucette-Stamm L, Weinstock K, Fechtel K, Weiss RB, Dunn DM, Green ED, Blakesley RW, Bouffard GG, De Jong PJ, Osoegawa K, Zhu B, Marra M, Schein J, Bosdet I, Fjell C, Jones S, Krzywinski M, Mathewson C, Siddiqui A, Wye N, McPherson J, Zhao S, Fraser CM, Shetty J, Shatsman S, Geer K, Chen Y, Abramzon S, Nierman WC, Havlak PH, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Li B, Liu Y, Qin X, Cawley S, Worley KC, Cooney AJ, D’Souza LM, Martin K, Wu JQ, Gonzalez-Garay ML, Jackson AR, Kalafus KJ, McLeod MP, Milosavljevic A, Virk D, Volkov A, Wheeler DA, Zhang Z, Bailey JA, Eichler EE, Tuzun E, Birney E, Mongin E, Ureta-Vidal A, Woodwark C, Zdobnov E, Bork P, Suyama M, Torrents D, Alexandersson M, Trask BJ, Young JM, Huang H, Wang H, Xing H, Daniels S, Gietzen D, Schmidt J, Stevens K, Vitt U, Wingrove J, Camara F, Mar Alba M, Abril JF, Guigo R, Smit A, Dubchak I, Rubin EM, Couronne O, Poliakov A, Hubner N, Ganten D, Goesele C, Hummel O, Kreitler T, Lee YA, Monti J, Schulz H, Zimdahl H, Himmelbauer H, Lehrach H, Jacob HJ, Bromberg S, Gullings-Handley J, Jensen-Seaman MI, Kwitek AE, Lazar J, Pasko D, Tonellato PJ, Twigger S, Ponting CP, Duarte JM, Rice S, Goodstadt L, Beatson SA, Emes RD, Winter EE, Webber C, Brandt P, Nyakatura G, Adetobi M, Chiaromonte F, Elnitski L, Eswara P, Hardison RC, Hou M, Kolbe D, Makova K, Miller W, Nekrutenko A, Riemer C, Schwartz S, Taylor J, Yang S, Zhang Y, Lindpaintner K, Andrews TD, Caccamo M, Clamp M, Clarke L, Curwen V, Durbin R, Eyras E, Searle SM, Cooper GM, Batzoglou S, Brudno M, Sidow A, Stone EA, Venter JC, Payseur BA, Bourque G, Lopez-Otin C, Puente XS, Chakrabarti K, Chatterji S, Dewey C, Pachter L, Bray N, Yap VB, Caspi A, Tesler G, Pevzner PA, Haussler D, Roskin KM, Baertsch R, Clawson H, Furey TS, Hinrichs AS, Karolchik D, Kent WJ, Rosenbloom KR, Trumbower H, Weirauch M, Cooper DN, Stenson PD, Ma B, Brent M, Arumugam M, Shteynberg D, Copley RR, Taylor MS, Riethman H, Mudunuri U, Peterson J, Guyer M, Felsenfeld A, Old S, Mockrin S, Collins F, Rat Genome Sequencing Project C Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Grisel JE, Metten P, Wenger CD, Merrill CM, Crabbe JC. Mapping of quantitative trait loci underlying ethanol metabolism in BXD recombinant inbred mouse strains. Alcohol Clin Exp Res. 2002;26:610–616. [PubMed] [Google Scholar]
- Guindalini C, Scivoletto S, Ferreira RG, Breen G, Zilberman M, Peluso MA, Zatz M. Association of genetic variants in alcohol dehydrogenase 4 with alcohol dependence in Brazilian patients. Am J Psychiatry. 2005;162:1005–1007. doi: 10.1176/appi.ajp.162.5.1005. [DOI] [PubMed] [Google Scholar]
- GWAS Catalog. https://www.ebi.ac.uk/gwas/. Accessed 04 Dec. 2017.
- Harrall KK, Kechris KJ, Tabakoff B, Hoffman PL, Hines LM, Tsukamoto H, Pravenec M, Printz M, Saba LM. Uncovering the liver’s role in immunity through RNA co-expression networks. Mamm Genome. 2016;27:469–484. doi: 10.1007/s00335-016-9656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley DP, Ruth JA, Petersen DR. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch Biochem Biophys. 1995;316:197–205. doi: 10.1006/abbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:83. doi: 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havlak P, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Weinstock GM, Gibbs RA. The Atlas genome assembly system. Genome Res. 2004;14:721–732. doi: 10.1101/gr.2264004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoog JO, Hedberg JJ, Stromberg P, Svensson S. Mammalian alcohol dehydrogenase - functional and structural implications. J Biomed Sci. 2001;8:71–76. doi: 10.1007/BF02255973. [DOI] [PubMed] [Google Scholar]
- Hoog JO, Ostberg LJ. Mammalian alcohol dehydrogenases–a comparative investigation at gene and protein levels. Chem Biol Interact. 2011;191:2–7. doi: 10.1016/j.cbi.2011.01.028. [DOI] [PubMed] [Google Scholar]
- Hotter D, Krabbe T, Reith E, Gawanbacht A, Rahm N, Ayouba A, Van Driessche B, Van Lint C, Peeters M, Kirchhoff F, Sauter D. Primate lentiviruses use at least three alternative strategies to suppress NF-kappaB-mediated immune activation. PLoS Pathog. 2017;13:e1006598. doi: 10.1371/journal.ppat.1006598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaki T, Wolfsegger MJ. Estimation of pharmacokinetic parameters with the R package PK. Pharmaceutical Statistics. 2010;10:284–288. [Google Scholar]
- Julia P, Farres J, Pares X. Characterization of three isoenzymes of rat alcohol dehydrogenase. Tissue distribution and physical and enzymatic properties. Eur J Biochem. 1987;162:179–189. doi: 10.1111/j.1432-1033.1987.tb10559.x. [DOI] [PubMed] [Google Scholar]
- Julia P, Pares X, Jornvall H. Rat liver alcohol dehydrogenase of class III. Primary structure, functional consequences and relationships to other alcohol dehydrogenases. Eur J Biochem. 1988;172:73–83. doi: 10.1111/j.1432-1033.1988.tb13857.x. [DOI] [PubMed] [Google Scholar]
- Kardon T, Noel G, Vertommen D, Schaftingen EV. Identification of the gene encoding hydroxyacid-oxoacid transhydrogenase, an enzyme that metabolizes 4-hydroxybutyrate. FEBS Lett. 2006;580:2347–2350. doi: 10.1016/j.febslet.2006.02.082. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002a;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002b;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna M, Ranasinghe C, Jackson R, Parish CR. Heparan sulfate as a receptor for poxvirus infections and as a target for antiviral agents. J Gen Virol. 2017 doi: 10.1099/jgv.0.000921. [DOI] [PubMed] [Google Scholar]
- Kim SY, Zhao J, Liu X, Fraser K, Lin L, Zhang X, Zhang F, Dordick JS, Linhardt RJ. Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry. 2017;56:1151–1162. doi: 10.1021/acs.biochem.6b01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyosov AA, Rashkovetsky LG, Tahir MK, Keung WM. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry. 1996;35:4445–4456. doi: 10.1021/bi9521093. [DOI] [PubMed] [Google Scholar]
- Konopka G, Bomar JM, Winden K, Coppola G, Jonsson ZO, Gao F, Peng S, Preuss TM, Wohlschlegel JA, Geschwind DH. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Sandell LL, Trainor PA, Koentgen F, Duester G. Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim Biophys Acta. 2012;1821:198–205. doi: 10.1016/j.bbalip.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunes J, Kren V, Pravenec M, Zicha J. Use of recombinant inbred strains for evaluation of intermediate phenotypes in spontaneous hypertension. Clin Exp Pharmacol Physiol. 1994;21:903–906. doi: 10.1111/j.1440-1681.1994.tb02463.x. [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Castellani LW, Zhou Z, Paul E, Davis R, Schadt EE, Lusis AJ, Horvath S, Mehrabian M. A systems genetic analysis of high density lipoprotein metabolism and network preservation across mouse models. Biochim Biophys Acta. 2012;1821:435–447. doi: 10.1016/j.bbalip.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M, Rushton G, Gallagher JT. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J Biol Chem. 1997;272:18000–18006. doi: 10.1074/jbc.272.29.18000. [DOI] [PubMed] [Google Scholar]
- Mackay TF, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- Matsumiya T, Stafforini DM. Function and regulation of retinoic acid-inducible gene-I. Crit Rev Immunol. 2010;30:489–513. doi: 10.1615/critrevimmunol.v30.i6.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney EM, Semendric L, Helbig KJ, Hinze S, Jones B, Weinman SA, Beard MR. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. J Infect Dis. 2008;198:1766–1775. doi: 10.1086/593216. [DOI] [PubMed] [Google Scholar]
- Mishra-Gorur K, Singer HA, Castellot JJ., Jr Heparin inhibits phosphorylation and autonomous activity of Ca(2+)/calmodulin-dependent protein kinase II in vascular smooth muscle cells. Am J Pathol. 2002;161:1893–1901. doi: 10.1016/S0002-9440(10)64465-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra JP, Mishra S, Gee K, Kumar A. Differential involvement of calmodulin-dependent protein kinase II-activated AP-1 and c-Jun N-terminal kinase-activated EGR-1 signaling pathways in tumor necrosis factor-alpha and lipopolysaccharide-induced CD44 expression in human monocytic cells. J Biol Chem. 2005;280:26825–26837. doi: 10.1074/jbc.M500244200. [DOI] [PubMed] [Google Scholar]
- Monneau Y, Arenzana-Seisdedos F, Lortat-Jacob H. The sweet spot: how GAGs help chemokines guide migrating cells. J Leukoc Biol. 2016;99:935–953. doi: 10.1189/jlb.3MR0915-440R. [DOI] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg A, Jones AW, Hahn RG, Gabrielsson JL. Role of variability in explaining ethanol pharmacokinetics: research and forensic applications. Clin Pharmacokinet. 2003;42:1–31. doi: 10.2165/00003088-200342010-00001. [DOI] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pares X, Cederlund E, Moreno A, Saubi N, Hoog JO, Jornvall H. Class IV alcohol dehydrogenase (the gastric enzyme). Structural analysis of human sigma sigma-ADH reveals class IV to be variable and confirms the presence of a fifth mammalian alcohol dehydrogenase class. FEBS Lett. 1992;303:69–72. doi: 10.1016/0014-5793(92)80479-z. [DOI] [PubMed] [Google Scholar]
- Perez-Enciso M, Quevedo JR, Bahamonde A. Genetical genomics: use all data. BMC Genomics. 2007;8:69. doi: 10.1186/1471-2164-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PhenoGen. http://phenogen.ucdenver.edu. Accessed 04 Dec. 2017.
- Plapp BV, Leidal KG, Murch BP, Green DW. Contribution of liver alcohol dehydrogenase to metabolism of alcohols in rats. Chem Biol Interact. 2015;234:85–95. doi: 10.1016/j.cbi.2014.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravenec M, Klir P, Kren V, Zicha J, Kunes J. An analysis of spontaneous hypertension in spontaneously hypertensive rats by means of new recombinant inbred strains. J Hypertens. 1989;7:217–221. [PubMed] [Google Scholar]
- Pravenec M, Zidek V, Landa V, Simakova M, Mlejnek P, Kazdova L, Bila V, Krenova D, Kren V. Genetic analysis of “metabolic syndrome” in the spontaneously hypertensive rat. Physiol Res. 2004;53(Suppl 1):S15–22. [PubMed] [Google Scholar]
- Rachamin G, MacDonald A, Wahid S, Clapp JJ, Khanna JM, Israel Y. Modulation of alcohol dehydrogenase and ethanol metabolism by sex hormones in the spontaneously hypertensive rat. Effect of chronic ethanol administration. Biochem J. 1980;186:483–490. doi: 10.1042/bj1860483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandani VA, Bosron WF, Li TK. Research advances in ethanol metabolism. Pathol Biol (Paris) 2001;49:676–682. doi: 10.1016/s0369-8114(01)00232-2. [DOI] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo F, Hashim A, Marchese G, Ravo M, Tarallo R, Nassa G, Giurato G, Rinaldi A, Cordella A, Persico M, Sulas P, Perra A, Ledda-Columbano GM, Columbano A, Weisz A. Timed regulation of P-element-induced wimpy testis-interacting RNA expression during rat liver regeneration. Hepatology. 2014;60:798–806. doi: 10.1002/hep.27267. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba LM, Flink SC, Vanderlinden LA, Israel Y, Tampier L, Colombo G, Kiianmaa K, Bell RL, Printz MP, Flodman P, Koob G, Richardson HN, Lombardo J, Hoffman PL, Tabakoff B. The sequenced rat brain transcriptome–its use in identifying networks predisposing alcohol consumption. FEBS J. 2015;282:3556–3578. doi: 10.1111/febs.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasisekharan R, Venkataraman G. Heparin and heparan sulfate: biosynthesis, structure and function. Curr Opin Chem Biol. 2000;4:626–631. doi: 10.1016/s1367-5931(00)00145-9. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, Linsley PS, Mao M, Stoughton RB, Friend SH. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Schenauer MR, Yu Y, Sweeney MD, Leary JA. CCR2 chemokines bind selectively to acetylated heparan sulfate octasaccharides. J Biol Chem. 2007;282:25182–25188. doi: 10.1074/jbc.M703387200. [DOI] [PubMed] [Google Scholar]
- Sellin S, Holmquist B, Mannervik B, Vallee BL. Oxidation and reduction of 4-hydroxyalkenals catalyzed by isozymes of human alcohol dehydrogenase. Biochemistry. 1991;30:2514–2518. doi: 10.1021/bi00223a031. [DOI] [PubMed] [Google Scholar]
- Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serin EA, Nijveen H, Hilhorst HW, Ligterink W. Learning from Co-expression Networks: Possibilities and Challenges. Front Plant Sci. 2016;7:444. doi: 10.3389/fpls.2016.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota G, Kanki K. Retinoids and their target genes in liver functions and diseases. J Gastroenterol Hepatol. 2013;28(Suppl 1):33–37. doi: 10.1111/jgh.12031. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-3.0 1996 [Google Scholar]
- Star Consortium. Saar K, Beck A, Bihoreau MT, Birney E, Brocklebank D, Chen Y, Cuppen E, Demonchy S, Dopazo J, Flicek P, Foglio M, Fujiyama A, Gut IG, Gauguier D, Guigo R, Guryev V, Heinig M, Hummel O, Jahn N, Klages S, Kren V, Kube M, Kuhl H, Kuramoto T, Kuroki Y, Lechner D, Lee YA, Lopez-Bigas N, Lathrop GM, Mashimo T, Medina I, Mott R, Patone G, Perrier-Cornet JA, Platzer M, Pravenec M, Reinhardt R, Sakaki Y, Schilhabel M, Schulz H, Serikawa T, Shikhagaie M, Tatsumoto S, Taudien S, Toyoda A, Voigt B, Zelenika D, Zimdahl H, Hubner N. SNP and haplotype mapping for genetic analysis in the rat. Nat Genet. 2008;40:560–566. doi: 10.1038/ng.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Svensson S, Stromberg P, Hoog JO. A novel subtype of class II alcohol dehydrogenase in rodents. Unique Pro(47) and Ser(182) modulates hydride transfer in the mouse enzyme. J Biol Chem. 1999;274:29712–29719. doi: 10.1074/jbc.274.42.29712. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Saba L, Printz M, Flodman P, Hodgkinson C, Goldman D, Koob G, Richardson HN, Kechris K, Bell RL, Hubner N, Heinig M, Pravenec M, Mangion J, Legault L, Dongier M, Conigrave KM, Whitfield JB, Saunders J, Grant B, Hoffman PL, State WISo, Trait Markers of A Genetical genomic determinants of alcohol consumption in rats and humans. BMC Biol. 2009;7:70. doi: 10.1186/1741-7007-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AL, Denniston MM, Klevens RM, McKnight-Eily LR, Jiles RB. Association of Hepatitis C Virus With Alcohol Use Among U.S. Adults: NHANES 2003-2010. Am J Prev Med. 2016;51:206–215. doi: 10.1016/j.amepre.2016.02.033. [DOI] [PubMed] [Google Scholar]
- The Complex Trait Consortium. Guidelines: The nature and identification of quantitative trait loci: a community’s view. Nature Reviews Genetics. 2003;4:911–916. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCSC Genome Browser. https://genome.ucsc.edu/. Accessed 15 April 2016.
- Vaglenova J, Martinez SE, Porte S, Duester G, Farres J, Pares X. Expression, localization and potential physiological significance of alcohol dehydrogenase in the gastrointestinal tract. Eur J Biochem. 2003;270:2652–2662. doi: 10.1046/j.1432-1033.2003.03642.x. [DOI] [PubMed] [Google Scholar]
- Vanderlinden LA, Saba LM, Kechris K, Miles MF, Hoffman PL, Tabakoff B. Whole brain and brain regional coexpression network interactions associated with predisposition to alcohol consumption. PLoS One. 2013;8:e68878. doi: 10.1371/journal.pone.0068878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Song D, Pedersen LC, Liu J. Mutational study of heparan sulfate 2-O-sulfotransferase and chondroitin sulfate 2-O-sulfotransferase. J Biol Chem. 2007;282:8356–8367. doi: 10.1074/jbc.M608062200. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29:245–254. [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.