

Abstract
Exfoliation syndrome (XFS) is an age-related disease involving the deposition of aggregated fibrillar material (XFM) at extracellular matrices in tissues that synthesize elastic fibers. Its main morbidity is in the eye, where XFM accumulations form on the surface of the ciliary body, iris and lens. Exfoliation glaucoma (XFG) occurs in a high proportion of persons with XFS and can be a rapidly progressing disease. Worldwide, XFG accounts for about 25% of open-angle glaucoma cases. XFS and XFG show a sharp age-dependence, similarly to the many age-related diseases classified as aggregopathies.
Progress in understanding the cellular bases for XFS/G has been slowed by a lack of experimental models. Working with primary human tenon fibroblasts (TF) derived from trabeculectomies of XFG patients and age-matched POAG controls, we found that TF from XFG cells display many of the functional features observed in cells from other protein aggregate diseases, such as Parkinson’s, Alzheimer’s, Huntington’s and age-related macular degeneration. We have documented defects in lysosomal positioning, microtubule organization, autophagy processing rate and mitochondrial health.
In regard to failure of lysosomal and autophagosome positioning in XFG cells, we have found that XFG TF are unable to establish the trans-nuclear microtubule organizing center that is required for efficient centripetal vesicular locomotion along microtubules. In regard to potential sources of the autophagy malfunction, we have directed our attention to a potential role of the lysyl oxidase-like 1 protein (LOXL1), the elastic fiber catalyst that displays variant-dependent association with risk for XFG. Our experiments show that a) in XFG cells, a substantial fraction of LOXL1 is processed for degradation by the autophagic system; b) most of the LOXL1 N-terminus domain exists in a highly disordered state, a condition known to greatly increase the frequency of polypeptide misfolding; c) that maximum misfolding occurs at amino acid position 153, the location of the high risk variant G153D; and d) that replacement of glycine (G) by aspartate (D) there results in a substantial decrease in disorder within the 20 amino acid surrounding domain. Finally, we show that clusterin, a protein that can be induced by the presence of intra-, or extra-cellular aggregates, is uniformly overexpressed in XFG TF. The implications of our results for a theory relating XFG to cellular aggregopathy are discussed.
Introduction
Exfoliation Syndrome (XFS) is characterized by the formation of aggregated protein deposits (exfoliation material, XFM) that appear as opaque flake-like material on the ciliary body, iris, lens and trabecular meshwork1. XFM incorporates many extracellular matrix proteins, proteases and protease inhibitors, including clusterin, ApoE, latent TGFβ binding proteins, fibrillins, fibulins, lysosomal hydrolases and most notably LOXL1, a crosslinking enzyme needed for elastin fiber formation2–4. While there are reports showing health impact in multiple organs, in particular in peri-cardiac vessels5, the most significant morbidity occurs in the eye6–8. Patients presenting with elevated intraocular pressure (IOP) and XFM have a significantly higher chance of developing glaucoma (XFG) with subsequent vision loss compared to those without the presence of XFM. XFS uncommonly presents before age 60 and increases in frequency of onset with age.
A common finding in many age-related diseases is dysfunctional cellular degradation involving impairments in degradation of a) misfolded proteins by the proteasome and/or b) toxic aggregates and organelles by the autophagy system9, 10. In addition, the mitophagy component of autophagy leads to increased oxidative stress and accumulation of depolarized mitochondria (or ‘effete’) when autophagy fails9–11. The latter effects combine to reduce the cell’s capacity for energy generation and thereby further compromise autophagy, a process that utilizes substantial amounts of ATP. The impact of autophagic/lysosomal disease are exemplified by the effect on AMD, where malfunction in both autophagy and lysosomes results in intracellular build-up of internal cellular waste12. Lysosomes become filled with lipofuscin and basolateral secretion of undigested lipoprotein aggregates is thought to promote the formation of Drusen (lipofuscin-containing) plaques in the RPE and Bruch’s membrane13, 14. Therefore lowering the pH of lysosomes has been suggested as a strategy to slow or prevent the progression of AMD14, 15.
A major clue to the etiology of XFS came when genome wide association studies (GWAS) identified that variants in the coding region and regulatory regions of LOXL1 gene are highly associated with XFS. The initial reporting study for the association of XFG with 3 missense/non-synonymous SNP in the LOXL1 gene in the Finish population calculated that the XFG risk is 100-fold higher in individuals carrying the homozygous genotype for the highest risk missense SNP than in individuals carrying only low-risk haplotypes16. Intriguingly, a great majority of the population carries one or the other of these SNPs17. This observation suggests that other factors are required for the development of XFS. However given the late age of onset of XFS, it is impossible to ascertain whether the additional contributor(s) to generating XFS pathology is a specific genetic variant without which the syndrome will not develop at any age or a multiplicity of inputs, such as the overall personal genomic complexion and its interaction with the environment (e.g., UV exposure) that accelerate or retard the rate of progress from covert to overt phenotype18, 19. Our recent data suggest that like other age-related protein aggregation diseases, dysfunctional autophagic clearance of waste plays a role in generating XFS pathology. Our hypothesis based on these studies is that a combination of a protein-folding defect in the LOXL1 protein resulting from LOXL1 gene variants in combination with an inability to degrade LOXL1 containing protein aggregates produces XFM (discussed below).
Tenon capsule fibroblasts; a model system for the study of XFS
Experimental options for the study of XFS are limited by difficulty of obtaining ocular human tissue from cadavers with a history of XFS and the lack of an animal model. The best available option consists of obtaining anterior tenon capsule tissue at the time of trabeculectomy or tube surgery. Fibroblasts are the only cellular component of the Tenon capsule that can be easily cultured from the tissue by enzymatic digestion or by outgrowth from explants. While the tenon tissue is not intraocular, the systemic nature of the syndrome and the fact that the samples derive from eyes with XFG makes these fibroblasts a potentially useful conduit to understand the drivers of XFG. Ritch and Schlötzer-Schrehardt have demonstrated that these cells produce XFM in cell culture and have made a number of important correlations between XFS and pro-fibrotic signaling cascades, increase in XFM protein components, and the risk factors associated with the XFS genotype20–23. Seeking further insight into XFS pathology using these cells, we obtained tenon tissue from XFG patients along with identically processed cultures from age matched primary open glaucoma (POAG) patients. The latter represents a reasonably good control because a) they are from eyes with high IOP and glaucoma and b) much like the XFS glaucoma samples, the patients have been previously exposed to IOP-lowering drugs. Furthermore, when comparing POAG TF to TF derived from young donors that underwent strabismus correction surgery, there were no differences in lysosomal distribution and microtubule nucleation in response to autophagy induction between young and old non-XFS donors, further supporting the idea that age itself is not the sole determinant for the XFS phenotype.
Initial finding in XFS tenon fibroblasts
Soon after these cultures were generated it was observed that at low density the XFS-derived TF had a larger surface area than the POAG TF, with many engorged vesicular structures and, as the cells approached confluence, they did not acquire the spindle shape oriented formations that characterize all normal fibroblast cultures17, 24. These results initiated a series of experiments that identified multiple cellular features of XFS TF that mirror features common to cells in many age related diseases, in particular the neurodegenerative diseases, Alzheimer’s, Parkinson’s and Huntington’s, which are characterized by the presence of perinuclear protein aggregates (Table 1) and the reason for which these diseases are referred to as aggregopathies. The cells of these diseases have a reduced capacity for autophagic flux, the ultimate process used by cells to digest macroscopic cellular detritus and greatly diminished mitochondrial function, again, two characteristics of the XFS cells24. In this review, we provide a general description on the commonalities of age related aggregopathies and describe our specific findings for XFS TF in this context.
Table 1.
| Age-dependent disease | Protein | References |
|---|---|---|
| Alzheimer’s | Presinilin | Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene25 |
| λ-secretase | Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability26 | |
| APP | The significance of the Swedish APP670/671 mutation for the development of Alzheimer’s disease amyloidosis27 | |
| Amyloid angiopathy | APP | Presentation of amyloidosis in carriers of the codon 692 mutation in the amyloid precursor protein gene (APP692)28 |
| Parkinson’s | SNCA, PARK2, PINK1, PARK7, and LRRK2 | Genetic etiology of Parkinson Disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 Genes: A Mutation Update29 |
| Frontotemporal dementia | Tau -microtubule associated protein | Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism30 |
| ALS | SOD1 | Amyotrophic lateral sclerosis: a new missense mutation in the SOD1 gene31 |
| Retinitis pigmentosa | Rhodopsin | Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa32; Novel rhodopsin mutation (M216R) in a Danish family with autosomal dominant retinitis pigmentosa33 |
| Late-onset fundus flavimaculatus (FFM) | photoreceptor ATP binding transporter gene | Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies34 |
| Emphysema | 1 Alpha anti-trypsin | Retarded protein folding of deficient human alpha 1-antitrypsin D256V and L41P variants35 |
Protein polymorphism and polypeptide misfolding in age-related disease
Protein aggregates are seen in many human diseases. The sources of aggregopathies differ from disease to disease and thus, cellular manifestations as well as the locale of pathological manifestation or morbidity vary widely. Many age-related aggregopathies show various degrees of genetic linkage to non-synonymous single nucleotide polymorphisms, i.e., to a single amino acid variant of a single protein (Table 1). These variants though, do not seem to prevent or largely affect protein function; carriers are asymptomatic for most of their life. Instead, a prevalent theory is that in most of these cases the single amino acid substitution may increase misfolding rates for the polypeptide chain during synthesis.
Misfolded or unfolded polypeptides represent a constant and present danger to cells; the unstructured sequences can adopt conformations that interact with the surface of other proteins and in this manner propagate the denaturation as done by prion proteins36, 37. Accordingly, cells have developed an armamentarium of simultaneous or sequential mechanisms to ensure that nascent misfolded proteins, proteins damaged by environmental inputs and naturally aging detritus, are degraded. As misfolding develops during protein synthesis, the unfolded protein response (UPR) signaling cascade is activated, leading to reduced protein translation in an effort to stop production of the misfolded protein, and an increase in gene expression of a specialized set of proteins (chaperones) which attempt to refold the deviant structure38 (Fig 1). Misfolded proteins that escape this first line of defense are retro-translocated from the ER by the endoplasmic reticulum (ER)-associated protein degradation system or ERAD,39. In the cytosol, these proteins are ubiquitinated, marking them for degradation by the proteosome (Fig 1). This path is shared with preexisting damaged proteins and many other normal proteins that are degraded as a way to maintain their numbers in check. Recent discoveries suggest that when the proteasome is dysfunctional, the misfolded proteins may be shuttled out of the cell through a misfolding-associated protein secretion pathway (MAPS), characterized by the newly identified deubiquitinase, USP1940. Paradoxically, while secreting misfolded proteins may protect the cell in the short term, in the long term expelling misfolded proteins is suggested to create extracellular prion-like aggregates41.
Figure 1. The ERAD and MAPS pathways.
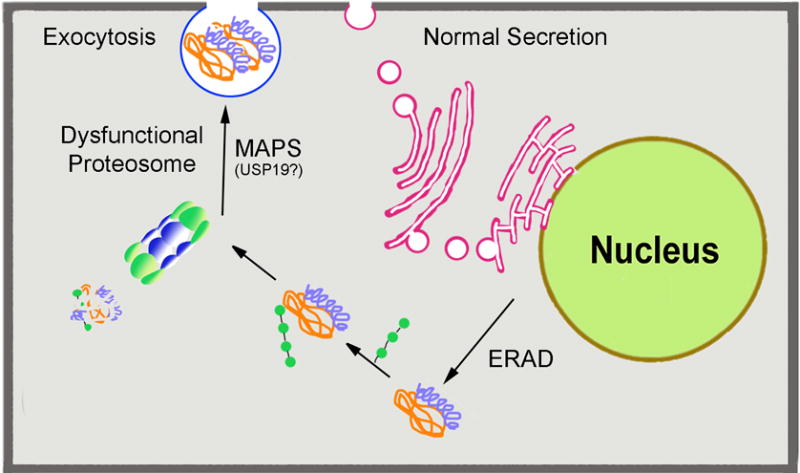
Misfolded proteins in the lumen of the ER activate the unfolded protein response (UPR) in which translation of some proteins is halted, while protein expression of chaperones is increased. If this response does not achieve refolding of the protein to a correct conformation, then the misfolded protein is retrotranslocated out of the ER, through the endoplasmic reticulum-associated degradation (ERAD) pathway. In the cytosol, ubiquitin ligases ubiquitinate the misfolded protein targeting it for degradation in the proteasome or the polypeptides are packed into aggresomes (described in Figure 2). Recently, it has been reported that when the the proteasome is dysfuctional, as occurs in many age-related diseases, misfolded proteins maybe disposed by secretion through a novel misfolding-associated protein secretion (MAPS) pathway. An isoform of the deubiquitinase, USP19 localized at the ER outer membrane plays a key role in MAPS.
Macroautophagy in age-related disease
A third mechanism that deals with the misfolded proteins not properly resolved by these two systems is macroautophagy (Fig 2). Macroautophagy is accomplished by extremely complex bio-mechanic cellular machinery designed to encapsulate structural detritus, in particular aging, decaying mitochondria (i.e., mitophagy), into a constantly and spontaneously forming organelle, the autophagosome. To join this path, misfolded and denatured proteins are packed by cellular machinery into aggresomes; these particulates can be then recognized and engulfed along with the large detritus in the autophagosome. All organellar components of autophagy are then transported toward, and concentrated into a juxta-nuclear location, the microtubule organizing center (MTOC), where they fuse with each other and finally with lysosomes to achieve their complete degradation. Current models of age related disease assume that early in life the vigor of the multiple mechanisms for misfolding correction and misfolded protein degradation keep misfolding inconsequential; disease arises when the combined capacity of these mechanisms weakens with age. The random nature of the aggresome composition (any of a very a large number of distinct misfolded polypeptides may be included in variable degree in each aggresome) implies the possibility that either these aggresomes interfere with the normal kinetic and chemical autophagy events and/or generate undegradable aggregates.
Figure 2. Macroautophagy.
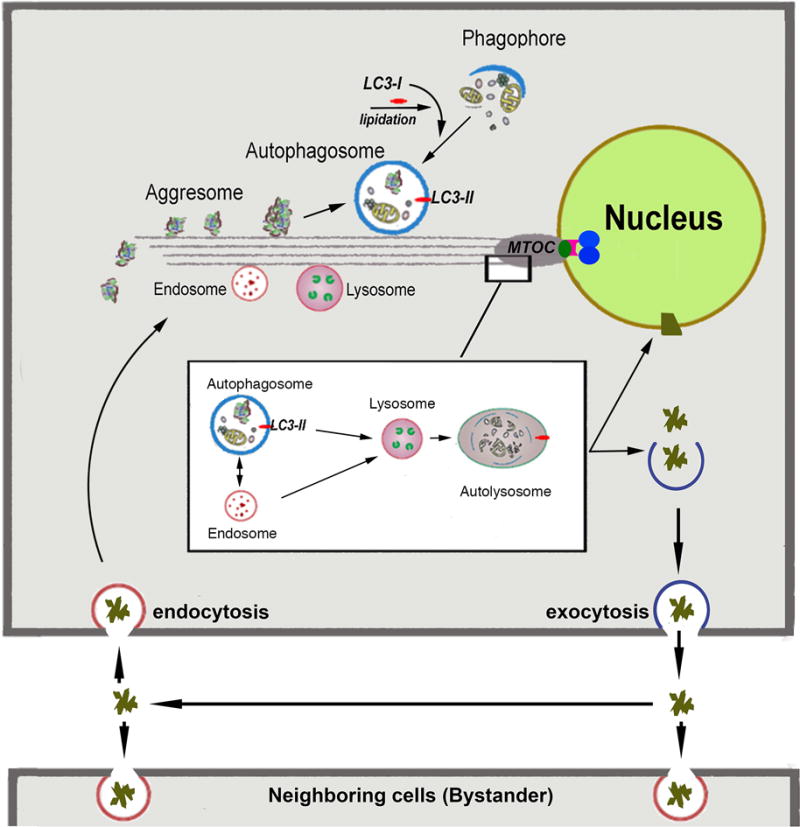
Macroautophagy involves biogenesis of autophagosomes from precursor phagophores within the cytosol. Attachment to microtubules is orchestrated by the lipidated form of LC3 (LC3-II). Autophagasomes engulf ‘macroscopic’ cellular detritus, naturally decaying organelles, in particular mitochondria (i.e., mitophagy) and misfolded or denatured proteins which have been ‘packed’ into particulates called aggresomes. These processess occur simultaneously with centripetal dyenin-dependent traffic of these organelles along microtubules towards the juxtanuclear microtubule organizing center (MTOC). The latter is formed by the anchoring of microtubules (composed of α and β tubulin) to a matching ‘ring’ made of γ-tubulin. This ring is held together by several other associated proteins to form a conical structure known as the γ-tubulin ring complex (γ-TuRC;
 ). γ-TuRCs, in turn, are densely packed by attachment to the cell centrosomes (
). γ-TuRCs, in turn, are densely packed by attachment to the cell centrosomes (
 ) via the adaptor protein ninein (NIN;
) via the adaptor protein ninein (NIN;
 ). Magnified MTOC detail: The insert depicts magnified details of the fusion events in the MTOC proximity. Early and late endosomes can fuse directly with lysosomes or with autophagasomes to form an amphisome. Both autophagosomes and amphisomes then fuse with the lysosome creating the autolysosome. In this transient structure the vesicular detritus cargo are lytically degraded into basic biochemical building blocks by the lysosome’s acidic hydrolases. Dense organelle accumulation at the MTOC greatly enhances these intervesicular fusions. Detritus that resists the sequential processing described above may accumulate in body inclusions (e.g., Lewy body in senile dementia), from where it may be exported. Re-uptake of these aggregates by the same (phagocytosis) will augment the load of undegredable protein in repetitive cycles. Furthermore, if exocytosis of aggregates or misfolded protein by the MAPS pathway described in Figure 1) is operative then the aggregopathy may propagate to adjacenct cells by particulate phagocytosis or endocytosis, as demonstrated for tau protein propagation in Alzheimer’s and α-synuclein protein in Parkinson’s.
). Magnified MTOC detail: The insert depicts magnified details of the fusion events in the MTOC proximity. Early and late endosomes can fuse directly with lysosomes or with autophagasomes to form an amphisome. Both autophagosomes and amphisomes then fuse with the lysosome creating the autolysosome. In this transient structure the vesicular detritus cargo are lytically degraded into basic biochemical building blocks by the lysosome’s acidic hydrolases. Dense organelle accumulation at the MTOC greatly enhances these intervesicular fusions. Detritus that resists the sequential processing described above may accumulate in body inclusions (e.g., Lewy body in senile dementia), from where it may be exported. Re-uptake of these aggregates by the same (phagocytosis) will augment the load of undegredable protein in repetitive cycles. Furthermore, if exocytosis of aggregates or misfolded protein by the MAPS pathway described in Figure 1) is operative then the aggregopathy may propagate to adjacenct cells by particulate phagocytosis or endocytosis, as demonstrated for tau protein propagation in Alzheimer’s and α-synuclein protein in Parkinson’s.
Autophagy dysfunction in tenon fibroblasts of XFG patients
Potential differences in autophagy between XFG and POAG TF became an intriguing line of study given the observed size and morphologic differences in conjunction with the significant body of literature on autophagy defects and age-related neurodegenerative diseases. Studies of autophagy are facilitated by the fact that in addition to serving as a backup mechanism for degradation of misfolded protein under steady conditions, in response to nutrient removal (starvation) the autophagic flux is greatly accelerated as a way to obtain fresh nutrients for the degradation process. In practice, for cells in culture just withdrawal of serum is enough to generate the starvation response24, 42. Comparing 4 XFS cell lines to 4 POAG cell lines, on induction of autophagy, XFS cells failed to properly centripetally stream endosomes, lysosomes and autophagosomes to the perinuclear area, the central organizing center for autophagic clearance. In fact, there was a gross defect in this process. LAMP-1 positive lysosomes were widely dispersed through the cell24 (Fig 3A-D). XFS also failed to develop a dense MTOC (microtubule organizing center, composed of the γ-TurC structure, gamma tubulin and Ninein) a feature that is consistent with the inability to concentrate and fuse vesicular structures at the juxtanuclear location24 (Fig 3E and F).
Figure 3. Salient phenotypic differences between TF derived from XFG and POAG donors.
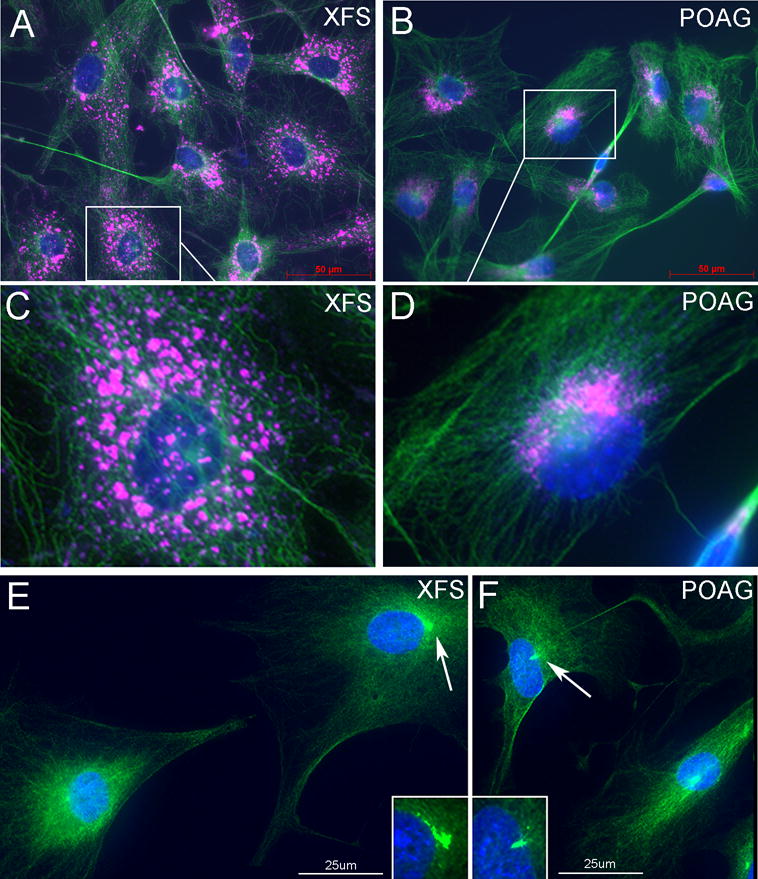
XFG and POAG TF were seeded under serum-free (starvation) conditions. A,B. Distribution of microtubules (β-tubulin, green) and lysosomes (LAMP1, pink), Bar=50um. C,D Magnified images. In XFG cells, with the exception of rare cells, the β-tubulin stain remains outside the nucleus and the lysosomes are minimally condensed. In the POAG cells lysosomes and β-tubulin staining is highly concentrated at a typical MTOC structure. E,F. In XFG cells the concentration of γ-tubulin remains outside the nucleus and is either small of amorphous γ-tubulin staining (arrow). In POAG cells, a large fraction of the stain is localized in a tubular or conical small structure consistent with the γ-TurC morphology that clearly penetrates deep into the nucleus (arrow). Bar = 25um. N=3.
Consistent with a dysfunction of lysosomal positioning, autophagic flux is compromised in XFS cells as well. This was demonstrated in two ways; first, a buildup of autophagasomes was demonstrated through the accumulation of the autophagosome marker, LC3-II, and second, autophagic flux through the cell, i.e., clearance of autophagasomes, was slowed in XFS cells24, 43. Interestingly, these data in XFS cells are consistent with data recently reported in neurons derived from late stage Alzheimer’s patients, in which autolysosomes (newly fused autophagosomes with lysosomes) were dispersed widely throughout the cell, LC3-II positive enlarged autophagosomes accumulated and autophagic flux, clearance of autophagosomes, was slowed44 again demonstrating a clear parallel between neurodegenerative pathological markers and XFS. Importantly, in XFS cells we also demonstrated that a sizable fraction of the XFS population contained failing depolarized mitochondria, consistent with reduced autophagy of dead mitochondria (mitophagy) activity24. The buildup of toxic mitochondria may interfere with cellular control of reactive oxygen species (ROS), which may further complicate autophagy by generating an additional oxidized and denatured protein load. Furthermore, a predictable lower energy output may impact the autophagy system, as the movement of vesicles for fusion requires ATP. Thus, a self-reinforcing loop of dysfunctional autophagy leading to increased oxidative stress and lowered energy supply and in turn, impacting autophagic flux may be an important contributor to a sudden onset of cellular pathology.
Autophagy inhibition suggests LOXL1 misfolding-XFS relationship
Given that autophagic flux in XFS TF is dysfunctional, testing whether LOXL1 protein is degraded in the autophagy pathway becomes an important first step to connect the production of XFM to the autophagy defect. Although autophagy is already compromised in XFS cells, total inhibition of autophagosome/lysosome fusion in both cell types will force the accumulation of LOXL1 only if the cell is attempting to degrade LOXL1-containing aggregates through the autophagy pathway. To test this, XFS and POAG TF were treated with Bafilomycin A1 (BafA) and Spautin-1. BafA is a lysosomal V-ATPase inhibitor that prevents autophagosome/lysosome fusion and degradation through the autophagy pathway45. The specific and potent autophagy inhibitor-1 (Spautin-1) inhibits the activity of Beclin-1, an autophagy initiation activator46. Preventing autophagic flux through the cells with these inhibitors demonstrated that, compared to XFS TF without inhibitors, XFS TF treated with BafA and Spautin-1 led to an increase in LOXL1 protein expression, 1.4 fold, p<0.001 (BafA), 1.63 fold, (Spautin-1), p<0.01. In POAG cells compared to POAG TF without inhibitors, POAG TF treated with BafA and Spautin-1 led to a decrease in LOXL1 protein expression, 11% p<0.05 (BafA), 40% p<0.05 (Spautin-1) (Fig 4). The finding that only in the XFS TF, the total amount of LOXL1 increases when autophagy is inhibited demonstrates that in these cells, LOXL1 polypeptide is being directed to the autophagy pathway. This observation would seem to support the presence of misfolded LOXL1 in XFS cells. The reason for the decrease in LOXL1 protein expression in POAG is not clear but we can hypothesize that blocking autophagy in POAG signaled an increase in proteasome degradation, stimulating the degradation of LOXL1. This may be the case in XFS TF as well, but like the deficiencies that were found in autophagic flux and mitochondrial function discussed above, it is likely that the proteasome is compromised as in other age-related diseases. Thus, blocking autophagy coupled with differences in proteasome health could explain the accumulation of misfolded proteins (in this case LOXL1) in XFS TF and a decrease in POAG TF.
Figure 4. Effect of Bafilomycin-A1 and Spautin-1 on LOXL1 protein.
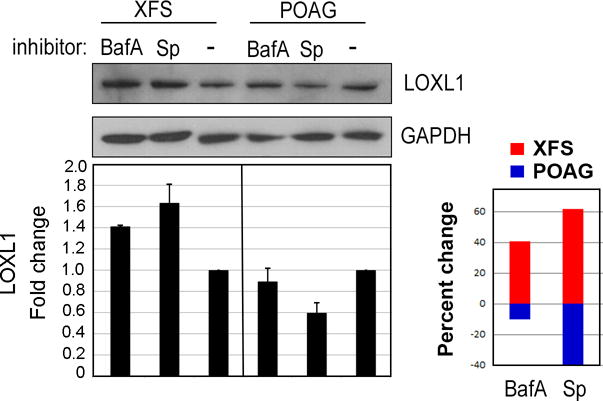
XFS and POAG TF were seeded in serum-free (starvation) conditions for 18 hours with 0.5uM Bafilomycin-A1 (BafA), 10uM Spautin-1 (Sp) or in control medium, without inhibitors. Western blot for LOXL1 shows that in XFG cells the amount of LOXL1 increases after treatment with autophagy inhibitors, whereas in POAG cells these inhibitors reduce LOXL1 amounts. Loading control, GAPDH. Statistical results for N = 4 are shown as the percent change (up or down) for XFG and POAG cells with each inhibitor.
Clusterin expression in XFS TF support the presence of aggregate material in XFS
Ocular XFM includes apolipoprotein J/clusterin (CLU). This is not surprising because CLU functions as a conformational stabilizer that maintains partially unfolded proteins in a state appropriate for subsequent refolding by chaperones. If XFS contains nascent misfolded or aggregated protein, the presence of these elements may trigger a higher CLU cellular concentration. Akin to the presence of clusterin in XFM extracellular aggregates, export of CLU out of the cell is well documented in Alzheimer’s, where it accumulates in extracellular beta amyloid plaques47. Using atomic force microscopy, an image of the buildup of clusterin in XFM on an XFS lens demonstrated a crosshatched appearance48. Interesting, using 3D self-synthesizing cultures49, a crosshatched appearance of clusterin in aggregates and along linear arrays was also visible (Fig 5A) which were completely absent in POAG cells (Fig 5B), magnified images (Fig 5C and D). Furthermore, an increase in clusterin protein expression was demonstrated by Western blot (Fig 5E, quantified in F, 1.9-fold increase in XFS p<0.005). It is important to consider that the excess cytosolic clusterin has potentially negative consequences. In Cos7 cells, ectopic overexpression of clusterin results in the formation of aggregates and damage to mitochondria, as determined by loss of membrane potential, resembling exactly the damage we have observed in the XFS TF50. Furthermore, our staining suggested that clusterin in not only present in large aggresomes but also present in smaller structures that seem to decorate microtubules. Highlighting the importance of clusterin to aggregopathies are the clinical studies now targeting clusterin with RNAi technology for neurodegenerative disease and cancer51.
Figure 5. Clusterin cellular distribution in 3D culture and protein expression in 2D culture.
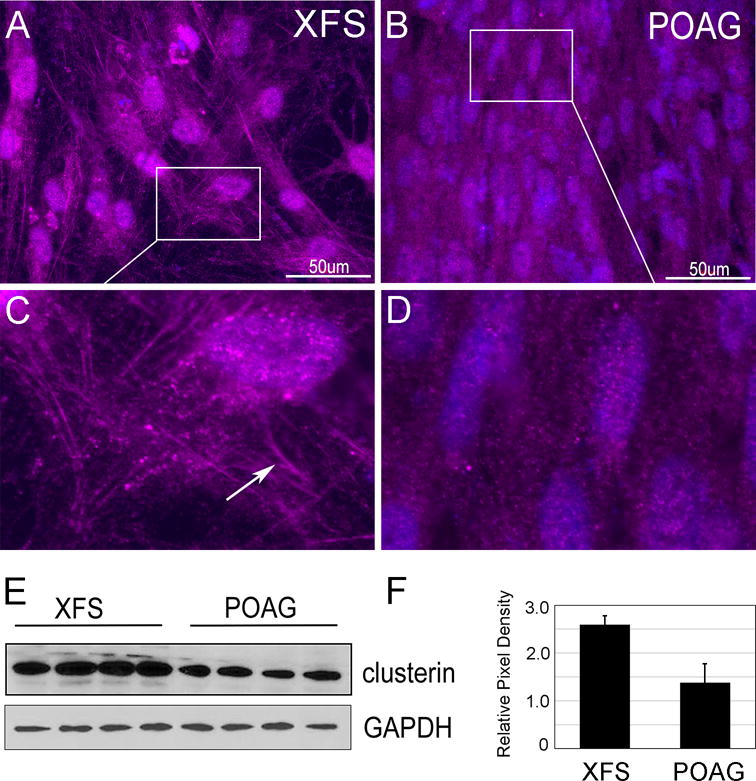
A-D) XFS and POAG TF were cultured in 1% FBS containing media with vitamin C for 1 month creating 3D self-synthesizing constructs. Cells were fixed and immunostained for clusterin. A,B bar = 50um. C,D Magnified images. N=3. In XFG cells, when compared with POAG cells, clusterin is in small globular structures and aligns with linear fibrillar arrays (arrow). E) Western blots of XFS and POAG cells cultured in starvation medium for two days. Loading control, GAPDH N=4. F) Quantification of Western blot.
Dysfunction of the γ-TurC MT anchoring structure may be at the root of MTOC disarray in XFS cells and contribute to poor organelle vectorial transit
As described in Figs 2 and 3, following induction of autophagy, the minus ends of microtubules are expected to coalesce into the juxtanuclear MTOC and indeed do so in the POAG cells but not in the XFS counterparts. This coalescence is mediated or supported by the anchoring or mounting of microtubules into a crown-, or Y-shaped structure, the γ-TurC, composed of γ-tubulin and a number of supplemental proteins and the γ-TurC in turn is anchored to the nuclear centrioles by Ninein (Fig 2). Staining POAG cells for γ-tubulin shows the expected intranuclear γ-TurC structures (Fig 3F). In contrast, in XFS cells either there is no discernable γ-TuRC or there is what appears to be a partial γ-TurC structure that a) does not display a triangulated shape and b) remains perinuclear (Fig 3E). The MTOC serves as a cap to α-β polymerization at the nuclear side (= minus end) of the microtubule, so that, in a cell with a well formed MTOC/γ-TuRC, polymerization is allowed only in the plus, outward growing end of the microtubule, whereas when the MTOC/γ-TuRC is not formed, there may be polymerization/depolymerization activity on both ends and there may be more but shorter microtubules. This lack of a rigid nucleation center appears an attractive option to explain the autophagic inefficiency in XFS cells. Microtubules with poor nucleation and capping at their minus end will have a higher degree of motion that the well nucleated microtubule of the POAG, in particular at the MTOC itself, where the autophagy organelles stream to fuse52. A looser or wobbling filament will interfere with the process by introducing dispersing forces. This may be the reason the increasing MT rigidity by davunetide, a peptide that stabilizes microtubules, has been found to improve autophagic flux and ameliorate neurodegenerative disease53, 54.
Disorder in LOXL1 protein
The studies above indicate that components of XFM such as LOXL1 may become the target of the autophagy system. Spontaneous misfolding of nascent polypeptides is one of the most important contributors to the cell’s denatured protein load.
Misfolding occurs and is an intrinsic feature of protein synthesis. As polypeptide synthesis proceeds, the local amino acid sequence favors the conformation with the lowest energy. However, at each step, alternative, incorrect configurations with very similar energies may exist, creating a finite possibility for misfolding in proportion to the energetic proximity between correct and incorrect conformations. It follows that highly ordered domains, which are stabilized by their very low energy state (−ΔG), will be less likely to misfold than less structured, more fluid domains. Unfortunately, many proteins need such low structure level or ‘disordered’ domains to be able to adapt to interactions with multiple partners. The proteins that generate toxic protein deposit in the main idiopathic neurodegenerative diseases, amyloid β (Aβ), tau and α-synuclein are all intrinsically disordered proteins (IDPs).
Thus, it has been proposed that IDP plays a critical role in the protein misfolding and/or post-translational denaturation that contributes to protein aggregation diseases 55. For this reason, to determine whether LOXL1 may have features that increase its chances for misfolding, we subjected LOXL1 and other related deaminases to disorder analysis by the Protein DisOrder prediction System (PrDOS; http://prdos.hgc.jp/cgi-bin/top.cgi). These analyses showed that the LOXL1 N-terminus contains multiple high disorder probability domains (Figure 6A). Maximal disorder occurs in a wide span between residues 150 and 170. Interestingly, this span includes position 153, where G homozygosity is found in 98% of XFG patients 56. Replacement of glycine by the negatively charged aspartic acid (D) at this position resulted in a substantial (8-10%) decrease in disorder probability over the 151-180 aa domain (Figure 6B). From Table 2 of the study by Wiggs and collaborators 56 it is possible to calculate that glycine-aspartic acid heterozygous individuals have a 16-fold lesser risk of developing XFS than individuals homozygous for glycine. Furthermore, that for individuals homozygous for expression of aspartic acid the decrease is approximately 26-fold. The C-terminus included a ‘deep well’ of low disorder probability (indicated by double asterisks) that was highly conserved throughout all the LOX protein family (Figure 6C, blue line). Finally, the comparative analysis of LOX proteins revealed that while the smaller LOX protein contains domains with disorder probability, none of the other LOX-like proteins displayed any disordered region (Figure 6C).
Figure 6. Structural disorder in LOXL1.
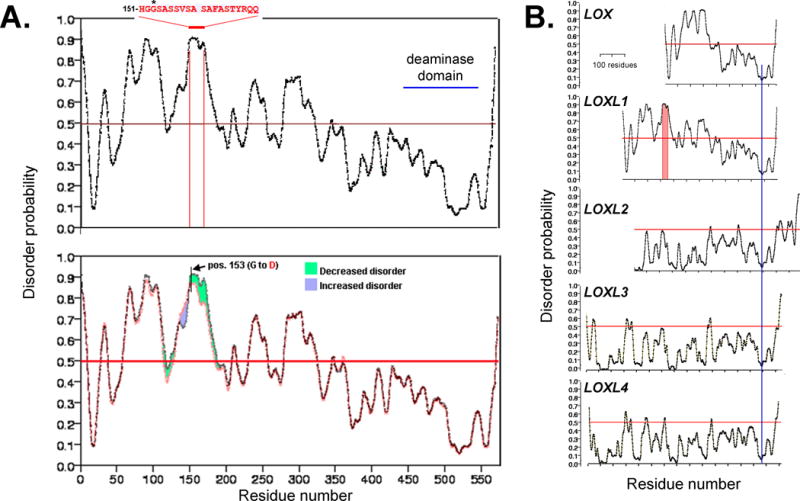
A. LOXL1 contains multiple disordered domains on its N-terminus. Particularly significant is a wide peak of disorder in the 151-170 amino acid domain. It includes the highest risk allele for XFG where homozygosity for glycine (G) at position 153 is associated with 98% of the XFG cases in a broad ethnic diversity USA population 56. The site of copper binding (site of deaminase activity) is indicated. B. Replacement of glycine by aspartic acid (D) at pos. 153 results in a ~ 8-10 % decrease in disorder probability in the 151-180 residue span. C. Comparison of disorder probability for the LOX family of proteins shows domains with high probability disorder exist only in the much smaller LOX protein; LOXL2-4 appear to be highly structured proteins. All sequences have been aligned using a preserved domain of very low disorder probability present at the C-terminus (blue line).
The potential pathological implications of this unique arrangement for LOXL1 deserve further study. Several pertinent questions that need examination are a) does LOXL1 misfold during synthesis at higher frequency than its LOXL-like congeners? b) Does the fully transcribed LOXL1 polypeptide denature more readily in unfavorable environments than its LOX congeners? c) do aa replacements at position 153 affect the frequency of misfolding? Additionally, since the C-terminus is highly ordered, the possibility that misfolding or denaturation of the N-terminus of LOXL1 occurs with preservation of deaminase activity does not appear far-fetched. If this happens, the various proteostatic intracellular waste disposal mechanisms dealing with misfolded and denatured protein may expose a variety of co-processed proteins to the crosslinking deaminase activity. Such random crosslinks may interfere with the ability of the waste disposal mechanism to complete their degradative functions.
Other genetic linkages to XFS and degradation pathways
Many genomic linkage studies have shown a high level of association of XFS with two SNP variants in the LOXL1 protein. The intersection of the individual genome with cumulative environmental insults, inflammation, UV exposure, etc., may also play a role in the pathologic outcome for LOXL1 variant carriers18, 57. For instance, linkage analysis has shown that a single amino acid variant of the calcium channel CACNA1A as an XFS precipitating factor58. Since this channel is essential for lysosome fusion59 it may be that the small variation causes a decrease in fusion ability and thus in autophagic capacity. In this context, our expanding study of disturbances of autophagy, presence of large cellular aggregates and unique secretion of LOXL1 aggregates in TF from XFG patients clearly suggests that XFS derives from a cellular aggregopathy. We believe these results may provide an important paradigm shift on the basic view on the causes of XFS and consequently on the focus of research that need to be pursued to cure XFS glaucoma.
Future Directions
Our studies demonstrate that autophagy is impeded in fibroblasts from XFG patients. The specific results including the increase in the amount of autophagosome-bound LC3 (LC3-II) and LC3II/LC3-I ratio upon acceleration of autophagy by starvation43 and the engorgement of the LAMP1-positive vesicles (Figure 3) are consistent with a reduced rate of autophagosome clearance from the cell24. The failure of lysosomes, autophagosomes and endosomes to concentrate at the MTOC in the starved condition, suggests that this reduction of vesicular clearance is at least in part from a lack of organelle proximity, a substantial factor for fusion frequency. As described above, inefficient centripetal organelle streaming may result from a lack of microtubule rigidity that is conferred by a well-organized MTOC and γ-TurC structure. Identifying the factors that interfere with the formation of these structures is challenging. This may result from a perturbation of signal transduction that modifies the behavior of the multiple proteins that are involved in the coalescence of the γ-TurC and its anchoring at the centrioles. More prosaically, a physical or steric hindrance may be the culprit. In this regard, the high accumulation of condensed clusterin (Fig 5) on microtubules provides an intriguing candidate. Although the expression of clusterin might be intended for extracellular protein housekeeping and as such a prosurvival agent, clusterin can become highly cytotoxic when certain spliced isoform accumulate in juxtanuclear aggresomes50. Microtubules may be unable to complete their nucleation at the MTOC because these aggresomes fail to readily detach from them at the minus end. Finally, it is important to remember that for control of misfolded and denatured proteins, autophagy serves as the last mechanism of defense. The observed deficiencies may derive not only from its intrinsic age-related functional decrease but may be compounded by an enhanced load secondary to age-related decreases in the upstream processing mechanisms. Given the parallels other age-related diseases and the accessibility of treating the eye, leveraging current therapies under consideration for other aggregopathies may prove to be beneficial to ameliorate or reverse XFS pathology.
Acknowledgments
This work was supported by The MYS Family U.S. Charitable Foundation, Inc., Research to Prevent Blindness, The Bright Focus Foundation, The Glaucoma Foundation, and a donation from Barbie and Tony Mayer (AMB and JMW), NIH-NEI R01 EY024942 and Lions District 20-Y (AMB) and NIH-NEI R01 EY018748 (JMW).
The authors gratefully acknowledge the contribution of the surgeons who supplied surgical specimens: Dr. Celso Tello, (Manhattan Eye and Ear and Throat Hospital); Dr. Sung Chul Park (Manhattan Eye and Ear and Throat Hospital); Dr. Jeffrey Liebmann (Columbia University Medical Center); Dr. Brian Campollataro (New York Eye and Ear Infirmary of Mount Sinai); Dr. Janet Serle and Dr. Donna Gagliuso (Icahn School of Medicine at Mount Sinai) and the support of the Icahn School of Medicine Microscopy CORE.
References
- 1.Ritch R, Schlötzer-Schrehardt U. Exfoliation syndrome. Surv Ophthalmol. 2001;45:265–315. doi: 10.1016/s0039-6257(00)00196-x. [DOI] [PubMed] [Google Scholar]
- 2.Ovodenko B, Rostagno A, Neubert TA, et al. Proteomic analysis of exfoliation deposits. Invest Ophthalmol Vis Sci. 2007;48:1447–1457. doi: 10.1167/iovs.06-0411. [DOI] [PubMed] [Google Scholar]
- 3.Schlötzer-Schrehardt U, Naumann GO. A histopathologic study of zonular instability in pseudoexfoliation syndrome. Am J Ophthalmol. 1994;118:730–743. doi: 10.1016/s0002-9394(14)72552-8. [DOI] [PubMed] [Google Scholar]
- 4.Schlötzer-Schrehardt U, Zenkel M, Küchle M, Sakai LY, Naumann GO. Role of transforming growth factor-beta1 and its latent form binding protein in pseudoexfoliation syndrome. Exp Eye Res. 2001;73:765–780. doi: 10.1006/exer.2001.1084. [DOI] [PubMed] [Google Scholar]
- 5.Katsi V, Pavlidis AN, Kallistratos MS, et al. Cardiovascular repercussions of the pseudoexfoliation syndrome. N Am J Med Sci. 2013;5:454–459. doi: 10.4103/1947-2714.117294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Streeten BW, Li ZY, Wallace RN, Eagle RC, Jr, Keshgegian AA. Pseudoexfoliative fibrillopathy in visceral organs of a patient with pseudoexfoliation syndrome. Arch Ophthalmol. 1992;110:1757–1762. doi: 10.1001/archopht.1992.01080240097039. [DOI] [PubMed] [Google Scholar]
- 7.Schlötzer-Schrehardt UM, Koca MR, Naumann GO, Volkholz H. Pseudoexfoliation syndrome. Ocular manifestation of a systemic disorder? Arch Ophthalmol. 1992;110:1752–1756. doi: 10.1001/archopht.1992.01080240092038. [DOI] [PubMed] [Google Scholar]
- 8.Bojic L, Ermacora R, Polic S, et al. Pseudoexfoliation syndrome and asymptomatic myocardial dysfunction. Graefes Arch Clin Exp Ophthalmol. 2005;243:446–449. doi: 10.1007/s00417-004-1074-9. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiselyov K, Jennigs JJ, Jr, Rbaibi Y, Chu CT. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy. 2007;3:259–262. doi: 10.4161/auto.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowald A, Kirkwood TB. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J Theor Biol. 2000;202:145–160. doi: 10.1006/jtbi.1999.1046. [DOI] [PubMed] [Google Scholar]
- 12.Golestaneh N, Chu Y, Xiao YY, Stoleru GL, Theos AC. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017;8:e2537. doi: 10.1038/cddis.2016.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaarniranta K, Hyttinen J, Ryhanen T, et al. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front Biosci (Elite Ed) 2010;2:1374–1384. doi: 10.2741/e198. [DOI] [PubMed] [Google Scholar]
- 14.Guha S, Liu J, Baltazar G, Laties AM, Mitchell CH. Rescue of compromised lysosomes enhances degradation of photoreceptor outer segments and reduces lipofuscin-like autofluorescence in retinal pigmented epithelial cells. Adv Exp Med Biol. 2014;801:105–111. doi: 10.1007/978-1-4614-3209-8_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guha S, Baltazar GC, Tu LA, et al. Stimulation of the D5 dopamine receptor acidifies the lysosomal pH of retinal pigmented epithelial cells and decreases accumulation of autofluorescent photoreceptor debris. J Neurochem. 2012;122:823–833. doi: 10.1111/j.1471-4159.2012.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorleifsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science. 2007;317:1397–1400. doi: 10.1126/science.1146554. [DOI] [PubMed] [Google Scholar]
- 17.Aboobakar IF, Johnson WM, Stamer WD, Hauser MA, Allingham RR. Major review: Exfoliation syndrome; advances in disease genetics, molecular biology, and epidemiology. Exp Eye Res. 2016;154:88–103. doi: 10.1016/j.exer.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Pasquale LR, Kang JH, Wiggs JL. Prospects for gene-environment interactions in exfoliation syndrome. Journal of glaucoma. 2014;23:S64–67. doi: 10.1097/IJG.0000000000000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiggs JL, Pasquale LR. Expression and regulation of LOXL1 and elastin-related genes in eyes with exfoliation syndrome. Journal of glaucoma. 2014;23:S62–63. doi: 10.1097/IJG.0000000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritch R, Hu D-N, SA M. Cultured Tenon’s capsule fibroblasts produce exfoliation material. Invest Ophthalmol Vis Sci. 1994;35(Suppl) [Google Scholar]
- 21.Zenkel M, Lewczuk P, Junemann A, Kruse FE, Naumann GO, Schlötzer-Schrehardt U. Proinflammatory cytokines are involved in the initiation of the abnormal matrix process in pseudoexfoliation syndrome/glaucoma. Am J Pathol. 2010;176:2868–2879. doi: 10.2353/ajpath.2010.090914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zenkel M, Krysta A, Pasutto F, Juenemann A, Kruse FE, Schlötzer-Schrehardt U. Regulation of lysyl oxidase-like 1 (LOXL1) and elastin-related genes by pathogenic factors associated with pseudoexfoliation syndrome. Invest Ophthalmol Vis Sci. 2011;52:8488–8495. doi: 10.1167/iovs.11-8361. [DOI] [PubMed] [Google Scholar]
- 23.Zenkel M, Schlötzer-Schrehardt U. Expression and regulation of LOXL1 and elastin-related genes in eyes with exfoliation syndrome. Journal of glaucoma. 2014;23:S48–50. doi: 10.1097/IJG.0000000000000120. [DOI] [PubMed] [Google Scholar]
- 24.Want A, Gillespie SR, Wang Z, et al. Autophagy and Mitochondrial Dysfunction in Tenon Fibroblasts from Exfoliation Glaucoma Patients. PLoS One. 2016;11:e0157404. doi: 10.1371/journal.pone.0157404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 26.De Jonghe C, Esselens C, Kumar-Singh S, et al. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum Mol Genet. 2001;10:1665–1671. doi: 10.1093/hmg/10.16.1665. [DOI] [PubMed] [Google Scholar]
- 27.Johnston J, O’Neill C, Lannfelt L, Winblad B, Cowburn RF. The significance of the Swedish APP670/671 mutation for the development of Alzheimer’s disease amyloidosis. Neurochem Int. 1994;25:73–80. doi: 10.1016/0197-0186(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 28.Roks G, Van Harskamp F, De Koning I, et al. Presentation of amyloidosis in carriers of the codon 692 mutation in the amyloid precursor protein gene (APP692) Brain. 2000;123(Pt 10):2130–2140. doi: 10.1093/brain/123.10.2130. [DOI] [PubMed] [Google Scholar]
- 29.Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31:763–780. doi: 10.1002/humu.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumanchin C, Camuzat A, Campion D, et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet. 1998;7:1825–1829. doi: 10.1093/hmg/7.11.1825. [DOI] [PubMed] [Google Scholar]
- 31.Tortelli R, Conforti FL, Cortese R, et al. Amyotrophic lateral sclerosis: a new missense mutation in the SOD1 gene. Neurobiol Aging. 2013;34:1709 e1703–1705. doi: 10.1016/j.neurobiolaging.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 32.Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 33.Haim M, Grundmann K, Gal A, Rosenberg T. Novel rhodopsin mutation (M216R) in a Danish family with autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 1996;17:193–197. doi: 10.3109/13816819609057893. [DOI] [PubMed] [Google Scholar]
- 34.Rozet JM, Gerber S, Souied E, et al. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet. 1998;6:291–295. doi: 10.1038/sj.ejhg.5200221. [DOI] [PubMed] [Google Scholar]
- 35.Jung CH, Na YR, Im H. Retarded protein folding of deficient human alpha 1-antitrypsin D256V and L41P variants. Protein Sci. 2004;13:694–702. doi: 10.1110/ps.03356604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker LC, Levine H, 3rd, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006;29:438–443. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prusiner SB. Shattuck lecture–neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 38.McCaffrey K, Braakman I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016;60:227–235. doi: 10.1042/EBC20160003. [DOI] [PubMed] [Google Scholar]
- 39.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- 40.He WT, Zheng XM, Zhang YH, et al. Cytoplasmic Ubiquitin-Specific Protease 19 (USP19) Modulates Aggregation of Polyglutamine-Expanded Ataxin-3 and Huntingtin through the HSP90 Chaperone. PLoS One. 2016;11:e0147515. doi: 10.1371/journal.pone.0147515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016;18:765–776. doi: 10.1038/ncb3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirkmajer S, Chibalin AV. Serum starvation: caveat emptor. Am J Physiol Cell Physiol. 2011;301:C272–279. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- 43.Wolosin JM, Ritch R, Bernstein AM. Is Autophagy dysfunction a key to exfoliation glaucoma? Journal of glaucoma. 2016 doi: 10.1097/IJG.0000000000000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bordi M, Berg MJ, Mohan PS, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–2483. doi: 10.1080/15548627.2016.1239003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calero M, Rostagno A, Frangione B, Ghiso J. Clusterin and Alzheimer’s disease. Sub-cellular biochemistry. 2005;38:273–298. [PubMed] [Google Scholar]
- 48.Doudevski I, Rostagno A, Cowman M, Liebmann J, Ritch R, Ghiso J. Clusterin and complement activation in exfoliation glaucoma. Invest Ophthalmol Vis Sci. 2014;55:2491–2499. doi: 10.1167/iovs.13-12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karamichos D, Guo XQ, Hutcheon AE, Zieske JD. Human corneal fibrosis: an in vitro model. Invest Ophthalmol Vis Sci. 2010;51:1382–1388. doi: 10.1167/iovs.09-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Debure L, Vayssiere JL, Rincheval V, Loison F, Le Drean Y, Michel D. Intracellular clusterin causes juxtanuclear aggregate formation and mitochondrial alteration. J Cell Sci. 2003;116:3109–3121. doi: 10.1242/jcs.00619. [DOI] [PubMed] [Google Scholar]
- 51.Wilson MR, Zoubeidi A. Clusterin as a therapeutic target. Expert Opin Ther Targets. 2016:1–13. doi: 10.1080/14728222.2017.1267142. [DOI] [PubMed] [Google Scholar]
- 52.Oakley BR, Paolillo V, Zheng Y. gamma-Tubulin complexes in microtubule nucleation and beyond. Mol Biol Cell. 2015;26:2957–2962. doi: 10.1091/mbc.E14-11-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Esteves AR, Gozes I, Cardoso SM. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson’s disease. Biochim Biophys Acta. 2014;1842:7–21. doi: 10.1016/j.bbadis.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 54.Magen I, Gozes I. Davunetide: Peptide therapeutic in neurological disorders. Curr Med Chem. 2014;21:2591–2598. doi: 10.2174/0929867321666140217124945. [DOI] [PubMed] [Google Scholar]
- 55.Breydo L, Redington JM, Uversky VN. Effects of Intrinsic and Extrinsic Factors on Aggregation of Physiologically Important Intrinsically Disordered Proteins. Int Rev Cell Mol Biol. 2017;329:145–185. doi: 10.1016/bs.ircmb.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 56.Fan BJ, Pasquale L, Grosskreutz CL, et al. DNA sequence variants in the LOXL1 gene are associated with pseudoexfoliation glaucoma in a U.S. clinic-based population with broad ethnic diversity. BMC Med Genet. 2008;9:5. doi: 10.1186/1471-2350-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miglior S, Bertuzzi F. Exfoliative glaucoma: new evidence in the pathogenesis and treatment. Prog Brain Res. 2015;221:233–241. doi: 10.1016/bs.pbr.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Aung T, Ozaki M, Mizoguchi T, et al. A common variant mapping to CACNA1A is associated with susceptibility to exfoliation syndrome. Nat Genet. 2015;47:387–392. doi: 10.1038/ng.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian X, Gala U, Zhang Y, et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS biology. 2015;13:e1002103. doi: 10.1371/journal.pbio.1002103. [DOI] [PMC free article] [PubMed] [Google Scholar]