

Abstract
Background
Glutathione-S-transferase A4-4 (GSTA4) is a key enzyme for removal of toxic lipid peroxidation products such as 4-hydroxynonenal (4-HNE). In the current study, we examined the potential role of GSTA4 on protein carbonylation and progression of alcoholic liver disease (ALD) by examining the development of liver injury in male wild type SV/J mice (WT) and SV/J mice lacking functional GSTA4 (GSTA4−/− mice).
Methods
Adult male WT and GSTA4−/− mice were fed chow (N = 10-12) or high fat Lieber-DeCarli liquid diets containing up to 28% calories as ethanol (EtOH) (N = 18-20) for 116 days. At the end of the study, half of the ethanol-fed mice were acutely challenged with an ethanol binge (3 g/kg given intragastrically) 12 hours before sacrifice. Carbonylation of liver proteins was assessed by immunohistochemical staining for 4-HNE adduction and by comprehensive liquid chromatography tandem mass spectrometry, LC-MS/MS, of purified carbonylated proteins.
Results
Chronic EtOH intake significantly increased hepatic 4-HNE adduction and protein carbonylation, including carbonylation of ribosomal proteins. EtOH intake also resulted in steatosis and increased serum ALTs. Hepatic infiltration with B-cells, T-cells, and neutrophils and mRNA expression of pro-inflammatory cytokines TNFα and IFNγ was modest in WT mice. However, an ethanol binge increased hepatic necrosis, hepatic cell proliferation and expression of TNFα mRNA (P<0.05). EtOH treatment of GSTA4−/− mice increased B-cell infiltration and increased mRNA expression of TNFα and IFNγ and of matrix remodeling markers MMP9, MMP13 and COL1A1 (P<0.05). GSTA4−/− mice exhibited panlobular rather than periportal distribution of 4-HNE-adducted proteins and increased overall 4-HNE staining after EtOH binge. Comprehensive liquid chromatography mass spectrometry of carbonylated proteins identified 1022 proteins of which 189 were unique to the GSTA4−/− group.
Conclusion
These data suggest long term adaptation to EtOH in WT mice does not occur in GSTA4−/− mice. Products of lipid peroxidation appear to play a role in inflammatory responses due to EtOH. and EtOH effects on B cell infiltration and autoimmune responses may be secondary to formation of carbonyl adducts.
Keywords: Ethanol, detoxification, protein carbonylation, liver, oxidative stress
BACKGROUND
Alcoholic liver disease (ALD) is a major contributor to liver failure in the United States today. A common phenotype of ALD is a hepatocellular environment characterized by pronounced lipid accumulation with enhanced oxidative stress (Shearn et al., 2014, Shearn et al., 2013). In this environment, increased lipid peroxidation occurs generating electrophilic short chain α/β unsaturated fatty acids such as 4-hydroxynonenal (4-HNE) and acrolein. The induction of an environment of increased lipid peroxidation and oxidative stress is mechanistically linked to the progression of ALD (Galligan et al., 2012b).
An important hepatic mechanism for removal of toxic lipid aldehydes is via conjugation with glutathione (Gallagher et al., 2007, Cheng et al., 2001). A primary enzyme that catalyzes this conjugation is Glutathione S-transferase A4-4 (GSTA4) (Gallagher et al., 2007). Increased expression of GSTA4 results in cellular resistance following exposure of HepG2 cells to lipid aldehydes including 4-HNE (Gallagher et al., 2007). In SV 129 mice, deletion of GSTA4 results in an increased propensity for obesity and increased 4-HNE levels, a phenotype not evident on the C57BL/6J background (Shearn et al., 2016a, Ronis et al., 2015, Ronis et al., 2017, Engle et al., 2004, Singh et al., 2008). This phenotype supports the contribution of GSTA4 in mitigating the accumulation of reactive aldehydes in the liver as well as supports the rationale for using the SV-129 background in this study. In GSTA4−/− mice, consumption of a high fat diet also results in increased adipocyte oxidative stress as evidenced by elevated concentrations of reactive aldehydes and mitochondrial damage (Curtis et al., 2010). Furthermore, when compared to WT SV 129 mice, lipid peroxidation/protein carbonylation is significantly elevated following intraperitoneal injection of carbon tetrachloride further highlighting the importance of GSTA4 in removal of lipid aldehydes (Dwivedi et al., 2006). Previously, using a 6-week (42 day) murine model of chronic ALD, we have determined that global deletion of the 4-HNE metabolizing protein GSTA4 resulted in an increase in inflammation and mitochondrial protein carbonylation (Ronis et al. 2015; Shearn et al. 2016a) ALD in humans is a disease that typically develops over many years of alcohol intake. In human alcoholics, binge drinking may also be the norm and contribute to the liver damage. In the current study, we have extended our previous research to examining the role of the GSTA4 gene on protein carbonylation and the progression of liver injury in a model consisting of long term (116 days) chronic EtOH consumption followed by a single EtOH binge.
METHODS
Animals and Experimental Design
All the animal studies described below were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences (UAMS). All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” at an American Association for Accreditation of Laboratory Animal Care-approved animal facility at UAMS. GSTA4-4−/− (GSTA4−/−) mice were generated on an SV/J-129 background as previously described (Singh et al. 2008). Groups of male wildtype (WT) SV/J and GSTA4−/− mice at age 13 weeks were fed chow (n = 10-12/group) or fed Lieber DeCarli liquid diets with or without ethanol (n = 18-20/group). The intake of the diet without ethanol was adjusted daily to give the pair-fed (PF) mice the same caloric intake as the ethanol-fed mice. The ethanol diet was Dyets Inc. #710260 ethanol diet with energy distribution of 18% protein (casein), 35% fat (1:3:10 mix of safflower oil, corn oil and olive oil), 18% carbohydrate (maltose dextrin) and 28% ethanol. The PF diet was Dyets Inc. #710027 PF control diet with 18% protein (casein), 35% fat (1:3:10 mix of safflower oil, corn oil and olive oil) and 46% carbohydrate (maltose dextrin). In the ethanol-fed groups, the ethanol concentration was increased from 0% to 5% (v/v) over 25 days by substituting carbohydrates (dextrose/maltodextrin) to a final ethanol level of 28% of total calories. The ethanol diet was kept at the maximal concentration that could be sustained without inducing morbid weight loss (3.6-5% (v/v)) for the remainder of the study. The average intake of ethanol diets in this period was 14 kcal/day/mouse with no significant differences between groups. Half of the mice fed the ethanol diet for 116 days were submitted to a subsequent ethanol binge or a sham binge (3 g/kg by gavage as a 30% solution in saline, sham = 30% saline) the evening before the dark period. After 12 hours, mice were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination.
Biochemical and Molecular Assessment of Liver Injury
Serum alanine aminotransferase (ALT) levels were assessed as a measure of liver damage by using the Infinity ALT liquid stable reagent (Thermo Electron, Waltham, MA). Triglycerides were extracted from liver homogenates with chloroform/methanol (2:1 v/v) and triglyceride concentration assayed using commercially available reagents (Cayman Chemical, Ann Arbor, MI).
Histopathological assessment of hepatic inflammation and oxidative stress
Formalin fixed slides were evaluated immunohistochemically using antibodies directed against Cyp2E1 (Millipore, Temecula, CA), 4-HNE (Shearn et al., 2014), B220 (BD Biosciences, San Jose, CA), CD3 (DAKO/Agilent Technologies, Santa Clara, CA) and myeloperoxidase ((MPO) R&D Systems Minneapolis, MN) as previously described (Shearn et al., 2016b, Shearn et al., 2015).
Sample preparation for mass spectrometry
Upon completion of the study, animals were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination. Livers were excised, weighed, and frozen for biochemical characterization and mRNA analysis or subjected to differential centrifugation for subcellular fractionation (cytosolic and mitochondrial) as previously described (Galligan et al., 2012a).
RT/PCR mRNA analysis of inflammation, fibrosis and proliferation
Hepatic markers of inflammation and macrophage infiltration, matrix remodeling and fibrosis were assessed by real time PCR (RT-PCR). Total RNA was isolated from tissue using Tri reagent (MRC, Cincinnati, OH) as per manufacturer’s protocol, and reversed transcribed using iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). 18S rRNA and transcripts of CD4, FoxP3 and Ki67 were measured with the Power SYBR Green RNA-to-CT 1-Step Kit (Applied Biosystems, Foster City, CA). Gene expression was determined by use of SYBR green and an ABI 7500 sequence detection system (Applied Biosystems) or a LightCycler® 480 II (Roche, Indianapolis, IN). Results were quantified using deltaCt method relative to 18 S rRNA for CD4, FoxP3 and Ki67 and relative to GAPDH for other targets. Data were normalized to levels in WT mice on the chow diet. Gene specific primers are presented in Table S1.
Biotin hydrazide purification of adducted proteins
From fresh tissue, 500 μg of aldehyde modified proteins from enriched cytosolic and mitochondrial fractions (Galligan et al., 2011) prepared from each mouse were derivatized using biotin hydrazide (BH)(ThermoFisher/Pierce, Waltham, MA)(5mM/2 hrs/RT/dark) followed by NaBH4 reduction (10mM/100mM NaOH 1hr/dark). Biotin hydrazide linked carbonylated proteins were incubated overnight using monoavidin columns (ThermoFisher/Pierce). Columns were washed 5X in phosphate buffered saline (PBS) pH 7.4, 5X in PBS 0.5M NaCl (pH 7.4) and 5X in PBS/2M Urea (pH 7.4) and eluted with 6×200 microliters of 0.2M NH4OH. Elutions were pooled and dried down using a roto-evaporator (Thermo Scientific/Savant) and boiled in 6X SDS PAGE loading buffer (15 minutes). Samples were loaded on 10% SDS PAGE gels and run at 160V for 20 min. Gels were removed and stained with Coomassie blue overnight (Imperial Stain, ThermoFisher/Pierce) followed by destaining in ddH2O O/N. Bands containing carbonylated proteins were excised, destained, and digested with trypsin as previously described (Shearn et al., 2015). Following digestion, peptides were extracted, dried down, and resuspended in 0.1% formic acid in ddH2O.
LC-MS/MS analysis of adducted peptides
For LC-MS/MS analysis, 8 μl of each peptide mixture was loaded on a Bruker Amazon Speed LC-MS/MS and a Bruker Maxis IMPACT LC-MS/MS and analyzed using Proteinscape V4.0 (Bruker) as previously described (Shearn et al., 2015). Following protein identification, all identified carbonylated proteins from each experimental condition were listed in Table S6.
Bioinformatic analysis of carbonylated proteins
Functional enrichment of carbonylated proteins for KEGG pathways (Kanehisa et al., 2017, Kanehisa, 2017) and Gene Ontology Terms (Gene Ontology, 2015) was tested using a one-sided Fisher Exact Test in each experimental group (2 genotypes × 3 treatment conditions = 6 experimental groups). Differential enrichment between experimental groups was examined using a two-sided Fisher Exact test. For KEGG pathway enrichment, proteins were mapped to pathways using their Uniprot ID and databases were downloaded from the KEGG API (downloaded 7/14/2017). For Gene Ontology Terms, gene annotations were downloaded directly from the Gene Ontology website (http://www.geneontology.org; GOC Validation Date – 08/28/2017) and Uniprot IDs were converted to gene symbols using the biomaRt package in R (Durinck et al., 2009). All analyses were executed in R (version 3.4.1).
Statistical Analysis
All statistical analysis was performed using SAS 9.4 software. Tables and column graphs present group means ± standard errors. Two-way ANOVA with Type 1 SS was performed. The first factor was genotype with two levels, GSTA4−/− and WT, and the second factor was diet with three levels, chow, ethanol, and ethanol + binge. We tested for the main effect of genotype, followed by main effect of diet, and the interaction between genotype and diet. ANOVA on qRT-PCR data were done on logarithm-transformed data. Using the ANOVA mean square error, a contrast for the main effect of binge between the ethanol and ethanol plus binge groups was tested for statistical significance (P<0.05). Pairwise comparisons between groups were made using Duncan’s Multiple Range test.
RESULTS
Pathological effects of pair-feeding simple carbohydrates
All groups that were PF or EtOH fed received a nutritional average of 14.0kcal/day with no significant differences between each group (Data not shown). Relative to ad libitum feeding of chow, chronic feeding of liquid diets high in fat and simple carbohydrates resulted in development of liver pathology independent of alcohol. A significant increase in hepatic triglycerides was accompanied by a decrease in serum triglycerides consistent with impairment of very low density lipoprotein secretion (P<0.0001 for the main effect of diet) (Table S2). Development of fatty liver was accompanied by significant increases (P<0.0001 for the main effect of diet) in expression of mRNAs encoding the inflammatory cytokines TNFα and IFNγ, in expression of mRNA encoding the B cell specific marker B220 (Table S3) and in mRNAs encoding matrix metalloproteinases MMP9 and MMP13 and the fibrotic protein collagen 1A1 (Table S4) (P<0.05). In the control liquid diet groups, no significant effects of the GSTA4−/− genotype were observed on these markers of liver pathology. Staining of lipid in the liver with Oil Red O revealed that pair-fed mice exhibited almost the same degree of hepatic steatosis as EtOH-fed mice, especially for the GSTA4−/− genotype (Figure S1). Since pair-feeding resulted in overt liver pathology with steatosis and induction of inflammation markers, the pair-feeding paradigm better describes the physiological status of humans with non-alcoholic steatohepatitis (NASH) rather than healthy human non-alcoholics. Therefore, we have chosen to compare the EtOH-fed groups against chow-fed controls in the results presented below to examine the effects of the GSTA4−/− genotype on liver pathology resulting from chronic EtOH exposure with or without a subsequent acute EtOH binge treatment. We recognize that effects observed between mice fed chow and the EtOH diet are a combination of effects caused by the ethanol in the diet and effects caused by exchanging solid chow for liquid diet high in fat and simple carbohydrates.
Effects of genotype and alcohol exposure on growth and liver pathology
There were no significant effects of neither genotype nor diet on body weight (Table 1). In contrast, the relative liver weight (liver weight/body weight ratio) was significantly decreased in the GSTA4−/− mice compared to WT mice for the EtOH and EtOH + Binge treatments. The binge increased the relative liver weight for both genotypes. In chow-fed mice, the hepatic triglyceride content and the serum triglyceride content was significantly higher for the GSTA4−/− than for the WT genotype. For both genotypes, chronic ethanol consumption significantly increased hepatic triglyceride content (P<0.05), while the addition of the binge decreased the hepatic triglyceride content (P<0.05). Conversely, chronic ethanol consumption decreased the concentration of serum triglycerides for both genotypes, and the addition of a single ethanol binge led to a significant increase in both genotypes. Two-way ANOVA of hepatic and serum triglycerides showed a significant interaction between genotypes and dietary treatments. This reflects that the GSTA4−/− genotype had a stronger effect in chow-fed animals than in the alcohol-exposed groups. It should also be noted that similar decreases in hepatic triglyceride concentration and increases in serum triglyceride concentration were observed in pair-fed animals receiving a sham binge with saline not containing ethanol (data not shown). Binge effects on hepatic and serum triglycerides thus seem to be a result of the stress of the gavage rather than the ethanol content of the gavage.
Table 1.
Effects of 116d EtOH +/− binge consumption on hepatocellular injury and triglycerides.
| Physiological Markers | ||||||
|---|---|---|---|---|---|---|
| Body Weight | Liver Weight | L/BW | ALT | Hepatic Triglycerides | Serum Triglycerides | |
| Group | (g) | (g) | % | (U/L) | (mg/g tissue) | (g/L) |
| WT + Chow | 27.96 ± 0.69a | 1.10 ± 0.00a,b | 3.94 ± 0.09b | 16.23 ± 1.43a | 27.18 ± 3.41a | 29.48 ± 3.10b |
| GSTA4 −/− + Chow | 29.80 ± 1.13a | 1.18 ± 0.06b | 3.96 ± 0.09b | 10.33 ± 0.56a | 75.36 ± 8.14c | 56.10 ± 9.29c |
| WT + EtOH | 26.63 ± 1.28a | 1.14 ± 0.04a,b | 4.30 ± 0.08a,b | 41.82 ± 8.53b | 86.77 ± 9.95c,d | 11.59 ± 4.39a |
| GSTA4 −/− + EtOH | 27.20 ± 0.71a | 0.99 ± 0.03a | 3.55 ± 0.11a | 26.83 ± 5.21a,b | 98.27 ± 7.59d | 9.12 ± 1.33a |
| WT + EtOH + Binge | 26.89 ± 1.35a | 1.20 ± 0.07b | 4.45 ± 0.14c | 61.79 ± 6.10c | 51.25 ± 2.46b | 30.22 ± 4.12b |
| GSTA4 −/− + EtOH + Binge | 26.44 ± 0.90a | 1.07 ± 0.04a,b | 4.05 ± 0.15b | 70.23 ± 4.48c | 44.56 ± 3.29a,b | 44.67 ± 2.42c |
| P values | ||||||
|
| ||||||
| Genotype | 0.6371 | 0.0359 | 0.0002 | 0.7632 | 0.0048 | 0.0074 |
| Diet | 0.1606 | 0.2552 | 0.0160 | <.0001 | <.0001 | <.0001 |
| Interaction | 0.6285 | 0.093 | 0.0299 | 0.1021 | 0.0021 | 0.0010 |
|
| ||||||
| P values | ||||||
|
| ||||||
| Main Binge Effect | 0.8081 | 0.0417 | 0.2286 | <.0001 | <.0001 | <.0001 |
Data are means ± SEM for n = 5-10 mice/group. Serum ALT, alanine aminotransferase activity, serum triglyceride and hepatic triglyceride concentrations were determined as described in Materials and Methods. L, Liver Weight, BW, body weight. Significance was determined by a 2-way ANOVA followed by Duncan’s Multiple Range post hoc analysis. Groups with no letter superscripts in common are significantly different from each other, P<0.05.
Serum alanine aminotransferase (ALT) activity was measured to assess hepatic necrotic injury. No differences in serum ALT was evident between the chow-fed groups. Chronic consumption of ethanol resulted in significantly elevated hepatocellular injury in WT animals. The addition of the ethanol binge resulted in a significant elevation of serum ALT activity for both genotypes. While there was no significant genotype difference in ALT for the EtOH + Binge groups, the increase in ALT caused by the binge was significantly higher (P<0.05) for the GSTA4−/− mice than for WT mice.
Effects of genotype and alcohol exposure on hepatic inflammation
In models of short term chronic alcohol feeding supplemented with alcohol binges, the addition of the binge causes more inflammation than observed after ethanol feeding without binges (Bertola et al., 2013b, Bertola et al., 2013a). To determine if GSTA4 deletion affects hepatic inflammation following alcohol exposures, we measured mRNA expression of proinflammatory cytokines TNFα, IFNγ and IL-6 and of CD138, B220, CD4 and FoxP3 as markers for plasma cells, B-lymphocytes, T helper cells and regulatory T-cells, respectively. As shown in Table 2, two-way ANOVA indicated significant main dietary effects (P<0.05) on expression of TNFα, IFNγ, and IL-6 reflecting that the alcohol exposures stimulate expression of these cytokines. There were also significant main dietary effects for B220, CD4 and FoxP3 mRNA with ethanol treatments stimulating B220 and FoxP3 expression and repressing CD4 expression. Importantly, we observed a significant main genotype effect for TNFα and IFNγ, reflecting that the GSTA4−/− genotype has enhanced hepatic expression of TNFα and IFN©mRNA (P<0.05), and a trend towards increased B220 mRNA (P<0.1) indicating that GSTA4−/− mice are predisposed towards inflammation and B cell infiltration.
Table 2.
Relative hepatic cytokine and chemokine mRNA expression in WT and GSTA4−/−mice following 116d EtOH +/− Binge consumption.
| Inflammatory Markers | |||||||
|---|---|---|---|---|---|---|---|
| Group | TNFα | IFNγ | IL-6 | CD138 | B220 | CD4 | FoxP3 |
| WT + Chow | 1.00 ± 0.32a | 1.00 ± 0.40a | 1.00 ± 0.55a | 1.00 ± 0.09a | 1.00 ± 0.27a | 1.00 ± 0.29b | 1.00 ± 0.13a |
| GSTA4 −/− + Chow | 2.15 ± 0.20a,b | 2.50 ± 1.15a,b,c | 2.11 ± 0.44a,b | 1.53 ± 0.07b | 1.09 ± 0.24a | 0.80 ± 0.17a,b | 0.94 ± 0.20a |
| WT + EtOH | 3.54 ± 1.47b,c | 1.41 ± 0.59a,b | 2.78± 0.53a,b | 1.26 ± 0.15a,b | 1.26 ± 0.29a,b | 0.60 ± 0.14a,b | 1.35 ± 0.22a |
| GSTA4 −/− + EtOH | 11.01 ± 7.22c,d | 4.92 ± 3.16b,c | 1.58 ± 0.64a | 0.97 ± 0.06a | 2.61 ± 0.98a,b | 0.53 ± 0.07a,b | 1.87 ± 0.27a |
| WT + EtOH + Binge | 10.70 ± 4.44c,d | 3.39 ± 2.02b,c | 5.70 ± 2.71b | 1.00 ± 0.07a | 2.30 ± 0.78a,b | 0.48 ± 0.20a,b | 6.74 ± 2.36b |
| GSTA4 −/− + EtOH + Binge | 15.40 ± 6.12d | 6.26 ± 2.58c | 2.93 ± 1.65a,b | 1.12 ± 0.14a | 3.07 ± 1.24b | 0.44 ± 0.08a | 11.11 ± 5.12b |
| P values | |||||||
|
| |||||||
| Genotype | 0.0125 | 0.0157 | 0.3818 | 0.5246 | 0.0912 | 0.3895 | 0.1442 |
| Diet | <.0001 | 0.0403 | 0.0254 | 0.3766 | 0.0189 | 0.0305 | <.0001 |
| Interaction | 0.558 | 0.6082 | 0.1809 | 0.0071 | 0.5769 | 0.9593 | 0.5639 |
|
| |||||||
| P values | |||||||
|
| |||||||
| Main Binge Effect | 0.0490 | 0.1122 | 0.0710 | 0.5861 | 0.1790 | 0.3480 | <.0001 |
Data are means ± SEM for n = 5-10 mice/group. Real-time qRT-PCR data are expressed relative to the WT + Chow group. Significance was determined by 2-way ANOVA followed by Duncan’s Multiple Range post hoc analysis. Groups with no letter superscripts in common are significantly different from each other, P <0.05.
To elucidate the effects of the ethanol treatments and the GSTA4−/− genotype on hepatic infiltration with inflammatory cells, immunohistochemistry was performed using antibodies directed against the T-lymphocyte marker CD3 (Figure 1, Panels A-F), the B cell marker B220 (Figure 1 arrows Panels G-L) and the neutrophil marker myeloperoxidase (MPO) (Figure 1 arrows Panels M-R) followed by quantification of positive cells per field (Table S5). Two-way ANOVA indicated highly significant (P<0.01) main dietary effects for all three inflammatory cell types, reflecting an increased infiltration with inflammatory cells for ethanol-treated groups compared to chow-fed animals. There was a main significant genotype effect for B220-positive cells with the GSTA4−/− genotype showing significantly higher numbers than the WT in all three dietary groups. The final ethanol binge showed a significant main stimulatory effect on the expression of TNFα mRNA. It did not however significantly impact infiltration with MPO-positive neutrophils (Table 2 and Table S5). The binge also showed complex significant effects for T-cells with a significant reduction of overall CD3-positive cells, but an apparent increase in regulatory T-cells as indicated by FoxP3 mRNA (Table 2 and Table S5).
Figure 1. Increased inflammation following 116d chronic EtOH consumption +/− Binge.
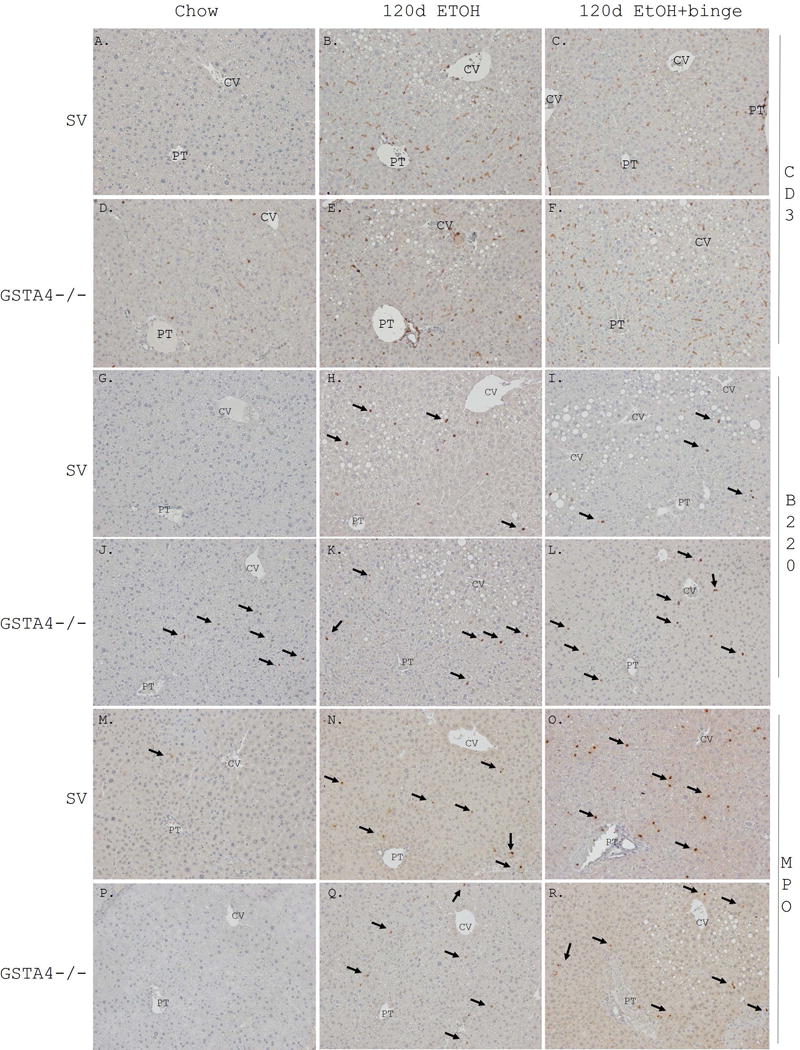
Formalin fixed tissue sections were analyzed immunohistochemically using polyclonal or monoclonal antibodies directed against Panels A-C SV CD3 chow, 116d EtOH, 116d EtOH +Binge, Panels D-F GSTA4−/− CD3 chow, 116d EtOH, 116d EtOH +Binge), Panels G-I SV B220 chow, 116d EtOH, 116d EtOH +Binge, Panels J-L GSTA4−/− B220 chow, 116d EtOH, 116d EtOH +Binge, Panels M-O SV MPO chow, 116d EtOH, 116d EtOH +Binge, P-R GSTA4−/− MPO chow, 116d EtOH, 116d EtOH +Binge. Arrows indicate B220 and MPO positive cells. Representative images are shown; n = at least 3 mice per genotype (CV, central vein, PT, portal triad).
Effects of genotype and alcohol exposure on markers of matrix remodeling, stellate cell activation and cellular proliferation
Chronic ethanol ingestion is known to induce fibrosis by activation of stellate cells. To determine potential effects of genotypes and dietary treatments on stellate cell activation and matrix remodeling, mRNA expression of matrix metalloproteinases 9 and 13 (MMP9 and MMP13) and collagen 1A1 (Col1A) was examined along with a marker of cellular proliferation (Ki67) (Table 3). A significant main dietary effect for MMP9 is indicative of increased expression in ethanol-treated mice. MMP9, MMP13 and Col1A all exhibited a significant main genotype effect with elevated expression in GSTA4−/− mice relative to WT mice (P<0.05). These data suggest that knockout of the Gsta4 gene makes the mice are more prone to fibrosis. Expression of Ki67 mRNA was strongly induced by the ethanol binge (P<0.001 for the main binge effect), but was not affected by genotype. The elevated hepatocyte proliferation may occur in response to the increased acute injury induced by the ethanol binge.
Table 3.
Relative mRNA expression of hepatic markers of matrix remodeling, stellate cell activation and cellular proliferation in WT and GSTA4−/− mice following 116d EtOH +/− Binge consumption.
| Fibrosis and Proliferation | ||||
|---|---|---|---|---|
| Group | MMP9 | MMP13 | Col1A1 | Ki67 |
| WT + Chow | 1.00 ± 0.20a | 1.00 ± 0.23a,b | 1.00 ± 0.55a | 1.00 ± 0.23a |
| GSTA4−/− + Chow | 1.64 ± 0.37a | 1.44 ± 0.40a,b | 2.94 ± 0.63b,c | 0.63 ± 0.25a |
| WT + EtOH | 2.57 ± 0.93a,b | 0.65 ± 0.16a | 1.15 ± 0.53a,b | 1.24 ± 0.56a |
| GSTA4−/− + EtOH | 7.13 ± 3.43b,c | 2.01 ± 1.14a,b | 3.53 ± 1.37c | 1.44 ± 0.31a |
| WT + EtOH + Binge | 6.49 ± 2.63b,c | 1.69 ± 0.77a,b | 3.06 ± 1.06b,c | 7.88 ± 3.98b |
| GSTA4−/− + EtOH + Binge | 7.76 ± 2.89c | 2.23 ± 0.80b | 3.17 ± 1.18b,c | 9.60 ± 5.28b |
| P values | ||||
|
| ||||
| Genotype | 0.0304 | 0.0478 | 0.0198 | 0.9652 |
| Diet | <0.0001 | 0.2651 | 0.2046 | <0.0001 |
| Interaction | 0.4152 | 0.4068 | 0.1868 | 0.6268 |
|
| ||||
| P values | ||||
|
| ||||
| Main Binge Effect | 0.1166 | 0.1199 | 0.1878 | <0.0001 |
Data are means ± SEM for n = 5-10 mice per group. Real-time qRT-PCR data are expressed relative to the WT + Chow group. Significance was determined by 2-way ANOVA. Groups with no letter superscripts in common are significantly different from each other, P <0.05.
Impact of genotype and alcohol exposure on hepatic Cyp2E1 expression and lipid peroxidation
Hepatic cytochrome 2E1 (Cyp2E1) which is an important enzyme for generation of reactive oxygen species is known to be induced by EtOH. To determine the effects of long term EtOH consumption and a subsequent single EtOH binge on induction of Cyp2E1 and formation of products generated as a consequence of lipid peroxidation, fixed paraffin embedded tissue sections were stained for Cyp2E1 (Figure 2 Panels A-F) and for protein aldehyde adduction by 4-HNE (Figure 2 Panels G-L) (Shearn et al., 2016b, Ronis et al., 2015). For both genotypes, EtOH feeding for 116 days induced Cyp2E1 expression, particularly in the centrilobular regions. No further induction was evident with the addition of the ethanol binge. In the chow-fed groups, deletion of GSTA4 resulted in increased 4-HNE staining when compared to the control group. Compared to chow-fed animals, 4-HNE staining increased for both genotypes following ethanol exposures (Figure 2 and Figure S2). In WT mice, 4-HNE staining was primarily periportal after chronic ethanol feeding and became more evenly distributed after the ethanol binge, whereas in the GSTA4−/− group, 4-HNE staining was panlobular under both conditions of ethanol exposure (arrows). Surprisingly, quantification of 4-HNE staining revealed a significant binge effect in the GSTA4−/− group but not the control group. It can be noted that inductions of Cyp2e1 and 4-HNE adducted proteins did not colocalize to the same hepatic regions, indicating that there is no simple direct link between these two phenomena.
Figure 2. Effects of 116d chronic EtOH consumption +/− Binge on Cyp2E1 expression and hepatic protein carbonylation.
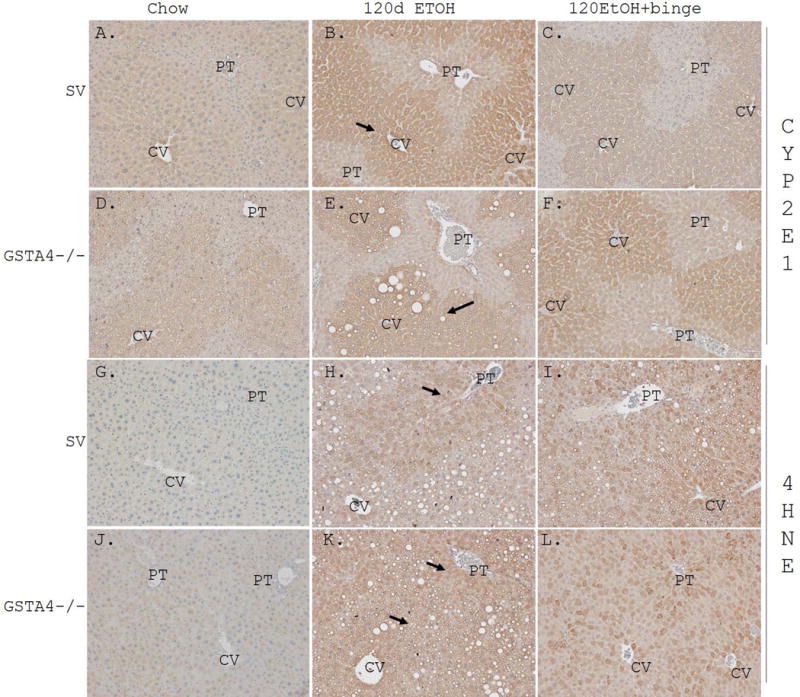
SV and GSTA4−/− mice. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against Panels A-C SV Cyp2E1 chow, 116d EtOH, 116d EtOH +Binge, Panels D-F GSTA4−/− Cyp2E1 chow, 116d EtOH, 116d EtOH +Binge, Panels G-I SV 4-HNE chow, 116d EtOH, 116d EtOH +Binge, Panels J-L GSSTA4−/− 4-HNE chow, 116d EtOH, 116d EtOH +Binge. Arrows demonstrate centrilobular Cyp2E1 expression and periportal accumulation of 4-HNE adducts in SV genotype and panlobular accumulation of 4-HNE adducts in GSTA4−/− genotype. Representative images are shown; n = at least 3 mice per genotype (CV, central vein, PT, portal triad).
Impact of genotype and alcohol exposure on hepatic protein carbonylation
To determine how the dietary treatments affect protein carbonylation for the two genotypes, LC-MS analysis of carbonylated proteins was performed using cytosolic and mitochondrial fractions prepared from each group (N=5 per genotype and dietary condition). Following LC-MS, identified proteins from both fractions were combined to reflect total proteins identified in each condition. In total, 1022 carbonylated proteins were identified (Table S6), of which 516 were previously noticed as carbonylated in a 6-week feeding study (Shearn et al., 2016a). VENN diagrams of carbonylated proteins shown in Figure 3A-C reveal that there is considerable overlap between groups. Thus, of the 484 carbonylated proteins identified from chow-fed animals, 83% and 80% were also observed in animals given chronic ethanol and chronic ethanol + binge, respectively. A breakdown of Table S6 and the VENN analysis depicting the protein specific impact of both genotype and diet is presented in Tables S7, S8 and S9. Compared to the chow-fed groups, significantly more carbonylated proteins were identified in the groups of ethanol-treated animals. Significant differences in total number of carbonylated proteins were not evident between genotypes. VENN analysis revealed definitive EtOH effects in both genotypes (Figure 3A-C). These effects were maintained with the addition of a single binge. A breakdown of Table S6 and the VENN analysis depicting the protein specific impact of both genotype and diet is presented in Tables S7, S8 and S9.
Figure 3. VENN analysis of carbonylated proteins identified following 116d EtOH +/− Binge in SV and GSTA4−/− mice.
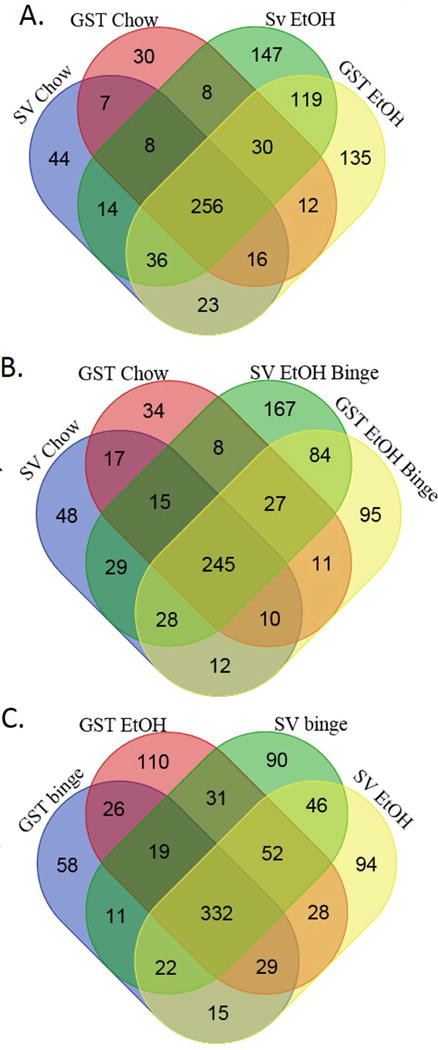
Data presented in Table S6 were subjected to VENN data analysis. A. Chow compared to 116d EtOH B. Chow compared to 116d EtOH + Binge C. 116d EtOH compared to 116d EtOH + Binge.
To determine if there were enriched pathways among the carbonylated proteins that could be explained by the genotype or dietary treatments, a bioinformatics approach using both Gene Ontology and KEGG analysis was utilized. The most consistent feature that we observed, was a statistically significant enrichment of carbonylated ribosomal proteins in the alcohol-exposed groups compared to chow-fed mice (Table S10 and S11). For each ribosomal protein from the enriched KEGG pathway, Ribosome (mmu03010), the percentages of animals in each group showing carbonylation of the ribosomal protein were calculated (Table S12) and a dendrogram constructed (Figure 4). Just 3 ribosomal proteins were detected as carbonylated in chow-fed animals, compared to 43 in the EtOH-exposed groups. To examine genotype differences in ribosomal carbonylation, (Table S12), relative percentages of carbonylation of each ribosomal protein were calculated. Although some proteins RSSA, RL8, RL11, RL12 were identified in a larger percent of animals in the GSTA4−/− groups, no overall genotype specific effects were evident. Among proteins that showed carbonylation in all four ethanol-treated groups but not in any of the two chow-fed groups, there were further three UDP glucuronosyl transferases (UD11, UD19 and UD17C) and glutathione S-transferase μ7 (GSTM7) that together represent an apparent enrichment of a Gene Ontology pathway (GO:0006805) for xenobiotic metabolism.
Figure 4. Impact of 116d EtOH +/− Binge on ribosomal protein carbonylation.
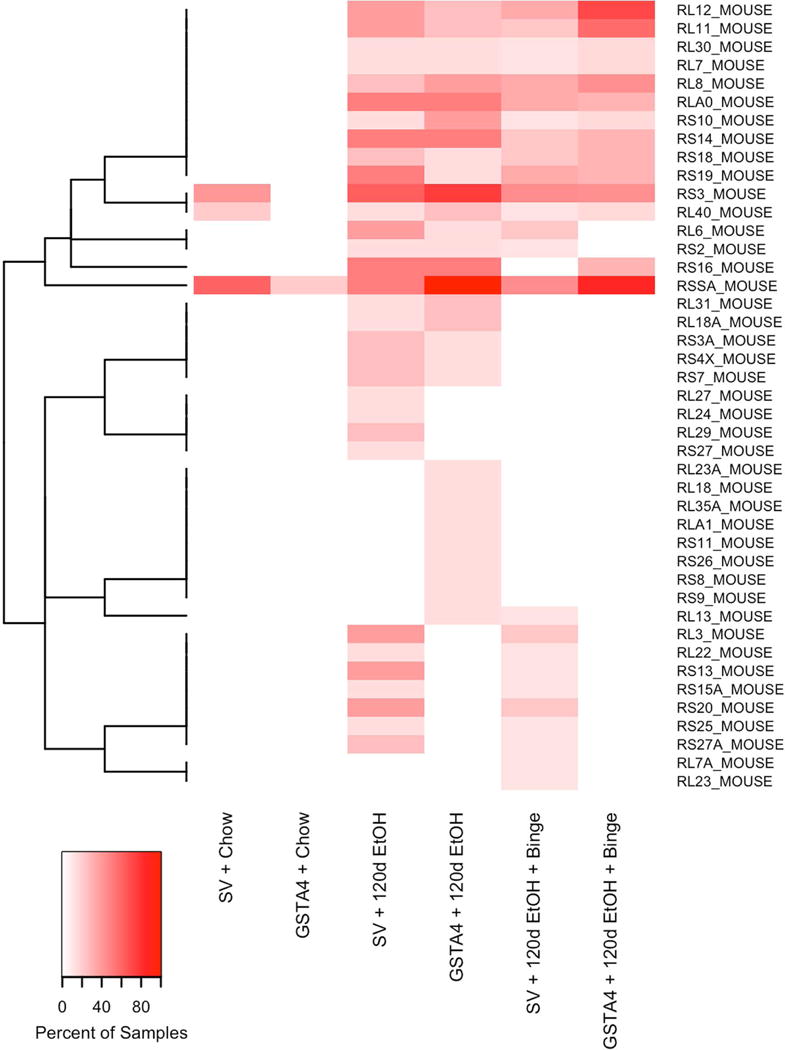
Percent of samples within each experimental group where a carbonylated protein from the enriched KEGG pathway. Each row represents a separate protein from the Ribosome KEGG pathway that was carbonylated in at least one sample and each column represents a different experimental group. The color gradient represents the percent of samples within the experimental group where the carbonylated protein was detected. White indicates the carbonylated protein was not detected in any sample within that experimental group. Proteins were clustered (dendrogram on left) based on binary indicators for detection in more than 10% of samples.
Addition of the EtOH binge had only minor effect on pathways with carbonylated proteins. Among 28 proteins with carbonylation in the two groups of ethanol-fed animals without a binge but not in the two groups receiving an ethanol binge, there was an apparent enrichment with 5 ribosomal proteins (Figure 4). However, there was no significant difference in the total number of carbonylated ribosomal proteins between animals receiving and not receiving the binge for either genotype (Table S10 and S11). A similar enrichment with 7 ribosomal proteins among 46 proteins that were carbonylated in alcohol-exposed WT mice but not GSTA4−/− mice was observed (Figure 4). Yet, there were no significant genotype differences in the number of total carbonylated ribosomal proteins (Table S10 and S11). Other genotype effects observed in alcohol-treated animals included 4 components (PRS7, PRS8, PSA1 and PSMD2) of the 26S proteasome complex that were carbonylated in WT and not in GSTA4−/− mice as well as 7 proteins involved in monocarboxylic acid metabolism (CP2DB, KAT1, MAOX, AL1A3, ACOT1, ACSM5 and ODP2 of pathway GO:0032787) that were carbonylated in GSTA4−/− mice and not in WT mice. The functional consequences of these differences in carbonylation are unknown.
Additional LC-MS/MS analysis was performed to further investigate the types of carbonylation and sites of carbonylation (Shearn et al., 2016a). As shown in Table S13, sites of protein carbonylation were identified on peptides originating from 22 different proteins. From the LC-MS/MS analysis, covalent 4-HNE modification was detected on histidine residue (His34) of albumin and to a lysine residue (Lys19,366) of titin. Covalent malondialdehyde (MDA) adduction was detected on titin (Lys6,321), regucalcin (Lys5) and meiosis inhibitor protein 1 (His1,000, His1,016). While our research focused on the role of GSTA4, carbonylation of other glutathione S-transferases was observed. For example, glutathione S-transferase μ1 (GSTM1) was carbonylated for both genotypes and under all dietary conditions (Table S6). In a previous study of ethanol feeding for 6 weeks, GSTM1 was also carbonylated in all experimental groups in both mitochondrial and cytosolic fractions (Shearn et al., 2016a). From Table S13, a heptenealdehyde adduct was identified on Heat Shock Protein 90 (Hsp90b, Lys559) as well as GSTM1 Lys51. Given the critical role that glutathione S-transferases play in mitigating EtOH-induced oxidative stress, we sought to determine the impact of carbonylation of Lys51 GSTμ by examining it location in relation to the active site using a previously reported crystal structure (pdb 1Xw6) (Patskovsky et al., 2006). As shown in Figure 5, Lys51 (Red) is located near the active site but is oriented away from where the GSH substrate (Blue) binds. Its close location to the active site suggests that carbonylation may affect its catalytic activity.
Figure 5. Identification of site of carbonylation on GSTμ following chronic EtOH consumption.
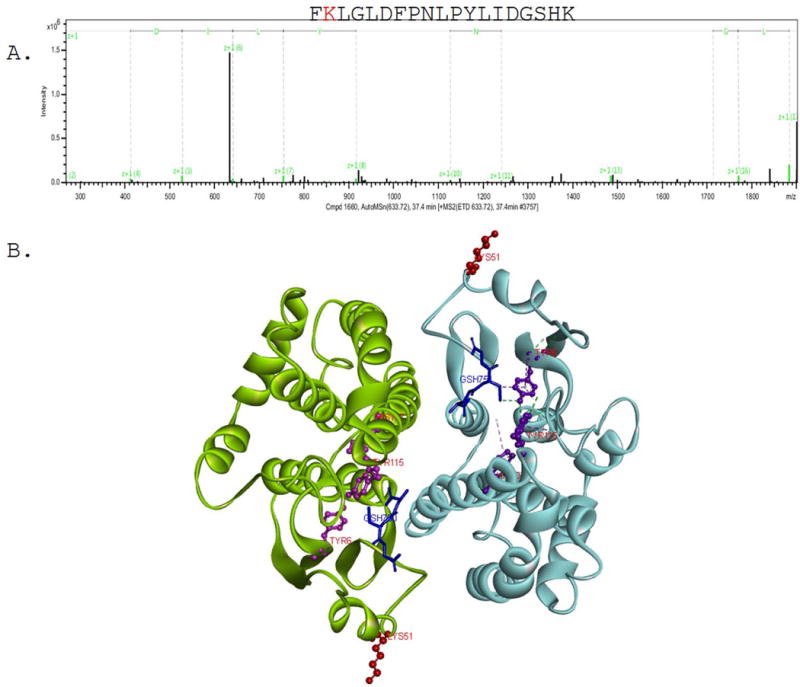
A. Representative electron-transfer dissociation (ETD) tandem mass spectra (MS/MS) of in vivo adducted GSTμ peptide isolated from SV EtOH, GSTA4−/− EtOH and SV EtOH + Binge groups. FK*LGLDFPNLPYLIDGSHK. B. Structural analysis of Lys51 GSTμ carbonylation using dimeric structure (PDB 1Xw6)(Patskovsky et al., 2006). Red is Lys51, Blue is GSH active site, Purple is Tyr6, His 107 and Tyr115 coordinate GSH (coordinating bonds shown on 1 dimer).
DISCUSSION
A major contributor to alcohol-induced liver injury is enhanced oxidative stress and the resulting increase in lipid peroxidation. In a 6-week mouse model of chronic ALD, global deletion of GSTA4 resulted in an increase in hepatic 4-HNE adduction and in mitochondrial protein carbonylation (Ronis et al. 2015; Shearn et al. 2016a). The phenotype of the GSTA4 deletion in combination with knock-out of the PPARα gene that renders mice highly steatotic, resulted in clear synergistic effects on fibrosis and necroinflammatory injury (Shearn et al., 2016a, Ronis et al., 2015). Thus, injury caused by GSTA4 deletion may depend on underlying steatosis. We hypothesized that ethanol exposure for a longer time period, 4 months rather than 6 weeks, combined with an ethanol binge would lead to greater hepatic injury. This may also be a better model of ALD in humans that is a disease typically appearing after many years of alcohol intake.
In our earlier 6-week study, deletion of GSTA4 in a PPARα knockout background significantly elevated serum ALT levels in ethanol-fed mice. While failing to reach statistical significance, ethanol-fed GSTA4−/− mice also had a higher mean serum ALT level compared to their respective WT controls (Ronis et al., 2015). Long-term ethanol feeding and a subsequent ethanol binge increased ALT levels in the current study. This was accompanied by an increase in hepatocellular proliferation measured by Ki67 expression in the binge group. Unexpectedly, differences in necrotic injury between genotypes were not apparent. We speculate that this may be due to compensatory responses that can occur during chronic ethanol consumption. Increased intestinal permeability and gut-derived endotoxin is a significant contributor to hepatic injury during ethanol consumption (Malaguarnera et al., 2014). Furthermore, data has indicated using acute murine models with combined endotoxin and ethanol exposure, injury is increased. This effect is mitigated by inhibition of the NADPH oxidase suggesting an oxidative stress component. In contrast, using chronic exposure models, when compared to ethanol alone, chronic ethanol plus endotoxin does not further increase injury. This effect was attributed to a decrease in CD14 receptor expression on Kupffer cells and an increase in anti-inflammatory cytokines including IL-10 indicating adaptation of the liver to continuous exposure to endotoxin (Jarvelainen et al., 1999).
In contrast to the previous 6-week feeding studies from our group, we observed clear main effects of the GSTA4 genotype on inflammatory markers (TNFα mRNA, IL-6 mRNA and infiltration with B220-positive cells) and matrix remodeling markers (MMP9 mRNA, MMP13 mRNA and Col1A1 mRNA) in the current study. The inflammatory parameters also exhibited significant dietary effects with higher levels in EtOH-exposed animals than in chow-fed animals. Likewise, MMP-9 expression was induced by alcohol feeding which is consistent with stellate cell activation downstream from increases in inflammatory responses after ethanol exposure.
Recently, it was reported that chronic murine models of ALD are associated with infiltration of macrophages whereas neutrophil infiltration occurs in the chronic/binge model (Bertola et al., 2013b, Bertola et al., 2013a). Using MPO as a marker for neutrophils, we find that 116d consumption of the EtOH diet induces hepatic neutrophil infiltration but infiltration is not enhanced by the addition of a binge. In murine models of ethanol exposure using the Tsukamoto-French intragastric method of EtOH administration, knockout of p47phox as well as the use of a NADPH oxidase inhibitor (diphenyleneiodonium sulfate) mitigate injury, validating the contribution of neutrophil-derived ROS produced from the NADPH oxidase complex in ethanol-induced injury (Kono et al., 2001, Kono et al., 2000). It should be noted however that the duration of EtOH administration in these studies was 4-weeks and the effects of an acute binge were not examined.
Effects caused by consumption of the ethanol diet for 116 days are combinations of effects mediated by the alcohol and effects mediated by replacing solid chow with a liquid high-fat diet. The effects of the final ethanol binge on hepatic injury and inflammatory status are more likely to be due to the ethanol per se. The inflammatory consequences of the binge are quite complex. While increased neutrophil infiltration and TNFα expression are indicative of increased inflammation, there are simultaneously fewer infiltrating T-cells and increased FoxP3 expression. FoxP3 is a marker for immunosuppressive regulatory T-cells. Thus, repair mechanisms operating to limit inflammation may also be activated by the ethanol binge.
Histologically, at 116d ETOH, the induction of Cyp2E1 is evident. One proposed consequence of EtOH-dependent elevation in Cyp2E1 is that during EtOH metabolism, as a byproduct, Cyp2E1 generates reactive species which would then contribute to observed increases in formation of products of lipid peroxidation. The data herein demonstrate that the generation of 4-HNE carbonylated proteins during EtOH consumption in the WT mouse is primarily peri-portal whereas Cyp2E1 expression is centri-lobular. In WT mice, the ethanol binge causes 4-HNE adduction to shift from a periportal a more evenly distributed phenomenon. The GSTA4−/− genotype had the intriguing effect of causing panlobular 4-HNE even in the absence of the ethanol binge. This may reflect that the GSTA4−/− genotype synthesizes more adducts even in the absence of an ethanol binge. The explanation for this is unclear, but may involve unknown breakdown or removal rates of 4-HNE adducts. Consistent with elevated 4-HNE adduction in alcohol-exposed animals compared to chow-fed animals, the total number of detected carbonylated proteins was higher in the ethanol-treated groups. There are other GST isoforms such as GSTπ and GSTμ that can utilize products of lipid peroxidation as substrates (Berhane et al., 1994). In an alcohol model, activation of GSTπ provided beneficial effects with respect to protein carbonylation (Chen et al., 2016). Further research will be necessary to determine how EtOH in combination with the GSTA4−/− genotype may impact other GST isoforms. Importantly though, 4HNE adducts are observed in the centrilobular region of the GSTA4−/− mouse following chronic EtOH indicating that chronic EtOH alone in this genotype is similar to the WT mouse following the combination of chronic EtOH plus a binge treatment. In other words, we suggest that the GSTA4−/− is already forming dangerous, pathologically important adducts even without the EtOH binge.
In both the 6 week study and the current 116d study, following EtOH consumption, GSTA4−/− mice exhibited an overall effect towards elevated expression of the pro-inflammatory cytokines TNFα and IFNγ as well as the B cell marker B220. This corresponded to an elevation of 4-HNE staining when compared to respective WT control groups indicating that elevated products of lipid peroxidation contributed to an adaptive immune response during early alcoholic liver disease (Ronis et al., 2015). Using a short-term (10 day) plus binge model of early ALD, flow cytometric analysis revealed increased B220+ cells and using immunohistochemistry, elevated IgA in the liver of EtOH fed mice (Moro-Sibilot et al., 2016). In the long term chronic EtOH model, deletion of GSTA4−/− resulted in significantly elevated B220 when compared to the WT 116d EtOH control. The addition of the binge further potentiated B220 expression and B cell numbers in the GSTA4−/− group but not in the WT group further supporting the contribution of products of lipid peroxidation in EtOH-induced B-cell responses. From our 2-way ANOVA analysis, both the GSTA4−/− genotype (elevated products of lipid peroxidation) and the addition of EtOH are driving the B-cell infiltration. As B-cells differentiate into plasma cells, there is progressive loss of B220 and elevation of CD138 (Shapiro-Shelef and Calame, 2005). However, we did not observe significant increases in expression of CD138 mRNA in the current study.
Bioinformatics analysis of carbonylated proteins indicates that compared to chow-fed animals, the most over-represented group of proteins that was significantly adducted after long term EtOH exposure were ribosomal proteins. This was not noted when comparing pair-fed and ethanol-fed animals in the previous 6-week feeding study (Shearn et al., 2016a). However, among 829 carbonylated proteins identified in the 6-week study, 23 were ribosomal proteins which represents a significant over-representation. Most of these carbonylated ribosomal proteins were detected in the microsomal fractions. However, 10 carbonylated ribosomal proteins were detected in the mitochondrial fraction (Shearn et al., 2016a). Previous reports have demonstrated a deficiency in mitochondrial protein synthesis in murine models of ALD (Karinch et al., 2008, Peters and Steele, 1982, Cahill and Cunningham, 2000, Cahill et al., 1996, Coleman and Cunningham, 1991). In addition, the structure of hepatic mitochondrial ribosomes is altered in murine models of ethanol (Patel and Cunningham, 2002). Using recombinant proteins, we have previously determined that protein exposure to reactive aldehydes can impact proteins both at the tertiary and quaternary structure impacting function (Shearn et al., 2014, Roede et al., 2008). Elevated carbonylation of ribosomal subunits during chronic EtOH consumption may impact ribosomal function by altering either overall ribosomal structure or ribosomal translation impacting protein synthesis. Future studies will need to be completed focusing on the impact of long term ethanol consumption on both mitochondrial protein synthesis as well as overall mitochondrial function. At the present, we can only speculate that ribosome carbonylation contributes to mitochondrial dysfunction. In turn, mitochondrial dysfunction and increased oxidative damage may ultimately lead to hepatic inflammation, as recently reviewed (Simões et al. 2018).
In the previous 6-week feeding study, GSTA4−/− mice compared to WT mice showed an increased propensity for carbonylated proteins in pathways of oxidative phosphorylation, glucose, fatty acid, glutathione and amino acid metabolic processes (Shearn et al., 2016a). Such pathways are also over-represented considering all the 733 carbonylated proteins identified in alcohol-exposed GSTA4−/− mice in the current study.
In summary, the GSTA4 deletion alters the pattern of 4-HNE adduction and induces hepatic inflammation after feeding alcohol as a component of a high fat liquid diet for 116 days.
The data presented here reinforce the hypothesis that elevated hepatic lipid peroxidation and protein carbonylation are sustained during long term ethanol consumption. Importantly, they provide additional correlative evidence that products of lipid peroxidation may be driving B-cell infiltration during chronic EtOH injury. When compared to our previous 6-week chronic EtOH study, the lack of severe pathology in WT mice following long term EtOH may result from adaptation in the inflammatory response perhaps as a result of development of tolerance to endotoxin.
Supplementary Material
Acknowledgments
This research was funded by a grant from the National Institutes of Health Institute of Alcohol Abuse and Alcoholism (NIAAA) 5R37 AA009300-22 (To D.R.P.). The authors also wish to acknowledge Joe Gomez and the Skaggs School of Pharmacy and Pharmaceutical Sciences Mass Spectrometry Core Facility for assistance in analysis of carbonylated proteins and E. Erin Smith, HTL(ASCP)CMQIHC of the University of Colorado Denver Cancer Center Research Histology Core for assistance in preparing histology slides. The UCDCCRHC is supported in part by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780 and the University of Colorado Cancer Center Grant (P30 CA046934).
Abbreviations
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- CID
collision-induced dissociation
- Cyp2E1
Cytochrome P4502E1
- ETD
electron transfer dissociation
- EtOH
ethanol
- GSTA4
glutathione S-transferase isoform A4
- HCC
hepatocarcinoma
- 4-HHE
4-hydroxy-2-hexenal
- 4-HNE
4-hydroxy-2-nonenal
- IFNγ
interferon gamma
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MDA
malondialdehyde
- MMP
matrix metalloproteinases
- MPO
Myeloperoxidase
- NASH
nonalcoholic steatohepatitis
- 4-ONE
4-oxo-2-nonenal
- PF
Pair-fed
- SNP
single nucleotide polymorphism
- TNFα
tumor necrosis factor alpha
- WCE
whole cell extract
Footnotes
DR. COLIN SHEARN (Orcid ID : 0000-0002-9618-9885)
Conflict of Interest
The authors have no conflicts of interest to report.
References
- Berhane K, Widersten M, Engstrom A, Kozarich JW, Mannervik B. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc Natl Acad Sci U S A. 1994;91:1480–1484. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013a;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013b;58:1814–1823. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill A, Baio DL, Ivester P, Cunningham CC. Differential effects of chronic ethanol consumption on hepatic mitochondrial and cytoplasmic ribosomes. Alcohol Clin Exp Res. 1996;20:1362–1367. doi: 10.1111/j.1530-0277.1996.tb01135.x. [DOI] [PubMed] [Google Scholar]
- Cahill A, Cunningham CC. Effects of chronic ethanol feeding on the protein composition of mitochondrial ribosomes. Electrophoresis. 2000;21:3420–3426. doi: 10.1002/1522-2683(20001001)21:16<3420::AID-ELPS3420>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Chen WY, Zhang J, Ghare S, Barve S, McClain C, Joshi-Barve S. Acrolein Is a Pathogenic Mediator of Alcoholic Liver Disease and the Scavenger Hydralazine Is Protective in Mice. Cell Mol Gastroenterol Hepatol. 2016;2:685–700. doi: 10.1016/j.jcmgh.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JZ, Singhal SS, Sharma A, Saini M, Yang Y, Awasthi S, Zimniak P, Awasthi YC. Transfection of mGSTA4 in HL-60 cells protects against 4-hydroxynonenal-induced apoptosis by inhibiting JNK-mediated signaling. Arch Biochem Biophys. 2001;392:197–207. doi: 10.1006/abbi.2001.2452. [DOI] [PubMed] [Google Scholar]
- Coleman WB, Cunningham CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim Biophys Acta. 1991;1058:178–186. doi: 10.1016/s0005-2728(05)80235-x. [DOI] [PubMed] [Google Scholar]
- Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Brestoff JR, Wiczer BM, Ilkayeva O, Cianflone K, Muoio DE, Arriaga EA, Bernlohr DA. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59:1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi S, Sharma R, Sharma A, Zimniak P, Ceci JD, Awasthi YC, Boor PJ. The course of CCl4 induced hepatotoxicity is altered in mGSTA4-4 null (−/−) mice. Toxicology. 2006;218:58–66. doi: 10.1016/j.tox.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, Yang Y, Awasthi S, Awasthi YC, Zimniak P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol Appl Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Gallagher EP, Huisden CM, Gardner JL. Transfection of HepG2 cells with hGSTA4 provides protection against 4-hydroxynonenal-mediated oxidative injury. Toxicol In Vitro. 2007;21:1365–1372. doi: 10.1016/j.tiv.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan JJ, Fritz KS, Tipney H, Smathers RL, Roede JR, Shearn CT, Hunter LE, Petersen DR. Profiling impaired hepatic endoplasmic reticulum glycosylation as a consequence of ethanol ingestion. J Proteome Res. 2011;10:1837–1847. doi: 10.1021/pr101101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan JJ, Smathers RL, Fritz KS, Epperson LE, Hunter LE, Petersen DR. Protein carbonylation in a murine model for early alcoholic liver disease. Chem Res Toxicol. 2012a;25:1012–1021. doi: 10.1021/tx300002q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan JJ, Smathers RL, Shearn CT, Fritz KS, Backos DS, Jiang H, Franklin CC, Orlicky DJ, Maclean KN, Petersen DR. Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease. J Toxicol. 2012b;2012:207594. doi: 10.1155/2012/207594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvelainen HA, Fang C, Ingelman-Sundberg M, Lindros KO. Effect of chronic coadministration of endotoxin and ethanol on rat liver pathology and proinflammatory and anti-inflammatory cytokines. Hepatology. 1999;29:1503–1510. doi: 10.1002/hep.510290508. [DOI] [PubMed] [Google Scholar]
- Kanehisa M. Enzyme Annotation and Metabolic Reconstruction Using KEGG. Methods Mol Biol. 2017;1611:135–145. doi: 10.1007/978-1-4939-7015-5_11. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karinch AM, Martin JH, Vary TC. Acute and chronic ethanol consumption differentially impact pathways limiting hepatic protein synthesis. Am J Physiol Endocrinol Metab. 2008;295:E3–9. doi: 10.1152/ajpendo.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Rusyn I, Uesugi T, Yamashina S, Connor HD, Dikalova A, Mason RP, Thurman RG. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1005–1012. doi: 10.1152/ajpgi.2001.280.5.G1005. [DOI] [PubMed] [Google Scholar]
- Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M. Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J Gastroenterol. 2014;20:16639–16648. doi: 10.3748/wjg.v20.i44.16639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, Traverse-Glehen A, Defrance T, Kaiserlian D, Dubois B. Mouse and Human Liver Contain Immunoglobulin A-Secreting Cells Originating From Peyer’s Patches and Directed Against Intestinal Antigens. Gastroenterology. 2016;151:311–323. doi: 10.1053/j.gastro.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Patel VB, Cunningham CC. Altered hepatic mitochondrial ribosome structure following chronic ethanol consumption. Arch Biochem Biophys. 2002;398:41–50. doi: 10.1006/abbi.2001.2701. [DOI] [PubMed] [Google Scholar]
- Patskovsky Y, Patskovska L, Almo SC, Listowsky I. Transition state model and mechanism of nucleophilic aromatic substitution reactions catalyzed by human glutathione S-transferase M1a-1a. Biochemistry. 2006;45:3852–3862. doi: 10.1021/bi051823+. [DOI] [PubMed] [Google Scholar]
- Peters JE, Steele WJ. Differential effect of chronic ethanol administration on rates of protein synthesis on free and membrane-bound polysomes in vivo in rat liver during dependence development. Biochem Pharmacol. 1982;31:2059–2063. doi: 10.1016/0006-2952(82)90421-x. [DOI] [PubMed] [Google Scholar]
- Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chem Res Toxicol. 2008;21:2289–2299. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- Ronis M, Mercer K, Engi B, Pulliam C, Zimniak P, Hennings L, Shearn C, Badger T, Petersen D. Global Deletion of Glutathione S-Transferase A4 Exacerbates Developmental Nonalcoholic Steatohepatitis. Am J Pathol. 2017;187:418–430. doi: 10.1016/j.ajpath.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronis MJ, Mercer KE, Gannon B, Engi B, Zimniak P, Shearn CT, Orlicky DJ, Albano E, Badger TM, Petersen DR. Increased 4-hydroxynonenal protein adducts in male GSTA4-4/PPAR-alpha double knockout mice enhance injury during early stages of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2015;308:G403–415. doi: 10.1152/ajpgi.00154.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- Shearn CT, Backos DS, Orlicky DJ, Smathers-McCullough RL, Petersen DR. Identification of 5′ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem. 2014;289:15449–15462. doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, Fritz KS, Shearn AH, Saba LM, Mercer KE, Engi B, Galligan JJ, Zimniak P, Orlicky DJ, Ronis MJ, Petersen DR. Deletion of GSTA4-4 results in increased mitochondrial post-translational modification of proteins by reactive aldehydes following chronic ethanol consumption in mice. Redox Biol. 2016a;7:68–77. doi: 10.1016/j.redox.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, Orlicky DJ, McCullough RL, Jiang H, Maclean KN, Mercer KE, Stiles BL, Saba LM, Ronis MJ, Petersen DR. Liver-Specific Deletion of Phosphatase and Tensin Homolog Deleted on Chromosome 10 Significantly Ameliorates Chronic EtOH-Induced Increases in Hepatocellular Damage. PLoS One. 2016b;11:e0154152. doi: 10.1371/journal.pone.0154152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, Orlicky DJ, Saba LM, Shearn AH, Petersen DR. Increased hepatocellular protein carbonylation in human end-stage alcoholic cirrhosis. Free Radic Biol Med. 2015;89:1144–1153. doi: 10.1016/j.freeradbiomed.2015.10.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, Petersen DR. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med. 2013;65:680–692. doi: 10.1016/j.freeradbiomed.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Niemczyk M, Saini D, Awasthi YC, Zimniak L, Zimniak P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry. 2008;47:3900–3911. doi: 10.1021/bi702124u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.