

Bansal and Simon discuss strategies to block glutathione synthesis and utilization pathways to inhibit tumor propagation and treatment resistance.
Abstract
Glutathione (GSH) is the most abundant antioxidant found in living organisms and has multiple functions, most of which maintain cellular redox homeostasis. GSH preserves sufficient levels of cysteine and detoxifies xenobiotics while also conferring therapeutic resistance to cancer cells. However, GSH metabolism plays both beneficial and pathogenic roles in a variety of malignancies. It is crucial to the removal and detoxification of carcinogens, and alterations in this pathway can have a profound effect on cell survival. Excess GSH promotes tumor progression, where elevated levels correlate with increased metastasis. In this review, we discuss recent studies that focus on deciphering the role of GSH in tumor initiation and progression as well as mechanisms underlying how GSH imparts treatment resistance to growing cancers. Targeting GSH synthesis/utilization therefore represents a potential means of rendering tumor cells more susceptible to different treatment options such as chemotherapy and radiotherapy.
Introduction
Reactive oxygen species (ROS; H2O2, O2-, OH-, etc.) are produced by aerobic cells under physiological conditions. These compounds, when produced in larger quantities under stress, can lead to extensive damage of DNA, proteins, and plasma/organelle membranes in the cell (Olinski et al., 1992; Ames et al., 1993; Shigenaga et al., 1994). Multiple repair mechanisms remove DNA lesions and ultimately degrade misfolded proteins (Ames et al., 1993; Shigenaga et al., 1994). However, ROS production is significantly increased in cancer cells because of mitochondrial dysfunction, altered metabolism, and frequent genetic mutations, resulting in an accumulation of large amounts of oxidized protein, DNA, and lipids (Demple and Harrison, 1994). Therefore, as an adaptive response, cancer cells harbor elevated levels of ROS-scavenging molecules. Antioxidant mechanisms include enhanced expression of molecules that can modulate ROS accumulation by both enzymatic and nonenzymatic means (Toyokuni et al., 1995; Gupta et al., 2014). Enzymatic antioxidant responses include increased abundance of superoxide dismutase or catalase (Sies, 1993; Ighodaro and Akinloye, 2017; Wang et al., 2018). In addition, other small redox protein-encoding genes that generate thioredoxins, heme oxygenases, and peroxiredoxins are also stimulated (Sies, 1993).
Glutathione (GSH) is an antioxidant that acts as a free radical scavenger and a detoxifying agent in cells. It is useful in a multitude of processes, cellular proliferation, cell division, and differentiation, and is the most commonly elevated metabolite detected during oxidative stress. Under physiological conditions, reduced GSH is the major form present with a concentration 10- to 100-fold higher than the oxidized species (GSH disulfide [GSSG]). Under oxidative stress, GSH is converted by GSH-dependent peroxidases into GSSG upon its reaction with ROS. GSH is synthesized in the cytosol and further distributed to different organelles. Apart from the classical antioxidant role, GSH also has several organelle-specific functions discussed in detail by Lushchak (2012). More than 10% of the synthesized GSH is found in mitochondria, where it reacts with ROS and prevents apoptosis. A highly reducing environment is maintained in subcellular compartments, e.g., the cytosol, nucleus, mitochondrial matrix, and peroxisome, to facilitate proper protein folding and activity. In contrast, the ER maintains a highly oxidized environment and increased levels of GSSG. This supports the functional conformation of peptides, especially for the addition of disulfide bonds to the nascent secretory and membrane proteins. Additionally, a high GSH/GSSG ratio in the nucleus ensures synthesis of deoxyribonucleotides from ribonucleotides and maintenance of sulfhydryl groups in proteins for proper nucleic acid biosynthesis and DNA repair. This ratio in each organelle also reflects the metabolic pathways and resulting oxidative burdens that they face throughout phases of cell growth. Furthermore, GSH is involved in (a) detoxification of xenobiotics, (b) maintenance of cysteine pools, (c) maturation of iron–sulfur clusters of diverse proteins, and (d) regulation of transcription factors related to redox signaling (Estrela et al., 2006; Traverso et al., 2013). GSH helps maintain protein sulfhydryl groups in a reduced form by supporting a balance of thiol redox potential in cells. As a result, disturbances in GSH homeostasis are often found in multiple pathologies, e.g., neurodegenerative disorders, cancer, cystic fibrosis, liver disorders, and diabetes (Forman et al., 2009).
With respect to cancer, GSH plays a dual role in its progression. It is crucial in the removal and detoxification of carcinogens, and alterations in this pathway can have a profound effect on cell survival. However, elevated levels of GSH in tumor cells are able to protect such cells in bone marrow, breast, colon, larynx, and lung cancers by conferring resistance to several chemotherapeutic drugs (Wu et al., 2004; Lu, 2009). The effect of GSH on oxidative stress and cancer initiation and progression is also complicated because of the dual role of ROS in these processes. Moderate ROS levels can support survival and proliferation by activating signaling pathways that can contribute to tumor growth in stressful tumor microenvironments (Fig. 1). However, excessive ROS accumulation, failure of proper scavenging mechanisms, or antioxidant scarcity results in severe damage of biomolecules, triggering cell death. Therefore, cancer cells need to maintain an intricate balance of antioxidant levels to survive. Additionally, ROS are able to regulate the potential of cancer cells to metastasize to distant locations. Data supporting this indicated that elevated GSH promotes metastasis in both melanoma and liver cancer (Carretero et al., 1999; Huang et al., 2001). In addition to GSH, other common antioxidants, N-acetyl cysteine (NAC) and the vitamin E analogue trolox, promote distant metastasis (Carretero et al., 1999; Huang et al., 2001; Sayin et al., 2014). These data emphasize the dual roles of ROS and GSH in cancer initiation and progression (Galadari et al., 2017).
Figure 1.
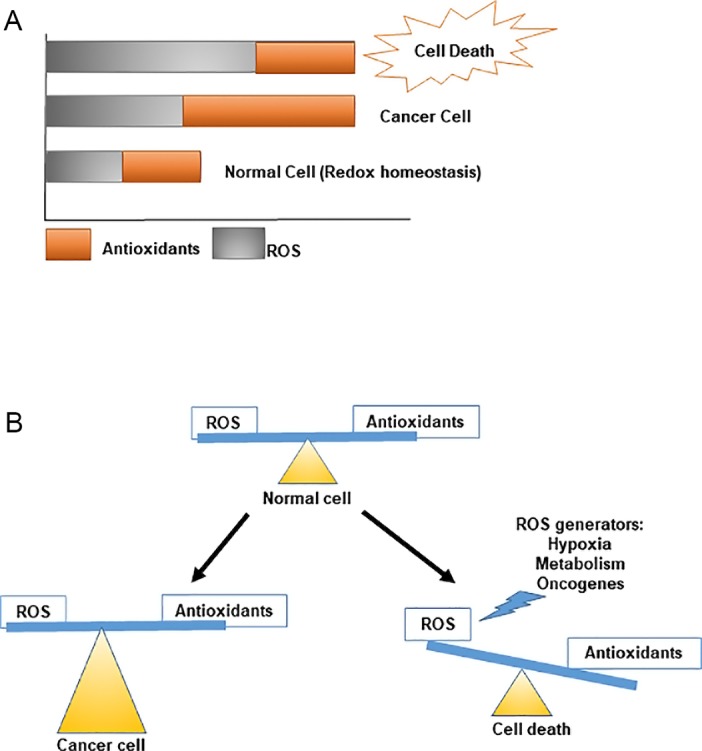
Maintenance of redox homeostasis in normal versus cancer cells. (A) Cells maintain a balance of antioxidants against ROS generated by the different physiological processes. (B) Cellular (Li et al., 2014a) redox states are maintained by modulating ROS production and elimination. Under normal conditions, cells maintain homeostasis by producing enough antioxidants to balance ROS production. However, because of hypoxia and metabolic alterations in cancer cells, increased amounts of ROS are generated, which are counteracted by enhanced antioxidant responses. However, when this balance is disrupted, high ROS levels can disrupt normal cellular machinery, triggering cell death.
GSH synthetic enzymes involved in tumor progression
GSH is the most prevalent nonprotein thiol in eukaryotic cells and exists in both thiol-reduced (GSH) and disulfide-oxidized (GSSG) forms (Kaplowitz et al., 1985). GSH is the predominant form, existing in millimolar concentrations in most cells (liver 5–10 mM; Wu et al., 2004). This tripeptide is produced by de novo biosynthesis involving a two-step, ATP-dependent enzymatic reaction (Fig. 2). The first is catalyzed by glutamate cysteine ligase (GCL), which ligates cysteine to glutamate to produce γ-glutamylcysteine. This dipeptide is then combined with glycine by GSH synthetase (GSS) to produce GSH (Lu, 2009). Besides the synthetic pathway described above, cells can convert GSSG to GSH (Lu, 2013) catalyzed by GSH reductase, which requires NADPH as a substrate and thus links NADPH levels directly to GSH synthesis. Therefore, high GSH/GSSG ratios in cancer cells can also be explained by increased flux through the pentose phosphate pathway (PPP), which produces NADPH (Li et al., 2014a; Zhang et al., 2016). Consistent with increased PPP flux, clear cell renal cell carcinoma (ccRCC) tumors display significant accumulation of reduced GSH and PPP-related metabolites (Li et al., 2014a).
Figure 2.

De novo biosynthesis of GSH. GSH is generated from glutamic acid, cysteine, and glycine in two successive ATP-dependent enzymatic steps. First, γ-glutamylcysteine is synthesized by a GCL, a reaction consuming glutamic acid and cysteine, and forming a γ-peptide bond. The second step is catalyzed by GSS, adding glycine to the C terminus of γ-glutamylcysteine, resulting in the final GSH product.
Regulation of GSH levels in the cells
Under physiological conditions, GCL expression, its enzymatic activity, and cysteine abundance constitute rate-limiting steps for GSH synthesis (Lu, 2009, 2013). GCL levels are controlled by the nuclear factor erythroid 2–related factor 2 (NRF2) transcription factor (discussed in more detail later in this review). GCL is a heterodimer consisting of a 73-kD catalytic subunit (GCLC) and 31-kD modulatory subunit (GCLM; Dickinson et al., 2004; Andringa et al., 2006; Lu, 2009). GCLM null mutant mice have <25% of cellular GSH yet are surprisingly viable. In contrast, high GCLM expression has been found to be associated with therapeutic resistance in breast cancer (Lu, 2009). Additionally, GCLM mutations result in delayed tumor onset in sarcoma, mammary, and lymphoma mouse models (Fujimori et al., 2004; Lu, 2009), making GCLM an effective pharmacologic target to combat chemotherapeutic resistance in these cancers. Under physiological conditions, the GCLC/GCLM heterodimer is regulated by feedback inhibition by its product GSH (Lu, 2009). However, under oxidative stress or disease conditions, GCL levels increase because of the action of several nuclear transcription factors: NRF2–Kelch-like ECH-associated protein 1 (KEAP1) and NFκB (Griffith and Mulcahy, 1999; Lu, 2013). GCL deficiency is a rare autosomal disease characterized by reduced GSH, hemolytic anemia, and neurological symptoms (Njålsson and Norgren, 2005; Ristoff and Larsson, 2007). High GCLC levels are found in patients suffering from melanoma and lung cancer (Fujimori et al., 2004). Moreover, increased GCLC enzymatic activity has been detected in renal cell carcinoma patients (Li et al., 2014b). GCLC expression is attenuated by Myc-induced miR-18a, and these tumors are highly susceptible to oxidative stress (Anderton et al., 2017). Myc also regulates GCLC expression in effector T cells, which can modulate immunotherapeutic responses in cancer patients (Park and Pan, 2015). Reduction of GSH production by the irreversible GCL inhibitor buthionine sulfoxime (BSO) promotes apoptosis in breast and ovarian cancer cells and attenuates cell growth in esophageal cancer (Andringa et al., 2006). Combinatorial therapy with BSO is being used in clinical trials to determine whether patients respond better to melphalan in resistant neuroblastoma patients (Villablanca et al., 2016; Anderton et al., 2017).
The enzyme GSS, involved in catalyzing the second step of de novo GSH biosynthesis, is not regulated by feedback inhibition by GSH. However, GSS activity is also important in certain disease conditions where an inactive enzyme results in γ-glutamylcysteine accumulation (Fig. 2). This intermediate can be converted to 5-oxoproline by γ-glutamylcyclotransferase and cause severe hemolytic anemia, metabolic acidosis, and central nervous system damage (Shi et al., 1996). Additionally, although increased GSS and GCLC are found in colon cancer patients (Kim et al., 2015), GSS variants are also correlated with the recurrence of bladder cancer and small cell lung cancer (Ke et al., 2015). However, roles of GSS in cancers have not been studied in detail, and few studies have been performed to discover inhibitors and regulators of GSS activity, which remains an area of active research that will help explore GSS as a possible therapeutic target in cancer cells.
Besides induction by common stressors like free radicals, peroxides, lipid peroxides, and heavy metals, increased GSH production can also be a byproduct of cancer cell metabolic reprogramming (Estrela et al., 2006). Oncogenic PI3K signaling has been recently shown to stimulate GSH production in breast cancer cells (Lien et al., 2016) specifically for tumor samples with mutations in this pathway. Additionally, in MYCN-amplified neuroblastomas and c-Myc–overexpressing Burkitt’s lymphomas, oncogenic Myc regulates the expression of amino acid transporters, which in turn modulate the availability of glutamine, which is the precursor of GSH synthesis. Moreover, if the cells encounter reduced amounts of cysteine, a unique cell death program known as ferroptosis is triggered. Ferroptosis is also triggered by the loss of GSH peroxidase 4 (GPX4), which leads to the accumulation of lipid peroxides (Yang and Stockwell, 2016; Yu and Long, 2016). Here GSH acts as a cofactor of GPX4; therefore, modulation of amino acid availability and in turn GSH production becomes an important factor for this form of cell death. GSH is also one of the most overrepresented metabolites in ccRCC, lung cancer, and melanoma patient samples relative to normal tissues (Gamcsik et al., 2012; Li et al., 2014a; Hakimi et al., 2016). Its increased levels are correlated with patients who relapsed after surgical removal of the primary tumor (Hakimi et al., 2016). In accordance with the data discussed earlier stating increased GSH levels found in cancers, BSO (commonly used as a synthetic inhibitor of GSH production) confers increased sensitivity to chemotherapeutics in myeloma and neck cancers (Tagde et al., 2014). Several other drugs known to reduce cellular GSH levels are currently being used in clinical trials to improve efficacy of targeted therapy; for example, disulfiram induces melanoma cell apoptosis by shifting the ratio of GSH/GSSG toward its oxidized state and is now being used in phase I/II clinical research in metastatic melanoma (Conticello et al., 2012). Similarly, arsenic trioxide chemotherapeutics combined with agents that deplete cellular GSH have been approved for the treatment of nonacute promyelocytic leukemia (Traverso et al., 2013). Therefore, modulating levels of GSH by inhibiting different steps of its de novo biosynthesis is a viable option either to increase the sensitivity of tumor cells to chemotherapeutics or for other targeted therapy.
γ Glutamyl cycle in cancer progression and therapeutic resistance
γ Glutamyl transferase (GGT) is a membrane-bound enzyme that catalyzes the degradation of extracellular GSH, favoring production of constituent glutamate and cysteine, needed for the synthesis of intracellular GSH (Fig. 3). This pathway, also known as the γ glutamyl cycle (Traverso et al., 2013), is an alternative to the de novo biosynthetic pathway and plays an important role in maintaining intracellular GSH and cysteine (Zhang and Forman, 2009). GGT levels are significantly up-regulated under oxidative stress, especially in highly metabolic cancer cells (Lu, 2009; Alanazi et al., 2015). GGT depletion in mice results in glutathionuria and cysteine deficiency because of decreased tissue GSH. These phenotypes can be rescued by addition of NAC treatment (Zhang et al., 2005). NAC acts as a source of external cysteine, implicating cysteine as a rate-limiting amino acid in this context (Zhang et al., 2005).
Figure 3.
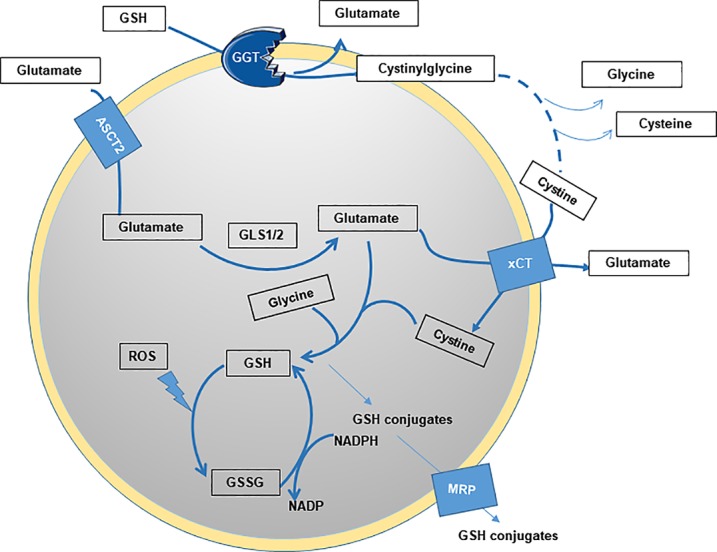
Steps of GSH production and utilization. GGT is a membrane-bound enzyme that catalyzes the degradation of extracellular GSH, favoring the production of constituent amino acids for the synthesis of intracellular GSH. It also catalyzes the transfer of the glutamyl moiety of GSH, linked through the glutamate γ-carboxylic acid, to acceptor molecules including amino acids and peptides. Other steps of regulation include the expression levels of transporters of glutamate (ASCT2) and cystine (xCT).
GGT is a heterodimeric glycoprotein consisting of two subunits, where molecular masses of the two chains are between 38 and 72 kD for the large subunit and 20 and 66 kD for the small subunit (Terzyan et al., 2015). These variations are explained by differential protein glycosylation on seven distinct N-linked glycosylation sites (Darbouy et al., 1991). GGT is most commonly expressed on the luminal surfaces of secretory cells, particularly in the bile ducts and renal proximal tubules (Darbouy et al., 1991). Previous research has reported that GGT is dysregulated in several tumor types, namely, ovary, colon, liver, astrocytic glioma, soft tissue sarcoma, melanoma, and leukemia (Corti et al., 2010).
GGT also catalyzes the transfer of the GSH glutamyl moiety, linked through the glutamate γ-carboxylic acid, to acceptor molecules like amino acids and peptides (Fig. 3). Cisplatin, a common chemotherapeutic, forms adducts with cysteinyl-glycine, a byproduct of GGT activity, much faster than with GSH (Pompella et al., 2007; Corti et al., 2010). Therefore, high GGT levels also correlate with therapeutic resistance and worse prognosis in breast cancer and sarcoma patients. TCGA analysis revealed that increased GGT is found in ccRCC tumor patient samples compared with adjacent normal tissues, and its expression in the serum of patients is correlated with worse survival rates (Cancer Genome Atlas Research Network, 2013; Hofbauer et al., 2014). GGT is also an important enzyme in the detoxifying renal mercapturic acid pathway (Zhang et al., 2005). Cisplatin is metabolized to a nephrotoxin when cleaved by GGT1 through this pathway, which can be inhibited by GGT knockdown (Zhang and Forman, 2009). Therefore, GGT1 is also overexpressed in cancer cells selected for resistance to alkylating agents or prooxidant therapeutics such as melphalan, oxaliplatin, and cisplatin.
GGT inhibition represents a novel approach to reduce intracellular GSH levels as a means of sensitizing them to different chemotherapeutics. The most common approach is the use of glutamate analogues like acivin, L azaserine, 6-diazo-5-oxo-l-norleucine, and boronate derivatives (Terzyan et al., 2015). However, high toxicity is seen in human patients. Novel classes of noncompetitive inhibitors like OU749, which have low toxicity, can also be used to make the cancer cells sensitive to therapy by lowering the GSH and cysteine availability (Corti et al., 2010).
Regulation of GSH levels by modulating precursor amino acids availability in cancer cells
Cysteine is a nonessential amino acid for protein synthesis, which stabilizes extracellular proteins by forming intraprotein disulfide bonds. Additionally, cysteine is commonly present in protease metal-binding enzymatic active sites (Gout et al., 1997). Multiple cellular pathways contribute to cysteine production. Apart from the γ glutamyl cycle discussed above, another source of cysteine is via transsulfuration of amino acid substrates. Methionine is converted to S-adenosyl methionine (SAM) by the enzyme methionine adenosyltransferase (Lu and Mato, 2012), which is then hydrolyzed to homocysteine and converted to cystathionine by two pyridoxal phosphate–dependent enzymes. This can then be cleaved to produce α-ketobutyrate and cysteine in subsequent reactions (Kredich, 2008). Metabolomic analyses of ccRCC tumors reveal increased amounts of serine, homocysteine, SAM, and S-adenosyl homocysteine (SAH) in low-stage tumors (Li et al., 2014a; Hakimi et al., 2016) compared with controls. High-stage or more aggressive tumors show increased accumulation of α-hydroxybutyrate and products of methionine metabolism, i.e., SAH, SAM, and homocysteine (Hakimi et al., 2016). These data indicate that there is preference for cysteine synthesis in early as well as late stage/aggressive ccRCC through a transsulfuration pathway.
Cysteine is also taken up from the tumor microenvironment by a sodium-independent cystine–glutamate antiporter that is chloride dependent, known as system Xc- or xCT, and encoded by the gene SLC7A11 (Shin et al., 2017). This transporter consists of light (xCT) and heavy (4F2) chains, linked by disulfide bridges. The light xCT chain also has 12 transmembrane domains and primarily imports cystine, the oxidized dimeric form of cysteine, in exchange for glutamate, contributing to tumor growth (Lo et al., 2008). Therefore, besides the biosynthetic pathway, tumor cells modulate cysteine levels to regulate its availability for GSH production as will be discussed.
SLC7A11 is not required for normal mouse development but has been shown to be important for the survival of prostate, breast, kidney, and colon cancer cells (Kredich, 2008). Moreover, perturbation of xCT levels in these cancers increases their sensitivity to chemotherapeutics, making it an appealing target for combinatorial therapy (Lo et al., 2008). Of note, cancer stem cells have been shown to up-regulate xCT levels, which increase intracellular GSH accumulation (Lewerenz et al., 2013). Sulfasalazine and erastin are the two most commonly used inhibitors of SLC7A11 and can be used as therapeutics to increase sensitivity to other agents (Lewerenz et al., 2013). These compounds induce a specific type of cell death, called ferroptosis (Yang and Stockwell, 2016), a nonapoptotic, peroxidation-driven mechanism dependent on cellular iron availability (Fujimori et al., 2004; Yu and Long, 2016). Suppression of cysteine desulfurase (NFS1) induces an iron starvation response, which can trigger this kind of cell death when exposed to ROS (Alvarez et al., 2017). Similarly, sulfasalazine also induces ferroptosis in diffuse large B cell lymphoma, which exhibits low expression of this cysteine transporter system (Gout et al., 2001). Finally, p53 suppresses xCT expression, indicating that xCT inhibitors could be effectively used to limit growth of p53 WT tumors (Jiang et al., 2015).
Besides cysteine, other amino acids like glutamine, glutamate, and glycine are also important in cell growth and GSH synthesis. Glutamine availability broadly impacts GSH production by three different mechanisms. First, glutamine is the primary source of glutamate, producing glutamate via two isozymes, glutaminase 1 and 2 (GLS1 and GLS2; Yu and Long, 2016; Zhang et al., 2017). GLS activity is tightly regulated to maintain appropriate amounts of intracellular glutamate for GSH production. Glutamine can also be transported by several different amino acid transporter systems. Among these, solute carrier family 1, member 5 (SLC1A5/ASCT2), is the most commonly overexpressed transporter protein in different human cancer cells addicted to excess extracellular glutamine (Yang et al., 2017). Glutamine, and consequently GSH levels, is reduced in cells with low ASCT2 expression, along with decreased GLS1 and GLS2. Interestingly, this system is also regulated by c-Myc in prostate cancer and lymphoma cells (Yang et al., 2017; Zhang et al., 2017). Second, glutamine contributes to the maintenance of GSH in its reduced form by supporting the production of NADPH through malic acid regulation (Zhang et al., 2017). This NADPH is crucial for the conversion of GSSG to GSH as discussed earlier. Third, the xCT transporter system described previously is also dependent on glutamate availability to the cells. Here, one molecule of glutamate is exchanged for every molecule of cysteine taken in. Collectively, abundances of glutamine and glutamate are crucial for the maintenance of proper cellular GSH (Zhang et al., 2017).
Glycine is another critical amino acid required for GSH production and maintenance of other metabolic processes, like purine biosynthesis in rapidly dividing cancer cells. Restricting the levels of serine and glycine reduced the levels of GSH synthesis and increased ROS in cancer cells in vitro. Serine can also be produced through transsulfuration pathway in some cases. In tumors where this pathway is interrupted, an increased accumulation of 3-phosphoglycerate has been detected (Locasale et al., 2011). Interestingly, glycine uptake and catabolism promote tumorigenesis and malignancy, suggesting that glycine metabolism could also be a target for therapeutic intervention (Jain et al., 2012). By assessing variations in the levels of >200 metabolites in the NCI-60 cell line series, both glycine consumption and expression of enzymes in the mitochondrial glycine biosynthetic pathway (but not the corresponding cytosolic enzymes) were found to correlate with cancer cell proliferation rates. This indicates that they might be dependent on GSH for their growth and survival. However, these notions need to be studied in more detail in the future (Jain et al., 2012).
Role of NRF2 and KEAP1 in the regulation of GSH abundance
NRF2 is regarded as a major contributor to cell survival and redox balance and is regulated by KEAP1, an adapter protein of Cullin 3 (Cul3) containing E3 ubiquitin ligases. Under basal conditions, KEAP1 binds to specific NRF2 motifs, leading to ubiquitination and subsequent NRF2 degradation (Kansanen et al., 2013). However, under oxidative stress, cysteine residues on KEAP1 are modified, altering its conformation and disrupting its association with NRF2 and subsequent NRF2 stabilization (Taguchi and Yamamoto, 2017). Free NRF2 then translocates to the nucleus, where it binds to antioxidant response elements throughout the genome, to activate downstream effector genes (Gupta et al., 2014; Taguchi and Yamamoto, 2017). In addition to the KEAP1-dependent pathway, NRF2 degradation is regulated by an additional β-TrCP–dependent degradation pathway, via NRF2 phosphorylation by GSK3 in a PI3K-dependent manner (Hayes et al., 2015). Phosphorylated NRF2 is then ubiquitinated by β-TrCP and degraded by the proteasome. In accordance with phenomena described above, simultaneous inactivation of phosphatase and tensin homolog (PTEN; a phosphoinositide 3-phosphatase) and KEAP1 has an additive effect on NRF2 activity (Taguchi and Yamamoto, 2017). The antioxidant response genes activated by NRF2 include genes responsible for (a) regulating GSH synthesis and metabolism, (b) antioxidant proteins like GPX, (c) drug-metabolizing enzymes and xenobiotic transporters, and (d) additional stress response proteins (Jaramillo and Zhang, 2013).
Recent studies have demonstrated that NRF2 promotes the survival of not only normal cells but also their malignant counterparts by increasing the expression of several cytoprotective genes (Magesh et al., 2012; Kansanen et al., 2013; Sayin et al., 2017; Taguchi and Yamamoto, 2017). NRF2 and KEAP1 are among the most frequently mutated genes in squamous cell lung cancers and hepatocellular carcinoma. Mutations in KEAP1 or NRF2 lead to metabolic rewiring of tumors toward increased glutamine consumption for GSH synthesis (Romero et al., 2017; Sayin et al., 2017). Several mechanisms appear to be responsible for increased NRF2 activity in cancers: somatic mutations in KEAP1, CUL3, or NRF2; epigenetic silencing of KEAP; transcriptional NRF2 up-regulation through oncogene-dependent signaling and modification of KEAP1 by metabolic alterations (Kansanen et al., 2013); and cysteine modification of KEAP1, leading to NRF2 accumulation. NRF2 and KEAP1 somatic mutations have been shown to correlate with poor clinical outcome in several cancers such as renal cell carcinoma and pancreatic cancer (Jaramillo and Zhang, 2013; Kansanen et al., 2013). NRF2 activation can also result in increased xCT accumulation, which eventually results in high GSH dependency of the affected cells.
Considering the important role NRF2 plays in tumor progression, thorough screens to identify potent NRF2 inhibitors have been performed by different groups. Small molecule inhibitors ML385 and CPUY192018 are the two compounds that have been discovered recently (Lu et al., 2016). The plant-derived compound brusatol also decreases NRF2 protein levels (Ren et al., 2011), and use of any of these compounds improves sensitivity to chemotherapeutics such as cisplatin and gemcitabine. Other small molecules that suppress NRF2 activity are retinoic acid, ascorbic acid, and luteolin (Wang et al., 2007; Gao et al., 2014). However, they are not very specific in their actions and also can promote NRF2 activity in some instances. Therefore, more thorough research needs to be performed to discover effective and highly selective inhibitors of NRF2 activity (Magesh et al., 2012).
GST and its role in therapeutic resistance
GST is primarily responsible for binding to xenobiotics and GSH, forming conjugates that are then secreted extracellularly (Zhang and Forman, 2009; Gamcsik et al., 2012; Lu, 2013). There are primarily three types of GSTs: cytosolic, mitochondrial, and nuclear. Specifically, these GSTs bind both the substrate and GSH at different sites of the enzyme, eventually activating the thiol group of GSH to enable nucleophilic attack on the substrate. Compounds that are targeted in this manner include pesticides, herbicides, carcinogens, and even certain chemotherapeutics. Hence, GST overexpression is known to be one of the underlying reasons for therapeutic resistance in some cancers (Gamcsik et al., 2012). The GSH–GST–drug conjugate formed is effluxed out of the cell via multiple resistance-associated protein (MRP1) transporters. MRP1 accumulation is often increased in drug-resistant cancer cells along with GST. Several inhibitors of GSTs have been developed to improve the response of human tumors to chemotherapeutics. Ethacraplatin, ethacrynic acid, and its analogues are currently being tested for their toxicity and tolerance in patients with invasive bladder cancer (Gamcsik et al., 2012). Collectively, a combination of inhibitors of GSH synthesis and/or its utilization with either chemotherapeutics or targeted treatment might increase the sensitivity of such drugs and provide viable options for patients suffering from therapy-resistant tumors.
Conclusion
The literature reviewed in our work indicates that GSH and GSH-related moieties play a significant role in tumor initiation, progression, and drug resistance. Although it has been known for some time that GSH is important in these processes, the distinct role it plays at each step is still being elucidated. Moreover, the interplay between metabolism and the microenvironment and their relationship to tumor GSH levels has not been studied. As discussed in this review, GSH also has antioxidant-independent functions in cancer cells. However, this has only recently begun to be investigated. A better understanding of these pathways and the role of GSH in them should aid the development of mechanism-based GSH inhibitors, which can then be combinatorically used with other drugs to effectively limit tumor growth.
Acknowledgments
This project is funded by National Institutes of Health grant PO1 CA 104838.
The authors declare no competing financial interests.
References
- Alanazi A.M., Mostafa G.A.E., and Al-Badr A.A.. 2015. Glutathione. Profiles Drug Subst. Excip. Relat. Methodol. 40:43–158. 10.1016/bs.podrm.2015.02.001 [DOI] [PubMed] [Google Scholar]
- Alvarez S.W., Sviderskiy V.O., Terzi E.M., Papagiannakopoulos T., Moreira A.L., Adams S., Sabatini D.M., Birsoy K., and Possemato R.. 2017. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 551:639–643. 10.1038/nature24637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames B.N., Shigenaga M.K., and Hagen T.M.. 1993. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA. 90:7915–7922. 10.1073/pnas.90.17.7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderton B., Camarda R., Balakrishnan S., Balakrishnan A., Kohnz R.A., Lim L., Evason K.J., Momcilovic O., Kruttwig K., Huang Q., et al. 2017. MYC-driven inhibition of the glutamate-cysteine ligase promotes glutathione depletion in liver cancer. EMBO Rep. 18:569–585. 10.15252/embr.201643068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andringa K.K., Coleman M.C., Aykin-Burns N., Hitchler M.J., Walsh S.A., Domann F.E., and Spitz D.R.. 2006. Inhibition of glutamate cysteine ligase activity sensitizes human breast cancer cells to the toxicity of 2-deoxy-D-glucose. Cancer Res. 66:1605–1610. 10.1158/0008-5472.CAN-05-3462 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network 2013. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 499:43–49. 10.1038/nature12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero J., Obrador E., Anasagasti M.J., Martin J.J., Vidal-Vanaclocha F., and Estrela J.M.. 1999. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin. Exp. Metastasis. 17:567–574. 10.1023/A:1006725226078 [DOI] [PubMed] [Google Scholar]
- Conticello C., Martinetti D., Adamo L., Buccheri S., Giuffrida R., Parrinello N., Lombardo L., Anastasi G., Amato G., Cavalli M., et al. 2012. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int. J. Cancer. 131:2197–2203. 10.1002/ijc.27482 [DOI] [PubMed] [Google Scholar]
- Corti A., Franzini M., Paolicchi A., and Pompella A.. 2010. Gamma-glutamyltransferase of cancer cells at the crossroads of tumor progression, drug resistance and drug targeting. Anticancer Res. 30:1169–1181. [PubMed] [Google Scholar]
- Darbouy M., Chobert M.N., Lahuna O., Okamoto T., Bonvalet J.P., Farman N., and Laperche Y.. 1991. Tissue-specific expression of multiple gamma-glutamyl transpeptidase mRNAs in rat epithelia. Am. J. Physiol. 261:C1130–C1137. 10.1152/ajpcell.1991.261.6.C1130 [DOI] [PubMed] [Google Scholar]
- Demple B., and Harrison L.. 1994. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 63:915–948. 10.1146/annurev.bi.63.070194.004411 [DOI] [PubMed] [Google Scholar]
- Dickinson D.A., Levonen A.-L., Moellering D.R., Arnold E.K., Zhang H., Darley-Usmar V.M., and Forman H.J.. 2004. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic. Biol. Med. 37:1152–1159. 10.1016/j.freeradbiomed.2004.06.011 [DOI] [PubMed] [Google Scholar]
- Estrela J.M., Ortega A., and Obrador E.. 2006. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 43:143–181. 10.1080/10408360500523878 [DOI] [PubMed] [Google Scholar]
- Forman H.J., Zhang H., and Rinna A.. 2009. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 30:1–12. 10.1016/j.mam.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori S., Abe Y., Nishi M., Hamamoto A., Inoue Y., Ohnishi Y., Nishime C., Matsumoto H., Yamazaki H., Kijima H., et al. 2004. The subunits of glutamate cysteine ligase enhance cisplatin resistance in human non-small cell lung cancer xenografts in vivo. Int. J. Oncol. 25:413–418. [PubMed] [Google Scholar]
- Galadari S., Rahman A., Pallichankandy S., and Thayyullathil F.. 2017. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 104:144–164. 10.1016/j.freeradbiomed.2017.01.004 [DOI] [PubMed] [Google Scholar]
- Gamcsik M.P., Kasibhatla M.S., Teeter S.D., and Colvin O.M.. 2012. Glutathione levels in human tumors. Biomarkers. 17:671–691. 10.3109/1354750X.2012.715672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B., Doan A., and Hybertson B.M.. 2014. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharmacol. 6:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout P.W., Kang Y.J., Buckley D.J., Bruchovsky N., and Buckley A.R.. 1997. Increased cystine uptake capability associated with malignant progression of Nb2 lymphoma cells. Leukemia. 11:1329–1337. 10.1038/sj.leu.2400739 [DOI] [PubMed] [Google Scholar]
- Gout P.W., Buckley A.R., Simms C.R., and Bruchovsky N.. 2001. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: A new action for an old drug. Leukemia. 15:1633–1640. 10.1038/sj.leu.2402238 [DOI] [PubMed] [Google Scholar]
- Griffith O.W., and Mulcahy R.T.. 1999. The enzymes of glutathione synthesis: Gamma-glutamylcysteine synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 73:209–267. [DOI] [PubMed] [Google Scholar]
- Gupta R.K., Patel A.K., Shah N., Chaudhary A.K., Jha U.K., Yadav U.C., Gupta P.K., and Pakuwal U.. 2014. Oxidative stress and antioxidants in disease and cancer: A review. Asian Pac. J. Cancer Prev. 15:4405–4409. 10.7314/APJCP.2014.15.11.4405 [DOI] [PubMed] [Google Scholar]
- Hakimi A.A., Reznik E., Lee C.-H., Creighton C.J., Brannon A.R., Luna A., Aksoy B.A., Liu E.M., Shen R., Lee W., et al. 2016. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell. 29:104–116. 10.1016/j.ccell.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J.D., Chowdhry S., Dinkova-Kostova A.T., and Sutherland C.. 2015. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem. Soc. Trans. 43:611–620. 10.1042/BST20150011 [DOI] [PubMed] [Google Scholar]
- Hofbauer S.L., Stangl K.I., de Martino M., Lucca I., Haitel A., Shariat S.F., and Klatte T.. 2014. Pretherapeutic gamma-glutamyltransferase is an independent prognostic factor for patients with renal cell carcinoma. Br. J. Cancer. 111:1526–1531. 10.1038/bjc.2014.450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.Z., Chen C., Zeng Z., Yang H., Oh J., Chen L., and Lu S.C.. 2001. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 15:19–21. 10.1096/fj.00-0445fje [DOI] [PubMed] [Google Scholar]
- Ighodaro O.M., and Akinloye O.A.. 2017. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med., in press. [Google Scholar]
- Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A.L., Kafri R., Kirschner M.W., Clish C.B., and Mootha V.K.. 2012. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 336:1040–1044. 10.1126/science.1218595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo M.C., and Zhang D.D.. 2013. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 27:2179–2191. 10.1101/gad.225680.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Kon N., Li T., Wang S.-J., Su T., Hibshoosh H., Baer R., and Gu W.. 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 520:57–62. 10.1038/nature14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen E., Kuosmanen S.M., Leinonen H., and Levonen A.-L.. 2013. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 1:45–49. 10.1016/j.redox.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplowitz N., Aw T.Y., and Ookhtens M.. 1985. The regulation of hepatic glutathione. Annu. Rev. Pharmacol. Toxicol. 25:715–744. 10.1146/annurev.pa.25.040185.003435 [DOI] [PubMed] [Google Scholar]
- Ke H.-L., Lin J., Ye Y., Wu W.-J., Lin H.-H., Wei H., Huang M., Chang D.W., Dinney C.P., and Wu X.. 2015. Genetic variations in glutathione pathway genes predict cancer recurrence in patients treated with transurethral resection and Bacillus Calmette-Guerin instillation for non-muscle invasive bladder cancer. Ann. Surg. Oncol. 22:4104–4110. 10.1245/s10434-015-4431-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A.D., Zhang R., Han X., Kang K.A., Piao M.J., Maeng Y.H., Chang W.Y., and Hyun J.W.. 2015. Involvement of glutathione and glutathione metabolizing enzymes in human colorectal cancer cell lines and tissues. Mol. Med. Rep. 12:4314–4319. 10.3892/mmr.2015.3902 [DOI] [PubMed] [Google Scholar]
- Kredich N.M. 2008. Biosynthesis of cysteine. Ecosal Plus. 3 10.1128/ecosalplus.3.6.1.11 [DOI] [PubMed] [Google Scholar]
- Lewerenz J., Hewett S.J., Huang Y., Lambros M., Gout P.W., Kalivas P.W., Massie A., Smolders I., Methner A., Pergande M., et al. 2013. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 18:522–555. 10.1089/ars.2011.4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Qiu B., Lee D.S.M., Walton Z.E., Ochocki J.D., Mathew L.K., Mancuso A., Gade T.P.F., Keith B., Nissim I., and Simon M.C.. 2014a Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 513:251–255. 10.1038/nature13557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Zhang Z., Yuan J., Zhang Y., and Jin X.. 2014b Altered glutamate cysteine ligase expression and activity in renal cell carcinoma. Biomed. Rep. 2:831–834. 10.3892/br.2014.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E.C., Lyssiotis C.A., Juvekar A., Hu H., Asara J.M., Cantley L.C., and Toker A.. 2016. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 18:572–578. 10.1038/ncb3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M., Wang Y.-Z., and Gout P.W.. 2008. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 215:593–602. [DOI] [PubMed] [Google Scholar]
- Locasale J.W., Grassian A.R., Melman T., Lyssiotis C.A., Mattaini K.R., Bass A.J., Heffron G., Metallo C.M., Muranen T., Sharfi H., et al. 2011. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43:869–874. 10.1038/ng.890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.C. 2009. Regulation of glutathione synthesis. Mol. Aspects Med. 30:42–59. 10.1016/j.mam.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.C. 2013. Glutathione synthesis. Biochim. Biophys. Acta. 1830:3143–3153. 10.1016/j.bbagen.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.C., and Mato J.M.. 2012. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 92:1515–1542. 10.1152/physrev.00047.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.-C., Ji J.-A., Jiang Y.-L., Chen Z.-Y., Yuan Z.-W., You Q.-D., and Jiang Z.-Y.. 2016. An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci. Rep. 6:26585 10.1038/srep26585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak V.I. 2012. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids. 2012:736837 10.1155/2012/736837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magesh S., Chen Y., and Hu L.. 2012. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 32:687–726. 10.1002/med.21257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njålsson R., and Norgren S.. 2005. Physiological and pathological aspects of GSH metabolism. Acta Paediatr. 94:132–137. 10.1080/08035250410025285 [DOI] [PubMed] [Google Scholar]
- Olinski R., Zastawny T., Budzbon J., Skokowski J., Zegarski W., and Dizdaroglu M.. 1992. DNA base modifications in chromatin of human cancerous tissues. FEBS Lett. 309:193–198. 10.1016/0014-5793(92)81093-2 [DOI] [PubMed] [Google Scholar]
- Park B.V., and Pan F.. 2015. Metabolic regulation of T cell differentiation and function. Mol. Immunol. 68:497–506. 10.1016/j.molimm.2015.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompella A., Corti A., Paolicchi A., Giommarelli C., and Zunino F.. 2007. γ-glutamyltransferase, redox regulation and cancer drug resistance. Curr. Opin. Pharmacol. 7:360–366. 10.1016/j.coph.2007.04.004 [DOI] [PubMed] [Google Scholar]
- Ren D., Villeneuve N.F., Jiang T., Wu T., Lau A., Toppin H.A., and Zhang D.D.. 2011. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA. 108:1433–1438. 10.1073/pnas.1014275108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristoff E., and Larsson A.. 2007. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2:16 10.1186/1750-1172-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R., Sayin V.I., Davidson S.M., Bauer M.R., Singh S.X., LeBoeuf S.E., Karakousi T.R., Ellis D.C., Bhutkar A., Sánchez-Rivera F.J., et al. 2017. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23:1362–1368. 10.1038/nm.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin V.I., Ibrahim M.X., Larsson E., Nilsson J.A., Lindahl P., and Bergo M.O.. 2014. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6:221ra15 10.1126/scitranslmed.3007653 [DOI] [PubMed] [Google Scholar]
- Sayin V.I., LeBoeuf S.E., Singh S.X., Davidson S.M., Biancur D., Guzelhan B.S., Alvarez S.W., Wu W.L., Karakousi T.R., Zavitsanou A.M., et al. 2017. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife. 6:e28083 10.7554/eLife.28083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.-Z., Habib G.M., Rhead W.J., Gahl W.A., He X., Sazer S., and Lieberman M.W.. 1996. Mutations in the glutathione synthetase gene cause 5-oxoprolinuria. Nat. Genet. 14:361–365. 10.1038/ng1196-361 [DOI] [PubMed] [Google Scholar]
- Shigenaga M.K., Hagen T.M., and Ames B.N.. 1994. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA. 91:10771–10778. 10.1073/pnas.91.23.10771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C.-S., Mishra P., Watrous J.D., Carelli V., D’Aurelio M., Jain M., and Chan D.C.. 2017. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 8:15074 10.1038/ncomms15074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H. 1993. Strategies of antioxidant defense. Eur. J. Biochem. 215:213–219. 10.1111/j.1432-1033.1993.tb18025.x [DOI] [PubMed] [Google Scholar]
- Tagde A., Singh H., Kang M.H., and Reynolds C.P.. 2014. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 4:e229 10.1038/bcj.2014.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K., and Yamamoto M.. 2017. The KeAP1–NRF2 System in Cancer. 7. Front. Oncol. 7:65 10.3389/fonc.2017.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzyan S.S., Burgett A.W.G., Heroux A., Smith C.A., Mooers B.H.M., and Hanigan M.H.. 2015. Human γ-glutamyl transpeptidase 1: Structures of the free enzyme, inhibitor-bound tetrahedral transition states, and glutamate-bound enzyme reveal novel movement within the active site during catalysis. J. Biol. Chem. 290:17576–17586. 10.1074/jbc.M115.659680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S., Okamoto K., Yodoi J., and Hiai H.. 1995. Persistent oxidative stress in cancer. FEBS Lett. 358:1–3. 10.1016/0014-5793(94)01368-B [DOI] [PubMed] [Google Scholar]
- Traverso N., Ricciarelli R., Nitti M., Marengo B., Furfaro A.L., Pronzato M.A., Marinari U.M., and Domenicotti C.. 2013. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013:972913 10.1155/2013/972913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villablanca J.G., Volchenboum S.L., Cho H., Kang M.H., Cohn S.L., Anderson C.P., Marachelian A., Groshen S., Tsao-Wei D., Matthay K.K., et al. 2016. A phase I new approaches to neuroblastoma therapy study of buthionine sulfoximine and melphalan with autologous stem cells for recurrent/refractory high-risk neuroblastoma. Pediatr. Blood Cancer. 63:1349–1356. 10.1002/pbc.25994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.J., Hayes J.D., Henderson C.J., and Wolf C.R.. 2007. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA. 104:19589–19594. 10.1073/pnas.0709483104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Branicky R., Noë A., and Hekimi S.. 2018. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 10.1083/jcb.201708007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Fang Y.-Z., Yang S., Lupton J.R., and Turner N.D.. 2004. Glutathione metabolism and its implications for health. J. Nutr. 134:489–492. 10.1093/jn/134.3.489 [DOI] [PubMed] [Google Scholar]
- Yang W.S., and Stockwell B.R.. 2016. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Venneti S., and Nagrath D.. 2017. Glutaminolysis: A hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 19:163–194. 10.1146/annurev-bioeng-071516-044546 [DOI] [PubMed] [Google Scholar]
- Yu X., and Long Y.C.. 2016. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 6:30033 10.1038/srep30033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., and Forman H.J.. 2009. Redox regulation of gamma-glutamyl transpeptidase. Am. J. Respir. Cell Mol. Biol. 41:509–515. 10.1165/rcmb.2009-0169TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Forman H.J., and Choi J.. 2005. γ-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 401:468–483. 10.1016/S0076-6879(05)01028-1 [DOI] [PubMed] [Google Scholar]
- Zhang J., Pavlova N.N., and Thompson C.B.. 2017. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 36:1302–1315. 10.15252/embj.201696151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.Z., Lee E.E., Sudderth J., Yue Y., Zia A., Glass D., Deberardinis R.J., and Wang R.C.. 2016. Glutathione depletion, pentose phosphate pathway activation, and hemolysis in erythrocytes protecting cancer cells from vitamin C-induced oxidative stress. J. Biol. Chem. 291:22861–22867. 10.1074/jbc.C116.748848 [DOI] [PMC free article] [PubMed] [Google Scholar]