

Abstract
Background and objectives
In-vitro data suggest that clearance of vilaprisan is mediated by cytochrome P450 3A4 (oxidation) and aldoketoreductases (reduction). To fully understand the elimination and biotransformation pathways of vilaprisan, a selective progesterone receptor modulator, and to quantify the impact of cytochrome P450 3A4 inhibition on the pharmacokinetics of vilaprisan, two clinical studies in healthy postmenopausal women were conducted.
Methods
In study 1, pharmacokinetics, mass balance, and metabolite patterns were determined after single oral administration of 5 mg of [14C]-labeled vilaprisan in six subjects. In study 2, pharmacokinetics were determined after single oral administration of 4 mg of vilaprisan without and with concomitant administration of the strong cytochrome P450 3A4 inhibitor itraconazole (200 mg/day) in 14 subjects. In addition, a microtracer dose of vilaprisan was given intravenously to determine absolute bioavailability, clearance, and volume of distribution.
Results
The dominant single compound in plasma was vilaprisan. No plasma metabolites exceeding 10% of total drug-related area under the concentration–time curve were detected. The absolute oral bioavailability of vilaprisan was ~ 60%. The mean clearance was ~ 7 L/h and the volume of distribution at steady state was ~ 360 L. Excretion occurred primarily via feces (73.5 ± 3.70% of dose; urine: 13.1 ± 1.71%; total recovery: 86.6 ± 2.81%), mostly in a metabolized form. Only small amounts of the parent drug were found in excreta. When vilaprisan was administered together with itraconazole, exposure to vilaprisan was increased 6.2-fold (90% confidence interval 5.4–7.2).
Conclusions
Vilaprisan is predominantly metabolized in the liver to a complex variety of metabolites, which are mainly excreted with feces. The pivotal role of cytochrome P450 3A4 in the metabolism of vilaprisan was confirmed.
Clinical Trial Registration
EudraCT numbers 2013-000707-16 (mass balance study) and 2014-004929-41 (drug–drug interaction/microtracer study); NCT02456129 (drug–drug interaction/microtracer study).
Electronic supplementary material
The online version of this article (10.1007/s40262-017-0607-4) contains supplementary material, which is available to authorized users.
Key points
| Vilaprisan, a new, promising, selective progesterone receptor modulator, exhibits an attractive pharmacokinetic profile with good absolute oral bioavailability, suitable for low-dose once-daily administration. |
| Vilaprisan is predominantly eliminated via hepatic biotransformation. |
| The pivotal role of cytochrome P450 3A4 in the elimination of vilaprisan was confirmed in a drug–drug interaction study with the strong cytochrome P450 3A4 inhibitor itraconazole. |
Introduction
Vilaprisan (VPR) is a potent and highly selective progesterone receptor modulator (SPRM) [1–3]. In a recently completed phase II study, treatment with VPR provided effective bleeding control for women with symptomatic uterine fibroids and was effective in reducing fibroid volume after only 3 months of treatment [4]. Based on these promising results, VPR is currently being developed in phase III for the long-term treatment of uterine fibroids.
A recently published pharmacokinetic (PK) study in humans has shown that the pharmacokinetics of VPR is linear in the expected therapeutic dose range [5]. This publication is supplemented by the current paper, which describes a mass balance (MB) study that was conducted to fully understand the elimination and biotransformation pathways of VPR. The results of this study suggest an important role of cytochrome P450 (CYP)3A4 in the metabolic clearance of VPR. This finding is supported by in-vitro investigations on CYP enzyme reaction phenotyping (data on file, Bayer AG) and has triggered the conduct of a drug–drug interaction (DDI) study to investigate the impact of the strong CYP3A4 inhibitor itraconazole (ITZ) on the pharmacokinetics of VPR. Itraconazole is listed in the European Medicines Agency and US Food and Drug Administration guidelines on DDI studies as a strong CYP3A4 inhibitor that may be used to investigate a drug’s PK interaction potential [6, 7].
In this study, multiple doses of ITZ were given to ensure continuous exposure to the inhibitor during the relatively long-lasting elimination of VPR from plasma. By giving an intravenous (IV) microdose of [14C]-labeled VPR on top of the oral dose [microtracer (MT)], the absolute bioavailability, clearance, and volume of distribution of VPR could be determined at clinically relevant exposure levels in a time and cost-saving way.1
Materials and Methods
Materials
Vilaprisan preparations for the two studies described in this paper were manufactured by Bayer AG (Berlin, Germany). Unlabeled and [14C]-labeled VPR for the MB study was provided as a powder for reconstitution in a macrogol-based solution. Vilaprisan for oral administration in the DDI/MT study was provided in the form of 4 mg of immediate-release tablets and [14C]VPR for IV microdosing was provided as a hydroxypropyl-beta-cyclodextrin-based solution for IV administration. Itraconazole (Sporanox® 10 mg/mL solution for oral administration; Janssen-Cilag Limited, High Wycombe, Buckinghamshire, UK) was purchased from a public pharmacy in the UK.
Study Population
Healthy postmenopausal women between 45 and 70 years of age (inclusive) and with a body mass index of ≥ 18 and ≤ 32 kg/m2 were eligible for the two studies. Follicle-stimulating hormone levels in serum were determined at screening to confirm the subject’s postmenopausal status. Specific exclusion criteria were specified to exclude subjects with conditions that might have had an impact on the aims of the study. Screenings for eligibility also included a gynecological examination. We intended six subjects to be included in the MB study and 14 subjects in the DDI/MT study (seven of these with administration of a microtracer).
Study Design and Conduct of the Study
The MB study was a single-center, open-label, non-randomized, non-controlled phase I study. Subjects received a single nominal dose of 5 mg of VPR containing approximately 2.3 MBq of [14C]-labeled VPR in 10 mL of PEG solution for oral administration, resulting in a committed effective radiation dose of ~ 0.613 mSv. The actual doses varied between 4.82 and 4.99 mg VPR (2.23–2.31 MBq).
The study drug was taken with water in the fasted state. After drug intake, subjects stayed at the clinical unit for 17 days and returned as outpatients until the following individual exit criteria were met: (1) total excretion of radioactivity in excreta > 90% of the administered dose or (2) excretion of radioactivity within two consecutive 24-h collection intervals < 1% of dose. Once an exit criterion was met, no further samples were collected.
The DDI/MT study was a single-center, open-label, fixed-sequence study with four periods: screening, treatment periods 1 and 2, and follow-up. In treatment period 1, subjects received a single oral dose of VPR 4 mg as a tablet and half the study population additionally received a single IV dose of [14C]VPR 15.7 µg (37 KBq) 1.5–2 h (30 min infusion, 1 mL/min) after administration of the oral dose, i.e., around the time of the expected maximum plasma concentration (Cmax) of the oral dose. After a washout period of ≥ 4 weeks, subjects received ITZ 200 mg as an oral solution once daily for 14 days in period 2. On the fourth day of ITZ administration, they received a single oral dose of VPR 4 mg, 1 h after intake of ITZ. Both VPR and ITZ were taken with water in the fasted state.
Sample Collection
In the MB study, blood samples for measurements of drug concentrations and [14C]-radioactivity and for metabolite profiling were taken before dosing and up to 21 days post-dose as specified in Table 1. Urine and feces samples for metabolite profiling and radio-analysis were collected pre-dose and in 12- or 24-h fractions post-dose. In the DDI/MT study, blood samples for measurements of drug concentrations were taken before and up to 11 or 13 days after oral administration of VPR (Table 1).
Table 1.
Sampling schedules for determination of vilaprisan (VPR) pharmacokinetics in plasma
| Study | Analyte (method) | Sampling schedule |
|---|---|---|
| MB study | VPR, [14C]-radioactivity (LC–MS/MS; LSC) | Pre-dose and 30 and 60 min, and 2, 3, 4, 6, 8, 12, 16, 24, 36, and 48 h, and 3, … 14 days post-dose |
| MT sub-study | [14C]VPR (AMS) | Pre-dose and 3, 10, 20, 30, 40, 60, 75, and 90 min, and 2, 3, 4, 5, 6.5, 10.5, 14.5, and 22.5 h, and 2, 3, 4, 5, 7, and 9 days after start of infusion (only period 1) |
| DDI study | VPR (LC–MS/MS) | Pre-dose and 30, 60, and 90 min, and 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 h, and 2, 3, … 11, and 13a days post-dose (periods 1 and 2) |
| DDI study | ITZ (LC–MS/MS) | Before 1st, 2nd, 3rd, 4th, 6th, 8th, and 12th ITZ dose and 0 and 60 min, and 2 and 4 h, and 11 days after administration of VPR (only period 2) |
AMS accelerator mass spectroscopy, DDI drug–drug interaction, ITZ itraconazole, LC–MS/MS liquid chromatography-tandem mass spectrometry, LSC liquid scintillation counting, MB mass balance, MT microtracer
aOnly in period 1
Assessments of Safety and Tolerability
Safety monitoring included the assessment of adverse events, standard clinical laboratory tests, and measurements of vital signs, electrocardiograms, and physical examinations.
Bioanalytics
Liquid chromatography-tandem mass spectrometry (LC–MS/MS) detection methods, which were validated according to the relevant European Medicines Agency and US Food and Drug Administration guidelines [8, 9], were used to determine unchanged VPR and ITZ in individual plasma samples. Furthermore, the primary VPR metabolite BAY 1139463 (M-4), which was identified as one of the minor metabolites in prior studies, was determined by LC–MS/MS in pooled plasma samples (DDI study). Total radioactivity in the samples collected in the MB study was determined by liquid scintillation counting. Plasma concentrations of [14C]VPR in the MT sub-study were determined using liquid chromatography fractionation with off-line accelerator MS quantification. For details, see the Electronic Supplementary Material (ESM). An overview of the performance of the bioanalytical methods is given in Table 2.
Table 2.
Bioanalytical method performance for vilaprisan (VPR)
| LLOQ | Calibrators > LLOQ | Calibrators at LLOQ | QC samples | ||||
|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | ||
| VPRa | 0.100 µg/L | 96.2–107 | ≤ 10.3 | 100 | 10.1 | 102–112 | 3.26–6.37 |
| VPRb | 0.100 µg/L | 98.8–101 | ≤ 4.4 | 100 | 3.9 | 96.9–98.3 | 3.3–4.8 |
| M-4c | 1.00 µg/L | 96.8–104 | ≤ 10.9 | 101 | 7.8 | 98.1–109 | 6.7–11.7. |
| [14C]VPRd | 0.502 ng/L | N/A | N/A | N/A | N/A | N/A | N/A |
| ITZe | 1.00 µg/L | 98.4–103 | ≤ 4.24 | 98.8 | 3.94 | 97.7–101 | 3.38–4.88 |
DDI drug–drug interaction, ITZ itraconazole, LC–MS/MS liquid chromatography-tandem mass spectrometry, LLOQ lower limit of quantitation, MB mass balance, M-4 BAY 1139463 (metabolite of vilaprisan), N/A not applicable, QC quality control, UHPLC ultra high-performance liquid chromatography
aMB study: LC–MS/MS; calibrators: 0.100–50.0 µg/L; QC samples: 0.250–40.0 µg/L
bDDI study: LC–MS/MS; calibrators: 0.100–50.0 µg/L; QC samples: 0.300–40.0 µg/L
cDDI study: LC–MS/MS; calibrators: 1.00–50.0 µg/L; QC samples: 1.00–40.0 μg/L
dDDI study: UHPLC + AMS; calibrators: 0.502–44.9 pg/mL
eDDI study: LC–MS/MS; calibrators: 1.00–1000 µg/L; QC samples: 3.00–750 µg/L
Metabolite Profiling and Identification
Plasma, urine, and feces samples from each time point and subject (MB study) were purified according to an appropriate extraction procedure, enabling recovery of radioactivity in the sample extract. Aliquots of the samples were subjected to high-performance LC with off-line radioactivity detection.
The structures of the parent drug and its metabolites represented by different peaks in the radiochromatograms were elucidated by investigating representative plasma, urine, and feces samples with combined radiometric and LC–MS/MS detection. For structure elucidation of metabolites, comparison with authentic reference compounds was used, if those were available. Structure proposals for further metabolites were made based on the determination of the molecular formula by high-resolution MS as well as a mass spectrometric fragmentation pattern determined by LC–MS/MS (see the ESM).
Pharmacokinetic Analysis
Standard PK parameters for VPR, M-4, and total radioactivity in plasma or blood were calculated from individual concentration–time data by non-compartmental analysis using the software WinNonlin (Pharsight Corporation, St. Louis, Missouri, USA), Version 4.1 (MB study) or 5.3 (DDI/MT study).
Maximum plasma concentration and time to reach Cmax were obtained directly from concentration data. The area under the plasma concentration–time curve from time zero to time t after administration (AUC0–t) was calculated by the mixed linear/logarithmic trapezoidal rule. The AUC from time zero to infinity was calculated as + (Clast,calc/ke), where Clast,calc was the calculated concentration value at tlast by regression of the terminal phase, and ke was the terminal rate constant estimated by log-linear regression analysis. The extrapolated fraction of an individual AUC value had to be below 20% to be evaluable. The terminal plasma half-life (t1/2) was calculated as t1/2 = ln2/ke.
The absolute bioavailability F was calculated as follows: . In addition, the whole blood-to-plasma ratio of [14C]-radioactivity was calculated for each sampling time point in the MB study. For ITZ, exposure was confirmed by analysis of trough concentrations. The amount of radioactivity recovered in the excreta was related (in percent) to the administered radioactive dose and tabulated by sampling interval. The PK parameters for VPR and its metabolites from the metabolite profiles were calculated from individual plasma concentrations by non-compartmental analysis using the software ToxKin (Entimo, Berlin, Germany).
Statistical Analysis
All data were evaluated by descriptive statistics. For the evaluation of the DDI potential, the primary PK parameters AUC0–11d and Cmax of VPR, which were assumed to be log-normally distributed, were analyzed by analysis of variance including treatment and subject effects. Based on these analyses, point estimates, 90% confidence intervals, and 95% prediction intervals for the ratio ‘VPR + ITZ/VPR’ were calculated by re-transformation of the logarithmic data using the intra-individual standard deviation of the analysis of variance. For the evaluation of the absolute bioavailability of VPR, point estimates and 90% confidence intervals were calculated for F, which was assumed to be log-normally distributed.
Results
Study Subjects
The demographic and baseline characteristics of the six subjects from the MB study, the 14 subjects from the DDI/MT study, and the subset of seven subjects from the DDI/MT study who received the microtracer are summarized in Table 3. All subjects completed the study as planned. No major protocol deviations were identified. Postmenopausal status was confirmed in all subjects by follicle-stimulating hormone measurements.
Table 3.
Demographic and baseline characteristics
| MB study N = 6 | DDI/MT study N = 14 | MT sub-study N = 7 | ||
|---|---|---|---|---|
| Sex: female | N | 6 | 14 | 7 |
| Race | ||||
| Asian | N | 0 | 1 | 0 |
| White | N | 6 | 13 | 7 |
| Age (years) | Mean (range) | 57.7 (49–63) | 58.9 (49–69) | 61.0 (55–69) |
| Weight (kg) | Mean ± SD | 62.1 ± 7.33 | 67.3 ± 8.02 | 68.4 ± 8.50 |
| BMI (kg/m2) | Mean ± SD | 23.1 ± 2.37 | 26.0 ± 2.66 | 26.4 ± 3.11 |
| FSH > 40 IU/L | N | 6 | 14 | 7 |
BMI body mass index, DDI drug–drug interaction, FSH follicle-stimulating hormone, MB mass balance, MT microtracer, SD standard deviation
Plasma Pharmacokinetics of Vilaprisan and [14C]-Radioactivity After Oral Administration (Mass Balance Study)
The mean concentrations of VPR and [14C]-radioactivity in plasma are shown in Fig. 1. The overall content of radioactivity in plasma was very low and VPR did not show any noteworthy tendency to bind tightly to plasma proteins as determined by standard in-vitro methods (data on file, Bayer AG). The geometric mean blood/plasma ratio of [14C]-radioactivity varied between 0.71 and 0.76 during the first 24 h post-dose, indicating that the radioactivity was mainly distributed in plasma at that time. Thereafter, the ratio increased to 0.79 at 48 h post-dose, the last evaluable time point.
Fig. 1.

Geometric mean vilaprisan (VPR) and [14C]-radioactivity concentrations in plasma following a single oral dose of 5 mg [14C]VPR (mass balance study). Geometric means ± geometric standard deviations (calculated if two thirds or more of the individual values were above the lower limit of quantitation, 0.1 µg/L), n = 6. Plasma samples beyond 168 h were not further investigated as the overall content of total radioactivity was too low
The geometric mean PK parameters of VPR in plasma are summarized in Table 4. The data show that VPR was rapidly absorbed. Maximum VPR concentrations were reached between 1 and 2 h post-dose and amounted to approximately 25 µg/L, on average. Post-maximum VPR concentrations declined with a biphasic profile and a geometric mean terminal half-life of approximately 32 h.
Table 4.
Pharmacokinetic parameters of vilaprisan (VPR) parent after single oral or intravenous administration of VPR without or with concomitant administration of itraconazole (ITZ) 200 mg/day
| Study | MB study VPRa Oral |
DDI study VPRb Oral |
DDI study VPR + ITZc Oral |
MT sub-study VPRd Intravenous |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment | |||||||||
| Route of administration | |||||||||
| Parameter | Unit | N = 6 | N = 14 | N = 14 | N = 7 | ||||
| gMean (CV%) | [Range] | gMean (CV%) | [Range] | gMean (CV%) | [Range] | gMean (CV%) | [Range] | ||
| AUC | µg·h/L | 324 (38.0) | [217–504] | 325 (42.3) | [191–607] | N/C | 2170 (35.9) | [1410–3500] | |
| AUC0–24 | µg·h/L | N/C | 146 (29.5) | [95.8–217] | 302 (18.9) | [238–388] | 980 (26.2) | [731–1330] | |
| AUC0–3d | µg·h/L | N/C | 247 (33.4) | [153–393] | 740 (18.1) | [586–970] | N/C | ||
| AUC0–11d | µg·h/L | N/C | 321 (41.3) | [190–580] | 2000 (18.4) | [1520–2740] | N/C | ||
| µg·h/L | 317 (38.3) | [212–495] | 317 (42.6) | [184–591] | 2000 (18.4) | [1520–2730] | 2100 (35.4) | [1340–3300] | |
| C max | µg/L | 24.9 (38.5) | [15.3–41.2] | 16.6 (32.3) | [9.14–24.5] | 29.5 (19.5) | [21.9–38.7] | 277 (29.7) | [168–389] |
| t 1/2 | h | 31.6 (24.7) | [22.6–44.1] | 38.9 (20.7) | [29.0–60.7] | N/C | 42.4 (20.7) | [28.6–56.7] | |
| t max | h | 1.00e | [1.00–2.00] | 2.50e | [1.42–4.00] | 2.25e | [1.50–3.00] | 0.483e | [0.333–0.483] |
| t last | h | 144e | [120–240] | 204e | [144–312] | 263e | [263–263] | 215e | [167–215] |
| CL/F or CL | L/h | 15.1 (38.0) | [9.7–22.4] | 12.3 (42.3) | [6.59–21.0] | N/C | 7.22 (35.9) | [4.47–11.1] | |
| Vz/F or Vz | L | 691 (27.4) | [492–978] | N/C | N/C | 442 (33.8) | [302–713] | ||
| V ss | L | – | – | – | 359 (23.4) | [277–479] | |||
| MRTIV | h | – | – | – | 49.7 (19.1) | [38.0–68.3] | |||
| F | % | – | – | – | 61.1 | [53.1–69.5] | |||
AUC0–t area under the concentration–time curve from time zero to time t, CL total body clearance of drug from plasma calculated after intravenous administration, CL/F total body clearance of drug from plasma calculated after oral administration (apparent oral clearance), Cmax maximum plasma concentration, CV% coefficient of variation [%], DDI drug–drug interaction, F absolute oral bioavailability, gMean geometric mean, MB mass balance, MRTIV mean residence time after intravenous administration, MT microtracer, N/C not calculated, t1/2 terminal half-life, tlast time of last concentration above lower limit of quantitation, tmax time to reach Cmax, Vss volume of distribution at steady state, Vz volume of distribution during terminal phase
Figures were rounded to three significant digits
F was calculated as follows:
a[14C]VPR 5 mg
bVPR 4 mg
cVPR 4 mg on the fourth day of concomitant treatment with ITZ 200 mg/day
dIntravenous microdose of [14C]VPR 15.7 µg (37 KBq) 1.5–2 h after oral administration of VPR 4 mg (30-min infusion)
eMedian (range)
The geometric mean exposure (AUC) to VPR was considerably lower than the mean exposure to [14C]-radioactivity (324 µg·h/L vs. 1180 µgEq·h/L) (Tables 4, 5), indicating that a major fraction of the circulating radioactivity can be attributed to metabolites of VPR, approximately 73% based on the ratio of AUCs. The fact that the mean terminal half-life of [14C]-radioactivity in plasma was markedly longer than the half-life of VPR (approximately 62 vs. 32 h) indicates the presence of circulating metabolites.
Table 5.
Pharmacokinetic parameters of [14C]-radioactivity in plasma and whole blood after single oral administration of 5 mg [14C]vilaprisan (mass balance study)
| Parameter | Unit | [14C]-radioactivity in plasma N = 6 | [14C]-radioactivity in whole blood N = 6 | ||
|---|---|---|---|---|---|
| gMean (CV%) | [Range] | gMean (CV%) | [Range] | ||
| AUC | µgEq·h/L | 1180 (16.9) | [953–1490] | 852 (28.0) | [647–1380] |
| µgEq·h/L | 923 (20.4) | [703–1200] | 498 (22.8) | [389–668] | |
| C max | µgEq/L | 46.9 (22.9) | [33.0–66.8] | 34.8 (20.9) | [26.0–49.1] |
| t max | h | 1.51a | [1.00–3.00] | 2.00a | [1.00–4.00] |
| t 1/2 | h | 62.2 (16.4) | [50.1–78.1] | 52.6 (42.5) | [31.6–97.0] |
| t last | h | 120 | [96.0–144] | 48.1 | [48.0–72.1] |
Figures were rounded to three significant digits
AUC area under the concentration–time curve from time zero to infinity, AUC(0–t) area under the concentration–time curve from time zero to time t, Cmax maximum plasma concentration, CV% coefficient of variation [%], gMean geometric mean, t1/2 terminal half-life, tlast time of last concentration above lower limit of quantitation, tmax time to reach Cmax
aMedian
Excretion of [14C]-Radioactivity (Mass Balance Study)
Figure 2 shows the cumulative amount of [14C]-radioactivity recovered in urine and feces during the 21-day collection period. In total, 86.6 ± 2.81% (mean ± standard deviation; range 82.0–90.7%) of the administered [14C]-radioactivity dose was recovered in excreta within 21 days post-dose.
Fig. 2.

Cumulative recovery of total [14C]-radioactivity in excreta following a single oral dose of [14C]vilaprisan 5 mg (mass balance study). Mean ± standard deviation (SD); n = 6
Excretion occurred mainly via feces (73.5 ± 3.70%; range 68.9–79.0%); only 13.1 ± 1.71% (range 11.4–15.9%) of the radioactivity dose administered was recovered in the urine. From 408 h post-dose onwards, only minimal amounts of radioactivity were recovered in excreta (< 0.1% of the administered radioactivity dose per day), indicating that excretion was almost complete after 17 days. No loss of radioactivity because of vomiting was observed in this study.
Metabolite Profiling in Plasma and Excreta (Mass Balance Study)
Vilaprisan is almost exclusively eliminated via metabolic processes. The main metabolic pathways are oxidations at the steroid skeleton as well as reductions in the 3-keto moiety as well as combinations of both. The primary reduction product at the 3-keto moiety is the alpha-hydroxy derivative M-4. Further oxidation of M-4 leads to the formation of various combination products of oxidation and reduction, e.g., metabolite M-7. The formation of oxidation products of both VPR and M-4 is mainly catalyzed by CYP3A4, the reduction at the 3-keto-moiety is catalyzed by aldoketoreductases.
In human plasma, only low amounts of radioactivity were determined and individual metabolite patterns were highly complex. Overall, VPR was the predominant component in plasma with a mean of 27.8% of AUC of total radioactivity. The amounts of the most prominent metabolites, M-4 (reduced at the 3-keto moiety of VPR) and M-7 (oxidation product of M-4) were estimated to be 5.2–6.6 and 5.2%, respectively, of total drug-related exposure. In addition, several other hydroxylated and reduced metabolites, i.e., M-1, M-5, M-6, M-9, M-12, M-16, and M-32, were detected in plasma, each at a low extent (example in Fig. 3). All metabolites identified in plasma are formed either via oxidation reactions or via combinations of oxidation and reduction reactions (Fig. 4).
Fig. 3.

Representative radio-chromatograms (mass balance study)
Fig. 4.
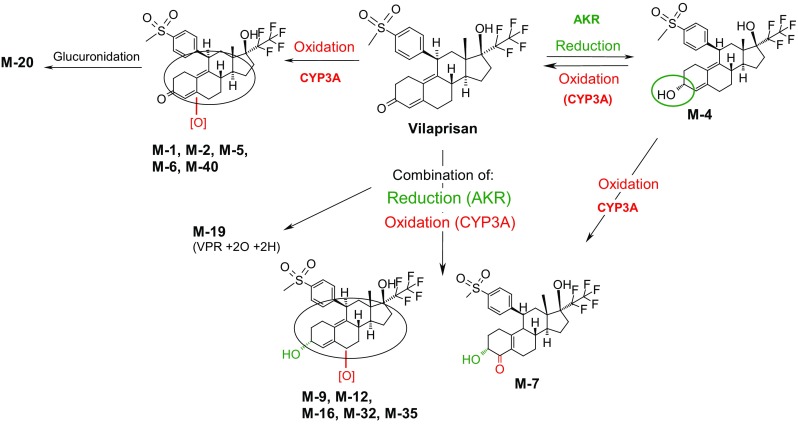
Proposed main metabolic pathways of vilaprisan in humans. AKR aldoketoreductase, CYP cytochrome P450, VPR vilaprisan
There was a high amount (about 45%) of drug-related [14C]-radioactivity exposure in plasma, which could not be assigned to single peaks and structures because of the very low amount of radioactivity in human plasma. Numerous metabolic pathways and combinations of them were observed as known from other compounds with a steroid skeleton.
The chemical structure and stereochemistry of metabolites M-4 and M-7 were verified by comparison with authentic reference compounds with a verified structure based on nuclear magnetic resonance spectroscopy. Structure proposals for further metabolites were made based on determination of a molecular formula by high-resolution MS as well as a mass spectrometric fragmentation pattern determined by LC–MS/MS and LC/MS.
Highly complex metabolite patterns were observed in excreta (Fig. 5a, b). In feces, oxidated metabolites M-1 and M-40 were identified as the main compounds accounting for, on average, 11.2 and 14.1% of the administered dose, respectively. In addition, metabolites M-2, M-5, M-6, M-7, M-12, M-16, and M-32 accounted each for 0.6–7.0% of the dose, on average. The parent drug accounted only for 1.1% of the dose. Overall, despite the complexity in metabolite patterns in the investigated excreta, 50.6% of the administered dose could be attributed to known metabolites and VPR.
Fig. 5.
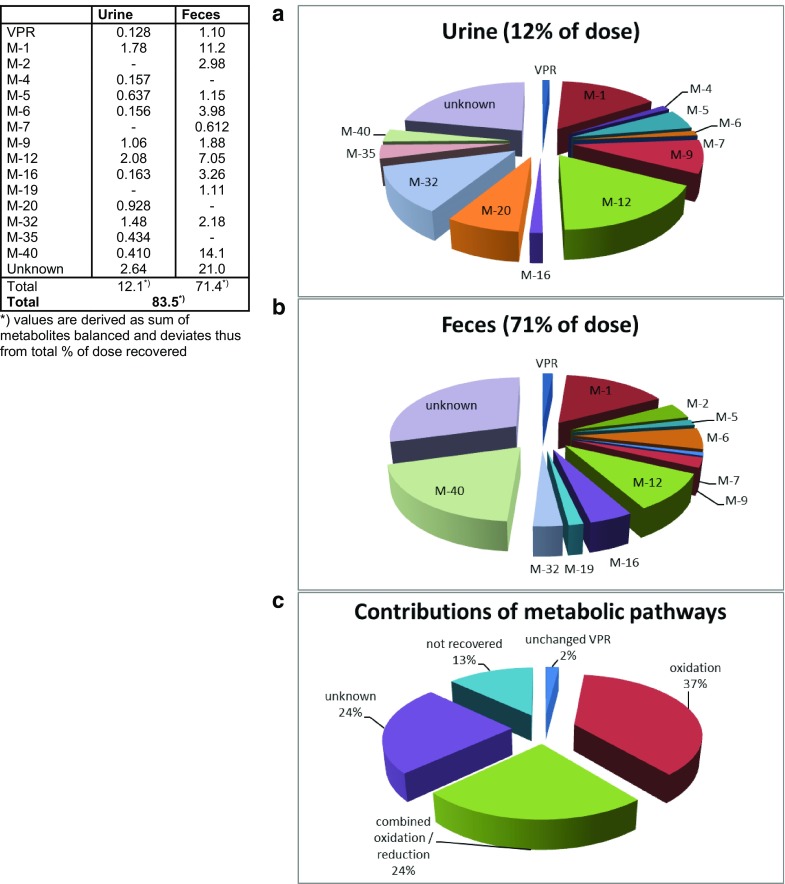
Metabolites in excreta [% of dose] and contributions of metabolic pathways to overall excretion (mean of six subjects). VPR vilaprisan
In urine, VPR accounted for an average of 0.1% of the dose in urine. Oxidated metabolite M-1 and metabolites M-12 and M-32, both formed by a combination of reduction at the 3-keto moiety and oxidation at the steroidal core, were found to be the main compounds accounting on average for 1.8, 2.1, and 1.5% of the administered dose, respectively. Despite the complexity in the profiles, 9.4% (out of a total of 13.1%) of the administered dose could be assigned to individual metabolites or VPR. In general, the complexity within the metabolite pattern in each subject remained largely unchanged over time.
Clearance, Volume of Distribution, and Absolute Bioavailability (Drug–Drug Interaction/Microtracer Study)
Following administration of the IV microtracer dose, VPR was eliminated from plasma with a mean clearance of ~ 7 L/h (Table 4). The terminal half-life of ~ 40 h was similar to that observed after oral administration of VPR (Fig. 6), indicating that absorption of the drug was completed during the terminal elimination phase. The large mean volume of distribution at steady state of ~ 360 L indicates pronounced distribution into tissues. The absolute oral bioavailability was approximately 60%.
Fig. 6.

Geometric mean concentrations of vilaprisan (± standard deviations) (VPR) and [14C]VPR in plasma after single oral and intravenous (i.v.) administration of VPR 4 mg (drug–drug interaction/microtracer study). All 14 subjects received a single oral dose of VPR 4 mg as a tablet and seven of these subjects additionally received a single i.v. dose of [14C]VPR 15.7 µg (37 KBq) [30-min infusion, 1 mL/min] starting 1.5–2 h after administration of the oral dose, i.e., around the time of the expected maximum plasma concentration of the oral dose
Impact of Itraconazole on the Pharmacokinetics of Vilaprisan (Drug–Drug Interaction/Microtracer Study)
Geometric mean ITZ trough concentrations above 530 µg/L during the blood sampling period for VPR showed that all subjects were sufficiently exposed to ITZ. When co-administered with ITZ, the concentrations of VPR were substantially higher and elimination was visibly slower than when the drug was administered alone (Fig. 7). This resulted, on average, in about twofold higher Cmax values and about sixfold higher AUC0–11d (Tables 4, 6; Fig. 8).
Fig. 7.

Geometric mean concentrations of vilaprisan (VPR) in plasma after single oral administration of VPR 4 mg with and without concomitant administration of itraconazole (ITZ) 200 mg/day (drug–drug interaction/microtracer study). Geometric means ± geometric standard deviations [calculated if two thirds or more of the individual values were above the lower limit of quantitation (LLOQ), 0.1 µg/L]; n = 14
Table 6.
Point estimators (least-squares means), two-sided 90% confidence intervals, and 95% prediction intervals for the ratio ‘vilaprisan + itraconazole to vilaprisan’ (drug–drug interaction study)
| Parameter | Unit | N | Geometric CV (%) | Estimated ratio | 90% confidence interval | 95% prediction interval |
|---|---|---|---|---|---|---|
| AUC0–11d | µg·h/L | 14 | 21.0 | 6.23 | 5.42–7.16 | 3.23–12.0 |
| C max | µg/L | 14 | 16.8 | 1.78 | 1.59–1.98 | 1.05–3.01 |
AUC0–t area under the concentration–time curve from time zero to time t, Cmax maximum plasma concentration, CV% coefficient of variation (%)
Fig. 8.

Exposure to vilaprisan after single oral administration of vilaprisan 4 mg with and without concomitant administration of itraconazole (ITZ) 200 mg/day (drug–drug interaction study). Left hand graph: box: 25th–75th percentile. Horizontal line: median. Vertical lines extend from the box as far as the data extend to a maximum distance of 1.5 interquartile ranges; any value more extreme is plotted separately. Right hand graph: time course of individual area under the concentration–time curve from time zero to time t (AUC0–11d) values; n = 14
Figure 9 and the PK parameters for M-4 in Table 7 show that co-administration of ITZ increased M-4 plasma concentrations similar to VPR. In particular, the extremely flat course of the terminal M-4 concentration–time curve following administration of VPR + ITZ is remarkable, corresponding to an elimination half-life of about 300 h. The exposure ratio of VPR/M-4 remained unchanged.
Fig. 9.

Concentrations of vilaprisan metabolite M-4 (BAY 1139463) in pooled plasma after single oral administration of vilaprisan 4 mg with and without concomitant administration of itraconazole (ITZ) 200 mg/day (drug–drug interaction study). M-4 concentrations in plasma were below the lower limit of quantitation after 12 h after administration of vilaprisan without concomitant administration of ITZ
Table 7.
Pharmacokinetic parameters of vilaprisan (VPR) metabolite M-4 (BAY 1139463) in pooled plasma samples of 14 subjects after single oral administration of VPR 4 mg with and without concomitant administration of itraconazole (ITZ) 200 mg/day (drug–drug interaction study)
| Parameter | Unit | VPR | VPR + ITZ |
|---|---|---|---|
| µg·h/L | 18.7 | 409 | |
| AUC0–24 | µg·h/L | 29.6 | 48.7 |
| C max | µg/L | 3.02 | 5.67 |
| t 1/2 | h | 26.4 | 303 |
| t last | h | 12 | 264 |
| t max | h | 2.00 | 2.00 |
AUC0–t area under the concentration–time curve from time zero to time t, Cmax maximum plasma concentration, t1/2 terminal half-life, tlast time of last concentration above lower limit of quantitation, tmax time to reach Cmax
Note: Pooled samples were used for exploratory quantitation of M-4. The extrapolated fraction of the AUC exceeded 50% for both treatments so that AUCs are not provided here
Safety Results
For all six subjects in the MB study, treatment-emergent adverse events (TEAEs) were reported and assessed as being related to the study drug VPR. The most frequently occurring VPR-related TEAEs were headache in three subjects (50.0%) and constipation in two subjects (33.3%). Further VPR-related events, which occurred in one subject each, were flatulence, nausea, and dizziness. These adverse events are in line with the side-effect profile already known for VPR from previous studies.
In the DDI/MT study, the only possibly VPR-related TEAE occurring in more than one subject was headache (three subjects, 21.4%). The frequency and intensity of TEAEs were not found to be increased with the combination VPR + ITZ in comparison to VPR alone. Clinically relevant changes in laboratory values, vital signs, or electrocardiograms were not reported neither of the studies. Overall, single doses of 4 or 5 mg of VPR were well tolerated in both studies and considered as safe.
Discussion
The main objective of the studies reported was to investigate the elimination and biotransformation pathways of VPR, focusing on the role of CYP3A4 in the metabolic clearance of VPR. The absolute oral bioavailability of VPR was found to be approximately 60%. The dominant single compound in plasma was VPR. No major plasma metabolites exceeding 10% of drug-related AUC were detected. The mean VPR clearance was ~ 7 L/h and the volume of distribution at steady state was ~ 360 L. Excretion occurred primarily via feces (73.5 ± 3.70% of dose; urine: 13.1 ± 1.71%; total recovery: 86.6 ± 2.81%), mostly as metabolites. Only small amounts of the parent drug were found in excreta.
Overall, excretion of administered radioactivity occurred slowly but was almost complete after 17 days. The presence of only marginal amounts of unchanged VPR in feces indicates that VPR was completely absorbed after oral administration. Similarly, the low amounts of unchanged VPR in urine suggest a negligible contribution of renal clearance to total elimination of VPR. Thus, the clearance of VPR is more likely to be affected by hepatic impairment than by renal dysfunction.
In plasma, low amounts of radioactivity and highly complex metabolite patterns were found. Overall, VPR was the predominant single component in plasma, representing about 30% of total drug-related radioactivity exposure, while the sum of a complex mixture of metabolites attributed to about 70%. The most prominent plasma metabolites, M-4 and M-7, were estimated to represent 5–7% of total drug-related exposure each. Hence, no ‘major metabolites,’ i.e., metabolites representing an exposure of more than 10% of total drug-related AUC [10, 11], were identified in this study. Nevertheless, both metabolites were further characterized in in-vitro transactivation assays investigating their pharmacological activity on the progesterone receptor. For both M-4 and M-7, a marginal antagonistic activity on the progesterone receptor (~ 1/100th and 1/100,000th of VPR, respectively) was observed demonstrating that M-4 and M-7 do not contribute to the pharmacological effect of VPR (data on file, Bayer AG).
In-vitro data suggest that clearance of VPR by oxidation or reduction reactions is mediated via CYP3A4 (oxidation) and via aldoketoreductases (reduction) (data on file, Bayer AG). Oxidation reactions occur at various positions of the steroidal core structure, whereas reduction is primarily observed at the 3-keto moiety of the steroid skeleton. Owing to the structural complexity of the steroid core, the exact position of oxidation and stereochemical assignment of the majority of the metabolites is unknown.
Re-conversion of M-4 into the parent compound, a process hampering the accurate determination of the relative contribution of the CYP3A4 and aldoketoreductase enzymes to total clearance, was observed in-vitro as well as in-vivo in animals. Furthermore, a majority of metabolites are formed by a combination of oxidation and reduction reactions, but the sequence of the two steps is not readily discernable. An examination of the pie chart depicting the contributions to metabolic pathways (Fig. 5c) determined in the MB study shows that oxidation plus the combination of oxidation and reduction contributed ~ 60% to the overall metabolic clearance. Approximately 25% of the radioactivity excreted could not be described by dedicated metabolites. Therefore, a reliable quantitative assessment of the contribution of CYP3A4 to the overall clearance of VPR could not be made. However, CYP3A4 was assumed to contribute predominantly to the biotransformation of VPR and thereby rendering the potential for a strong interaction with CYP3A4 inhibitors.
This assumption was confirmed in the DDI study with the strong CYP3A4 inhibitor ITZ. After co-administration of ITZ 200 mg/day, a sixfold higher exposure to VPR was observed. A similar increase in exposure was also seen for M-4 suggesting that the systemic metabolic clearance of VPR and M-4 is reduced by inhibition of CYP3A4.
The data collected in the DDI study were further analyzed by a physiologically based pharmacokinetic model, which was previously established using single and multiple oral dose clinical PK data and further qualified with IV data from this study. The final model was able to describe all prior clinical studies with the implemented assumptions that (1) the hepatic metabolism is not saturable, (2) metabolism is exclusively via CYP3A4, the pharmacokinetics of VPR can be accurately described without considering the transformation of VPR to M-4, and (3) no active transport is involved. The interaction of VPR with ITZ could be adequately described with this model when coupled with an ITZ physiologically based PK model [12], thereby supporting the assumption that CYP3A4 plays a pivotal role.
A comparison of the PK properties of VPR with those of other well-characterized SPRMs such as mifepristone and ulipristal acetate reveals some features they have in common and some features that are specific to VPR. All three compounds are eliminated from plasma mainly via metabolism in the liver with subsequent biliary excretion of the metabolites [13, 14], whereas renal excretion is negligible. More specifically, the pivotal role of CYP3A4 in hepatic metabolism has been demonstrated for all three SPRMs [14, 15]. As a consequence, all three SPRMs are potentially prone to DDIs with CYP3A4 inhibitors as recently shown, for example, for ulipristal acetate, where a strong interaction with ketoconazole was observed (5.8-fold increase in exposure) [14], similar to the interaction of VPR with ITZ. However, the metabolites formed differ markedly between VPR on the one hand and mifepristone and ulipristal acetate on the other hand. For mifepristone and ulipristal acetate, oxidative N-demethylation of the para dimethylamino group (Fig. 10) is the primary metabolic reaction resulting in high exposure concentrations of the respective mono- and di-N-desmethyl metabolites in plasma with significantly more than 10% of drug-related exposure [16, 17].
Fig. 10.

Structure of vilaprisan in comparison to mifepristone and ulipristal acetate
The introduction of a 4-(methylsulfonyl) group instead of the dimethylamino group in VPR, in contrast, leads to a metabolically stable moiety at this position. The position of biotransformation is shifted to the steroid skeleton with formation of various metabolites by oxidation reactions as well as reductions of the 3-keto group. This results in a complex metabolite pattern in plasma and excreta with rather low drug exposures of less than 10% to single metabolites in plasma.
Furthermore, the mono-N-demethylated metabolites of mifepristone and ulipristal acetate still exhibit relevant antiprogestogenic activity in-vitro similar to that of the parent compound and thus likely contribute to the overall clinical efficacy [18]. In contrast, the two (low-level) main metabolites of VPR in plasma elicit only marginal pharmacological activity at the progesterone receptor, not contributing to clinical efficacy. The absence of active metabolites for VPR facilitates the evaluation of a safety and exposure–response relationship, as besides the parent compound no other active components have to be considered.
Based on the major role of CYP3A4 seen in the present DDI study, a clinically relevant interaction of VPR with strong CYP3A4 inducers is also likely. Therefore, the concomitant use of strong CYP3A4 inducers is also not recommended owing to a potential loss of efficacy. A DDI study with a strong CYP3A4 inducer has been recently initiated to investigate the interaction potential with inducers.
Following IV administration of the microtracer dose, VPR showed a rapid and extensive distribution into tissues as indicated by a high volume of distribution at steady state of approximately 360 L, which is typical of highly lipophilic compounds such as VPR. Vilaprisan shows a good oral absolute bioavailability of about 60%, suggesting that the impact of ITZ on VPR clearance can be predominantly attributed to inhibition of CYP3A4 in the liver, which is also in line with the observed prolonged systemic elimination in the presence of ITZ.
The excellent safety profile of VPR has been proven in several phase I studies with oral doses of ≤ 30 mg of VPR given daily for 28 days. Furthermore, VPR was well tolerated and safe in a phase II dose-finding study with daily administration of up to 4 mg of VPR over 3 months in patients with uterine fibroids [4]. The anticipated exposure to VPR after multiple administrations of the therapeutic dose together with multiple administrations of strong CYP3A4 inhibitors is well within the range already tested in humans. The respective anticipated exposure to M-4 was investigated in pivotal long-term systemic tolerance studies in animals. Thus, the available preclinical and clinical data suggest no safety concern with VPR even in the presence of strong CYP3A4 inhibitors.
Nevertheless, strong CYP3A4 inhibitors are not recommended to be used together with VPR and will be excluded in the further clinical development program, whereas mild and moderate CYP3A4 inhibitors will be acceptable, allowing the collection of further clinical PK and safety data for these co-medications. Furthermore, it is worth mentioning that, despite higher exposure to VPR when ITZ was used concomitantly, the frequency and intensity of TEAEs were not found to be increased with the combination of VPR and ITZ in comparison to VPR alone.
Conclusion
A comprehensive characterization of the clinical PK properties of VPR has been achieved. Following almost complete oral absorption, VPR is predominantly metabolized in the liver to a complex variety of metabolites, which are mainly excreted with feces. The pivotal role of CYP3A4 in the metabolism of VPR has been confirmed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The clinical parts of the two studies reported, including clinical laboratory analyses, were performed at Covance Clinical Research Unit, Leeds, UK. Ashley Brooks was the principal investigator in the mass balance study and Jim Bush was the principal investigator in the drug-drug interaction/microtracer study. Total radioactivity in whole blood, plasma, urine and feces was analyzed by Covance Laboratories Ltd., Harrogate, UK. Bioanalytical analyses of vilaprisan in plasma were performed at Bayer AG, Wuppertal, Germany. AMS measurements were performed at Xceleron Inc. QC.
Author contributions
All authors participated in the research design and contributed to the interpretation of the data and the writing of the manuscript.
Compliance with Ethical Standards
Funding
This study was sponsored by Bayer AG, Berlin, Germany. Medical writing support was provided by C. Hilka Wauschkuhn, Bonn and funded by Bayer AG.
Disclosure of potential conflicts of interest
Marcus-Hillert Schultze-Mosgau, Joachim Höchel, Olaf Prien, Torsten Zimmermann, and Antje Rottmann are employees of Bayer AG. Ashley Brooks and Jim Bush have received consulting fees from Bayer AG.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the studies.
Footnotes
Clinical pharmacokinetic profiling of VPR has been completed with a second DDI study using a strong CYP3A4 inducer, the results of which are reported separately. The key results of the above MB study and the DDI study were presented at the Congress of the Society for Reproductive Investigation, 2016 (poster no. 117) and at the Marbach DDI Workshop, 2017, respectively.
Electronic supplementary material
The online version of this article (10.1007/s40262-017-0607-4) contains supplementary material, which is available to authorized users.
References
- 1.Wagenfeld A, Bone W, Schwede W, Fritsch M, Fischer OM, Moeller C. BAY 1002670: a novel, highly potent and selective progesterone receptor modulator for gynaecological therapies. Hum Reprod. 2013;28:2253–2264. doi: 10.1093/humrep/det247. [DOI] [PubMed] [Google Scholar]
- 2.Schütt B, Schultze Mosgau M-H, Draeger C, et al. Effect of the novel selective progesterone receptor modulator vilaprisan on ovarian activity in healthy women. J Clin Pharmacol. 2017. 10.1002/jcph.998. [DOI] [PMC free article] [PubMed]
- 3.Schütt B, Kaiser A, Schultze-Mosgau M-H, et al. Pharmacodynamics and safety of the novel selective progesterone receptor modulator vilaprisan: a double-blind, randomized, placebo-controlled phase 1 trial in healthy women. Hum Reprod. 2016;31:1703–1712. doi: 10.1093/humrep/dew140. [DOI] [PubMed] [Google Scholar]
- 4.Bradley L, Ren X, Groettrup-Wolfers E, Petersdorf K, Seitz C. Results of the asteroid (assess safety and efficacy of vilaprisan in patients with uterine fibroids) 1 study: a phase 2, placebo-controlled dose finding study. Fertil Steril. 2016;106:e95–e96. doi: 10.1016/j.fertnstert.2016.07.278. [DOI] [Google Scholar]
- 5.Schultze-Mosgau M-H, Schuett B, Hafner F-T, et al. Pharmacokinetics and safety of the selective progesterone receptor modulator vilaprisan in healthy postmenopausal women. Int J Clin Pharmacol Ther. 2017;55:16–24. doi: 10.5414/CP202756. [DOI] [PubMed] [Google Scholar]
- 6.European Medicines Agency (EMA). Guideline on the investigation of drug interactions. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed 24 July 2017.
- 7.US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for industry: drug interaction studies: study design, data analysis, implications for dosing, and labeling recommendations. http://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf. Accessed 24 July 2017.
- 8.European Medicines Agency (EMA). Guideline on bioanalytical method validation. 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Accessed 26 May 2016.
- 9.US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM). Guidance for industry: bioanalytical method validation (2001). http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070107.pdf. Accessed 24 July 2017.
- 10.European Medicines Agency (EMA). ICH topic M 3(R2) non-clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf. Accessed 24 July 2017.
- 11.US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for industry: safety testing of metabolites. Revision 1. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079266.pdf. Accessed 24 July 2017.
- 12.Kanacher T, Frechen S, Wendl T, et al. Towards a detailed understanding of the impact of drug-drug interactions on vilaprisan exposure by PBPK modeling and simulation: Poster presented at the DDI Workshop at Marbach Castle; May 27–29, 2017.
- 13.Sarkar NN. Mifepristone: bioavailability, pharmacokinetics and use-effectiveness. Eur J Obstet Gynecol Reprod Biol. 2002;101:113–120. doi: 10.1016/S0301-2115(01)00522-X. [DOI] [PubMed] [Google Scholar]
- 14.Pohl O, Zobrist RH, Gotteland J-P. The clinical pharmacology and pharmacokinetics of ulipristal acetate for the treatment of uterine fibroids. Reprod Sci. 2015;22:476–483. doi: 10.1177/1933719114549850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang GR, Wrighton SA, Benet LZ. Identification of CYP3A4 as the principal enzyme catalyzing mifepristone (RU 486) oxidation in human liver microsomes. Biochem Pharmacol. 1996;52:753–761. doi: 10.1016/0006-2952(96)00357-7. [DOI] [PubMed] [Google Scholar]
- 16.Pohl O, Osterloh I, Gotteland J-P. Ulipristal acetate: safety and pharmacokinetics following multiple doses of 10–50 mg per day. J Clin Pharm Ther. 2013;38:314–320. doi: 10.1111/jcpt.12065. [DOI] [PubMed] [Google Scholar]
- 17.Shi YE, Ye ZH, He CH, et al. Pharmacokinetic study of RU 486 and its metabolites after oral administration of single doses to pregnant and non-pregnant women. Contraception. 1993;48:133–149. doi: 10.1016/0010-7824(93)90004-Q. [DOI] [PubMed] [Google Scholar]
- 18.Attardi BJ, Burgenson J, Hild SA, Reel JR. In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone. J Steroid Biochem Mol Biol. 2004;88:277–288. doi: 10.1016/j.jsbmb.2003.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.