

Abstract
The absence of a histidine biosynthesis pathway in humans, coupled with histidine essentiality for survival of the important human pathogen Mycobacterium tuberculosis (Mtb), underscores the importance of the bacterial enzymes of this pathway as major antituberculosis drug targets. However, the identity of the mycobacterial enzyme that functions as the histidinol phosphate phosphatase (HolPase) of this pathway remains to be established. Here, we demonstrate that the enzyme encoded by the Rv3137 gene, belonging to the inositol monophosphatase (IMPase) family, functions as the Mtb HolPase and specifically dephosphorylates histidinol phosphate. The crystal structure of Rv3137 in apo form enabled us to dissect its distinct structural features. Furthermore, the holo-complex structure revealed that a unique cocatalytic multizinc–assisted mode of substrate binding and catalysis is the hallmark of Mtb HolPase. Interestingly, the enzyme–substrate complex structure unveiled that although monomers possess individual catalytic sites they share a common product-exit channel at the dimer interface. Furthermore, target-based screening against HolPase identified several small-molecule inhibitors of this enzyme. Taken together, our study unravels the missing enzyme link in the Mtb histidine biosynthesis pathway, augments our current mechanistic understanding of histidine production in Mtb, and has helped identify potential inhibitors of this bacterial pathway.
Keywords: Mycobacterium tuberculosis, tuberculosis, histidine, phosphatase, inhibitor
Introduction
Tuberculosis (TB),4 caused by the pernicious pathogen Mycobacterium tuberculosis (Mtb), remains a leading agent of morbidity and mortality worldwide largely due to the widespread emergence of multidrug-resistant and extremely drug–resistant strains of Mtb. To combat this global threat, there is a research priority to identify newer drugs with novel mechanisms of action. In this regard, inhibition of the enzymes belonging to the central metabolic pathways, particularly those involved in macromolecular biosynthesis, is a major focus of current research interests (1). A recent study has demonstrated that tryptophan biosynthesis by Mtb is required to confer protection from T-cell–mediated killing (2). Furthermore, inhibition of tryptophan synthase by small-molecule inhibitors offers a new avenue for TB treatment (3). Unlike humans, Mtb biosynthesizes amino acids that serve as the building blocks for protein synthesis and as nutrients for various metabolic pathways (4). Studies pertaining to high-density mutagenesis and starvation survival response have shown that the histidine biosynthesis pathway is essential for Mtb growth (5, 6).
Bacteria, fungi, lower eukaryotes, and plants synthesize histidine de novo from the metabolite phosphoribosyl pyrophosphate in a similar manner with subtle differences mainly in the sixth and the eighth steps of the 10-step pathway. In prokaryotes such as Escherichia coli (7), Salmonella enterica serovar Typhimurium (8), and Azospirillum brasilense (9), the conversion of imidazole glycerol phosphate to imidazole acetol phosphate (sixth step) is catalyzed by the dehydratase domain (C-terminal domain) of a bifunctional enzyme, HisB. The N-terminal domain of this enzyme functions as the histidinol phosphate phosphatase (HolPase) which catalyzes the 8th step, the conversion of l-histidinol phosphate (HOLP) to l-histidinol (HOL). However, in organisms such as Mtb (4, 10), fungi (11), and plants (12, 13), these two steps are separately catalyzed by two different monofunctional enzymes, imidazoleglycerol-phosphate dehydratase and HolPase, respectively. Monofunctional HolPase is often referred to as HisN. Of note, the much awaited Mtb genome sequence was deciphered in 1998 (4) and clearly annotated the genes encoding histidine biosynthesis pathway enzymes, barring that of HolPase. In the context of better understanding this important pathway, we pursued studies intending to identify the Mtb HolPase and obtain a comprehensive mechanistic understanding of it. We show herein that Rv3137 of Mtb, like the actinobacterial HolPases of Streptomyces coelicolor (14) and Corynebacterium glutamicum (15), is monofunctional and belongs to the inositol monophosphatase (IMPase) family. Furthermore, we determined its crystal structure, illuminating the first three-dimensional structural insights of an IMPase family HolPase from the bacterial kingdom, both in apo and holo forms. The enzyme exhibits a unique mode of substrate binding mediated by cocatalytic Zn2+ ions. The enzyme substrate complex was captured in both the pre- and the post-reaction states, which provided insights into the underlying mechanism of its action and the product exit. In an effort to design anti-TB molecules by inhibiting the function of this important enzyme, we have identified novel small-molecule inhibitors of Rv3137 by high-throughput screening. The most potent inhibitor, NSC311153, displayed a submicromolar in vitro efficiency.
Results
Rv3137 is the Mtb HolPase and not an IMPase
To identify the gene that encodes for HolPase, we used a bioinformatics approach. A multiple sequence analysis of histidinol phosphatase sequences retrieved from the Swiss-Prot database (16) followed by a phylogenetic analysis of 50 unique genera revealed a divergent evolution of bifunctional HisB and monofunctional HisN/HolPase encoded phosphatases (Fig. 1A). The domain possessing the phosphatase activity was conserved within the bifunctional HisB of modern bacterial clades such as Gammaproteobacteria (Escherichia and Salmonella), whereas in clades such as Actinomycetales (Mycobacteria, Corynebacterium, and Streptomyces), the phosphatase activity was performed by a unique monofunctional enzyme. Interestingly, Rv0114, which is annotated as a d-glycero-α-d-mannoheptose 1,7-bisphosphate phosphatase (gmhB) (17), also speculated to be a probable HolPase (18) has diverged separately from both the bifunctional HisB and monofunctional HisN, suggesting a possibly different role of this protein other than that of a HolPase (Fig. 1A). The amino acid sequence of Rv3137 showed high degrees of sequence identities with the reported monofunctional HolPases from other Actinomycetales, including Corynebacterium (66%) and Streptomyces (58%), suggesting that Rv3137 is the potential HolPase of Mtb (Fig. 1C).
Figure 1.

Conservation of HisN (Mtb HolPase) in clade Actinomycetales. A, an unrooted phylogenetic tree depicting the conservation of HisN among the clade Actinomycetales (in the red circle). Also, clear divergent evolution is evident within HisB (bifunctional) and HisN (monofunctional) encoded phosphatases. Enzymes on the right side of the dashed purple line are monofunctional HisN, whereas those on the left are bifunctional HisB except for GmhB (underlined in gray), which has diverged from both HisB and HisN. B, an ESPript representation of sequence alignments of Rv3137 and the evolutionarily closer HisN for the represented genera showing conservation of key residues and motifs. C, sequence identities and query coverage for Rv3137 against known HolPases.
A few conserved stretches of active-site residues of other homologs and key residues such as GEE, DPIDGT, and AGG were observed among Rv3137 as well (Fig. 1B). A search for conserved domains revealed that Rv3137 possesses a bacterial family of Mg2+-dependent phosphatase related to bacterial IMPase type 1 domain (E value, 3.11e−94). The ScanProsite results together with ProRule-based predicted intradomain features identified the presence of inositol monophosphatase family signature 1, PS00629 ((F/W/V)X0,1(L/I/V/M)DP(L/I/V/M)D(S/G)(S/T)X2(F/Y/A)X0,1(H/K/R/N/S/T/Y)), within amino acids 80–91 (WIVDPIDGTKNFVR) in the Rv3137 sequence (19). However, another signature conserved in IMPases, PS00630, was absent in Rv3137. The most characteristic motif of HolPase of the IMPase family that distinguishes it from other IMPases (20) was conserved in Rv3137 within amino acids 185–190 (RAYGDF) with Asp189 being unique to this class. Briefly, this motif consists of a highly conserved Arg followed by a Gly/Ala and an aromatic amino acid. Furthermore, a highly conserved Gly and a strictly conserved Asp follow in the sequence. In actinobacterial HolPase, an aromatic amino acid follows this Asp (20). Taken together, these observations suggest that Rv3137 is likely the gene encoding for Mtb HolPase, which is monofunctional and like other known actinobacterial HolPases belongs to the IMPase family.
To confirm that Rv3137 is indeed the Mtb HolPase, we carried out relevant biochemical assays. First, the enzyme was cloned using gene-specific primers (Table 1) into the Mycobacterium smegmatis (Msg)–E. coli shuttle expression vector pYUB1062 and overexpressed in Msg expression system mc24517. Then, the enzyme was purified to homogeneity using nickel-nitrilotriacetic acid affinity followed by size-exclusion chromatography. The enzyme exists as a dimer in solution as is evident from the size-exclusion chromatogram (Fig. S1). The molecular mass of the recombinant Rv3137 is 28.62 kDa. The protein eluted at 60-ml volume, which is in between the elution volume of 75- and 44-kDa molecular mass markers, closer to the latter. Phosphatase activity results (Fig. 2 and Table 2) revealed that in the presence of Mg2+ Rv3137 was able to specifically dephosphorylate HOLP and not IMP. Mg2+ was used as a cofactor for this enzyme based on the metal preference of its conserved domain. The optimal Mg2+ concentration, pH, and temperature for the enzyme's maximal activity were determined to be 1 mm, 8.0, and 310 K, respectively.
Table 1.
Sequences of the primers used in the study
| Primer | Sequence |
|---|---|
| Rv3137_Fpa | 5′-CACCCATATGCACCATCATCATCATCATGTGAGCCACGACGATCTAATG-3′ |
| Rv3137_Rpa | 5′-TATAAGCTTTTACCCGGCGTTGAGCCGTGT-3′ |
| Asp44-Ala_Fp | 5′-CGACCGCCCGAGCGGCGTCGGTC-3′ |
| Asp44-Ala_Rp | 5′-GACCGACGCCGCTCGGGCGGTCG-3′ |
| Glu67-Ala_Fp | 5′-CCGCCGAACTCCGCGCCCAAGACGC-3′ |
| Glu67-Ala_Rp | 5′-GCGTCTTGGGCGCGGAGTTCGGCGG-3′ |
| Asp83-Ala_Fp | 5′-CAGTGGATCGTAGCCCCGATCGACGGC-3′ |
| Asp83-Ala_Rp | 5′-GCCGTCGATCGGGGCTACGATCCACTG-3′ |
| Thr88-Ala_Fp | 5′-GCACAAAGTTTTTGGCGCCGTCGATCGGGTC-3′ |
| Thr88-Ala_Rp | 5′-GACCCGATCGACGGCGCCAAAAACTTTGTGC-3′ |
| Asp213-Ala_Fp | 5′-CAGTGCCGCCAGAGCCCATACCGACAC-3′ |
| Asp213-Ala_Rp | 5′-GTGTCGGTATGGGCTCTGGCGGCACTG-3′ |
a Underlined sequences represent restriction sites. Italic sequences represent His6 tag sequence. Sequences in bold are Rv3137-specific N- and C-terminal sequences. Fp and Rp stand for forward and reverse primer, respectively.
Figure 2.

Enzyme kinetics. Saturation kinetics for Mtb HolPase with Mg2+ as a cofactor are shown. Data are replicate values from triplicate experiments and were fitted to the Michaelis–Menten equation using GraphPad Prism version 6 software. Error bars represent S.D.
Table 2.
Kinetic parameters of Mtb HolPase
| HOLP (Zn2+) | HOLP (Mg2+) | IMP | |
|---|---|---|---|
| Km (μm) | 27.66 ± 2.39 | 31.98 ± 4.81 | No activity |
| kcat (s−1) | 0.99 ± 0.04 | 0.99 ± 0.07 | No activity |
| kcat/Km (m−1 s−1) | 35.79 ± 4.51 × 103 | 30.96 ± 6.8 × 103 | No activity |
To examine the catalytic efficiency of the bacterial and plant IMPase family HolPases, we compared Mtb HolPase kinetic parameters with those of its counterpart from Medicago truncatula (Mt). Rv3137 followed Michaelis–Menten kinetics and exhibited a Km value of 31.98 ± 4.81 μm (Fig. 2 and Table 2), which is ∼8-fold lower than the reported Km (263 ± 28 μm) for Mt HolPase (21). The catalytic constants (kcat) of Mtb HolPase and Mt HolPase are 0.99 ± 0.07 and 3.6 ± 0.14 s−1, respectively. Mtb HolPase catalyzes the dephosphorylation of HOLP more efficiently than that of Mt HolPase as its specificity constant (kcat/Km) is 30.96 ± 6.8 × 103 m−1 s−1, which is ∼2-fold higher than that of the latter (13.7 ± 1.9 × 103 m−1 s−1). Neither the Mtb HolPase nor any of its mutants listed in Table 1 exhibited any detectable activity with IMP, substantiating that Rv3137 is the Mtb HolPase of the IMPase family and is not an IMPase.
The 3D structure of Mtb HolPase reveals its structural features, mode of dimer assembly, and metal binding in the active site
To derive mechanistic insights into the function of Mtb HolPase, we determined its three-dimensional structure at 1.95-Å resolution by X-ray crystallography (Table 3). Analysis of the 3D structure showed that the polypeptide chain folds as two distinct structural domains, named here as the N-terminal (amino acids 1–136) and the C-terminal (amino acids 152–260) domains. The tertiary structure comprises 10 helices (seven α-type and three 310-type), 12 β-strands, and 21 loops (Fig. 3A). The N-terminal domain folds into a mixed six-stranded β-sheet underneath of which lie two long α-helices (Fig. 3A). Conversely, the C-terminal domain folds as a globular structure comprising a mixed five-stranded β-sheet sandwiched between six helices. Both domains are connected by a 15-residue–long loop. The biological functional unit of Mtb HolPase is a homodimer as observed both in the solution and crystal structures. The crystal asymmetric unit of the native crystal contains two copies of such dimer. The monomers of the biological unit are assembled in an inverted manner and are related to each other by a 2-fold rotational symmetry perpendicular to the plane of the paper (Fig. 3B). The dimer interface buries a surface area of about 1817 Å2, ∼16% of the total accessible surface area of a monomer as calculated using the PISA server (22). The dimer is stabilized largely by hydrogen bonds, salt bridges, and Van der Waals interactions. Asp189, Val149, Arg183, Asp202, Ala119, Arg171, Asn90, Arg93, Ile31, Arg122, and Ala186 of one monomer form hydrogen-bonding interactions with Arg185, Ile31, Asn90, Arg93, Arg122, Asp179, Tyr187, Gly199, Ser148, Gln121, and Ala186, respectively, of the other monomer and vice versa (Fig. 3C). Analysis of the electrostatic surface potential of the residues lining the interface clearly shows that charge and shape complementarities contribute in the formation of a stable dimer assembly (Fig. 3D). Neither intrasubunit nor intersubunits disulfide bridges are present in the molecule.
Table 3.
Data collection and refinement statistics
Values in parentheses are for the highest resolution range. r.m.s., root mean square.
| Native (5ZON) | HOLP complex (5YHT) | |
|---|---|---|
| Data collection | ||
| Space group | P1 | P21212 |
| Unit cell dimensions | ||
| a (Å) | 49.03 | 92.66 |
| b (Å) | 71.06 | 142.45 |
| c (Å) | 91.59 | 48.72 |
| α (°) | 92.16 | 90 |
| β (°) | 96.16 | 90 |
| γ (°) | 101.94 | 90 |
| Temperature (K) | 100 | 100 |
| Resolution (Å) | 50.00–1.95 | 50.00–2.90 |
| Highest resolution range | 2.02–1.95 | 3.00–2.90 |
| Unique reflections | 85,945 | 14,904 |
| 〈I/σ(I)〉 | 10.21 (2.51) | 12.75 (2.0) |
| Completeness (%) | 97.6 (96.7) | 99.2 (98.7) |
| Redundancy | 2.9 (2.9) | 4.7 (3.9) |
| Rmerge (%)a | 10.2 (50.0) | 12.40 (50.8) |
| Refinement | ||
| Rworkb | 0.2120 | 0.2293 |
| Rfreeb | 0.2540 | 0.2788 |
| Average B-factor (Å2) | ||
| Protein | 27.10 | 64.51 |
| Metal ions | 31.12 | 58.06 |
| Ligand | 47.51 | 78.95 |
| Solvent | 42.14 | 48.92 |
| Number of atoms | ||
| Protein | 7,425 | 3,425 |
| Metal ions | 8 | 4 |
| Ligand | 52 | 49 |
| Solvent | 971 | 23 |
| r.m.s. deviations from ideal | ||
| Bond lengths (Å) | 0.0099 | 0.0100 |
| Bond angle (°) | 1.4889 | 1.5025 |
| Ramachandran plot analysis (%) | ||
| Preferred regions | 97.0 | 94.0 |
| Allowed regions | 3.0 | 6.0 |
| Disallowed regions | 0.0 | 0.0 |
a Rmerge(I) = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl) for n independent reflections and i observations of a given reflection. 〈I(hkl)〉 is the average intensity of the i observations.
b Rwork and Rfree = Σh‖F(h)o| − |F(h)c‖/Σh|F(h)o| where F(h)o and F(h)c are the observed and calculated structure-factor amplitudes, respectively. Rfree was calculated using 5% of data.
Figure 3.

Presentation of the 3D structure and mode of dimerization of native Mtb HolPase. A, three-dimensional structure of Mtb HolPase is depicted in cartoon representation. Helices (green, α-type; blue, 310-type) and β-strands are numbered. B, cartoon representation of a physiological dimer showing the inverted mode arrangement of the monomers. The line of symmetry passes between the two monomers and is represented as a black ellipse. Monomers A and B are represented in lime green and salmon colors, respectively. C, hydrogen-bonding interactions between the two monomers are shown by dotted lines. D, electrostatic potential surface map shows that a charge complementarity exists at the dimer interface.
The general location of the active-site pocket of the Mtb HolPase was deduced by overlaying its 3D structure with that of its Mt homolog. Analysis of the electron density (ed) maps in the active-site pocket of each monomer revealed the presence of two distinct spherical 2|Fo| − |Fc| and |Fo| − |Fc| ed peaks, implying that the enzyme harbors two metals in each monomer (Fig. 4A). As mentioned earlier, CDD predicted that the enzyme possessed a Mg2+-dependent domain. Therefore, Mg2+ atoms were modeled into the ed peaks, and their positions were refined. However, residual difference ed peaks at the Mg2+ metal positions (Fig. 4B) indicated the presence of metals heavier than Mg2+. To identify the probable metals that correspond to these ed peaks, we carried out a 4-(2-pyridylazo)resorcinol (PAR) assay. The PAR assay can detect the presence of copper, manganese, cobalt, nickel, and zinc as they form colored complexes with PAR (23). The peak at 500 nm confirmed the formation of (PAR)2Me(II) complex (Fig. S2A). Of those metals that can be detected using the PAR assay, the presence of zinc in the active site of a few bacterial HolPases has been reported (24). The presence of Zn2+ in the purified protein was further confirmed using a zinc assay kit (BioVision Inc.) (Fig. S2B). Based on this, we modeled Zn2+ atoms in those ed peaks. These Zn2+ atoms did fit satisfactorily in the ed peaks in both monomers, leaving no residual difference ed (Fig. 4C).
Figure 4.
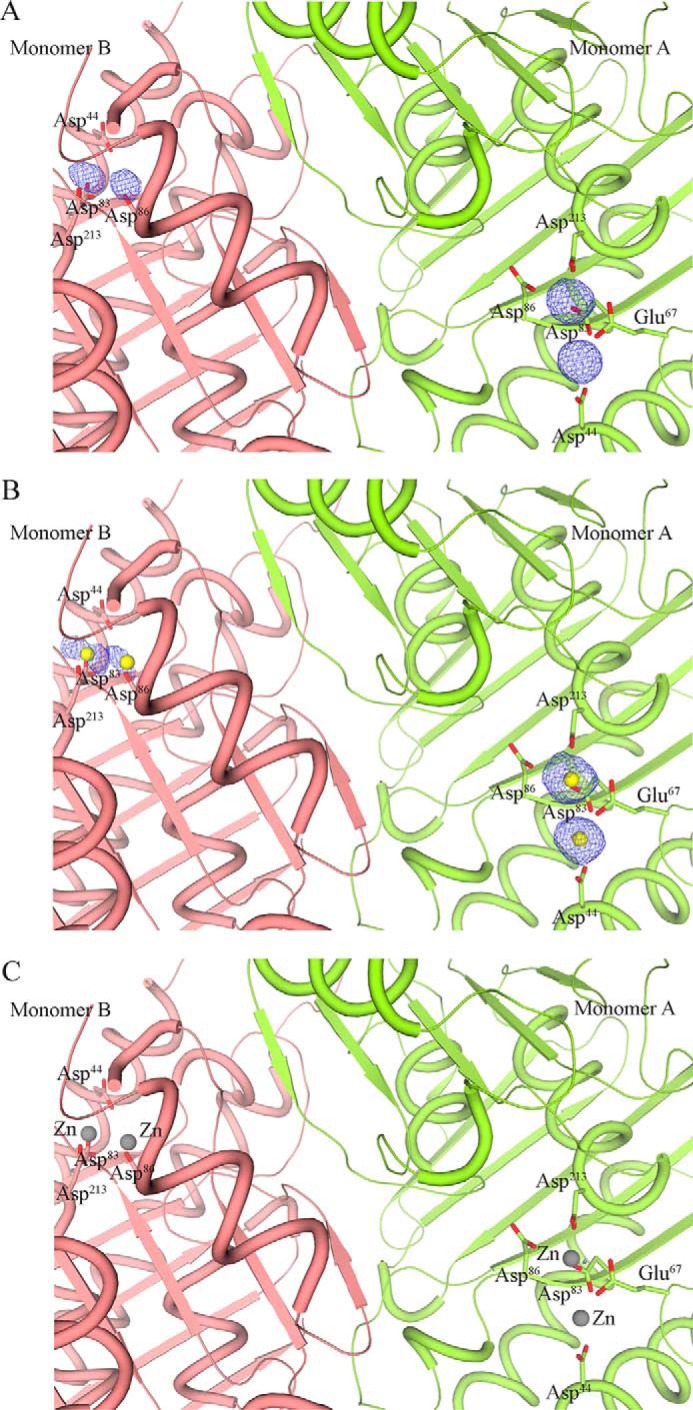
Metal binding in the active site of Mtb HolPase. A, two distinct |Fo| − |Fc| spherical ed peaks (contoured at 5.0σ) were observed in the active-site pocket of each monomer, indicating the presence of metal atoms. B, residual difference (|Fo| − |Fc|) ed peaks (2.5σ) at all the metal positions after incorporating and refining Mg2+ atoms in those positions. C, no residual difference ed peaks (2.5σ) were observed in the dimer after incorporating Zn2+ in those positions. In all cases, 2|Fo| − |Fc| ed peaks (at 1σ) were observed.
Superimposition of these monomers using PDBeFOLD (25) (root mean square deviation (r.m.s.d.) value of 0.70 Å and Q score of 0.91 for 246 Cα atoms) showed that the spatial positions of Zn2+ ions are different in the two monomers. The dimer possesses four Zn2+ ions, two in each monomer, at three different positions (Fig. 5A). They are bound at positions 1 and 2 in monomer A and at positions 1 and 3 in monomer B (Fig. 5, B and C). Position 1 is a common preference for Zn2+ in both monomers. For reference, we denote Zn2+ atoms at 1-, 2-, and 3-positions as Zn2+/1, Zn2+/2, and Zn2+/3, respectively. Furthermore, examination of the interactions between these metal ions and the enzyme shows that both Zn2+/1 and Zn2+/2 in monomer A possess complete octahedral coordination spheres (Fig. 5B). Zn2+/1 interacts with Glu67, Asp83, Asp213, Pi, and two water molecules, whereas Asp44, Glu67, Glu68, Asp83, Pi, and a water molecule are coordinated with Zn2+/2. However, Zn2+/1 and Zn2+/3 of monomer B do not have complete coordination geometries (Fig. 5C). In this monomer, Zn2+/1 interacts with Asp83, Asp213, and Pi; Zn2+/3 interacts with Asp86, Asp189, and Pi. Even the water molecules closest to these Zn2+ ions are beyond 3-Å distance. Although the relatively few interactions of Zn2+/3 with the enzyme is likely linked to its solvent-exposed position, a similar scenario for Zn2+/1 is due to the absence of the L3 loop in this monomer. Loop L3, from which Glu67 and Glu68 protrude, is a dynamic region with an instability index of 40.96, classifying this region as unstable, as calculated using the ExPASy ProtParam tool (26). L3 is ordered in the crystal structure only when a Zn2+ is present at position 2 (Fig. 5, A, B, and C). These observations indicate that either the presence of a Zn2+ ion at position 2 stabilizes the loop or vice versa. Zn2+/1 and Zn2+/3 in monomer B exhibit weak interactions, thereby their typical coordination is missing, so we refer to this conformation as a “relaxed” state. In contrast, Zn2+/1 and Zn2+/2 in monomer A have complete coordination spheres and were found to be favorable for substrate binding in the cocrystal structure as described later in the text. This conformation we refer to as a “ready” state.
Figure 5.
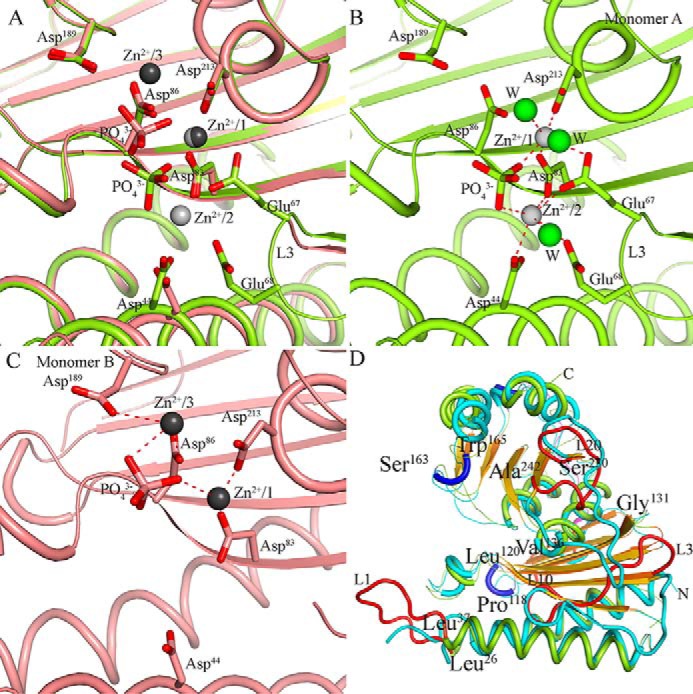
Metal-binding positions in Mtb HolPase monomers and comparison of Mtb HolPase structure with Mt equivalent. A, superposition of two monomers of a native crystal dimer. Monomer A shown in lime green color consists of two Zn2+ (light gray spheres) ions at positions 1 and 2, and monomer B in salmon color consists of two Zn2+ (dark gray spheres) ions at positions 1 and 3. Metal position 1 is common in both monomers. Although the majority of residues superpose, loop L3 containing Glu67 and Glu68 is not complete in monomer B where position 2 is empty. B and C, metal binding in their respective monomers. B shows Zn2+ interactions at positions 1 and 2 in monomer A. C depicts Zn2+ interactions at positions 1 and 3 in monomer B. Water molecules (W) are represented as green spheres. D, an overlay of the backbone structures between Mtb HolPase (colored in lime green, red, blue, and orange) and Mt HolPase (colored in cyan and gold), highlighting the distinct secondary structures of Mtb HolPase.
As the crystal structure showed that the enzyme harbors Zn2+ ions, we carried out the enzyme kinetics in the presence of this metal also. The optimum concentration of cofactor Zn2+ and pH of the buffer were determined to be 1 mm and 7.0, respectively. As shown in Table 2, the kinetic constants were largely similar in enzymatic reaction buffer containing either of the two (Zn2+ or Mg2+) metal ions. Like some of the enzymes that show activity with a variety of divalent metals in vitro (27), Rv3137 also shows in vitro activity with both Zn2+ and Mg2+. The specificity constant (kcat/Km) with Zn2+ is 1.16-fold higher (Fig. 6 and Table 2).
Figure 6.

Enzyme kinetics with Zn2+ cofactor. Saturation kinetics for Mtb HolPase with Zn2+ as the cofactor are shown. Data are replicate values from triplicate experiments and were fitted to the Michaelis–Menten equation using GraphPad Prism version 6 software. Error bars represent S.D.
IMPase family HolPases: Similarity and differences in overall 3D structure
The availability of the structure of an IMPase family HolPase from Mt (21) enabled us to dissect the similarities and differences between the bacterial and plant HolPases of this class. Both enzymes share a common fold (Fig. 5D) and overlay with a moderate r.m.s.d. value of 1.40 Å (for 233 Cα atoms) and a Q score of 0.68, largely because of the features that both enzymes are functionally similar and belong to the same family. However, they exhibit marked structural differences in certain regions. Mt HolPase contains a 62-residue-long N-terminal signal peptide, whereas its Mtb counterpart possesses no such signal peptide sequence. The reported 3D structure of Mt HolPase is of a signal peptide–truncated construct (21). In Mtb HolPase, loop L1 (Leu26–Leu37) that connects helix1 and helix2 is well-ordered (Fig. 5D). Ile31 protruding from this loop is involved in the formation of the biological functional unit of this enzyme. The corresponding structural elements in the Mt HolPase crystal structure are incomplete as residues 91–97 are missing, probably because of the dynamicity of the region. The instability index of this seven-amino acid–long stretch, computed using the ExPASy ProtParam tool (26), is 60.03, suggesting that this stretch is unstable. Although residues Pro118–Leu120 form a 310 helix in Mtb HolPase, the corresponding residues (180–183) of Mt HolPase form a turn in the loop connecting two β-strands. Ala119 protruding from this 310 helix of Mtb HolPase forms hydrogen bonds with Arg122 of the partner monomer at the dimer interface and contributes toward the dimer stability (Fig. 3C). Perhaps because of its role in dimer formation, it arises from a comparatively stable structural element. In Mtb HolPase, residues Gly131–Val136 form a β-strand, whereas the equivalent region (194–200) in the Mt counterpart makes up a loop structure (Fig. 5D). Residues Ser163–Trp165 make up a 310 helix in Mtb HolPase, whereas the corresponding region (221–223) in Mt HolPase contributes to a loop structure. Leu222 from this loop of Mt HolPase is involved in binding of the HOL moiety of the substrate HOLP. Furthermore, in Mt HolPase, 287–305 is an extra-long loop for which the corresponding loop region (230–242) in Mtb HolPase is shorter. Although the biological functional units of both Mt and Mtb HolPases are dimers with a similar mode of organization, the distinct structural differences between them attribute to the differences in their secondary structural elements and may exert an influence on their enzymatic activities and regulation.
Cocrystal structure of Mtb HolPase with HOLP reveals a unique mode of substrate binding, a multizinc active-site pocket, and a product-exit channel
To derive the underlying mechanism of the action of Mtb HolPase, we determined its cocrystal structure with its substrate (Table 3). The difference Fourier electron density map in Fig. 7, A, B, C, and D, clearly shows that the enzyme is bound with HOLP and two Zn2+ ions in monomer A in the active-site pocket situated between the N- and C-terminal domains. In addition to this crystallographic evidence, the presence of zinc was confirmed by a zinc assay kit (BioVision Inc.). Notably, the substrate-binding pocket is bounded by a large number of aspartates and glutamates and is exposed to the bulk solvent environment. The electrostatic potential surface of the active-site area is largely negative, providing a favorable chemical environment for the binding of two Zn2+ ions (Fig. 7E). These Zn2+ ions, at a distance of ∼4.0 Å from each other, occupy metal-binding sites 1 and 2 and are tetrahedrally coordinated with the atoms of the substrates and the active-site residues (Fig. 7, B and D). In particular, Zn2+/2 interacts with Oϵ2 of Glu67, Oδ1 of Asp83, Oδ2 of Asp44, and OP1 of HOLP (Fig. 7F). Similarly, Oδ1 of Asp213, Oϵ1 of Glu67, Oδ2 of Asp83, and OP3 of HOLP are linked to Zn2+/1 (Fig. 7F). Notably, the representatives of such a cocatalytic zinc motif possess two or three metals in close proximity, of which two are bridged by a side-chain moiety of a single amino acid residue, such as Asp, Glu, or His, and sometimes a water molecule (28). Mtb HolPase is unique as it possesses two bridges between Zn2+/1 and Zn2+/2, both of which are contributed by side chains of amino acid residues Glu67 and Asp83 (Fig. 7F).
Figure 7.

Substrate recognition by Mtb HolPase. A, the omit |Fo| − |Fc| electron density map at 2.5σ contour level evidenced HOLP and zinc binding in monomer A at the active site of the enzyme. The final refined HOLP and Zn2+ ions are superimposed on the electron density map and are shown in stick and sphere representations, respectively. B, zoomed-in view of a portion of A, showing residues that interact with the two Zn2+ ions. C, the omit |Fo| − |Fc| electron density map contoured at 5σ clearly shows the binding of zinc in the active site. D, zoomed-in view of a portion of C. E, electrostatic potential surface of the active-site area with HOLP and Zn2+ ions shows the charge complementarity involved in metal and substrate binding. F, atomic interactions among the enzyme, HOLP, and Zn2+ ions are depicted.
Notably, the substrate binds to the monomer A, which possesses Zn2+ at positions 1 and 2 where they complete their respective tetrahedral coordination sphere by interacting with corresponding amino acids and the phosphate moiety of the substrate (Fig. 7, A and B). These positions of Zn2+ ions are similar to those in monomer A of the native enzyme crystal structure (Fig. 5B). We refer to this state of the enzyme as a ready state, which can readily accept the substrate. The partner monomer B in the complex structure possesses Zn2+/1 and Zn2+/3 and is in a relaxed state (Fig. 7A) as was observed in monomer B of the native structure (Fig. 5C). The crystal asymmetric unit of the HolPase–HOLP complex consists of a functional dimer, represented here as monomers A and B. As discussed above, the positions of Zn2+ ions in these monomers are the same as in their counterparts in the native structure. Interestingly, although in the active-site pocket of monomer A the substrate HOLP is bound, monomer B possesses only a phosphate (Fig. 8A). A careful examination of the ed maps pinpointed the presence of the product HOL in a channel at the dimer interface (Fig. 8A). HOL was modeled in the observed ed with a real-space correlation coefficient (RSCC) value of 0.91 according to the Protein Data Bank validation report. This implies that the dephosphorylation reaction has already occurred in monomer B. The atomic interactions of Zn2+ ions in monomer A and monomer B are depicted in Fig. 8, B and C, respectively. Although Glu67 interacts with the Zn2+ at position 1 in both monomers, the dynamic loop L3 is disordered in monomer B where the reaction is complete (Fig. 8, C and D). This indicates that, upon completion of the reaction, the Zn2+ ions move to positions 1 and 3, and as position 2 is empty loop L3 loses its stability. We hypothesize that the completion of the reaction in one monomer switches it to a relaxed state, which probably prompts the partner monomer to adopt a ready state. The existence of the two monomers of a physiological dimer in two different states, ready and relaxed, in the native as well as in the substrate-bound structures clearly indicates that the monomers act one at a time. The same is observed in the cocrystal structure where the reaction is complete in monomer B and substrate is bound in monomer A. This finding represents another case of half-of-the-sites reactivity that some other enzymes also exhibit (29–31).
Figure 8.

Comprehensive view of the protein dimer with its substrate binding clefts and product exit points. The Mtb HolPase exists as a dimer in the solution as well as in the crystal structure. A, surface representation of the cartoon diagram of the dimer depicting catalytic sites of both monomers. The product HOL is visible at the dimer interface. B and C, front view of the catalytic sites of monomers A (wheat color) and B (pale cyan color). B shows that, in addition to Zn2+/1 and Zn2+/2, monomer A has the substrate HOLP bound in its catalytic pocket. The ring of HOLP is headed toward “cleft A.” C depicts monomer B with bound phosphate in its catalytic pocket along with Zn2+/1 and Zn2+/3. Interacting residues at positions 1, 2, and 3 are shown in sticks. D, superimposition of the two monomers of Mtb HolPase showing the pre- and the post-reaction states in the catalytic sites. As is evident from the figure, the ring of the HOLP in monomer A occupies metal-binding position 3, and its phosphate is closer to its corresponding Zn2+/1 and Zn2+/2. Loop L3 is stable only when a metal occupies position 2. The free phosphate molecule and Zn2+/1 and Zn2+/3 in monomer B are shifted toward the cleft. E, surface representation of the cartoon diagram of the protein dimer with product HOL in the center and catalytic sites in the rear view. The product lies at the perfect symmetric position in a product-exit channel with two exit points and hence can exit from either “Exit 1” or “Exit 2.” F and G, front views of Exit 1 and Exit 2, respectively.
Superposition of monomers A and B (r.m.s.d. of 0.1 Å and Q score of 0.96 for 246 Cα atoms) visualizes the pre- and post-dephosphorylation differences in the positions of the entities present in the catalytic pocket (Fig. 8D). The position occupied by the histidine ring of the HOLP in monomer A is occupied by Zn2+/3 in the partner monomer B (Fig. 8D). It appears that following the reaction the product HOL likely enters into a cleft (Fig. 8, B and C), leaving the phosphate and the Zn2+ ions behind (Fig. 8C), as is evident from the shift in the positions of Zn2+ ions and the phosphate molecule (Fig. 8D). The product ultimately reaches the channel at the dimer interface where it can exit from the enzyme through either of the two symmetric exit points (Fig. 8, E, F, and G). However, the product got trapped in this case.
The presence of the product (in the channel at the dimer interface) and the substrate in the active site of one of the monomers raised a pertinent question: what triggered the active enzyme to become inactive? In an effort to address this, we examined the effects of the reagents used to grow crystals on the activity of the enzyme. First, we checked the activity of the enzyme in the cocrystallization buffer (20 mm Tris, 200 mm NaCl, pH 7.5, 0.1 mm ZnCl2) in which the enzyme–substrate complex was prepared prior to setting up the crystallization experiment. Then, the activity of the enzyme in this buffer in the presence of varying concentrations of the crystallization precipitant (1 m ammonium sulfate), the condition that yielded diffraction-quality crystals, was measured. As shown in Fig. S3, A and B, although the enzyme exhibited relatively slowed down activity in the cocrystallization buffer compared with the standard conditions used for kinetics experiment, it lost any detectable activity as soon as an equal volume of the precipitant was added. This observation clearly demonstrates that the presence of 1 m ammonium sulfate in the crystallization droplet (1:1 ratio) locks the otherwise active enzyme in an inactive state. The situation that is observed in the crystal structure is likely a result of the slow activity of the enzyme (in cocrystallization mixture), leading to the product formation followed by enzyme inactivation (in crystallization droplet) that can still bind the substrate but cannot catalyze it.
Mutational studies confirmed the catalytic residues and the presence of a cocatalytic site
To identify the residues involved in the dephosphorylation reaction, structure-guided mutational studies were carried out. In this context, mapping the interactions between the enzyme and the substrate was helpful. Various mutations involving different active-site residues listed in Table 1 were made, and the activity of each mutant enzyme was checked. Asp44, Glu67, Asp83, Thr88, and Asp213 were individually mutated to alanine. The activity results showed that even a single mutation abolished the activity of Mtb HolPase (Fig. S4, A and B). It is evident from the structure that Asp44, Glu67, Asp83, and Asp213 are directly involved in the interactions with Zn2+ ions, with Glu67 and Asp83 being the bridging residues (Fig. 7F), and that they play important role in HOLP dephosphorylation. Mutating any of the four residues involved in the coordination with either of the two Zn2+ ions leads to loss of any detectable activity (Fig. S4, A and B). This suggests that both Zn2+ ions are critically important for the activity of Mtb HolPase, confirming that these two ions form a cocatalytic zinc motif. The participating Zn2+ ions are in close proximity (∼4 Å) and function as a catalytic unit, typical of this motif. It can be hypothesized that these Zn2+ ions coordinate to maintain the active-site conformation and to initiate the reaction. This site is “cocatalytic” as both Zn2+ ions play crucial roles in catalysis, although only one of them activates the attacking water (32, 33). However, Thr88 makes no interaction with either of the Zn2+ ions; instead its Oγ1 and nitrogen are involved in making hydrogen bonds with the phosphate oxygen ions OP4 and OP2 of the HOLP, respectively (Fig. 7F). These interactions suggest that Thr88 is involved in positioning the substrate in a correct orientation for facilitating the dephosphorylation reaction.
Plant and bacterial HolPases: Differential substrate binding
To map the differences and similarities in the mode of substrate binding and disposition of the active-site residues between the plant and bacterial HolPases, their substrate-bound structures were superimposed (r.m.s.d. of 1.46 Å and Q score of 0.65 for 229 Cα atoms), and the active sites were examined (Fig. 9, A and B). Both the HolPases show a similar catalytic pocket with a shift in the position of the bound substrates (Fig. 9, A and B). Presence of a cocatalytic site with two Zn2+ ions in Mtb HolPase creates a positively charged environment to accommodate the phosphate group of HOLP, leading to the displacement of HOLP toward the metal ions compared with the HOLP in Mt HolPase. This position of HOLP in Mtb would not have been possible without the two metal ions as otherwise the whole environment is largely made up of negatively charged residues (Asp44, Glu67, Asp83, and Asp213) that would repel the phosphate group of HOLP. In Mt HolPase, there is a single Mg2+ ion behind the carboxylic groups of Asp146 and Asp270, corresponding to Asp83 and Asp213, respectively, of Mtb HolPase. It is obvious that this metal alone cannot hold the substrate as close and tight as can the two Zn2+ ions. However, Mg2+ is indispensable for HOLP to bind in the active site in Mt HolPase. It neutralizes the phosphate, and it has been shown that Mg2+ enters the active site in complex with HOLP, and both of them bind together (21). Mg2+ is bound to Mt HolPase without a typical octahedral coordination sphere and therefore easily escapes the catalytic center post-reaction (21). In contrast, both Zn2+ ions along with the released phosphate bind in the Mtb HolPase active site even in the absence of the substrate. It can be hypothesized that the phosphate moiety of incoming HOLP replaces the free phosphate molecule. It is then tightly held by the two Zn2+ ions at positions 1 and 2, thereby completing their respective coordination spheres.
Figure 9.
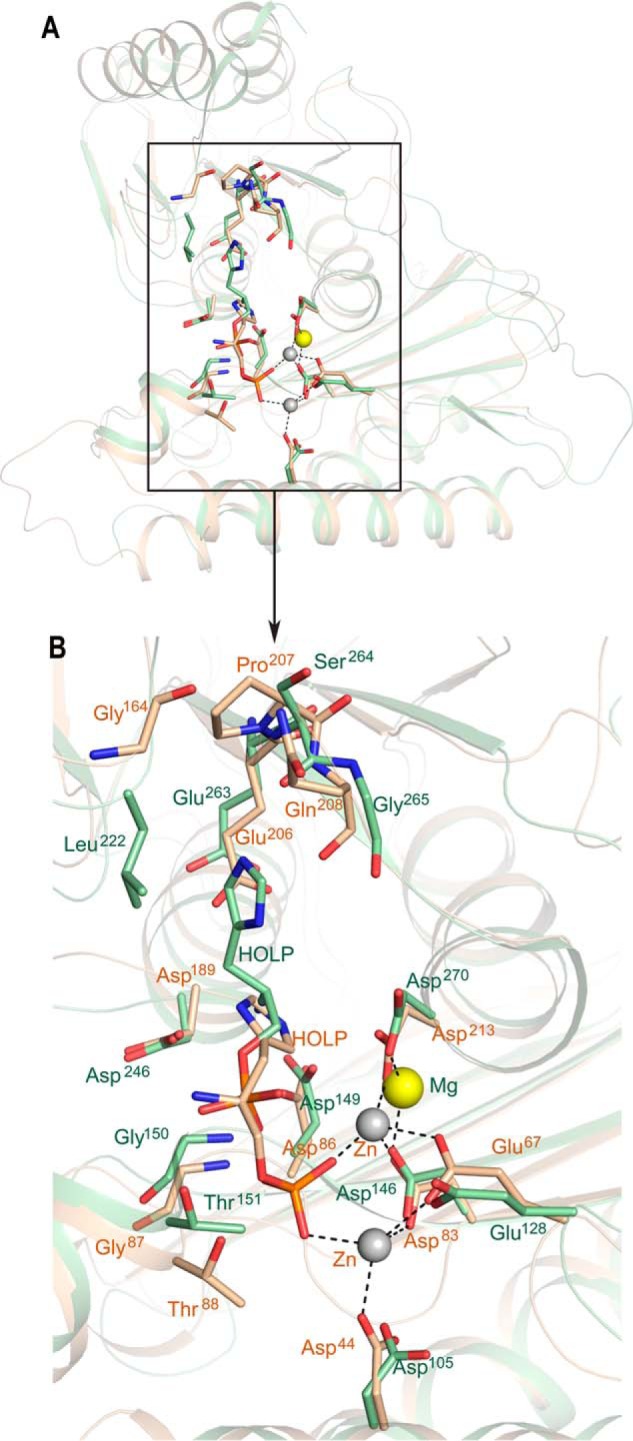
Mode of substrate binding in Mtb and Mt HolPases. A, superimposition of the substrate-bound structures of Mtb and Mt HolPases shows the difference in substrate- and metal-binding positions. The residues involved in the interactions with substrate in both the Mtb and Mt HolPases are depicted. B, zoomed-in view of the portion of A shown in the rectangular box. Entities belonging to Mtb are represented in wheat, and those to Mt are in green.
In Mt HolPase, it is reported that the Nϵ atom of HOLP via a water molecule interacts with the amide nitrogen atom of Ser264 and with the carbonyl oxygen of Leu222. Nδ forms a water-bridged hydrogen bond with the carbonyl oxygen of Gly265. The nitrogen atom of HOLP forms hydrogen bonds with the carboxyl oxygen atoms of Asp149 and Asp246 and a water-bridged interaction with the carboxyl oxygen of Glu263. The oxygen atom of HOLP, where the hydrolysis occurs, interacts with the Asp246 by a water molecule. The phosphate moiety interacts with amide nitrogen of Gly150, Thr151 (OP1 atom of phosphate), and oxygen of Thr151 (OP2) (21). However, in Mtb HolPase, the Nϵ atom of HOLP forms a direct hydrogen bond with Oδ1 of Asp86 (corresponding to Asp149 of Mt) and van der Waals interactions with Oδ2 of Asp213 (Asp270 of Mt). The Nδ of HOLP forms van der Waals interactions with Oδ1 of Asp189 (Asp246 of Mt). The nitrogen atom of HOLP interacts indirectly via a water molecule with oxygen of Thr88 (Thr151 of Mt), and the OP2 of HOLP interacts with the amide nitrogen of Thr88 (Fig. 9, A and B). The phosphate moiety directly interacts with the two Zn2+ ions and is held close to them. Although the major interacting residues significantly superpose and are more or less the same in both Mtb and Mt HolPases, the substrate atoms with which the corresponding residues of the two HolPases interact are different. This is attributed to the difference in the spatial positions of the substrates caused mainly by the presence of a cocatalytic multizinc site unique to Mtb HolPase. Because of this, the dephosphorylation mechanism of Mtb HolPase is different and is discussed later.
It is evident from the detailed comparison that remarkable differences exist between the Mt HolPase and the Mtb HolPase. Such observations were not surprising as these enzymes belong to two entirely different genera of life. What is quite interesting here is that nature has given rise to differences in the catalytic pockets despite conserving the catalytic residues.
Elucidation of the dephosphorylation mechanism of Mtb HolPase
Primarily based on the enzyme–substrate cocrystal structure and kinetic data, we propose a reaction mechanism underlying the action of Rv3137 (Fig. 10). The enzyme possesses a cocatalytic Zn2+ motif and catalyzes the hydrolysis of HOLP to form HOL and phosphate. The reaction is initiated by the activation of a water molecule in the active-site pocket by Zn2+/1, leading to the ionization of the water molecule (Fig. 10A). In the second step, the hydroxyl ion generated in the process initiates a nucleophilic attack on the scissile bond between phosphorus and oxygen of HOLP (Fig. 10B). This is followed by electron delocalization and donation of a proton to the leaving HOL, ultimately leading to the dephosphorylation of the substrate. The source of the general acid that ultimately donates a proton to the leaving group alcohol of the product might be one of the interacting aspartates, most likely Asp213 (Fig. 10B). Following the leaving of HOL, the two Zn2+ ions move from sites 1 and 2 (ready state) to sites 1 and 3 (relaxed state), holding the free phosphate in proximity, and L3 loses its stability (Fig. 10C).
Figure 10.

Elucidation of the dephosphorylation reaction mechanism. Schematic diagrams show various steps involved in the dephosphorylation reaction. A, the water molecule is positioned appropriately and is activated by Zn2+/1. B, the hydroxyl ion attacks the phosphate moiety of the substrate HOLP, and the product HOL is released. C, Zn2+ ions move to positions 1 and 3 from positions 1 and 2. Free phosphate is held close to the two Zn2+ ions.
The Mtb HolPase possesses aspartate- and glutamate-bound Zn2+ that activates the water molecule and initiates the dephosphorylation of HOLP. However, in Mt HolPase, the water molecule is activated indirectly by magnesium-bound aspartates and not directly by Mg2+. The weakly bound Mg2+ of Mt HolPase leaves the active site along with the phosphate, leaving HOL behind (21). However, in Mtb HolPase, HOL leaves the active site, but the phosphate and Zn2+ ions are left behind. The Zn2+ ions move from sites 1 and 2 to the relaxed-state sites 1 and 3, and the phosphate moves accordingly.
Compound library screening identified inhibitors against Mtb HolPase
To identify Mtb HolPase inhibitors, high-throughput screening was performed using an end-point 96-well assay system. The screening experiments were performed in the following assay conditions: 50 mm Tris, pH 7.4, 5 mm DTT, 5 mm MgCl2, 100 μm l-histidinol phosphate, 0.5 μm recombinant Mtb HolPase. The amount of Pi released in the enzymatic assay was measured using malachite green reagent according to the manufacturer's recommendations. The small molecules belonging to Diversity Set V of the National Cancer Institute Developmental Therapeutics Program (NCI-DTP) library were screened to identify novel inhibitors of Mtb HolPase enzyme. The screening performed at 100 μm concentration identified five compounds that inhibited HisN-dependent dephosphorylation activity by >40% (Fig. 11A). NSC311153, the most potent inhibitor in our in vitro Mtb HolPase inhibition assays, displayed an IC50 value of 94.25 μm (Fig. 11B). This compound will be explored further for its efficacy in vitro, ex vivo, and in vivo in future studies in our laboratory.
Figure 11.

Mtb HolPase inhibitors. A, histogram showing percent inhibition of Mtb HolPase by five different compounds belonging to Diversity Set V of the NCI-DTP. B, varying concentrations of the most potent inhibitor, NSC311153, were used in the phosphatase assay to obtain activity curves for Mtb HolPase. Data were plotted in GraphPad Prism version 6 and fitted into a four-parameter dose-response curve to obtain IC50 value. Error bars represent S.D.
Discussion
Mtb HolPase as established in the present study has a direct lineage from the IMPase family. Although the enzyme retains the IMPase fold, it dephosphorylates HOLP specifically. Mtb harbors four IMPase homologs. The proteins encoded by three of these (cysQ, suhB, and impA) have been reported to show IMPase activity (34). However, the fourth encoded by impC (Rv3137) was shown to be essential for the survival of Mtb in culture medium, but its function was unclear (34). We show that Rv3137 specifically dephosphorylates HOLP (Table 2 and Figs. 2 and 6). Because the other three paralogs of Rv3137 have been analyzed and their mutants have been characterized (34), it is likely that this is the sole enzyme responsible for HOLP hydrolysis in Mtb. The other probable candidate that shows some degree of sequence similarity with the HolPase-encoding segment of bifunctional hisB of E. coli is Rv0114, which has been annotated as gmhB. Because it has diverged independently from both HisB and HisN (Fig. 1A), its role in compensating HolPase activity is questionable. Also, it is not feasible to delete Rv3137 if a histidine-free medium is used during the required selection steps (34). If any of the enzymes could substitute its role, making an Rv3137 mutant would have been possible. Notably, HolPases of the bacterial kingdom have evolved from three unrelated families, haloacid dehalogenase-like hydrolase (HAD), polymerase HolPase (PHP), and IMPase (20). The third lineage, i.e. the IMPase family HolPases, although found in two plant species (21, 35), are largely restricted to the notable Actinomycetales in the bacterial kingdom (14, 15).
The solution and crystallographic data clearly show that the functional unit of Mtb HolPase is a dimer. The enzyme–substrate complex structure revealed that, in the dimer, the phosphorylation reaction occurs in one of the monomers while the uncleaved substrate is bound in the other monomer (Fig. 8, A, B, and C). The enzyme exhibits half-of-the-sites reactivity because of the different orientations of Zn2+ ions in the two monomers of the physiological dimer. In the presented native and complex structures of Rv3137, one monomer adopts a relaxed state, whereas the other is in a ready state (Figs. 5 and 8). The ready state favors substrate binding and switches to a relaxed conformation upon completion of the reaction.
The enzyme–substrate complex crystal structure revealed a product molecule (HOL) at the dimer interface trapped in a channel. Although there are two catalytic sites in a dimer, there is only one product-exit channel shared by the two monomers at the dimer interface. It is yet another indication of half-of-the-sites reactivity. Its regulation might be an attractive strategy for drug development. It is therefore reasonable to hypothesize that blocking the product-exit channel would lock down the enzyme in a state of suspended activity.
Furthermore, the cocrystal structure of Mtb HolPase along with mutational studies revealed unique features of this enzyme. The enzyme possesses a cocatalytic Zn2+ motif in which the two Zn2+ ions are bridged by side chains of two amino acid residues (Asp83 and Glu67) (Fig. 7F). Notably, the majority of enzymes possessing a cocatalytic site have no more than one bridging amino acid. Mtb HolPase might be a pioneer of a new subclass of cocatalytic sites with two bridging amino acid residues. It has been suggested that as the amino acid residues interacting with these sites often come from nearly the entire length of the protein, the metals in these sites along with their bridging residues may therefore be important to the overall fold of the protein as well as catalytic function. Mutation of any of the residues involved in the interaction with either of the two Zn2+ ions abrogated the enzyme activity, underscoring the importance of cooperative action of both Zn2+ ions. Dissociation of the bridging residue from one or both Zn2+ ions can alter its charge and hence influence catalysis. Mtb is an ever-evolving bacterium, and the presence of two bridges provides it with more options of modulating the charge of the metal ion and hence regulating catalysis. Mutation of any of the bridging residues leads to the complete loss of enzyme activity.
Histidine pathway enzymes are among the attractive drug targets. As the three-dimensional structure of the enzymes is central to a molecular-level understanding of their function and for enabling structure-guided drug discovery, the Mtb HolPase structure opens up an opportunity in this regard. Thus far, structures of five of the His pathway enzymes, HisA (36), HisB (10), HisC (37), HisE (38), and HisG (39), have been elucidated. Moreover, development of small-molecule inhibitors against HisB (10) and HisG (40) through a structure-guided approach sounds promising. Elucidation of the structures of the other histidine pathway enzymes will augment the current mechanistic understanding of the pathway, which in turn will be helpful to develop new alternative antitubercular compounds. Importantly, we have identified, through a small-molecule library screening, a few inhibitors of Mtb HolPase in vitro. The most potent inhibitor exhibits an IC50 value of <100 μm. In conclusion, the present study provides detailed mechanistic insights into Mtb HolPase and expands the list of anti-TB targets.
Experimental procedures
In silico analysis
Protein sequences were retrieved from the Swiss-Prot domain of the UniProt database with a search term of “histidinol phosphatase” signifying protein name (16). Multiple sequence alignments were performed using ClustalW (41). Evolutionary distances were measured with respect to amino acid substitutions, and gaps were computed using Poisson correction in MEGA (42). Alignments were converted to NEXUS format for phylogenetic analysis using MEGA with an evolutionary tree being inferred using a neighbor-joining method (bootstrap correction with >300 replicates). An unrooted phylogenetic tree was generated using iTOL with the NEXUS tree as the input (43). Alignment for representative purposes was created using ESPript 3.0 with a Clustal alignment as the input (44). Sequence identities and query coverages for selected sequences were retrieved using NCBI BLAST. Motif and conserved domain searches were carried out using PROSITE (45) and NCBI Conserved Domains (46), respectively.
Amplification and cloning of Rv3137
The ORF corresponding to Rv3137 was amplified by PCR using Mtb H37Rv genomic DNA as the template, gene-specific primers (Table 1), deoxynucleotides, MgCl2 and Phusion polymerase (Finnzymes, Finland). The reaction was carried out for 30 cycles, each consisting of denaturation at 371 K for 2 min, annealing at 339.5 K for 1 min, and extension at 345 K for 1 min. The amplified PCR product was purified using a miniprep kit (Qiagen, Germany) and then inserted directionally into the entry vector pENTR following the manufacturer's protocol (Invitrogen). The entry clone was isolated, digested with NdeI and HindIII, and purified using a gel extraction kit (Qiagen). The purified insert was ligated into the NdeI- and HindIII-digested Msg–E. coli shuttle expression vector pYUB1062 using T4 DNA ligase. The successful integration of the insert into the expression vector and its directionality were confirmed by DNA sequencing (Macrogen Ltd.).
Site-directed mutagenesis was carried out using an Agilent QuikChange II XL site-directed mutagenesis kit. Briefly, the original Rv3137pYUB plasmid that was used for overexpression served as the template. Different pairs of primers were used for corresponding mutations (Table 1). Following amplification, the parental methylated and hemimethylated DNA was digested using DpnI enzyme. The mutated molecule was then transformed in highly efficient E. coli DH5α competent cells. The sequences of the resulting plasmids were verified by DNA sequencing.
Overexpression and purification
The construct containing the expression sequence (an N-terminal methionine and hexa-His tag followed by the Rv3137-specific amino acids except incorporating valine instead of the starting methionine immediately after the histidine tag) was overexpressed in Msg strain mc24517. The molecular masses of the native and recombinant version of Rv3137 are 27,693 and 28,615 Da, respectively. For overexpression, Rv3137-pYUB1062 expression vector was electroporated into Msg competent cells. A single transformed colony was revived in 10 ml of Luria Bertani (LB) broth supplemented with 0.05% Tween 80, 0.2% glycerol, and the antibiotics kanamycin (25 μg/ml) and hygromycin B (100 μg/ml). The culture was grown at 310 K, 180 rpm for 24 h. 1 ml of the primary culture was inoculated into 50 ml of the same medium and grown at 310 K, 180 rpm until A600 nm reached 0.6–0.8. Subsequently, the culture was diluted 30-fold into 1.5 liters of the same medium to make a secondary culture and grown to midexponential phase (A600 nm ∼ 0.8) at 310 K, 200 rpm and then induced with 0.03% acetamide. After 24 h of induction, the cells were harvested by centrifugation at 10,000 × g for 20 min. The cell pellet was resuspended into 50 ml of 20 mm Tris, pH 7.5, 200 mm NaCl, 5% glycerol, 20 mm imidazole buffer with one Complete Mini, EDTA-free protease inhibitor tablet (Roche Applied Science).
The cells were lysed at 277 K at high pressure (25,000 p.s.i.) using a cell disrupter (Constant Systems Ltd., UK). The lysate was centrifuged at 10,000 × g for 45 min at 277 K to remove unbroken cells and inclusion bodies. The supernatant was then loaded on an equilibrated nickel-nitrilotriacetic acid affinity column. The column was washed with 20 mm Tris, pH 7.5, 200 mm NaCl, 5% glycerol, 50 mm imidazole to wash away nonspecifically bound proteins. Subsequently, Rv3137 was eluted from the column using 300 mm imidazole in the same buffer. The eluted protein was concentrated and further purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare) in 20 mm Tris, pH 7.5, 200 mm NaCl buffer. The degree of purity of the Rv3137 was examined by 15% SDS-PAGE. The identity of the sample as Rv3137 was confirmed by mass spectrometric analysis of the purified protein gel band (Central Mass Spectrometry Facility, National Institute of Immunology). Protocols used for the overexpression and purification of the mutant proteins were the same as those for the WT enzyme.
Biochemical assays
The activity of Rv3137 was determined by a phosphate-release detection method using the classical malachite green assay as described elsewhere (47). Reactions were carried out in 200-μl volumes in Tris-HCl buffer either of pH 7 or 8 depending upon the metal in the reaction buffer, Zn2+ or Mg2+, respectively, at 310 K for 1 min. The concentration of metal in each case was 1 mm. Eight different concentrations of the substrate (5, 10, 20, 30, 40, 50, 60, and 70 μm) in the reaction mixture containing ZnCl2 and seven concentrations (5, 10, 20, 30, 40, 50, and 60 μm) in the reaction mixture containing MgCl2 were used. The reaction was initiated by the addition of Mtb HolPase to a final concentration of 175 nm and stopped after 1 min by adding 24 μl of 3 n NaOH. This was followed by the addition of 56 μl of color reagent. The color was allowed to develop for 10 min at 303 K. All assays were performed in triplicate. The absorbance was measured at 630 nm using a spectrophotometer (TCC-240A, Shimadzu Corp.). Standard reactions to optimize pH, temperature, and metal ion concentration were carried out using 50 μm l-histidinol phosphate lithium salt (Sigma). The amount of product, i.e. the released phosphate, was calculated using a molar extinction coefficient of 90,000 m−1 cm−1 for the malachite green–phosphomolybdate complex. In addition to taking a reference blank with enzyme, any kind of interference arising from organophosphate detection was nullified by taking another blank with an equal amount of substrate for each reaction. No activity was observed with IMP (d-myo-inositol 1-phosphate sodium salt, Cayman Chemical).
Biochemical assays for checking the enzyme activity in cocrystallization buffer were carried out in a similar manner as described for kinetic studies but with some modifications. The buffer used in this case was protein purification buffer (20 mm Tris, pH 7.5, 200 mm NaCl) containing 0.1 mm ZnCl2. The reaction was carried out for 40 min, and the product formation was monitored at regular intervals of 10 min. For checking the effect of crystallization solution ammonium sulfate, it was either added to the reaction mixture in 1:1 ratio after 10 min or was kept in buffer solution from the beginning in different concentrations in a different set of reactions.
For the inhibition experiment, varying concentrations of the compound NSC311153 were added to the reaction mixture containing 0.1 m buffer, 175 nm enzyme, 50 μm substrate, and 1 mm Zn2+. The absorbance was measured at 630 nm. The Michaelis–Menten, inhibition curve, and other assay data were plotted using Prism version 6 (GraphPad Software).
The PAR assay was carried out using a protocol described elsewhere (48). Briefly, a standard curve was prepared with 50 μm PAR in 4 m guanidine hydrochloride and known zinc concentrations. The protein was denatured with 4 m guanidine hydrochloride, and 50 μm PAR was added to the sample. The absorbance at 500 nm, due to the formation of the (PAR)2Me(II) complex, was monitored and compared with the standard. The zinc colorimetric assay was performed according to the manufacturer's protocol. Briefly, a standard curve was prepared with known zinc concentrations in 7% TCA in 20 mm Tris, pH 7.5, 200 mm NaCl buffer. Zinc reagent was added at a 2:1 ratio by volume to the sample, and the color was developed for 10 min. A560 nm was measured. The protein (in 20 mm Tris, pH 7.5, 200 mm NaCl buffer) was denatured and precipitated using 7% TCA followed by centrifugation at 13,000 rpm. Supernatant was collected and used for zinc detection as described for the standard. The concentration of zinc was interpreted from the standard curve.
Crystallization and structure determination
Crystals of Mtb HolPase were grown at 296 K by the hanging-drop vapor-diffusion method. Experiments were set up in 24-well plates with a drop size of 3.75 μl (1.5 μl of protein, 1.5 μl of crystallization reservoir solution, and 0.75 μl of additive). The crystallization reservoir solution consisted of 1 m ammonium sulfate, and the additive was praseodymium (III) acetate hydrate. Diffraction quality crystals of Rv3137, besides tiny clusters of salt crystals, grew after about 35–40 days. Crystals of the enzyme–substrate complex were grown by a cocrystallization method in the same condition that yielded native enzyme crystals except that the protein solution contained 7.5 mm HOLP and 0.1 mm ZnCl2. The mixture was incubated for 10 min followed by manual plate setup. Crystals grew after about a month.
For X-ray data collection, crystals were mounted on CryoLoops (Hampton Research), rinsed in cryoprotectant solution (33% (v/v) glycerol in reservoir solution) and flash cooled directly in a nitrogen stream at 100 K. Complete data sets were collected at 100 K for the native crystal and the enzyme–substrate cocrystals on beamlines BM14 and ID30B, European Synchrotron Radiation Facility (France), respectively. The data sets were indexed, integrated, and scaled using HKL-2000 (49). Data collection statistics for the best-diffracting crystals for both the native and the cocrystal are summarized in Table 3. Matthew's coefficient (50) and solvent content with four molecules in the native crystal asymmetric unit were 2.77 and 55.56%, respectively, whereas the asymmetric unit of enzyme–substrate cocrystals contains two molecules, and the corresponding Matthew's coefficient and solvent content of the unit cell are 2.87 and 57.19%, respectively.
The structure of Mtb HolPase was solved by the molecular replacement method using as the search model the crystal structure of its Mt counterpart (Protein Data Bank code 5EQ7) that shares 33% sequence identity (21). The program Phaser (51) of CCP4 (52) used in solving the structure yielded a model (solution) comprising four molecules in the crystal asymmetric unit. To start with, the model was subjected to 50 cycles of rigid-body refinement. Subsequently, 100 cycles of restrained-coordinate refinement were carried out using a maximum likelihood target function. At this stage, the Mtb Rv3137-specific amino acids were substituted into the electron density using the model-building program Coot (53). After every round of model building, positional and isotropic B-factor refinements were carried out. Water molecules were incorporated in the model based on the peak heights (2|Fo| − |Fc| at 1σ and |Fo| − |Fc| at 3σ contour level) in the electron density maps. In the active site of Rv3137, indigenously bound metal ions were modeled based on the Fourier electron density maps. The Mtb HolPase–HOLP complex cocrystal structure was solved using the refined native Rv3137 structure as the model. The HOLP and Zn2+ atoms were incorporated into their respective positions on the basis of the difference electron density map (|Fo| − |Fc|). Subsequently, the complex structure was refined in a manner similar to that used for the apo structure. The data collection, data processing, and refinement statistics are tabulated in Table 3. The stereochemical acceptability of the structures was validated using the program PROCHECK (54). The rotational symmetry was checked using the program LABELIT before submission to the Protein Data Bank (55). The secondary structural elements were assigned using the program DSSP (56). Figs. 3–5 and 7–9 were prepared using PyMOL (57).
Compound library screening
The Pi release assay was adapted for a high-throughput screen to identify novel inhibitors of Mtb HolPase. This end-point assay was performed in a final volume of 50 μl in assay buffer in the presence of 1 μm Mtb HolPase. The small-molecule library from the NCI-DTP (https://dtp.cancer.gov/repositories/default.htm) comprising 1360 structurally diverse compounds was assayed at a final concentration of 100 μm in assay buffer (containing 1 μm enzyme) for preliminary screening. All reaction plates included proper controls such as buffer only, no substrate, and no enzyme control. The enzyme-scaffold mixture was incubated at room temperature for 10 min, and the reaction was initiated by addition of 100 μm l-histidinol phosphate. After incubation for a further 10 min, the formation of Pi in the enzyme reaction with or without inhibitor was monitored by measuring absorbance at 630 nm using a Quantichrome phosphate assay kit (Bioassay Systems) according to the manufacturer's recommendations. The half-maximal inhibitory (IC50) concentration determination protocol is described above under “Biochemical assays.”
Author contributions
B. J. and B. K. B. conceptualization; B. J., D. K., A. S., and A. D. data curation; B. J., D. K., A. S., A. D., R. S., and B. K. B. formal analysis; B. J., D. K., and B. K. B. validation; B. J. methodology; B. J. writing-original draft; B. J., R. S., and B. K. B. writing-review and editing; R. S. and B. K. B. resources; R. S. and B. K. B. supervision; B. K. B. software; B. K. B. funding acquisition; B. K. B. investigation; B. K. B. project administration.
Supplementary Material
Acknowledgments
Mtb H37Rv genomic DNA was obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources, Manassas, VA). We are grateful to Prof. William R. Jacobs of the Department of Microbiology and Immunology and the Howard Hughes Medical Institute, Albert Einstein College of Medicine, Bronx, NY, for providing the expression vector pYUB1062 and Msg mc24517 expression system. The in-house X-ray diffraction facility, RIGAKU FR-E+ SuperBright microfocus rotating anode dual-wavelength (Cu and Cr) X-ray generator mounted with RAXIS IV++ detectors, was established with the financial support from the Department of Biotechnology, Government of India. We are grateful to the staff at the European Synchrotron Radiation Facility for providing assistance in using beamlines BM14 and ID30B. We sincerely thank Ravi Kant Pal for help in crystal screening at the home source and Shanta Sen for help in protein confirmation by MS. We sincerely acknowledge Drs. Khundrakpam Herojit Singh and Savita Yadav for useful discussions.
This work was supported in part by the Department of Biotechnology (DBT), Government of India, for X-ray data collection at synchrotron beamlines BM14 and ID30B of the European Synchrotron Radiation Facility, Grenoble, France. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
The atomic coordinates and structure factors (codes 5ZON and 5YHT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TB
- tuberculosis
- Mtb
- M. tuberculosis
- HolPase
- histidinol phosphate phosphatase
- IMPase
- inositol monophosphatase
- HOLP
- l-histidinol phosphate
- HOL
- l-histidinol
- Msg
- M. smegmatis
- Mt
- M. truncatula
- ed
- electron density
- PAR
- 4-(2-pyridylazo)resorcinol
- r.m.s.d.
- root mean square deviation.
References
- 1. Murima P., McKinney J. D., and Pethe K. (2014) Targeting bacterial central metabolism for drug development. Chem. Biol. 21, 1423–1432 10.1016/j.chembiol.2014.08.020 [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y. J., Reddy M. C., Ioerger T. R., Rothchild A. C., Dartois V., Schuster B. M., Trauner A., Wallis D., Galaviz S., Huttenhower C., Sacchettini J. C., Behar S. M., and Rubin E. J. (2013) Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155, 1296–1308 10.1016/j.cell.2013.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wellington S., Nag P. P., Michalska K., Johnston S. E., Jedrzejczak R. P., Kaushik V. K., Clatworthy A. E., Siddiqi N., McCarren P., Bajrami B., Maltseva N. I., Combs S., Fisher S. L., Joachimiak A., Schreiber S. L., et al. (2017) A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 13, 943–950 10.1038/nchembio.2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., and Chillingworth T. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 5. Sassetti C. M., Boyd D. H., and Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 10.1046/j.1365-2958.2003.03425.x [DOI] [PubMed] [Google Scholar]
- 6. Parish T. (2003) Starvation survival response of Mycobacterium tuberculosis. J. Bacteriol. 185, 6702–6706 10.1128/JB.185.22.6702-6706.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiariotti L., Nappo A. G., Carlomagno M. S., and Bruni C. B. (1986) Gene structure in the histidine operon of Escherichia coli. Identification and nucleotide sequence of the hisB gene. Mol. Gen. Genet. 202, 42–47 10.1007/BF00330514 [DOI] [PubMed] [Google Scholar]
- 8. Staples M. A., and Houston L. L. (1979) Proteolytic degradation of imidazoleglycerolphosphate dehydratase-histidinol phosphatase from Salmonella typhimurium and the isolation of a resistant bifunctional core enzyme. J. Biol. Chem. 254, 1395–1401 [PubMed] [Google Scholar]
- 9. Bazzicalupo M., Fani R., Gallori E., Turbanti L., and Polsinelli M. (1987) Cloning of the pyrimidine and cysteine genes of Azospirillum brasilense: expression of pyrimidine and three clustered histidine genes in Escherichia coli. Mol. Gen. Genet. 206, 76–80 10.1007/BF00326539 [DOI] [Google Scholar]
- 10. Ahangar M. S., Vyas R., Nasir N., and Biswal B. K. (2013) Structures of native, substrate-bound and inhibited forms of Mycobacterium tuberculosis imidazoleglycerol-phosphate dehydratase. Acta Crystallogr. D Biol. Crystallogr. 69, 2461–2467 10.1107/S0907444913022579 [DOI] [PubMed] [Google Scholar]
- 11. Parker A. R., Moore T. D., Edman J. C., Schwab J. M., and Davisson V. J. (1994) Cloning, sequence analysis and expression of the gene encoding imidazole glycerol phosphate dehydratase in Cryptococcus neoformans. Gene 145, 135–138 [DOI] [PubMed] [Google Scholar]
- 12. Mano J., Hatano M., Koizumi S., Tada S., Hashimoto M., and Scheidegger A. (1993) Purification and properties of a monofunctional imidazoleglycerol-phosphate dehydratase from wheat. Plant Physiol. 103, 733–739 10.1104/pp.103.3.733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tada S., Volrath S., Guyer D., Scheidegger A., Ryals J., Ohta D., and Ward E. (1994) Isolation and characterization of cDNAs encoding imidazoleglycerolphosphate dehydratase from Arabidopsis thaliana. Plant Physiol. 105, 579–583 10.1104/pp.105.2.579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marineo S., Cusimano M. G., Limauro D., Coticchio G., and Puglia A. M. (2008) The histidinol phosphate phosphatase involved in histidine biosynthetic pathway is encoded by SCO5208 (hisN) in Streptomyces coelicolor A3(2). Curr. Microbiol. 56, 6–13 10.1007/s00284-007-9014-7 [DOI] [PubMed] [Google Scholar]
- 15. Mormann S., Lömker A., Rückert C., Gaigalat L., Tauch A., Pühler A., and Kalinowski J. (2006) Random mutagenesis in Corynebacterium glutamicum ATCC 13032 using an IS6100-based transposon vector identified the last unknown gene in the histidine biosynthesis pathway. BMC Genomics 7, 205 10.1186/1471-2164-7-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. The UniProt Consortium (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 10.1093/nar/gkw1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lew J. M., Kapopoulou A., Jones L. M., and Cole S. T. (2011) TubercuList—10 years after. Tuberculosis 91, 1–7 10.1016/j.tube.2010.09.008 [DOI] [PubMed] [Google Scholar]
- 18. Lunardi J., Nunes J. E., Bizarro C. V., Basso L. A., Santos D. S., and Machado P. (2013) Targeting the histidine pathway in Mycobacterium tuberculosis. Curr. Top. Med. Chem. 13, 2866–2884 10.2174/15680266113136660203 [DOI] [PubMed] [Google Scholar]
- 19. de Castro E., Sigrist C. J., Gattiker A., Bulliard V., Langendijk-Genevaux P. S., Gasteiger E., Bairoch A., and Hulo N. (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365 10.1093/nar/gkl124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kulis-Horn R. K., Rückert C., Kalinowski J., and Persicke M. (2017) Sequence-based identification of inositol monophosphatase-like histidinol-phosphate phosphatases (HisN) in Corynebacterium glutamicum, Actinobacteria, and beyond. BMC Microbiol. 17, 161 10.1186/s12866-017-1069-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruszkowski M., and Dauter Z. (2016) Structural studies of Medicago truncatula histidinol phosphate phosphatase from inositol monophosphatase superfamily reveal details of penultimate step of histidine biosynthesis in plants. J. Biol. Chem. 291, 9960–9973 10.1074/jbc.M115.708727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 23. Högbom M., Ericsson U. B., Lam R., Bakali H M. A., Kuznetsova E., Nordlund P., and Zamble D. B. (2005) A high throughput method for the detection of metalloproteins on a microgram scale. Mol. Cell. Proteomics 4, 827–834 10.1074/mcp.T400023-MCP200 [DOI] [PubMed] [Google Scholar]
- 24. Ghodge S. V., Fedorov A. A., Fedorov E. V., Hillerich B., Seidel R., Almo S. C., and Raushel F. M. (2013) Structural and mechanistic characterization of L-histidinol phosphate phosphatase from the polymerase and histidinol phosphatase family of proteins. Biochemistry 52, 1101–1112 10.1021/bi301496p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 26. Gasteiger E., Hoogland C., Gattiker A., Duvaud S. E., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein Identification and Analysis Tools on the ExPASy Server, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 27. Jiang J., Zhou Z., Dong Y., Jiang B., Chen Z., Yang A., Wang B., Guan X., Gao S., and Sun H. (2016) The in vitro effects of divalent metal ions on the activities of immune-related enzymes in coelomic fluid from the sea cucumber Apostichopus japonicus. Aquac. Res. 47, 1269–1276 10.1111/are.12586 [DOI] [Google Scholar]
- 28. Auld D. S. (2001) Zinc coordination sphere in biochemical zinc sites. Biometals 14, 271–313 10.1023/A:1012976615056 [DOI] [PubMed] [Google Scholar]
- 29. Li W., To D., Lee J., Crane E. J., and Sazinsky M. (2016) Investigating the half-site reactivity of a member of the PNDOR family of proteins. FASEB J. 30, (suppl.) 834.21 (abstr.) [Google Scholar]
- 30. Seelig G. F., and Folk J. E. (1980) Half-of-the-sites and all-of-the-sites reactivity in human plasma blood coagulation factor XIIIa. J. Biol. Chem. 255, 9589–9593 [PubMed] [Google Scholar]
- 31. Castellani M., Covian R., Kleinschroth T., Anderka O., Ludwig B., and Trumpower B. L. (2010) Direct demonstration of half-of-the-sites reactivity in the dimeric cytochrome bc1 complex enzyme with one inactive monomer is fully active but unable to activate the second ubiquinol oxidation site in response to ligand binding at the ubiquinone reduction site. J. Biol. Chem. 285, 502–510 10.1074/jbc.M109.072959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vallee B. L., and Auld D. S. (1993) Cocatalytic zinc motifs in enzyme catalysis. Proc. Natl. Acad. Sci. U.S.A. 90, 2715–2718 10.1073/pnas.90.7.2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCall K. A., Huang C., and Fierke C. A. (2000) Function and mechanism of zinc metalloenzymes. J. Nutr. 130, 1437S–1446S 10.1093/jn/130.5.1437S [DOI] [PubMed] [Google Scholar]
- 34. Movahedzadeh F., Wheeler P. R., Dinadayala P., Av-Gay Y., Parish T., Daffé M., and Stoker N. G. (2010) Inositol monophosphate phosphatase genes of Mycobacterium tuberculosis. BMC Microbiol. 10, 50 10.1186/1471-2180-10-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Petersen L. N., Marineo S., Mandalà S., Davids F., Sewell B. T., and Ingle R. A. (2010) The missing link in plant histidine biosynthesis: Arabidopsis myoinositol monophosphatase-like2 encodes a functional histidinol-phosphate phosphatase. Plant Physiol. 152, 1186–1196 10.1104/pp.109.150805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Due A. V., Kuper J., Geerlof A., von Kries J. P., and Wilmanns M. (2011) Bisubstrate specificity in histidine/tryptophan biosynthesis isomerase from Mycobacterium tuberculosis by active site metamorphosis. Proc. Natl. Acad. Sci. U.S.A. 108, 3554–3559 10.1073/pnas.1015996108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nasir N., Anant A., Vyas R., and Biswal B. K. (2016) Crystal structures of Mycobacterium tuberculosis HspAT and ArAT reveal structural basis of their distinct substrate specificities. Sci. Rep. 6, 18880 10.1038/srep18880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Javid-Majd F., Yang D., Ioerger T. R., and Sacchettini J. C. (2008) The 1.25 A resolution structure of phosphoribosyl-ATP pyrophosphohydrolase from Mycobacterium tuberculosis. Acta Crystallogr. D Biol. Crystallogr. 64, 627–635 10.1107/S0907444908007105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cho Y., Sharma V., and Sacchettini J. C. (2003) Crystal structure of ATP phosphoribosyltransferase from Mycobacterium tuberculosis. J. Biol. Chem. 278, 8333–8339 10.1074/jbc.M212124200 [DOI] [PubMed] [Google Scholar]
- 40. Cho Y., Ioerger T. R., and Sacchettini J. C. (2008) Discovery of novel nitrobenzothiazole inhibitors for Mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J. Med. Chem. 51, 5984–5992 10.1021/jm800328v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., and Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 10.1093/nar/gkg500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumar S., Stecher G., and Tamura K. (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Letunic I., and Bork P. (2007) Interactive Tree of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 10.1093/bioinformatics/btl529 [DOI] [PubMed] [Google Scholar]
- 44. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sigrist C. J., de Castro E., Cerutti L., Cuche B. A., Hulo N., Bridge A., Bougueleret L., and Xenarios I. (2013) New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–D347 10.1093/nar/gks1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marchler-Bauer A., Derbyshire M. K., Gonzales N. R., Lu S., Chitsaz F., Geer L. Y., Geer R. C., He J., Gwadz M., Hurwitz D. I., Lanczycki C. J., Lu F., Marchler G. H., Song J. S., and Thanki N. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226 10.1093/nar/gku1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baykov A. A., Evtushenko O. A., and Avaeva S. M. (1988) A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 171, 266–270 10.1016/0003-2697(88)90484-8 [DOI] [PubMed] [Google Scholar]
- 48. Sydor A. M., Jost M., Ryan K. S., Turo K. E., Douglas C. D., Drennan C. L., and Zamble D. B. (2013) Metal binding properties of Escherichia coli YjiA, a member of the metal homeostasis-associated COG0523 family of GTPases. Biochemistry 52, 1788–1801 10.1021/bi301600z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 50. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 10.1016/0022-2836(68)90205-2 [DOI] [PubMed] [Google Scholar]
- 51. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., and Powell H. R. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 54. Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 55. Poon B. K., Grosse-Kunstleve R. W., Zwart P. H., and Sauter N. K. (2010) Detection and correction of underassigned rotational symmetry prior to structure deposition. Acta Crystallogr. D. Biol. Crystallogr. 66, 503–513 10.1107/S0907444910001502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 57. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.