

Abstract
Pendrin and prestin belong to the solute carrier 26 (SLC26) family of anion transporters. Prestin is unique among the SLC26 family members in that it displays voltage-driven motor activity (electromotility) and concurrent gating currents that manifest as nonlinear cell membrane electrical capacitance (nonlinear capacitance (NLC)). Although the anion transport mechanism of the SLC26 proteins has begun to be elucidated, the molecular mechanism of electromotility, which is thought to have evolved from an ancestral ion transport mechanism, still remains largely elusive. Here, we demonstrate that pendrin also exhibits large NLC and that charged residues present in one of the extracellular loops of pendrin and prestin play significant roles in setting the voltage-operating points of NLC. Our results suggest that the molecular mechanism responsible for sensing voltage is not unique to prestin among the members of the SLC26 family and that this voltage-sensing mechanism works independently of the anion transport mechanism.
Keywords: membrane protein, molecular motor, electrophysiology, anion transport, cell motility, electromotility, SLC26
Introduction
The solute carrier 26 (SLC26)4 proteins are ubiquitously expressed in a wide variety of species from bacteria to humans and constitute the SLC26/SulP family of ion transporters and channels (1). Although most of the members of this large gene family function as anion exchangers or channels, only prestin (SLC26A5) exhibits voltage-dependent motor activity referred to as electromotility. Electromotility of prestin and the anion transport mediated by pendrin (SLC26A4) are essential for normal development and operation of mammalian inner ears (2–4). Prestin-mediated electromotility confers a rapid voltage-induced force-generating cell length change on cochlear outer hair cells (5), which is indispensable for the frequency selectivity and sensitivity of mammalian hearing (4). Recent structural and biochemical studies (6, 7) have provided significant insights into the ion transport mechanism of the SLC26 proteins. However, the molecular mechanism of the voltage-driven motor activity, which is thought to be unique to prestin among the SLC26 family members, still remains largely unknown. Electromotility accompanies a movement of prestin-associated charge. Because this charge movement is rapid, it manifests as nonlinear electrical capacitance of the cell membrane (nonlinear capacitance (NLC)) (8, 9). Thus, NLC is regarded as the electric signature of electromotility, and its measurement often substitutes for direct electromotility measurement.
Because of the high degree of similarity in amino acid sequences among the SLC26 family members, it is conceivable that the tertiary structures of SLC26 proteins are quite similar among the family members and that a common molecular mechanism underlies their diverse physiological functions. Both pendrin and prestin are expressed in the mammalian inner ear, and they share 36% homology at the amino acid level. Using SLC26 models that were generated based on the solved structure of a bacterial SLC26 protein (see Fig. 1) (7), we carried out a comprehensive mutational analysis combined with whole-cell patch-clamp recordings and anion transport assays to understand the molecular basis for the functional difference between pendrin, a well-characterized anion transporter, and prestin, a voltage-driven motor. We found that pendrin also retains voltage-sensing ability, which becomes evident with addition of cyclodextrin to depolarize the peak voltage-operating point. We also found that the electrostatic property of the characteristic extracellular loop of pendrin and prestin (termed “L2” in this study; see Fig. 1) influences the voltage-operating points of these proteins. Furthermore, the anion transport activity of pendrin was not affected by the alterations in operating voltage range, suggesting that the voltage-sensing and anion transport mechanisms are independent of each other. Our results suggest that the extracellular loop of prestin has adopted the optimal electrostatic property, which affects the membrane surface electric potential, so that the voltage-operating point resides within the physiologically relevant voltage range, and that the electromotility may have evolved independently of its anion transport mechanism.
Figure 1.
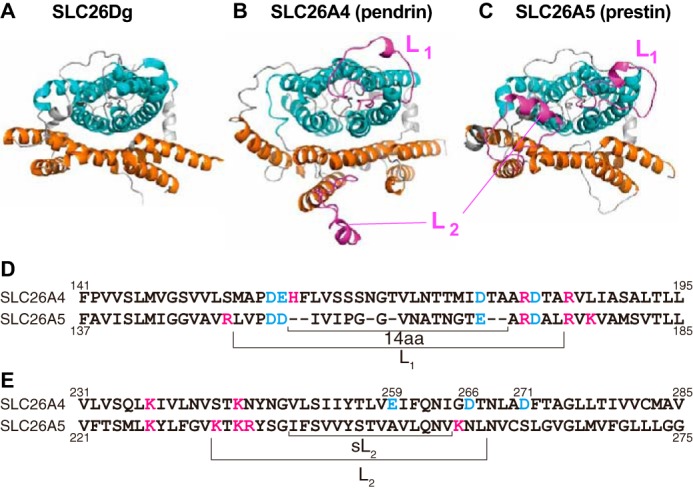
Structural models of pendrin and prestin. A–C, extracellular views of pendrin and prestin models generated based on the solved structure of SLC26Dg. The N- and C-terminal cytosolic regions are not shown. The core and the gate domains are shown in cyan and orange, respectively. The two prominent extracellular loops (L1 and L2), which are not present in the bacterial SLC26 structure, are highlighted in magenta. D and E, partial amino acid sequences of SLC26A4 (pendrin) and SLC26A5 (prestin) that contain the L1 (D) and L2 (E) regions. Characters shown in cyan and magenta indicate acidic and basic residues, respectively. L1, L2, 14aa, and sL2 indicate the segments that were swapped between pendrin and prestin in this study.
Results
The extracellular loops of pendrin and prestin
Recent structural and biochemical studies have provided significant insights into the ion transport mechanism of the SLC26 proteins (6, 7, 10). Because the basic architecture of the transmembrane (TM) region that accommodates the putative ion translocation pathway is likely quite similar among the SLC26 family members, it is possible that the diverse physiological functions of the SLC26 family of proteins have arisen from modifications of a common ancestral ion transport mechanism. Structural information often facilitates efforts to identify regions in proteins responsible for the expressions of their diverse physiological functions. Because the structure of the TM region has only been solved for a bacterial SLC26 protein, SLC26Dg (Fig. 1A) (7), we modeled the TM structures of pendrin and prestin based on the solved structure of SLC26Dg (Fig. 1, B and C). Two prominent extracellular loops, which are referred to as loop-1 (L1) and loop-2 (L2) in this study, are present in pendrin and prestin but not in the bacterial SLC26 protein (Fig. 1, B and C, magenta). Because many disease-associated pendrin mutations have been identified in the L1 (F161I, V163I, V163L, S166N, and T178P) and L2 (V250A, S252P, I253V, L257P, and D266N) regions (11–19), these extracellular loops may play significant roles in defining the physiological functions of pendrin (HCO3−/I−/Cl− exchanger) and prestin (voltage-operated motor).
The extracellular loops modulate the voltage-operating point of prestin
To examine the contributions of L1 and L2 to the function of prestin, we generated several prestin-based constructs whose L1 and L2 are replaced with those of pendrin individually (A5-L1 and A5-L2) or in combination (A5-L1/L2) (Figs. 1 and 2) and measured NLC in HEK293T cell lines expressing these constructs (Fig. 2). Replacement of L1 did not abrogate NLC but resulted in significant hyperpolarization of the voltage-operating points (Vpk) (Fig. 2, B and E). Likewise, replacement of L2 resulted in significant Vpk hyperpolarization but to a much greater degree compared with L1 replacement (Fig. 2, C and E). No further hyperpolarization was observed upon double (L1/L2) replacement (Fig. 2, D and E). The voltage sensitivity (α) was not significantly affected by any of these loop replacements (one-way analysis of variance (ANOVA), p = 0.14) (Fig. 2F). These results suggest that the extracellular loops of prestin modulate its operating voltage range without affecting its voltage sensitivity.
Figure 2.

NLC measurements for prestin and prestin-based constructs. A–D, schematic diagrams of WT prestin (WT-A5) (A) and prestin-based constructs (A5-L1 (B), A5-L2 (C), and A5-L1/L2 (D)) along with their representative NLCs. The parts of prestin and pendrin are indicated by magenta and dark blue, respectively, in the schematic diagrams. The magnitudes of NLCs (Cm − Clin) are corrected for cell size (Clin) because larger cells tend to express greater amounts of prestin in their cell membranes (NLCsp ≡ (Cm − Clin)/Clin). Different colors indicate individual recordings. A two-state Boltzmann model (see “Experimental procedures”) was used to interpret the NLC data (solid lines). E, the Vpk values (mean ± S.D.) are as follows: WT-A5, −91 ± 24 mV (n = 9); A5-L1, −132 ± 31 mV (n = 13); A5-L2, −169 ± 26 mV (n = 9); A5-L1/L2, −170 ± 24 mV (n = 9). The asterisks indicate the degree of statistical significance (**, p < 0.01; ****, p < 0.0001). Error bars represent S.D. ns, not significant. F, the α values (mean ± S.D.) are as follows: WT-A5, 0.031 ± 0.006 mV−1 (n = 9); A5-L1, 0.026 ± 0.007 mV−1 (n = 13); A5-L2, 0.025 ± 0.005 mV−1 (n = 9); A5-L1/L2, 0.026 ± 0.005 mV−1 (n = 9). Error bars represent S.D.
Pendrin possesses voltage-sensing ability
The voltage-driven motor function of prestin is thought to be unique among the members of the SLC26 family; however, the veracity of this presumption has not been tested rigorously. Given the high degree of similarity in amino acid sequences among the SLC26 family members, it is possible that many, if not most, other SLC26 proteins also retain a voltage-sensing mechanism, but it is not readily detectable within the experimentally measurable voltage range (typically ±150 mV). We noticed that some HEK293T cells expressing the WT pendrin construct tend to exhibit a gradual increase in cell membrane capacitance below ∼0 mV, hinting at the presence of large NLCs (Fig. 3A). We speculated that pendrin also retains voltage-sensing ability, but its Vpk is extremely hyperpolarized and thus not easily detected. To test this possibility, we repeated capacitance measurement for the WT pendrin construct in the presence of methyl-β-cyclodextrin (MβCD), which is known to depolarize Vpk of prestin by removing cholesterol from the cell membrane (20). As shown in Fig. 3B, NLCs of WT pendrin became quite evident upon application of MβCD due to its anticipated depolarizing effect, which brought the otherwise barely detectable NLC of pendrin into the experimentally measurable voltage range. We also performed NLC measurement in Sf9 cells (Fig. S1). The low membrane cholesterol level in this cell line (21) allowed detection of large NLC of pendrin without the use of MβCD.
Figure 3.

NLC measurements for pendrin and pendrin-based constructs. Representative NLC recordings for WT-A4 (A), WT-A4 + MβCD (B), A4-L1 (C), A4-L1(14aa) (D), A4-L2 and A4-L1/L2 (E), A4-sL2 (F), and A4-L1/sL2 and A4-L1(14aa)/sL2 (G) are shown. Schematic diagrams of these constructs are also shown above the NLC data. The parts of prestin and pendrin are indicated by magenta and dark blue, respectively. The magnitudes of NLCs are corrected for cell size as in Fig. 2. Different colors indicate individual recordings. A two-state Boltzmann model was used to interpret the NLC data (solid lines) for WT-A4 + MβCD (B) and A4-sL2 (F). The Vpk and α values are summarized in Fig. 5, J and L.
The importance of the extracellular loops of pendrin for membrane targeting and modulation of the voltage-operating point
We examined the contributions of L1 and L2 to the voltage-sensing function of pendrin as we did for prestin. To this end, we generated several pendrin-based constructs whose L1 and L2 are replaced with those of prestin individually (A4-L1 and A4-L2) or in combination (A4-L1/L2) (Figs. 1 and 3). We anticipated finding large NLCs for A4-L1 because the L1 region contains the “14-aa” segment that was reported to confer large NLC and electromotility on pendrin (22). However, the effect of L1 replacement was ambiguous (Fig. 3C) as NLCs of A4-L1 (and WT pendrin (WT-A4)) were only partially measurable to confidently estimate the NLC parameters. Qualitatively, the L1 replacement did not seem to significantly affect Vpk of pendrin (Fig. 3, A and C). We also generated a construct in which only the 14-aa segment used in the previous study (22) was replaced (termed “A4-L1(14aa)” in this study; Fig. 1D). Unexpectedly, however, we could not detect NLC for this construct (Fig. 3D). The reason for the discrepancy is unclear. At the least, the absence of NLC in A4-L1(14aa) should not be solely ascribed to impaired membrane targeting of the construct because the HCO3−/Cl− antiport activity was detected for A4-L1(14aa) (Fig. 4A). Although the transport activity of A4-L1(14aa) was smaller than that of WT-A4 (p = 0.0016) (Fig. 4A), it was comparable with that of A4-sL2 (p > 0.999; Fig. 4A), which exhibited a large NLC (Fig. 3F; see below).
Figure 4.
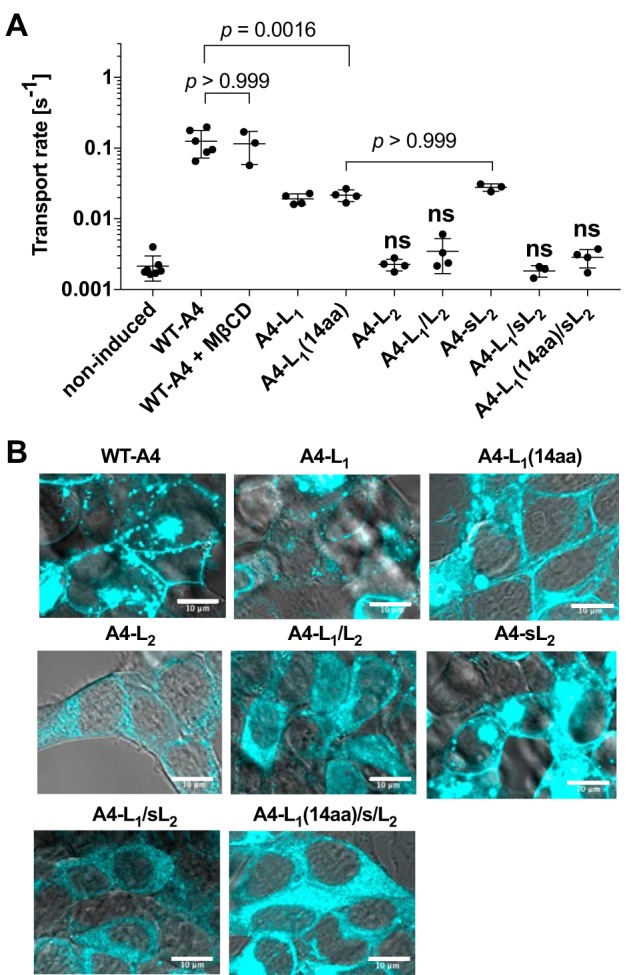
A summary of anion transport assay and qualitative assessment of subcellular localization for the pendrin-based constructs that did not show transport activity. A, HCO3−/Cl− antiport activities of noninduced negative control (n = 7), WT-A4 (n = 6), WT-A4 + MβCD (n = 3), A4-L1 (n = 4), A4-L1(14aa) (n = 4), A4-L2 (n = 4), A4-L1/L2 (n = 4), A4-sL2 (n = 3), A4-L1/sL2 (n = 3), and A4-L1(14aa)/sL2 (n = 4). Error bars represent S.D. The transport activities of A4-L2, A4-L1/L2, A4-L1/sL2, and A4-L1(14aa)/sL2 were indistinguishable from that of the noninduced negative control (determined by Student's t test and indicated by “ns”). The results of the other constructs that showed significant transport activities (compared with the noninduced negative control) were further analyzed by one-way ANOVA and the Tukey–Kramer multiple comparison test to determine the adjusted p values. Only selected p values that are mentioned in the main text (under “Results”) are shown in the figure. B, the subcellular localizations of the pendrin constructs were microscopically examined. A4-L2, A4-L1/L2, A4-L1/sL2, and A4-L1(14aa)/sL2, which did not show significant transport activity (A), exhibited predominantly cytosolic localizations, indicating severely impaired membrane targeting. A result for WT-A4 is also included as a positive control. Scale bars, 10 μm.
Replacement of L2 (A4-L2) or both L1/L2 (A4-L1/L2) resulted in loss of NLC (Fig. 3E) and HCO3−/Cl− antiport activity (Fig. 4A). This is very likely due to the lack of membrane targeting as they are predominantly cytosolic and are qualitatively very different from WT-A4 (Fig. 4B). To circumvent this membrane targeting issue, we generated additional pendrin constructs with a shorter L2 (“sL2”; Fig. 1E) segment replaced (termed “A4-sL2,” “A4-L1/sL2,” and “A4-L1(14aa)/sL2” Fig. 3). The replacement of the shorter L2 segment (A4-sL2; Fig. 3F) resulted in the depolarization of Vpk (as in MβCD-treated WT-A4) (Fig. 3F), indicating that this segment similarly modulates the voltage-operating point in pendrin as in prestin. However, we did not detect any NLC in A4-L1/sL2 and A4-L1(14aa)/sL2 (Fig. 3G). Because the HCO3−/Cl− antiport activity was not detected for A4-L1/sL2 and A4-L1(14aa)/sL2 either (Fig. 4A), we examined the expression of A4-L1/sL2 and A4-L1(14aa)/sL2 and found that they failed to target the plasma membrane (Fig. 4B). These negative observations on A4-L1/L2, A4-L1/sL2, and A4-L1(14aa)/sL2 imply the importance of pendrin's extracellular loops for maintaining its structural integrity and/or membrane targeting.
The role of the charged residues in loop-2 in establishing Vpk
The L2 swapping between prestin and pendrin resulted in hyperpolarization for prestin and depolarization for pendrin (Figs. 2 and 3). This inverse effect of the L2 swapping can be explained by the electrostatic property of the L2 region that is presumably expected to be exposed to the extracellular surface of the membrane. For example, the presence of acidic residues would decrease the surface charge density (σo), which should result in concomitant drops (hyperpolarization) of the membrane surface potential (Φo) (Fig. 5A). The opposite (depolarization of Φo) would be expected if basic residues were present in L2 (Fig. 5B). If the inner surface potential (Φi) remained unchanged, hyperpolarization of Φo should result in hyperpolarization of Vpk and vice versa. This interpretation is compatible with the fact that the net calculated charges of L2 are −1 and +4 for pendrin and prestin, respectively (Fig. 1E).
Figure 5.

The effects of the charged residues in L2 on Vpk. A and B, schematic representations of the cell membranes (yellow) and surrounding electrolyte solutions. σo and σi are charge densities at the outer and inner surfaces of the cell membrane, respectively. Φo and Φi are electric potentials at the outer and inner surfaces of the cell membrane, respectively. C–I, representative NLC recordings for A4-E259Q (C), A4-E259R (D), A4-E259K (E), A4-D266N (F), A4-E259Q/D266N (G), A4-E259R/D266N (H), and A4-E259K/D266N (I). The magnitudes of NLCs are corrected for cell size as in Figs. 2 and 3. Different colors indicate individual recordings. A two-state Boltzmann model was used to interpret the NLC data (solid lines). J, the Vpk values (mean ± S.D.) are as follows: WT-A4 + MβCD, −166 ± 26 mV (n = 12); A4-sL2, −173 ± 22 mV (n = 9); A4-E259Q, −184 ± 24 mV (n = 10); A4-E259R, −154 ± 19 mV (n = 9); A4-E259K, −134 ± 20 mV (n = 10); A4-D266N, −189 ± 20 mV (n = 11); A4-E259Q/D266N, −157 ± 16 mV (n = 11); A4-E259R/D266N, −132 ± 21 mV (n = 13); A4-E259KR/D266N, −149 ± 20 mV (n = 12). Error bars represent S.D. K, correlation between the net charge in L2 versus Vpk. The means and standard deviations of the Vpk data shown in J are plotted against the calculated net charge in L2 (at pH 7.3). Error bars represent S.D. L, the α values (mean ± S.D.) are as follows: WT-A4 + MβCD, 0.012 ± 0.002 mV−1 (n = 12); A4-sL2, 0.010 ± 0.001 mV−1 (n = 9); A4-E259Q, 0.010 ± 0.002 mV−1 (n = 10); A4-E259R, 0.011 ± 0.002 mV−1 (n = 9); A4-E259K, 0.010 ± 0.002 mV−1 (n = 10); A4-D266N, 0.011 ± 0.002 mV−1 (n = 11); A4-E259Q/D266N, 0.010 ± 0.001 mV−1 (n = 11); A4-E259R/D266N, 0.011 ± 0.001 mV−1 (n = 13); A4-E259K/D266N, 0.012 ± 0.002 mV−1 (n = 12). Error bars represent S.D.
Glu259 is the sole charged residue present in sL2 of pendrin, whereas none is present in that of prestin (Fig. 1E). To test whether the significant Vpk depolarization found in A4-sL2 (Fig. 3F) can be attributed to neutralization of the negative charge pertaining to Glu259, additional pendrin constructs harboring E259Q, E259R, or E259K mutation were generated, and their NLCs were measured. NLCs became clearly measurable upon neutralization of Glu259 (E259Q) (Fig. 5, C and J) and resemble those found in A4-sL2 (Fig. 3F). Polarity reversal of Glu259 (E259R and E259K) also made NLC of pendrin clearly detectable within the measurable voltage range due to Vpk depolarization (Fig. 5, D, E, and J). We also examined the effect of the negative charge pertaining to Asp266, which is not located within the sL2 region but located in L2 (Fig. 1E). We found that neutralization of this negative charge (D266N) also depolarizes Vpk and makes large NLC of pendrin readily detectable (Fig. 5, F and J). Simultaneous introduction of charge-affecting mutations to Glu259 and Asp266 (A4-E259Q/D266N, A4-E259R/D266N, and A4-E259K/D266N) also depolarized Vpk but to greater extents (Fig. 5, G–J). These results suggest that the effects of these charge manipulations are additive. In fact, we found a statistically significant linear correlation (r = 0.85, p = 0.008) between the calculated net charge in L2 versus Vpk (Fig. 5K). The α value and its variation were similar among the pendrin constructs (one-way ANOVA, p = 0.39) (Fig. 5L), which argues against the possibility that the Vpk values were significantly over- or underestimated. These observations are uniformly explained by depolarization of Φo as a result of reduction of the negative surface charge present in sL2 or L2 (Fig. 5, A and B).
Charge manipulations of Asp271 (D271N, D271R, and D271K), which is located in the transmembrane helix that immediately follows L2 (Fig. 1), significantly impaired the targeting of pendrin to the cell membrane (Fig. S2A). Consistently, these mutants showed little or no NLC and transport activity (Fig. S2, B and E), implying the importance of Asp271 for maintaining the structural integrity of the transmembrane region.
The relationship between the voltage-sensing ability of pendrin and its anion transport function
The voltage-driven motor function of prestin has been presumed to stem from a common anion transport mechanism that is likely shared among the members of the SLC26 family (23, 24). To define the molecular mechanism underlying electromotility, it is important to examine whether the anion transport activity of prestin is affected by voltage. Unfortunately, prestin is not suitable for pursuing this possibility because we could not detect HCO3−/Cl− antiport activity for prestin (WT-A5) or any of our prestin-based constructs. We therefore examined the NLC–anion transport relationship in pendrin because we identified large NLC in pendrin (Fig. 3). The resting membrane potentials of HEK293T cells expressing WT pendrin (mean ± S.D.) were −15.0 ± 5.8 (n = 12) and −12.3 ± 4.4 mV (n = 16) in the presence and absence of MβCD, respectively, with no statistically significant difference among the groups (p = 0.17). If the voltage-sensing mechanism were intimately related to the anion transport mechanism, augmentation of the anion transport activity would be expected upon application of MβCD due to a significant shift of Vpk toward the resting membrane potential of the pendrin-expressing cells (Fig. 3, A and B). However, we did not find a statistically significant change in the anion transport activity upon application of MβCD (p > 0.999; Fig. 4A), suggesting independence of the transport and voltage-sensing mechanisms.
NLC1 and NLC2 segments of pendrin and prestin are not mutually interchangeable
Previous efforts to identify a segment(s) in the prestin molecule that is essential for the expression of electromotility have been inconsistent. One study (25) identified such segments, termed “NLC1” and “NLC2,” in the region forming the presumed anion translocation pathway (6), whereas other studies identified a short segment (termed “14aa” in the present study) located in L1 (22, 26). These results are apparently irreconcilable because the short 14-aa segment is not a part of the large NLC1/NLC2 segments. Because we could not confirm the importance of the 14-aa short segment for the expression of NLC (Fig. 3D), we also generated eight chimeric constructs in which the NLC1 and NLC2 segments were swapped between pendrin and prestin (A4A5Ch1–A4A5Ch8) (Fig. 6A), which are similar to those generated by Schaechinger et al. (25) using electromotile and nonelectromotile prestin orthologues. Unfortunately, none of our pendrin/prestin chimeras showed NLC or HCO3−/Cl− antiport activity (Fig. S2, C–E), probably due to the lack of membrane targeting (Fig. 6B), suggesting a difference in the intramolecular interactions within the TM domain between pendrin and prestin.
Figure 6.

Eight NLC1/NLC2–focused pendrin/prestin chimeras used in this study. A, both schematic diagram and tertiary structural models (lateral and extracellular views) are shown for prestin-based (A4A5Ch1, A4A5Ch2, A4A5Ch3, and A4A5Ch4) and pendrin-based (A4A5Ch5, A4A5Ch6, A4A5Ch7, and A4A5Ch8) chimeras. The parts of prestin and pendrin are indicated by magenta and dark blue, respectively. The amino acids and their residue numbers at the boundaries of A4 and A5 are also provided. The C termini of all the constructs were tagged with ECFP (for prestin-based constructs, A4A5Ch1–A4A5Ch4) or mTurquoise2 (for pendrin-based constructs, A4A5Ch5–A4A5Ch8). B, qualitative assessments of the subcellular localizations of the eight chimeras (A4A5Ch1–8) heterologously expressed in HEK293T cells. Results for WT-A5 and WT-A4 are also included as positive controls. The scale bars indicate 10 μm. All eight chimeras show predominantly cytosolic localization, indicating severely impaired membrane targeting.
Discussion
The electromotile function of prestin (SLC26A5) is thought to be unique among the members of the SLC26 family. Although not all prestin orthologues exhibit motor activity, they all show NLC (27–30). NLC indicates the presence of a rapid voltage-sensing mechanism, and it is directly coupled to concurrent motor activity in electromotile prestin orthologues (31–34). In the present study, we show that pendrin (SLC26A4) also exhibits NLC but with extremely hyperpolarized Vpk. Given the high degree of similarity in amino acid sequences among the SLC26 proteins, the presence of NLC in other members in the family would not be surprising, but this possibility has not been thoroughly examined heretofore. A previous study also reported NLC parameters for pendrin-expressing cells (22). However, the magnitude of the NLCs were much smaller than those observed in the present study despite their apparent Vpk (∼−40 mV) located within an easily measurable voltage range (22). In addition, the small NLCs found in the previous study were qualitatively very different from those reported in this study as the Vpk values were depolarized, but not hyperpolarized, with respect to Vpk of prestin (22). We claim that the NLCs detected in the present study for the pendrin-expressing cells are indeed mediated by pendrin because we found that the Vpk of the NLCs responds to cholesterol depletion by MβCD (Fig. 3) in the same way as previously demonstrated for prestin (20) and that swapping of L2 that contains opposite electrostatic property between pendrin (net negative) and prestin (net positive) results in Vpk shifts toward mutually opposing directions for pendrin (depolarization; Fig. 3) and prestin (hyperpolarization; Fig. 2). Successful identification of large NLCs in pendrin-expressing Sf9 cells without the use of any genetic or chemical manipulations further authenticates our claim (Fig. S1).
There is controversy over the identity of the voltage-sensing charge (6, 35–38). An early study proposed an incomplete transport model to explain the voltage-sensing mechanism of prestin where intracellular Cl− or HCO3− is used as an extrinsic voltage sensor (35). The basic notion of this model does not disagree with the fact that the HCO3−/Cl− antiport activity of prestin is very low (39) or virtually undetectable. However, later studies found that electromotility can be conferred on nonelectromotile SLC26 proteins without diminishing or abrogating the transport activities (25, 26). Our finding that pendrin exhibits both significant HCO3−/Cl− antiport activity and NLC (Figs. 3 and 4) underscores that the anion transport function and voltage-sensing ability are not mutually exclusive. The identification of large NLC in pendrin provided a fortuitous opportunity to examine the functional relationship between voltage-sensing and anion transport mechanisms, which could not be pursued in prestin due to its extremely small anion transport activity to be compared with NLC. We found that the HCO3−/Cl− antiport activity of pendrin is not affected by a significant depolarizing shift of NLC toward the resting membrane potentials of pendrin-expressing cells upon MβCD application (Figs. 3 and 4). This observation suggests that voltage-sensing and anion transport mechanisms may not be intimately related, which is in agreement with a previous observation suggesting separation of voltage-sensing and anion transport functions for prestin (38).
It is possible that most, if not all, members of the SLC26 family retain voltage-sensing ability. It is important to note that the absence of measurable NLC does not necessarily mean inability of an SLC26 protein to sense voltage. Because the experimentally measurable range of membrane potential is limited, NLC with extremely hyper-/depolarized Vpk is difficult to identify. In the present study, we demonstrate that this is, in fact, the case for pendrin (Fig. 3A). Fast voltage-sensing kinetics is another important factor for a successful NLC recording because detection of capacitive currents is the basis of NLC measurement. If voltage-sensing charge movement is very slow, it would not manifest as NLC. For example, NLC of zebrafish prestin shows significant stimulus voltage frequency dependence, and it becomes unmeasurable at high stimulus frequencies due to its relatively slow voltage-sensing kinetics (27). It is conceivable that the voltage-sensing kinetics of some SLC26 members may be too slow to exhibit its voltage-sensing ability as NLC. Collectively, absence or abrogation of NLC by genetic or pharmacological manipulation needs to be concluded carefully.
For physiological electromotile function of prestin orthologues, it is important to set their Vpk within the physiological Vm range. Our study demonstrates that the charged residues present in L2 greatly contribute to set distinct Vpk for prestin and pendrin (Figs. 2, 3, and 5) and that the directions of the Vpk shift induced by charge-affecting mutations in L2 can be simply explained by changes in the membrane surface charge density (σo) that concomitantly affect membrane surface electrical potential (Φo) (Fig. 5, A and B). Furthermore, all previously reported Vpk shifts of prestin that were induced by charge-affecting mutations introduced to L2, i.e. T234K (depolarization) (40), K233Q/K235Q/R236Q (hyperpolarization) (35), S238D (hyperpolarization) (41), and K255Q (hyperpolarization) (38), can be explained by their anticipated effects on the membrane surface potential. On the contrary, L1 does not seem to contribute to confer distinct Vpk on pendrin (Fig. 3C). Although significant Vpk hyperpolarization was observed in prestin upon L1 swapping (Fig. 2B), no additive effect on Vpk was found upon L1/L2 double swapping (Fig. 2D). In any case, the hyperpolarizing Vpk shift of prestin upon L1 swapping (Fig. 2B) cannot be explained by the electrostatic property of L1 because the net charge of L1 is almost the same between prestin and pendrin (Fig. 1D). Also, hyperpolarizing Vpk shifts of prestin induced by D154N and D155N mutations (35) cannot be explained by an L1-associated surface charge effect. These irregular and unpredictable consequences of L1 swapping may imply a dynamic role of the L1 region in prestin.
Both single (L1 or L2) and double (L1 and L2) loop swapping of the extracellular loops does not deprive prestin of its function (Fig. 2). Pendrin also tolerates single (L1 or L1(14aa)) swapping, but neither single L2- nor double L1/L2-swapped pendrin constructs (A4-L2 and A4-L1/L2) showed NLC or HCO3−/Cl− antiport activity, likely due to impaired membrane targeting (Fig. 4B). We eluded the membrane targeting issue for A4-L2 by shortening the swapped L2 region (sL2) (Fig. 3F). However, double loop-swapped pendrin constructs with sL2 (A4-L1/sL2 and A4-L1(14aa)/sL2) still fail to target the membrane (Fig. 4B). These observations, together with our negative observation on the eight pendrin/prestin chimeras (Fig. 6), suggest a difference in the intramolecular interactions between pendrin and prestin or may imply a difference in the TM domain architecture between pendrin and prestin.
It is noteworthy that the voltage sensitivity of pendrin, which is reflected in its α value, is 2–3 times smaller than that of prestin (Figs. 2F and 5L) and that the α values of nonelectromotile prestin orthologues are also smaller than those of electromotile prestin orthologues (27, 28). Because α values would indicate the magnitude and extent of voltage-induced conformational change (see “Experimental procedures”), it is interesting to examine whether and how the magnitudes of α correlate with the expression of electromotility. Although electrophysiological characterization has already been conducted for various prestin orthologues (27–30), similar efforts have not been expanded to the other members in the SLC26 family. Thorough comparative studies across the SLC26 family would facilitate efforts in defining the molecular mechanisms underlying the uniqueness and generality of electrophysiological characteristics among the SLC26 family members.
Experimental procedures
Generation of stable cell lines that express various SLC26 protein constructs
The pendrin (UniProt accession number O43511)- and prestin (UniProt accession number Q9JKQ2)-based constructs were generated by multistep PCR (A4-L1, A4-L2, A4-L1/L2, A5-L1, A5-L2, and A5-L1/L2) (42), site-directed mutagenesis (A4-E259Q, A4-E259R, A4-E259K, A4-D266N, A4-E259Q/D266N, A4-E259R/D266N, and A4-E259K/D266N), and de novo DNA synthesis combined with template vector PCR (A4-sL2, A4-L1/sL2, A4-L1(14aa), A4-L1(14aa)/sL2, and A4A5Ch1–A4A5Ch8). The complete DNA sequences of all these constructs, along with the PCR primers and synthesized dsDNA information, are provided in the supporting information. These constructs with C-terminally attached ECFP (for prestin-based constructs) or mTurquoise2 (for pendrin-based constructs) were cloned into a pSBtet-Pur vector (Addgene, Cambridge, MA) (43). The functions of pendrin and prestin are not abrogated by a C-terminally fused fluorescent protein (32, 44). Stable cell lines that express these recombinant protein constructs were established in HEK293T cells as described previously (43). Expressions of the pendrin/prestin constructs were induced by application of doxycycline (1 μg/ml) to the cell culture medium 1–3 days prior to experiments.
Protein structure modeling
The structural models of the transmembrane regions of pendrin and prestin were generated by Phyre2 (45) based on the SLC26Dg structure (Protein Data Bank code 5DA0) (7) using the partial amino acid sequences of pendrin (Pro76–Arg512) and prestin (Pro72–Arg502). The models were rendered by PyMOL (Fig. 1) or CueMol software (http://www.cuemol.org/)5 (Fig. 6).
Electrophysiology
Whole-cell recordings were performed at room temperature using an Axopatch 200A/B amplifier (Molecular Devices, Sunnyvale, CA). Recording pipettes were pulled from borosilicate glass to achieve initial bath resistances averaging 3–4 megaohms. Whole-cell NLC recordings were performed using a 0-mV holding potential and a sinusoidal voltage stimulus (2.5-Hz, 120–150-mV amplitude) superimposed with two higher frequency stimuli (390.6 and 781.2 Hz, 10-mV amplitude). Recording pipettes were filled with an intracellular solution containing 140 mm CsCl, 2 mm MgCl2, 10 mm EGTA, and 10 mm HEPES (pH 7.3 adjusted at room temperature). Cells were bathed in an extracellular solution containing 120 mm NaCl, 20 mm tetraethylammonium chloride, 2 mm CoCl2, 2 mm MgCl2, and 10 mm HEPES (pH 7.3 adjusted at room temperature). Osmolarity was adjusted to 310 mosmol liter−1 with glucose (34, 46). Intracellular pressure was kept at 0 mmHg, and current data were collected by jClamp (SciSoft Co., New Haven, CT) using a fast Fourier transform–based admittance analysis to determine NLC (47). The resting cell membrane potentials were determined in Hanks' balanced salt solution (14025, Thermo Fisher Scientific, Waltham, MA) using recording pipettes filled with an intracellular solution containing 140 mm KCl, 2 mm MgCl2, 10 mm EGTA, and 10 mm HEPES (pH 7.3 adjusted at room temperature).
NLC data analysis
Voltage-dependent cell membrane electric capacitance data were analyzed using the following two-state Boltzmann equation,
| (1) |
where α is the slope factor of the voltage dependence of charge transfer, Qmax is the maximum charge transfer, Vm is the membrane potential, Vpk is the voltage at which the maximum charge movement is attained, and Clin is the linear capacitance (34, 46, 48, 49). In this two-state Boltzmann model, α is defined as zappe/kBT where zapp is the apparent valence of charge movement, e is electron charge, kB is the Boltzmann constant, and T is absolute temperature. The magnitude of zapp reflects the valence (z) and the traveling distance (δ) along the direction of the electric field of a voltage-sensing charge.
Anion transport assay
A ratiometric fluorescent pH indicator, SNARF-5F (S23923, Thermo Fisher Scientific), was loaded into cells in a high-chloride buffer containing 140 mm NaCl, 4.5 mm KCl, 1 mm MgCl2, 2.5 mm CaCl2, and 20 mm HEPES (pH 7.4 adjusted at room temperature, 320 mosmol liter−1) for 30 min in the presence of 5% CO2 at room temperature. The cells were washed with the high-chloride buffer once and resuspended in 100 μl of the high-chloride buffer. Portions of the cell suspensions (50 μl) were transferred to wells in a 96-well plate (∼1.5 × 105 cells/well). The HCO3−/Cl− antiport assay was initiated by an automated injection of 200 μl of a low-chloride buffer containing 125 mm sodium gluconate, 5 mm potassium gluconate, 1 mm MgCl2, 1 mm CaCl2, 20 mm HEPES, and 25 mm NaHCO3 (pH 7.4 at room temperature under 5% CO2) in a Synergy2 plate reader (BioTek, Winooski, VT). The fluorescence intensity of SNARF-5F was measured in a time-dependent manner. The excitation and emission filters used were 504/12 (excitation) (Semrock, Rochester, NY), 572/28 (F1) (Semrock), and 709/167 (F2) (Semrock), respectively. The fluorescence ratio, F2/F1, was converted into the intracellular H+ concentration using a pH calibration curve. The HCO3−/Cl− antiport activity (s−1) was determined from exponential curve fitting.
Confocal imaging
The stable cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM, high glucose, GlutaMAX, pyruvate, Thermo Fisher Scientific) with 10% FBS (French origin, BioWest, Riverside, MO) at 37 °C (5% CO2) in glass-bottom dishes (D11130H, Matsunami Glass, Kishiwada, Japan). When cell confluence reached 20–30%, expressions of the pendrin/prestin constructs were induced by 1 μg/ml doxycycline hyclate (LKT Laboratories, St. Paul, MN) included in the medium for 2–3 days. Fluorescence images of the cells were acquired using an LSM710 confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a 63× Plan-Apochromat objective lens (numerical aperture, 1.40 oil) and a filter set (BP 395–440 excitation filter, FT460 dichroic mirror, and LP470 emission filter). The cells were illuminated with a 30-milliwatt 405-nm diode laser (laser power, 2%) at room temperature, and the images were taken with ZEN software (Carl Zeiss). The pinhole size and pixel dwell time were set to be 1 airy unit (490 nm) and 1.27 μs, respectively. Four sequentially scanned images were averaged and then processed with a despeckle algorithm and contrast/brightness adjustment using Fiji software (50).
Statistical analyses
Statistical analyses were performed using Prism (GraphPad Software). Student's t test was used for comparisons between two groups. One-way ANOVA combined with the Tukey–Kramer test was used for multiple comparisons. p < 0.05 was considered statistically significant.
Author contributions
M. F. K., K. W., and K. H. formal analysis; M. F. K., K. W., S. T., J. B., T. K., and K. H. investigation; T. K., S. U., J. Z., T. S., and K. H. resources; T. K., S. U., T. S., and K. H. supervision; T. K., S. U., T. S., and K. H. funding acquisition; T. S. and K. H. conceptualization; K. H. writing-original draft; K. H. writing-review and editing.
Supplementary Material
Acknowledgments
We appreciate Dr. Mikiko C. Siomi (The University of Tokyo) for the use of the confocal microscope. Connor Divelbiss (Plainfield Central High School, Plainfield, IL) contributed to NLC data collection.
This work was supported in part by National Institutes of Health Grant DC014553 (to K. H.); Japan Science and Technology Agency, Core Research for Evolutional Science and Technology Grant JPMJCR14W1 (to S. U.); Ministry of Education, Culture, Sports, Science and Technology, Japan Grants 15K18514 (to T. S.) and 17K15100 (to T. K.); and the Hugh Knowles Center. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and other supporting information.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- SLC26
- solute carrier 26
- NLC
- nonlinear capacitance
- TM
- transmembrane
- L
- loop
- ANOVA
- analysis of variance
- MβCD
- methyl-β-cyclodextrin
- aa
- amino acid
- σo
- surface charge density
- Φo
- membrane surface potential
- Φi
- inner surface potential
- ECFP
- enhanced cyan fluorescent protein.
References
- 1. Alper S. L., and Sharma A. K. (2013) The SLC26 gene family of anion transporters and channels. Mol. Aspects Med. 34, 494–515 10.1016/j.mam.2012.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Everett L. A., Belyantseva I. A., Noben-Trauth K., Cantos R., Chen A., Thakkar S. I., Hoogstraten-Miller S. L., Kachar B., Wu D. K., and Green E. D. (2001) Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum. Mol. Genet. 10, 153–161 10.1093/hmg/10.2.153 [DOI] [PubMed] [Google Scholar]
- 3. Liberman M. C., Gao J., He D. Z., Wu X., Jia S., and Zuo J. (2002) Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419, 300–304 10.1038/nature01059 [DOI] [PubMed] [Google Scholar]
- 4. Dallos P., Wu X., Cheatham M. A., Gao J., Zheng J., Anderson C. T., Jia S., Wang X., Cheng W. H. Y., Sengupta S., He D. Z., and Zuo J. (2008) Prestin-based outer hair cell motility is necessary for mammalian cochlear amplification. Neuron 58, 333–339 10.1016/j.neuron.2008.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brownell W. E., Bader C. R., Bertrand D., and de Ribaupierre Y. (1985) Evoked mechanical responses of isolated cochlear outer hair cells. Science 227, 194–196 10.1126/science.3966153 [DOI] [PubMed] [Google Scholar]
- 6. Gorbunov D., Sturlese M., Nies F., Kluge M., Bellanda M., Battistutta R., and Oliver D. (2014) Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nat. Commun. 5, 3622 10.1038/ncomms4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geertsma E. R., Chang Y.-N., Shaik F. R., Neldner Y., Pardon E., Steyaert J., and Dutzler R. (2015) Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat. Struct. Mol. Biol. 22, 803–808 10.1038/nsmb.3091 [DOI] [PubMed] [Google Scholar]
- 8. Ashmore J. F. (1990) Forward and reverse transduction in the mammalian cochlea. Neurosci Res. Suppl. 12, S39–S50 10.1016/0921-8696(90)90007-P [DOI] [PubMed] [Google Scholar]
- 9. Santos-Sacchi J. (1991) Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 11, 3096–3110 10.1523/JNEUROSCI.11-10-03096.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang Y.-N., and Geertsma E. R. (2017) The novel class of seven transmembrane segment inverted repeat carriers. Biol. Chem. 398, 165–174 10.1515/hsz-2016-0254 [DOI] [PubMed] [Google Scholar]
- 11. Sloan-Heggen C. M., Babanejad M., Beheshtian M., Simpson A. C., Booth K. T., Ardalani F., Frees K. L., Mohseni M., Mozafari R., Mehrjoo Z., Jamali L., Vaziri S., Akhtarkhavari T., Bazazzadegan N., Nikzat N., et al. (2015) Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J. Med. Genet. 52, 823–829 10.1136/jmedgenet-2015-103389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chai Y., Huang Z., Tao Z., Li X., Li L., Li Y., Wu H., and Yang T. (2013) Molecular etiology of hearing impairment associated with nonsyndromic enlarged vestibular aqueduct in East China. Am. J. Med. Genet. A 161A, 2226–2233 10.1002/ajmg.a.36068 [DOI] [PubMed] [Google Scholar]
- 13. Yao G., Chen D., Wang H., Li S., Zhang J., Feng Z., Guo L., Yang Z., Yang S., Sun C., Zhang X., and Ma D. (2013) Novel mutations of SLC26A4 in Chinese patients with nonsyndromic hearing loss. Acta Otolaryngol. 133, 833–841 10.3109/00016489.2013.777160 [DOI] [PubMed] [Google Scholar]
- 14. Park H.-J., Lee S.-J., Jin H.-S., Lee J. O., Go S.-H., Jang H. S., Moon S.-K., Lee S.-C., Chun Y.-M., Lee H.-K., Choi J.-Y., Jung S.-C., Griffith A. J., and Koo S. K. (2005) Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin. Genet. 67, 160–165 10.1111/j.1399-0004.2004.00386.x [DOI] [PubMed] [Google Scholar]
- 15. Kim M., Kim J., Kim S. H., Kim S. C., Jeon J. H., Lee W. S., Kim U.-K., Kim H. N., and Choi J. Y. (2011) Hemorrhage in the endolymphatic sac: a cause of hearing fluctuation in enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 75, 1538–1544 10.1016/j.ijporl.2011.09.002 [DOI] [PubMed] [Google Scholar]
- 16. Dai P., Stewart A. K., Chebib F., Hsu A., Rozenfeld J., Huang D., Kang D., Lip V., Fang H., Shao H., Liu X., Yu F., Yuan H., Kenna M., Miller D. T., et al. (2009) Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S. patients with nonsyndromic hearing loss. Physiol. Genomics 38, 281–290 10.1152/physiolgenomics.00047.2009 [DOI] [PubMed] [Google Scholar]
- 17. Park H.-J., Shaukat S., Liu X.-Z., Hahn S. H., Naz S., Ghosh M., Kim H.-N., Moon S.-K., Abe S., Tukamoto K., Kabra M., Erdenetungalag R., Radnaabazar J., Khan S., Pandya A., et al. (2003) Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J. Med. Genet. 40, 242–248 10.1136/jmg.40.4.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yuan Y., Guo W., Tang J., Zhang G., Wang G., Han M., Zhang X., Yang S., He D. Z., and Dai P. (2012) Molecular epidemiology and functional assessment of novel allelic variants of SLC26A4 in non-syndromic hearing loss patients with enlarged vestibular aqueduct in China. PLoS One 7, e49984 10.1371/journal.pone.0049984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sloan-Heggen C. M., Bierer A. O., Shearer A. E., Kolbe D. L., Nishimura C. J., Frees K. L., Ephraim S. S., Shibata S. B., Booth K. T., Campbell C. A., Ranum P. T., Weaver A. E., Black-Ziegelbein E. A., Wang D., Azaiez H., et al. (2016) Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet 135, 441–450 10.1007/s00439-016-1648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rajagopalan L., Greeson J. N., Xia A., Liu H., Sturm A., Raphael R. M., Davidson A. L., Oghalai J. S., Pereira F. A., and Brownell W. E. (2007) Tuning of the outer hair cell motor by membrane cholesterol. J. Biol. Chem. 282, 36659–36670 10.1074/jbc.M705078200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marheineke K., Grünewald S., Christie W., and Reiländer H. (1998) Lipid composition of Spodoptera frugiperda (Sf9) and Trichoplusia ni (Tn) insect cells used for baculovirus infection. FEBS Lett. 441, 49–52 10.1016/S0014-5793(98)01523-3 [DOI] [PubMed] [Google Scholar]
- 22. Tang J., Pecka J. L., Tan X., Beisel K. W., and He D. Z. (2011) Engineered pendrin protein, an anion transporter and molecular motor. J. Biol. Chem. 286, 31014–31021 10.1074/jbc.M111.259564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dallos P., and Fakler B. (2002) Prestin, a new type of motor protein. Nat. Rev. Mol. Cell Biol. 3, 104–111 10.1038/nrm730 [DOI] [PubMed] [Google Scholar]
- 24. Ashmore J. (2008) Cochlear outer hair cell motility. Physiol. Rev. 88, 173–210 10.1152/physrev.00044.2006 [DOI] [PubMed] [Google Scholar]
- 25. Schaechinger T. J., Gorbunov D., Halaszovich C. R., Moser T., Kügler S., Fakler B., and Oliver D. (2011) A synthetic prestin reveals protein domains and molecular operation of outer hair cell piezoelectricity. EMBO J. 30, 2793–2804 10.1038/emboj.2011.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan X., Pecka J. L., Tang J., Lovas S., Beisel K. W., and He D. Z. (2012) A motif of eleven amino acids is a structural adaptation that facilitates motor capability of eutherian prestin. J. Cell Sci. 125, 1039–1047 10.1242/jcs.097337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Albert J. T., Winter H., Schaechinger T. J., Weber T., Wang X., He D. Z., Hendrich O., Geisler H.-S., Zimmermann U., Oelmann K., Knipper M., Göpfert M. C., and Oliver D. (2007) Voltage-sensitive prestin orthologue expressed in zebrafish hair cells. J. Physiol. 580, 451–461 10.1113/jphysiol.2007.127993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan X., Pecka J. L., Tang J., Okoruwa O. E., Zhang Q., Beisel K. W., and He D. Z. (2011) From zebrafish to mammal: functional evolution of prestin, the motor protein of cochlear outer hair cells. J. Neurophysiol. 105, 36–44 10.1152/jn.00234.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang J., Pecka J. L., Fritzsch B., Beisel K. W., and He D. Z. Z. (2013) Lizard and frog prestin: evolutionary insight into functional changes. PLoS One 8, e54388 10.1371/journal.pone.0054388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beurg M., Tan X., and Fettiplace R. (2013) A prestin motor in chicken auditory hair cells: active force generation in a nonmammalian species. Neuron 79, 69–81 10.1016/j.neuron.2013.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kakehata S., and Santos-Sacchi J. (1995) Membrane tension directly shifts voltage dependence of outer hair cell motility and associated gating charge. Biophys. J. 68, 2190–2197 10.1016/S0006-3495(95)80401-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ludwig J., Oliver D., Frank G., Klöcker N., Gummer A. W., and Fakler B. (2001) Reciprocal electromechanical properties of rat prestin: the motor molecule from rat outer hair cells. Proc. Natl. Acad. Sci. U.S.A. 98, 4178–4183 10.1073/pnas.071613498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X., Yang S., Jia S., and He D. Z. (2010) Prestin forms oligomer with four mechanically independent subunits. Brain Res. 1333, 28–35 10.1016/j.brainres.2010.03.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Homma K., and Dallos P. (2011) Evidence that prestin has at least two voltage-dependent steps. J. Biol. Chem. 286, 2297–2307 10.1074/jbc.M110.185694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oliver D., He D. Z., Klöcker N., Ludwig J., Schulte U., Waldegger S., Ruppersberg J. P., Dallos P., and Fakler B. (2001) Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science 292, 2340–2343 10.1126/science.1060939 [DOI] [PubMed] [Google Scholar]
- 36. Rybalchenko V., and Santos-Sacchi J. (2003) Cl− flux through a non-selective, stretch-sensitive conductance influences the outer hair cell motor of the guinea-pig. J. Physiol. 547, 873–891 10.1113/jphysiol.2002.036434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rybalchenko V., and Santos-Sacchi J. (2008) Anion control of voltage sensing by the motor protein prestin in outer hair cells. Biophys. J. 95, 4439–4447 10.1529/biophysj.108.134197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bai J.-P., Surguchev A., Montoya S., Aronson P. S., Santos-Sacchi J., and Navaratnam D. (2009) Prestin's anion transport and voltage-sensing capabilities are independent. Biophys. J. 96, 3179–3186 10.1016/j.bpj.2008.12.3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mistrík P., Daudet N., Morandell K., and Ashmore J. F. (2012) Mammalian prestin is a weak Cl−/HCO3− electrogenic antiporter. J. Physiol. 590, 5597–5610 10.1113/jphysiol.2012.241448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oliver D., Schächinger T., and Fakler B. (2006) Interaction of prestin (SLC26A5) with monovalent intracellular anions. Novartis Found. Symp. 273, 244–253; discussion 253–260, 261–264 [PubMed] [Google Scholar]
- 41. Deák L., Zheng J., Orem A., Du G.-G., Aguiñaga S., Matsuda K., and Dallos P. (2005) Effects of cyclic nucleotides on the function of prestin. J. Physiol. 563, 483–496 10.1113/jphysiol.2004.078857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grandori R., Struck K., Giovanielli K., and Carey J. (1997) A three-step PCR protocol for construction of chimeric proteins. Protein Eng. 10, 1099–1100 10.1093/protein/10.9.1099 [DOI] [PubMed] [Google Scholar]
- 43. Kowarz E., Löscher D., and Marschalek R. (2015) Optimized sleeping beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 10, 647–653 10.1002/biot.201400821 [DOI] [PubMed] [Google Scholar]
- 44. Taylor J. P., Metcalfe R. A., Watson P. F., Weetman A. P., and Trembath R. C. (2002) Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J. Clin. Endocrinol. Metab. 87, 1778–1784 10.1210/jcem.87.4.8435 [DOI] [PubMed] [Google Scholar]
- 45. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 46. Homma K., Miller K. K., Anderson C. T., Sengupta S., Du G.-G., Aguiñaga S., Cheatham M., Dallos P., and Zheng J. (2010) Interaction between CFTR and prestin (SLC26A5). Biochim. Biophys. Acta 1798, 1029–1040 10.1016/j.bbamem.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Santos-Sacchi J., Kakehata S., and Takahashi S. (1998) Effects of membrane potential on the voltage dependence of motility-related charge in outer hair cells of the guinea-pig. J. Physiol. 510, 225–235 10.1111/j.1469-7793.1998.225bz.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Homma K., Duan C., Zheng J., Cheatham M. A., and Dallos P. (2013) The V499G/Y501H mutation impairs fast motor kinetics of prestin and has significance for defining functional independence of individual prestin subunits. J. Biol. Chem. 288, 2452–2463 10.1074/jbc.M112.411579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keller J. P., Homma K., Duan C., Zheng J., Cheatham M. A., and Dallos P. (2014) Functional regulation of the SLC26-family protein prestin by calcium/calmodulin. J. Neurosci. 34, 1325–1332 10.1523/JNEUROSCI.4020-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.