

Abstract
Structural characterization of glycosaminoglycans remains a challenge but is essential for determining structure–function relationships between glycosaminoglycans and the biomolecules with which they interact and for gaining insight into the biosynthesis of glycosaminoglycans. We have recently reported that xyloside-primed chondroitin/dermatan sulfate derived from a human breast carcinoma cell line, HCC70, has cytotoxic effects and shown that it differs in disaccharide composition from nontoxic chondroitin/dermatan sulfate derived from a human breast fibroblast cell line, CCD-1095Sk. To further investigate the structural requirements for the cytotoxic effect, we developed a novel LC–MS/MS approach based on reversed-phase dibutylamine ion-pairing chromatography and negative-mode higher-energy collision dissociation and used it in combination with cell growth studies and disaccharide fingerprinting. This strategy enabled detailed structural characterization of linkage regions, internal oligosaccharides, and nonreducing ends, revealing not only differences between xyloside-primed chondroitin/dermatan sulfate from HCC70 cells and CCD-1095Sk cells, but also sialylation of the linkage region and previously undescribed methylation and sulfation of the nonreducing ends. Although the xyloside-primed chondroitin/dermatan sulfate from HCC70 cells was less complex in terms of presence and distribution of iduronic acid than that from CCD-1095Sk cells, both glucuronic acid and iduronic acid appeared to be essential for the cytotoxic effect. Our data have moved us one step closer to understanding the structure of the cytotoxic chondroitin/dermatan sulfate from HCC70 cells primed on xylosides and demonstrate the suitability of the LC–MS/MS approach for structural characterization of glycosaminoglycans.
Keywords: cancer, chondroitin sulfate, dermatan sulfate, glycosaminoglycan, mass spectrometry (MS), glycan structure, xyloside
Introduction
Glycosaminoglycans (GAGs)3 are complex linear polysaccharides important in many cellular processes, both in health and disease (1–5). They are commonly linked to a core protein as part of a proteoglycan and are produced by virtually all mammalian cells. Chondroitin/dermatan sulfate (CS/DS) and heparan sulfate (HS) are the two major classes of GAGs composed of repeating N-hexosamine (HexNAc; N-acetylgalactosamine (GalNAc) or N-acetylglucosamine (GlcNAc)) and hexuronic acid units (HexUA or UA; glucuronic acid (GlcUA) or iduronic acid (IdoUA)): [-3GalNAcβ1–4GlcUAβ1-] in CS, [-3GalNAcβ1-4IdoUAα1-] in DS, and [-4GlcNAcα1-4GlcUAβ1/IdoUAα1-] in HS (Fig. 1A) (1, 6). Notably, DS is commonly found as CS/DS copolymers with varying proportions of IdoUA-containing disaccharide units.
Figure 1.

Structure of CS/DS, cleavage sites of GAG-degrading bacterial enzymes, and xylosides included in the study. A, established structure of CS/DS, including modifications in the chain and linkage region shown together with the theoretical cleavage sites of the bacterial CS/DS-degrading enzymes. B, chemical structures of 2-naphthyl β-d-xylopyranoside (XylNap, left) and 2-(6-hydroxylnaphthyl) β-d-xylopyranoside (XylNapOH, right).
The biosynthesis of both CS/DS and HS is initiated by the attachment of xylose (Xyl) to a serine residue of a core protein, followed by stepwise assembly of a common linkage region tetrasaccharide [-4GlcUAβ1–3Galβ1–3Galβ1–4Xylβ1-]. The linkage region can be modified by sulfation (7, 8), phosphorylation (9), and as recently shown, also by sialylation and fucosylation (Fig. 1A) (10). The phosphorylation is involved in the regulation of the GAG chain elongation during biosynthesis (11), but the functional roles of the other modifications are as yet unknown. The critical addition of GalNAc or GlcNAc to the linkage region tetrasaccharide directs the biosynthesis to either CS/DS or HS, respectively. Further extension of the GAG chains is mediated by class-specific GAG polymerases generating the carbohydrate backbone, epimerases converting GlcUA to IdoUA, N-deacetylases/N-sulfotransferases (HS only), and O-sulfotransferases adding sulfates at position 2 of GlcUA and IdoUA in both CS/DS and HS, at positions 4 and 6 of GalNAc in CS/DS, and at position 6 and, more rarely, at position 3 of GlcNAc in HS (Fig. 1A). GAGs are commonly extensively modified and extended to a total length of dp50–dp200 (dp; degree of polymerization, i.e. the number of monosaccharide residues), corresponding to 25–100 kDa in size (12).
Accumulating data indicate that specific sulfation and epimerization patterns are required for a number of GAG–protein interactions (13). However, because of the size and heterogeneity of GAGs, structural characterization has proven to be particularly difficult. Disaccharide fingerprinting, entailing enzymatic GAG degradation, disaccharide labeling, and detection by HPLC or LC–MS/MS, is a common analytical approach used to obtain an overview of the sulfation pattern of the GAGs expressed by a certain cell type or tissue (14–17). For GAG sequencing, various mass spectrometric approaches represent promising methods (18–22). The challenges associated with these approaches include LC–MS/MS compatible chromatography, alkali adduct formation, in-source sulfate loss, and complex data analysis, although progress to minimize and circumvent these issues have been made during the past few years (23–25). The field is moving fast forward, yet only a few successful attempts of sequencing intact GAGs have been reported (26–28). Thus, novel LC–MS/MS approaches with improved separation, capacity, sensitivity, specificity and higher mass accuracy, in addition to more efficient bioinformatics tools are needed.
The cellular assembly of GAG chains onto core proteins can be perturbed by a group of compounds called β-d-xylopyranosides or xylosides in short, comprising a Xyl in β-linkage to an aglycon (29, 30). They can act as acceptor substrates for GAG biosynthesis, thereby inducing the formation and secretion of xyloside-primed GAGs and concurrently inhibiting the formation of GAGs on core proteins. The xyloside concentration, type of xyloside, and cell type have been shown to influence the amount and composition of the GAGs produced (31–35), but detailed knowledge about the structure of xyloside-primed GAGs is lacking. We have recently reported a cytotoxic effect of CS/DS derived from human breast carcinoma cells, HCC70, primed on either 2-naphthyl β-d-xylopyranoside (XylNap, Fig. 1B) or 2-(6-hydroxynaphthyl) β-d-xylopyranoside (XylNapOH, Fig. 1B), and shown that the effect can be inhibited by the corresponding HS primed on XylNap (34). Moreover, the XylNap- and XylNapOH-primed CS/DS was shown to differ in disaccharide composition when compared with the noncytotoxic XylNap- and XylNapOH-primed CS/DS derived from a human breast fibroblast cell line, CCD-1095Sk, indicating that the detailed structures of these CS/DS GAGs are important for the cytotoxic effect.
To further investigate the structural requirements for the cytotoxic effect, we have here developed a novel LC–MS/MS approach based on reversed-phase dibutylamine ion-pairing chromatography and negative-mode higher-energy collision dissociation (HCD), and we used it in combination with disaccharide fingerprinting and cell growth studies. Our approach enabled us to, in one single LC–MS/MS run, characterize internal disaccharides, oligosaccharides, linkage regions, and nonreducing ends (NREs) of CS/DS from HCC70 cells and CCD-1095Sk cells primed on XylNap generated after degradation with established GAG-degrading enzymes.
Results
Both GlcUA- and IdoUA-containing structures are important for the cytotoxic effect of XylNap-primed CS/DS from HCC70 cells
To investigate whether the cytotoxic effect of XylNap-primed CS/DS from HCC70 cells could be linked specifically to GlcUA- or IdoUA-containing structures of the CS/DS, XylNap-primed GAGs were isolated from culture media of HCC70 cells after treatment with 100 μm XylNap and degraded enzymatically. Chondroitinase ABC and heparinase II and III cleave CS/DS and HS, respectively, to disaccharides and linkage regions (36, 37). Chondroitinase AC-I and -II, however, cleave at GlcUA in CS/DS, whereas chondroitinase B cleaves at IdoUA in CS/DS, generating disaccharides, oligosaccharides (dp3–dp10), polysaccharides (>dp10), and linkage regions depending on the distribution of GlcUA and IdoUA in the GAG chain (Fig. 1A) (37). The XylNap-primed GAGs were degraded using mixtures of heparinase II and III and chondroitinase AC-I and -II or heparinase II and III and chondroitinase B before they were used to treat HCC70 cells. Their effects were compared with those of XylNap-primed GAGs degraded with heparinase II and III alone or with a mixture of heparinase II and III and chondroitinase ABC.
Treatment of HCC70 cells with heparinase II- and III-degraded XylNap-primed GAGs resulted in a growth-reducing effect, with an IC50 value of 6 μg/ml (Fig. 2). This was in accordance with previously published data (34), where we showed that the growth reduction was mediated by induction of apoptosis and the effect was thereby concluded to be cytotoxic. In this study, the XylNap-primed GAGs degraded with a mixture of heparinase II and III and chondroitinase ABC were found to lack a distinct effect on the cell growth, indicating that the disaccharides and linkage region-containing structures obtained after this degradation were nontoxic. The XylNap-primed GAGs degraded both with the mixture of heparinase II and III and chondroitinase AC-I and -II, and the mixture of heparinase II and III and chondroitinase B, in contrast, had growth-reducing effects on the HCC70 cells, corresponding to IC50 values of ∼9 μg/ml in each case (Fig. 2). This suggests that both GlcUA- and IdoUA-containing structures are important for the cytotoxic effect of the XylNap-primed CS/DS from HCC70 cells.
Figure 2.
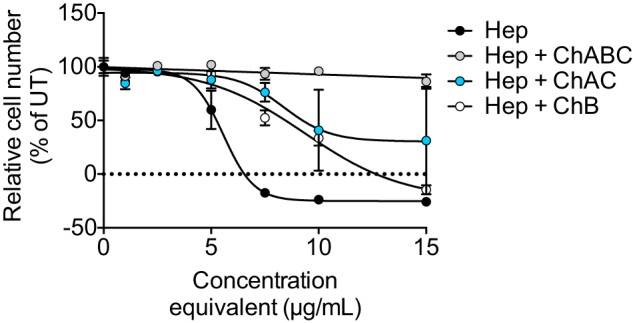
Growth of HCC70 cells in the presence of enzymatically degraded XylNap-primed CS/DS from HCC70 cells. The growth of HCC70 cells after 96 h of treatment with XylNap-primed GAGs degraded with heparinase II and III (Hep; black), heparinase II and III and chondroitinase ABC (Hep + ChABC; gray), heparinase II and III and chondroitinase AC-I and -II (Hep + ChAC; blue), or heparinase II and III and chondroitinase B (Hep + ChB; white). The concentrations of the GAGs administered to the cells were de facto lower than the indicated concentrations, as the indicated concentrations correspond to the concentrations of the GAGs before enzymatic degradation. The data points are the means ± S.D., in which n = 3. UT, untreated.
XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells differ in CS/DS and HS proportions and disaccharide composition
As an initial step in the structural characterization of the xyloside-primed GAGs, disaccharide fingerprinting using HPLC was performed. Xyloside-primed GAGs were isolated from culture media of HCC70 cells and CCD-1095Sk cells after incubation with 100 μm XylNap or XylNap deuterated in the naphthyl moiety (2-naphthyl-1,3,4,5,6,7,8-d7) β-d-xylopyranoside (XylNap-d7). XylNap-d7-primed GAGs were included to enable relative quantification of linkage region structures using the LC–MS/MS approach. The XylNap- and XylNap-d7-primed GAGs were degraded using heparinase II and III, chondroitinase ABC, chondroitinase AC-I and -II, or chondroitinase B, and these samples were used throughout the study unless otherwise stated. Because the enzymes cleave between HexNAc and UA through an eliminative mechanism resulting in 4,5-unsaturated UA (ΔUA) disaccharide units, discrimination between terminal and internal structures was possible. Disaccharide fingerprinting was performed as described under “Experimental procedures,” and the relative proportions of HS and CS/DS were calculated based on the data (see Equation 2–5), including the relative proportions of IdoUA present in blocks and as alternating or single IdoUA-containing disaccharide units.
The XylNap-primed GAGs from both HCC70 cells and CCD-1095Sk cells were primarily composed of CS/DS (69 and 98%, respectively) (Fig. 3A). The xyloside-primed CS/DS from HCC70 cells corresponded to 11% of IdoUA-containing disaccharide units (7% in blocks and 4% as alternating or single ones) (Fig. 3A). The disaccharides generated after chondroitinase ABC degradation were ΔUA-GalNAc,6S (63%), ΔUA-GalNAc,4S (30%), ΔUA-GalNAc,4S,6S (5%), ΔUA-GalNAc (2%), and ΔUA,2S-GalNAc,6S (<1%) (Fig. 3B). In contrast, the XylNap-primed CS/DS from CCD-1095Sk cells corresponded to 42% of IdoUA-containing disaccharide units (27% in blocks and 15% as alternating or single ones) (Fig. 3A), and the disaccharides generated after chondroitinase ABC degradation were ΔUA-GalNAc,4S (59%), ΔUA-GalNAc,6S (30%), ΔUA,2S-GalNAc,6S (5%), ΔUA,2S-GalNAc,4S (4%), ΔUA-GalNAc (1%), and ΔUA-GalNAc,4S,6S (1%) (Fig. 3C). The combined disaccharide proportions obtained after chondroitinase AC-I and -II and chondroitinase B degradation did not completely correspond to that obtained after the chondroitinase ABC degradation. After chondroitinase AC-I and -II and chondroitinase B degradation, 7% of the XylNap-primed CS/DS from HCC70 cells (ΔUA-GalNAc,4S and ΔUA-GalNAc,4S,6S disaccharide units) and 28% of the XylNap-primed CS/DS from CCD-1095Sk cells (ΔUA-GalNAc,4S, ΔUA,2S-GalNAc,6S, and ΔUA-GalNAc,6S disaccharide units) were lacking from the proportion of disaccharides obtained after chondroitinase ABC degradation (Fig. 3, B and C). This suggests that XylNap-primed CS/DS from both cell lines contains IdoUA distributed as alternating or single disaccharide units, which may be found as oligosaccharides (dp4 or larger) after chondroitinase AC-I and -II and chondroitinase B degradation. The GlcUA and IdoUA proportions and disaccharide composition of XylNap-d7-primed CS/DS was similar to those of the XylNap-primed CS/DS (summarized in Tables S1 and S2).
Figure 3.

Relative proportions of CS/DS and HS and disaccharide composition of XylNap-primed CS/DS from HCC70 cells and CCD-1095Sk cells. A, relative proportions of GlcUA in CS/DS (CS/DSGlcUA), IdoUA in CS/DS distributed as alternating or single IdoUA-containing units (CS/DSIdoUA_Alt/single), IdoUA in CS/DS distributed as blocks (CS/DSIdoUA_ChB), and HS of XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells, respectively. B and C, disaccharide composition of XylNap-primed GAGs from HCC70 cells (B) and CCD-1095Sk cells (C) after depolymerization with chondroitinase ABC (total), chondroitinase AC-I and -II (ChAC), chondroitinase B (ChB), and the remaining disaccharides degraded by chondroitinase ABC but not by chondroitinase AC-I and -II and chondroitinase B (Ch(ABC-AC-B)). The data are the means of two independent experiments. Raw data can be found in Tables S1 and S2.
Taken together, disaccharide fingerprinting of XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells showed that the relative proportions of CS/DS and HS and the specific disaccharide composition of CS/DS differed substantially between the two cell lines. In particular, the lower proportion of IdoUA and disulfated disaccharides in XylNap-primed CS/DS from HCC70 cells suggests the presence of less complex structures than in the XylNap-primed CS/DS from CCD-1095Sk cells.
XylNap-primed CS/DS from HCC70 cells and CCD-1095Sk cells differs not only in disaccharide composition but also in linkage region modifications
To study the structure of the XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells in more detail, the nonlabeled GAG products generated after the different enzymatic degradations were subjected to LC–MS/MS. The LC step was inspired by Kuberan et al. (38) and comprised reversed-phase ion-pairing chromatography on a C18 column with dibutylamine as the ion-pairing agent. Dibutylamine was used to enable glycan separation, circumvent metal ion adduct formation, and improve the ionization (38, 39). The MS/MS setup was developed from our previous work on glycopeptides (10, 40) adapted to GAGs. Because of the highly anionic nature of GAGs, negative-mode was chosen instead of positive mode, and fragmentation was performed using HCD at the normalized collision energy of 80%. At this energy level, high intensity glycosidic and cross-ring fragment ions were generated (Fig. S1).
Commercially available unsaturated CS/DS disaccharide standards showed limited separation on the LC level but distinct MS2 fragmentation patterns, allowing for discrimination between the different variants (Fig. 4, A–F). The fragment ion at m/z 300.04, corresponding to [HexNAc + sulfate]−, dominated for ΔUA-GalNAc,4S, whereas the fragment ion at m/z 282.03, corresponding to [HexNAc + sulfate − H2O]−, dominated for ΔUA-GalNAc,6S (Fig. 4, B and C) (18, 41). In addition, the fragmentation patterns of the disulfated ΔUA,2S-GalNAc,4S and ΔUA,2S-GalNAc,6S disaccharides both displayed a diagnostic ion at m/z 236.97, corresponding to [ΔUA + sulfate − H2O]−, and a relatively high intensity fragment ion at m/z 157.01, corresponding to [ΔUA − H2O]− (Fig. 4, E and F).
Figure 4.

LC–MS/MS of disaccharide standards and chondroitinase ABC-degraded XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells. A, base peak chromatograms of the analyzed disaccharide standards, of which the HCD-MS2 spectra are displayed in B–F; ΔUA-GalNAc,4S (B); ΔUA-GalNAc,6S (C); ΔUA-GalNAc,4S,6S (D); ΔUA,2S-GalNAc,4S (E); and ΔUA,2S-GalNAc,6S (F). G, base peak chromatogram of the chondroitinase ABC-degraded sample from HCC70 cells. H and I, average HCD-MS2 spectra of precursor ions corresponding to monosulfated ΔUA-GalNAc (H) and disulfated ΔUA-GalNAc (I). J, base peak chromatogram of the chondroitinase ABC-degraded sample from CCD-1095Sk cells. K and L, average HCD-MS2 spectra of precursor ions corresponding to monosulfated ΔUA-GalNAc (K) and disulfated ΔUA-GalNAc (L). The m/z peaks at retention times 40.3 and 40.5 min in G and J, respectively, correspond to monosulfated linkage region hexasaccharides (L6S1 in Table 1). The nomenclature of the fragment ion at m/z 198.99 in B was based on that described by Domon and Costello (60). *, ΔUA,2S-GalNAc,6S was analyzed at a later stage than the other standards. A different gradient was used then, resulting in the longer retention time.
For the chondroitinase ABC-degraded XylNap-primed GAGs from both cell lines (Fig. 4, G and J), the base peak chromatograms displayed three major peaks eluting at 18, 20–21, and 40–48 min, corresponding to monosulfated disaccharides, disulfated disaccharides, and linkage region oligosaccharides, respectively. The average fragmentation pattern of the monosulfated disaccharides of the XylNap-primed GAGs generated after chondroitinase ABC degradation showed that they were composed of co-eluting ΔUA-GalNAc,4S and ΔUA-GalNAc,6S disaccharides in the samples from both cell lines (Fig. 4, H and K). In accordance with the results from the disaccharide fingerprinting, ΔUA-GalNAc,6S dominated in the sample from HCC70 cells (Fig. 4H), whereas ΔUA-GalNAc,4S dominated in the sample from CCD-1095Sk cells (Fig. 4K). Furthermore, the disulfated disaccharides were identified as primarily ΔUA-GalNAc,4S,6S in the sample from HCC70 cells (Fig. 4I) and as a mixture of ΔUA,2S-GalNAc,4S and ΔUA,2S-GalNAc,6S in the sample from CCD-1095Sk cells (Fig. 4L). This was also in agreement with the disaccharide fingerprinting data.
The linkage region oligosaccharides in the samples from both cell lines appeared as broad tailoring peaks in the chromatograms (Fig. 4, G and J), from which [M − 2H]2− precursor ions at m/z 567.66, m/z 607.64, and m/z 647.62 corresponded to an unsulfated hexasaccharide (L6; Table 1), a monosulfated hexasaccharide (L6S1; Fig. 5, A and B), and a disulfated hexasaccharide (L6S2; Fig. 5, C and D), respectively. In addition, a precursor ion at m/z 753.19 [2−] was detected, corresponding to a monosulfated hexasaccharide carrying N-acetylneuraminic acid (Neu5Ac; L6S1SA1; Fig. 5, E and F) as indicated by the presence of a diagnostic ion derived from the Neu5Ac residue (m/z 290.09). The position of Neu5Ac was not assigned due to lack of diagnostic ions. The presence of fragment ions at m/z 282.03 and m/z 300.04 for the monosulfated hexasaccharides indicated that the sulfate group was located on the GalNAc residue (Fig. 5, A, B, E, and F). Similarly, one of the sulfate groups of the disulfated hexasaccharide was assigned to the GalNAc residue (Fig. 5, C and D), whereas the second sulfate was assigned to one of the Gal residues. The ratio of m/z 282.03 to m/z 300.04 was roughly constant for the linkage region variants from HCC70 cells; m/z 282.03 was consistently dominating, suggesting that the GalNAc was primarily sulfated at position 6. In contrast, the ratio differed between the linkage region variants from CCD-1095Sk cells going from an equal relative abundance for L6S1 toward a dominating m/z 282.03 for L6S2. This suggests that the linkage regions were sulfated either at position 4 or 6 and that the position differed between the linkage region variants and between HCC70 cells and CCD-1095Sk cells.
Table 1.
Summary of oligosaccharides and linkage regions of XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells identified by LC-MS/MS after degradation with chondroitinase AC-I and -II (AC), or chondroitinase B (B)
| Cell line | Degradation enzyme | Precursor ion mass | Negative charge | Glycan compositiona | |
|---|---|---|---|---|---|
| HCC70 | CCD-1095Sk | AC; B | 458.06 | 2 | dp4S2 |
| HCC70 | CCD-1095Sk | AC | 498.04 | 2 | dp4S3 |
| HCC70 | CCD-1095Sk | AC | 562.61 | 2 | dp4S3b |
| CCD-1095Sk | B | 562.61 | 2 | dp4S3b | |
| HCC70 | CCD-1095Sk | AC; B | 458.06 | 3 | dp6S3 |
| HCC70 | CCD-1095Sk | AC; B | 752.17 | 2 | dp6S3b |
| HCC70 | CCD-1095Sk | AC | 757.22 | 1 | L4 |
| HCC70 | CCD-1095Sk | AC; B | 837.17 | 1 | L4S1 |
| HCC70 | CCD-1095Sk | AC | 523.65 | 2 | L4SA1 |
| HCC70 | CCD-1095Sk | AC | 563.63 | 2 | L4S1SA1 |
| HCC70 | B | 563.63 | 2 | L4S1SA1 | |
| HCC70 | CCD-1095Sk | AC | 567.66 | 2 | L6 |
| HCC70 | CCD-1095Sk | AC; B | 607.64 | 2 | L6S1 |
| HCC70 | CCD-1095Sk | AC; B | 647.61 | 2 | L6S2 |
| CCD-1095Sk | B | 687.59 | 2 | L6S3 | |
| HCC70 | CCD-1095Sk | AC; B | 753.19 | 2 | L6S1SA1 |
| HCC70 | AC | 793.16 | 2 | L6S2SA1 | |
| HCC70 | CCD-1095Sk | AC; B | 797.19 | 2 | L8S1 |
| HCC70 | CCD-1095Sk | B | 837.17 | 2 | L8S2 |
| HCC70 | AC | 837.17 | 2 | L8S2 | |
| CCD-1095Sk | B | 557.78 | 3 | L8S2 | |
| CCD-1095Sk | AC; B | 941.73 | 2 | L8S3* | |
| HCC70 | CCD-1095Sk | B | 982.72 | 2 | L8S2SA1 |
| HCC70 | AC | 982.72 | 2 | L8S2SA1 | |
| CCD-1095Sk | B | 986.75 | 2 | L10S1 | |
| HCC70 | CCD-1095Sk | B | 1026.73 | 2 | L10S2 |
| CCD-1095Sk | B | 684.15 | 3 | L10S2 | |
| CCD-1095Sk | AC | 710.80 | 3 | L10S3 | |
| HCC70 | B | 710.80 | 3 | L10S3 | |
| HCC70 | CCD-1095Sk | AC; B | 1131.28 | 2 | L10S3b |
| CCD-1095Sk | B | 1235.84 | 2 | L10S4b | |
| HCC70 | B | 807.83 | 3 | L10S3SA1 | |
| CCD-1095Sk | B | 1216.28 | 2 | L12S2 | |
| CCD-1095Sk | B | 810.52 | 3 | L12S2 | |
| HCC70 | CCD-1095Sk | B | 837.17 | 3 | L12S3 |
| HCC70 | CCD-1095Sk | B | 1320.84 | 2 | L12S3b |
| CCD-1095Sk | B | 647.62 | 4 | L12S4 | |
| CCD-1095Sk | B | 1425.39 | 2 | L12S4b | |
| HCC70 | B | 906.87 | 3 | L12S4b | |
| HCC70 | B | 1003.90 | 3 | L12S4SA1b | |
| CCD-1095Sk | B | 963.54 | 3 | L14S3 | |
| HCC70 | CCD-1095Sk | B | 1033.24 | 3 | L14S4b |
| HCC70 | CCD-1095Sk | B | 1102.95 | 3 | L14S5b |
| CCD-1095Sk | B | 1063.26 | 3 | L16S2 | |
| CCD-1095Sk | B | 1159.61 | 3 | L16S4b | |
| CCD-1095Sk | B | 1229.32 | 3 | L16S5b | |
| HCC70 | CCD-1095Sk | B | 1299.02 | 3 | L16S6b |
a Glycans were identified using the algorithm described under “Experimental procedures.” Glycans were ordered according to their glycan compositions and characterized by their precursor ion masses and net charges. Dp is degree of polymerization; L is linkage region; n is number of repeats; S is sulfate; SA is Neu5Ac.
b These are dibutylamine adducts.
Figure 5.

Linkage region products generated after chondroitinase ABC degradation of XylNap-primed CS/DS from HCC70 cells and CCD-1095Sk cells. A–F, average HCD-MS2 spectra of the precursor ions at m/z 607.64 [2−] corresponding to a naphthyl-containing hexasaccharide linkage region modified with one sulfate group (A and B; L6S1 in Table 1), at m/z 647.62 [2−] corresponding to one with two sulfate groups (C and D; L6S2 in Table 1), and at m/z 753.19 [2−] corresponding to one carrying one sulfate group and one Neu5Ac residue (E and F; L6S1SA1 in Table 1) from chondroitinase ABC-degraded XylNap-primed GAGs from HCC70 cells (A, C, and E) and CCD-1095Sk cells (B, D, and F). G and H, relative abundance ratios of precursor ions of the observed linkage region variants primed on XylNap (light) and XylNap-d7 (heavy) generated after chondroitinase ABC degradation of 1:1 mixed samples from HCC70 cells (red) and CCD-1095Sk cells (blue). G, HCC70 sample was heavy; H, CCD-1095Sk sample was heavy.
To quantify the relative abundances of the linkage region variants, the chondroitinase ABC-degraded XylNap (light)- and XylNap-d7 (heavy)-primed GAGs from HCC70 cells and CCD-1095Sk cells were mixed in equal amounts and analyzed by LC–MS/MS. Extracted ion chromatograms were generated for the different linkage region variants, L6, L6S1, L6S2, and L6S1SA1, and their relative areas were computed (Fig. 5, G and H). Independently of the order of labeling (light and heavy, respectively), the results indicated that the Neu5Ac modification was considerably more abundant in the XylNap-primed CS/DS from HCC70 cells than in that from CCD-1095Sk cells, whereas the abundances of the other linkage region variants were similar between the two cell lines.
To summarize, the disaccharide data obtained using the LC–MS/MS approach was in agreement with the disaccharide fingerprinting data, showing differences in sulfation pattern between the CS/DS from HCC70 cells and CCD-1095Sk cells. Furthermore, using this approach, the CS/DS linkage regions were discovered to carry not only sulfate groups but also Neu5Ac, in particular those from HCC70 cells.
Amount and distribution of GlcUA and IdoUA differ between the XylNap-primed CS/DS from HCC70 cells and CCD-1095Sk cells
To better understand the differences between the GAGs from the two cell lines, the relative amount of CS/DS and the distribution of GlcUA and IdoUA in CS/DS in the XylNap-primed GAGs from each cell line were investigated using the LC–MS/MS approach. This was performed by comparing the products generated after heparinase II and III, chondroitinase AC-I and -II, and chondroitinase B degradations (displayed as total ion chromatograms (TICs) in Fig. 6, A–F). The LC–MS/MS identification of the linkage region structures and internal saccharides generated after chondroitinase AC-I and -II and chondroitinase B degradation are described below, and the data are summarized in Table 1.
Figure 6.

Total ion chromatograms of enzymatically degraded XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells and MS1 of intact XylNap-primed CS/DS. A–F, degradation products of XylNap-primed GAGs from HCC70 cells (A, C, and E) and CCD-1095Sk cells (B, D, and F) generated after heparinase II and III (A and B), chondroitinase AC-I and -II (C and D), or chondroitinase B (E and F) treatment. G–I, full MS spectra of the peaks in A eluting at 41.81–42.40 min (G), 43.61–44.11 min (H), and 45.99–46.98 min (I), corresponding to intact XylNap-primed CS/DS (L11 to L19) carrying five to seven sulfate groups (S5–S7) and/or one Neu5Ac (SA1). DBA, dibutylamine.
The TICs of the heparinase II- and III-degraded XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells displayed one minor peak eluting at 16–18 min, three minor peaks eluting at 41–47 min, and one dominating peak eluting at 50 min (Fig. 6, A and B). The minor peak at 16–18 min corresponded to HS disaccharides, which were not further explored in this study. The precursor ion masses of the three minor peaks eluting at 41–47 min corresponded to nondegraded, or intact, XylNap-primed CS/DS of L11 to L19 carrying various numbers of sulfate groups (Fig. 6, G–I). The major chromatographic peak at 50 min observed in the samples from both cell lines contained co-eluting intact XylNap-primed GAGs with a chain length above L19 (the previously estimated average chain length is 30 disaccharides (34)), but they were not possible to resolve further at the precursor ion level. Because intact GAGs were present as dominating peaks after the heparinase II and III degradation in both cell lines (Fig. 6, A and B), the majority of the XylNap-primed GAGs was suggested to be CS/DS, which was completely in line with the disaccharide fingerprinting data.
The TICs of the chondroitinase AC-I– and -II–degraded XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells displayed several peaks eluting at 16–42 min, indicating complex mixtures of sulfated disaccharides, IdoUA-containing oligosaccharides, and linkage region structures (Fig. 6, C and D). The virtual absence of intact XylNap-primed GAGs implies that all CS/DS from both cell lines were susceptible to chondroitinase AC-I and -II degradation, and therefore contained GlcUA to some extent. The TICs of the chondroitinase B–degraded XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells (Fig. 6, E and F) displayed several peaks eluting at 16–47 min, indicating the presence of complex degradation products, but also a peak eluting at 50 min, possibly corresponding to CS polysaccharides, or near-intact or intact XylNap-primed CS (compare Fig. 6, E and F with A and B). As indicated by the relative intensities of the peaks corresponding to degradation products and CS polysaccharides or intact CS, respectively, the sample from HCC70 cells, which was composed of only a small proportion of IdoUA, constituted mainly CS polysaccharides or intact CS GAGs. In contrast, the sample from CCD-1095Sk cells, which instead was composed of a considerably higher proportion of IdoUA, constituted mainly CS/DS co-polymeric structures.
To further elucidate the main products of the XylNap-primed CS/DS from both cell lines generated after chondroitinase AC-I and -II and chondroitinase B degradation, the LC-MS chromatograms were displayed as base peak chromatograms (Fig. 7). The major internal saccharide products generated after chondroitinase AC-I and -II and chondroitinase B degradation were identified as di-, tetra-, and hexasaccharides substituted with 1–3 sulfate groups, respectively (dp2S1–dp6S3 in Fig. 7), suggesting that the stretches of consecutive IdoUA- and GlcUA-containing disaccharide units in the CS/DS are short and primarily alternating or two consecutive units. Base peak chromatograms differ from TICs by showing only the most abundant products within a sample; thus, the apparent absence of larger products (>dp6 and >L10, Fig. 7), such as those observed at retention times of 40–50 min in the TICs (Fig. 6, A, B, E, and F), may be due to their greater heterogeneity. The double peak of dp2S1 observed in the sample from CCD-1095Sk cells after chondroitinase B degradation (Fig. 7B) gave identical MS2 fragmentation spectra, where the first peak corresponded to the disaccharide alone, and the second peak corresponded mainly to an adduct composed of two disaccharides and one dibutylamine.
Figure 7.

Base peak chromatograms of chondroitinase AC-I– and -II–degraded and chondroitinase B–degraded XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells. A and B, degradation products of XylNap-primed GAGs from HCC70 cells (A) and CCD-1095Sk cells (B) generated after chondroitinase AC-I and -II (gray) or chondroitinase B (pink) treatment, containing disaccharides (dp2), oligosaccharides (dp4 and dp6), and linkage regions (L4 to L10) carrying one to three sulfate groups (S1–S3) and/or one Neu5Ac (SA1).
The linkage region structures generated after the two enzymatic degradations differed; tetrasaccharide linkage regions were the most abundant products detected after chondroitinase AC-I and -II degradation (L4 in Fig. 7), whereas hexa-, octa-, and decasaccharide linkage regions substituted with 1–3 sulfates, respectively, were the most abundant products detected after chondroitinase B degradation (L6S1, L8S2, and L10S3 in Fig. 7, HCD-MS2 spectra of L6S1 in Fig. 5, A and B). This indicates that IdoUA is present primarily close to the reducing end in the CS/DS GAGs from both cell lines, i.e. 1–3 disaccharides away from the linkage region tetrasaccharide. Interestingly, a distinct chromatographic peak eluting at 35–37 min was observed in the sample from HCC70 cells, but not in the sample from CCD-1095Sk cells. This peak, with a precursor ion of m/z 523.65 [2−], was consistent with a tetrasaccharide linkage region modified with one Neu5Ac residue (L4SA1; Fig. 7A). Thus, the Neu5Ac modification in the linkage region observed in XylNap-primed CS/DS from HCC70 cells after chondroitinase ABC degradation described above was confirmed with the chondroitinase AC-I and -II degradation.
Taken together, the data showed that the CS/DS co-polymeric structures from both HCC70 cells and CCD-1095Sk cells contain primarily alternating or two consecutive GlcUA and IdoUA units. The overall CS/DS from HCC70 cells, which was composed only to a small proportion of IdoUA, contained primarily CS polysaccharides or intact XylNap-primed CS, whereas in the CS/DS from CCD-1095Sk cells, which was composed to a similar degree of IdoUA and GlcUA, the CS/DS co-polymeric structures were dominating. Furthermore, the Neu5Ac residue observed in the linkage region of XylNap-primed CS/DS from HCC70 cells after chondroitinase ABC degradation was observed also after chondroitinase AC-I and -II degradation.
Presence of Neu5Ac in the linkage region impacts the structure of xyloside-primed GAGs
To investigate whether Neu5Ac in the linkage region of xyloside-primed CS/DS influenced the biosynthesis and thereby the structures of xyloside-primed GAGs, XylNapOH-primed GAGs were isolated from culture media of HCC70 cells and CCD-1095Sk cells after treatment with or without the general sialyltransferase inhibitor, 3Fax-peracetyl Neu5Ac (42). In these experiments, XylNapOH was chosen as primer to verify that the Neu5Ac modification is not restricted to GAGs primed on XylNap. Treatment with the inhibitor resulted in the complete absence of Neu5Ac in the linkage region structures of the XylNapOH-primed GAGs from both cell lines, as confirmed by MS (Fig. S2). The most pronounced difference was the reduction in the chain length of XylNapOH-primed GAGs from HCC70 cells upon sialyltransferase inhibition (Fig. 8A). In contrast, the XylNapOH-primed GAGs from CCD-1095Sk cells, where the Neu5Ac modification was much less abundant, did not differ in chain length upon sialyltransferase inhibition (Fig. 8B). Furthermore, a higher proportion of HS was observed in the XylNapOH-primed GAGs from HCC70 cells upon inhibition (Fig. 8C; raw data in Table S3), whereas there was little or no difference in the proportion of HS in the GAGs from CCD-1095Sk cells. Finally, minor differences due to sialyltransferase inhibition were observed in the disaccharide composition of the XylNapOH-primed CS/DS from both HCC70 cells and CCD-1095Sk cells (Fig. 8D; raw data in Table S4). The proportion of ΔUA-GalNAc,4S was lower in the CS/DS lacking the Neu5Ac modification from both cell lines (∼1 and ∼3% when from HCC70 cells and CCD-1095Sk cells, respectively). Furthermore, the proportion of ΔUA-GalNAc,4S,6S was higher in the XylNapOH-primed CS/DS lacking Neu5Ac from HCC70 cells (∼1%), and the proportion of the ΔUA-GalNAc,6S was higher in the XylNapOH-primed CS/DS lacking Neu5Ac from CCD-1095Sk cells (∼3%; Fig. 8D). Taken together, the data indicate that a Neu5Ac modification in the linkage region of CS/DS indeed had an impact on the biosynthesis of XylNapOH-primed GAGs.
Figure 8.

Effects of sialyltransferase inhibition on the structure and composition of XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells. A and B, chromatograms from size-exclusion HPLC on two serially coupled G2000SWxl columns of XylNapOH-primed GAGs from HCC70 cells (A) and CCD-1095Sk cells (B) either untreated (solid line) or treated with the sialyltransferase inhibitor 3Fax-peracetyl Neu5Ac (Neu5Ac inhib; dashed line). F.I., fluorescence intensity. C, proportion of HS in XylNapOH-primed GAGs from HCC70 cells and CCD-1095Sk cells treated with or without sialyltransferase inhibitor. D, relative differences in disaccharide composition (%) after chondroitinase ABC degradation between XylNapOH-primed GAGs from HCC70 cells (red) and CCD-1095Sk cells (blue) treated or untreated with sialyltransferase inhibitor (Δdisaccharide composition = disaccharide compositionNeu5Ac inhib − disaccharide compositionuntreated). The bars are the means ± S.D., where n = 3.
Terminal nonreducing ends of XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells contain cell-specific and novel modifications
To identify terminal NRE structures, the LC–MS/MS data analyses of products obtained after chondroitinase ABC degradation were extended by searching for fragment ions at m/z 282.03 and m/z 300.04, diagnostic for sulfated GalNAc residues (18). This resulted in the identification of several spectra that deviated from the HCD-MS2 spectra of the available disaccharide standards, and manual interpretation of the spectra demonstrated precursor ions and fragmentation patterns that were annotated as terminal mono- and disaccharides. However, because of the lack of appropriate standards, the position of the sulfate on GalNAc could not be assigned. Common for the samples from both HCC70 cells and CCD-1095Sk cells, terminal NREs were identified as a 4,6-disulfated GalNAc residue (Fig. 9A) and a saturated GlcUA bound to a sulfated GalNAc residue (Fig. 9B). Interestingly, in addition to these two previously described NRE products (43–45), a precursor ion at m/z 490.09 [1−] was detected consistent with a methylated saturated GlcUA bound to a sulfated GalNAc residue, as indicated by diagnostic ions at m/z 189.04 and m/z 207.05 (Fig. 9C).
Figure 9.

NREs common to XylNap-primed GAGs from both HCC70 cells and CCD-1095Sk cells. A–C, average HCD-MS2 spectra of the NRE precursor ions at m/z 380.00 [1−] (A), 476.07 [1−] (B), and 490.09 [1−] (C) observed in chondroitinase ABC-degraded XylNap-primed GAGs from both HCC70 cells and CCD-1095Sk cells. D, incorporation of [3H]methyl in XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells expressed as radioactivity in dpm per μg of GAGs. E, average HCD-MS2 spectrum of the NRE precursor ion at m/z 556.01 [1−] observed in chondroitinase AC-I– and -II–degraded XylNap-primed GAGs from HCC70 cells. The nomenclature of the fragment ion at m/z 198.99 was based on that described by Domon and Costello (60).
To our knowledge, methylation of mammalian-derived glycans has not previously been observed, and therefore, verification of the observed methylation was also performed biochemically. XylNap-primed GAGs were isolated from HCC70 cells and CCD-1095Sk cells treated with and without [3H]methyl-methionine, and the amount of radioactivity in the XylNap-primed GAGs was determined. In both cell lines, there was a distinct amount of radioactivity in the XylNap-primed GAGs from the cells treated with [3H]methyl-methionine compared with the GAGs from the untreated cells (Fig. 9D), not only confirming the LC–MS/MS data but also suggesting the general methyl donor S-adenosylmethionine as the donor for this modification.
In addition to the above described NREs, a precursor ion at m/z 556.03 [1−] was detected in the XylNap-primed CS/DS from HCC70 cells after chondroitinase AC-I and -II degradation. Its fragmentation pattern was consistent with a sulfated saturated GlcUA bound to a sulfated GalNAc residue, as indicated by the presence of a diagnostic ion at m/z 254.98 corresponding to [GlcUA + sulfate-H2O]− (Fig. 9E). Such modification has not previously been observed in the NRE of GAGs extended beyond the linkage region tetrasaccharide (46).
Discussion
We have recently shown a cytotoxic effect of CS/DS from a human breast carcinoma cell line, HCC70, primed on XylNap or XylNapOH, and linked the effect to the GAG structure, as the disaccharide composition differed from that of nontoxic CS/DS from a human breast fibroblast cell line, CCD-1095Sk, primed on the same xylosides (34). Here, by using a novel LC–MS/MS approach, we have in detail characterized the linkage region structures, internal saccharides, and NREs of the CS/DS from HCC70 cells and CCD-1095Sk cells primed on XylNap. The aim was to, in one single LC–MS/MS run, identify qualitative differences in the structures of the CS/DS from these cells and, as for traditional disaccharide analysis, exploit the efficiency and cleavage specificities of available GAG degradation enzymes to generate classes of defined products. To acquire the reversed-phase chromatographic separation and improve ionization of the glycan products, dibutylamine was used as a ion-pairing agent (38). This strategy enabled separation of glycan products ranging from dp2S1 and dp2S2 to L19S7 (Figs. 6 and 7). Similar separation capacities using reversed-phase ion-pairing chromatography have been demonstrated for heparosan and heparin, but without performing MS/MS of the glycan products (38, 47). For the MS/MS analysis, we used negative-mode HCD on an Orbitrap spectrometer to accomplish high mass accuracies of precursor and fragment ions, which provided excellent fragmentation spectra (Figs. 4, 5, and 9). Thus, HCD appears suitable not only for detailed characterization of linkage regions on glycopeptides, as shown previously (10, 40), but also for GAGs. The presence of the xyloside aglycon made it straightforward to identify linkage region structures, including cell-specific sulfate and Neu5Ac modifications. Because the MS/MS analyses were run in negative mode, the efficiency of the method to identify single glycan products depends on the presence of negative charges provided by sulfate and carboxyl groups. Quantitative estimation and comparison of the samples were performed with caution, because there are currently no oligosaccharide or linkage region standards available. Optimized LC separation and additional standards are also required for identification of longer structures (>L19) and characterization of oligosaccharides with sulfate groups at various positions and different mixtures of GlcUA and IdoUA (>dp2 and >L6). Nevertheless, a substantial number of CS/DS oligosaccharides were identified using the LC–MS/MS approach, providing information about structural differences between XylNap-primed CS/DS from HCC70 cells and CCD-1095Sk cells.
In this study, we showed that although CS/DS was dominating the GAGs from both HCC70 cells and CCD-1095Sk cells primed on XylNap, the presence and distribution of GlcUA and IdoUA differed markedly between the CS/DS from the two cell lines. In accordance with the disaccharide fingerprinting data, the MS data showed that the proportion of IdoUA was low in the CS/DS from HCC70 cells, whereas GlcUA and IdoUA were in similar proportions in the CS/DS from CCD-1095Sk cells. Based on the base peak chromatograms (Fig. 7), the CS/DS co-polymeric structures from HCC70 cells appeared limited in length, as the dominating products after chondroitinase B degradation were linkage region variants rather than internal saccharides (Fig. 10A). In the samples from CCD-1095Sk cells, however, there was a balance in the relative abundances between the internal saccharide structures and the linkage region structures suggesting that the CS/DS co-polymeric structures may be longer than those from HCC70 cells (Fig. 10). The TICs displayed that in addition to internal saccharides up to dp6 and linkage region structures up to L10, the samples degraded with chondroitinase B, in particular that from HCC70 cells, contained larger products corresponding to CS polysaccharides and/or intact XylNap-primed CS. Based on our previous publication (34), the average length of the CS/DS from the two cell lines was shown to be similar. Thus, the CS/DS from HCC70 cells may contain shorter CS/DS co-polymeric structures extended with CS polysaccharides (Fig. 10). In the CS/DS from both cell lines, the internal saccharides generated after chondroitinase AC-I and -II and chondroitinase B degradation were hexasaccharides or shorter, implying that the distribution of the IdoUA- and GlcUA-containing disaccharide units were primarily different combinations of two consecutive units and alternating units throughout the co-polymeric chain (Fig. 10). IdoUA was primarily detected 1–3 disaccharides away from the linkage region tetrasaccharide in the CS/DS from both cell lines, indicating that IdoUA tends to be formed close to the linkage region in this type of CS/DS. Although less abundant, additional linkage region variants were detected (Table 1), indicating that the CS/DS is heterogeneous. Both the disaccharide fingerprinting and LC–MS/MS provided detailed data on the disaccharide composition; nevertheless, an estimation of the sulfation pattern along the GAG chains requires additional studies. Taken together, future investigations will focus on further details in the characterization of these complex GAGs.
Figure 10.

Proposed distribution of GlcUA and IdoUA in CS/DS from HCC70 cells and CCD-1095Sk cells primed on XylNap. A and B, proposed structures of XylNap-primed CS/DS from HCC70 cells (A) and CCD-1095Sk cells (B). The distribution of IdoUA and GlcUA was based on the MS data of the main products generated after chondroitinase AC-I and -II and chondroitinase B degradation. Two parallel lines indicate that the chain either ends or continues with the following polysaccharide. B, different co-polymeric CS/DS variants illustrate possible GlcUA and IdoUA distributions as repeating patterns rather than established structures. The length of the GAGs was based on previous results (34), corresponding to an average length of 30 disaccharides. Sulfation was not taken into consideration.
The modifications in the linkage region of GAGs have been suggested to influence the GAG structure (48). However, the mechanisms directing the biosynthesis toward HS, CS/DS, or CS formation have not yet been well-established. In accordance with previously published data on proteoglycan-derived CS/DS (10), the linkage region structures observed in XylNap-primed GAGs from HCC70 cells and CCD-1095Sk cells were nonsulfated or sulfated to a varying degree. The sulfate group at the first GalNAc was not limited to position 4 as suggested previously for bikunin and decorin (26, 27), rather the opposite; in general, sulfation at position 6 was dominating. However, differences between HCC70 cells and CCD-1095Sk cells were observed, which may be related to differences in their CS/DS structures. In addition to sulfated linkage region variants, we discovered linkage region variants modified with a Neu5Ac residue. The Neu5Ac modification was clearly observed in the linkage region structures of CS/DS from HCC70 cells after degradation with chondroitinase ABC or AC-I and -II but not after degradation with chondroitinase B, suggesting that the modification may be associated with certain CS structures. Thus, the lower proportion and indicated distribution of GlcUA in the CS/DS from CCD-1095Sk cells may explain the difference in the proportion of Neu5Ac between the CS/DS from the two cell lines. Interestingly, sialyltransferase inhibition influenced the GAG structure in more than one way; the most pronounced impact was on the chain lengths of the GAGs from HCC70 cells, which were shorter in the absence of Neu5Ac. There are a few reports of Neu5Ac modifications in GAGs (10, 11, 49–52); however, whether Neu5Ac in the linkage region could influence the formation of CS or CS/DS remains to be elucidated, as well as identification of the transferase or transferases responsible for the sialylation and the impact of Neu5Ac in the linkage region on the biosynthesis of proteoglycan-derived GAGs.
Although little is known about GAG termination, it is not unreasonable that different terminal NREs may be related to certain structures or chain lengths. In addition to previously reported terminal NREs (43–45), a methylated GlcUA bound to a sulfated GalNAc residue was identified in CS/DS from both cells. Methylation has previously not been observed in glycans from mammalian cells, but based on our present results we speculate that it may serve as a mechanism for termination of GAG elongation. Whether proteoglycan-derived GAGs can be methylated in a corresponding manner remains to be elucidated. However, as xyloside stimulation of cultured cells tends to generate considerably higher amounts of GAGs than originally produced (30, 34, 53), methylation may be more abundant in xyloside-primed GAGs than in proteoglycan-derived GAGs. In line with this, there are examples of modifications, such as α-GalNAc-capping and Neu5Ac-capping, which were initially discovered in xyloside-primed oligosaccharides but later also in proteoglycan-derived GAGs (49, 54). A terminal sulfated GlcUA linked to a sulfated GalNAc residue was observed the CS/DS from HCC70 cells after chondroitinase AC-I and -II degradation. It was not possible to assign the position of the sulfate group on GlcUA; however, human natural killer-1 sulfotransferase (HNK-1ST), the enzyme responsible for the 3-O-sulfation of the truncated linkage region of α-thrombomodulin, may be involved (55). Although terminal 3-O-sulfation of GlcUA in mammals has hitherto only been observed in this context, it is noteworthy that recombinant HNK-1ST has shown activity also toward the terminal GlcUA residues in chondroitin of various lengths.
The cytotoxic effect of CS/DS from HCC70 cells primed on XylNap appeared to be dependent on both GlcUA and IdoUA, because the CS/DS degraded with either chondroitinase AC-I and -II or chondroitinase B had a growth-reducing effect on HCC70 cells, although not as explicit as that of the nondegraded CS/DS. The disaccharides and linkage region structures generated after heparinase II and III and chondroitinase ABC degradation did not have any effect on cell growth, implying that saccharides larger than disaccharides and/or the linkage region structures larger than hexasaccharides are required for a growth-reducing effect. Mere structural complexity did not appear as a prerequisite for the cytotoxic effect, as the CS/DS from HCC70 cells was less complex than that from CCD-1095Sk cells; the CS/DS from HCC70 cells had lower proportions of IdoUA and disulfated disaccharides and a different distribution of IdoUA than the CS/DS from CCD-1095Sk cells. Although the modifications of the linkage regions and terminal NREs themselves may not be responsible for the cytotoxic effect, they may be associated with a specific substructure that is responsible for the effect. In addition to the differences in presence and distribution of GlcUA and IdoUA in the CS/DS from the two cell lines, both the disaccharide fingerprinting and the MS analyses revealed differences in the sulfation patterns of the CS/DS. This may also be crucial for the cytotoxic effect of the CS/DS from HCC70 cells. To obtain a better understanding of the structure–function relationship and to which extent the cytotoxic effect applies to many types of cancer cells, future studies will in parallel with further structural characterization of the GAGs focus on specific interactions between the GAGs and proteins.
In conclusion, we have shown that GlcUA and IdoUA are both required for the cytotoxic effect on HCC70 cells of CS/DS from HCC70 cells primed on XylNap. Furthermore, using a novel LC–MS/MS approach, we have identified possible candidates for the cytotoxic effect, including internal saccharides (tetra- and hexasaccharides) and linkage region structures (octa- to decasaccharides). The XylNap-primed CS/DS from HCC70 cells appeared structurally less complex and diverse than the CS/DS from CCD-1095Sk cells, suggesting that mere structural complexity or diversity was not essential for the cytotoxic effect. Detailed characterization of the modifications of the CS/DS from the two cell lines revealed both shared and cell-specific modifications, some previously observed in proteoglycan-derived GAGs and others previously undescribed. Although the LC–MS/MS approach still needs some optimization as well as bioinformatic development for handling all of the spectral data, it appears as a promising method for structural characterization of GAGs.
Experimental procedures
Xylosides
(2-Naphthyl-1,3,4,5,6,7,8-d7) β-d-xylopyranoside 1,2,3,4-tetra-O-acetyl-β-d-xylopyranose (158 mg, 0.50 mmol) was added to CH2Cl2 (6 ml) under N2 while stirring. 2-Naphthol-1,3,4,5,6,7,8-d7 (50 mg, 0.33 mmol) and Et3N (0.023 ml, 0.17 mmol) were added, followed by BF3·OEt2 (0.082 ml, 0.66 mmol). After 50 min, NaHCO3 (saturated aqueous solution) was added to quench the reaction. The aqueous phase was extracted three times with CH2Cl2, and the combined organic phases were washed with brine and concentrated in vacuo. The residue was purified by chromatography. The crude product (134 mg) was dissolved in NaOMe/MeOH (20 ml, 0.05 m). After 1.5 h, the reaction mixture was neutralized with AcOH and concentrated in vacuo. The crude product was purified by chromatography to give (2-naphthyl-1,3,4,5,6,7,8-d7) β-d-xylopyranoside (56 mg, 0.025 mmol, 60% over two steps). 1H NMR was (CD3OD) as follows: δ 8.55 (s, 0.06H, Ar-H), 7.79 (s, 0.06H, Ar-H), 7.42 (s, 0.44H, Ar-H), 5.03 (d, 1H, J = 7.1 Hz, H-1), 3.97 (dd, 1H, J = 11.4, 5.4 Hz, H-5), 3.64–3.56 (m, 1H, H-4), 3.53–3.41 (m, 3H, H-2, H-3, H-5). 13C NMR (CD3OD) was as follows: δ 156.6, 135.7, 131.1, 111.8, 102.9, 77.7, 74.8, 71.1, 67.0 Optical rotation was: [α]D20 −29 (c 1.0, MeOH). 2-Naphthyl β-d-xylopyranoside and 2-(6-hydroxynaphthyl) β-d-xylopyranoside were synthesized as described previously (32, 33).
Isolation of xyloside-primed GAGs
The procedure has been described previously (34). Briefly, HCC70 cells and CCD-1095Sk cells cultured in T75 flasks to 70% confluence were pre-incubated in serum-free DME/F-12 medium (Sigma) for 24 h and then treated with 100 μm xyloside in fresh medium. After 48 h of incubation, the media were collected and subjected to ion-exchange chromatography and hydrophobic interaction chromatography. The GAGs were precipitated and further purified by size-exclusion HPLC using a Superose 12 HR 10/30 column (GE Healthcare). The xyloside-primed GAGs were collected based on fluorescence, freeze-dried, and quantified using the 1,9-dimethylmethylene blue method (56).
Enzymatic degradation of GAGs for cell growth assay, disaccharide fingerprinting, and LC–MS/MS
Approximately 10 μg of XylNap- and XylNap-d7-primed GAGs were degraded using either chondroitinase ABC (EC 4.2.2.20) (Seikagaku), chondroitinase AC-I and -II (EC 4.2.2.5) (Seikagaku), chondroitinase B (EC 4.2.2.19) (R&D Systems), or heparinase II (no EC number) and heparinase III (EC 4.2.2.8) (from Flavobacterium heparinum overexpressed in Escherichia coli, a gift from Prof. Jian Liu, University of North Carolina). Chondroitinase ABC degradations were performed in 100 μl of 50 mm NH4OAc, pH 8.0, containing 100 milliunits of enzyme at 37 °C for 16 h. Chondroitinase AC-I and -II degradations were performed in 100 μl of 50 mm NH4OAc, pH 8.0, containing 10 milliunits of each enzyme at 37 °C for 16 h. Chondroitinase B degradations were performed in 100 μl of 50 mm NH4OAc, 4 mm CaCl2, pH 7.4, containing 50 milliunits of enzyme at 37 °C for 16 h. Heparinase II and III degradations were performed in 100 μl of 50 mm NH4OAc, 4 mm CaCl2, pH 7.4, containing 50 milliunits of each enzyme at 37 °C for 16 h. After degradation, the samples were boiled for 10 min and centrifuged at 10,000 × g before the supernatants were dried by centrifugal evaporation. Commercially available CS A, CS B, and heparin (all from Sigma) were included as controls.
Cell growth determination using the crystal violet method
The procedure has been described previously (34, 57). Briefly, HCC70 cells were dissociated and seeded in 96-well plates at plating densities of 5000 cells/well. After 24 h of plating, the cells were allowed to grow in DME/F-12 medium supplemented with increasing concentrations of XylNap-primed GAGs degraded using either heparinase II and III, heparinase II and III and chondroitinase ABC, heparinase II and III and chondroitinase AC-I and -II, or heparinase II and III and chondroitinase B. The completion of each degradation was verified by disaccharide fingerprinting. Untreated cells and blanks only containing medium were included as well as a plate containing the number of cells at the time for the initiation of the experiment (day 0). After 96 h of incubation, the cell density was measured using the crystal violet method, and the relative cell number (in % of untreated (UT)) was calculated according to Equation 1,
| (1) |
where the Abs is absorbance (in absorbance units). The inhibitory concentrations of the GAGs are expressed as IC50 values and calculated after Boltzmann sigmoidal fitting (applied when effect was observed) or linear fitting (applied when no effect was observed) to the data points.
Disaccharide fingerprinting
The procedure has been described previously (34, 58). The disaccharides generated as described above were labeled with 2-aminoacridone and separated on an XBridge BEH Shield RP18 (2.1 × 100 mm, 2.5 μm) column (Waters). Identification and quantification were performed using disaccharide standards (Iduron) subjected to the corresponding labeling and separation. HS and CS/DS proportions were calculated using Equations 2–5, also described in Ref. 34)
| (2) |
| (3) |
| (4) |
| (5) |
where m is mass (in nanograms) calculated based on the disaccharide data after degradation with the indicated enzymes, and CS/DSIdoUA_ChB and CS/DSIdoUA_Alt/single correspond to the proportion of IdoUA in blocks and IdoUA as alternating or single disaccharide units, respectively. Equation 3 and 4 were generated based on the cleavage sites of chondroitinase B and chondroitinase AC-I and -II (37). All equations are based on the assumption that the degradations have gone to completion.
LC–MS/MS analysis
LC–MS/MS setup, inspired by Kuberan et al. (38), consisted of the Dionex Ultimate 3000 RS chromatography system (ThermoFisher Scientific, Germering, Germany) equipped with the in-house–made flow split and coupled to an Orbitrap Elite mass spectrometer (ThermoFisher Scientific, Bremen, Germany). Glacial AcOH (for analysis) and LC-MS grade MeOH were purchased from Merck (Darmstadt, Germany), HPLC-grade di-n-butylamine was purchased from Acros Organics (Geel, Belgium). Water was deionized.
Dried samples were reconstituted in 30 μl of H2O. Two to six μl of the sample was injected onto the Acquity BEH C18 column (300-Å pore size, 1.7-μm particle size, 300 μm × 150 mm column dimensions) purchased from Waters. Solvent A was 5 mm di-n-butylamine and 8 mm AcOH in H2O; solvent B was 5 mm di-n-butylamine and 8 mm AcOH in 70% MEOH. All methods used stepwise isocratic elution at ∼2 μl/min flow on column. To separate the whole range of the digested and intact XylNap-primed GAGs, the products were eluted at 100% A for 13 min, at 30% B for 15 min, then at 60% B for 10 min, and at 100% B for 19 min.
The electrospray source was operated in negative ionization mode at 3.5 kV. Precursor ion mass spectra were recorded at 60–000 resolution in the m/z range 350–2000. The 10 most intense precursor ions were selected with an isolation window of 5.0 Thomsons without a dynamic exclusion, fragmented using HCD at the normalized collision energy of 80%, and the MS2 spectra were recorded at a resolution of 15,000 with the first mass 100; precursors with unassigned charge states were rejected. The MS data for the disaccharides and chondroitinase ABC-degraded samples have been deposited to the ProteomeXchange Consortium via the PRIDE (59) partner repository with the dataset identifier PXD009652.
The following combinatorial algorithm was introduced into SweetNET (24) to estimate the glycan composition of deconvoluted single charged MS1 peaks.
1) For each peak, the running mass, mrunning, was set to the following two values: (a) for linkage regions: mrunning =mXylNap/XylNapOH + (mHex × 2) + mUA; (b) for internal fragments: mrunning = mHexNAc + mUA, where mXylNap/XylNapOH is the mass of one XylNap or XylNapOH residue; mHex is the mass of one hexose residue; mUA is the mass of one UA residue; and mHexNAc is the mass of one HexNAc residue.
The following steps were performed for both mrunning values.
2) The Δmass (Δm) was calculated as the difference between total precursor mass, mtotal and mrunning: Δm = mtotal − mrunning. 3) Given that Δm > 0, the mass of the following components were added to mrunning one by one in a combinatorial fashion using nested loops: sulfate, hexose, UA, HexNAc, Neu5Ac, adducts. 4) After each step in the loop, the values of mrunning and subsequently Δm were updated to accommodate the newly added residues. 5) New compositions were written only when running mass became equal to the total precursor mass within the tolerance value of 0.01 Da.
The list of new compositions was manually scrutinized to output only the logically possible combinations.
Sialyltransferase inhibition
HCC70 cells and CCD-1095Sk cells cultured in T75 flasks to 60% confluence were treated with or without 100 μm of the sialyltransferase inhibitor, 3Fax-peracetyl Neu5Ac (Millipore), in serum-free DME/F-12 medium for 48 h before changing to fresh medium supplemented with 100 μm XylNapOH and 100 μm 3Fax-peracetyl Neu5Ac and incubating for another 48 h. The GAGs were isolated and purified as described above. GAG chain length determination was performed by size-exclusion HPLC using a TSKgel G2000SWxl column (Tosoh) under isocratic conditions (20% MeCN, 80% 60 mm NH4OAc, pH 5.6). The XylNapOH-primed GAGs were detected by fluorescence.
Incorporation of [3H]methyl-methionine
HCC70 cells and CCD-1095Sk cells were cultured and pre-incubated as described above, and then treated with or without [3H]methyl-methionine (PerkinElmer Life Sciences, 10 μCi/ml medium) and 100 μm XylNap in fresh medium for 48 h. The GAGs were isolated and purified as described above. Radioactivity was determined using a liquid scintillation counter (PerkinElmer Life Sciences).
Author contributions
A. P., A. G. T., K. M., and G. L. conceptualization; A. P., A. G. T., E. V., W. N., D. W., F. N., U. E., K. M., J. N., and G. L. data curation; A. P., A. G. T., E. V., W. N., F. N., J. N., and G. L. formal analysis; A. P., U. E., K. M., and G. L. funding acquisition; A. P., A. G. T., E. V., W. N., F. N., J. N., and G. L. investigation; A. P., A. G. T., and J. N. visualization; A. P., A. G. T., E. V., W. N., F. N., J. N., and G. L. methodology; A. P., A. G. T., and G. L. writing-original draft; A. P., A. G. T., and G. L. project administration; A. P., A. G. T., E. V., W. N., D. W., F. N., U. E., K. M., J. N., and G. L. writing-review and editing; A. G. T. and W. N. software; K. M. and G. L. resources; J. N. supervision.
Supplementary Material
Acknowledgments
We thank Carina Sihlbom and Anders Malmström for valuable discussions and the Proteomics Core Facility at the University of Gothenburg for analyzing the samples.
This work was supported by Governmental grants to the Sahlgrenska University Hospital, Gothenburg, the Fund of the Hedda and John Forssman Foundation, Lund University, the Medical Faculty at Lund University, Swedish Cancer Society Grant 17 0255 (to K. M.), Swedish Research Council Grants K2014-68X-08266-27-4 (to G. L.) and K2015-66X-22693-01-4 (to K. M.), and the Åhlen Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
The mass spectrometric raw data and spectral libraries associated with this manuscript are available from ProteomeXchange with the accession number PXD009652.
 This article contains Tables S1–S4 and Figs. S1 and S2.
This article contains Tables S1–S4 and Figs. S1 and S2.
- GAG
- glycosaminoglycan
- CS/DS
- chondroitin sulfate/dermatan sulfate
- HS
- heparan sulfate
- UA
- hexuronic acid
- GlcUA
- glucuronic acid
- IdoUA
- iduronic acid
- dp
- degree of polymerization
- XylNap
- 2-naphthyl β-d-xylopyranoside
- XylNapOH
- 2-(6-hydroxynaphthyl) β-d-xylopyranoside
- HCD
- higher-energy collision dissociation
- NRE
- nonreducing end
- XylNap-d7
- (2-naphthyl-1,3,4,5,6,7,8-d7) β-d-xylopyranoside
- Neu5Ac
- N-acetylneuraminic acid
- TIC
- total ion chromatogram
- DME
- Dulbecco's modified Eagle's.
References
- 1. Mikami T., and Kitagawa H. (2013) Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta 1830, 4719–4733 10.1016/j.bbagen.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 2. Malmström A., Bartolini B., Thelin M. A., Pacheco B., and Maccarana M. (2012) Iduronic acid in chondroitin/dermatan sulfate: biosynthesis and biological function. J. Histochem. Cytochem. 60, 916–925 10.1369/0022155412459857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bishop J. R., Schuksz M., and Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 10.1038/nature05817 [DOI] [PubMed] [Google Scholar]
- 4. Afratis N., Gialeli C., Nikitovic D., Tsegenidis T., Karousou E., Theocharis A. D., Pavão M. S., Tzanakakis G. N., and Karamanos N. K. (2012) Glycosaminoglycans: key players in cancer cell biology and treatment. FEBS J. 279, 1177–1197 10.1111/j.1742-4658.2012.08529.x [DOI] [PubMed] [Google Scholar]
- 5. Kamhi E., Joo E. J., Dordick J. S., and Linhardt R. J. (2013) Glycosaminoglycans in infectious disease. Biol. Rev. 88, 928–943 10.1111/brv.12034 [DOI] [PubMed] [Google Scholar]
- 6. Sugahara K., and Kitagawa H. (2002) Heparin and heparan sulfate biosynthesis. IUBMB Life 54, 163–175 10.1080/15216540214928 [DOI] [PubMed] [Google Scholar]
- 7. Sugahara K., Masuda M., Harada T., Yamashina I., de Waard P., and Vliegenthart J. F. (1991) Structural studies on sulfated oligosaccharides derived from the carbohydrate-protein linkage region of chondroitin sulfate proteoglycans of whale cartilage. Eur. J. Biochem. 202, 805–811 10.1111/j.1432-1033.1991.tb16436.x [DOI] [PubMed] [Google Scholar]
- 8. Kitagawa H., Tsutsumi K., Ikegami-Kuzuhara A., Nadanaka S., Goto F., Ogawa T., and Sugahara K. (2008) Sulfation of the galactose residues in the glycosaminoglycan-protein linkage region by recombinant human chondroitin 6-O-sulfotransferase-1. J. Biol. Chem. 283, 27438–27443 10.1074/jbc.M803279200 [DOI] [PubMed] [Google Scholar]
- 9. Koike T., Izumikawa T., Sato B., and Kitagawa H. (2014) Identification of phosphatase that dephosphorylates xylose in the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 289, 6695–6708 10.1074/jbc.M113.520536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gomez Toledo A., Nilsson J., Noborn F., Sihlbom C., and Larson G. (2015) Positive mode LC–MS/MS analysis of chondroitin sulfate modified glycopeptides derived from light and heavy chains of the human inter-α-trypsin inhibitor complex. Mol. Cell. Proteomics 14, 3118–3131 10.1074/mcp.M115.051136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wen J., Xiao J., Rahdar M., Choudhury B. P., Cui J., Taylor G. S., Esko J. D., and Dixon J. E. (2014) Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 111, 15723–15728 10.1073/pnas.1417993111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turnbull J., Powell A., and Guimond S. (2001) Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 11, 75–82 10.1016/S0962-8924(00)01897-3 [DOI] [PubMed] [Google Scholar]
- 13. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 10.1146/annurev-biochem-060713-035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Volpi N., Galeotti F., Yang B., and Linhardt R. J. (2014) Analysis of glycosaminoglycan-derived, precolumn, 2-aminoacridone–labeled disaccharides with LC-fluorescence and LC-MS detection. Nat. Protoc. 9, 541–558 10.1038/nprot.2014.026 [DOI] [PubMed] [Google Scholar]
- 15. Li G., Li L., Tian F., Zhang L., Xue C., and Linhardt R. J. (2015) Glycosaminoglycanomics of cultured cells using a rapid and sensitive LC–MS/MS approach. ACS Chem. Biol. 10, 1303–1310 10.1021/acschembio.5b00011 [DOI] [PubMed] [Google Scholar]
- 16. Lawrence R., Olson S. K., Steele R. E., Wang L., Warrior R., Cummings R. D., and Esko J. D. (2008) Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 283, 33674–33684 10.1074/jbc.M804288200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Skidmore M. A., Guimond S. E., Dumax-Vorzet A. F., Yates E. A., and Turnbull J. E. (2010) Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using UV or high-sensitivity fluorescence (BODIPY) detection. Nat. Protoc. 5, 1983–1992 10.1038/nprot.2010.145 [DOI] [PubMed] [Google Scholar]
- 18. Zaia J., and Costello C. E. (2001) Compositional analysis of glycosaminoglycans by electrospray mass spectrometry. Anal. Chem. 73, 233–239 10.1021/ac000777a [DOI] [PubMed] [Google Scholar]
- 19. Wolff J. J., Laremore T. N., Busch A. M., Linhardt R. J., and Amster I. J. (2008) Electron detachment dissociation of dermatan sulfate oligosaccharides. J. Am. Soc. Mass Spectrom. 19, 294–304 10.1016/j.jasms.2007.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zaia J., and Costello C. E. (2003) Tandem mass spectrometry of sulfated heparin-like glycosaminoglycan oligosaccharides. Anal. Chem. 75, 2445–2455 10.1021/ac0263418 [DOI] [PubMed] [Google Scholar]
- 21. Miller R. L., Guimond S. E., Prescott M., Turnbull J. E., and Karlsson N. (2017) Versatile separation and analysis of heparan sulfate oligosaccharides using graphitised carbon liquid chromatography and electrospray mass spectrometry. Anal. Chem. 89, 8942–8950 10.1021/acs.analchem.7b01417 [DOI] [PubMed] [Google Scholar]
- 22. Miller R. L., Wei W., Schwörer R., Zubkova O. V., Tyler P. C., Turnbull J. E., and Leary J. A. (2015) Composition, sequencing and ion mobility mass spectrometry of heparan sulfate-like octasaccharide isomers differing in glucuronic and iduronic acid content. Eur. J. Mass Spectrom. 21, 245–254 10.1255/ejms.1337 [DOI] [PubMed] [Google Scholar]
- 23. Nilsson J., Noborn F., Gomez Toledo A., Nasir W., Sihlbom C., and Larson G. (2017) Characterization of glycan structures of chondroitin sulfate-glycopeptides facilitated by sodium ion-pairing and positive mode LC–MS/MS. J. Am. Soc. Mass Spectrom. 28, 229–241 10.1007/s13361-016-1539-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nasir W., Toledo A. G., Noborn F., Nilsson J., Wang M., Bandeira N., and Larson G. (2016) SweetNET: a bioinformatics workflow for glycopeptide MS/MS spectral analysis. J. Proteome Res. 15, 2826–2840 10.1021/acs.jproteome.6b00417 [DOI] [PubMed] [Google Scholar]
- 25. Palaniappan K. K., and Bertozzi C. R. (2016) Chemical glycoproteomics. Chem. Rev. 116, 14277–14306 10.1021/acs.chemrev.6b00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao X., Yang B., Solakyildirim K., Joo E. J., Toida T., Higashi K., Linhardt R. J., and Li L. (2013) Sequence analysis and domain motifs in the porcine skin decorin glycosaminoglycan chain. J. Biol. Chem. 288, 9226–9237 10.1074/jbc.M112.437236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ly M., Leach F. E. 3rd., Laremore T. N., Toida T., Amster I. J., and Linhardt R. J. (2011) The proteoglycan bikunin has a defined sequence. Nat. Chem. Biol. 7, 827–833 10.1038/nchembio.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu Y., Duan J., Leach F. E. 3rd., Toida T., Higashi K., Zhang H., Zhang F., Amster I. J., and Linhardt R. J. (2017) Sequencing the dermatan sulfate chain of decorin. J. Am. Chem. Soc. 139, 16986–16995 10.1021/jacs.7b10164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okayama M., Kimata K., and Suzuki S. (1973) The influence of p-nitrophenyl β-d-xyloside on the synthesis of proteochondroitin sulfate by slices of embryonic chick cartilage. J. Biochem. 74, 1069–1073 [PubMed] [Google Scholar]
- 30. Schwartz N. B., Galligani L., Ho P.-L., and Dorfman A. (1974) Stimulation of synthesis of free chondroitin sulfate chains by β-d-xylosides in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 71, 4047–4051 10.1073/pnas.71.10.4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cöster L., Hernnäs J., and Malmström A. (1991) Biosynthesis of dermatan sulphate proteoglycans. The effect of β-d-xyloside addition on the polymer-modification process in fibroblast cultures. Biochem. J. 276, 533–539 10.1042/bj2760533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mani K., Havsmark B., Persson S., Kaneda Y., Yamamoto H., Sakurai K., Ashikari S., Habuchi H., Suzuki S., Kimata K., Malmström A., Westergren-Thorsson G., and Fransson L. Å. (1998) Heparan/chondroitin/dermatan sulfate primer 2-(6-hydroxynaphthyl)-O-β-d-xylopyranoside preferentially inhibits growth of transformed cells. Cancer Res. 58, 1099–1104 [PubMed] [Google Scholar]
- 33. Fritz T. A., Lugemwa F. N., Sarkar A. K., and Esko J. D. (1994) Biosynthesis of heparan sulfate on β-d-xylosides depends on aglycone structure. J. Biol. Chem. 269, 300–307 [PubMed] [Google Scholar]
- 34. Persson A., Tykesson E., Westergren-Thorsson G., Malmström A., Ellervik U., and Mani K. (2016) Xyloside-primed chondroitin sulfate/dermatan sulfate from breast carcinoma cells with a defined disaccharide composition has cytotoxic effects in vitro. J. Biol. Chem. 291, 14871–14882 10.1074/jbc.M116.716829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chua J. S., and Kuberan B. (2017) Synthetic xylosides: probing the glycosaminoglycan biosynthetic machinery for biomedical applications. Acc. Chem. Res. 50, 2693–2705 10.1021/acs.accounts.7b00289 [DOI] [PubMed] [Google Scholar]
- 36. Linhardt R. J., Turnbull J. E., Wang H. M., Loganathan D., and Gallagher J. T. (1990) Examination of the substrate specificity of heparin and heparan sulfate lyases. Biochemistry 29, 2611–2617 10.1021/bi00462a026 [DOI] [PubMed] [Google Scholar]
- 37. Linhardt R. J., Avci F. Y., Toida T., Kim Y. S., and Cygler M. (2006) CS lyases: structure, activity, and applications in analysis and the treatment of diseases. Adv. Pharmacol. 53, 187–215 10.1016/S1054-3589(05)53009-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuberan B., Lech M., Zhang L., Wu Z. L., Beeler D. L., and Rosenberg R. D. (2002) Analysis of heparan sulfate oligosaccharides with ion pair-reverse phase capillary high performance liquid chromatography-microelectrospray ionization time-of-flight mass spectrometry. J. Am. Chem. Soc. 124, 8707–8718 10.1021/ja0178867 [DOI] [PubMed] [Google Scholar]
- 39. Lawrence R., Kuberan B., Lech M., Beeler D. L., and Rosenberg R. D. (2004) Mapping critical biological motifs and biosynthetic pathways of heparan sulfate. Glycobiology 14, 467–479 10.1093/glycob/cwh057 [DOI] [PubMed] [Google Scholar]
- 40. Noborn F., Gomez Toledo A., Sihlbom C., Lengqvist J., Fries E., Kjellén L., Nilsson J., and Larson G. (2015) Identification of chondroitin sulfate linkage region glycopeptides reveals prohormones as a novel class of proteoglycans. Mol. Cell. Proteomics 14, 41–49 10.1074/mcp.M114.043703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Desaire H., and Leary J. A. (2000) Detection and quantification of the sulfated disaccharides in chondroitin sulfate by electrospray tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 11, 916–920 10.1016/S1044-0305(00)00168-9 [DOI] [PubMed] [Google Scholar]
- 42. Rillahan C. D., Antonopoulos A., Lefort C. T., Sonon R., Azadi P., Ley K., Dell A., Haslam S. M., and Paulson J. C. (2012) Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 8, 661–668 10.1038/nchembio.999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plaas A. H., Wong-Palms S., Roughley P. J., Midura R. J., and Hascall V. C. (1997) Chemical and immunological assay of the nonreducing terminal residues of chondroitin sulfate from human aggrecan. J. Biol. Chem. 272, 20603–20610 10.1074/jbc.272.33.20603 [DOI] [PubMed] [Google Scholar]
- 44. Otsu K., Inoue H., Tsuzuki Y., Yonekura H., Nakanishi Y., and Suzuki S. (1985) A distinct terminal structure in newly synthesized chondroitin sulphate chains. Biochem. J. 227, 37–48 10.1042/bj2270037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laremore T. N., Ly M., Zhang Z., Solakyildirim K., McCallum S. A., Owens R. T., and Linhardt R. J. (2010) Domain structure elucidation of human decorin glycosaminoglycans. Biochem. J. 431, 199–205 10.1042/BJ20100788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakagawa N., Izumikawa T., Kitagawa H., and Oka S. (2011) Sulfation of glucuronic acid in the linkage tetrasaccharide by HNK-1 sulfotransferase is an inhibitory signal for the expression of a chondroitin sulfate chain on thrombomodulin. Biochem. Biophys. Res. Commun. 415, 109–113 10.1016/j.bbrc.2011.10.023 [DOI] [PubMed] [Google Scholar]
- 47. Thanawiroon C., Rice K. G., Toida T., and Linhardt R. J. (2004) Liquid chromatography/mass spectrometry sequencing approach for highly sulfated heparin-derived oligosaccharides. J. Biol. Chem. 279, 2608–2615 10.1074/jbc.M304772200 [DOI] [PubMed] [Google Scholar]
- 48. Prydz K. (2015) Determinants of glycosaminoglycan (GAG) structure. Biomolecules 5, 2003–2022 10.3390/biom5032003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Freeze H. H., Sampath D., and Varki A. (1993) α- and β-xylosides alter glycolipid synthesis in human melanoma and Chinese hamster ovary cells. J. Biol. Chem. 268, 1618–1627 [PubMed] [Google Scholar]
- 50. Dickenson J. M., Huckerby T. N., and Nieduszynski I. A. (1990) Two linkage-region fragments isolated from skeletal keratan sulphate contain a sulphated N-acetylglucosamine residue. Biochem. J. 269, 55–59 10.1042/bj2690055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hopwood J. J., and Robinson H. C. (1974) The alkali-labile linkage between keratan sulphate and protein. Biochem. J. 141, 57–69 10.1042/bj1410057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Otsuka Y., and Sato T. (2018) Comparative quantification method for glycosylated products elongated on β-xylosides using a stable isotope-labeled saccharide primer. Anal. Chem. 90, 5201–5208 10.1021/acs.analchem.7b05438 [DOI] [PubMed] [Google Scholar]
- 53. Vassal-Stermann E., Duranton A., Black A. F., Azadiguian G., Demaude J., Lortat-Jacob H., Breton L., and Vivès R. R. (2012) A new C-xyloside induces modifications of GAG expression, structure and functional properties. PLoS One 7, e47933 10.1371/journal.pone.0047933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salimath P. V., Spiro R. C., and Freeze H. H. (1995) Identification of a novel glycosaminoglycan core-like molecule. II. α-GalNAc-capped xylosides can be made by many cell types. J. Biol. Chem. 270, 9164–9168 10.1074/jbc.270.16.9164 [DOI] [PubMed] [Google Scholar]
- 55. Hashiguchi T., Mizumoto S., Nishimura Y., Tamura J., Yamada S., Sugahara K. (2011) Involvement of human natural killer-1 (HNK-1) sulfotransferase in the biosynthesis of the GlcUA(3-O-sulfate)-Gal-Gal-Xyl tetrasaccharide found in α-thrombomodulin from human urine. J. Biol. Chem. 286, 33003–33011 10.1074/jbc.M111.279174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Farndale R. W., Sayers C. A., and Barrett A. J. (1982) A direct spectrophotometric microassay for sulfated glycosaminoglycans in cartilage cultures. Connect. Tissue Res. 9, 247–248 10.3109/03008208209160269 [DOI] [PubMed] [Google Scholar]
- 57. Westergren-Thorsson G., Onnervik P.-O., Fransson L.-A., Malmström A. (1991) Proliferation of cultured fibroblasts is inhibited by l-iduronate-containing glycosaminoglycans. J. Cell. Physiol. 147, 523–530 10.1002/jcp.1041470319 [DOI] [PubMed] [Google Scholar]
- 58. Stachtea X. N., Tykesson E., van Kuppevelt T. H., Feinstein R., Malmström A., Reijmers R. M., and Maccarana M. (2015) Dermatan sulfate-free mice display embryological defects and are neonatal lethal despite normal lymphoid and non-lymphoid organogenesis. PLoS One 10, e0140279 10.1371/journal.pone.0140279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Domon B., and Costello C. E. (1988) A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 5, 397–409 10.1007/BF01049915 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.