

Abstract
α-Galactosidases (EC 3.2.1.22) are retaining glycosidases that cleave terminal α-linked galactose residues from glycoconjugate substrates. α-Galactosidases take part in the turnover of cell wall–associated galactomannans in plants and in the lysosomal degradation of glycosphingolipids in animals. Deficiency of human α-galactosidase A (α-Gal A) causes Fabry disease (FD), a heritable, X-linked lysosomal storage disorder, characterized by accumulation of globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3). Current management of FD involves enzyme-replacement therapy (ERT). An activity-based probe (ABP) covalently labeling the catalytic nucleophile of α-Gal A has been previously designed to study α-galactosidases for use in FD therapy. Here, we report that this ABP labels proteins in Nicotiana benthamiana leaf extracts, enabling the identification and biochemical characterization of an N. benthamiana α-galactosidase we name here A1.1 (gene accession ID GJZM-1660). The transiently overexpressed and purified enzyme was a monomer lacking N-glycans and was active toward 4-methylumbelliferyl-α-d-galactopyranoside substrate (Km = 0.17 mm) over a broad pH range. A1.1 structural analysis by X-ray crystallography revealed marked similarities with human α-Gal A, even including A1.1's ability to hydrolyze Gb3 and lyso-Gb3, which are not endogenous in plants. Of note, A1.1 uptake into FD fibroblasts reduced the elevated lyso-Gb3 levels in these cells, consistent with A1.1 delivery to lysosomes as revealed by confocal microscopy. The ease of production and the features of A1.1, such as stability over a broad pH range, combined with its capacity to degrade glycosphingolipid substrates, warrant further examination of its value as a potential therapeutic agent for ERT-based FD management.
Keywords: glycoside hydrolase, plant, human, enzyme inhibitor, glycolipid, fluorescence, protein expression, enzyme purification, enzyme structure, α-galactosidase, enzyme-replacement therapy, Fabry disease, glycosphingolipids, Nicotiana benthamiana, homologue, lysosomal storage disorder, recombinant enzyme, glycosphingolipid
Introduction
α-Galactosidases (EC 3.2.1.22) occur widely in plants, animals, and microorganisms. Based on primary and secondary structures, they are classified into the three glycoside hydrolase families 4, 27, and 36. Family 27 contains mainly eukaryotic α-galactosidases removing terminal α-galactosyl moieties from glycoconjugates (1–3). Plant α-galactosidases are so far known to participate in the catabolism of galactosyl-sucrose oligosaccharides, raffinose family of oligosaccharides, and cell wall galactomannans primarily during germination of seeds (1, 4–7). In man, a single α-galactosidase occurs in lysosomes, α-galactosidase A (α-Gal A),2 and is primarily responsible for the metabolism of glycosphingolipids (8, 9).
A well-established classification of the plant enzymes is based on their pH optimum for enzymatic activity: acidic ones with broad pH optima from 4.5 to 6.5, and alkaline ones with pH optimum between 7.0 and 7.5 (1, 10). These two classes of plant α-galactosidases might differ in localization. The acidic α-galactosidases most likely locate inside the vacuoles and apoplast, whereas alkaline α-galactosidases act in the cytoplasm with a neutral pH, where they might catalyze removal of terminal galactose residues of substrates (1, 10). One of the first plant α-galactosidases to be cloned and biochemically characterized was an enzyme from coffee beans (11). It occurs as two different isoforms with molecular masses of 28 and 36.5 kDa showing slightly different pH optima (pH 5.3 and 6.3) and isoelectric points (12, 13). Similar heterogeneity of α-galactosidases exists in other plant species, for example in rice (6). Interestingly, the acidic plant α-galactosidases are most homologous to the human enzyme (1). For example, rice α-galactosidase shows 37% homology in amino acid sequence to human α-Gal A (5).
The human α-Gal A enzyme is able to cleave α-1,4-linked galactosyl moieties from glycosphingolipids such as globotriaosylceramide (Gb3; ceramide trihexoside) and galabiosylceramide and from blood groups B, B1, and P1 antigens. The hydrolase is encoded by the GLA gene (gene ID, 2717) at locus Xq22 (14). Its mature form lacking the signal sequence contains 398 amino acids with three N-glycans, naturally forming homodimers (14–17). Mannose 6-phosphate moieties (Man-6-P) on the three N-linked glycans of α-Gal A mediate the transport of newly formed enzyme to lysosomes by Man-6-P receptors (MPRs). Alternative sorting via the mannose receptor pathway was suggested by Sakuraba et al. (18) and Shen et al. (19).
Dysfunction or absence of α-Gal A leads to Fabry disease (FD), an X-linked lysosomal disorder characterized by accumulation of glycosphingolipids with terminal galactosyl moieties in tissues and body fluids of FD patients (9, 20). The classic manifestation of FD in males involves development of acroparasthesias, corneal clouding, and neuronopathic pain followed by later-onset renal and cardiac disease and strokes. Female carriers may develop an attenuated disease. In so-called atypical manifestations of FD, associated with missense mutations in α-Gal A, pathology is to a single organ like heart or kidney and only develops late in life (9).
Enzyme-replacement therapy (ERT) with an MPR-targeted recombinant enzyme is used to treat FD (15, 21). Two therapeutic enzymes (agalsidase α, Replagal®, and agalsidase β, Fabrazyme®) produced in mammalian cells are in use, and a plant-produced enzyme is being developed (18, 22). The efficacy of present ERT interventions is considered to be poor (23). Unfortunately, most male FD patients lack the α-Gal A protein and consequently develop neutralizing antibodies against the therapeutic recombinant enzymes that might contribute to the noted poor responses to current treatments (24). Indeed, it was recently reported that compared with agalsidase inhibition-negative men, agalsidase inhibition-positive men showed greater left ventricular mass and substantially lower renal function (25). Additionally, these patients presented more often with symptoms such as diarrhea, fatigue, and neuropathic pain. Despite the lack of residual α-Gal A, even in classic male FD patients, Gb3 accumulation is leveling with age. In Fabry patients, accumulating Gb3 is alternatively metabolized in lysosomes by acid ceramidase to globotriaosylsphingosine (lyso-Gb3) (26). In plasma of male FD patients and mice, lyso-Gb3 is several hundredfold elevated, an abnormality that can be exploited for diagnosis and monitoring of disease progression and therapeutic correction (27–30). Excessive lyso-Gb3 is toxic for nociceptive neurons and podocytes, which might explain the development of neuronopathic pain and renal failure in FD patients (31, 32). Again, the formation of neutralizing antibodies in male FD patients receiving ERT is reported to impair reduction in plasma lyso-Gb3 (24).
Recently, novel chemical tools have been developed to study different retaining glycosidases, including α-galactosidases (33, 34). These activity-based probes (ABPs) are mechanism-based irreversible inhibitors functionalized with a bio-orthogonal tag such as a fluorophore or biotin. The first of these, ABPs, was developed for retaining β-glucosidases such as the human lysosomal glucocerebrosidase (GBA). The natural glucosyl-configured suicide inhibitor cyclophellitol covalently binds the catalytic nucleophile residue, Glu-340, in the enzymatic pocket of GBA (35). Equipped with a fluorescent reporter, the C-6 functionalized cyclophellitol permits specific and sensitive visualization of active enzyme molecules. Their amphiphilic nature renders the fluorescent ABPs membrane-permeable and allows in situ detection of active glucocerebrosidase in cells and organisms. Subsequently, cyclophellitol aziridine–type probes were developed to label, again in a mechanism-based manner, a broad range of β-glucosidases (36, 37). Next, the approach was extended to other retaining glycosidases by variation of the cyclophellitol configuration, yielding ABPs for α-galactosidases, β-galactosidases, α-fucosidases, and β-glucuronidases (38–41). Equipping the cyclophellitol scaffold with a biotin allows streptavidin-mediated enrichment and subsequent chemical proteomics experiments using LC-MS/MS. The successful application of biotin-tagged ABPs in proteomics profiling of β-glucosidases from different plant species was demonstrated (42). A potent fluorescent α-galactosyl–configured cyclophellitol aziridine ABP was developed and shown to label human α-Gal A as well as α-Gal B, the homologous N-acetyl galactosaminidase arisen by a gene duplication (39).
In this study, we used fluorescent α-galactosyl-configured cyclophellitol-aziridine ABPs and synthesized a biotinylated cyclophellitol-aziridine ABP to search for α-galactosidases in Nicotiana benthamiana. With these tools, we were able to identify an abundant apoplast α-galactosidase. We successfully cloned the gene and transiently overexpressed the protein, which we named A1.1, in N. benthamiana leaves. The recombinant enzyme was purified, crystallized, and biochemically characterized. We here report on the outcome of the investigation, including a comparison of the plant α-galactosidase with the human enzyme, which is deficient in FD patients.
Results
Screening for α-galactosidases in N. benthamiana leaf extracts and apoplast fractions
N. benthamiana leaf extracts and apoplast samples thereof were prepared and examined for α-galactosidase activity using 4-methylumbelliferyl–α-d-galactose (4MU–α-Gal) as substrate. Enzyme activity, with a broad pH optimum of 5.0–6.7, was detected in both samples (Fig. 1A). Next, we incubated the leaf and apoplast samples with Cy5-functionalized TB474 ABP at different pH values. In the case of the leaf extract, Cy5-ABP–labeled proteins with apparent molecular mass of ∼39 and 45 kDa were detected (Fig. 1B). The apoplast fraction contained mainly the ∼39-kDa labeled protein (Fig. 1B). To assess whether the plant α-galactosidases of interest are glycosylated, their binding to concanavalin A (ConA)-Sepharose beads was examined (Fig. 1, C and D). After incubation of samples with the lectin beads, ABP labeling and 4MU–α-Gal activities were performed. The ∼39-kDa protein did not bind to the lectin beads, in contrast to the ∼45 kDa protein (Fig. 1C). Approximately 50–55% of the 45-kDa protein is bound to the lectin beads, as judged by quantification of the band intensity of Fig. 1C. This band corresponds to 5–10% of the total 4-MU–α-Gal activity, suggesting that the 45-Da protein is not as active as the 39 kDa protein. More than 80% of the total 39-kDa labeled protein in both lysate and apoplast fractions was found in the unbound material. Likewise, more than 70% of the total activity of both fractions was not precipitated by ConA beads (Fig. 1D). The 39-kDa protein is highly present in the apoplast fraction. Thus, a discrete α-galactosidase, possibly lacking high-mannose N-glycans, is secreted to the apoplast space of N. benthamiana leaves.
Figure 1.

Screening for α-galactosidases in N. benthamiana. Plant leaf lysates and apoplast samples were tested on their α-galactosidase activity via 4MU–Gal assays and ABP in vitro labeling, following in-gel detection of the labeled proteins. A, 4MU–α-Gal activities present in plant leaf lysates and apoplast samples were first examined at different pH values of 3–8. (n = 2, error bars indicate mean ± S.D.) B, 45 μg of plant leaf lysate and 12 μg of apoplast sample were incubated with 0.25 μm TB474 for 30 min at pH 4–7 at room temperature. Labeled proteins were detected via in-gel fluorescent scanning (ABP), and the gels were stained with Coomassie Brilliant Blue (CBB) to show equal total protein loading. C, plant leaf lysates and apoplast samples were incubated with ConA-Sepharose beads for 2 h at 4 °C. After incubation, the samples were centrifuged, and different fractions were tested for α-galactosidase activity via ABP labeling and (D, left panel) 4MU–α-Gal assays. (n = 2, error bars indicate mean ± S.D.) In addition, D, right panel, quantification of the band intensity of C is presented. Samples tested: starting material = sample prior to ConA-Sepharose bead incubation, containing all proteins. Unbound = supernatant after incubation with the beads and centrifugation. This sample contains all material not bound to the beads. Wash = wash of the beads. Beads = pellet after incubation with the beads and centrifugation, containing the proteins attached to the ConA-Sepharose beads.
Identification of potential N. benthamiana α-galactosidases
Newly designed biotinylated ABP ME741 (Fig. 2A) was found to completely compete labeling of plant α-galactosidase in the apoplast fraction and leaf extracts by the Cy5-TB474 ABP (Fig. 2B). Conversely, labeling of plant α-galactosidase by biotinylated ME741 (visualized by Western blotting with streptavidin-HRP) is blocked by prior incubation with TB474 ABP. Thus, both probes recognize the same α-galactosidases. Next, we conducted large-scale pull downs of the α-galactosidases present in leaf and apoplast samples using biotinylated ME741. After the pulldown, the target proteins were bound to streptavidin beads following bead tryptic digestion. Tryptic peptides were analyzed by nanoscale LC coupled to tandem MS (nano-LC-MS/MS) (43). Peak lists were then searched against the Swiss-Prot (version June, 2017) database, and the identified peptides were manually curated. In this manner, peptides from known, well-annotated plant α-galactosidases, similar in sequence to potential N. benthamiana α-galactosidases, were identified (Table S1). The identified peptides belonged to Coffea arabica α-galactosidase (protein accession code Q42656) and Arabidopsis thaliana α-galactosidase 1 and 2 (protein accession codes Q9FT97 and Q8RX86, respectively). Next, we conducted alignment analysis of the cDNA sequence encoding for C. arabica α-galactosidase, as it is a well-characterized protein, known not to be N-glycosylated, toward the cDNA sequence of N. benthamiana (Sol Genomics Network). Based on the alignment, we designed specific primers to amplify the gene encoding for that most similar to Coffee, N. benthamiana α-galactosidase, which we named A1.1. An additional peptide search was then conducted, after the insertion to the Swiss-Prot database of this new potential N. benthamiana enzyme, and the results were even more prominent (Table S2). We could identify more peptides, with better scores and higher protein coverage, indicating that this is the protein we were looking for and not the homologues from the other organisms.
Figure 2.

Pulldown of plant α-galactosidases using ME741 and their identification by proteomics. A, i, α-galactosidase-configured aziridine scaffold; ii, Cy5 ABP, TB474; iii, biotin ABP, ME741. B, to prove that the same α-galactosidases are recognized by both ABPs, ME741 and TB474, competition experiments were performed. α-Galactosidases present in apoplast and plant leaf lysates were incubated with 0.25 μm ME741, at pH 5.5, for 30 min at room temperature, with or without subsequent incubation with 0.25 μm TB474, under the same conditions. Similarly, the samples were incubated first with TB474 and then with ME741. The labeled proteins were detected via in-gel fluorescent scanning and Western blot analysis using streptavidin-HRP (dilution 1:4000). The blot was then stained with CBB to show equal protein loading. The black arrow indicates the 45-kDa band; the open arrow indicates the 29-kDa protein, and the gray arrow indicates unspecific biotinylated plant proteins.
N. benthamiana A1.1 purification and characterization
To verify that A1.1 is truly an active α-galactosidase, we transiently overexpressed the enzyme via the Agrobacterium tumefaciens infiltration of N. benthamiana leaves. The p19 RNAi silencing inhibitor was used to ensure optimum protein expression levels (44). Leaves were harvested at different days post-infiltration (dpi), and A1.1 expression in the total leaf as well as in apoplast fluid was detected by 4MU–α-Gal activities, TB474 ABP labeling, and Coomassie Brilliant Blue staining of SDS-polyacrylamide gels (Fig. 3, A and B). Optimal starting materials for purification were apoplast fractions, collected at 4 dpi, showing the highest 4MU–α-Gal activity. The α-galactosidase activity per μg of protein in apoplast fractions was 20–40-fold higher in the case of leaves overexpressing A1.1, as compared with those treated with empty vector or nontreated plant leaves. A1.1 α-galactosidase was purified to homogeneity in three purification steps with high recovery (>30%) (Table 1). The presence of the enzyme in fractions was monitored by enzymatic assay with 4MU–α-Gal substrate. As the first purification step, we used ConA column chromatography, collecting the unbound material (flow-through fraction). In this step, other α-galactosidases were removed by binding to the lectin column. Next, cation-exchange chromatography was applied, resulting in a further modest purification (recovery 40%). Finally, the sample was subjected to gel-filtration chromatography, revealing that A1.1 behaves as monomer of about 30 kDa (Fig. S1). The final preparation of A1.1 was apparently pure as judged by SDS-PAGE analysis following silver staining of the gel (see Fig. 3B for an overview of the entire purification).
Figure 3.

Overexpression and biochemical characterization of the newly identified N. benthamiana α-galactosidase, A1.1. A, transient overexpression of A1.1 in N. benthamiana leaves via A. tumefaciens transformation assays. Infiltrated leaves and apoplast samples were harvested on different days post-infiltration (dpi): 2, 4, and 6. The expression levels of the enzyme were first tested via 4MU–α-Gal activities in lysates and apoplast fractions of different dpi. n = 2. (A1.1 = leaf overexpressing A1.1; E. = empty vector; N. = nontreated plant leaf.) B, next, the expression levels of active enzyme molecules were tested via in-gel ABP labeling. CBB staining of the gels followed to ensure the presence of the overexpressed enzyme. (P. = plant lysate; A. = apoplast sample; E. = empty vector; N. = nontreated plant leaf.) C, purification overview. SDS-PAGE analysis following silver staining of fractions obtained during different purification steps. (St. M. = starting material; ConA = fraction not bound to concanavalin A column; D. = sample obtained after 48 h of dialysis; C.E. = pooled collected eluate from cation-exchange chromatography; G.F. = final pooled fraction obtained after gel filtration.) 2 μg of total protein were loaded in each lane, except in G.F., where 1 μg was loaded. D, SDS-PAGE analysis following Coomassie Brilliant Blue staining of 1 μg of pure A1.1 and Fabrazyme after treatment with PNGase F and EndoH (C = untreated pure enzyme; P = enzyme treated with PNGase F; E = enzyme treated with EndoH), shows that A1.1 is not likely N-glycosylated, whereas Fabrazyme is, due to the difference in molecular weight after treatment.
Table 1.
Overview of A1.1 purification
| Purification steps | Protein concentration | Volume | Total protein | Total activity | Total specific activity | Purification fold | Yield = recovery activity |
|---|---|---|---|---|---|---|---|
| mg/ml | ml | mg | μmol/h/ml | μmol/h/mg | % | ||
| Starting material | 1.3 | 20 | 26 | 63,338 | 2436 | 1 | 100 |
| ConA | 0.44 | 45 | 20 | 50,171 | 2534 | 1 | 79 |
| Dialysis | 0.38 | 45 | 17 | 49,550 | 2905 | 1.2 | 78 |
| Ion exchange | 1.2 | 4.5 | 5.4 | 25,977 | 4810 | 2 | 41 |
| Gel filtration | 5 | 0.6 | 3 | 20,879 | 6959 | 2.9 | 33 |
A1.1 seems not to carry N-glycosyl groups as demonstrated by the lack of effect of PNGase and EndoH on its molecular weight (Fig. 3C). The molecular weight of A1.1, in contrast to that of Fabrazyme, is not influenced after the endoglycanase treatments (Fig. 3C). Consistently, following incubation of A1.1 with PNGase A, no released N-glycans were detected by MALDI-TOF MS analysis (Fig. S2).
Structural features of A1.1 α-galactosidase determined by crystallography
Pure N. benthamiana A1.1 was crystallized. The protein structure was determined at a resolution of 2.8 Å using the molecular replacement method with the rice α-Gal A (PDB code 1UAS) as search model. The statistics data of the collection and refinement are listed in Table 2. The protein model is a monomer, consisting of 363 amino acids (not including the first 57 amino acid signal-peptide sequence) and being separated into two domains. The N-terminal domain or catalytic domain (1–278) contains a TIM (β/α)8-barrel, a common motif among glycosidases (5), and the C-terminal domain (279–363) contains eight β-strands forming a “Greek key” motif (Fig. 4A). The active site of the enzyme is found by prediction as the final model obtained without a bound ligand, at the C-terminal end of the catalytic domain. Similar to other glycoside hydrolases of family 27, two aspartic acid residues (Asp-181 and Asp-236) were observed to serve as the catalytic amino acids, one acting as the nucleophile and the other as the acid/base, taking part in the double displacement reaction mechanism of α-galactosidases (45). A1.1 is negatively charged at the site of its active site and more neutral at the opposite site (Fig. 4B). The pI of the enzyme is at around 5.32. Amino acid sequence alignment of A1.1, human (protein accession code P06280), rice (protein accession code Q9FXT4), and coffee (protein accession code Q42656) α-Gals reveals great secondary structure identity of all enzymes (Fig. 4C). A1.1 shares 42% amino acid identity with the human enzyme, showing overall highly conserved secondary structures at the catalytic domain and not as much at the C-terminal domain. No Asn-Xaa-(Ser/Thr/Cys) motifs are present in amino acid sequences of A1.1, revealing that the protein is most likely not N-glycosylated. In contrast, four potential N-glycosylation sites are present in human enzyme. A common amino acid sequence pattern (CEW, at positions 212–214 in Fig. 4C) occurs in all aligned galactosidases, as described previously by Motabar et al. (46).
Table 2.
Data collection and refinement statistics (PBD code 6F4C)
| Data collection | |
| Space group | P 41 21 2 |
| Unit-cell parameters a, b, c, α, β, γ | 74.04Å, 74.04Å, 133.31Å |
| 90.00, 90.00, 90.00 | |
| Resolution (Å) | 25.00 2.80 |
| 24.77 2.80 | |
| % Data completeness (in resolution range) | 98.4 (25.00–2.80) |
| 99.9 (24.77–2.80) | |
| 〈I = (I)〉 | 2.53 (at 2.80Å) |
| Refinement statistics | |
| Refinement program REFMAC 5.8.0158 | |
| R, Rfree | 0.216, 0.287 |
| 0.225, 0.282 | |
| Rfree test set | 486 reflections (5.30%) |
| Fo, Fc correlation | 0.88 |
| Total no. of atoms | 2861 |
| Average B, all atoms (Å2) | 4.0 |
| Ramachandran plot (%) | |
| Preferred regions | 96 |
| Allowed regions | 3 |
| Outliers | 1 |
Figure 4.
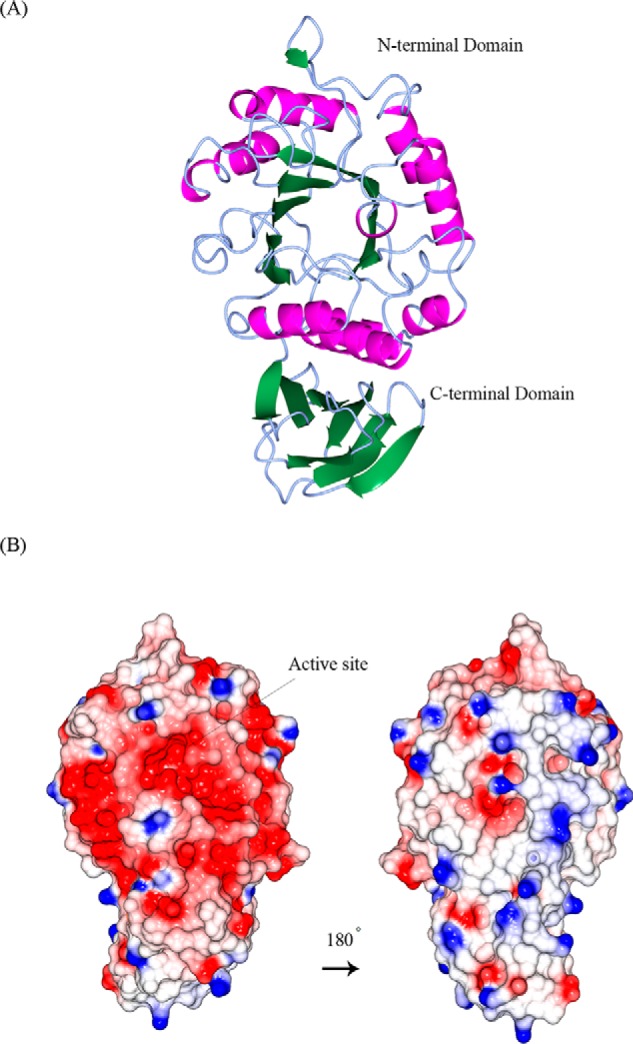


Structural similarities of α-galactosidases from different species. A, stereo view of the ribbon model of A1.1 (PDB code 6F4C). Catalytic domain contains a (β/α)8-barrel. C-terminal domain forms a Greek key. β-Strands are represented in green; α-helices are represented in pink; and loops are represented in light blue. B, electrostatic map of A1.1: red indicates the − charges; blue indicates the + charges. C, multiple amino acid sequence alignment of A1.1: human, rice, and coffee galactosidases. The sequence alignment showing structural details of A1.1 was achieved using ESPript3.0. α-Helices are shown as coils labeled α; β-strands are shown as arrows labeled β, and β-turns are labeled TT. Identical residues are shown on a red background; conserved residues are shown in red, and conserved regions are shown as blue boxes. The catalytic residues are indicated with an asterisk. D, right panel, stereo view of the superimposed models of A1.1 with the human α-galactosidase. 348 residues were aligned, having a root-mean-square deviation of 1.3358 Å of their C-α atoms. The A1.1 model (PDB code 6F4C) is shown in green; the human α-Gal model (PDB code 3HG2) is shown in gray. The backbone of a modeled galactose molecule is shown in white, and its oxygen atoms are shown in red. Left panel, a closer look at the amino acids around the active site of both enzymes, presented as fat bonds.
The overall structure of A1.1 is very similar to that of human (PDB code 3HG2) α-Gal A, as visualized by the superimposed models, consistent with their 42% sequence identity (Fig. 4D, left panel). In addition, a closer look at the active site of both enzymes shows great conservation of the majority of the amino acids (Fig. 4D, right panel).
Catalytic features of A1.1
We first examined substrate specificity of pure A1.1 using artificial 4-MU–glycoside substrates (Table 3). The enzyme hydrolyzes with considerable affinity 4MU–α-Gal substrate but is inactive toward β-d-glucopyranoside, β-d-galactopyranoside, α-d-mannopyranoside, α-l-fucopyranoside, N-acetyl-α-d-galactosaminide, and N-acetyl-β-d-glucosaminide. The pH optimum of A1.1 with 4MU–α-Gal substrate is broad, ranging from pH 5.0 to 6.5 (Fig. 5A). The apparent Km with 4-MU–α-Gal is 0.17 mm and kcat 81 s−1 (Fig. 5B). Comparable kinetic values are also obtained with Fabrazyme, although its pH optimum toward 4MU–α-Gal substrate is rather acidic at pH 4.5 (Fig. 5, A and B).
Table 3.
Activity of A1.1 toward different 4MU substrates
| 4MU substrates | Activity |
|---|---|
| nmol/h/μg | |
| β-d-Glucopyranoside, pH 5.2 | 0 |
| β-d-Glucopyranoside, pH 5.8 | 8 |
| β-d-Glucopyranoside, pH 7 | 0 |
| α-d-Galactopyranoside, pH 4.6 | 905 |
| α-d-Galactopyranoside, pH 6 | 1645 |
| β-d-Galactopyranoside, pH 4.3 | 8 |
| α-d-Mannopyranoside, pH 4 | 3 |
| α-l-Fucopyranoside, pH 5.5 | 6 |
| N-Acetyl-α-d-galactosaminide, pH 4.6 | 8 |
| N-Acetyl-β-d-glucosaminide, pH 4.4 | 2 |
Figure 5.

Activity of N. benthamiana A1.1 and Fabrazyme toward 4MU-α-GAL and human glycosphingolipids. A, 4MU–α-Gal activities of pure A1.1 and Fabrazyme at different pH values (3.8–8). B, Michaelis-Menten kinetic values of both enzymes toward 4MU–α-Gal, NBD-Gb3, and lyso-Gb3 substrates. Data represent mean values, n ≥ 2. C, degradation of 5 μm NBD-Gb3 to NBD-LacCer by 50 ng of A1.1 or Fabrazyme at varying pH values (4.5–7.5). Quantification of HPTLC plates were conducted more than once, showing one gel quantification.
The structural resemblance of A1.1 with human α-Gal A prompted us to examine its activity toward glycosphingolipid substrates. First, we tested the activity of A1.1 toward artificial NBD-labeled Gb3. A1.1 was found to convert NBD-Gb3 to NBD-lactosylceramide by removal of the terminal α-linked galactose. The activity of A1.1 toward NBD-Gb3 is in several aspects comparable with that of Fabrazyme (recombinant human α-Gal A). The pH optimum is similarly acidic at pH 4.5 (Fig. 5C). The affinity of A1.1 for NBD-Gb3 is high with an apparent Km of 32 μm, similar again to that of Fabrazyme. However, the kcat value is ∼4-fold lower than that of human α-galactosidase (Fig. 5B).
Next, we tested natural glycosphingolipids as substrates. A1.1 degrades natural C18-Gb3 by converting it to C18-LacCer as measured by HPLC analysis (47). Its activity is again comparable with that of human enzyme: 96.3% of C18-Gb3 is converted to LacCer upon overnight incubation with 3 μg of A1.1 at pH 4.5 versus 98.3% conversion with the same amount of Fabrazyme at the same condition. In addition, A1.1 hydrolases best this lipid substrate (C18-Gb3) again at acidic conditions, pH 4.6, the same as for the human enzyme (Fig. S3, A and B). Finally, we determined by LC-MS/MS the ability of A1.1 to degrade lyso-Gb3, the toxic base of Gb3, to lactosylsphingosine (30). A1.1 degrades lyso-Gb3, showing a Km of 37 μm, quite comparable with that of Fabrazyme. Its kcat value, however, is ∼2-fold lower than that of the human enzyme (Fig. 5B). The noted activity of A1.1 toward glycosphingolipid substrates in vitro resembles the earlier observed ability of rice α-galactosidase to hydrolyze Gb3 in vitro (48).
A1.1 reduces lyso-Gb3 in intact FD fibroblasts
At first the uptake of A1.1 from lysosomes of two different FD patients' fibroblasts was evaluated. Different amounts of pre-labeled enzyme with TB474 A1.1 were applied overnight to the cells. After extensive washing (Fig. S4), the cells were lysed, and the uptake of A1.1 was studied via SDS-PAGE analysis (Fig. 6A). The cells from both FD patients were able to uptake A1.1. An increase in the uptake depending on the concentration of the pre-labeled protein added to the culture media was observed. To estimate the amount of the internalized enzyme, 1% of the input using 150 and 300 μg/ml were loaded on gel, together with 13–17% of the total lysates after treatment with the prelabeled enzyme (Fig. 6B). The quantification of the gels revealed that 0.3–0.6% of the 150 μg/ml entered the cells, and 0.5–1% of the 300 μg/ml (Fig. 6C).
Figure 6.

Internalization of TB474-labeled A1.1 from Fabry fibroblasts. A, Fabry fibroblasts from two different patients were grown in 12-well plates. The cells were incubated with different amounts pre-labeled with TB474, A1.1 (0, 5, 10, 70, 150, and 300 μg per 1 ml of culture media). Lysis of the cells took place, and samples were measured for Cy5 signal, through SDS-PAGE. Left top panel, results were obtained when using patient 1 cell line, SDS-polyacrylamide gel image, following fluorescent scanning and quantification of signal, left bottom panel. Right panel, results obtained from patient 2 cell line. The gels were conducted more than once. The uptake is gradually increased while increasing the amount of pre-labeled protein in both cell lines. 10 μg of total fibroblast protein was added in each well. Coomassie staining of the cells took place to show equal loading. ABP = activity-based probe signal; CBB = Coomassie Brilliant Blue signal; P1 = patient 1; P2 = patient 2. B, estimation of the % of the internalization of pre-labeled A1.1 (150 and 300 μg per ml of culture media) from the same FD fibroblasts. Loaded on gel, 1% of the pre-labeled A1.1 input, together with 10 μg of total fibroblast protein, accounted for 13–17% of the total cell lysate. C, quantification of signal obtained in B. The uptake of 150 μg of A1.1 accounts to 0.3–0.5% of the total input and 0.5–1% when using 300 μg. ABP = activity-based probe signal; P1 = patient 1; P2 = patient 2. D, overnight uptake of A1.1, pre-labeled with TB474, by FD fibroblasts monitored by confocal microscopy. Cells were grown overnight on glass coverslips and incubated with 150 μg of pre-labeled A1.1 per ml of culture medium (here shown cells from patient 1). Panel in red, visualization of the internalized TB474-labeled A1.1. Panel in green, lysosomal labeling using anti-LAMP-1 antibody. Panel in blue, cellular nuclei stained with DAPI. D, overlay image. The white arrowheads indicate examples of co-localization of internalized TB474-labeled A1.1 with the late endosomal/lysosomal marker LAMP-1. Scale bar 25 μm.
In addition, the uptake of the enzyme was studied by confocal microscopy. For this, A1.1 was again labeled with TB474 and subsequently applied to the FD fibroblasts. After a 16-h uptake, punctate fluorescence was observed in some cells, which was reminiscent of a lysosomal pattern, possibly through fluid-phase endocytosis. Immunofluorescent staining of the late endosomal/lysosomal marker LAMP-1 occasionally showed co-localization with TB474-A1.1, indicating lysosomal delivery of the enzyme (Fig. 6D).
To examine whether A1.1 is also able to degrade glycosphingolipid in vivo, we incubated the same cultured FD fibroblasts with 300 μg/ml A1.1. After overnight incubation, the cells were first washed and lysed, and the levels of both lyso-Gb3 and Gb3 were determined by LC-MS/MS. In addition, the 4MU–α-GAL cellular activity was measured. The elevated lyso-Gb3 and Gb3 in both FD fibroblasts were found to be significantly reduced by the exposure to A1.1 (Fig. 7A). In addition, the 4MU–α-Gal cellular activity was significantly increased after exposure to the recombinant protein (Fig. 7B). Activities were also measured at washing steps 1 and 5 to ensure that no protein remained outside of the cells prior to lysis (Fig. S5).
Figure 7.

Correction of FD lipid abnormalities in Fabry fibroblasts via A1.1 treatment. A, lyso-Gb3 and Gb3 levels measured by LC-MS/MS in Fabry fibroblasts from two different patients, with or without overnight treatment with 300 μg of A1.1 per ml of culture medium. B, cellular 4MU–α-Gal activity found in the same lysates that were used for lipid measurements, before and after treatment with A1.1. Data are represented as mean ± S.D., n ≥ 3. Asterisk(s) indicate significant differences as measured by a standard t test: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
We earlier used N. benthamiana for expression of WT and mutagenized human α-galactosidase and α-galactosaminidase (49). The N. benthamiana plant is a convenient production platform for recombinant proteins given the ease of cultivation and transfection with A. tumefaciens (49). In our previous study (49), we employed a cyclophellitol-type activity-based probe (TB474) to visualize the recombinant human α-galactosidases. In the course of the experiments, we noted the presence of two endogenous enzymes (39 and 45 kDa) labeled by TB474. We focused attention to the 39-kDa enzyme (named A1.1) that is present in relatively high concentrations in the apoplast. Using biotin-containing ABP (ME741) and streptavidin pulldown, the identity of the 39 kDa protein in apoplast fluid was determined by proteomics. The corresponding gene (accession ID: GJZM-1660) was cloned from a N. benthamiana cDNA library and transiently overexpressed in N. benthamiana leaves using infiltrations with A. tumefaciens harboring the appropriate expression vectors. A high yield of A1.1 in the apoplast fluid was obtained, comprising 11.5% of total soluble apoplastic protein. Next, exploiting the lack of binding of A1.1 to ConA beads, the protein was purified with 33% recovery to homogeneity by sequential ConA chromatography, cation-exchange chromatography, and gel filtration. The molecular mass of 39 kDa of the purified enzyme is similar to that reported for rice and coffee α-galactosidases (3, 5, 6, 48).
We characterized A1.1 regarding structural features. The mature form of the enzyme consists of 363 amino acids and has a molecular mass of 39 kDa. The purified enzyme is an active monomer, based on gel filtration behavior. This feature is common to rice α-galactosidase, but the human enzyme occurs as a homodimer (49). As predicted by the absence of appropriate Asn-Xaa-(Ser/Thr/Cys) motifs, A1.1 is not N-glycosylated, and endoglycanase digestion points to no N-linked glycans. Similarly, rice and coffee α-galactosidases lack N-glycans (5, 50). Based on amino acid sequence alignments, the enzyme has 82% identity with coffee α-galactosidase, 68% identity with rice α-galactosidase, and 42% identity with human α-galactosidase A, also revealing great secondary structural conservation.
Crystals of pure A1.1 were obtained, and the structure was resolved by X-ray diffraction at a resolution of 2.8 Å. The enzyme contains a C-terminal domain with Greek key motif and N-terminal domain with a (β/α)8-barrel, the common catalytic domain among the hydrolases of family 27 (5). The active site of retaining α-galactosidases contains a catalytic nucleophile and acid/base residue mediating double-displacement as a catalytic mechanism (51, 52). In the active site of A1.1, other amino acids appear involved in substrate recognition, such as Trp-67, Asp-102, Tyr-144, and Lys-179. Two disulfide bonds occur close to the catalytic pocket, Cys-72/104 and Cys-152/183, as also present in human α-galactosidase A (17, 51). Notably, the existence of a second ligand-binding site in human α-galactosidase A, centered on Tyr-329, was postulated by Guce et al. (45). This putative site, speculated to bind β-galactose, is located between the two domains of the human monomer enzyme, being exposed at the surface. However, in A1.1, instead of the Tyr-329, a Lys-330 is present, and the overall second binding-site region is different from the human enzyme. The C-terminal domain of A1.1 contains eight β-strands forming a Greek key motif. The C-terminal amino acid sequences of human and A1.1 α-galactosidases are different. Coffee α-galactosidase, however, contains a C-terminal domain that is similar to A1.1 and is thought to essentially contribute to the overall structure of the enzyme, as its deletion results in loss of enzymatic activity (3).
A1.1 shows strong specificity regarding the substrate sugar. It hydrolyzes 4MU–α-galactopyranoside liberating fluorescent methylumbelliferone, but it does not correspond to β-d-glucopyranoside, β-d-galactopyranoside, α-d-mannopyranoside, α-l-fucopyranoside, N-acetyl-α-d-galactosaminide, or N-acetyl-β-d-glucosaminide. The broad pH optimum of A1.1, pH 5.0–6.5, is also reported for coffee and rice enzymes (12, 48). The activity of A1.1 toward 4MU–α-galactopyranoside (Km of 0.17 mm) is quite comparable with that of human α-galactosidase A (Km of 2.4 mm). For the rice enzyme, a high affinity toward pNP–α-Gal substrate was earlier reported by Chien et al. (48) (Km of 0.47 mm). For coffee α-galactosidase, a pH optimum of 6.5 with a Km of 0.29 mm toward 4MU–α-Gal substrate was observed by Maranville and Zhu (3).
α-Galactosidases from rice and coffee have been shown to remove terminal α1–3-linked galactose residues from type B glycolipid antigens on red blood cells, converting them to type O structures (48, 53). The ability of A1.1 to hydrolyze terminal α-galactoses from artificial and natural glycosphingolipid substrates was therefore studied. A1.1 was found to be well able to hydrolyze NBD-Gb3, C18-Gb3, and lyso-Gb3 in vitro. Of note, the presence of Gb3 in N. benthamiana leaves has not been reported. We could not detect Gb3 with a regular sphingolipid base (data not shown). A similar observation was earlier made for rice α-galactosidase; Gb3 was shown to be converted to LacCer, but kinetic parameters were not determined (48). We observed a considerable affinity of pure A1.1 for lipids as substrates (Km values of 32 μm for NBD-Gb3 and 38 μm for lyso-Gb3), almost equal to those observed for human α-galactosidase A.
Presently, two different recombinant human α-galactosidase A preparations are used to treat FD, an X-linked disorder with lysosomal Gb3 accumulation in various cell types and affecting heart and kidney (54). The therapeutic enzymes use Man-6-P containing N-linked glycans that are aimed to correct the lysosomal enzyme deficiency in all cell types following uptake and delivery to lysosomes by the ubiquitous MPR at the surface of cells (54). Exposure of cultured fibroblasts to recombinant human α-galactosidase A (Fabrazyme) results in a reduction of excessive Gb3 and is deacylated metabolite lyso-Gb3 (54). We investigated whether recombinant A1.1 also manages to degrade the toxic lyso-Gb3 accumulating in cells of FD patients. Indeed, an overnight incubation of patient fibroblasts with 300 μg/ml A1.1 resulted in significant reduction of both lyso-Gb3 and Gb3 accumulating glycosphingolipids, reaching levels found in normal fibroblasts (∼4 pmol/mg and ∼15 nmol/mg for lyso-Gb3 and Gb3, respectively). It should be kept in mind that because A1.1 lacks glycans, no lectin-mediated uptake of the enzyme by cells occurs, in contrast to that of Man-6-P–rich Fabrazyme. The ability of the latter enzyme to correct lyso-Gb3 in cultured fibroblasts is therefore far superior than that of A1.1. A same reduction in cellular lyso-Gb3 of FD fibroblasts was obtained, reaching levels found in normal fibroblasts, with 60-fold less Fabrazyme than A1.1 (Fig. S6).
Unfortunately, the present enzyme replacement therapies meet limited clinical success. Although storage lipid is cleared from endothelial cells, complications in heart and kidney may nevertheless develop in FD patients, possibly due to the use of insufficient therapeutic enzyme to penetrate these organs. High costs of the present α-galactosidase A preparations hamper the use of significantly higher doses. The elevated circulating lyso-Gb3, considered to be toxic for podocytes and nociceptive neurons, is not completely corrected by present ERT (30, 31). Finally, a complicating factor with the present ERT of FD proves to be the antigenicity of recombinant human α-galactosidase A in the majority of male FD patients that lack any endogenous enzyme (25). In these individuals, neutralizing antibodies against therapeutic enzyme develop quickly, resulting in a relapse in the reduction of circulating toxic lyso-Gb3 (24). The clinical outcome of ERT in antibody-positive FD patients is reported to be poorer (24). In view of all this, the use of a plant α-galactosidase to treat male FD patients deserves consideration. First, plant α-galactosidase could be produced in N. benthamiana or other plant production platforms at considerable lower costs than the present human recombinant enzymes, allowing use of higher doses. Second, A1.1 is able to degrade toxic lyso-Gb3. The enzyme can in principle be further engineered to increase stability and desired enzymatic activity. The lack of N-glycans on a plant-derived α-galactosidase might even be beneficial, preventing a lectin receptor-mediated sink in the endothelium and liver as observed with present therapeutic human glycoprotein enzymes. Finally, we observed that N. benthamiana A1.1 does not cross-react with neutralizing antibodies directed against human α-galactosidase A that are present in serum of male FD patients treated with therapeutic enzyme (Fig. S7). Antigenicity of a plant protein might be a concern, but it should be kept in mind that this also exists with the human enzyme preparations in most male FD patients. The considerable progress made in induction of tolerance against foreign proteins may be exploited (55). It is presently unclear how antigenic a plant α-galactosidase will be: the absence of glycans might reduce C-type lectin receptor-mediated presentation by dendritic cells.
In conclusion, further research on the optimization of plant α-galactosidases such as A1.1 to reduce toxic lyso-Gb3 in FD should be considered to meet the need for an affordable treatment of this devastating disorder.
Experimental procedures
Chemicals
All chemicals were obtained from Sigma (Germany) if not indicated otherwise. Fluorescent NBD-lipids and pure lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Antibodies purchased from Abcam (Cambridge, MA).
Plants
N. benthamiana plants were grown at 21 °C and 60–70% humidity in the Unifarm greenhouses of Wageningen University (56).
Patient materials
Fibroblast cell lines from classical FD individuals were obtained from the Lysosomal Outpatient Clinic of the Academic Medical Center in Amsterdam (AMC). The cells were cultured in Dulbecco's modified Eagle's medium/Nutrient Mixture F-12 (DMEM/F-12, Sigma) media, supplemented with 10% fetal calf serum and 1% penicillin/streptomycin, at 37 °C with 5% CO2 in a humidified incubator. Plasma specimens from FD individuals were obtained from the Lysosomal Outpatient Clinic of the Academic Medical Center in Amsterdam (see supporting information). All patient materials were obtained after approval of the Academic Medical Center's review board and abide by the Declaration of Helsinki principles.
ABP ME741, synthesis
The α-galactopyranose-configured cyclophellitol aziridine grafted with Cy5 as fluorophore ABP (TB474) was synthesized as described previously (49). The biotinylated α-galactopyranoside cyclophellitol aziridine (ME741) was synthesized by copper-catalyzed click chemistry of azido cyclophellitol aziridine intermediate with biotin alkyne (Scheme S1). The α-galactose-configured azido cyclophellitol aziridine (39) (14.6 mg, 0.043 mmol) synthesized in 15 steps form d-xylose, and the desired biotin-alkynes (18.5 mg, 0.047 mmol) were dissolved in N,N-dimethylformamide (2 ml). CuSO4 (0.043 ml, 1 m in H2O) and sodium ascorbate (0.043 ml, 1 m in H2O) were added, and the reaction mixture was stirred at room temperature under argon atmosphere for 18 h. Then the solution was diluted with CH2Cl2, washed with H2O, dried over MgSO4, and concentrated under reduced pressure. The crude was purified by semi-preparative reversed-phase HPLC (linear gradient: 15–24% B in A, 12 min, solutions used A, 50 mm NH4HCO3 in H2O, and B, MeCN) and lyophilized to yield biotinylated ABP ME741 as a white powder (8.0 mg, 11 μmol, 26%). 1H NMR (600 MHz, CD3OD) contained the following: δ 7.85 (d, J = 3.7 Hz, 1H, CH); 4.90 (s, 1H, NH); 4.49 (dd, J = 7.9, 4.9 Hz, 1H, CH); 4.42 (d, J = 2.3 Hz, 2H, CH2); 4.38 (td, J = 7.1, 2.5 Hz, 2H, CH2); 4.30 (dd, J = 7.9, 4.5 Hz, 1H, CH); 4.08 (dd, J = 8.6, 3.9 Hz, 1H, CH); 3.85–3.87 (m, 1H, CH); 3.81–3.68 (m, 2H, CH2); 3.36 (dd, J = 8.6, 1.7 Hz, 1H, CH); 3.24–3.17 (m, 1H, CH); 3.16 (td, J = 7.0, 2.5 Hz, 2H, CH2); 2.92 (dd, J = 12.7, 5.0 Hz, 1H, 1/2CH2); 2.70 (d, J = 12.7 Hz, 1H, 1/2CH2); 2.61 (d, J = 6.1 Hz, 1H, CH); 1.91–1.86 (m, 4H, 2CH2); 1.75–1.56 (m, 4H, 2CH2); 1.51 (p, J = 7.2 Hz, 2H, CH2); 1.43 (p, J = 8.8, 8.2 Hz, 2H, CH2); 1.39–1.26 (m, 4H, 2CH2). 13C NMR (151 MHz, CD3OD) contained the following: δ 188.7, 176.0, 176.0, 166.1, 146.2, 146.2, 74.3, 74.2, 73.2, 73.1, 70.5, 69.5, 63.4, 62.8, 61.6, 57.0, 51.4, 51.3, 44.8, 43.3, 41.1, 40.2, 39.4, 37.1, 36.8, 36.8, 35.6, 31.3, 31.2, 30.4, 30.1, 30.0, 29.9, 29.8, 29.7, 29.5, 27.5, 27.4, 27.3, 27.2, 26.9, 26.5, 25.8 ppm; HRMS was calculated for C34H56N8O8S [M + H]+ 737.40201 and found was 737.40146.
N. benthamiana leaf extracts
Leaves of 5–6-week-old N. benthamiana plants were collected, snap-frozen in liquid nitrogen, and homogenized by grinding with a mortar and a pestle. To 1 g of leaf, 4 ml of ice-cold extraction buffer (30 mm citrate/phosphate buffer, pH 6, containing 2% w/v polyvinylpolypyrrolidone, 0.1% v/v Tween 20, 0.15 m NaCl, and protease inhibitor by Roche Applied Science, EDTA-free) was added, and the material was again homogenized. The homogenates were centrifuged at 16,000 rcf, at 4 °C, for 10 min, and the supernatant containing soluble proteins was collected.
Isolation of apoplast proteins
For the isolation of apoplast proteins, N. benthamiana leaves were gently submerged in ice-cold extraction buffer (50 mm PBS, pH 6, 0.1 m NaCl, and 0.1% v/v Tween 20) and exposed to vacuum for 10 min. Then the vacuum was released very slowly to ensure infiltration of the apoplast. The leaves were then collected and carefully placed in 10-ml syringes plugged in 50-ml tubes. The samples were centrifuged for 10 min at 2000 rcf, and apoplast fluid was collected (44).
Small-scale ABP labeling of α-galactosidases in N. benthamiana plant leaf extracts and apoplast fluid
10 μl of N. benthamiana plant leaf extracts (4.5 mg of total soluble protein/ml) and 20 μl of apoplast sample (0.21 mg of total soluble protein/ml) were incubated with 0.25 μm fluorescent TB474 in 150 mm citrate/phosphate buffer at different pH values, for 30 min at room temperature. Gel loading buffer (with additional β-mercaptoethanol) was added to samples, followed by incubation for 5 min at 95 °C. The proteins in the samples were separated by 10% polyacrylamide gels. Labeled proteins were visualized by fluorescence scanning as described earlier (34).
Analysis of N-glycosylation by concanavalin A lectin binding
Concanavalin A-Sepharose 4B beads (ConA beads) were first washed three times in 0.1 m sodium acetate, 0.1 m NaCl, 1 mm MgCl2, 1 mm CaCl2, 1 mm MnCl2, pH 6.0, washing buffer using brief centrifugation at 2000 rcf for 2 min. Next, 150 μl of beads were mixed with 300 μl of plant leaf extracts or 150 μl of apoplast sample and incubated for 2 h at 4 °C while rotating. After this incubation, the mixture was centrifuged at 16,000 rcf at 4 °C for 10 min. The supernatant was collected, and the beads were washed three times with washing buffer. The samples were stored for a short period at 4 °C until further use.
Competitive ABPP using ME741 ABP
The ability of biotinylated ME741 to label α-galactosidases was first established by performing competition experiments. Homogenates were first labeled with Cy5 functionalized TB474 at 0.25 μm for 30 min at room temperature, followed by labeling the homogenate with biotinylated ME741 ABP at 0.25 μm and the other way around. Subsequently, SDS-PAGE and Western blot analysis with HRP-streptavidin was performed as described previously (41, 43).
In vitro biotin pulldown of bound targets, followed by on-bead tryptic digestion and LC-MS/MS identification
Plant leaf extracts (4.5 mg of total soluble protein/ml) and apoplast samples (0.21 mg of total soluble protein/ml (no. 1) and 0.6 mg of total soluble protein/ml (no. 2)) were pre-incubated or not with 5 μm TB474 for 2 h at room temperature, following overnight incubation with 5 μm ME741, room temperature at a final volume of 500 μl. Then the reaction was stopped by the addition of 120 μl of 10% (w/v) SDS and subsequent incubation of the sample at 95 °C for 5 min. Preparation of proteins for on-bead and tryptic digestion following LC-MS/MS identification was performed exactly as described previously (43). Prior to LC-MS analysis, peptides were desalted by Stagetips as described by Rappsilber et al. (57). EmporeTM C18 47-mm extraction discs were used to fabricate the StageTips. Typically, two discs were placed on top of each other to make StageTips with two layers of column material inserted in a yellow pipette tip.
MS acquisition
The Synapt G2Si mass spectrometer (Waters) operating with Masslynx for acquisition and ProteinLynx Global Server (PLGS) for peptide identification was used for analysis. The following settings in positive resolution mode were used: source temperature of 80 °C; capillary voltage 3.0 kV; nano flow gas of 0.25 bar; purge gas 250 liters/h; trap gas flow 2.0 ml/min; cone gas 100 liters/h; sampling cone 25 V; source offset 25; lock mass acquisition was done with a mixture of Leu-enkephalin (556.2771) and Glu-fibrinogen (785.84265) and lockspray voltage 3.5 kV; and Glu-fibrinogen fragmentation was used as calibrant. An UDMSe data-independent acquisition method was used for analysis. Briefly, the mass range is set from 50 to 2000 Da with a scan time of 0.6 s in positive resolution mode. The collision energy is set to 4 V in the trap cell for low-energy MS mode. For the elevated energy scan, the transfer cell collision energy is ramped to higher collision energies, and data are recorded. The lock mass is sampled every 30 s and used for accurate determination of parent ions mass after peak picking. Peak lists containing parent and daughter ions were compiled in .mgf format and searched against the Swiss-Prot (version June 2017) database. The identified peptides were manually curated.
Plant expression vector for overexpression of A1.1
The N. benthamiana α-galactosidase A1.1 (gene accession ID: GJZM-1660), was amplified from an N. benthamiana cDNA library made by our laboratory using the InvitrogenTM CloneMinerTM II cDNA library construction kit (Thermo Fisher Scientific). The gene was amplified with its native signal peptide, using Phusion® High Fidelity PCR Master Mix (BioLabs), with the following primers: sense, 5′-CCCATGGGTTTGCCACCAATTTTAAAGCTGCTACTAT-3′, and antisense, 5′-GGGTACCTTATTTTTTAGGAGTCAGAACATACATCCTGCA-3′. The gene was flanked between NcoI and Acc65I restriction sites, and it was cloned into a pGEM®-T Easy Vector System (Promega). Confirmation of sequences was done by sequencing (Macrogen, the Netherlands), and the complete ORF of the gene was inserted into the pHYG plant expression vector, as described previously (58). The construct was under the control of cauliflower mosaic virus 35S constitutive promoter, with duplicate enhancer and the nopaline synthase terminator (49). The pHYG vectors harboring the genes were used for transformation of A. tumefaciens strain MOG101, following N. benthamiana plant leaf infiltrations.
A. tumefaciens transient transformation assay and plant infiltrations
A. tumefaciens cultures were grown as described previously (56, 58). The constructs were co-expressed with the tomato bushy stunt virus silencing inhibitor p19 to ensure optimum expression levels (44, 49). The inoculated bacterial cultures were used for infiltration of 5–6-week-old N. benthamiana plants, as described previously (56).
Isolation of apoplast overexpressing A1.1
The isolation of apoplast fluid was performed as described previously (44), using as extraction buffer 30 mm citrate/phosphate, pH 6, 0.5 m NaCl, and 0.1% (v/v) Tween 20. The sample was passed through a G-25 Sephadex column for desalting and snap-frozen in liquid nitrogen. The sample was stored at −80 °C until further use.
Enzyme purification
Chromatography with 5 ml of concanavalin A-Sepharose column (GE Healthcare) was used as a first step for purification. The column was equilibrated with 40 ml of washing buffer (0.1 m sodium acetate, 0.1 m NaCl, 1 mm MgCl2, 1 mm CaCl2, 1 mm MnCl2, pH 6.0). Then, 40 ml of apoplast fluid overexpressing A1.1 was applied to the column, 1:1, diluted in washing buffer (1 ml/min loading conditions). The enzyme was present in the (unbound) flow-through fractions, ensuring separation from bound plant glycoproteins. About 40 ml of sample was collected and tested for enzymatic activity. Next, the sample was extensively dialyzed in 20 mm sodium acetate buffer, pH 5.5, 4 °C, for 2 days. Subsequently, the dialyzed fraction was subjected to chromatography on two HiTrap SP HP columns, 1 ml each, plugged on top of each other (GE Healthcare), and equilibrated with 30 ml of binding buffer: 20 mm sodium acetate buffer, pH 5.0. The sample, 40 ml, was applied on the columns, which were extensively washed with 30 ml of binding buffer afterward. Then, protein was eluted using a 15-ml gradient of 0–400 mm NaCl. Fractions containing the highest protein activity and purity were pooled. Next, the pooled sample (4.5 ml) was applied to a HiLoadTM 16/600 SuperdexTM 200 preparation grade column (GE Healthcare). The column was first equilibrated with 500 ml of 20 mm sodium acetate buffer with 150 mm NaCl, pH 5.5, and the sample was applied to the column at a flow rate of 1 ml/min. Four samples of 2 ml each, with the highest purity and activity, were collected and concentrated using Centricon Plus-20, 15 ml with 10-kDa molecular cutoff (Millipore, Bedford, MA) until 0.8 ml. The final material was snap-frozen in liquid nitrogen and stored at −80 °C until further use.
Protein determination
Total soluble protein content from cell lysates was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific), using BSA as a standard, according to the manufacturer's protocol. Pure protein concentrations were measured in NanoDrop 2000c (Thermo Fisher Scientific).
Enzymatic assays and determination of kinetic parameters
Different 4-MU substrates were used to test the specificity of A1.1. Tested substrates were the following glycosides linked to 4-MU: β-d-glucopyranoside, α-d-galactopyranoside, β-d-galactopyranoside, α-d-mannopyranoside, α-l-fucopyranoside, N-acetyl-α-d-galactosaminide, and N-acetyl-β-d-glucosaminide at the following assay concentrations and pH conditions: 1.36 mg/ml, pH 5.2, 5.8, and 7; 1.25 mg/ml, pH 4.6 and 6; 0.144 mg/ml, pH 4.3; 2.7 mg/ml, pH 4; 0.26 mg/ml, pH 5.5; 0.4 mg/ml, pH 4.6; 1.52 mg/ml, pH 4.4, respectively. All substrates were dissolved in 150 mm citrate/phosphate (McIlvaine) buffer, at the appropriate pH for each enzyme, supplemented with 0.1% (w/v) BSA. Released 4-MU was fluorometrically quantified as described earlier (60, 61). Km and kcat values were determined using 4-MU–α-GAL substrate. Reactions were performed for 25 min at 37 °C at 10 different 4-MU–α-GAL concentrations in 150 mm citrate/phosphate buffer, pH 6, for A1.1 or pH 4.6 for Fabrazyme supplemented with 0.1% (w/v) BSA. The 4-MU–α-GAL concentrations in the assays ranged from 0.074 to 4.72 mm. Protein concentrations in the assays were as follows: A1.1, 1.6 and 4 ng/ml; Fabrazyme, 2 and 5 ng/ml. GraphPad Prism7 was used for all calculations.
Activity toward lipid substrates
For activity measurement, 50 ng of A1.1 or Fabrazyme were incubated with 5 μm NBD-C12–globotriaosylceramide, (NBD-Gb3) for 1 h at 37 °C in 150 mm citrate/phosphate buffers at different pH values (4.5–7.5), containing 0.05% (v/v) Triton X-100 and 0.2% (w/v) sodium taurocholate. The lipids were next extracted by the Bligh and Dyer protocol and subjected to HPTLC as described earlier (62). The plate was scanned for fluorescent lipids with a Typhoon FLA 9500. Kinetics parameters Km and kcat were determined using NBD-Gb3 substrate, at pH 4.5.
Alternatively, HPLC analysis and LC-MS/MS were used to study hydrolysis of C18-Gb3 (Matreya, State College, PA) and C18–lyso-Gb3 (Avanti, Alabaster, AL) by A1.1, respectively. For the degradation of C18-Gb3, 5 μm natural lipid were incubated with 3 μg of pure A1.1 or Fabrazyme overnight at 37 °C. Following extraction (Bligh and Dyer protocol), neutral glycosphingolipids were analyzed by HPLC using C17-sphinganine as internal standard (28). For lyso-Gb3, eight different concentrations of the lipid, ranging from 2 to 200 μm, were incubated with 2.5 μg/ml of A1.1 and Fabrazyme for 45 min at 37 °C. LC-MC/MS was used to measure glycosphingoid bases (lyso-glycosphingolipids), as described previously (30, 49). As internal standard [13C]lyso-Gb3 was used (30). Kinetics parameters Km and kcat were determined at pH 4.5, using GraphPad Prism7 for calculations. The mature form of A1.1, having a mass of 39 kDa, and the mature monomer human α-galactosidase (Fabrazyme), 51 kDa, adjusted to the carbohydrate mass, were used to calculate kcat values.
Uptake of TB474-labeled A1.1 by FD fibroblasts and confocal microscopy
Fabry fibroblasts from two different patients were grown in 12-well plates. Different amounts of pre-labeled TB747 with A1.1 were added to 500 μl of the culture media. The amounts were 0, 2.5, 5, 35, 75, and 150 μg. After overnight incubation, the cells were extensively washed with PBS and lysed in 100 μl of ice-cold 50 mm phosphate buffer, pH 6.5, supplemented with 0.1% Triton X-100. The total protein content of the cells was measured. Then 10 μg of total protein of the lysates, accounting to 15–20% of total cell lysate, were analyzed in SDS-PAGE. Quantification of the gels took place using ImageJ program.
The pre-labeling of the enzyme was done for 4 h, at room temperature, in 150 mm citrate/phosphate buffer, pH 5.5, at a final TB474 concentration of 0.25 μm, 100-μl volume, followed by removal of unbound TB474 by passing the sample over a 0.7-ml PierceTM polyacrylamide spin desalting column 7000 MWCO (Thermo Fisher Scientific), according to the manufacturer's protocol.
For the confocal microscopy experiments, the same Fabry fibroblasts were grown in 12-well plates containing coverslips, until they reached 70–80% confluency. On the day of the uptake, the culture medium was removed, and 500 μl of fresh culture medium was placed in each well. 75 μg of A1.1 pre-labeled with TB474 was applied to the fibroblasts overnight.
After the incubation, the fibroblasts were washed extensively with PBS and subsequently were fixed with 4% (w/v) formaldehyde/PBS for 25 min at room temperature. After the fixation, the cells were washed three times quickly with PBS and incubated for 10 min with 2% (w/v) BSA and 0.1% (w/v) saponin in PBS (“permeabilization buffer”). To stain for LAMP-1, the cells were incubated for 1 h at room temperature with rabbit anti-LAMP-1 antibody (Abcam) at a dilution of 1:400 in permeabilization buffer. The cells were washed three times in permeabilization buffer and incubated for 1 h at room temperature with Alexa Fluor 488-coupled donkey anti-rabbit IgG (Invitrogen), 1:500 in permeabilization buffer. After the incubation, the cells were further washed in permeabilization buffer and quickly with distilled water. The coverslips were mounted on a microscope slide (VWR) with ProLong Diamond antifade reagent containing DAPI (Molecular Probes). Imaging of the cells was performed with a Leica SP8 confocal microscope with a ×63/1.40 NA HC Plan Apo CS2 oil immersion objective and equipped with a hybrid detector. TB474-labeled A1.1 was imaged with excitation at 638 nm and emission at 650–700 nm; LAMP-1 with excitation at 488 nm and emission at 500–540 nm; and DAPI at 405 nm excitation and emission at 420–480 nm.
A1.1 treatment of FD fibroblasts
FD fibroblasts were grown in 12-well plates in a humidified incubator at 37 °C with 5% CO2. To 500 μl of the culture medium, 150 μg of A1.1 were added, followed by overnight incubation. Next, cells were extensively washed in PBS, collected, and lysed in 100 μl of ice-cold 50 mm phosphate buffer, pH 6.5, supplemented with 0.1% Triton X-100. Protein determination of lysates took place, and the lysates were used for lipid extractions, following LC-MS/MS analysis and 4MU–α-GAL activity assays.
Crystallization conditions and data collection
Purified A1.1 was concentrated to 5 mg/ml and incubated with 10 μm TB474 for 30 min at room temperature. After incubation, crystallization conditions were screened by sitting-drop vapor diffusion using the JCSG+ kit Premier (Molecular Dimensions). The screenings were performed by the NT8 robot (Formulatrix) at 20 °C, using 200 nl drops, with a well volume of 70 μl. After 12 h the crystals were formed at the H9 condition of the JCSG+ screening. The condition consisted of 0.1 m BisTris buffer, pH 5.5, 0.2 m LiSO4, 25% w/v PEG 3350. The crystal had a rhombus shape with 30.7/37-μm size. The crystal was flash-frozen in liquid nitrogen using 20% glycerol as a cryo-protectant. X-ray data collection was performed at the ESRF (Grenoble, France) on beamline ID30A-3, using a PIXEL, Eiger_4M (DECTRIS), X-ray detector. A total of 1900 images were collected, with an oscillation of 0.05° and exposure time of 0.01 s (total of 19 s). Then the data were processed by XDS and scaled by AIMLESS. The structure was solved using the molecular replacement method (MOLREP), having 1UAS as search model, through the CCP4 suite and further refined using REFMAC. Manual model building was done using Coot. Images of structures were obtained using CCP4mg, and amino acid alignments were performed using ClustalOmega and ESPript3.0.
The superposition of A1.1 and human proteins was performed by secondary structure-matching (59), using the A monomer of human α-galactosidase (PDB 3HG2) and our structure. Secondary structure matching aligned 348 residues with a root-mean-square deviation of 1.3358 Å of the C-α atoms of the aligned residues.
Author contributions
K. K., J. B., and J. M. A. conceptualization; K. K., M. A., E. v. M., B. I. F., and N. P. data curation; K. K., M. A., E. v. M., G. F. M., B. I. F., C. H. H., and N. P. formal analysis; K. K., E. v. M., G. F. M., N. G., H. S. O., A. S., D. B., and J. M. A. supervision; K. K., J. B., M. A., E. v. M., B. I. F., C. H. H., N. P., and J. M. A. validation; K. K. and J. M. A. investigation; K. K., M. A., E. v. M., R. H. W., G. F. M., N. G., M. J. F., R. K., P. V., B. I. F., and N. P. methodology; K. K. and J. M. A. writing-original draft; K. K. and J. M. A. project administration; K. K., J. B., M. A., E. v. M., R. H. W., A. S., D. B., and J. M. A. writing-review and editing; R. H. W., A. S., D. B., N. P., and J. M. A. resources; B. I. F. and N. P. software.
Supplementary Material
Acknowledgments
We thank Mina Mirzaian, Kimberley Zwiers, and Rafaella Tassoni for their technical support and discussion. We also thank Thomas J. M. Beenakker for the synthesis of TB474 ABP.
This work was supported by NWO ZonMW Grant 91116025. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7, Tables S1 and S2, and Scheme S1.
The atomic coordinates and structure factors (code 6F4C) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The gene sequence reported in this paper has been submitted to the Solcyc Database under accession no. GJZM-1660.
- α-GAL A
- human α-galactosidase
- FD
- Fabry disease
- ERT
- enzyme-replacement therapy
- α-GAL
- any α-galactosidase, including plant enzymes
- 4MU
- 4-methylumbelliferyl
- ABP
- activity-based probe
- CBB
- Coomassie Brilliant Blue
- ConA
- concanavalin A
- Gb3
- globotriaosylceramide
- lyso-Gb3
- globotriaosylsphingosine
- LacCer
- lactosylceramide
- lyso-LacCer
- lactosylsphingosine
- GBA
- human lysosomal glucocerebrosidase
- MPR
- mannose 6-phosphate receptor
- Man-6-P
- mannose 6-phosphate moiety
- HRP
- horseradish peroxidase
- NBD
- nitrobenzoxadiazole
- NBD-Gb3
- nitrobenzoxadiazole-C12-globotriaosylceramide
- DAPI
- 4′,6-diamidino-2-phenylindole
- TB474
- Cy5-labeled α-galactosidase activity-based probe
- ME741
- biotinylated α-galactosidase activity-based probe
- A1.1
- N. benthamiana α-galactosidase, identified in this study
- PDB
- Protein Data Bank
- dpi
- days post-infiltration
- rcf
- relative centrifugal force
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PNGase F
- peptide:N-glycosidase F
- EndoH
- endoglycanase H.
References
- 1. Carmi N., Zhang G., Petreikov M., Gao Z., Eyal Y., Granot D., and Schaffer A. A. (2003) Cloning and functional expression of alkaline α-galactosidase from melon fruit: similarity to plant SIP proteins uncovers a novel family of plant glycosyl hydrolases. Plant J. 33, 97–106 10.1046/j.1365-313X.2003.01609.x [DOI] [PubMed] [Google Scholar]
- 2. Henrissat B., and Romeu A. (1995) Families, superfamilies and subfamilies of glycosyl hydrolases. Biochem. J. 311, 350–351 10.1042/bj3110350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maranville E., and Zhu A. (2000) The carboxyl terminus of coffee bean α-galactosidase is critical for enzyme activity. Arch. Biochem. Biophys. 373, 225–230 10.1006/abbi.1999.1532 [DOI] [PubMed] [Google Scholar]
- 4. Gao Z., and Schaffer A. A. (1999) A novel alkaline α-galactosidase from melon fruit with a substrate preference for raffinose. Plant Physiol. 119, 979–988 10.1104/pp.119.3.979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fujimoto Z., Kaneko S., Momma M., Kobayashi H., and Mizuno H. (2003) Crystal structure of rice α-galactosidase complexed with d-galactose. J. Biol. Chem. 278, 20313–20318 10.1074/jbc.M302292200 [DOI] [PubMed] [Google Scholar]
- 6. Kim W.-D., Kobayashi O., Kaneko S., Sakakibara Y., Park G.-G., Kusakabe I., Tanaka H., and Kobayashi H. (2002) α-Galactosidase from cultured rice (Oryza sativa L. var. Nipponbare) cells. Phytochemistry 61, 621–630 10.1016/S0031-9422(02)00368-0 [DOI] [PubMed] [Google Scholar]
- 7. Zha H.-G., Flowers V. L., Yang M., Chen L.-Y., and Sun H. (2012) Acidic α-galactosidase is the most abundant nectarin in floral nectar of common tobacco (Nicotiana tabacum). Ann. Bot. 109, 735–745 10.1093/aob/mcr321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murali R., Ioannou Y. A., Desnick R. J., and Burnett R. M. (1994) Crystallization and preliminary X-ray analysis of human α-galactosidase A complex. J. Mol. Biol. 239, 578–580 10.1006/jmbi.1994.1397 [DOI] [PubMed] [Google Scholar]
- 9. Desnick R. J. (1995) α-Galactosidase A deficiency: Fabry disease. Metab. Mol. Basis Inherit. Dis. [Google Scholar]
- 10. Toumi D. S., Reustle I., Krczal G. M., Ghorbel G., Mliki A., and Höfer A. (2012) Molecular cloning and characterisation of a cDNA encoding a putative alkaline α-galactosidase from grapevine (Vitis vinifera L.) that is differentially expressed under osmotic stress. Acta Physiol. Plant. 34, 891–903 10.1007/s11738-011-0887-5 [DOI] [Google Scholar]
- 11. Carchon H., and DeBruyne C. K. (1975) Purification and properties of coffee-bean α-d-galactosidase. Carbohydr. Res. 41, 175–189 10.1016/S0008-6215(00)87017-2 [DOI] [PubMed] [Google Scholar]
- 12. Marraccini P., Rogers W. J., Caillet V., Deshayes A., Granato D., Lausanne F., Lechat S., Pridmore D., and Pétiard V. (2005) Biochemical and molecular characterization of α-d-galactosidase from coffee beans. Plant Physiol. Biochem. 43, 909–920 10.1016/j.plaphy.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 13. Haibach F., Hata J., Mitra M., Dhar M., Harmata M., Sun P., and Smith D. (1991) Purification and characterization of a Coffea canephora α-d-galactosidase isozyme. Biochem. Biophys. Res. Commun. 181, 1564–1571 10.1016/0006-291X(91)92117-3 [DOI] [PubMed] [Google Scholar]
- 14. Ioannou Y. A., Zeidner K. M., Gordon R. E., and Desnick R. J. (2001) Fabry disease: preclinical studies demonstrate the effectiveness of α-galactosidase A replacement in enzyme-deficient mice. Am. J. Hum. Genet. 68, 14–25 10.1086/316953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schiffmann R., Kopp J. B., Austin H. A. 3rd., Sabnis S., Moore D. F., Weibel T., Balow J. E., and Brady R. O. (2001) Enzyme-replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285, 2743–2749 10.1001/jama.285.21.2743 [DOI] [PubMed] [Google Scholar]
- 16. Hamers M. N., Westerveld A., Khan M., and Tager J. M. (1977) Characterization of α-galactosidase isoenzymes in normal and Fabry human-Chinese hamster somatic cell hybrids. Hum. Genet. 36, 289–297 10.1007/BF00446279 [DOI] [PubMed] [Google Scholar]
- 17. Bishop D. F., Kornreich R., and Desnick R. J. (1988) Structural organization of the human α-galactosidase A gene: further evidence for the absence of a 3′ untranslated region. Proc. Natl. Acad. Sci. U.S.A. 85, 3903–3907 10.1073/pnas.85.11.3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakuraba H., Murata-Ohsawa M., Kawashima I., Tajima Y., Kotani M., Ohshima T., Chiba Y., Takashiba M., Jigami Y., Fukushige T., Kanzaki T., and Itoh K. (2006) Comparison of the effects of agalsidase α and agalsidase β on cultured human Fabry fibroblasts and Fabry mice. J. Hum. Genet. 51, 180–188 [DOI] [PubMed] [Google Scholar]
- 19. Shen J.-S., Busch A., Day T. S., Meng X.-L., Yu C. I., Dabrowska-Schlepp P., Fode B., Niederkrüger H., Forni S., Chen S., Schiffmann R., Frischmuth T., and Schaaf A. (2016) Mannose receptor-mediated delivery of moss-made α-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J. Inherit. Metab. Dis. 39, 293–303 10.1007/s10545-015-9886-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sweeley C. C., and Bernard Klionsky (1963) Fabry's disease: classification as a sphingolipidosis and partial characterization of a novel glycolipid. J. Biol. Chem. 238, 3148–3150 [PubMed] [Google Scholar]
- 21. Eng C., Muenzer J., Wraith E., Beck M., Giugliani R., Harmatz P., Vellodi A., Martin R., Ramaswami U., Calikoglu M., Vijayaraghavan S., Puga A., Ulbrich B., Shinawi M., Cleary M., and Wendt S. (2007) 39 Formation of a lysosomal disease testing network to enhance the delivery of diagnostic services to patients with lysosomal storage disorders. Mol. Genet. Metab. 92, 20 10.1016/j.ymgme.2007.08.044 [DOI] [Google Scholar]
- 22. Kizhner T., Azulay Y., Hainrichson M., Tekoah Y., Arvatz G., Shulman A., Ruderfer I., Aviezer D., and Shaaltiel Y. (2015) Characterization of a chemically modified plant cell culture expressed human α-galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metab. 114, 259–267 [DOI] [PubMed] [Google Scholar]
- 23. Arends M., Biegstraaten M., Hughes D. A., Mehta A., Elliott P. M., Oder D., Watkinson O. T., Vaz F. M., van Kuilenburg A. B. P., Wanner C., and Hollak C. E. M. (2017) Retrospective study of long-term outcomes of enzyme-replacement therapy in Fabry disease: Analysis of prognostic factors. PLoS ONE 12, e0182379 10.1371/journal.pone.0182379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Linthorst G. E., Hollak C. E., Donker-Koopman W. E., Strijland A., and Aerts J. M. (2004) Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase α and β. Kidney Int. 66, 1589–1595 10.1111/j.1523-1755.2004.00924.x [DOI] [PubMed] [Google Scholar]
- 25. Lenders M., Stypmann J., Duning T., Schmitz B., Brand S.-M., and Brand E. (2016) Serum-mediated inhibition of enzyme-replacement therapy in Fabry disease. J. Am. Soc. Nephrol. 27, 256–264 10.1681/ASN.2014121226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferraz M. J., Marques A. R., Appelman M. D., Verhoek M., Strijland A., Mirzaian M., Scheij S., Ouairy C. M., Lahav D., Wisse P., Overkleeft H. S., Boot R. G., and Aerts J. M. (2016) Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 590, 716–725 10.1002/1873-3468.12104 [DOI] [PubMed] [Google Scholar]
- 27. Aerts J. M., Groener J. E., Kuiper S., Donker-Koopman W. E., Strijland A., Ottenhoff R., van Roomen C., Mirzaian M., Wijburg F. A., Linthorst G. E., Vedder A. C., Rombach S. M., Cox-Brinkman J., Somerharju P., Boot R. G., et al. (2008) Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. U.S.A. 105, 2812–2817 10.1073/pnas.0712309105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gold H., Mirzaian M., Dekker N., Joao Ferraz M., Lugtenburg J., Codée J. D., van der Marel G. A., Overkleeft H. S., Linthorst G. E., Groener J. E., Aerts J. M., and Poorthuis B. J. (2013) Quantification of globotriaosylsphingosine in plasma and urine of Fabry patients by stable isotope ultraperformance liquid chromatography tandem mass spectrometry. Clin. Chem. 59, 547–556 10.1373/clinchem.2012.192138 [DOI] [PubMed] [Google Scholar]
- 29. Mirzaian M., Wisse P., Ferraz M. J., Marques A. R. A., Gabriel T. L., van Roomen C. P. A. A., Ottenhoff R., van Eijk M., Codée J. D. C., van der Marel G. A., Overkleeft H. S., and Aerts J. M. (2016) Accurate quantification of sphingosine-1-phosphate in normal and Fabry disease plasma, cells and tissues by LC-MS / MS with 13C-encoded natural S1P as internal standard. Clin. Chim. Acta. 459, 36–44 10.1016/j.cca.2016.05.017 [DOI] [PubMed] [Google Scholar]
- 30. Mirzaian M., Wisse P., Ferraz M. J., Marques A. R. A., Gaspar P., Oussoren S. V., Kytidou K., Codée J. D. C., van der Marel G., Overkleeft H. S., and Aerts J. M. (2017) Simultaneous quantitation of sphingoid bases by UPLC-ESI-MS/MS with identical 13C-encoded internal standards. Clin. Chim. Acta 466, 178–184 10.1016/j.cca.2017.01.014 [DOI] [PubMed] [Google Scholar]
- 31. Choi L., Vernon J., Kopach O., Minett M. S., Mills K., Clayton P. T., Meert T., and Wood J. N. (2015) The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci. Lett. 594, 163–168 10.1016/j.neulet.2015.01.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanchez-Niño M. D., Carpio D., Sanz A. B., Ruiz-Ortega M., Mezzano S., and Ortiz A. (2015) Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 24, 5720–5732 10.1093/hmg/ddv291 [DOI] [PubMed] [Google Scholar]
- 33. Witte M. D., van der Marel G. A., Aerts J. M., and Overkleeft H. S. (2011) Irreversible inhibitors and activity-based probes as research tools in chemical glycobiology. Org. Biomol. Chem. 9, 5908–5926 10.1039/c1ob05531c [DOI] [PubMed] [Google Scholar]
- 34. Willems L. I., Jiang J., Li K. Y., Witte M. D., Kallemeijn W. W., Beenakker T. J. N., Schröder S. P., Aerts J. M. F. G., Van Der Marel G. A., Codée J. D. C., and Overkleeft H. S. (2014) From covalent glycosidase inhibitors to activity-based glycosidase probes. Chemistry 20, 10864–10872 10.1002/chem.201404014 [DOI] [PubMed] [Google Scholar]
- 35. Witte M. D., Kallemeijn W. W., Aten J., Li K.-Y., Strijland A., Donker-Koopman W. E., van den Nieuwendijk A. M., Bleijlevens B., Kramer G., Florea B. I., Hooibrink B., Hollak C. E., Ottenhoff R., Boot R. G., van der Marel G. A., et al. (2010) Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 6, 907–913 10.1038/nchembio.466 [DOI] [PubMed] [Google Scholar]
- 36. Kallemeijn W. W., Witte M. D., Voorn-Brouwer T. M., Walvoort M. T., Li K.-Y., Codée J. D., van der Marel G. A., Boot R. G., Overkleeft H. S., and Aerts J. M. (2014) A sensitive gel-based method combining distinct cyclophellitol-based probes for the identification of acid/base residues in human retaining β-glucosidases. J. Biol. Chem. 289, 35351–35362 10.1074/jbc.M114.593376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kallemeijn W. W., Scheij S., Hoogendoorn S., Witte M. D., Herrera Moro Chao D., van Roomen C. P., Ottenhoff R., Overkleeft H. S., Boot R. G., and Aerts J. M. (2017) Investigations on therapeutic glucocerebrosidases through paired detection with fluorescent activity-based probes. PLoS ONE 12, e0170268 10.1371/journal.pone.0170268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Willems L. I., Beenakker T. J., Murray B., Gagestein B., van den Elst H., van Rijssel E. R., Codée J. D. C., Kallemeijn W. W., Aerts J. M., van der Marel G. A., and Overkleeft H. S. (2014) Synthesis of α- and β-galactopyranose-configured isomers of cyclophellitol and cyclophellitol aziridine. Eur. J. Org. Chem. 2014, 6044–6056 10.1002/ejoc.201402589 [DOI] [Google Scholar]
- 39. Willems L. I., Beenakker T. J., Murray B., Scheij S., Kallemeijn W. W., Boot R. G., Verhoek M., Donker-Koopman W. E., Ferraz M. J., van Rijssel E. R., Florea B. I., Codée J. D., van der Marel G. A., Aerts J. M., and Overkleeft H. S. (2014) Potent and selective activity-based probes for GH27 human retaining α-galactosidases. J. Am. Chem. Soc. 136, 11622–11625 10.1021/ja507040n [DOI] [PubMed] [Google Scholar]
- 40. Jiang J., Kallemeijn W. W., Wright D. W., van den Nieuwendijk A. M. C. H., Rohde V. C., Folch E. C., van den Elst H., Florea B. I., Scheij S., Donker-Koopman W. E., Verhoek M., Li N., Schürmann M., Mink D., Boot R. G., et al. (2015) In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 α-l-fucosidases. Chem. Sci. 6, 2782–false 10.1039/C4SC03739A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu L., Jiang J., Jin Y., Kallemeijn W. W., Kuo C.-L., Artola M., Dai W., van Elk C., van Eijk M., van der Marel G. A., Codee J. D. C., Florea B. I., Aerts J. M. F. G., Overkleeft H. S., and Davies G. J. (2017) Activity-based probes for functional interrogation of retaining β-glucuronidases. Nat. Chem. Biol. 13, 867–873 10.1038/nchembio.2395 [DOI] [PubMed] [Google Scholar]
- 42. Chandrasekar B., Colby T., Emran Khan Emon A., Jiang J., Hong T. N., Villamor J. G., Harzen A., Overkleeft H. S., and van der Hoorn R. A. (2014) Broad-range glycosidase activity profiling. Mol. Cell. Proteomics 13, 2787–2800 10.1074/mcp.O114.041616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jiang J., Kuo C.-L., Wu L., Franke C., Kallemeijn W. W., Florea B. I., van Meel E., van der Marel G. A., Codée J. D., Boot R. G., Davies G. J., Overkleeft H. S., and Aerts J. M. (2016) Detection of active mammalian GH31 α-glucosidases in health and disease using in-class, broad-spectrum activity-based probes. ACS Cent. Sci. 2, 351–358 10.1021/acscentsci.6b00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilbers R. H., Westerhof L. B., van Noort K., Obieglo K., Driessen N. N., Everts B., Gringhuis S. I., Schramm G., Goverse A., Smant G., Bakker J., Smits H. H., Yazdanbakhsh M., Schots A., and Hokke C. H. (2017) Production and glyco-engineering of immunomodulatory helminth glycoproteins in plants. Sci. Rep. 7, 45910 10.1038/srep45910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guce A. I., Clark N. E., Salgado E. N., Ivanen D. R., Kulminskaya A. A., Brumer H. 3rd., and Garman S. C. (2010) Catalytic mechanism of human α-galactosidase. J. Biol. Chem. 285, 3625–3632 10.1074/jbc.M109.060145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Motabar O., Liu K., Southall N., Marugan J. J., Goldin E., Sidransky E., and Zheng W. (2010) High throughput screening for inhibitors of α-galactosidase. Curr. Chem. Genomics 4, 67–73 10.2174/1875397301004010067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Groener J. E., Poorthuis B. J., Kuiper S., Helmond M. T., Hollak C. E., and Aerts J. M. (2007) HPLC for simultaneous quantification of total ceramide, glucosylceramide, and ceramide trihexoside concentrations in plasma. Clin. Chem. 53, 742–747 10.1373/clinchem.2006.079012 [DOI] [PubMed] [Google Scholar]
- 48. Chien S.-F., Chen S.-H., and Chien M.-Y. (2008) Cloning, expression, and characterization of rice α-galactosidase. Plant Mol. Biol. Rep. 26, 213–224 10.1007/s11105-008-0035-6 [DOI] [Google Scholar]
- 49. Kytidou K., Beenakker T. J. M., Westerhof L. B., Hokke C. H., Moolenaar G. F., Goosen N., Mirzaian M., Ferraz M. J., de Geus M., Kallemeijn W. W., Overkleeft H. S., Boot R. G., Schots A., Bosch D., and Aerts J. M. F. G. (2017) Human α-galactosidases transiently produced in Nicotiana benthamiana leaves: new insights in substrate specificities with relevance for Fabry disease. Front. Plant Sci. 8, 1026 10.3389/fpls.2017.01026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu A., Wang Z.-K., and Goldstein J. (1995) Identification of tyrosine 108 in coffee bean α-galactosidase as an essential residue for the enzyme activity. Biochim. Biophys. Acta 1247, 260–264 10.1016/0167-4838(94)00228-9 [DOI] [PubMed] [Google Scholar]
- 51. Garman S. C., Hannick L., Zhu A., and Garboczi D. N. (2002) The 1.9 Å structure of α-N-acetylgalactosaminidase. Structure 10, 425–434 10.1016/S0969-2126(02)00726-8 [DOI] [PubMed] [Google Scholar]
- 52. Mathew C. D., and Balasubramaniam K. (1987) Mechanism of action of α-galactosidase. Phytochemistry. 26, 1299–1300 10.1016/S0031-9422(00)81798-7 [DOI] [PubMed] [Google Scholar]
- 53. Zhu A., and Goldstein J. (1994) Cloning and functional expression of a cDNA encoding coffee bean α-galactosidase. Gene 140, 227–231 10.1016/0378-1119(94)90548-7 [DOI] [PubMed] [Google Scholar]
- 54. Ferraz M. J., Kallemeijn W. W., Mirzaian M., Herrera Moro D., Marques A., Wisse P., Boot R. G., Willems L. I., Overkleeft H. S., and Aerts J. M. (2014) Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta 1841, 811–825 10.1016/j.bbalip.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 55. Rezende R. M., and Weiner H. L. (2017) History and mechanisms of oral tolerance. Semin. Immunol. 30, 3–11 10.1016/j.smim.2017.07.004 [DOI] [PubMed] [Google Scholar]
- 56. Westerhof L. B., Wilbers R. H., van Raaij D. R., Nguyen D.-L., Goverse A., Henquet M. G., Hokke C. H., Bosch D., Bakker J., and Schots A. (2014) Monomeric IgA can be produced in planta as efficient as IgG, yet receives different N-glycans. Plant Biotechnol. J. 12, 1333–1342 10.1111/pbi.12251 [DOI] [PubMed] [Google Scholar]
- 57. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro purification, enrichment, pre fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 10.1038/nprot.2007.261 [DOI] [PubMed] [Google Scholar]
- 58. Westerhof L. B., Wilbers R. H., Roosien J., van de Velde J., Goverse A., Bakker J., and Schots A. (2012) 3D domain swapping causes extensive multimerisation of human interleukin-10 when expressed in planta. PLoS ONE 7, e46460 10.1371/journal.pone.0046460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 60. Aerts J. M., Donker-Koopman W. E., Koot M., Barranger J. A., Tager J. M., and Schram A. (1986) Deficient activity of glucocerebrosidase in urine from patients with type 1 Gaucher disease. Clin. Chim. Acta 158, 155–163 10.1016/0009-8981(86)90231-7 [DOI] [PubMed] [Google Scholar]
- 61. Blom D., Speijer D., Linthorst G. E., Donker-Koopman W. G., Strijland A., and Aerts J. M. (2003) Recombinant enzyme therapy for Fabry disease: absence of editing of human α-galactosidase A mRNA. Am. J. Hum. Genet. 72, 23–31 10.1086/345309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marques A. R., Mirzaian M., Akiyama H., Wisse P., Ferraz M. J., Gaspar P., Ghauharali-van der Vlugt K., Meijer R., Giraldo P., Alfonso P., Irún P., Dahl M., Karlsson S., Pavlova E. V., et al. (2016) Glucosylated cholesterol in mammalian cells and tissues: formation and degradation by multiple cellular β-glucosidases. J. Lipid Res. 57, 451–463 10.1194/jlr.M064923 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.