

Abstract
Ribonucleotide reductases (RNRs) convert ribonucleotides to deoxynucleotides, a process essential for DNA biosynthesis and repair. Class Ia RNRs require two dimeric subunits for activity: an α2 subunit that houses the active site and allosteric regulatory sites and a β2 subunit that houses the diferric tyrosyl radical cofactor. Ribonucleotide reduction requires that both subunits form a compact α2β2 state allowing for radical transfer from β2 to α2. RNR activity is regulated allosterically by dATP, which inhibits RNR, and by ATP, which restores activity. For the well-studied Escherichia coli class Ia RNR, dATP binding to an allosteric site on α promotes formation of an α4β4 ring-like state. Here, we investigate whether the α4β4 formation causes or results from RNR inhibition. We demonstrate that substitutions at the α–β interface (S37D/S39A-α2, S39R-α2, S39F-α2, E42K-α2, or L43Q-α2) that disrupt the α4β4 oligomer abrogate dATP-mediated inhibition, consistent with the idea that α4β4 formation is required for dATP's allosteric inhibition of RNR. Our results further reveal that the α–β interface in the inhibited state is highly sensitive to manipulation, with a single substitution interfering with complex formation. We also discover that residues at the α–β interface whose substitution has previously been shown to cause a mutator phenotype in Escherichia coli (i.e. S39F-α2 or E42K-α2) are impaired only in their activity regulation, thus linking this phenotype with the inability to allosterically down-regulate RNR. Whereas the cytotoxicity of RNR inhibition is well-established, these data emphasize the importance of down-regulation of RNR activity.
Keywords: allosteric regulation, nucleoside/nucleotide biosynthesis, mutagenesis mechanism, ribonucleotide reductase, protein–protein interaction, α4β4 ring, allosteric inhibition, radical-based chemistry, tyrosyl radical cofactor
Introduction
Ribonucleotide reductases (RNRs)4 are essential enzymes for all living organisms due to their role in providing deoxynucleotides for DNA synthesis and repair (1–4). In particular, RNRs use radical-based chemistry to convert ribonucleotides (ribonucleoside diphosphates in the case of class Ia RNRs) into deoxyribonucleotides (Fig. 1A). The prototypic enzyme for understanding RNR chemistry and regulation is the class Ia RNR from Escherichia coli. This enzyme consists of two dimeric subunits, α2 and β2. α2 contains the catalytic machinery for nucleotide reduction as well as two allosteric nucleotide-binding sites: an allosteric specificity site at the dimer interface that modulates the affinity of the enzyme for pyrimidine versus purine substrates (5–7) and an allosteric activity site at an N-terminal cone domain that modulates overall activity (Fig. 1B) (8–10). β2 contains the diferric tyrosyl radical cofactor that is essential to RNR's catalytic mechanism (Fig. 1B) (11–14). Enzyme activity requires the formation of a compact α2β2 state that is capable of transferring the radical from β2 to α2 to generate a catalytically essential Cys-439 thiyl radical species (Fig. 1, C and D) (15). Residues responsible for the radical transfer (RT) from β2 to α2 include Tyr-356, which is part of the C-terminal tail of β2 that is disordered in all crystal structures, and Tyr-730–Tyr-731 of the α2 subunit (Fig. 1D) (4). The C-terminal tail of β2 also provides much of the binding affinity of β2 for α2 (16), and although no high resolution structures of the α2β2 state have been determined, the binding site on α2 for the last 13 residues of the C-terminal tail of β2 has been visualized by crystallization of α2 with a β2 peptide (Fig. 1B) (8).
Figure 1.
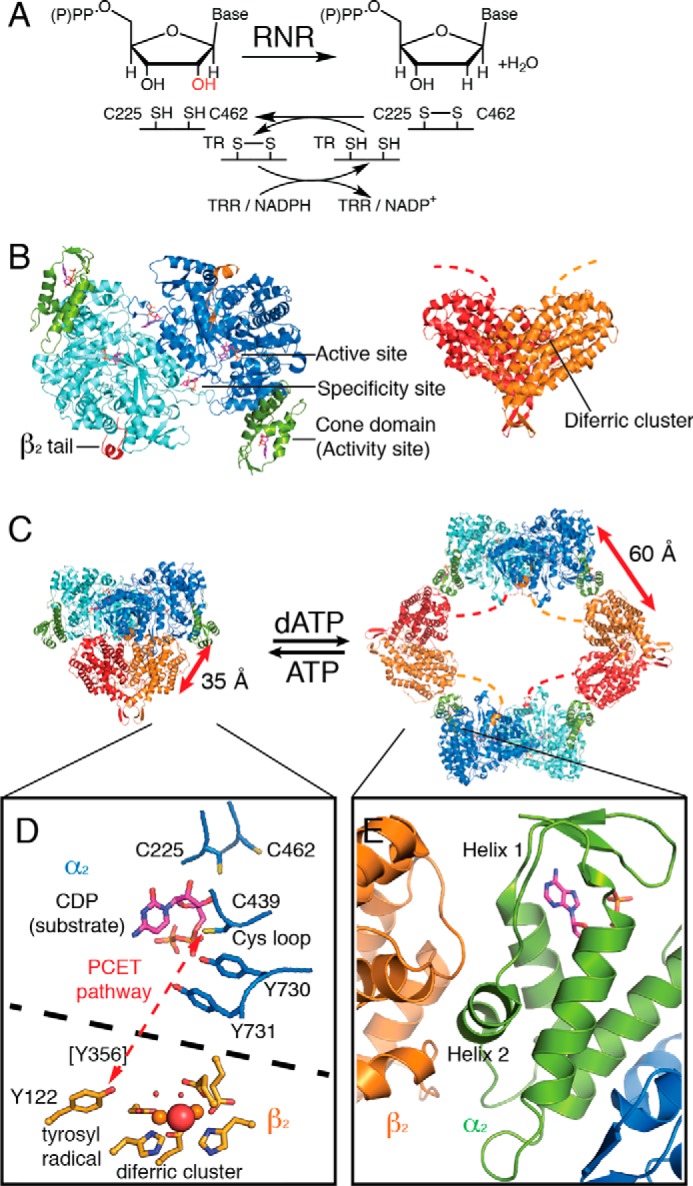
Oligomeric transitions in E. coli class Ia RNR influence proton-coupled electron transfer between subunits. A, RNR reduces ribonucleoside di- (class Ia) or triphosphates into deoxyribonucleoside di- or triphosphates. For E. coli class Ia RNR, the reaction is accompanied with disulfide bond formation between Cys-225 and Cys-462, which can be re-reduced in vivo by NADPH through a series of disulfide exchange reactions (4). B, two proteins are required for class Ia RNR activity: α2, the reductase subunit (blue/cyan/green); and β2, the radical subunit (red/orange). Dashed lines represent disordered C-terminal residues of β2 that were not resolved in crystal structures. C, during turnover, a transient α2β2 complex is formed that is capable of radical transfer. No high resolution structure is available for this enzyme state; the figure shown is a docking model based on shape-complementarity (66), spectroscopic data (67, 68), negative-stain EM (10), and SAXS (10). High levels of dATP result in the formation of an inactive α4β4-ring structure (Protein Data Bank code 5CNS (7)) in which the N-terminal cone domain of α2 (green) forms an interface with β2. The radical transfer pathway is interrupted in this oligomeric state. Not only is the distance between the diferric tyrosyl radical cofactor in β2 and the catalytic Cys-439 in α2 now ∼60 Å instead of ∼35 Å, but the radical pathway is disrupted by the presence of a large (∼100 Å) solvent-filled cavity in the center of the α4β4 ring. D, proton-coupled electron transfer (PCET) pathway in the α2β2 active complex (red dashed line). Tyr-356 is present on the C-terminal tail of β2 and is essential for radical propagation, but it has never been visualized in a crystal structure. E, zoom-in of secondary structure at the α–β interface. Helices 1 and 2 of the cone domain are responsible for contacting β. The activity regulator (hydrolyzed from dATP to dADP in this structure), shown as magenta sticks, is not in direct contact with helix 2.
Once a compact α2β2 state is formed and a thiyl radical species is generated on Cys-439, catalysis occurs via radical-based substrate intermediates. The hydroxyl group of the ribonucleotide substrate is lost in the form of water with two cysteines in the active site (Cys-225 and Cys-462, Fig. 1D) providing the reducing equivalents (17, 18). Following product formation, the radical species returns to β2, and the compact α2β2 complex disassembles (19), allowing for re-reduction of Cys-225 and Cys-462 via a series of disulfide exchange reactions that utilize thioredoxin (TR), thioredoxin reductase (TRR), and NADPH (4) (Fig. 1A).
An improper balance in the ratio of deoxyribonucleotides to ribonucleotides in a cell can be toxic (20–25), and RNRs have been proposed (3) to play an important role in maintaining this balance by sensing and responding to the ratio of dATP to ATP in the cell (26). When dATP concentrations are too high, RNR activity is inhibited; restoration of the appropriate ratio of dATP to ATP reverses the RNR inhibition (5, 8, 9, 27). The molecular mechanism of dATP-induced inhibition for the E. coli class Ia RNR appears to involve an oligomeric state change from the compact α2β2 state to an α4β4 state (Fig. 1C) (10, 28). In particular, dATP binding to the N-terminal cone domain of α leads to the establishment of a small (∼580 Å2) α–β interface and the formation of an α4β4 ring-like structure, which has been captured by small-angle X-ray scattering (SAXS), electron microscopy (EM), and crystallography (7, 10, 29). In this α4β4 state, the C-terminal tail of β also contacts α, maintaining the interaction observed in the β peptide-bound α2 structure (8, 29) (Fig. 1, B and C). Reversal of E. coli class Ia RNR inhibition occurs when dATP/ATP ratio decreases, which leads to the displacement of dATP by ATP in the cone domain, and results in a shift of the equilibrium away from the α4β4 state toward the active α2β2 state (10, 30).
Although it is known that dATP inhibits RNR activity and leads to α4β4 ring formation in E. coli class Ia RNR, there is no experimental evidence that ring formation causes inhibition rather than being a result of the inhibition. We have previously proposed that the α4β4 state is an inactive form of RNR, one that is incapable of RT between β and α subunits due to the lengthened RT distance (60 Å) and the abrogated RT pathway (10). Here, we seek to obtain experimental evidence whether ring formation is causative of inactivity. We reason that if ring formation is indeed responsible for dATP-induced RNR inhibition, then mutations that disrupt ring stability would result in an RNR that is insensitive to dATP. Here, we test this hypothesis by employing site-directed mutagenesis to prepare RNR mutant proteins that have substitutions at the α–β interface, including two RNR variants that were previously found to cause mutator phenotypes in vivo (31). We then used negative stain EM and analytical ultracentrifugation (AUC) to identify RNR mutant proteins that cannot form the α4β4 oligomeric state and subjected those enzymes to dATP-inhibition studies. We find that RNRs that cannot adopt the α4β4 oligomeric state also cannot be inhibited by dATP. In other words, formation of the α4β4 state is required for dATP allosteric inhibition. The implications of these findings are also discussed.
Results
Select number of residues make contact across the α4β4 interface
The α residues from helices 1 and 2 of the cone domain contact β (Fig. 1E) with the majority of interactions being made by residues from helix 2. Four residues on helix 2 (Ser-37, Ser-39, Glu-42, and Arg-44) and His-23 on helix 1 form hydrogen-bonding interactions, including a salt bridge, across the α–β interface (Fig. 2A). Hydrophobic residues from both α and β also contribute to the interface packing. For example, Ile-297 of β fits into a hydrophobic pocket composed of Leu-19 of α helix 1 and Leu-43, Ile-47 and Phe-49 of α helix 2 (Fig. 2B). To test the hypothesis that E. coli class Ia RNR inhibition depends upon α4β4 ring formation, mutations of residues at the interface were prepared. In particular, five RNR mutants with substitutions in α helix 2 were generated as follows: four single mutations, S39R-α2, S39F-α2, E42K-α2, and L43Q-α2, and a double mutation, S37D/S39A-α2 (Table 1). All residues chosen are involved in the α–β interface interactions, shown in Fig. 2. All single substitutions are to larger residues that could sterically block formation of the interface. Four substitutions change the charge of residues that are involved in at least a hydrogen bond (S39R, S39F, E42K, and S37D/S39A), and one substitution, L43Q, is expected to alter the hydrophobic pocket on α that is occupied by Ile-297 of β. Finally, two substitutions, S39F and E42K, were chosen because these substitutions in E. coli class Ia RNR were previously shown to disrupt nucleotide pools in E. coli (31).
Figure 2.
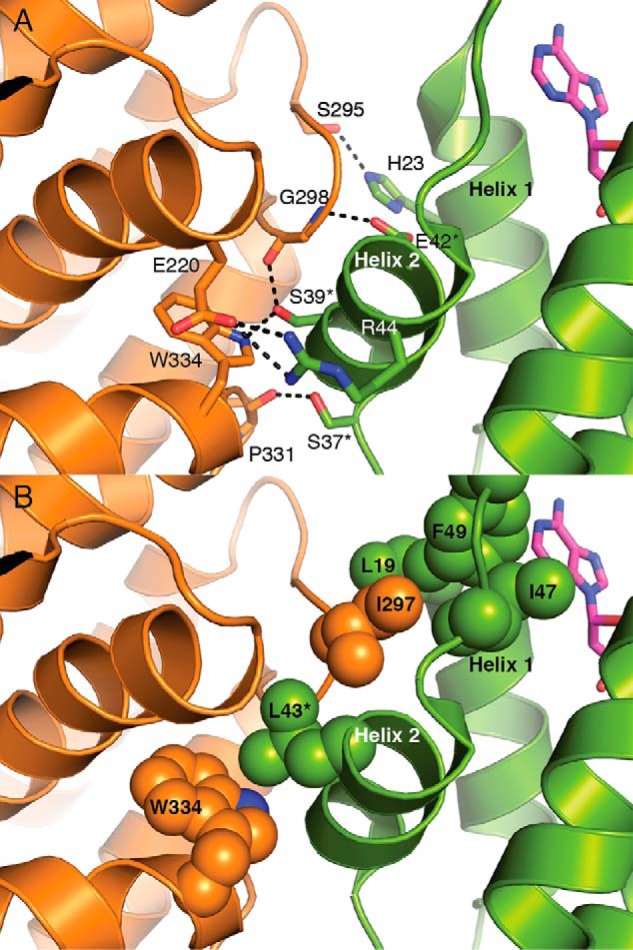
Residues along helix 2 of the cone domain are targets for disrupting the α4β4 interface. A, α–β interface in the 2.97-Å resolution structure of dATP-inhibited E. coli class Ia RNR in the α4β4 state (7). Colors are as in Fig. 1E. Deoxyadenosine moiety of the activity regulator is shown as magenta sticks. Interface residues that form hydrogen bonds between the cone domain and β are also shown as sticks. Ser-37, Ser-39, Arg-44, and Glu-42 belong to helix 2; His-23 belongs to helix 1. B, α–β interface also shows a series of hydrophobic residues that form hydrophobic contacts. These residues include β residue Ile-297, which inserts into a hydrophobic groove between helix 1 and helix 2. Leu-43, Ile-47, and Phe-49 belong to helix 2; Leu-19 belongs to helix 1. Residues mutated in this study are labeled with an asterisk.
Table 1.
E. coli class Ia RNR α2 mutant proteins generated in this study and a brief summary of results
| Helix 2 mutant proteins | Disrupted contacts with β2 | Ring formation upon addition of dATP observed via EM or AUC | Effect of addition of dATP on activity as compared with active conditions |
|---|---|---|---|
| WT | None | Yes (EM, AUC) | ∼ 7% of maximal activity |
| S37D/S39A | Pro-331, Gly-298, and Trp-334 (hydrogen bonds) | No (AUC) | No detectable loss in activity |
| S39R | Gly-298 and Trp-334 (hydrogen bonds) | No (AUC) | No detectable loss in activity |
| S39F | Gly-298 and Trp-334 (hydrogen bonds) | No (EM, AUC) | No detectable loss in activity |
| E42K | Gly-298 (hydrogen bond) | No (EM, AUC) | No detectable loss in activity |
| L43Q | Trp-334 (hydrophobic) | No (EM, AUC) | ∼ 85% of maximal activity |
Mutation of interface residues leads to a dramatic reduction in α4β4-ring formation
Negative stain EM provides a visual method for assessing the propensity of E. coli class Ia RNR to form α4β4 rings (10) at concentrations at or below physiological levels. As shown previously (10), α4β4 rings form in the presence of a physiological concentration of the allosteric effector dATP (175 μm), substrate CDP (1 mm), α2 (150 nm), and met-β2 (150 nm) (met-β2 is β2 in which the radical has been quenched by hydroxyurea) (Fig. 3A). In contrast, when α2 and met-β2 are mixed (150 nm) in the presence of 1 mm CDP and allosteric activity effector ATP (3 mm), the physiological concentration of ATP, mostly dissociated particles (free α2 and free β2) and some associated α2β2 are observed (Fig. 3B). The results for the mutant proteins are dramatically different from wildtype (WT). No α4β4 rings are observed in the presence of 175 μm dATP and 1 mm CDP when met-β2 is mixed with α2 mutant proteins S39F, E42K, or L43Q at the same concentrations used above (Fig. 3C). Because of small particle dimensions of the individual subunits and/or α2β2 complex, no attempt was made to extract and classify the particles, but we inspected over 1,000 α2β2 equivalents for each mutant protein with no sign of α4β4 rings.
Figure 3.
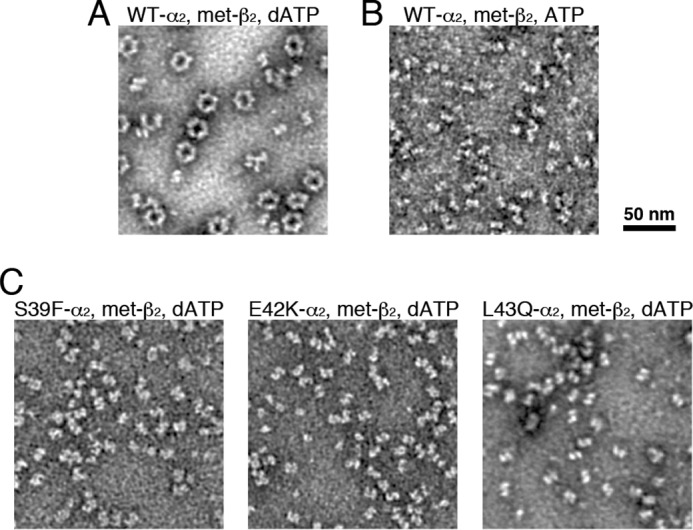
Negative stain EM reveals the inability of RNR interface mutant proteins to form α4β4 rings in the presence of dATP. A, WT α2 (150 nm) and met-β2 (150 nm) with 1 mm CDP substrate and 175 μm dATP effector. The β2 used in all EM experiments was met-β2, β2 in which the radical species has been quenched with hydroxyurea. B, WT α2 (150 nm) and met-β2 (150 nm) with 1 mm CDP substrate and 3 mm ATP effector. C, 150 nm RNR mutant proteins S39F, E42K, and L43Q and met-β2 (150 nm) with 1 mm CDP substrate and 175 μm dATP effector.
The enzyme concentrations used for EM (150 nm) are somewhat below the estimated RNR concentration in vivo, 0.5–2 μm (32), and similar to those used in in vitro activity assays, 50–100 nm. To assess ring formation at high enzyme concentrations, we turned to AUC. At high RNR enzyme concentrations, EM grids become too crowded, but higher enzyme concentrations are not problematic for AUC. We previously used AUC to show that the dATP-induced α4β4 ring formation was independent of protein concentration in the 1–10 μm range for WT α2 and met-β2 (10). The α4β4 ring corresponds to a peak with a sedimentation coefficient of 15.6 S, which is the dominant peak at both 1 and 10 μm WT α2/met-β2 in the presence of 175 μm dATP (Fig. 4A, upper traces). In the presence of ATP (3 mm), the dominant peak at 1 μm of protein is ∼10 S, whereas at 10 μm the dominant peak is ∼15 S, close to a value that corresponds to α4β4 rings (15.6 S) (Fig. 4A, lower traces). Additionally, in the presence of ATP, the peaks are broadened. Both the protein concentration-dependent peak shift and the peak broadening suggest that in the presence of ATP, an equilibrium mixture exists composed of both α2β2 and α4β4 states, which is driven toward α4β4 by an increase in protein concentration.
Figure 4.
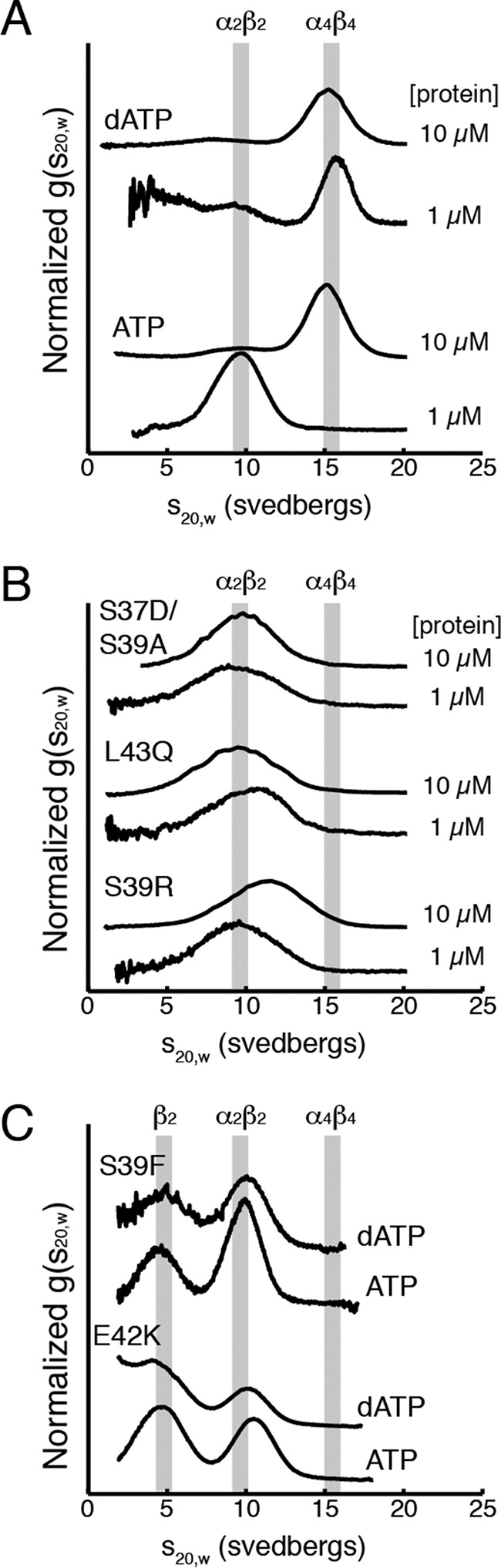
Interface mutant RNRs are insensitive to dATP levels even at high enzyme concentrations, as judged by AUC. A, WT E. coli class Ia RNR in the presence of 175 μm dATP (top traces) or 3 mm ATP (bottom traces) at a physiological protein concentration (1 μm) and above physiological concentrations (10 μm). These results agree well with previously reported behavior of the WT enzyme (10). B, sedimentation of interface mutant proteins S37D/S39A, L43Q, and S39R with 175 μm dATP. C, sedimentation of the previously identified RNR mutant proteins S39F and E42K with 175 μm dATP or 3 mm ATP. A and B, experiments were performed at 1 or 10 μm α2/met-β2. C, 1 μm α2 and a 5-fold excess of met-β2 (5 μm) were used to avoid the α2β2 species overlapping with free α2, sedimenting at 8 S (10). The expected peak positions for α2β2 and α4β4 (and β2 where appropriate) are shown as gray bars.
In stark contrast to the behavior of WT E. coli class Ia RNR, for interface mutant proteins S39D/S37A, L43Q, and S39R, the 15.6 S peak that corresponds to the α4β4 ring is not observed in the presence of dATP. Instead, a single ∼10 S species is observed at both 1 and 10 μm protein concentration (Fig. 4B). This sedimentation value is consistent with an α2β2 complex, as observed for 1 μm WT class Ia RNR with 3 mm ATP (Fig. 4A, lower traces). Even at 10 μm enzyme in the presence of dATP, no discernable shift in the sedimentation values is observed for S37D/S39A and L43Q, indicating the inability of the mutant proteins to form the α4β4 ring cannot be overcome by increasing the protein concentration. Although S39R showed a slight shift toward a larger species, it is apparent that the predominant form for this mutant protein is still the α2β2 species. The ∼10 S species observed for the interface mutant proteins with dATP is slightly broader than the ∼10 S species formed at 1 μm WT α2 and met-β2 with ATP, which we attribute to the presence of a slight excess of α2 in these experiments. We conducted similar experiments with the S39F and E42K interface mutant proteins and observed no difference in the protein size distribution between conditions with 3 mm ATP and 175 μm dATP (Fig. 4C). For these experiments, a 5-fold excess of met-β2 was used to eliminate the likelihood of any excess α2, which led to the appearance of a peak corresponding to free β2, and more importantly, it led to sharper peaks at the same sedimentation coefficient (∼10 S), indicating that these RNR mutant proteins also form α2β2 species (Fig. 4C). Taken together, the EM and AUC experiments show that mutation of interface residues in all five cases result in RNR mutant proteins that are no longer able to form α4β4 rings (Table 1).
When the α4β4 ring cannot form, dATP cannot inhibit RNR activity
We next asked whether dATP inhibits E. coli class Ia RNR mutant proteins that are incapable of forming α4β4 rings. We used a spectrophotometric assay that measures NADPH consumption by TRR in a coupled assay with TR and RNR. The specific activity for CDP reduction by the interface mutant proteins under active conditions (3 mm ATP) was comparable with that of WT (Fig. 5), indicating that the mutant proteins were properly folded and competent to form the active α2β2 complex. In contrast, the WT and mutant proteins differed greatly in their activities under inactivating conditions (175 μm dATP as the allosteric specificity and activity effector). In the presence of 175 μm dATP, the CDP reduction activity of WT enzyme decreased to 7% of the activity under activating conditions (3 mm ATP), whereas the activity of interface mutant proteins is not at all, or only very slightly, diminished under the same conditions (Fig. 5 and Table 1).
Figure 5.
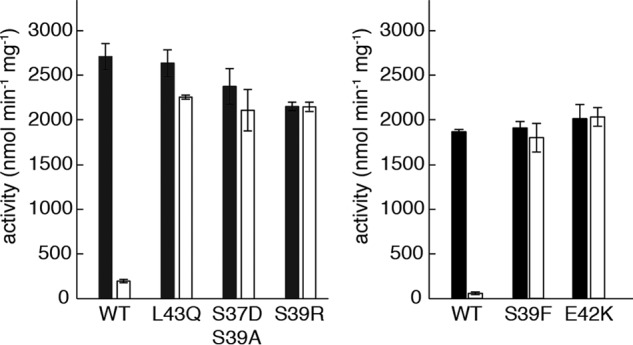
Substitutions of interface residues abolish dATP-induced inhibition of E. coli class Ia RNR. Active conditions (black) contain 3 mm ATP. Inactive conditions (white) contain 175 μm dATP. Each experiment was performed in triplicate. The data in the left and right panels were collected on different days and reflect normal variation in the assay. RNR activity is defined as nanomoles of NADPH consumed in the RNR/TR/TRR-coupled assay, per min per mg of α2 after subtraction for basal oxidation by TR/TRR. Typical reported WT enzyme activities are 2000–2500 nmol min−1 mg−1 (63).
Discussion
In this study, we have characterized a number of mutations in the N-terminal cone domain of the E. coli class Ia RNR α subunit. In particular, we have investigated substitutions of residues located on helix 2, the α-helix that has the largest number of contacts between the cone domain and β in the α4β4 state. None of the residues that were altered directly contact dATP (Fig. 2A) and thus are not designed to impair dATP binding. Instead, mutations were designed to disrupt the α–β interface at the cone domain and prevent α4β4 ring formation. Incredibly, our EM and AUC data show that changing any of the targeted residues is enough to prevent the formation of the α4β4 state. Notably, at high protein concentrations, which were shown in WT RNR to shift the natural equilibrium between α2β2 and α4β4 to the α4β4 state even in the absence of dATP (10), the RNR mutant proteins fail to form α4β4 rings. Thus, high WT RNR protein concentrations can override the requirement for dATP in ring formation. However, this propensity for ring formation appears completely lost in the RNR mutant proteins. Notably, we did not change the other α–β interface in these studies, i.e. the interface between the C-terminal tail of β and the α subunit that is shown in Fig. 1B. Therefore, we can conclude that the β tail interaction is insufficient to allow formation of α4β4 when the cone domain interface is not intact.
It is also interesting that each one of the substitutions in the cone domain yielded the same result (Table 1). Mutation at three different positions (Ser-37, Glu-42, and Leu-43) disrupted ring formation, regardless of whether the change affected a hydrophobic contact or hydrophilic one. Taken together, these results suggest that the α–β interface at the cone domain is quite fragile and relatively easily disrupted. This finding is consistent with the small area of this interface (∼580 Å2). It also explains why α and β subunits of human class Ia RNR do not form α4β4-ring structures despite having highly homologous structures; one amino acid change is enough to abrogate ring formation, and the α–β interface residues are not conserved between human and E. coli class Ia RNR.
With this series of RNR mutant proteins that are incapable of forming α4β4 rings, we have been able to interrogate whether ring formation is required for allosteric activity inhibition, and we find that it is. In the absence of ring formation, dATP no longer inhibits activity. In contrast, all of the mutant proteins retain normal activity in the presence of ATP. These data support a model in which ring formation is causative of RNR inhibition, presumably by positioning β such that RT is abrogated. It is a fascinating feature of RNRs that RT is modulated to control enzyme activity rather than disrupting substrate binding or a catalytic step. Although human class Ia RNR does not form α4β4 rings, allosteric regulation of activity is also proposed to involve an oligomeric state change that affects RT (9, 33–35), suggesting that controlling RT may be a common theme in the allosteric regulation of class Ia enzymes.
Importantly, two of the RNR substitutions studied here, S39F and E42K, were previously shown to increase each dNTP concentration in E. coli by 2.3–25-fold, leading to mutator phenotypes (31). These mutants were identified in in vivo studies before the structure of the dATP-inhibited α4β4 complex was available, although authors did consider the possibility that these residue substitutions might affect allosteric regulation due to their location in the cone domain (31). Following the α4β4 structure determination (10, 29), the position of these residues could be mapped to the α–β interface, and here we show that these substitutions do prevent α4β4-ring formation and lead to an RNR that cannot be turned off by dATP (Table 1). The data presented here also indicate that these two mutant enzymes are not impaired outside of their ability to be allosterically regulated by dATP; they are soluble and show WT levels of activity (Fig. 5). Taken together, this study and the previous study (31) provide insight into the question of how important allosteric regulation of RNR activity is to cell survival. What happens to a cell when RNR cannot be turned off? The observation that S39F and E42K mutant proteins have mutator phenotypes indicates that allosteric regulation of activity is critical for cellular health, at least in E. coli. Similarly, a mutation in a mammalian RNR gene that generates an RNR enzyme that is insensitive to dATP levels has been implicated in a mutator phenotype (36). In the latter case, the amino acid substitution is of a residue in the dATP-binding site and not at an oligomeric interface (36, 37). Importantly, different classes and different species of RNRs are regulated to varying degrees in different systems at the level of transcription (38, 39), translation (40), cellular localization (41, 42), and degradation (43). How these modes of regulation work together to maintain nucleotide pools is not yet established. Not all RNRs have a cone domain (2), whereas others appear to have two or more cone domains (44–47). Thus, the overall importance of allosteric regulation of activity is likely to differ in different systems. However, for mammalian and E. coli cells expressing class Ia RNRs, allosteric regulation of activity is clearly important.
It is interesting to consider these finding in terms of RNR inhibitor design. Given their important role in DNA replication and repair, RNRs are targets for anti-cancer, antibacterial, and antiviral therapies. Gemcitabine is a Food and Drug Administration (FDA)-approved prodrug used clinically in the treatment of various forms of cancer (48). In its phosphorylated state, gemcitabine-diphosphate is an RNR mechanism-based inhibitor (49, 50). In contrast, FDA-approved hydroxyurea, used in the treatment of HIV (51, 52), inactivates class Ia RNRs by quenching the tyrosyl-radical species on the β2 subunit (53, 54). More recently, it has been suggested that FDA-approved clofarabine in its phosphorylated form may inhibit human RNR by binding to the cone domain in the α subunit and locking the enzyme in an inactive α6 state that mimics the dATP-inhibited α6 state of the human RNR (34, 35, 55, 56). Thus, we have three RNR inhibitors that appear to work in different ways, targeting the active site (gemcitabine-diphosphate), the radical site (hydroxyurea), and an allosteric activity regulatory site (clofarabine di- and triphosphate). This work suggests that designing small molecules to bind to the cone domain to prevent dATP-induced inhibition could also yield successful therapeutics. In other words, a small molecule that targets the cone domain could be therapeutically valuable regardless of whether it induces a tighter inhibited complex or prevents complex formation. It is problematic for a cell to have an RNR that is either always off or always on. Although a number of RNR-based therapies are used clinically (25), the promise of RNR as an antibacterial target has not been fully realized. The allosteric activity site of bacterial RNR offers another prospect. Given that mammalian and bacterial RNRs appear to employ different oligomeric states (9, 10, 28, 33–35, 57), side effects due to human RNR disruption are unlikely for small molecules that are designed to bind at or near a bacterial RNR α4β4 interface. Targeting allosteric regulation also has the benefit of not directly killing cells, but instead elevating mutation rates through disrupting nucleotide metabolism. Such strategies may be less susceptible to resistance and have fewer deleterious effects on off-target microbial populations, such as gut commensals.
In summary, we have provided evidence that allosteric regulation of activity for the best-studied class Ia RNR directly involves dATP-induced α4β4 ring formation, which subsequently causes enzyme inactivation. We still do not know how dATP binding to the cone domain results in formation of the observed α–β interface, given that the dATP binding is near to, but does not directly contact, the residues involved in interface formation. Also, we do not know the molecular basis by which ATP replacing dATP in this site serves to disrupt the interface. We do know that the affinity of this allosteric site is tuned such that small changes in the ratio of dATP to ATP can be sensed, shifting the oligomeric states of the enzyme and the enzyme activity, appropriately. It is fascinating that this RNR has adopted such an unusual mechanism for regulating activity. Instead of moving a loop or a side chain, the whole β2 subunit is held at arms length from α2 so that RT, and thus ribonucleotide reduction, cannot occur until more deoxynucleotides are needed.
Experimental procedures
Protein production
The E. coli class Ia RNR (nrdA) gene with an N-terminal hexahistidine (His6) tag was previously cloned into a pET28a vector under the control of a T7 promoter (58). All E. coli α2 mutants were constructed using QuikChange mutagenesis technology (Agilent) with primers from Integrated DNA Technologies, and the sequence of each mutant was confirmed through DNA sequencing (Genewiz). Primer sequences for building each construct are listed in Table 2. His6-tagged WT E. coli α2 was overexpressed and purified based on previously described methods (58). Briefly, cells were harvested and resuspended in Buffer A (50 mm HEPES, pH 7.6, 300 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine) lysed by sonication and clarified by centrifugation at 29,000 × g. Lysate was applied to a 5-ml HisTrap HP column (GE Healthcare), washed with Buffer A supplemented with 30 mm imidazole, and eluted with Buffer A supplemented with 300 mm imidazole. Protein was further purified on a Superdex 200 16/60 size-exclusion column (GE Healthcare) in a final buffer of 20 mm HEPES, pH 7.6, 100 mm NaCl, 5% (w/v) glycerol. A final yield of ∼25–50 mg/liter of culture for WT α2 was typical. The purification for the mutant proteins was identical, with similar yields. All proteins were judged as purified to homogeneity by SDS-PAGE. None of the interface mutant proteins showed any indication of instability or propensity to aggregate during purification.
Table 2.
DNA sequences of primers used to build E. coli class Ia RNR α mutants
| Mutants | Primers | Sequences |
|---|---|---|
| S37D/S39A | Forward | 5′-CTGCATAACGTTGATATTGCCCAGGTCGAGCTGCG-3′ |
| Reverse | 5′-CGCAGCTCGACCTGGGCAATATCAACGTTATGCAG-3′ | |
| S39R | Forward | 5′-GACTGCATAACGTTTCCATTCGCCAGGTGGAGCTGCGCTCCC-3′ |
| Reverse | 5′-GGGAGCGCAGCTCCACCTGGCGAATGGAAACGTTATGCAGTC-3′ | |
| S39F | Forward | 5′-CGTTTCGATTTTCCAGGTAGAGCTGCG-3′ |
| Reverse | 5′-CGCAGCTCTACCTGGAAAATCGAAACG-3′ | |
| E42K | Forward | 5′-CGATTTCCCAGGTCAAGCTGCGATCCC-3′ |
| Reverse | 5′-GGGATCGCAGCTTGACCTGGGAAATCG-3′ | |
| L43Q | Forward | 5′-CATAACGTTTCCATTTCCCAGGTGGAGCAGCGCTCCCACATTC-3′ |
| Reverse | 5′-GAATGTGGGAGCGCTGCTCCACCTGGGAAATGGAAACGTTATG-3′ |
Untagged E. coli β2 was purified, as described previously (59), and contained ∼1.2 radicals per dimer as estimated by UV-visible spectroscopy of the Tyr-122 radical (ϵ411 of 1760 mm−1 cm−1 after drop-line subtraction of the diferric cluster absorbance) (60). The met-β2 was produced by reduction of active β2 by hydroxyurea as described previously (10). All proteins were buffer-exchanged into buffer containing 50 mm HEPES, pH 7.5, 100 mm NaCl, 1 mm EDTA, 5 mm DTT, with a PD-10 column (GE Healthcare). E. coli TR and TRR for coupled activity assays were produced as described previously (7, 61, 62).
Activity assays
Activities of α2 mutant proteins were determined by a spectrophotometric assay using E. coli TR and TRR and NADPH as a terminal electron donor (63). NADPH consumption was monitored at 340 nm with a Cary 300 Bio spectrophotometer (Varian/Agilent). Reaction mixtures contained 100 nm WT or mutant α2, 500 nm β2, 30 μm TR, 0.5 μm TRR, 200 μm NADPH, 1 mm CDP as substrate, and either 3 mm ATP or 175 μm dATP as allosteric effectors. The assay buffer contained 50 mm HEPES, pH 7.6, 15 mm MgCl2, 1 mm EDTA. Data analysis was performed in the Cary WinUV Kinetics program (Varian/Agilent) and MATLAB (Mathworks). Initial rates were determined by linear fitting of the NADPH consumption 30–60 s after initiation of the reaction by addition of β2. The α4β4 on-rate has not been directly reported, but surface plasmon resonance data suggest a very fast association (64) such that the α4β4 complex should form within the mixing time of the assay. Basal NADPH oxidation was determined over 30 s prior to addition of β2.
AUC
All AUC experiments were performed in a Beckman XL-1 centrifuge at 42,000 × g and 25 °C. Interference optics were used due to high UV absorption by nucleotides. Protein solutions at the specified concentration and reference solutions were prepared with 175 μm dATP or 3 mm ATP as effector, 1 mm CDP, and 5 mm DTT in the same buffer used for assays. Data were analyzed in DCDT+ (65). The sedimentation coefficient distribution, g(s*), was generated based on 50–80 scans. Sedimentation coefficient values were adjusted for buffer composition to standard values using the previously described parameters (10): solvent viscosity of 1.042 centipoise, solvent density of 1.00348 g ml−1, and partial specific volume of 0.7335 and 0.7346 ml g−1 for α2 and β2, respectively. The distributions were fit by least-squares analysis in DCDT+ to obtain the temperature and solvent-corrected sedimentation coefficient s20, w, molecular weight, and an approximate concentration.
Electron microscopy (EM)
α2 and met-β2 at 150 nm each were mixed with 1 mm CDP and 175 μm dATP or 3 mm ATP and incubated for 5 min, and 5 μl of the mixture was applied to a carbon film on a copper/rhodium EM grid (Ted Pella, Inc.). The protein was immediately blotted off, and the grid was stained by repeated application and blotting of 5 μl of 2% uranyl acetate. The grid was covered with a second carbon layer prior to drying. The image of the grid with ATP was acquired from the same equipment previously described (10). The other images were taken on a Tecnai Spirit (FEI) instrument with an XR16 camera (AMT) operated at 120 kV at 68,000 × magnification. 5–10 grid squares were examined, and a representative field was shown for each protein sample.
Author contributions
P. Y.-T. C., M. A. F., and C. L. D. conceptualization; P. Y.-T. C., M. A. F., and C. L. D. formal analysis; P. Y.-T. C., M. A. F., and E. J. B. investigation; P. Y.-T. C. visualization; P. Y.-T. C., M. A. F., E. J. B., and C. L. D. writing-review and editing; M. A. F. and C. L. D. writing-original draft; C. L. D. supervision; C. L. D. funding acquisition; C. L. D. validation; C. L. D. project administration.
Acknowledgments
We thank Deborah Pheasant for assistance in collecting analytical ultracentrifugation data and Samuel Thompson for assistance with the site-directed mutagenesis. Dr. Nozomi Ando and Dr. Christina M. Zimanyi provided valuable discussions. Analytical ultracentrifugation data were collected at the MIT Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems, supported by National Science Foundation Grant NSF-0070319. EM images were collected at the W. M. Keck Microscopy Facility (Whitehead Institute).
This work was supported in part by National Science Foundation Graduate Research Fellowship under Grant 0645960 (to M. A. F.) and MIT Center for Environmental Health Science (CEHS) Grant P30-ES002109 from the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- RNR
- ribonucleotide reductase
- SAXS
- small-angle X-ray scattering
- TR
- thioredoxin
- TRR
- thioredoxin reductase
- RT
- radical transfer
- AUC
- analytical ultracentrifugation
- FDA
- Food and Drug Administration.
References
- 1. Licht S., and Stubbe J. (1999) Mechanistic investigations of ribonucleotide reductases. Compr. Nat. Prod. Chem. 5, 163–203 [Google Scholar]
- 2. Nordlund P., and Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 10.1146/annurev.biochem.75.103004.142443 [DOI] [PubMed] [Google Scholar]
- 3. Hofer A., Crona M., Logan D. T., and Sjöberg B. M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 10.3109/10409238.2011.630372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minnihan E. C., Nocera D. G., and Stubbe J. (2013) Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 46, 2524–2535 10.1021/ar4000407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown N. C., and Reichard P. (1969) Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol. 46, 39–55 10.1016/0022-2836(69)90056-4 [DOI] [PubMed] [Google Scholar]
- 6. von Döbeln U., and Reichard P. (1976) Binding of substrates to Escherichia coli ribonucleotide reductase. J. Biol. Chem. 251, 3616–3622 [PubMed] [Google Scholar]
- 7. Zimanyi C. M., Chen P. Y.-T., Kang G., Funk M. A., and Drennan C. L. (2016) Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. eLife 5, e07141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eriksson M., Uhlin U., Ramaswamy S., Ekberg M., Regnström K., Sjöberg B. M., and Eklund H. (1997) Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure 5, 1077–1092 10.1016/S0969-2126(97)00259-1 [DOI] [PubMed] [Google Scholar]
- 9. Fairman J. W., Wijerathna S. R., Ahmad M. F., Xu H., Nakano R., Jha S., Prendergast J., Welin R. M., Flodin S., Roos A., Nordlund P., Li Z., Walz T., and Dealwis C. G. (2011) Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 18, 316–322 10.1038/nsmb.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ando N., Brignole E. J., Zimanyi C. M., Funk M. A., Yokoyama K., Asturias F. J., Stubbe J., and Drennan C. L. (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 21046–21051 10.1073/pnas.1112715108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Atkin C. L., Thelander L., Reichard P., and Lang G. (1973) Iron and free radical in ribonucleotide reductase. Exchange of iron and Mössbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme. J. Biol. Chem. 248, 7464–7472 [PubMed] [Google Scholar]
- 12. Sjöberg B. M., Reichard P., Gräslund A., and Ehrenberg A. (1978) The tyrosine free radical in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 253, 6863–6865 [PubMed] [Google Scholar]
- 13. Stubbe J., and Riggs-Gelasco P. (1998) Harnessing free radicals: formation and function of the tyrosyl radical in ribonucleotide reductase. Trends Biochem. Sci. 23, 438–443 10.1016/S0968-0004(98)01296-1 [DOI] [PubMed] [Google Scholar]
- 14. Cotruvo J. A., and Stubbe J. (2011) Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 80, 733–767 10.1146/annurev-biochem-061408-095817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stubbe J., Nocera D. G., Yee C. S., and Chang M. C. (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 103, 2167–2201 10.1021/cr020421u [DOI] [PubMed] [Google Scholar]
- 16. Climent I., Sjöberg B. M., and Huang C. Y. (1992) Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2. Effects on catalytic activity and subunit interaction. Biochemistry 31, 4801–4807 10.1021/bi00135a009 [DOI] [PubMed] [Google Scholar]
- 17. Thelander L. (1974) Reaction mechanism of ribonucleoside diphosphate reductase from Escherichia coli oxidation-reduction-active disulfides in the B1 subunit. J. Biol. Chem. 249, 4858–4862 [PubMed] [Google Scholar]
- 18. Mao S. S., Holler T. P., Yu G. X., Bollinger J. M. Jr., Booker S., Johnston M. I., and Stubbe J. (1992) A model for the role of multiple cysteine residues involved in ribonucleotide reduction: amazing and still confusing. Biochemistry 31, 9733–9743 10.1021/bi00155a029 [DOI] [PubMed] [Google Scholar]
- 19. Minnihan E. C., Ando N., Brignole E. J., Olshansky L., Chittuluru J., Asturias F. J., Drennan C. L., Nocera D. G., and Stubbe J. (2013) Generation of a stable, aminotyrosyl radical-induced α2β2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 110, 3835–3840 10.1073/pnas.1220691110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kunz B. A., Kohalmi S. E., Kunkel T. A., Mathews C. K., McIntosh E. M., and Reidy J. A. (1994) Deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat. Res. 318, 1–64 10.1016/0165-1110(94)90006-X [DOI] [PubMed] [Google Scholar]
- 21. Wheeler L. J., Rajagopal I., and Mathews C. K. (2005) Stimulation of mutagenesis by proportional deoxyribonucleoside triphosphate accumulation in Escherichia coli. DNA Repair 4, 1450–1456 [DOI] [PubMed] [Google Scholar]
- 22. Mathews C. K. (2006) DNA precursor metabolism and genomic stability. FASEB J. 20, 1300–1314 10.1096/fj.06-5730rev [DOI] [PubMed] [Google Scholar]
- 23. Gon S., Napolitano R., Rocha W., Coulon S., and Fuchs R. P. (2011) Increase in dNTP pool size during the DNA damage response plays a key role in spontaneous and induced-mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 19311–19316 10.1073/pnas.1113664108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar D., Abdulovic A. L., Viberg J., Nilsson A. K., Kunkel T. A., and Chabes A. (2011) Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 39, 1360–1371 10.1093/nar/gkq829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aye Y., Li M., Long M. J., and Weiss R. S. (2015) Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34, 2011–2021 10.1038/onc.2014.155 [DOI] [PubMed] [Google Scholar]
- 26. Bochner B. R., and Ames B. N. (1982) Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J. Biol. Chem. 257, 9759–9769 [PubMed] [Google Scholar]
- 27. Larsson A., and Reichard P. (1966) Enzymatic synthesis of deoxyribonucleotides IX. Allosteric effects in the reduction of pyrimidine ribonucleotides by the ribonucleoside diphosphate reductase system of Escherichia coli. J. Biol. Chem. 241, 2533–2539 [PubMed] [Google Scholar]
- 28. Rofougaran R., Crona M., Vodnala M., Sjöberg B. M., and Hofer A. (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 283, 35310–35318 10.1074/jbc.M806738200 [DOI] [PubMed] [Google Scholar]
- 29. Zimanyi C. M., Ando N., Brignole E. J., Asturias F. J., Stubbe J., and Drennan C. L. (2012) Tangled up in knots: structures of inactivated forms of E. coli class Ia ribonucleotide reductase. Structure 20, 1374–1383 10.1016/j.str.2012.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brignole E. J., Ando N., Zimanyi C. M., and Drennan C. L. (2012) The prototypic class Ia ribonucleotide reductase from Escherichia coli: still surprising after all these years. Biochem. Soc. Trans. 40, 523–530 10.1042/BST20120081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahluwalia D., Bienstock R. J., and Schaaper R. M. (2012) Novel mutator mutants of E. coli nrdAB ribonucleotide reductase: insight into allosteric regulation and control of mutation rates. DNA Repair 11, 480–487 10.1016/j.dnarep.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eriksson S., Sjöberg B. M., and Hahne S. (1977) Ribonucleoside diphosphate reductase from Escherichia coli. An immunological assay and a novel purification from an overproducing strain lysogenic for phage λdnrd. J. Biol. Chem. 252, 6132–6138 [PubMed] [Google Scholar]
- 33. Ando N., Li H., Brignole E. J., Thompson S., McLaughlin M. I., Page J. E., Asturias F. J., Stubbe J., and Drennan C. L. (2016) Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer. Biochemistry 55, 373–381 10.1021/acs.biochem.5b01207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brignole E. J., Tsai K.-L., Chittuluru J., Li H., Aye Y., Penczek P. A., Stubbe J., Drennan C. L., and Asturias F. (2018) 3.3-Å resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. eLife 7, e31502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aye Y., Brignole E. J., Long M. J., Chittuluru J., Drennan C. L., Asturias F. J., and Stubbe J. (2012) Clofarabine targets the large subunit (α) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol. 19, 799–805 10.1016/j.chembiol.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caras I. W., and Martin D. W. Jr. (1988) Molecular cloning of the cDNA for a mutant mouse ribonucleotide reductase M1 that produces a dominant mutator phenotype in mammalian cells. Mol. Cell. Biol. 8, 2698–2704 10.1128/MCB.8.7.2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Birgander P. L., Kasrayan A., and Sjöberg B. M. (2004) Mutant R1 proteins from Escherichia coli class Ia ribonucleotide reductase with altered responses to dATP inhibition. J. Biol. Chem. 279, 14496–14501 10.1074/jbc.M310142200 [DOI] [PubMed] [Google Scholar]
- 38. Gon S., Camara J. E., Klungsøyr H. K., Crooke E., Skarstad K., and Beckwith J. (2006) A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. EMBO J. 25, 1137–1147 10.1038/sj.emboj.7600990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Torrents E., Grinberg I., Gorovitz-Harris B., Lundström H., Borovok I., Aharonowitz Y., Sjöberg B. M., and Cohen G. (2007) NrdR controls differential expression of the Escherichia coli ribonucleotide reductase genes. J. Bacteriol. 189, 5012–5021 10.1128/JB.00440-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abid M. R., Li Y., Anthony C., and De Benedetti A. (1999) Translational regulation of ribonucleotide reductase by eukaryotic initiation factor 4E links protein synthesis to the control of DNA replication. J. Biol. Chem. 274, 35991–35998 10.1074/jbc.274.50.35991 [DOI] [PubMed] [Google Scholar]
- 41. Engström Y., and Rozell B. (1988) Immunocytochemical evidence for the cytoplasmic localization and differential expression during the cell cycle of the M1 and M2 subunits of mammalian ribonucleotide reductase. EMBO J. 7, 1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pontarin G., Fijolek A., Pizzo P., Ferraro P., Rampazzo C., Pozzan T., Thelander L., Reichard P. A., and Bianchi V. (2008) Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 105, 17801–17806 10.1073/pnas.0808198105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. D'Angiolella V., Donato V., Forrester F. M., Jeong Y.-T., Pellacani C., Kudo Y., Saraf A., Florens L., Washburn M. P., and Pagano M. (2012) Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149, 1023–1034 10.1016/j.cell.2012.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aravind L., Wolf Y. I., and Koonin E. V. (2000) The ATP-cone: an evolutionarily mobile, ATP-binding regulatory domain. J. Mol. Microbiol. Biotechnol. 2, 191–194 [PubMed] [Google Scholar]
- 45. Torrents E., Westman M., Sahlin M., and Sjöberg B. M. (2006) Ribonucleotide reductase modularity: atypical duplication of the ATP-cone domain in Pseudomonas aeruginosa. J. Biol. Chem. 281, 25287–25296 10.1074/jbc.M601794200 [DOI] [PubMed] [Google Scholar]
- 46. Jonna V. R., Crona M., Rofougaran R., Lundin D., Johansson S., Brännström K., Sjöberg B. M., and Hofer A. (2015) Diversity in overall activity regulation of ribonucleotide reductase. J. Biol. Chem. 290, 17339–17348 10.1074/jbc.M115.649624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johansson R., Jonna V. R., Kumar R., Nayeri N., Lundin D., Sjöberg B. M., Hofer A., and Logan D. T. (2016) Structural mechanism of allosteric activity regulation in a ribonucleotide reductase with double ATP cones. Structure 24, 906–917 10.1016/j.str.2016.03.025 [DOI] [PubMed] [Google Scholar]
- 48. Hertel L. W., Boder G. B., Kroin J. S., Rinzel S. M., Poore G. A., Todd G. C., and Grindey G. B. (1990) Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res. 50, 4417–4422 [PubMed] [Google Scholar]
- 49. van der Donk W. A., Yu G., Pérez L., Sanchez R. J., Stubbe J., Samano V., and Robins M. J. (1998) Detection of a new substrate-derived radical during inactivation of ribonucleotide reductase from Escherichia coli by gemcitabine 5′-diphosphate. Biochemistry 37, 6419–6426 10.1021/bi9729357 [DOI] [PubMed] [Google Scholar]
- 50. Wang J., Lohman G. J., and Stubbe J. (2007) Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. U.S.A. 104, 14324–14329 10.1073/pnas.0706803104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lori F., Malykh A., Cara A., Sun D., Weinstein J. N., Lisziewicz J., and Gallo R. C. (1994) Hydroxyurea as an inhibitor of human immunodeficiency virus-type 1 replication. Science 266, 801–805 10.1126/science.7973634 [DOI] [PubMed] [Google Scholar]
- 52. Rutschmann O. T., Opravil M., Iten A., Malinverni R., Vernazza P. L., Bucher H. C., Bernasconi E., Sudre P., Leduc D., Yerly S., Perrin L. H., and Hirschel B. (1998) A placebo-controlled trial of didanosine plus stavudine, with and without hydroxyurea, for HIV infection. AIDS 12, F71–77 10.1097/00002030-199808000-00003 [DOI] [PubMed] [Google Scholar]
- 53. Lassmann G., Thelander L., and Gräslund A. (1992) EPR stopped-flow studies of the reaction of the tyrosyl radical of protein R2 from ribonucleotide reductase with hydroxyurea. Biochem. Biophys. Res. Commun. 188, 879–887 10.1016/0006-291X(92)91138-G [DOI] [PubMed] [Google Scholar]
- 54. Karlsson M., Sahlin M., and Sjöberg B. M. (1992) Escherichia coli ribonucleotide reductase. Radical susceptibility to hydroxyurea is dependent on the regulatory state of the enzyme. J. Biol. Chem. 267, 12622–12626 [PubMed] [Google Scholar]
- 55. Aye Y., and Stubbe J. (2011) Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. U.S.A. 108, 9815–9820 10.1073/pnas.1013274108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wisitpitthaya S., Zhao Y., Long M. J., Li M., Fletcher E. A., Blessing W. A., Weiss R. S., and Aye Y. (2016) Cladribine and fludarabine nucleotides induce distinct hexamers defining a common mode of reversible RNR inhibition. ACS Chem. Biol. 11, 2021–2032 10.1021/acschembio.6b00303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rofougaran R., Vodnala M., and Hofer A. (2006) Enzymatically active mammalian ribonucleotide reductase exists primarily as an α6β2 octamer. J. Biol. Chem. 281, 27705–27711 10.1074/jbc.M605573200 [DOI] [PubMed] [Google Scholar]
- 58. Minnihan E. C., Seyedsayamdost M. R., Uhlin U., and Stubbe J. (2011) Kinetics of radical intermediate formation and deoxynucleotide production in 3-aminotyrosine-substituted Escherichia coli ribonucleotide reductases. J. Am. Chem. Soc. 133, 9430–9440 10.1021/ja201640n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salowe S. P., and Stubbe J. (1986) Cloning, overproduction, and purification of the B2 subunit of ribonucleoside-diphosphate reductase. J. Bacteriol. 165, 363–366 10.1128/jb.165.2.363-366.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bollinger J. M. Jr., Edmondson D. E., Huynh B. H., Filley J., Norton J. R., and Stubbe J. (1991) Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science 253, 292–298 10.1126/science.1650033 [DOI] [PubMed] [Google Scholar]
- 61. Russel M., and Model P. (1985) Direct cloning of the trxB gene that encodes thioredoxin reductase. J. Bacteriol. 163, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chivers P. T., Prehoda K. E., Volkman B. F., Kim B.-M., Markley J. L., and Raines R. T. (1997) Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry 36, 14985–14991 10.1021/bi970071j [DOI] [PubMed] [Google Scholar]
- 63. Ge J., Yu G., Ator M. A., and Stubbe J. (2003) Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry 42, 10071–10083 10.1021/bi034374r [DOI] [PubMed] [Google Scholar]
- 64. Kasrayan A., Birgander P. L., Pappalardo L., Regnström K., Westman M., Slaby A., Gordon E., and Sjöberg B. M. (2004) Enhancement by effectors and substrate nucleotides of R1-R2 interactions in Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 279, 31050–31057 10.1074/jbc.M400693200 [DOI] [PubMed] [Google Scholar]
- 65. Philo J. S. (2006) Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal. Biochem. 354, 238–246 10.1016/j.ab.2006.04.053 [DOI] [PubMed] [Google Scholar]
- 66. Uhlin U., and Eklund H. (1994) Structure of ribonucleotide reductase protein R1. Nature 370, 533–539 10.1038/370533a0 [DOI] [PubMed] [Google Scholar]
- 67. Seyedsayamdost M. R., Chan C. T., Mugnaini V., Stubbe J., and Bennati M. (2007) PELDOR spectroscopy with DOPA-β2 and NH2Y-α2s: distance measurements between residues involved in the radical propagation pathway of E. coli ribonucleotide reductase. J. Am. Chem. Soc. 129, 15748–15749 10.1021/ja076459b [DOI] [PubMed] [Google Scholar]
- 68. Yokoyama K., Smith A. A., Corzilius B., Griffin R. G., and Stubbe J. (2011) Equilibration of tyrosyl radicals (Y356·, Y731·, Y730·) in the radical propagation pathway of the Escherichia coli class Ia ribonucleotide reductase. J. Am. Chem. Soc. 133, 18420–18432 10.1021/ja207455k [DOI] [PMC free article] [PubMed] [Google Scholar]