

Transcriptional reporters can be powerful tools for assessing when a particular regulator is active; however, native promoters may not be ideal for this purpose. Optimal reporters should be specific to the regulator being examined and should maximize the difference between the “on” and “off” states; however, these properties are distinct from the selective pressures driving the evolution of natural promoters. Synthetic promoters offer a promising alternative, but our understanding often does not enable fully predictive promoter design, and the large number of alternative sequence possibilities can be intractable. In a synthetic promoter region with over 34 billion sequence variants, we identified bases correlated with favorable performance by screening only 83 candidates, allowing us to rationally constrain our design. We thereby generated an optimized reporter that is induced by PhoB and used it to explore the low-phosphate response of V. fischeri. This promoter design strategy will facilitate the engineering of other regulator-specific reporters.
KEYWORDS: photobacterium, Aliivibrio, synthetic biology
ABSTRACT
Transcriptional reporters are common tools for analyzing either the transcription of a gene of interest or the activity of a specific transcriptional regulator. Unfortunately, the latter application has the shortcoming that native promoters did not evolve as optimal readouts for the activity of a particular regulator. We sought to synthesize an optimized transcriptional reporter for assessing PhoB activity, aiming for maximal “on” expression when PhoB is active, minimal background in the “off” state, and no control elements for other regulators. We designed specific sequences for promoter elements with appropriately spaced PhoB-binding sites, and at 19 additional intervening nucleotide positions for which we did not predict sequence-specific effects, the bases were randomized. Eighty-three such constructs were screened in Vibrio fischeri, enabling us to identify bases at particular randomized positions that significantly correlated with high-level “on” or low-level “off” expression. A second round of promoter design rationally constrained 13 additional positions, leading to a reporter with high-level PhoB-dependent expression, essentially no background, and no other known regulatory elements. As expressed reporters, we used both stable and destabilized variants of green fluorescent protein (GFP), the latter of which has a half-life of 81 min in V. fischeri. In culture, PhoB induced the reporter when phosphate was depleted to a concentration below 10 μM. During symbiotic colonization of its host squid, Euprymna scolopes, the reporter indicated heterogeneous phosphate availability in different light-organ microenvironments. Finally, testing this construct in other members of the Proteobacteria demonstrated its broader utility. The results illustrate how a limited ability to predict synthetic promoter-reporter performance can be overcome through iterative screening and reengineering.
IMPORTANCE Transcriptional reporters can be powerful tools for assessing when a particular regulator is active; however, native promoters may not be ideal for this purpose. Optimal reporters should be specific to the regulator being examined and should maximize the difference between the “on” and “off” states; however, these properties are distinct from the selective pressures driving the evolution of natural promoters. Synthetic promoters offer a promising alternative, but our understanding often does not enable fully predictive promoter design, and the large number of alternative sequence possibilities can be intractable. In a synthetic promoter region with over 34 billion sequence variants, we identified bases correlated with favorable performance by screening only 83 candidates, allowing us to rationally constrain our design. We thereby generated an optimized reporter that is induced by PhoB and used it to explore the low-phosphate response of V. fischeri. This promoter design strategy will facilitate the engineering of other regulator-specific reporters.
INTRODUCTION
Malcolm Casadaban's pioneering use of the lac operon as a transcriptional reporter revolutionized the study of gene regulation, paving a new way to assess the activity of a transcriptional promoter by fusing it to a gene encoding a readily screened and measured phenotype, such as β-galactosidase activity (1). Ultimately, transcriptional reporters became a mainstay of bacterial genetics and have led to countless discoveries of conditions and regulators that activate or repress genes of interest. In a twist on this original application, when a particular transcriptional regulator controls a promoter, a corresponding promoter-reporter fusion is sometimes used to assess the activity of that regulator (2–4). However, native promoters are not necessarily ideal for the purpose of measuring the activity of a specific transcription factor.
An ideal readout of regulator activity would have minimal expression in the “off” state, high-level “on” expression, and specificity for the regulator in question. Unfortunately, these attributes do not necessarily reflect the evolutionary forces shaping native promoters. For example, some leaky expression in the “off” state is selected for in some cases (e.g., the lac operon), and native promoters are often regulated by multiple transcription factors simultaneously (5–7). Such coregulation has been addressed by avoiding or removing regulatory elements other than those of the transcription factor being assessed (2, 8–12); however, given the complexity of gene regulation, and the challenge of cataloging all the regulatory mechanisms at a natural promoter, this approach may not always be effective for generating a regulator-specific reporter.
Synthetic promoters offer a powerful alternative approach for generating regulator-specific reporters. Such promoters can be engineered to incorporate sequence motifs consistent with regulation by one mechanism while avoiding binding sites for other regulators. Online tools can aid in this process (13–15), but promoter-reporter performance is not entirely predictable. Sequences with no known function, other than spacing between established regulatory elements, can affect output (15), hampering the rational design of such transcriptional reporters.
We sought to develop a transcriptional reporter to monitor the activation state of PhoB in Vibrio fischeri. PhoB is the response regulator portion of a two-component regulatory system that activates the expression of several genes under low-phosphate conditions in various bacteria (16, 17). In Escherichia coli, PhoB is activated upon phosphorylation by its cognate sensor kinase PhoR in response to low (<4 μM) phosphate concentrations (18). Phosphorylated PhoB then binds to a “Pho box” sequence consisting of two well-conserved 7-bp direct repeats (5′-CTGTCAT-3′) separated by 4 bp of AT-rich sequence (18–20). PhoB binding can compensate for a poor −35 promoter element and activate transcription. In V. fischeri, PhoB appears to indirectly activate bioluminescence, which is a colonization factor in V. fischeri's light-organ symbiosis with the Hawaiian bobtail squid, Euprymna scolopes (21–23). Moreover, mutations in phoB or the phosphate uptake system that it controls reduce colonization competitiveness, suggesting a symbiotic role for the PhoB regulon and prompting our interest in investigating the PhoB-dependent response to low phosphate levels in V. fischeri (21, 24). Previously, PhoB-dependent reporters have been generated by using native promoters for pstS and phoA (17, 25–28), but these may have shortcomings, such as high background levels or shared control with other regulators (29, 30).
Here we describe the generation of a synthetic PhoB-dependent transcriptional promoter, which we linked to gfp as a reporter gene. We also expanded the utility of green fluorescent protein (GFP) for assessing dynamic changes in gene expression in V. fischeri by evaluating destabilized GFP variants. Our constructs are useful for assessing the low-phosphate response in V. fischeri and other bacteria, and our general method of synthetic reporter design should be effective for studying other regulators in a variety of bacteria.
RESULTS
Generation and screening of semirandomized variants of a synthetic promoter region.
To generate a PhoB-dependent and PhoB-specific synthetic transcriptional reporter, we began with a framework based on canonical attributes of PhoB-activated promoters, such as those upstream of phoB and pstS in E. coli (31), and randomized the sequences between the set sequences relevant to PhoB activation. In this initial semirandomized sequence (Fig. 1A), we placed two closely spaced Pho boxes, with one overlapping the −35 promoter region, which lacks a strong canonical −35 sequence element, consistent with multiple Pho boxes and their arrangements in native promoters (27, 32–35). As noted above, the canonical Pho box includes 7-bp direct repeats; however, an additional 3′ adenine may play a role (20). Of the four repeats in the two Pho boxes that we used, none deviates from the 8-bp sequence 5′-CTGTCATA-3′ by more than a single mismatch. Within each Pho box, the A/T richness of sequences between the direct repeats appears to affect relative PhoB binding (36), and this spacer between repeats was set at 5′-AAT-3′ in the Pho box distal to the reporter and semirandomized as A or T (W) in the Pho box overlapping the −35 region (positions 4 to 6) (Fig. 1A). The three nucleotides between Pho boxes were completely randomized (positions 1 to 3) (Fig. 1A), as were positions upstream and downstream of the −10 promoter element (positions 7 to 11 and 12 to 19, respectively) (Fig. 1A). Altogether, this promoter has 34,359,738,368 (416 × 23) possible sequence variants.
FIG 1.

Design and screening of synthetic PhoB-dependent promoter variants. (A) Synthetic constructs included four PhoB-binding sites (gray boxes) as well as a −10 promoter element. The −35 promoter region is indicated, although it does not resemble a −35 consensus. Randomized or semirandomized positions are numbered, corresponding to the numbering in panel C. “N” indicates a randomized position (labeled 1 to 3 and 7 to 19). Positions 4 to 6 were restricted to A or T (W). Eighty-three variants of the sequence were cloned upstream of gfp and screened for activity. (B) GFP output for four representative promoter variants. GFP was measured for constructs in ES114 and JLS9 (ΔphoB) grown in FMM medium with a high (378 μM) or low (37.8 μM) added PO4 concentration. Strains with the promoterless parent vector (labeled “None”) show background fluorescence. Values were taken at an OD595 of 1.0 and normalized to red fluorescence. Error bars indicate standard deviations (n = 3). Data from one representative experiment of three are shown. (C) Each variable position (positions 1 to 19) was subjected to a Mann-Whitney U nonparametric test to compare the average GFP output associated with each individual nucleotide (A, T, C, or G) against the average value for constructs with the other nucleotides in that position. Pairs of nucleotides were also compared at each position (A/T versus G/C, C/T versus A/G, G/T versus A/C, and C/G versus A/T). Only comparisons with P values of <0.05 are shown. Values indicate the mean GFP output for all constructs with the nucleotide (or nucleotide pair) at the indicated position minus the mean value for the other constructs. For each of the four strain-medium combinations, nucleotides that correlated with significantly higher or lower GFP levels are indicated by green or red, respectively; *, **, and *** indicate P values of <0.05, <0.01, and < 0.001, respectively. The right column indicates how randomization was further constrained for the next set of variants (round 2). † indicates that the C at position 2 was underrepresented in the screened variants (in only 1 of the 83 variants) and therefore was included in round 2.
We cloned and sequenced 83 variants of this construct (Fig. 1A) upstream of a promoterless gfp in a vector that also expresses mCherry constitutively. The GFP expression level for each construct was measured in V. fischeri ES114 (WT) and its ΔphoB derivative JLS9 grown in fischeri minimal medium (FMM) with low or high PO4 levels (for sequences and fluorescence from all 83 constructs, see the supplemental material). Figure 1B shows GFP expression for a promoterless negative control and four promoter variants that represent the wide range of variation in GFP outputs observed among the 83 variants screened. Although most clones displayed some of the desired low-PO4- and phoB-dependent activation, the degree of activation varied, as did the level of undesirable background GFP expression in the ΔphoB mutant and/or under high-PO4 conditions (e.g., see Fig. 1B). In some constructs, PhoB-dependent activation under low-PO4 conditions was too weak to be useful (e.g., variant 1) (Fig. 1B). In others, the background GFP expression level, determined relative to the promoterless control, was higher than optimal (e.g., variant 2) (Fig. 1B). Some variants displayed significant activation with reasonably low background levels (e.g., variants 3 and 4) (Fig. 1B); however, assessing 83 clones out of over 34 billion possibilities had not come close to saturating the screen.
Optimization of the synthetic promoter.
To assess the prospects for improving the PhoB-dependent promoter without screening an intractably large number of clones, we analyzed whether the identity of any of the 19 variable nucleotides (Fig. 1A) correlated with GFP expression. Specifically, at each of the 19 positions, we used the Mann-Whitney U test to compare the reporter output corresponding to each nucleotide to the output from constructs with the other three possible nucleotides (A versus C, G, or T; C versus A, G, or T; G versus A, C, or T; and T versus A, C, or G). We likewise made comparisons based on possible pairs of nucleotides at a position, for example, an A or C at a position versus a G or T at the same position. Variable nucleotide positions 4, 5, and 6 were limited to A or T (W) by design (Fig. 1A), so only those two comparisons were made in those cases.
Figure 1C summarizes the significant (P < 0.05, P < 0.01, or P < 0.001) correlations between nucleotide identity at each of the 19 variable positions in the promoter and the reporter output. We averaged the GFP expression level (normalized to mCherry) corresponding to each nucleotide (or nucleotide pair) at each position and subtracted the average GFP expression level of the constructs with other nucleotides at that position, to yield the numbers reported in Fig. 1C. The output for each reporter was analyzed for each of the four strain-medium combinations tested (wild type or ΔphoB mutant under high- or low-phosphate conditions). Given that 460 comparisons were tested for significance, some of those indicated in Fig. 1C may appear significant by chance, particularly at the level of a P value of <0.05. On the other hand, in some positions, all three background conditions (wild type under high-PO4 conditions or ΔphoB mutant under high- or low-PO4 conditions) yielded similar results, suggesting reproducibility and value in this approach. We did not perform a multiple-testing correction because the P values are used only to select the most promising candidates for further experiments. Our goal was to determine how we might rationally constrain our sequence design to optimize output and not to definitively state a role for the sequence at any position, and the results suggested a rational approach to further optimization might be possible.
Our data pointed to further sequence constraints that might contribute to desirable reporter properties, specifically, high-PhoB-dependent activation under low-PO4 conditions or low background levels under the other conditions. For example, a C or a T at position 1 was correlated with above- or below-average background levels, respectively (Fig. 1C), and therefore, we defined this position as a T in our second-round construct. As another example, a C at position 12 was correlated with below-average expression in the wild type under low-PO4 conditions, so this position was constrained to A, T, or G in the second-round construct. Using this approach, most positions were defined more narrowly in the second round of screening. Three of the 19 positions (positions 6, 13, and 18) showed no correlation with promoter activity, and three others (positions 5, 11, and 15) had a weak correlation, as significance was seen under only one of three background conditions at the level of a P value of <0.05. These six positions were not constrained further but rather were left as they were in the initial screen. For the most part, the nucleotide representation at each randomized position had been evenly distributed, but only 1 of the 83 promoters had a C at position 2, so C was included again at this position in the second round of promoter design.
A second round of synthetic inserts was engineered to contain the same conserved sequences as those in round 1 (Fig. 1A), along with the newly constrained positions (Fig. 1C, right). The resulting constructs were assayed in the same manner as for the first-round reporters. Many of the 99 resulting promoters that were screened exhibited an increase in GFP expression in the “on” state (wild type under low-PO4 conditions) and/or decreased background levels under other conditions relative to earlier constructs. Figure 2 illustrates the optimal second-round reporter (pJLS1088) compared to the best promoter from round 1 (pEW6AQ). The newer reporter exhibited significantly higher-level “on” activation and lower background levels (P < 0.05 by Student's t tests). The difference in GFP expression levels between the wild type and the phoB mutant in low-PO4 medium was 11-fold for cells carrying pJLS1088, compared to 3-fold induction for the best first-round construct, and the PhoB-independent background was eliminated (Fig. 2).
FIG 2.
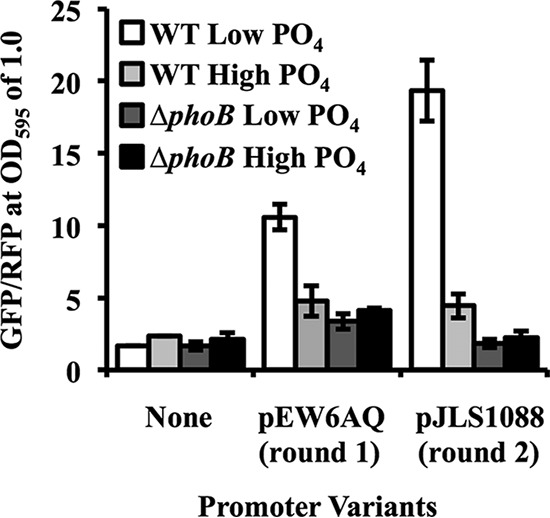
Iterative optimization of PhoB-dependent reporter performance. A representative of the best promoters from the first set of semirandomized variants (pEW6AQ) (numbered 4 in Fig. 1B) was compared to an optimal variant (pJLS1088) generated after constraining additional positions, as indicated in Fig. 1C. Reporters were evaluated in the ES114 and ΔphoB strains and were grown in minimal medium amended with high or low concentrations of PO4. The promoterless parent vector pJLS27 (labeled “None”) is included to show the background. Fluorescence values were taken at an OD595 of 1.0, and error bars indicate standard deviations (n = 3). Data show results from one representative experiment of three performed.
Reporter responsiveness to extracellular phosphate.
To evaluate the level of PO4 at which the PhoB-specific reporter was activated, we measured both GFP expression from pJLS1088 and phosphate levels during the growth of wild-type strain ES114 in batch cultures, using the empty vector (pJLS27) as a negative control (Fig. 3). Due to the rapid depletion of PO4 during the exponential growth phase, measuring the exact PO4 concentration when GFP was first detectable proved difficult. However, GFP was consistently first detected when PO4 levels were depleted to between 1 and 10 μM (Fig. 3), similar to the activation of PhoB in E. coli below an environmental concentration of 4 μM PO4 (18).
FIG 3.
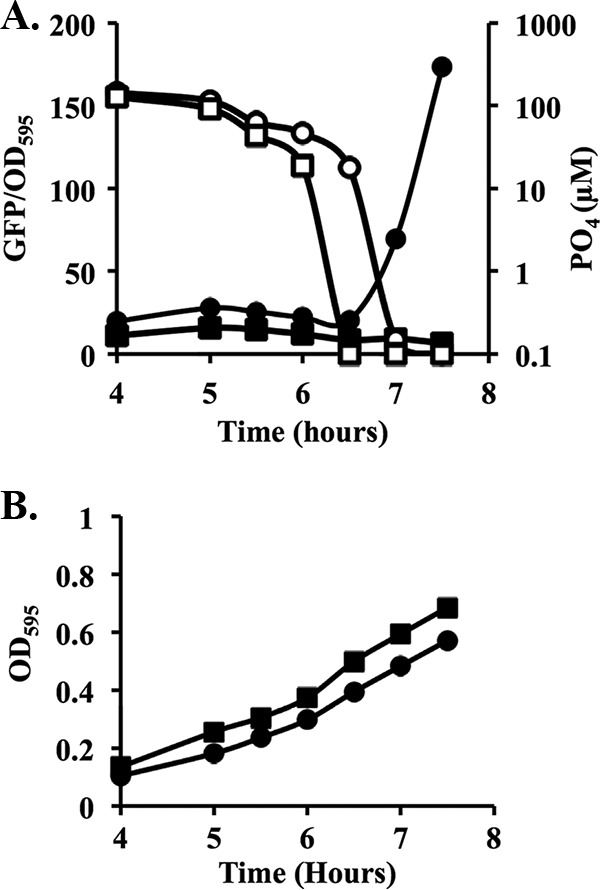
A PhoB-dependent reporter is induced upon PO4 depletion in batch culture. (A) GFP/OD595 values and supernatant phosphate levels were measured over time in cultures of wild-type cells containing either the promoterless parent vector (pJLS27) (squares) or the optimized PhoB reporter (pJLS1088) (circles) grown in batch cultures in FMM amended at the outset with 37.8 μM phosphate for a final concentration of 100 μM total phosphate. Filled shapes represent the GFP/OD595 fluorescence, whereas open shapes represent the extracellular PO4 concentration. (B) Growth of the cultures from panel A shown as cell density (OD595) over time. Data from one representative experiment of three are shown.
The PhoB-dependent reporter responds to low PO4 levels in other proteobacteria.
Because the Pho regulon is well conserved among proteobacteria (37), we tested whether the optimized reporter functions in bacteria other than V. fischeri. The reporter plasmid pJLS1088 and its parent, pJLS27, each have two origins of replication: the R6K gamma origin (38) and the origin from pES213, which replicates well in members of the Vibrionaceae without the need to maintain antibiotic selection (39, 40). For the plasmid to replicate in a variety of backgrounds, we added the pir gene to the parent vector and reporter plasmid, which should enable replication from the R6K origin in a broad range of non-Vibrio hosts (41).
We tested the GFP induction of our reporter in Vibrio cholerae, Escherichia coli, Salmonella enterica, and Ruegeria pomeroyi, each of which encodes PhoB. Figure 4 shows the red and green fluorescence of colonies grown on solid media with relatively high or low PO4 levels. V. fischeri colonies are included for comparison. Red fluorescence is the result of constitutive mCherry expression from the plasmids, whereas green fluorescence corresponds to reporter GFP expression, with pJLS27 and pJLS71 serving as promoterless gfp negative controls. Each strain displayed green fluorescence only when grown with the reporter on low-phosphate plates (Fig. 4). Moreover, GFP expression in V. cholerae was eliminated in a phoB mutant, just as we observed in V. fischeri (Fig. 4).
FIG 4.

Response of reporters to low PO4 concentrations in other proteobacteria. Red and green fluorescence from colonies of V. fischeri, V. cholerae, E. coli, S. enterica, or R. pomeroyi carrying a promoterless parent vector (pJLS27 for vibrios or pJLS71 for nonvibrios) or a vector containing the PhoB-activated promoter (pJLS1088 for vibrios or pJLS137 for nonvibrios) are shown. Strains include V. fischeri ES114 and JLS9 (ΔphoB), V. cholerae AC2764 (ΔtcpA) and AC3236 (ΔphoB), E. coli MG1655, S. enterica MS1868, and R. pomeroyi DSS-3. Strains were grown on agar plates with defined medium containing low or high concentrations of added PO4 (see Materials and Methods). Colonies of similar sizes were imaged by using a Nikon Eclipse E600 microscope with a 51005v2 filter, which enabled the simultaneous visualization of both the constitutive red fluorescence and the green fluorescence of the reporter.
Testing of GFP variants with reduced half-life in V. fischeri.
The stability of the GFP protein can lead to its accumulation and render this reporter nonideal for assessing dynamic changes in gene expression. Specifically, the downregulation of the promoter is difficult to detect if the reporter protein (in this case GFP) does not turn over. In some bacteria, adding an SsrA tag to the C terminus of GFP increases its recycling via an AAA+ protease (42–44), such as ClpAP or ClpXP (45). Variations in the last 3 amino acids of the 11-residue SsrA peptide sequence (AANDENYALAA) can alter the efficiency with which the protein is recycled (42). We generated modified versions of gfp, encoding C-terminal SsrA tags terminating in the tripeptide LAA, ASV, or AAV, and expressed them from an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible promoter in wild-type strain ES114 and three transposon mutants with insertions in clpA, clpX, or clpS, the last of which encodes an adapter that delivers proteins to ClpAP (46, 47). The stable parental GFP and the *ASV SsrA variant produced high-level fluorescence in all backgrounds (Fig. 5). The *LAA and *AAV SsrA-tagged GFP variants yielded reduced or nearly undetectable fluorescence in the wild type as well as the clpA and clpS mutants. In the clpX mutant background, the level of fluorescence from the *AAV and *LAA variants was higher than that from the wild type (Fig. 5). These results suggested that SsrA tagging directed GFP turnover by ClpXP in V. fischeri; however, the level of fluorescence from the *LAA and *AAV derivatives was too low in the wild type to be useful, apparently because they are degraded quickly by ClpX.
FIG 5.

Effects of clpX and specific SsrA tags on GFP fluorescence in V. fischeri. Specific fluorescence associated with GFP variants is shown for wild-type strain ES114 or mutants with transposon insertions disrupting clpA, clpX, or clpS. pJLS153 (“stable”) has gfp without an ssrA tag. pJLS150 (*ASV), pJLS151 (*LAA), and pJLS152 (*AAV) all have gfp with a modified ssrA tag, exchanging the last three C-terminal amino acids, as indicated. Strains were grown in SWTO medium with 2 mM IPTG to induce gfp expression. GFP/OD595 values were taken at an OD595 of 1.0, and error bars indicate standard deviations (n = 3). Data from one representative experiment of three performed are shown.
The *ASV variant of GFP yielded fluorescence values similar to those of the parental GFP when IPTG was added to induce their expression (Fig. 5); however, when IPTG was washed away, the fluorescence from the parental and *ASV-tagged GFPs diverged (Fig. 6). The parental GFP was remarkably stable, whereas fluorescence from the *ASV-tagged GFP decayed noticeably (Fig. 6). The slope constant, μ, and half-life (T1/2 = −ln 2/μ) for fluorescence were determined for each biological replicate (a total of 12 across four experiments) and averaged. The fluorescence values for the parental GFP were unchanged for the duration of the experiment, so a half-life could not be calculated, but fluorescence from the ASV SsrA-tagged GFP had an estimated half-life of 81 min. Taken together, our results suggest that GFP*ASV should be a useful reporter in V. fischeri, displaying sufficient fluorescence to be detected when expressed but decaying quickly enough to capture some changes in gene expression that would be missed by using the parental GFP.
FIG 6.
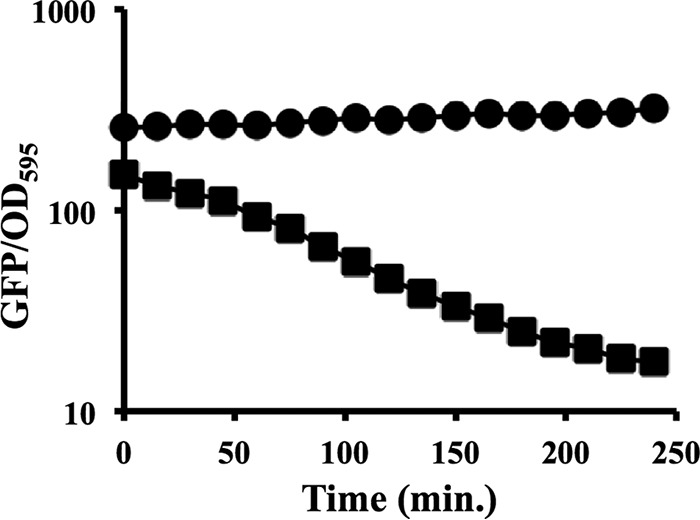
Addition of the *ASV SsrA tag to GFP increases the turnover rate in V. fischeri. ES114 cells carrying pJLS150 (GFP*ASV) (squares) or pJLS153 (parental GFP with no SsrA tag) (circles) were grown in SWTO medium plus 2 mM IPTG or SWTO medium with no inducer to an OD595 of 1.5 before being washed and resuspended in SWTO medium without IPTG. GFP/OD595 values were taken every 15 min for 4 h. Values under noninduced conditions (no IPTG added) were subtracted as the background from values under induced conditions. Error bars indicating standard deviations (n = 3) are smaller than the symbols. Data from one representative experiment of four performed are shown.
Variability in expression of the PhoB-dependent reporter within symbiont populations.
Initially, to begin assessing the state of PhoB in the juvenile squid light organ, we infected juveniles with either ES114 or JLS9 (ΔphoB) harboring the best reporter from round 1, pEW6AQ (Fig. 3), or its parent vector, pJLS27. In aposymbiotic (i.e., uninoculated and uncolonized) squid, the light organ has minimal green or red fluorescence (Fig. 7A), and juveniles infected with JLS9 or ES114 carrying the empty vector display red, but not green, fluorescence (Fig. 7A and data not shown). Juvenile squid infected with ES114 carrying pEW6AQ displayed distinct and in some cases heterogeneous reporter expression. For example, some light organs displayed homogenous red fluorescence (Fig. 7B, top), while in other juvenile light organs, symbiont fluorescence showed distinct areas of red or green fluorescence (Fig. 7B, bottom). These results suggested spatial and/or temporal heterogeneity in the induction of the PhoB response. Using an epifluorescence microscope, however, we were unable to distinguish the microenvironments being colonized, and the stable GFP used in these experiments could potentially compromise our ability to distinguish temporal regulation.
FIG 7.

Expression of PhoB-dependent reporters in symbiotic V. fischeri cells. (A and B) Juvenile squid were left uninoculated and aposymbiotic (A, left) or were infected with V. fischeri containing either the promoterless vector (pJLS27) (A, right) or the first-round reporter pEW6AQ (B). Light organs (∼250 μm across; outlined with dotted white lines) were visualized by epifluorescence microscopy with a red/green filter at 24 h postinoculation. (C) Confocal microscopic image of a juvenile squid light organ showing one-half of the organ from a juvenile colonized with ES114 cells harboring the second-round PhoB-dependent promoter driving the expression of destabilized GFP (pJLS298). The squid tissue (blue) and the pores, antechambers, and crypts are labeled when visible.
To better define the expression patterns observed in our initial studies, we combined our round 2 PhoB-dependent promoter with the destabilized GFP*ASV variant and used this construct to assess PhoB activation in symbiotic V. fischeri cells using confocal microscopy. Within the bilobed E. scolopes light organ, symbionts colonize distinct microenvironments. Six pores on the organ surface lead through ducts and antechambers to six epithelium-lined crypts, three in each lobe, designated crypts 1, 2, and 3 based on their progression in development (48). Using confocal microscopy, we were able to delineate the individual crypt spaces. Again, in animals infected with JLS9 (ΔphoB) carrying the reporter pJLS298, all crypt spaces displayed only red fluorescence from constitutive mCherry (data not shown). Also, similar to the results using an epifluorescence microscope, some of the animals infected with ES114 carrying the reporter pJLS298 that were viewed by using confocal microscopy had red fluorescent symbionts within the crypts yet had no visible GFP fluorescence (data not shown); however, some displayed heterogeneous GFP expression. Where heterogeneous expression was seen within a crypt, the population of GFP-expressing cells was clustered together rather than dispersed throughout the crypt space. Typically, cells in crypt 2 displayed the strongest GFP fluorescence, while crypt 1 had at most a smaller and fractional population of the cells expressing GFP (Fig. 7C and data not shown). Crypt 3, which is the smallest crypt and the last to develop embryologically, was sometimes not yet colonized, and in only one animal did we see GFP-expressing symbionts in this crypt (data not shown).
DISCUSSION
Synthetic regulator-specific transcriptional reporters offer a promising approach to assess regulator activity under different conditions. When the goal is to examine the activity of a particular regulator, synthetic reporters may be superior to native promoters if they can be optimized in terms of specificity (response to a specific transcription factor) and the signal-to-noise ratio (high-level expression in the “on” state and low-level background expression in the “off” state). However, this synthetic approach is hindered by our limited ability to predict the effects of specific sequences in the promoter region and by the absence of high-throughput technology that would allow the testing of all possible construct variants, which could include billions of possibilities. One approach to overcome the intractable number of sequence possibilities is to screen a subset of sequence variants, identify positions where a specific nucleotide(s) correlates with enhanced performance, and thereby experimentally evolve a better synthetic reporter.
We used such an approach to study the PhoBR two-component regulatory system in V. fischeri, both in culture and in symbiosis with the Hawaiian bobtail squid, E. scolopes. First, we generated an improved PhoB-dependent reporter that has high-level expression when activated by PhoB but low-level background expression. We began with a rationally designed sequence (Fig. 1A), based on known elements of PhoB-regulated transcriptional promoters; however, our results underscored our limited ability to predict promoter function and effectiveness as a reporter. We randomized, or semirandomized, nucleotide positions for which we did not foresee a sequence-specific role in promoter optimization, yet constructs with different nucleotides at these positions had highly variable performances (e.g., see Fig. 1B). While some of the 83 constructs initially screened worked reasonably well, others did not. Had we designed a single defined sequence for a reporter, it would most likely have been only marginally effective.
Our inability to predictively design an optimal reporter highlights gaps in our understanding of how specific sequences influence a PhoB-dependent promoter or promoters in general. As noted above, the individual nucleotide identities highlighted in Fig. 1C should be viewed cautiously, particularly at the level of a P value of <0.05, given the number of comparisons being made, but in aggregate, the data make a compelling case that positions that we did not predict would influence the reporter were important. Even in retrospect, some of the nucleotide identities that appear to impact promoter performance (Fig. 1C) seem inexplicable, especially for the positions outside the Pho boxes or the promoter elements. In one notable example, a guanosine at position 10, in the gap between Pho boxes and the −10 element, significantly correlated with decreased fluorescence under all three background conditions (P < 0.01 to P < 0.001). Although there is precedence for the promoter spacer region sequence affecting transcription (49–51), to our knowledge, it remains difficult to predict such effects in a synthetic promoter.
Similarly, the mechanisms underlying the differences in promoter-reporter performance are unknown. The sequence-specific effects that we observed may be due to subtle changes in DNA topology or context-specific effects on RNA polymerase (RNAP) or PhoB binding and/or interactions. Additionally, there may be alternative weak promoters and transcriptional start sites aside from the promoter highlighted in Fig. 1A, and changes that minimize their activity would decrease the PhoB-independent background. Although we cannot rule out the possibility that we have created binding sites for additional regulators, this mechanism seems improbable given the sequence specificity of most regulators and the lack of any apparent binding sites for known regulators. Moreover, a non-PhoB activator of the reporter would itself have to be activated by PhoB, because activation is specifically PhoB dependent.
A second round of screening, narrowed by informative results from the first round, enabled us to increase the reporter output under low-phosphate conditions and to decrease background expression (Fig. 2). While a similar combination of rational and randomized nucleotide screening methods to design promoters for reporter use was used previously (15), this study shows that by screening only 83 unique promoters out of over 34 billion possible sequence combinations, rational changes could be made to optimize reporter performance. The very best possible variant was likely not screened, and some aspects of the optimized sequence may be too dependent on the sequence context to be identified by our approach, but nonetheless, we showed that improvements could be made by screening a manageable number of clones. Importantly, the round 2 promoters essentially eliminated the PhoB-independent background of the reporters (Fig. 2). This elimination of the background is particularly important for applications where an obvious qualitative distinction between “active” or “inactive” is beneficial. Notably, although background fluorescence can be subtracted readily in comparisons of batch cultures, in examining symbiotic cells in situ, a low background level allows the sensitive detection of the “on” state of a reporter.
In addition to providing a proof of principle for optimizing a synthetic promoter, our reporter proved a useful tool for investigating the PhoB-mediated response to low phosphate levels in V. fischeri. The E. coli PhoBR system is activated upon sensing environmental phosphate levels of less than 4 μM (18), and we similarly observed the PhoB-dependent activation of the reporter to occur at concentrations of between 1 and 10 μM phosphate (Fig. 3). In the future, more samples could be taken from batch cultures, or phosphate could be defined under chemostat conditions, to more precisely determine the threshold phosphate concentration required for activation. Furthermore, the threshold for reporter activation may not be constant for all bacteria. Among various proteobacteria, different concentrations of phosphate in minimal media were required to visualize reporter activation (or a lack thereof) (Fig. 4). Therefore, it could be useful to determine the threshold phosphate concentration below which the reporter turns “on” in different organisms. Such differences could possibly be utilized for determining bioavailable phosphate levels in various environments and samples by using different species, or engineered strains of V. fischeri, that induce their PhoB response at different phosphate concentrations. It is worth noting that the PhoB-activated pst operon, which encodes the proteins for a high-affinity phosphate importer (18, 52), is often used in metatranscriptomic data sets as an indication of bacteria coming from phosphate-limiting conditions (53). Reporter strains added to such samples may help refine our understanding of bioavailable phosphate.
In this study, we also extended the utility of GFP as a reporter in V. fischeri. The use of GFP reporters can lead to an accumulation of a fluorescent signal that remains even after transcription has turned off, due to GFP's stability (42). To address this problem, we tagged gfp with the three variants used previously in E. coli (42–44), and our results are consistent with the SsrA tag targeting proteins to the ClpXP protease in V. fischeri (Fig. 5). Even in the clpX mutant background, the GFP variant with the LAA-SsrA tag showed attenuated fluorescence (Fig. 5), which could suggest that either (i) the protein is also recycled by another protease or (ii) it is not properly folded, resulting in less fluorescence. Regardless, and more importantly, we determined the half-life of the ASV SsrA variant to be 81 min, and it showed a useful balance between fluorescence under inducing conditions (Fig. 5) and the decay of the signal once the inducer was removed (Fig. 6). Our results parallel data from studies of E. coli where the LAA SsrA variant had the shortest half-life, followed by the AAV and ASV variants (42). This decreased-half-life GFP variant should be particularly useful in in situ experiments assessing symbiotic V. fischeri, and it should be able to capture major changes in gene expression over the diurnal symbiotic cycle (54, 55).
Different reports suggest that PhoB in symbiotic V. fischeri might be either active (indicative of low phosphate availability) or inactive during colonization with E. scolopes. Mutants with a transposon insertion in phoB or the genes for the high-affinity phosphate importer, pstA and pstS, have decreased competitive fitness when competed against the wild type for symbiotic colonization (24, 56), suggesting that phosphate might be limiting. However, in a separate study looking at the transcriptome of V. fischeri cells vented from the light organ by the host, the pst genes were not upregulated, suggesting that PhoB was not in an activated state and that the light organ was not a phosphate-limited environment (57). A way to reconcile these two data sets could be if PhoB was activated in specific microenvironments of the light organ, as is the case with the lux operon (39). Sycuro et al. found that the crypts of the light organ are not vented equally (58), but rather, most vented cells are from crypt 1. Therefore, if PhoB is activated more significantly in crypts 2 and 3, which are not vented as completely, the Pho regulon may not appear induced in the transcriptomic data set.
We found that the PhoB-dependent reporter has heterogeneous expression between and within juvenile squid light organs at 24 h (Fig. 7B and C). Likewise, in infected animals displaying symbionts with green fluorescence, we saw a noticeable partitioning of GFP expression within and between the crypts. For example, in Fig. 7C, crypt 1 is shown in three regions, with GFP being detectable in only a portion of one of the regions, yet crypt 2 shows that most cells are expressing GFP, with only a small section of cells with undetectable green fluorescence. The clustered nature of the GFP-expressing cells may indicate that there was a localized low-phosphate microenvironment in the host. Still, some squid were well colonized by V. fischeri based on the homogenous mCherry fluorescence (and symbiotic bioluminescence) yet showed little to no green fluorescence, indicating that these symbionts in the light organ are not inducing a low-phosphate response (Fig. 7B, top, and data not shown). These data suggest that select regions of the light organ are phosphate limited at some point, which is consistent with the competitive defect seen when phoB, pstA, or pstS is disrupted. As crypt 1 tends to be the most completely vented each morning and could account for most cells in the light-organ ventate (58), these data are also consistent with the results of the transcriptomic study of vented cells, as many crypt 1 symbionts did not display PhoB-dependent reporter activation. Therefore, our data illustrate the power of considering the microenvironments of the squid light-organ crypts in expression studies. Future studies should help to further define the spatial and temporal pattern of gene expression in symbiotic V. fischeri.
MATERIALS AND METHODS
Media and growth conditions.
Vibrio fischeri strain ES114 was used as the wild type and parent for strain construction (59). Escherichia coli strains DH5α and DH5αλpir (40) were used as hosts for plasmids. V. fischeri was grown at either 24°C or 28°C in either lysogeny broth (LB) salt (LBS) medium (60), seawater tryptone (SWT) medium (59), seawater tryptone osmolarity (SWTO) medium (61), or modified fischeri minimal medium (FMM) (62), with 37.8 μM K2HPO4 or 378 μM K2HPO4 added for “low”- and “high”-phosphate conditions, respectively. Plasmids were maintained in E. coli grown in LB (63) at 37°C. Solid media were prepared by adding 15 g liter−1 agar. For the selection of plasmids in E. coli, kanamycin (Kn), chloramphenicol (Cm), erythromycin (Erm), or trimethoprim (Tmp) was added to LB at a final concentration of 40, 20, 150, or 10 μg ml−1, respectively. To maintain selection in V. fischeri on LBS medium, Kn, Cm, Erm, or Tmp was added at 100, 2, 5, or 10 μg ml−1, respectively.
E. coli strain MG1655, Salmonella enterica serovar Typhimurium strain MS1868, and Vibrio cholerae strains AC3236 and AC2764 were maintained and transformed by using LB medium, and their response to PO4 was assessed by using modified MOPS (morpholinepropanesulfonic acid) minimal medium at 37°C (64). V. cholerae strain AC2764 has a deletion of tcpA, reducing strain virulence. To generate a base for the MOPS medium, we added no phosphate and used 1 g/liter Casamino Acids. For low-phosphate MOPS medium, the base minimal medium was used with no added phosphate other than that in Casamino Acids and other components, whereas 800 μM K2HPO4 was added to the base medium for high-phosphate conditions. R. pomeroyi strain DSS-3 was grown at 30°C on half-strength yeast tryptone sea salts (1/2YTSS) medium (65), and to manipulate PO4 availability, it was grown in modified marine basal medium (MBM) (66), with either 0.2 mM K2HPO4 or 2 mM K2HPO4 added for low- and high-phosphate conditions, respectively. A total of 80 μg ml−1 Kn was included to maintain plasmid selection in DSS-3 (65, 66).
Plasmid and strain construction.
Plasmids were mobilized from E. coli to V. fischeri by triparental mating using helper plasmid pEVS104 maintained in CC118λpir (67), as previously described (68). Plasmids were moved into E. coli MG1655 and S. enterica MS1868 by transformation. Plasmids were moved into V. cholerae AC3236 and AC2764 by conjugation using helper plasmid pEVS104 and by selecting for recipients on LB supplemented with Kn (to select for the plasmid) and 2 μg ml−1 potassium tellurite (to select against E. coli donor cells). Plasmids were similarly moved into R. pomeroyi DSS-3 via conjugation using helper plasmid pEVS101 and by selection on 1/2YTSS medium with Kn and 2 μg ml−1 potassium tellurite.
JLS9, an in-frame ΔphoB deletion mutant, was constructed through allelic exchange (22) and verified by PCR. To generate the ΔphoB allele, 2 kb upstream and downstream of phoB was PCR amplified (using primer pair PhoBupF and PhoBupR and primer pair PhoBdnF and PhoBdnR, respectively) and cloned into pCR-Blunt TOPO, generating pSJB2 and pDC6, respectively. To generate the in-frame deletion, two small self-annealed oligonucleotides were inserted at the AscI sites of pSJB2 and pDC6, generating an NdeI site and yielding pJLS21 and pJLS22, respectively. pJLS22 was fused to pEVS118 at the KpnI site, generating pJLS23, which was subsequently digested with ApaI and self-ligated to remove the ColE1 origin of replication and generate pJLS24. The sequences upstream and downstream of phoB were joined by fusing pJLS21 and pJLS24 at the NdeI site. The resulting construct, pJLS25 was mobilized into ES114 to generate ΔphoB mutant strain JLS9 via allelic exchange.
To generate the semirandomized promoter regions for reporter plasmid screening, oligonucleotides JLSPhoBPF2 and JLSPhoBPR (round 1) or JLSPhoBPF3 and JLSPhoBPR2 (round 2) (Table 1) were annealed together and filled in by using the DNA polymerase I Klenow fragment to generate a blunt-ended double-stranded product. The filled-in product was digested with SphI and SalI, ligated into similarly digested pJLS27, and transformed into DH5αλpir. Transformants were screened by visualizing plates under an epifluorescence microscope, using a red/green filter, and the green or yellow colonies were picked. Fluorescence intensities of V. fischeri were later screened by using a Synergy 2 plate reader (BioTek, Winooski, VT), with excitation/emission wavelengths for GFP of 485 nm/528 nm and for mCherry of 530 nm/590 nm, normalizing GFP to mCherry (GFP/mCherry) or GFP to the optical density at 595 nm (OD595), as indicated.
TABLE 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Genotype, description, or sequence (5′–3′)a | Reference or source |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| CC118λpir | Δ(ara-leu) araD Δlac74 galE galK phoA20 thi-1 rpsE rpsB argE(Am) recA λpir | 67 |
| DH5α | ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 72 |
| DH5αλpir | λpir derivative of DH5α | 40 |
| MG1655 | F− λ− ilvG rfb-50 rph-1 | 73 |
| Ruegeria pomeroyi DSS-3 | Wild-type isolate from coastal seawater, Georgia (USA) | 74 |
| Salmonella enterica serovar Typhimurium MS1868 | leuA414(Am) hsdSB(r− m+) Fels− | 75 |
| Vibrio cholerae | ||
| AC2764 | E7946 ΔtcpA | A. Camilli |
| AC3236 | E7946 ΔphoB | 76 |
| Vibrio fischeri | ||
| ES114 | Wild-type isolate from E. scolopes light organ | 59 |
| JLS9 | ES114 ΔphoB | This study |
| JLS38 | ES114 ΔphoU | This study |
| VFS008C4 | ES114 clpA::mini-Tn5 Ermr | C. Whistler |
| VFS024A1 | ES114 clpS::mini-Tn5 Ermr | C. Whistler |
| VFS025G1 | ES114 clpX::mini-Tn5 Ermr | C. Whistler |
| Select plasmidsb | ||
| pEVS101 | Conjugative helper plasmid; oriVColE1 oriTRP4 Ermr | 68 |
| pEVS104 | Conjugative helper plasmid; oriVR6Kγ oriTRP4 Knr | 68 |
| pEW6AQ | oriVR6Kγ oriTRP4 pES213 mCherry Knr PphoB-Cmr-gfp (round 1) | This study |
| pJLS25 | ΔphoB allele; oriVColE1 oriVR6Kγ oriTRP4 Knr Cmr | This study |
| pJLS27 | oriVR6Kγ oriTRP4 pES213 mCherry Knr promoterless Cmr-gfp | 15 |
| pJLS70 | oriVR6Kγ oriTRP4 pES213 mCherry Knr pir PphoB-Cmr-gfp (round 1) | This study |
| pJLS71 | oriVR6Kγ oriTRP4 pES213 mCherry Knr pir promoterless Cmr-gfp | This study |
| pJLS137 | oriVR6Kγ oriTRP4 pES213 mCherry Knr pir PphoB-Cmr-gfp (round 2) | This study |
| pJLS149 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr lacIq-Ptac | This study |
| pJLS150 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr lacIq-Ptac-gfp-ssrA-ASV | This study |
| pJLS151 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr lacIq-Ptac-gfp-ssrA-LAA | This study |
| pJLS152 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr lacIq-Ptac-gfp-ssrA-AAV | This study |
| pJLS153 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr lacIq-Ptac-gfp | This study |
| pJLS198 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr promoterless gfp-ssrA-ASV | This study |
| pJLS203 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr PphoB-gfp-ssrA-ASV | This study |
| pJLS298 | oriVR6Kγ oriTRP4 pES213 mCherry Pcon-Tmpr PphoB-gfp-ssrA-ASV | This study |
| pJLS1088 | oriVR6Kγ oriTRP4 pES213 mCherry Knr PphoB-Cmr-gfp (round 2) | This study |
| pRK12 | Knr oriVR6Kγ promoterless Cmr-gfp-ssrA | R. Kaul |
| pVSV33 | Knr oriVR6Kγ promoterless Cmr-gfp | 39 |
| pVSV102 | oriVR6Kγ oriTRP4 pES213 Knr gfp | 39 |
| Oligonucleotides | ||
| PhoBupF | GGC GCC TAG AGT GTT GTC TGG ACG | This study |
| PhoBupR | ATG GCG CGC CGG ATC CTT CTA GCC ATT CTC | This study |
| PhoBdnR | GGC GTA TCC ATA GGT GCC AGA GAC TGA G | This study |
| PhoBdnF | ATG GCG CGC CGG TAT AAA GGT AAT GGT TGA GCG TC | This study |
| NdeIprimer1 | CGC GAA ACA TAT GAA A | This study |
| NdeIprimer2 | CGC GTT TCA TAT GTT T | This study |
| JLSMCS1 | GCA TGC TGT AAA ACG ACG GCC AGT ACG TGC TAT GCG AGC TCG GGC CCG C | This study |
| JLSMCS2 | GTC GAC GCT AGC CAT TGC GCA GCG CGC TCT AGA TAG CGG GCC CGA GCT CGC ATA GC | This study |
| JLSphoBPF2 | TAG CAT GCC TGT CAT AAA TCT GTC ATA NNN CTG ACA TAW WWC TGT CAC ATG TT | This study |
| JLSphoBPR | TAG TCG ACT GNN NNN NNN AAA ATA NNN NNA ACA TGT GAC AG | This study |
| JLSphoBPF3 | TAG CAT GCC TGT CAT AAA TCT GTC ATA TSR CTG ACA TAT WWC TGT CAC ATG TT | This study |
| JLSphoBPR2 | TAG TCG ACT GDN CNH AAA ATA NCV DVA ACT AGT GAC AG | This study |
| JLSpirF3 | TAC CGC GGT TGA CTC TCA TGT TAT TGG CG | This study |
| JLSpirR3 | TAC CGC GGA CGC GTT CAC CCC TTA GCT TTT TTG GGA GG | This study |
| DSgfpP1 | ACA CTA GTC ACT ACT CTG TGC TAT GG | This study |
| DSgfpP2 | AGCTGCCAATGCGTAGTTTTCGTCGTTTGCGACGTT GTA CAG TTC ATC CAT GCC ATG | This study |
| DSgfpP3 | GTC GCA AAC GAC GAA AAC TAC GCA TTG GCA GCT TGA GGA TCC CCG GGA ATT C | This study |
| DSgfpP4 | ACC CGC GGG GAT CTT AGG | This study |
| JLSgfpF | ATG GCT AGC AAA GGA GAA GAA CTC T | This study |
| JLSssrA-ASV | TAT GGA TCC TCA AAC TGA TGC TGC GTA GTT TTC GTC GTT TGC GAC | This study |
| JLSssrA-LAA | TAT GGA TCC TCA AGC TGC CAA TGC GTA GTT TTC GTC GTT TGC GAC | This study |
| JLSssrA-AAV | TAT GGA TCC TCA AAC TGC TGC TGC GTA GTT TTC GTC GTT TGC GAC | This study |
| JLSgfpR | GCA GGA TGG TCA GTT GTA CAG TTC ATC CA | This study |
| pJLS198mCherryPcon | 475-bp synthetic DNA fragmentc | This study |
Drug resistance abbreviations: Cmr, chloramphenicol resistance (cat); Ermr, erythromycin resistance; Knr, kanamycin resistance (aph); Tmpr, trimethoprim resistance (dfr). Nonstandard nucleotides are as follows: N (A, C, G, or T), W (A or T), D (A, G, or T), H (A, C, or T), V (A, C, or G), S (C or G), and R (A or G).
The plasmids listed may contain the RP4 origin of transfer (oriTRP4). Replication origins are denoted ColE1, pES213, and/or R6Kγ.
Designed DNA “gene block” ordered from Integrated DNA Technologies (Coralville, IA); the sequence is available upon request.
To increase the bacterial host range of the optimized reporter, we amplified the pir gene from pGRG36pir (69) with primers JLSpirF3 and JLSpirR3 and cloned the SacII-digested product into the “round 1” and “round 2” optimized reporters pEW6AQ and pJLS1088, generating pJLS70 and pJLS137, respectively. To generate an isogenic promoterless vector, we digested pJLS70 with SalI and ligated-in a fragment consisting of oligonucleotides JLSMCS1 and JLSMCS2 annealed together, which created a multiple-cloning site and generated pJLS71.
To generate destabilized GFP variants, an ssrA tag (VANDENYALAA) was added at the end of the gfp coding sequence. The gfp-ssrA variant was constructed by using splicing by overhang extension (SOE) PCR with primers DSgfp1, DSgfp2, DSgfp3, and DSgfp4 to amplify gfp and add an ssrA tag to the end of the protein just before the stop codon. The PCR product was digested with SacI and SpeI and ligated into pVSV102 cut with the same enzymes to generate pRK12. A new vector (pJLS149; GenBank accession number MG752896) was constructed to enable the IPTG induction of GFP variants in V. fischeri, with mCherry expression from a consensus sigma-70 promoter. To obtain different variants of the unstable GFP, primers JLSgfpf and either JLSssrA-ASV, JLSssrA-LAA, or JLSssrA-AAV were used to amplify gfp from pRK12, and the subsequent fragments were digested with NheI and BamHI and ligated into pJLS149 cut with the same enzymes, generating pJLS150, pJLS151, and pJLS152, respectively. A stable variant was constructed in the same manner, using primers JLSgfpF and JLSgfpR to PCR amplify gfp from pVSV33 to generate plasmid pJLS153. To use the PhoB-dependent reporter from pJLS1088 (Fig. 2) in combination with the destabilized GFP*ASV variant, we generated pJLS298 (GenBank accession number MG752897), which is mobilizable and stable in V. fischeri and encodes constitutively expressed mCherry.
Sequencing and statistical analyses.
Sequences of over 100 semirandomized promoter regions were determined at the University of Michigan DNA Sequencing Core Facility. To determine whether nucleotide identity at a particular position was significantly correlated with above- or below-average fluorescence from the reporter, a Mann-Whitney U test was utilized. The reporter data analyzed were the green/red fluorescence values at an OD of 1.0 from ES114 and JLS9 cultures grown in FMM with 10 μM or 200 μM KH2PO4. The background fluorescence from strains with the promoterless parent vector was subtracted from each of the 83 promoter variants for each combination of strain and medium. The fluorescence and sequence data for the 83 variants are provided in the supplemental material. At each (semi)randomized nucleotide position, each nucleotide was compared to the other three nucleotides, and similarly, pairs of nucleotides were compared at each position against the other pairs (see Results).
Fluorescence assays.
To measure levels of GFP and mCherry, cultures were grown in 96-well black clear-bottom plates (Greiner Bio-One, Monroe, NC). Plates were placed into a Synergy 2 plate reader (BioTek) and incubated without shaking, and OD595, GFP (480/20-nm excitation filter and 528/20-nm emission filter), and mCherry (530/25-nm excitation filter and 590/35-nm emission filter) measurements were taken every 30 min for 12 h. OD595 readings were divided by 0.46 so that the value corresponds to the OD595 over a 1-cm path length. Green and red fluorescence measurements were compared at a corrected OD595 of around 1.0.
Phosphate concentration assays.
The concentration of inorganic phosphate in the culture medium was measured by using the ascorbic acid method (70). Briefly, 1 ml of cells was removed at different times during growth, and the cells were removed by centrifugation. The supernatant was assayed for inorganic phosphate directly, or after dilution in distilled water, by amending a 500-μl sample with 10 μl of a solution containing 11 N sulfuric acid, 40 μl AM-APT (described below), and 20 μl fresh ascorbic acid. AM-APT was made by dissolving 8 g ammonium molybdate and 0.2 g antimony potassium tartrate in a final volume of 1 liter. Ascorbic acid was prepared by adding 60 g to a final volume of 1 liter of water and adding 2 ml acetone. After mixing the reagents well and incubation at room temperature for 5 min, the absorbance of the samples at an OD650 was determined and compared to a standard phosphate curve.
Microscopy.
Bacteria were grown on FMM, MOPS medium, or MBM, as indicated above, with either no K2HPO4 added or the amount of K2HPO4 indicated, for 24 to 48 h at 28°C, 30°C, or 37°C as appropriate for the bacterial species being assessed. Colonies of similar sizes were imaged by using a Nikon Eclipse E600 microscope with filter set 51005v2 to visualize simultaneously both the constitutive red fluorescence and the green fluorescence of the reporter. To assess V. fischeri strains in the juvenile squid light organ, strains were grown in static SWT cultures to an OD600 of between 0.4 and 0.7 before dilution to between 3,000 and 7,000 CFU ml−1 in ocean water or Instant Ocean (Spectrum Brands, Blacksburg, VA) mixed to 36 ppt; in either case, water was sterilized by passing it through a 0.22-μm filter. Juvenile squid were exposed to the prepared inocula for 24 h before either dissection and imaging by epifluorescence microscopy or fixation overnight in 4% paraformaldehyde in marine phosphate-buffered saline (mPBS) (71) for confocal imaging. Animals used for confocal imaging were then washed three times for 30 min each in mPBS, dissected to expose their light organ, and stained overnight with a nuclear dye (Hoechst 33342). Dissected animals were mounted in Vectashield (Vector Laboratories, Burlingame, CA) to preserve fluorophores before imaging. Confocal microscopy was performed by using a Zeiss LSM 710 confocal microscope. Samples were excited at 405 nm, 488 nm, and 561 nm and visualized at an emission wavelength of 462 nm (Hoechst), 473 nm (GFP), or 610 nm (mCherry). Images were processed by using FIJI (ImageJ).
Accession number(s).
Plasmid data are deposited in GenBank under accession numbers MG752896 and MG752897.
Supplementary Material
ACKNOWLEDGMENTS
We thank Margaret McFall-Ngai for helpful suggestions, Alecia Septer and Reni Kaul for generating pRK12, Hannah Bullock and William Whitman for assisting with growth and manipulation of R. pomeroyi DSS-3, and Andrew Camilli and Anna Karls for sharing strains.
The National Science Foundation supported this research under grants IOS-1121106, IOS-1557964, and MCB-1716232. The research in this article was partially supported by a grant jointly funded by the University of Georgia Franklin College of Arts and Sciences, the Office of the Vice President for Research, and the Department of Microbiology.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00603-18.
REFERENCES
- 1.Casadaban MJ. 1975. Fusion of the Escherichia coli lac genes to the ara promoter: a general technique using bacteriophage Mu-1 insertions. Proc Natl Acad Sci U S A 72:809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch AS, Lin EC. 1996. Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J Bacteriol 178:6238–6249. doi: 10.1128/jb.178.21.6238-6249.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimada T, Fujita N, Yamamoto K, Ishihama A. 2011. Novel roles of cAMP receptor protein (CRP) in regulation of transport and metabolism of carbon sources. PLoS One 6:e20081. doi: 10.1371/journal.pone.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muir RE, Gober JW. 2005. Role of integration host factor in the transcriptional activation of flagellar gene expression in Caulobacter crescentus. J Bacteriol 187:949–960. doi: 10.1128/JB.187.3.949-960.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Antonio A, Collado-Vides J. 2003. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr Opin Microbiol 6:482–489. doi: 10.1016/j.mib.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Balleza E, Lopez-Bojorquez LN, Martinez-Antonio A, Resendis-Antonio O, Lozada-Chavez I, Balderas-Martinez YI, Encarnacion S, Collado-Vides J. 2009. Regulation by transcription factors in bacteria: beyond description. FEMS Microbiol Rev 33:133–151. doi: 10.1111/j.1574-6976.2008.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Y, Cronan JE. 2010. Overlapping repressor binding sites result in additive regulation of Escherichia coli FadH by FadR and ArcA. J Bacteriol 192:4289–4299. doi: 10.1128/JB.00516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotter PA, Melville SB, Albrecht JA, Gunsalus RP. 1997. Aerobic regulation of cytochrome d oxidase (cydAB) operon expression in Escherichia coli: roles of Fnr and ArcA in repression and activation. Mol Microbiol 25:605–615. doi: 10.1046/j.1365-2958.1997.5031860.x. [DOI] [PubMed] [Google Scholar]
- 9.Iuchi S, Matsuda Z, Fujiwara T, Lin EC. 1990. The arcB gene of Escherichia coli encodes a sensor-regulator protein for anaerobic repression of the Arc modulon. Mol Microbiol 4:715–727. doi: 10.1111/j.1365-2958.1990.tb00642.x. [DOI] [PubMed] [Google Scholar]
- 10.Cotter PA, Gunsalus RP. 1992. Contribution of the fnr and arcA gene products in coordinate regulation of cytochrome o and d oxidase (cyoABCDE and cydAB) genes in Escherichia coli. FEMS Microbiol Lett 70:31–36. doi: 10.1111/j.1574-6968.1992.tb05179.x. [DOI] [PubMed] [Google Scholar]
- 11.Bekker M, Alexeeva S, Laan W, Sawers G, Teixeira de Mattos J, Hellingwerf K. 2010. The ArcBA two-component system of Escherichia coli is regulated by the redox state of both the ubiquinone and the menaquinone pool. J Bacteriol 192:746–754. doi: 10.1128/JB.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawers G, Kaiser M, Sirko A, Freundlich M. 1997. Transcriptional activation by FNR and CRP: reciprocity of binding-site recognition. Mol Microbiol 23:835–845. doi: 10.1046/j.1365-2958.1997.2811637.x. [DOI] [PubMed] [Google Scholar]
- 13.Estrada J, Ruiz-Herrero T, Scholes C, Wunderlich Z, DePace AH. 2016. SiteOut: an online tool to design binding site-free DNA sequences. PLoS One 11:e0151740. doi: 10.1371/journal.pone.0151740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilman J, Love J. 2016. Synthetic promoter design for new microbial chassis. Biochem Soc Trans 44:731–737. doi: 10.1042/BST20160042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colton DM, Stoudenmire JL, Stabb EV. 2015. Growth on glucose decreases cAMP-CRP activity while paradoxically increasing intracellular cAMP in the light-organ symbiont Vibrio fischeri. Mol Microbiol 97:1114–1127. doi: 10.1111/mmi.13087. [DOI] [PubMed] [Google Scholar]
- 16.Yang C, Huang TW, Wen SY, Chang CY, Tsai SF, Wu WF, Chang CH. 2012. Genome-wide PhoB binding and gene expression profiles reveal the hierarchical gene regulatory network of phosphate starvation in Escherichia coli. PLoS One 7:e47314. doi: 10.1371/journal.pone.0047314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lubin EA, Henry JT, Fiebig A, Crosson S, Laub MT. 2016. Identification of the PhoB regulon and role of PhoU in the phosphate starvation response of Caulobacter crescentus. J Bacteriol 198:187–200. doi: 10.1128/JB.00658-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wanner BL. 1996. Signal transduction in the control of phosphate-regulated genes of Escherichia coli. Kidney Int 49:964–967. doi: 10.1038/ki.1996.136. [DOI] [PubMed] [Google Scholar]
- 19.Okamura H, Hanaoka S, Nagadoi A, Makino K, Nishimura Y. 2000. Structural comparison of the PhoB and OmpR DNA-binding/transactivation domains and the arrangement of PhoB molecules on the phosphate box. J Mol Biol 295:1225–1236. doi: 10.1006/jmbi.1999.3379. [DOI] [PubMed] [Google Scholar]
- 20.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701–713. doi: 10.1016/S0969-2126(02)00761-X. [DOI] [PubMed] [Google Scholar]
- 21.Lyell NL, Dunn AK, Bose JL, Stabb EV. 2010. Bright mutants of Vibrio fischeri ES114 reveal conditions and regulators that control bioluminescence and expression of the lux operon. J Bacteriol 192:5103–5114. doi: 10.1128/JB.00524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bose JL, Rosenberg CS, Stabb EV. 2008. Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch Microbiol 190:169–183. doi: 10.1007/s00203-008-0387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visick KL, Foster J, Doino J, McFall-Ngai M, Ruby EG. 2000. Vibrio fischeri lux genes play an important role in colonization and development of the host light organ. J Bacteriol 182:4578–4586. doi: 10.1128/JB.182.16.4578-4586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyell NL, Stabb EV. 2013. Symbiotic characterization of Vibrio fischeri ES114 mutants that display enhanced luminescence in culture. Appl Environ Microbiol 79:2480–2483. doi: 10.1128/AEM.03111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gebhard S, Cook GM. 2008. Differential regulation of high-affinity phosphate transport systems of Mycobacterium smegmatis: identification of PhnF, a repressor of the phnDCE operon. J Bacteriol 190:1335–1343. doi: 10.1128/JB.01764-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munoz-Martin MA, Mateo P, Leganes F, Fernandez-Pinas F. 2011. Novel cyanobacterial bioreporters of phosphorus bioavailability based on alkaline phosphatase and phosphate transporter genes of Anabaena sp. PCC 7120. Anal Bioanal Chem 400:3573–3584. doi: 10.1007/s00216-011-5017-0. [DOI] [PubMed] [Google Scholar]
- 27.Makino K, Shinagawa H, Amemura M, Kimura S, Nakata A, Ishihama A. 1988. Regulation of the phosphate regulon of Escherichia coli. Activation of pstS transcription by PhoB protein in vitro. J Mol Biol 203:85–95. doi: 10.1016/0022-2836(88)90093-9. [DOI] [PubMed] [Google Scholar]
- 28.Cardemil CV, Smulski DR, Larossa RA, Vollmer AC. 2010. Bioluminescent Escherichia coli strains for the quantitative detection of phosphate and ammonia in coastal and suburban watersheds. DNA Cell Biol 29:519–531. doi: 10.1089/dna.2009.0984. [DOI] [PubMed] [Google Scholar]
- 29.Spira B, Yagil E. 1999. The integration host factor (IHF) affects the expression of the phosphate-binding protein and of alkaline phosphatase in Escherichia coli. Curr Microbiol 38:80–85. doi: 10.1007/s002849900407. [DOI] [PubMed] [Google Scholar]
- 30.Taschner NP, Yagil E, Spira B. 2006. The effect of IHF on sigmaS selectivity of the phoA and pst promoters of Escherichia coli. Arch Microbiol 185:234–237. doi: 10.1007/s00203-005-0082-4. [DOI] [PubMed] [Google Scholar]
- 31.Aoyama T, Oka A. 1990. A common mechanism of transcriptional activation by the three positive regulators, VirG, PhoB, and OmpR. FEBS Lett 263:1–4. doi: 10.1016/0014-5793(90)80691-B. [DOI] [PubMed] [Google Scholar]
- 32.Diniz MM, Goulart CL, Barbosa LC, Farache J, Lery LM, Pacheco AB, Bisch PM, von Kruger WM. 2011. Fine-tuning control of phoBR expression in Vibrio cholerae by binding of PhoB to multiple Pho boxes. J Bacteriol 193:6929–6938. doi: 10.1128/JB.06015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SK, Kimura S, Shinagawa H, Nakata A, Lee KS, Wanner BL, Makino K. 2000. Dual transcriptional regulation of the Escherichia coli phosphate-starvation-inducible psiE gene of the phosphate regulon by PhoB and the cyclic AMP (cAMP)-cAMP receptor protein complex. J Bacteriol 182:5596–5599. doi: 10.1128/JB.182.19.5596-5599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura S, Makino K, Shinagawa H, Amemura M, Nakata A. 1989. Regulation of the phosphate regulon of Escherichia coli: characterization of the promoter of the pstS gene. Mol Gen Genet 215:374–380. doi: 10.1007/BF00427032. [DOI] [PubMed] [Google Scholar]
- 35.Kasahara M, Makino K, Amemura M, Nakata A, Shinagawa H. 1991. Dual regulation of the ugp operon by phosphate and carbon starvation at two interspaced promoters. J Bacteriol 173:549–558. doi: 10.1128/jb.173.2.549-558.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao R, Stock AM. 2015. Temporal hierarchy of gene expression mediated by transcription factor binding affinity and activation dynamics. mBio 6:e00686-15. doi: 10.1128/mBio.00686-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan Z-C, Zaheer R, Morton R, Finan TM. 2006. Genome prediction of PhoB regulated promoters in Sinorhizobium meliloti and twelve proteobacteria. Nucleic Acids Res 34:2686–2697. doi: 10.1093/nar/gkl365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolter R, Inuzuka M, Helinski DR. 1978. trans-Complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell 15:1199–1208. doi: 10.1016/0092-8674(78)90046-6. [DOI] [PubMed] [Google Scholar]
- 39.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol 72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134. doi: 10.1016/j.plasmid.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Wild J, Czyz A, Rakowski S, Filutowicz M. 2004. γ origin plasmids of R6K lineage replicate in diverse genera of Gram-negative bacteria. Ann Microbiol 54:471–480. [Google Scholar]
- 42.Andersen JB, Sternberg C, Poulsen LK, Bjorn SP, Givskov M, Molin S. 1998. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol 64:2240–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller WG, Leveau JH, Lindow SE. 2000. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact 13:1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- 44.Keiler KC, Waller PR, Sauer RT. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 45.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA. 2004. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivera-Rivera I, Roman-Hernandez G, Sauer RT, Baker TA. 2014. Remodeling of a delivery complex allows ClpS-mediated degradation of N-degron substrates. Proc Natl Acad Sci U S A 111:E3853–E3859. doi: 10.1073/pnas.1414933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dougan DA, Reid BG, Horwich AL, Bukau B. 2002. ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9:673–683. doi: 10.1016/S1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- 48.Montgomery MK, McFall-Ngai M. 1993. Embryonic development of the light organ of the sepiolid squid Euprymna scolopes berry. Biol Bull 184:296–308. doi: 10.2307/1542448. [DOI] [PubMed] [Google Scholar]
- 49.Jensen PR, Hammer K. 1998. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl Environ Microbiol 64:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawley DK, McClure WR. 1983. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res 11:2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kammerer W, Deuschle U, Gentz R, Bujard H. 1986. Functional dissection of Escherichia coli promoters: information in the transcribed region is involved in late steps of the overall process. EMBO J 5:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wanner BL. 1993. Gene regulation by phosphate in enteric bacteria. J Cell Biochem 51:47–54. doi: 10.1002/jcb.240510110. [DOI] [PubMed] [Google Scholar]
- 53.Konopka A, Wilkins MJ. 2012. Application of meta-transcriptomics and -proteomics to analysis of in situ physiological state. Front Microbiol 3:184. doi: 10.3389/fmicb.2012.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boettcher KJ, Ruby EG, McFall-Ngai MJ. 1996. Bioluminescence in the symbiotic squid Euprymna scolopes is controlled by a daily biological rhythm. J Comp Physiol A 179:65–73. doi: 10.1007/BF00193435. [DOI] [Google Scholar]
- 55.Wier AM, Nyholm SV, Mandel MJ, Massengo-Tiasse RP, Schaefer AL, Koroleva I, Splinter-Bondurant S, Brown B, Manzella L, Snir E, Almabrazi H, Scheetz TE, Bonaldo MDF, Casavant TL, Soares MB, Cronan JE, Reed JL, Ruby EG, McFall-Ngai MJ. 2010. Transcriptional patterns in both host and bacterium underlie a daily rhythm of anatomical and metabolic change in a beneficial symbiosis. Proc Natl Acad Sci U S A 107:2259–2264. doi: 10.1073/pnas.0909712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hussa EA, O'Shea TM, Darnell CL, Ruby EG, Visick KL. 2007. Two-component response regulators of Vibrio fischeri: identification, mutagenesis, and characterization. J Bacteriol 189:5825–5838. doi: 10.1128/JB.00242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thompson LR, Nikolakakis K, Pan S, Reed J, Knight R, Ruby EG. 2017. Transcriptional characterization of Vibrio fischeri during colonization of juvenile Euprymna scolopes. Environ Microbiol 19:1845–1856. doi: 10.1111/1462-2920.13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sycuro LK, Ruby EG, McFall-Ngai M. 2006. Confocal microscopy of the light organ crypts in juvenile Euprymna scolopes reveals their morphological complexity and dynamic function in symbiosis. J Morphol 267:555–568. doi: 10.1002/jmor.10422. [DOI] [PubMed] [Google Scholar]
- 59.Boettcher KJ, Ruby EG. 1990. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J Bacteriol 172:3701–3706. doi: 10.1128/jb.172.7.3701-3706.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stabb EV, Reich KA, Ruby EG. 2001. Vibrio fischeri genes hvnA and hvnB encode secreted NAD(+)-glycohydrolases. J Bacteriol 183:309–317. doi: 10.1128/JB.183.1.309-317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bose JL, Kim U, Bartkowski W, Gunsalus RP, Overley AM, Lyell NL, Visick KL, Stabb EV. 2007. Bioluminescence in Vibrio fischeri is controlled by the redox-responsive regulator ArcA. Mol Microbiol 65:538–553. doi: 10.1111/j.1365-2958.2007.05809.x. [DOI] [PubMed] [Google Scholar]
- 62.Septer AN, Wang Y, Ruby EG, Stabb EV, Dunn AK. 2011. The haem-uptake gene cluster in Vibrio fischeri is regulated by Fur and contributes to symbiotic colonization. Environ Microbiol 13:2855–2864. doi: 10.1111/j.1462-2920.2011.02558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 64.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez JM, Covert JS, Whitman WB, Henriksen JR, Mayer F, Scharf B, Schmitt R, Buchan A, Fuhrman JA, Kiene RP, Moran MA. 2003. Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int J Syst Evol Microbiol 53:1261–1269. doi: 10.1099/ijs.0.02491-0. [DOI] [PubMed] [Google Scholar]
- 66.Reisch CR, Moran MA, Whitman WB. 2008. Dimethylsulfoniopropionate-dependent demethylase (DmdA) from Pelagibacter ubique and Silicibacter pomeroyi. J Bacteriol 190:8018–8024. doi: 10.1128/JB.00770-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herrero M, de Lorenzo V, Timmis KN. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J Bacteriol 172:6557–6567. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stabb EV, Ruby EG. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol 358:413–426. doi: 10.1016/S0076-6879(02)58106-4. [DOI] [PubMed] [Google Scholar]
- 69.Kvitko BH, Bruckbauer S, Prucha J, McMillan I, Breland EJ, Lehman S, Mladinich K, Choi K-H, Karkhoff-Schweizer R, Schweizer HP. 2012. A simple method for construction of pir(+) enterobacterial hosts for maintenance of R6K replicon plasmids. BMC Res Notes 5:157. doi: 10.1186/1756-0500-5-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.US Environmental Protection Agency Water Quality Office. 1971. Methods for chemical analysis of water and wastes. US Government Printing Office, Cincinnati, OH. [Google Scholar]
- 71.Troll JV, Adin DM, Wier AM, Paquette N, Silverman N, Goldman WE, Stadermann FJ, Stabb EV, McFall-Ngai MJ. 2009. Peptidoglycan induces loss of a nuclear peptidoglycan recognition protein during host tissue development in a beneficial animal-bacterial symbiosis. Cell Microbiol 11:1114–1127. doi: 10.1111/j.1462-5822.2009.01315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 73.Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev 53:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gonzalez JM, Kiene RP, Moran MA. 1999. Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Appl Environ Microbiol 65:3810–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grana D, Youderian P, Susskind MM. 1985. Mutations that improve the ant promoter of Salmonella phage P22. Genetics 110:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pratt JT, Ismail AM, Camilli A. 2010. PhoB regulates both environmental and virulence gene expression in Vibrio cholerae. Mol Microbiol 77:1595–1605. doi: 10.1111/j.1365-2958.2010.07310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.