

In human cells, the integrity of genetic information is constantly challenged by endogenous and exogenous chemical agents that can induce DNA damage. To counteract the deleterious effects of DNA lesions, cells are equipped with multiple repair mechanisms, such as base-excision repair (BER), nucleotide-excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end joining (NHEJ), to remove DNA lesions from the genome. Unrepaired DNA adducts may accumulate in cells and lead to deleterious biological consequences through perturbation of DNA replication and transcription.1–4
A comprehensive understanding about the human health impact of DNA lesions requires the assessment about their occurrence, repair, and biological consequences, where chemical analysis has played important roles. In this vein, significant advances have been made in the chemical analysis of DNA damage in the past two years. These include the increased application of high-resolution mass analyzers (Orbitrap and, to a lesser degree, time-of-flight) in liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based analysis of DNA adducts, the analysis of DNA adducts using an omics approach (i.e., DNA adductomics), and the adoption of next-generation sequencing (NGS) for mapping the genome-wide distribution of DNA adducts.
In this Review, we will discuss chemical analysis of common DNA damage products, including those induced by reactive oxygen species (ROS), alkylating agents, and heterocyclic aromatic amines (HAAs), with emphasis being placed on studies published in the last two years. We will summarize briefly the formation of various types of DNA lesions, and we will review analytical methods for the quantitative measurements of DNA lesions, including LC-MS/MS, optical, and electrochemical methods. We will also review recent shuttle vector-based studies for exploring quantitatively how DNA lesions modulate the efficiencies and fidelities of cellular DNA replication and transcription and recent NGS-based approaches for mapping the genome-wide occupancy of DNA lesions.
FORMATION OF COMMON TYPES OF DNA LESIONS
As a result of endogenous metabolism or exposure to environmental toxicants, such as ionizing radiation and heavy metal ions, ROS constitute a major type of DNA damaging agents.5–7 For instance, electron leakage from the electron transport chain during mitochondrial respiration can lead to the generation of superoxide anion radical (O0–•), which can be transformed to hydrogen peroxide (H2O2) via the action of superoxide dismutase (SOD).8 H2O2 may react with transition metal ions, e.g., Fe2+ and Cu+, to yield hydroxyl radical (•OH) via the Fenton reactions.9
ROS, especially the highly reactive •OH, can directly react with DNA and lead to the formation of a variety of single-nucleobase lesions and bulky lesions. Common single-nucleobase lesions include 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine-2′-deoxynucleoside (Fapy-dG), 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxo-dA), 4,6-diamino-5-formamidopyrimidine 2′-deoxynucleoside (Fapy-dA), guanidinohydantoin 2′-deoxynucleoside (dGh), spiroiminodihydantoin 2′-deoxynucleoside (dSp), and 5,6-dihydroxy-5,6-dihydrothymidine (thymidine glycol).6 Common ROS-induced bulky lesions include 8,5′-cyclopurine-2′-deoxynucleosides (cPus) and nucleobase–nucleobase intrastrand cross-links.6
ROS can also lead to the formation of DNA lesions indirectly through peroxidation of polyunsaturated fatty acids (PUFA) to generate reactive aldehydes that form adducts with DNA.10,11 For example, malondialdehyde may react with guanine in DNA to give the exocyclic pyrimido-[1,2-α]purine-10(3H)-one-2′-deoxyribose (M1dG).10 Other α,β-unsaturated lipid peroxidation (LPO) products, such as acrolein, crotonaldehyde, and 4-hydroxy-2-nonenal (HNE), can conjugate with the N2 amino group of guanine to yield exocyclic six-membered ring 1,N2-propano-dG products (i.e., Acr-dG, Cro-dG, and HNE-dG, respectively).12 HNE may also induce DNA adducts with an unsaturated five-membered exocyclic ring being attached to nucleobases, such as 1,N6-etheno-2′-deoxyadenosine (εdA), 3,N4-etheno-2′-deoxycytidine (εdC), 1,N2-etheno-2′-deoxyguanosine (1,N2-εdG), and N2,3-etheno-2′-deoxyguanosine (N2,3-εdG).13–16
Apart from ROS, alkylating agents represent another important class of DNA damaging agents.17,18 Because of the existence of multiple nucleophilic sites in nucleobases, including the N3, N7, O6, C8, and N2 of guanine, the N1, N3, and N7 of adenine, the O2 and O4 of thymine, and the O2 and N4 of cytosine, DNA is susceptible to attack from reactive electrophiles.19–21 For example, DNA methylating agents can attack the nitrogen or oxygen atoms of nucleobases, leading to the formation of a plethora of methylated DNA lesions.17,18,22–24
Besides simple methylation, more complex alkylated lesions can also be induced. For example, the exposure of DNA to some endogenous and exogenous N-nitroso compounds (NOCs) and the pancreatic carcinogen azaserine may also lead to DNA carboxymethylation in vitro and in vivo,25–28 which may contribute to the pathogenesis of human colorectal and stomach cancers.27,28 Polycyclic aromatic hydrocarbons (PAHs), e.g., benzo[a]pyrene (B[a]P), after absorption via lung, gut, and/or skin and metabolic activation by cytochrome P450-dependent monooxygenases (CYPs) and peroxidases, are able to yield reactive intermediates such as diol-epoxides and radical cations, which give rise to the formation of a series of bulky PAH adducts mainly at the N2, N7, and C8 positions of guanine, along with the N6 and N7 positions of adenine.29–31 In addition to reacting with nucleobases, alkylating chemical species can also attack the oxygen atoms of the phosphodiester linkages in DNA to generate alkyl phosphotriesters (PTEs).32–34
The cytotoxic properties of alkylated DNA lesions have also been exploited for cancer chemotherapy for several decades. In this regard, bifunctional alkylating agents (e.g., nitrogen mustards) are among the oldest and most commonly prescribed chemotherapeutic agents for treating cancer.35,36 The bifunctional agents can covalently react with nucleobases on both strands of DNA, leading to the formation of DNA interstrand cross-links (ICLs). The unrepaired interstrand cross-links can prohibit DNA strand separation, thereby blocking DNA replication and/or DNA transcription and resulting in cell death.37
Carcinogenic heterocyclic aromatic amines (HAAs), emanating from tobacco combustion or high-temperature cooking of meat, fish, and poultry, may also lead to the formation of DNA adducts.38–40 These compounds, such as 2-amino-9H-dipyrido-[2,3-b]indole (AαC), 2-amino-3,4-dimethylimidazo[4,5-f ]-quinoxaline (MeIQx), 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), and 2-amino-3-methylimidazo[4,5-f ]-quinoline (IQ), can undergo metabolic activation by CYP1A2 in liver and by CYP1A1- and CYP1B1-mediated N-oxidation in extrahepatic tissues, respectively.39,41,42 After N-hydroxylation of the exocyclic amino group, the resultant arylnitrenium ion is the crucial intermediate involved in HAA–DNA adduct formation at the C8 and N2 positions of guanine.39,43
RECENT DEVELOPMENTS IN LC-MS-BASED ANALYSIS OF DNA ADDUCTS
LC-MS/MS coupled with the stable isotope dilution technique represents the most powerful technique for the unambiguous identification and accurate quantification of DNA lesions in cells and tissues. The method provides structural information for the confirmation of known DNA adducts and for the identification of unknown DNA adducts. DNA adducts involving modifications of a single nucleoside are mainly quantified in their 2′-deoxyribonucleoside form generated from enzymatically digested DNA or, to a lesser extent, in the free nucleobase form isolated from urine or blood samples. Sample preparation and cleanup procedures are essential for the subsequent LC-MS detection due to the existence of salts, enzymes, and other impurities in samples that are not amenable to direct MS analysis. In this vein, the readers should consult a recent review19 for typical approaches of DNA hydrolysis, sample cleanup, and analyte enrichment.
Recent advances in LC-MS instrumentation also enable high-throughput quantification of low levels of DNA lesions in cellular and tissue samples.19–21,44 For example, small internal-diameter columns (e.g., 75 μm I.D.) coupled with nanoflow (e.g., 300 nL/min) LC-nanoelectrospray ionization (nanoLC-NSI)-MS/MS or ultra-performance liquid chromatography (UPLC)-NSI-MS/MS have been employed for simultaneous and sensitive quantification of multiple DNA lesions, owing to the high ionization and ion transmission efficiency provided by NSI45 and high separation efficiency afforded by UPLC.46–48 Meanwhile, the use of high-resolution Orbitrap mass analyzer provided high mass accuracy, thereby offering high specificity for analysis. In the following section, we discuss recent developments in LC-MS-based analysis of DNA adducts, and we organize this section according to the types of DNA lesions.
Oxidative Stress-Induced DNA Damage
Several novel LC-MS-based analytical methods were reported for the quantification of 8-oxo-dG, a biomarker for oxidative stress (Figure 1). A column-switching LC-MS/MS method was developed for simultaneous quantification of monohydroxybutenylmercapturic acid (MHBMA) and N-acetyl-S-(3,4-dihydroxybutyl)cysteine (DHBMA), which are biomarkers for 1,3-butadiene exposure, and 8-oxo-dG in human urine.49 Urine samples were first loaded onto a column for the extraction and cleanup of analytes, followed by another column for analyte separation. An improvement in limit of detection (LOD) by 100-fold was observed for 8-oxo-dG (0.17 vs 28.2 ng/mL) with this method, compared to a previously reported column-switching LC-MS/MS method.49,50
Figure 1.
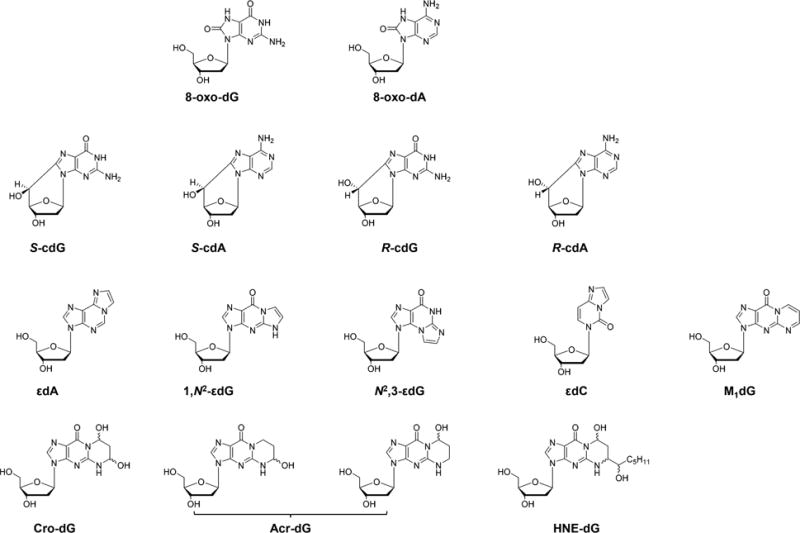
Chemical structures of selected ROS- and lipid peroxidation byproduct-induced DNA lesions.
Ma et al.51 reported a sensitive nanoLC-NSI-MS/MS system to quantify simultaneously 8-oxo-dG and 8-oxo-dA (Figure 1) in human retina mitochondrial DNA (mtDNA) with the use of nanogram quantities of DNA. This method reached an on-column limit-of-quantitation (LOQ) of 0.1 fmol for 8-oxo-dG and 0.02 fmol for 8-oxo-dA. In addition, the use of 8-hydroxyquinoline as an antioxidant during the DNA isolation process completely diminished oxidation-induced artificial generation of 8-oxo-dG, providing reliable measurements of 8-oxodG and 8-oxo-dA.51
The UPLC-MS/MS method was also used for sensitive and accurate measurement of 8-oxo-dG in newborn mouse brain tissues, with the LOD and LOQ values being 0.09 and 0.29 nmol/L, respectively.52 Similarly, UPLC-ESI-MS/MS in combination with sample cleanup using SPE was employed to determine 8-oxo-dG levels in urine samples from 60 patients with early stage breast cancer (stage I, II). Markedly elevated levels of 8-oxo-dG were found for the breast cancer group than the non-cancerous group.53
A novel reusable three-enzyme cascade capillary monolithic bioreactor with immobilized deoxyribonuclease I (DNase I), snake venom phosphodiesterase, and alkaline phosphatase was introduced by Yin et al.,54 achieving fast and efficient digestion of genomic DNA (to 100%) into mononucleosides in 45 min. This technique, in conjunction with UPLC-MS/MS, enabled sensitive detection of 8-oxo-dG in cellular DNA, with the LOD and LOQ being 0.3 and 1.0 nM, respectively. In addition, artificial generation of 8-oxo-dG was reduced due to the short incubation time for digestion, compared with a traditional 2–6 h digestion system.54
Recently, new techniques were also developed for quantitative analysis of the bulky cPu lesions (Figure 1) in cells and tissues. By employing nanoLC-NSI coupled with MS/MS, Yu et al.55 reported a method with improved sensitivity in simultaneous measurements of cPus and LPO-induced εdA (Figure 1) and εdG (Figure 1) lesions in liver and brain tissues of Long-Evans Agouti (LEA) and Long-Evans Cinnamon (LEC) rats. It was found that the oxidative stress, owing to aberrant copper accumulation, leads to preferential accumulation of direct ROS-induced cPus over the lipid peroxidation-induced etheno adducts in liver tissues of LEC rats.55 Another LC-MS/MS method was applied for the quantification of the levels of all four diastereomers of cPus in estrogen receptor-α positive (ER-α) MCF-7 and triple-negative MDA-MB-231 breast cancer cell lines with or without exposure to 5 Gy of γ-rays or 300 μM hydrogen peroxide.56 The quantification results indicated that both breast cancer cell lines were highly susceptible to oxidative stress-induced DNA damage, with elevated levels of cPus being observed in cells exposed with γ-rays or hydrogen peroxide.56
To explore the role of red meat consumption in the initiation and progression of human colorectal cancer, Hemeryck et al.57 developed a new SPE cleanup coupled with a UPLC-high resolution MS (HRMS) method for the simultaneous quantifications of O6-MeG (Figure 2), O6-carboxymethyl-G (O6-CMG, Figure 2), M1G (Figure 1), and Cro-G (Figure 1). The UPLC-HRMS detection techniques enabled accurate identification and sensitive quantification of analytes based on chemical composition and exact compound mass. The method was validated and optimized by detecting the above-mentioned lesions in calf thymus DNA treated with potassium diazoacetate, malondialdehyde, and crotonaldehyde, with LOQ being 2.82–22.2 adducts per 108 nucleosides in the MS/MS mode.57 Quantification results revealed that O6-CMG could be detected and quantified in 8 out of 10 colon tumor samples, and the mean level of this lesion was 8.15 adducts per 106 nucleotides, with a relatively high inter-individual variability (range from <LOD to 16.3 adducts per 106 nucleotides). However, M1G, Cro-G, and O6-MeG could not be detected in tumor samples.57
Figure 2.
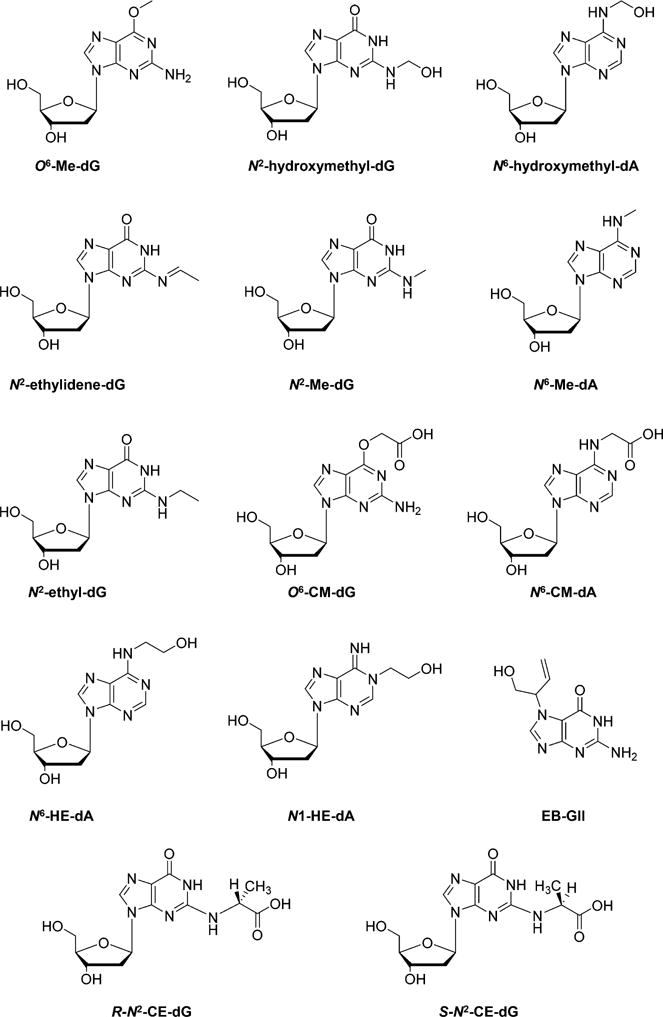
Chemical structures of selected alkylated DNA lesions.
LPO-induced lesions were also used as biomarkers for evaluating oxidative stress induced by exposure to chemical toxicants or tobacco smoke. Lung DNA samples extracted from mice after inhalation exposure to atmospheric ethylene oxide (EO) at concentrations of 0, 100, and 200 ppm and the LC/ESI-MS/MS measurement results showed that the levels of 8-oxo-dG and Cro-dG were 39–46 and 0.15–0.19 lesions per 106 dG, respectively.58 Another SPE-LC-MS/MS method was developed to investigate the levels of Acr-dG (Figure 1) and Cro-dG in DNA extracted from human saliva samples, though no significant difference in levels of these two lesions was found between non-smokers and smokers.59
Oxidative stress induced by polychlorinated biphenyls (PCBs) was also evaluated.60 Female Sprague–Dawley rats were treated with 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126), 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB 153), and a binary mixture (PCB 126 + 153) for 14, 31, and 53 weeks, respectively.60 Multiple oxidatively induced DNA lesions (8-oxo-dG, εdA, N2,3-εdG, 1,N2-εdG, M1dG, Acr-dG, Cro-dG, and HNE-dG, Figure 1) were enriched from the enzymatic digestion mixture of DNA using offline HPLC and quantified by LC-MS/MS. It was revealed that the levels of these DNA lesions were significantly elevated after treatment with PCBs, especially in groups treated with the binary mixture for 53 weeks.60 A similar sample preparation strategy was employed for simultaneous quantifications of etheno-DNA adducts in human white blood cells,61 as well as tissues of LEA and LEC rats.55 Li et al.61 reported an ultrasensitive UPLC-ESI-MS/MS method for the analyses of εdA and εdC in white blood cells of human subjects with occupational exposure to benzene. The measurement data revealed significant increases in levels of these two lesions in benzene-exposed workers compared with those in non-benzene-exposed workers.
Recently, Zhang et al.62,63 assessed different mobile phases for UPLC-MS/MS quantification of Cro-dG in cellular DNA or in human urine samples. Four aqueous mobile phases, i.e., ammonium bicarbonate, ammonium formate, ammonium acetate, and formic acid, were evaluated, and 2 mM ammonium bicarbonate was found to provide the best sensitivity and separation for Cro-dG by UPLC-MS/MS analysis. Moreover, ammonium bicarbonate can enhance the protonation efficiency of Cro-dG and suppress the generation of undesirable [M + Na]+ and [M + K]+ ions of Cro-dG, which further improved the detection sensitivity.62,63 This approach also provided new perspectives for optimizing the sensitivity of LC-MS-based detection methods for other DNA lesions in the future.
Alkylated DNA Lesions
There are a number of recent studies about the quantification of the alkylated DNA lesions induced by dietary alkylating agents, tobacco combustion products, and occupational chemical exposure. UPLC-MS provides a powerful approach in quantification of alkylated DNA lesions. With this technique, Ma et al.64 investigated the formation of O6-Me-dG in the liver tissues of female Wistar rats treated, for 15 days, with drinking water containing various concentrations of Cr(VI) and/or a DNA methylating agent, N-nitrosodimethylamine (NDMA). Dose-dependent induction of O6-Me-dG was observed in the hepatic DNA samples of the NMDA-treated rats, especially in the groups co-treated with Cr(VI). Moreover, Cr(VI) exposure led to decreased glutathione content in rat liver tissues, which can reduce Cr(VI) to Cr(III) and detoxify the NDMA-derived methylating metabolites in vivo.65 This result provided a possible mechanism for the potentiated induction of O6-Me-dG by Cr(VI) coexposure.64 DNA lesions induced by formaldehyde and acetaldehyde present in tobacco smoke, i.e., N2-hydroxymethyl-dG, N6-hydroxymethyl-dA, and N2-ethylidene-dG (Figure 2), were also measured with UPLC-MS/MS.59 In this report, saliva samples from smokers and nonsmokers were collected and, due to their lack of chemical stability, these aldehyde induced-DNA adducts were first reduced to their alkyl forms, i.e., N2-Me-dG, N6-Me-dA, and N2-ethyl-dG (Figure 2), prior to LC-MS/MS detection.59
Other approaches were developed for improving the sensitivity and specificity for the quantification of the methylated DNA lesions. For example, a UPLC-HRMS/MS method using a quadrupole-Orbitrap instrument was developed to detect O6-Me-dG and O6-CM-dG in colon biopsies for evaluating DNA damage emanating from diet-related N-nitroso compounds.57 The excellent separation of analytes from impurities by UPLC, in combination with high mass accuracy afforded by HRMS, provided improved selectivity and sensitivity for the quantifications of these two lesions. In addition, Yu et al.66 applied a novel nanoLC-NSI coupled with LC-MS3 method for the simultaneous detection of O6-CM-dG, O6-Me-dG, and N6-CM-dA (Figure 2) in azaserine-treated GM04429 human skin fibroblasts and HCT-116 human colon carcinoma cells. Offline HPLC enrichment was also employed, and nanoLC-NSI gave better sensitivity than the previously reported method with a normal ESI source. Furthermore, the MS3 detection mode on the linear ion trap mass spectrometer further enhanced specificity for quantification, compared to MS/MS.66
Reactive epoxides, and chemical pollutants that can be metabolically activated into epoxides, can also alkylate DNA. For instance, ethylene oxide induced N6-(2-hydroxyethyl)-2′-deoxyadenosine (N6-HE-dA) (Figure 2) and 1-(2-hydroxyethyl)-2′-deoxyadenosine (N1-HE-dA) (Figure 2) were quantified with an LC-ESI-MS/MS method.58 A dose-dependent increase of these two lesions in mouse lung DNA was observed after a 12-week inhalation exposure to EO.58
Another epoxide, 3,4-epoxy-1-butene (EB), which is a reactive intermediate from metabolic activation of 1,3-butadiene (BD), was also investigated. Sangaraju et al.67 reported an isotope dilution nanoLC/ESI+-HRMS3 method for the quantitation of EB-induced N7-(1-hydroxy-3-buten-2-yl)guanine (EB-GII) (Figure 2) adduct in urine samples, with the LOD and LOQ being 0.25 and 1.0 fmol/mL, respectively. EB-GII adducts in urine samples were enriched by SPE, followed by an offline HPLC enrichment. All analyses were performed on a nano2D-LC (trapping column + analytical column) system coupled with an LTQ Orbitrap Velos mass spectrometer for high-resolution detection. It was demonstrated that the levels of these adducts increased with the concentration of BD exposure, and very low levels of EB-GII (14.5–87.0 pg/mL in urine) were also detectable in urine of control rats.67 A significantly higher level (1.25 pg/mg creatinine) of EB-GII was found in urine of workers occupationally exposed to 0.1–2.2 ppm of BD than in urine of administrative controls exposed to <0.01 ppm of BD (0.22 pg/mg creatinine), suggesting that EB-GII is a useful biomarker for monitoring DNA damage induced by BD exposure.67
Methylglyoxal, a hyperglycemia-induced advanced glycation end-product (AGE), can conjugate with DNA and lead to the formation of N2-(1-carboxyethyl)-2′-deoxyguanosine (N2-CE-dG) (Figure 2). Jaramillo et al.68 employed UPLC-MS/MS coupled with the isotope dilution method and quantified the levels of (R,S)-N2-CE-dG in the urine, blood, and tissues (liver, kidney, pancreas, and colon) of hyperglycemic and normoglycemic mice. N2-CE-dG is significantly elevated in urine of hyperglycemic mice treated with fasting plasma glucose (FPG) ≥200 mg/dL (11 mM) compared to that in mice treated with FPG <200 mg/dL, i.e., (240 ± 110) vs (16 ± 12) pmol CE-dG/24 h. Average tissue-derived N2-CE-dG was also higher in hyperglycemic mice (18.4 per 106 dG) than normoglycemic mice (4.4 per 106 dG).68 This finding is in line with previous observations in cultured human cells, where exposure to increasing concentration of glucose or methylglyoxal led to augmented formation of N2-CE-dG lesions in cellular DNA.69
Extensive studies have been performed to assess the lesions generated from tobacco smoke exposure. Different from lung and upper aerodigestive tract tissues, oral cavity cells from cigarette smokers are relatively easy to obtain and highly susceptible to tobacco-induced DNA damage. For this reason, DNA adduct analysis in oral cells and saliva could potentially identify those individuals carrying relatively high levels of cancer-prone DNA adducts and provide preventive information for cigarette smokers.70 O6-POB-dG (Figure 3) was quantified in DNA isolated from tissues of F344 rats exposed with (R)-, (S)-, and racemic N′-nitrosonornicotine (NNN), using an HPLC enrichment coupled with LC-NSI-HRMS/MS method.71 The HRMS/MS mode of Orbitrap Fusion Tribrid mass spectrometer pronouncedly reduced the interference from the sample matrix. Moreover, the nanoLC (0.3 μL/min in a 75 μm i.d. column), when coupled with NSI, increased sensitivity by ∼100-fold (LODs of 6.5 amol for diluted standard and 100 amol for spiked calf thymus DNA samples) compared to that of the low mass resolution LC-ESI-MS/MS method (LODs of 0.6 fmol for pure standard and 3 fmol for rat DNA samples).71
Figure 3.
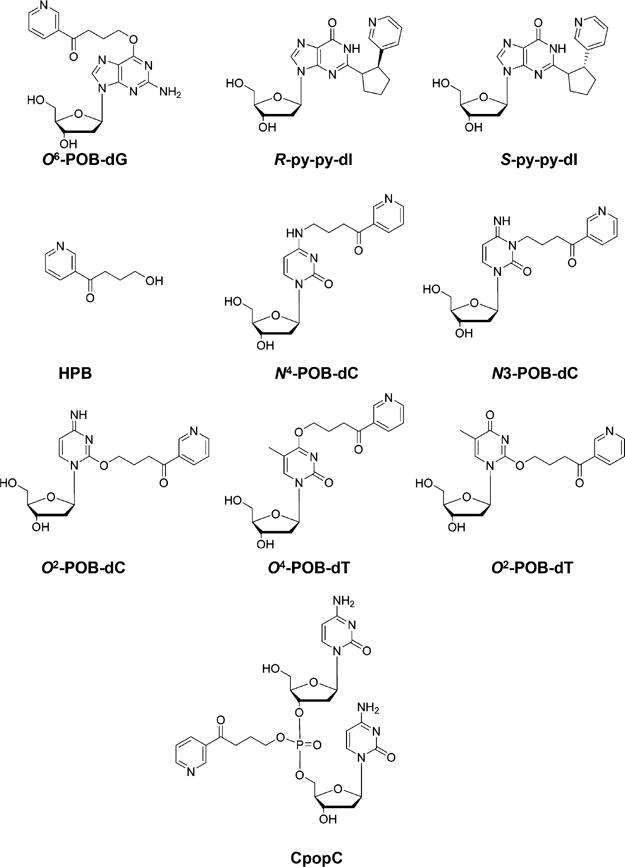
Chemical structures of DNA lesions induced by tobacco-specific nitrosamines.
2-(2-(3-pyridyl)-N-pyrrolidinyl)-2′-deoxyinosine (py-py-dI) (Figure 3), a DNA adduct induced by the 5′-hydroxylation product of NNN, was also quantified in NNN-treated human liver S9 fraction and human and rat hepatocytes, as well as rat tissues using similar LC-ESI-MS/MS or nanoLC-NSI-HRMS/MS methods.72 A clear dose-responsive relationship was observed for py-py-dI induction in all tissues, especially in lung, nasal respiratory mucosa, and nasal olfactory mucosa, of rats treated with racemic NNN. Additionally, in vitro assays demonstrated a preferential formation of py-py-dI lesion induced by 5′-hydroxylation of NNN relative to the corresponding POB lesion.72
Given the fact that the majority of POB–DNA adducts decompose to release 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) (Figure 3) upon hydrolysis with strong acid, a rapid and sensitive nanoLC-NSI-HRMS/MS method was developed for monitoring NNN exposure in oral cells of smokers, with an LOD of 5 amol.73 HPB moieties of various POB–DNA adducts were easily hydrolyzed by hydrochloric acid at 80 °C within 3 h and rapidly purified by HyperSep Hypercarb cartridges without any time-consuming enzymatic digestion procedures. The method obviates the need for the syntheses of stable isotope-labeled standards of all possible NNN-induced POB–DNA lesions except [pyridine-d4]-HPB.73 Nevertheless, conjugation of POB to different positions in nucleobases and phosphate backbone in DNA is likely to confer distinct biological consequences; thus, the method falls short in the risk assessment of the POB lesions induced by NNN or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK).
Apart from guanine lesions, metabolic activation of the carcinogenic tobacco-specific NNK and NNN can also result in the formation of POB-dC and POB-dT adducts.74–76 In this vein, 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc) could be hydrolyzed by esterase to generate an alkylating intermediate, which can further react with calf thymus DNA, leading to the formation of three newly identified POB DNA adducts, N4-POB-dC, 3-POB-dC, and O4-POB-dT, along with the previously reported O2-POB-C and O2-POB-dT (Figure 3).74,76 It was also observed that O2-POB-dT and, to a lower extent, O4-POB-dT, but not O6-POB-dG, are substrates for NER in mammalian cells.76
In addition to the aforementioned NNN-,NNK-, or NNKOAc-induced POB-nucleobase lesions, Ma et al.34 explored the possible formation of POB–PTEs in NNKOAc-treated calf thymus DNA or liver and lung tissues of NNK-treated rats. Ten different combinations of NNKOAc-derived PTEs, including 32 possible isomers, were considered in LC-NSI-HRMS/MS analysis. The 15N3-labeled internal standard was synthesized for one of the most abundant phosphate adducts, dCp[4-oxo-4-(3-pyridyl)butyl]dC (CpopC) (Figure 3), to quantify CpopC and to estimate the levels of other PTE adducts.34 An in vitro assay revealed the levels of two isomers of CpopC to be 340 and 670 adducts per 108 nucleosides, respectively, in NNKOAc-treated calf thymus DNA, accounting for 18% of the total phosphate adducts. All 10 combinations of POB–PTEs were detected in liver DNA from rats acutely exposed to NNK (0.1 mmol/kg per day for 4 days) and in both liver and lung DNA in the chronic treatment group.34 Future approaches are required to characterize further the formation and repair of these POB–PTEs in vivo.
Several MS-based quantification methods were also introduced for monitoring exposure to PAHs and the corresponding DNA damage. Sun et al.77 investigated tissue distribution, excretion, and pharmacokinetics of the environmental pollutant dibenzo[def,p]chrysene (DBP) in mice by combining radiolabeled [3H]-DBP as well as LC-MS/MS quantification of (–)-anti-trans-DBPDE-dA (Figure 4) adducts in targeted and non-targeted tissues. After a 24 h treatment with [3H]-DBP, tissues in the digestive tract including the stomach and intestine had the highest radioactivity; however, after 1 week, radioactivity in the stomach and intestine drastically decreased to below 2% of that at 24 h, which is different from ovary, mammary glands, lung, and liver where more than 20% of the radioactivity was retained, suggesting different rates of metabolism of [3H]-DBP and/or removal of DBP-induced DNA adducts in various tissues. LC-MS quantification data revealed that the level of (–)-anti-trans-DBPDE-dA adducts in the ovary (8.91 ± 0.08 adducts/107 dA) was significantly higher than those in the kidney (0.69 ± 0.09 adducts/107 dA) and liver (0.63 ± 0.11 adducts/107 dA), a finding that is in keeping with the preferential carcinogenicity of DBP in the ovary.
Figure 4.
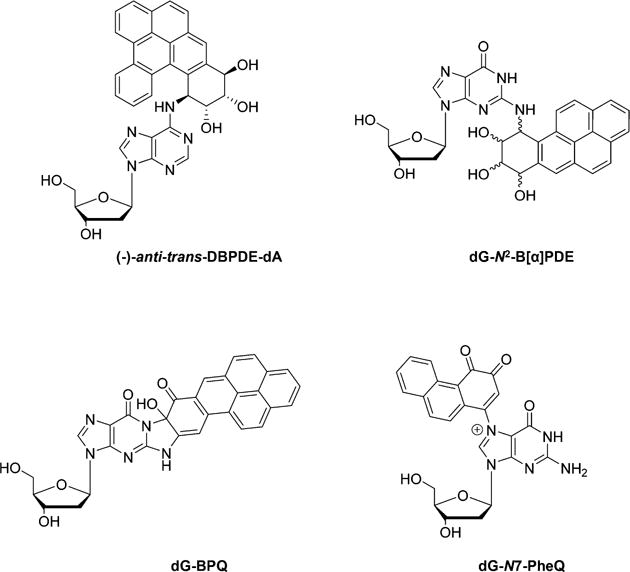
Chemical structures of selected DNA lesions induced by polycyclic aromatic hydrocarbons.
In another study, Klaene et al.78 described a comprehensive investigation of ion signal suppression owing to individual steps in the sample preparation process for the analysis of benzo[a]pyrene-induced 10-(deoxyguanosin-N2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene (dG-N2-B[a]PDE) (Figure 4) adduct. By optimizing the analytical steps, the authors concluded that butanol extraction of enzymes used for DNA digestion, followed by online SPE enrichment, can significantly improve sample throughput, increase analyte recovery, and elevate sensitivity of the LC-MS/MS method.78
Due to the lack of fresh-frozen tissues, the readily accessible formalin-fixed paraffin-embedded (FFPE) tissues may constitute very useful resources for DNA adduct and human exposure studies. A method was developed for the retrieval of DNA from FFPE tissues, and it was found that dG-N2-B[a]PDE could be retrieved from FFPE tissues of liver, lung, bladder, pancreas, and colon of rodents in high yield and quantified by UPLC/ESI-MS3, with comparable levels as those measured in fresh-frozen tissues.79
Comprehensive investigation of B[a]P-induced DNA lesions was also achieved by using a rapid non-targeted screening of DNA adducts in follicular cells exposed to B[a]P and cigarette smoke condensate (CSC) for 13 days of culture. A scan based on a constant neutral loss (CNL) of 116 Da (2-deoxyribose moiety) was employed to identify possible unknown DNA lesions, which were further analyzed by full-scan MS/MS to elucidate corresponding structures of detected DNA adducts from the CNL scan.80 Three DNA adducts, dG-N2-B[a]PDE, phenanthrene 1,2-quinone-dG (dG-N7-PheQ), and B[a]P-7,8-quinone-dG (dG-BPQ) (Figure 4), were identified in the B[a]P-treated follicular cells, and dose-dependent formation of these lesions was observed.80
DNA damage induced by chemotherapeutic agents was also widely explored in recent years. Corte-Rodríguez et al.81 compared the biological behaviors of three platinum-containing agents, cisplatin, oxaliplatin, and pyrodach-2, in three different cell lines, i.e., A549 lung adenocarcinoma, A2780 cisplatin-sensitive ovarian carcinoma, and A2780cis cisplatin-resistant ovarian carcinoma cells. Dose-dependent Pt incorporation in calf thymus DNA and DNA from the treated cells was observed for all three Pt-containing drugs. Moreover, Pt incorporation into DNA was more efficient for cisplatin than other Pt drugs in all three cell lines, especially the drug-sensitive A2780 cells.81 Further complementary HPLC-ICPMS and HPLC-ESI-Q-TOF MS characterizations illustrated that the main target of Pt drugs in DNA was the N7 of the guanine (Figure 5).81
Figure 5.
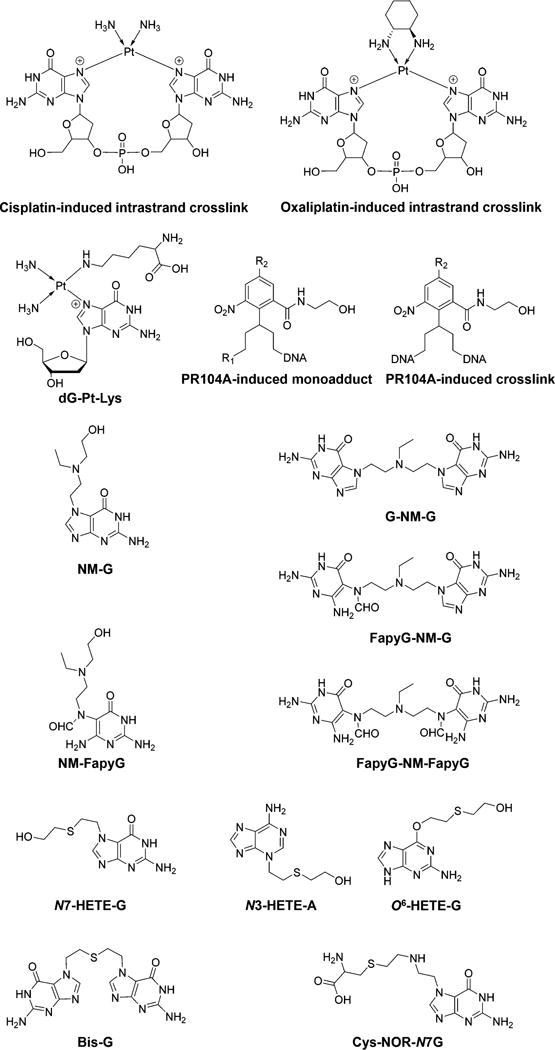
Chemical structures of selected DNA lesions induced by chemotherapeutic drugs.
In addition to the DNA nucleobase–nucleobase cross-linking adducts, Pt agents can also induce the formation of bulky and helix-distorting DNA–protein cross-links (DPCs). In this vein, Ming et al.82 reported an MS-based proteomic study of cisplatin-induced DNA–protein cross-links in human fibrosarcoma HT1080 cells. In that study, DPCs in cisplatin-treated cells were isolated using a modified phenol/chloroform DNA extraction procedure. Over 250 nuclear proteins in DPCs were released from DNA strands and identified by MS-based proteomics and Western blot analysis.82 In addition, detection of the conjugate formed between the N7 of guanine and the ε-amino group of lysine, 1,1-cis-diammine-2-(5-amino-5-carboxypentyl)amino-2-(2′-deoxyguanosin-7-yl)-platinum(II) (dG-Pt-Lys) (Figure 5), was achieved using a triple-quadrupole mass spectrometer, providing evidence for DPC formation in vivo upon Pt agents exposure.82
A CNL-MSn data-dependent scan was developed for an untargeted DNA adductomic approach in the investigation of DNA lesions induced by PR104A, an experimental DNA alkylating nitrogen mustard prodrug, in human colon adenocarcinoma HT-29 cells.83 Analysis was performed in real time by the instrument software with repeated full-scan detection in the Orbitrap detector at a resolution of 60 000, followed by MS2 acquisition and constant neutral loss triggering of MS3 fragmentation in the ion trap on the three most abundant full-scan ions listed in a parent mass list (targeted approach) or on the three most abundant full-scan ions from the full-scan spectra (untargeted approach) with Orbitrap detection. The MS2 parent mass list comprised 298 masses of the protonated ions of anticipated mono and cross-linked DNA adducts induced in the four nucleobases by PR104A. Finally, HCD fragmentation followed by MS3 acquisition in the Orbitrap was triggered upon observation of neutral losses of m/z 116.0474 (2′-deoxyribose), m/z 151.0494 (guanine), m/z 135.0545 (adenine), m/z 126.0429 (thymine), and m/z 111.0433 (cytosine) between the parent ion and one of the 50 most abundant product ions found in MS/MS.83 A total of 14 mono and cross-linked DNA adducts induced by PR104A were detected (Figure 5), and dose-dependent formation of adducts was observed in HT-29 cells upon PR104A treatment.83
UPLC-MS3 or MS2-analysis was also introduced for the quantification of DNA lesions in human cells induced by nitrogen mustard bis(2-chloroethyl)-ethylamine or sulfur mustard 2,2′-dichloroethyl sulfide.84,85 In particular, the nitrogen mustard-induced N7-guanine (NM-G) adduct and its cross-link (G-NM-G), the ring-opened formamidopyrimidine monoadduct (NM-FapyG), and cross-links in which one (FapyG-NM-G) or both (FapyG-NM-FapyG) guanines underwent ring-opening to FapyG units were identified (Figure 5). Moreover, sulfur mustard-induced N7-[2-[(2-hydroxyethyl)-thio]-ethyl]guanine (N7-HETE-G), bis(2-ethyl-N7-guanine)-thioether (Bis-G), N3-(2-hydroxyethylthioethyl)adenine (N3-HETE-A), and O6-[2-[(2-hydroxyethyl)thio]-ethyl]guanine (O6-HETE-G) (Figure 5) were also observed.84,85
Similar to Pt agents, N,N-bis(2-chloroethyl)-phosphorodiamidic acid (phosphoramide mustard, PM) can spontaneously dephosphoramidate to N,N-bis(2-chloroethyl)-amine (nornitrogen mustard, NOR), inducing the formation of DPCs.86 A nanoLC-NSI-MS/MS analysis was used to quantify the N-[2-[cysteinyl]ethyl]-N-[2-(guan-7-yl)ethyl]amine (Cys-NOR-N7G) (Figure 5) conjugates, which corresponds to cross-linking between the N7-position of guanine and sulfhydryl moiety of cysteine.86 Dose-dependent induction of Cys-NOR-N7G adduct was observed in NOR-treated HT1080 human fibrosarcoma cells. Moreover, both NER-deficient xeroderma pigmentosum XPA cells and FANCD2-deficient PD20 cells were more sensitive to PM treatment than the corresponding repair-competent cells, indicating the involvement of Fanconi anemia pathway and NER in the removal of PM-induced DNA damage.86
DNA alkylation induced by chemical carcinogens arising from high-temperature food processing and herbal plant secondary metabolites was also investigated. For example, Høie et al.87 reported an isotope-dilution coupled with UPLC-MS/MS method (with LOD of 0.34 adducts/108 nucleosides) to evaluate the formation of DNA adducts N2-((furan-2-yl)methyl)-2′-deoxyguanosine (N2-MF-dG) (Figure 6) in wild-type and transgenic FVB/N (FVB) mice after oral exposure to furfuryl alcohol (FFA, 250 mg/kg body weight), a carcinogen formed during thermal and acid-catalyzed dehydration of pentoses. The levels of N2-MF-dG were measured in liver, kidney, and proximal and distal small intestine as well as colon of FAA-treated mice. The detection of N2-MF-dG in the untreated mice indicated unknown internal source of this lesion in vivo. Significant higher adduct levels were observed in colonic and hepatic DNA of FFA-treated mice than untreated mice.87
Figure 6.
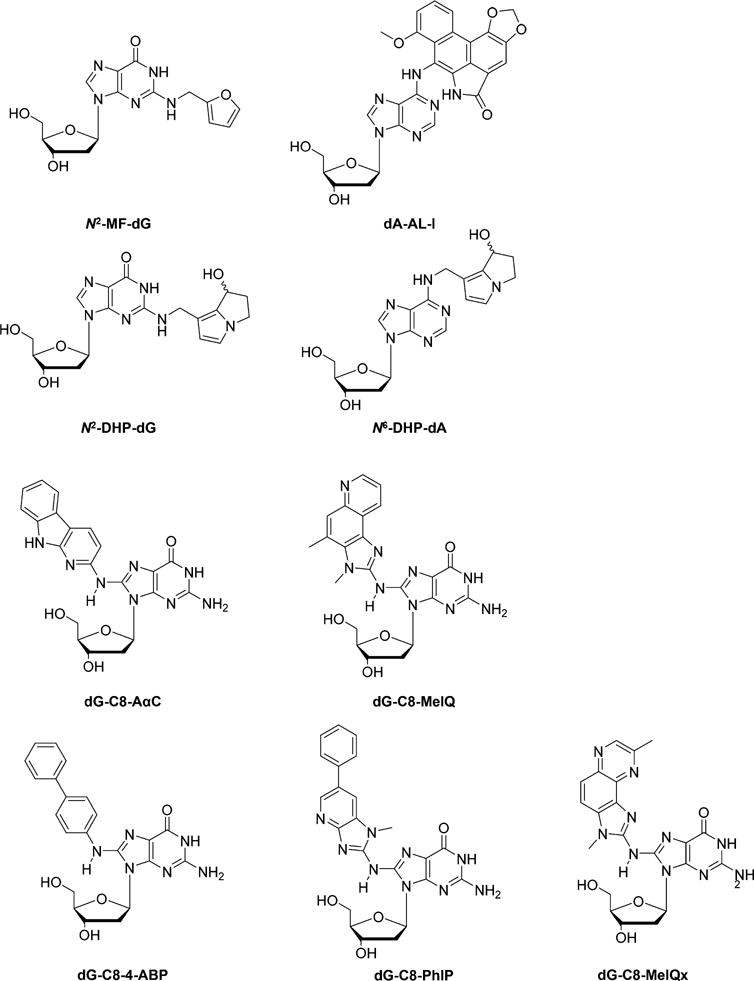
Chemical structures of selected DNA lesions induced by food-related chemical carcinogens, herbal plant secondary metabolites, and heterocyclic aromatic amines.
Carcinogenic plant secondary metabolites, such as aristolochic acid (AA) and pyrrolizidine alkaloids (PA), may also react with DNA to yield bulky adducts. By using a newly reported UPLC-NSI/MS3 analysis, Turesky et al.88 studied human exposure to AA in Romania and its implications in renal cell carcinoma (RCC). The levels of AA-induced DNA lesion, 7-(deoxyadenosin-N6-yl) aristolactam I (dA-AL-I) (Figure 6), were quantified in non-tumor renal tissues of 14 RCC cases from Romania and 15 cases from Czech republic, United Kingdom, and Russia.88 The quantification results showed that dA-AL-I was detected at levels of 0.7–26.8 adducts per 108 nucleobases in all the 14 Romanian cases, consistent with levels reported in Asian and Balkan populations exposed to AA-contaminated herbal remedies or food. No detectable dA-AL-I was observed in the 15 cases from the three countries without reported AA exposure.88
PA-containing plants are among the most common poisonous plants affecting livestock, wildlife, and humans. PA can be metabolically activated to dehydropyrrolizidine alkaloids, giving rise to the formation of (±)-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine (DHP)-derived DNA adducts. To measure these tumorigenic DNA adducts, Zhu et al.89 developed a UPLC-ESI-MS/MS method for accurate quantifications of N2-DHP-dG (7-hydroxy-9-(deoxyguanosin-N2-yl)-dehydrosupinidine) and N6-DHP-dA (7-hydroxy-9-(deoxyadenosin-N6-yl)dehydrosupinidine (Figure 6), the representative retronecine-type of hepatocarcinogenic PA, in retrorsine-treated male ICR mice. N2-DHP-dG and N6-DHP-dA were quantified in multiple-reaction monitoring (MRM) mode, with the levels of these two lesions increasing with retronecine dose (4.98 to 83.25 per 108 nucleosides in total).89 In this vein, the same group also investigated the formation of these DHP–DNA adducts in livers of cattle poisoned with Heliotropium europaeum.90 LC-ESI-MS/MS analysis in the selected-reaction monitoring (SRM) mode revealed that the DHP-induced DNA adducts could be detected in liver tissues of cows exposed to the contaminated feed, suggesting that these DNA adducts can be considered as sensitive biomarkers for PA exposure and poisoning.90
HAA-Induced DNA Lesions
Recently Turesky and co-workers79,91–95 investigated HAA-induced DNA damage. First, they explored the roles of CYP450 in the liver and intestine in bioactivation of AαC in male B6 wild-type (WT) mice, liver-specific CYP450 reductase (Cpr)-null (LCN) mice, and intestinal epithelium-specific Cpr-null (IECN) mice.91 A capillary UPLC coupled with the NSI-MS3 on an LTQ Velos linear ion-trap mass spectrometer was used for the quantifications of N-(deoxyguanosin-8-yl)-2-amino-9H-pyrido-[2,3-b]indole (dG-C8-AαC) (Figure 6).91 No significant difference in the levels of dG-C8-AαC formed in liver of WT, LCN, and IECN mice was observed. However, the higher levels of dG-C8-AαC in the colon and lung of LCN mice, together with higher metabolic rate of AαC in liver tissues of WT than LCN mice, illustrated that the formation of dG-C8-AαC in mice was independent of hepatic microsomal or intestinal CYP450; however, CYP450 played a crucial role in the detoxification of AαC.91
This UPLC-NSI-MS3 method was later applied for the measurement of dG-C8-AαC and N-(deoxyguanosin-8-yl)-2-amino-3,4-dimethylimidazo[4,5-f ]quinoline (dG-C8-MeIQ) (Figure 6) in tissues of AαC- and MeIQ-treated male A/J mice.92 The levels of dG-C8-AαC were significantly higher than dG-C8-MeIQ in both liver (470.1 vs 70.3 per 107 nucleosides) and colon (50.9 vs 8.2 per 107 nucleosides) tissues of the treated mice. In contrast, after normalization with the numbers of DNA adducts, dG-C8-MeIQ was more potent than dG-C8-AαC in inducing colonic aberrant crypt foci (ACF).92 dG-C8-AαC was also quantified in AαC-treated human hepatocytes originated from three donors, with the highest adduct levels being observed in the hepatocytes with the highest activities of CYP450 1A1 and 1A2.93
The nanoflow UPLC-NSI-MS3 method was also extended to the simultaneous detection of other HAA-derived DNA lesions, i.e., the 4-aminobiphenyl (4-ABP)-induced N-(deoxyguanosin-8-yl)-4-aminobiphenyl (dG-C8-4-ABP) and the PhIP-induced N-(deoxyguanosin-8-yl)-PhIP (dG-C8-PhIP) (Figure 6), in the FFPE tissues of rodents treated with these procarcinogens, with no significant difference relative to the levels found in fresh-frozen tissues.79 Furthermore, an improved method with higher sensitivity and selectivity was developed by using nanoflow UPLC-NSI coupled with an Orbitrap Fusion Tribrid mass spectrometer for the analysis of HAA–DNA adducts, including dG-C8-PhIP, dG-C8-4-ABP, dG-C8-AαC, and the MeIQx-induced dG-C8-MeQIx (Figure 6), with LOQs ranging from 1.3 to 2.2 adducts per 109 nucleotides when 2.5 μg of DNA was used for measurement.95
HAA-induced DNA lesions were also explored by other research groups. By using a previously reported online column-switching LC-ESI-MS/MS method,96 Krais et al.97 studied the potential role of p53 in PhIP metabolism in vivo by treating Trp53+/+, Trp53+/−, and Trp53−/− mice with PhIP. Levels of dG-C8-PhIP were found to be significantly lower in liver, forestomach, glandular stomach, and colon and markedly higher in kidney and bladder of Trp53−/− mice compared to Trp53+/+ mice, suggesting that p53 can modulate PhIP metabolism in vivo in a tissue-dependent manner.97 Similarly, Høie et al.98 employed an isotope dilution coupled with a UPLC-MS/MS technique for quantification of dG-C8-PhIP in male FVB mice. Significantly higher levels of dG-C8-PhIP were observed in PhIP-treated mice expressing human sulfotransferase (hSULT) 1A1 and 1A2 compared to those in wild-type mice for all organs tested (liver, kidney, small intestine, and colon), indicating the role of hSULTs in the metabolic activation of PhIP in vivo.98
Interestingly, dietary consumption of cruciferous vegetables (CRU) and apiaceous vegetables (API) was found to reduce PhIP-induced DNA adducts in rats.99,100 The levels of dG-C8-PhIP in colonic tissues of PhIP-treated rats were quantified by using an online column switching LC-ESI-MS3 method.100–102 A 20.4% reduction in levels of dG-C8-PhIP was found in the API-fed rats compared to the control group.100 However, results from a further rat-feeding experiment demonstrated that only phenethyl isothiocyanate and indole-3-carbinol from CRU can reduce the formation of dG-C8-PhIP in rat colon by 43.5%, indicating the complexity of metabolic activation of phytochemicals and detoxification of PhIP in vivo.99
DNA Adductomics
The above-mentioned targeted DNA adduct quantification is not adequate in fulfilling the goal of comprehensively exploring DNA damaging agent-induced DNA lesions and their implications in human diseases. This is because, in current targeted DNA adduct measurements, only certain types of chemically modified 2′-deoxynucleosides or nucleobases were considered for analysis. For this reason, untargeted DNA adduct profiling, the DNA adductomics, is essential for the simultaneous assessment of all possible known and unknown, directly and indirectly induced lesions in genomic DNA after exposure to certain chemicals or a mixture of DNA damaging agents, such as diet-related carcinogens or tobacco smoke.
The CNL of 116 Da in LC-MS/MS analysis was commonly monitored to screen for possible covalently modified nucleobase adducts possessing the common 2′-deoxyribose (dR) moiety in nucleosides.103,104 Different analytical strategies, such as CNL, pseudo-CNL, data-dependent MS/MS (DD-MS/MS) acquisition, and data-dependent-CNL-MS3 (DD-CNL-MS3) analysis, have been developed, as discussed in a previous review.103 For example, pseudo CNL on a triple-quadrupole mass spectrometer was used for exploring acrolein-induced DNA adducts in soil bacterium Sphingobium sp. strain KK22.105 In this study, 100 transitions were monitored over the m/z range of protonated nucleosides ([M + H]+) from transitions of m/z 250 → 134 to m/z 350 → 234. The acquisition results were processed by using MassLynx software to produce DNA adductome maps, revealing numerous putative DNA lesions in both treated and untreated groups or exclusively in one group.105
A CNL-based untargeted screening coupled with targeted analysis for covalently modified 2′-deoxynucleosides was developed, which was validated by evaluating DNA lesions generated from reaction of 20-mer oligodeoxyribonucleotides (ODNs) with phenyl glycidyl ether (PGE) and styrene-7,8-oxide (SO).106 In the CNL mode on a Q-Trap mass spectrometer, quadrupole 1 (Q1) and quadrupole 3 (Q3) were scanned simultaneously to monitor compounds with the neutral loss of a 2-deoxyribose, with 10 and 9 unknown DNA adducts found in the PGE- and SO-treated ODN, respectively. The discovered unknown DNA lesions were further characterized in the product-ion scan mode, providing structural information for the identifications of these unknown modified nucleobases.106
In addition to the triple quadrupole/Q-trap-based pseudo-CNL/CNL methods, the recent use of nanoLC-NSI or UPLC coupled with a high-resolution mass spectrometer, especially the hybrid Orbitrap instruments, provided both high sensitivity and selectivity for DNA adductome analysis in a DD-MS/MS or DD-CNL-MS3 mode, as discussed above for the nitrogen mustard PR104A-induced DNA lesions.83
Taking advantage of the new benchtop Q-Exactive hybrid quadrupole-Orbitrap high resolution instrument, Hemeryck et al.57,107 systematically studied the diet-related DNA adductome. Untargeted analysis of ten colon biopsies from colon tumor patients was performed in parallel with the assistance of a DNA adductome database. A series of typical DNA adducts induced by alkylating agents and oxidative stress were observed, including carboxymethyl-G, butyl-G, hydroxyethyl-G, carboxyethyl-A, carboxymethyl-A, methyl-T, dimethyl-T or ethyl-T, carboxy-T, Fapy-G, Fapy-A, thymine glycol, hydroxy-T, 1,N2-εG, N2,3-εG, 1,N6-εA, malondialdehyde-induced M3C, and methoxymethyl-G.57 Using the same method, possible genotoxic effects, arising from the consumption of red meat compared to white meat and the interfering role of dietary fat, was investigated in Sprague–Dawley rats, where the liver, duodenum, and colon DNA adductomes were analyzed.107 Quantitative data revealed that 22 DNA adducts, arising from alkylation, nitrosation, and/or oxidation processes, were significantly increased upon the consumption of beef and/or fat-supplemented beef or chicken relative to the chicken-only diet. This result is in line with the current hypothesis that NOCs and LPOs can arise from red meat and high fat intake and can give rise to DNA adducts.107
RECENT DEVELOPMENT OF NON-LC-MS-BASED ANALYSIS OF DNA ADDUCTS
Next-Generation Sequencing-Based Analysis of DNA Damage
LC-MS-based methods provide accurate and sensitive quantification of DNA damage products; they, however, do not offer information about the distribution of DNA damage in the genome. Recent developments in NGS techniques have afforded new opportunities for the genome-wide mapping of DNA lesions (Figure 7). Two key issues to be addressed during NGS-based method development are the enrichment of lesion-containing DNA fragments and the PCR amplification of the resulting fragments. With the use of lesion-specific antibodies or proteins involved in excision repair or DNA damage recognition, methods have been developed for sequencing and mapping DNA lesions induced by oxidative stress, UV irradiation, DNA alkylating agents, and platinum drugs.
Figure 7.
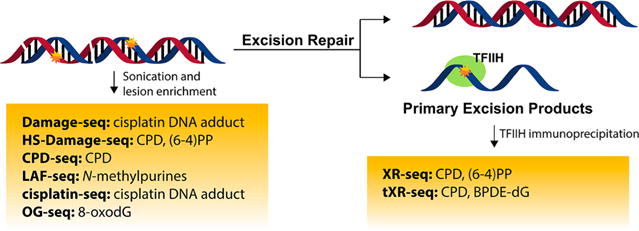
Next-generation sequencing for mapping the genome-wide distributions of DNA lesions.
It was suggested recently that 8-oxo-dG might assume an epigenetic-like role.108 To further explore the mutagenic and epigenetic-like properties of 8-oxo-dG, a sequencing approach termed “OG-seq” was developed to map 8-oxo-dG sites at ∼0.15-kb resolution.109 To overcome the low yield of lesion-containing DNA fragments by immunoprecipitation, the authors employed a strategy of labeling 8-oxo-dG with K2IrBr6 and amine-terminated biotin with a polyethylene glycol linker followed by affinity purification using streptavidin-coated magnetic beads. The complementary strands were subsequently released and subjected to NGS. OG-seq was applied to wild-type and Ogg1−/− mouse embryonic fibroblasts and revealed enrichment of 8-oxo-dG in regions of the genomes that encode for gene promoters and untranslated regions, suggesting the potential role of 8-oxo-dG in epigenetic gene regulation.
For UV-induced DNA damages, such as cyclobutane pyrimidine dimers (CPDs) and pyrimidine(6–4)pyrimidone photoproducts [(6–4)PPs], Hu et al.110 developed “XR-seq” that mapped CPDs and (6–4)PPs at single-nucleotide resolution in human skin fibroblasts. Primary excision repair products were first isolated by TFIIH immunoprecipitation and then with CPD and (6–4)PP antibodies. The lesions were subsequently repaired by DNA photolyases to render the oligomers amenable for PCR amplification and NGS analysis. This method has been used for exploring the relationship between DNA excision repair and chromatin state in normal and Cockayne syndrome B mutant human skin fibroblasts.111
XR-seq is limited to those lesions that can be repaired by photolyases. To further expand the method for mapping other lesions that cannot be reversed by photolyases, Li et al.112 employed translesion synthesis (TLS) DNA polymerases for generating PCR amplifiable DNA oligomers. This optimized method, dubbed as “tXR-seq”, used human DNA polymerases η and κ to convert the excised ssDNA oligomer that contains CPD or dG-N2-B[a]PDE to the dsDNA form that can be amplified by PCR and subjected to NGS analysis.112 The authors successfully mapped the repair of CPD and dG-N2-B[a]PDE in GM12878 human lymphocytes and revealed sequence specificity of nucleotide excision repair of the dG-N2-B[a]PDE. This method can be applicable to other NER substrates, though it should be noted that it depends on the accuracy and efficiency of TLS polymerases as well as neighboring sequence effect of TLS.
“HS-Damage-seq” (high-sensitivity damage sequencing) was developed for mapping the genome-wide distribution of CPD and (6–4)PP based on “Damage-seq” (described below for cisplatin adducts).113,114 This method used a different DNA library preparation kit, which reduced the amount of starting material from 5 to 1 μg. It also included a step of subtractive hybridization with an oligomer identical to the 5′ sequence of the first adaptor. These optimizations increased the detection limit and enabled the analysis of samples with relatively low damage levels. Since XR-seq and HS-Damage-seq capture different features regarding DNA damage, a combination of the two can provide a better picture about DNA damage formation and repair. The authors used both sequencing methods for generating repair and damage maps of CPD and (6–4)PP. Their results showed that, although the UV-induced damage is uniformly distributed throughout the genome, the repair is modulated by chromatin states, transcription activity, and transcription factor binding.
Another method, “CPD-seq”, was introduced by Mao et al.115 for mapping CPDs in the yeast genome. In this method, 3′-OH groups of fragmented DNA were blocked by ligation with an adaptor, followed by T4 endonuclease V (T4 endoV) and apurinic/apyrimidinic endonuclease (APE1) treatment to generate a free 3′-OH terminus upstream of the CPD lesion. A second adaptor was then ligated to the 3′-OH terminus, followed by purification of the ligated DNA, PCR amplification, and sequencing.
For alkylated DNA lesions, Li et al.116 reported Lesion-Adjoining Fragment Sequencing (LAF-seq) for mapping N-methylpurines in human cells. Briefly, apurinic/apyrimidinic (AP) sites were generated through depurination of N-methylpurines and cleaved by spontaneous β elimination or β and δ eliminations. Isolated DNA was digested with restriction enzyme, and the remaining unsaturated sugar or phosphate group at lesion sites was further processed by endonuclease IV. DNA fragments from regions of interest were subsequently enriched by biotinylated oligomers. In theory, LAF-seq can be applied to DNA lesions as long as they can be converted to DNA strand breaks, though it is more suitable for investigating specific regions rather than genome-wide mapping.
A method coined as “cisplatin-seq” by Shu et al.117 was reported for the genome-wide mapping of cisplatin–DNA adduct at single-base resolution. The authors employed domain A of HMGB1 protein to pull down cisplatin–DNA adducts for high-throughput sequencing. Adduct-containing DNA fragments were ligated with the first adaptor that only ligates with the 3′ end of DNA. Primer extension is stalled at cisplatin cross-linking sites during the first strand DNA synthesis, and the second adaptor was ligated after the synthesis of the first DNA strand. This method can localize platinum modifications even when they are clustered within short stretches of DNA (12 bp). It was revealed that mitochondrial DNA is a preferential target of cisplatin, and in nuclear genomes, cisplatin–DNA adducts are enriched within promoters and transcription termination sites.
Another method, “Damage-seq”, also took advantage of the fact that bulky lesions block DNA replication.114 Fragmented DNA was ligated with first adaptor on both ends. Pt–DNA adduct-containing strands were enriched with antibodies recognizing the Pt adduct. A biotinylated primer was annealed and extended until it is blocked by the Pt adduct. The extended primers were purified before ligation with the second adaptor, followed by PCR amplification, sequencing, and genome alignment. In this case, both damaged and undamaged strands were amplified by PCR, generating numerous unnecessary reads, which was later addressed by HS-Damage-seq.113 Using damage-seq in combination with XR-seq, it was found that cisplatin-induced DNA damage is uniformly distributed throughout the human genome, and incidence of damage is largely dependent upon the underlying G–G sequence. The repair efficiency is, however, heterogeneous in the genome and correlates with transcription and chromatin states.
Other Sequencing-Based Analysis of DNA Damage
Aside from the aforementioned NGS-based methods, Sanger sequencing has also been used for the detection of DNA damage owing to its high accuracy.118 For example, DNA glycosylases were employed to first remove lesions located at codon 12 of the KRAS gene to generate a gap in DNA. This gap is then filled with unnatural base pairs (UBP)119 or ligated120 so that the DNA can be amplified by PCR. The PCR products are subsequently subjected to Sanger sequencing. In the first example, UBP (i.e., dNaM:d5SICS) was incorporated at the lesion sites and amplified by PCR in the presence of the artificial nucleotides so that PCR products retain these marks.119 Sanger sequencing comes to a stop at the UBP site; thus, the site of DNA lesion can be recognized. The method was capable of correctly locating a dU lesion in mixtures that consisted of dU-containing strand and non-damaged strand at a ratio of 1:10 or 1:100. In the second case, the gap was ligated and the PCR products, which are of different lengths, were sequenced to produce two sequence readouts that were easily observed as a pair of peaks starting at the modification site.120 Since there was no abrupt stop of Sanger sequencing, this method could identify the position of two uracils separated by as few as 9 nucleotides. This method was also found to be capable of identifying an 8-oxo-dG in a plasmid DNA. Although these methods are not amenable for high-throughput genome-wide mapping of DNA lesions, they can be useful for monitoring DNA damage within specific genes, especially in single cells. Currently, the methods are limited to those lesions that could be processed by glycosylases, such as 8-oxo-dG, dSp, 2′-deoxyuridine (dU), and AP sites, and their application to cellular and/or tissue DNA samples awaits further testing.
As an emerging high-throughput sequencing technique, α-hemolysin (α-HL) nanopore has also been utilized for DNA damage detection. Riedl et al.119 reported that Sanger sequencing stalls when DNA polymerase encounters unnatural base pairs. The stalling sites indicate the locations of 8-oxo-dG, with the shortfall that multiple lesion sites on the same strand cannot be detected due to the sequencing stop. As an alternative approach, the authors utilized α-HL for the detection of multiple lesions and were able to detect two marker nucleotides on the same DNA strand. Liu et al.121 employed α-HL nanopore for the detection of 8-oxo-dG in ssDNA. In their work, 8-oxo-dG was first coupled with 1,12-dodecanediamine and then incubated with cucurbit[7]uril to form a host–guest complex-modified DNA hybrid. Translocation of this DNA hybrid generates current signatures reflective of the presence of 8-oxo-dG. Although the reported method did not work well when there were multiple 8-oxo-dG sites on the same DNA strand, it could measure the lesion-containing DNA in a DNA mixture quantitatively. These studies showed that nanopore sequencing is a potential high-throughput screening method for DNA lesions.
Optical Detection Methods
Capillary electrophoresis laser-induced fluorescence (CE-LIF) is an alternative method for selective and sensitive detection of DNA lesions. With the use of quantum dot (QD), CE-LIF has been used for the analysis of UV light- or oxidative stress-induced DNA lesions. For example, Li and Wang122 used BER glycosylase to cleave oxidatively modified nucleosides generated by Fenton reaction or UV irradiation to yield AP sites. A biotinylated ddNTP was incorporated into the nucleotide gap, and the biotin is labeled with a QD–streptavidin conjugate. The DNA-bound and -unbound QDs were subsequently separated by CE and detected by LIF. The authors found that the use of formamidopyrimidine DNA glycosylase (FPG) and endonuclease VIII enables the detection of oxidized purine and pyrimidine bases, respectively. The detection limit with synthesized standard DNA was 1.1 × 10–19 mol in mass and 2.9 pM in concentration. In addition, Lee et al.123 reported the analysis of ROS-induced DNA damage at single-molecule level. Damaged DNA was first processed by three types of DNA glycosylases to generate AP sites, and DNA polymerase I was subsequently employed to incorporate Alexafluor-647-labeled dUTPs into AP sites. Damage sites on a single λ bacteriophage DNA molecule can be counted. Since the damage sites can be visualized on a single molecule, it provides information about the locations of DNA damage as a different strategy for “mapping” the DNA damage.
Taking a different approach of labeling DNA with QD, Guthrie et al.124 used CPD or (6–4)PP antibodies to label the lesion-containing DNA, followed by secondary antibodies conjugated with fluorescent QD reporters that could attach to primary antibodies. The unbound antibodies were separated from lesion-bound antibodies by CE, and fluorescence signals from QD were analyzed. This method provides simultaneous detection of CPD and (6–4)PP with as little as 6 ng of DNA, though the levels of DNA damage were not quantified.
Förster (or fluorescence) resonance energy transfer (FRET) has also been employed for measuring UV-induced DNA damage. In this vein, carbon dots (C-dots) functionalized with amine moieties were prepared for DNA damage detection.125 These C-dots have a high affinity to DNA, and their binding to DNA resulted in changes in the photoluminescence and absorption spectra of C-dots. In this FRET-based method, C-dots transfer excited-state energy to ethidium bromide (EtBr). The fluorescence of EtBr could distinguish native DNA from damaged DNA. The sensor was used for the detection of UV irradiation-induced or oxidative stress (Fenton reaction)-induced DNA damage in isolated DNA as well as genomic DNA isolated from cultured human cells or tissues.125
In addition to the aforementioned PCR amplification method,119 unnatural nucleobases are also utilized in other methods for the detection of DNA damage. Trantakis et al.126 synthesized a nucleoside analog that, when incorporated into DNA, can form stable DNA duplexes with O6-Me-dG-containing DNA. The authors used this strategy to quantify O6-Me-dG within mutational hotspots of the human KRAS gene in the presence of human genomic DNA, and the limit of detection for O6-Me-dG-containing DNA with genomic DNA as background was 0.24%.
To evaluate the accessibility of DNA 5hmC in chromatins, Zhong et al.127 developed a method using anti-5-hydroxymethylcytosine (5hmC) antibody under non-denaturing conditions. Overexpression of TET1 catalytic domain in MCF-7 human breast cancer cells resulted in a significant increase in antibody-accessible 5hmC along with an increase in total 5hmC sites. This method could provide a landscape of accessible 5hmC within chromatin contexts. To selectively detect 5hmC in DNA, 5hmC was oxidized to 5-formylcytosine (5fC) with KRuO4 and subsequently labeled with N-(4-aminobutyl)-N-ethylisoluminol.128 A thiolated ssDNA probe was self-assembled on a gold surface, and it could hybridize with target DNA, resulting in electrogenerated chemiluminescence (ECL) whose intensity is proportional to the number of 5hmC residues. The detection limit of 5hmC-DNA was 0.14 × 10–13 M, and this method was used for quantification of 5hmC in serum samples.
Some analytical methods were also developed for the investigation of repair processes or potential toxicity from exogenous exposures by characterizing ICLs or AP sites. Evison et al.129 prepared 8-propargyloxypsoralen (8-POP) that could efficiently generate DNA ICLs in cells upon UVA irradiation. 8-POP harbors an alkyne handle, which allows post-labeling with an azide-tagged fluorescent reporter via click chemistry. This approach allows for the detection and quantification of 8-POP-induced ICLs by fluorescence microscopy and flow cytometry, and it can be used in DNA repair studies. AP sites are often generated after removal of damaged nucleobases by glycosylases. Condie et al.130 developed a near-infrared (NIR) fluorescent probe that could bind to AP site and showed that it can detect AP sites in DNA from chemotherapy-treated DLD1 colon cancer cells. Since NIR, when compared to UV and visible light, has deeper tissue penetration and less tissue-induced light scattering, this method could potentially permit cost-effective imaging in live animals, thus providing information on DNA damage and repair in vivo.
Electrochemical Methods
Various electrochemical sensors have been developed for simple, cost-effective, and sensitive detection of DNA damage in complex samples such as human urine, thereby allowing for potential clinical applications. The majority of these sensors target 8-oxo-dG in urine or serum samples as a biomarker of oxidative DNA damage.
Pan et al.131 developed an electrochemical immunosensor for the determination of 8-oxo-dG. They immobilized poly(indole-5-carboxylic acid) and chitosan onto a glassy carbon electrode, followed by modification of the electrode with protein A and anti-8-oxo-dG antibody. The linear range of differential pulse voltammetry peak currents for 8-oxo-dG detection was from 0.353 nM to 35.3 μM, with the detection limit being 105.9 pM. This immunosensor displayed good accuracy when tested on human urine samples with standard addition.
Another biosensor assembly employed the molecular imprinting technology with the use of 8-oxo-dG as the template molecule.132 Electrochemical impedance spectroscopy (EIS) characterization of this biosensor showed a linear relationship for 8-oxo-dG over a concentration range of 3.5–3500 pM, and the sensor exhibited good selectivity in complex sample matrices such as human urine samples spiked with 8-oxo-dG.
Instead of detecting the oxidation current of 8-oxo-dG, Fan et al.133 exploited electrochemical signal obtained from the reduction of polyaniline (PANI) under the catalysis of hemin/G-quadruplexes. They used a tetrahedral DNA nanostructure (TDN) to anchor 8-oxo-dG aptamer onto the surface of a gold electrode. 8-oxo-dG induced the formation of hemin/G-quadruplexes from its aptamer. The hemin/G-quadruplexes triggered PANI deposition and supplied a deposition environment together with the TDN structure, which greatly improved the detection sensitivity. The linear range for 8-oxo-dG detection is from 10 pM to 2 nM with a detection limit of 1 pM. The method improved sensitivity by almost 300-fold compared with the method using oxidation current of 8-oxo-dG. The method has been applied for the quantification of 8-oxo-dG in urine and human serum samples and showed potentials in clinical diagnosis.
Song et al.134 reported a microfluidic electrochemical sensor array for the simultaneous detection of various DNA damage products. The sensors were coated with osmium or ruthenium bipyridyl-poly(vinylpyridine) chloride (OsPVP, RuPVP) metal-lopolymers along with DNA and human liver microsomes. OsPVP selectively detects guanine oxidation products on the DNA strands, and RuPVP detects DNA adducts. This array directly measures unhydrolyzed DNA and requires only 22 ng of DNA. The mass detection limit is 15 pg of 8-oxo-dG (or 672 modifications per 106 bases). Although this method requires a very small amount of DNA, its application for monitoring DNA damage in biological samples necessitates further improvement in sensitivity since 8-oxo-dG is present at much lower frequencies in cellular DNA.6
RECENT DEVELOPMENTS IN SHUTTLE VECTOR-BASED METHODS FOR THE ASSESSMENT OF BIOLOGICAL CONSEQUENCES AND REPAIR OF DNA ADDUCTS
With the development of analytical methods and their applications in biological analysis, in the last few decades, there has been significant progress in understanding the biological consequences of DNA adducts. Previously, extensive colony picking and Sanger sequencing procedures have been required for assessing how DNA lesions compromise the efficiency and fidelity of DNA replication/transcription and for evaluating the roles of DNA polymerases in bypassing the lesion sites.3,135–137 Recently, mass spectrometry-based strategies and next-generation sequencing facilitated accurate and high-throughput analysis of the genetic perturbations elicited by DNA adducts.4 In this section, we will describe the use of MS in the replication and transcription studies in cells, and we will briefly discuss the application of next-generation sequencing in assessing the biological endpoints of chemically modified nucleosides.
LC-MS-Based Replication Studies in E. coli Cells
Initially introduced by Delaney and Essigmann,138 and further developed by our laboratory, the competitive replication and adduct bypass (CRAB) assay is employed to assess the genetic perturbations induced by lesions during DNA replication in E. coli cells using single-stranded M13 bacteriophage (Figure 8a).139–143
Figure 8.
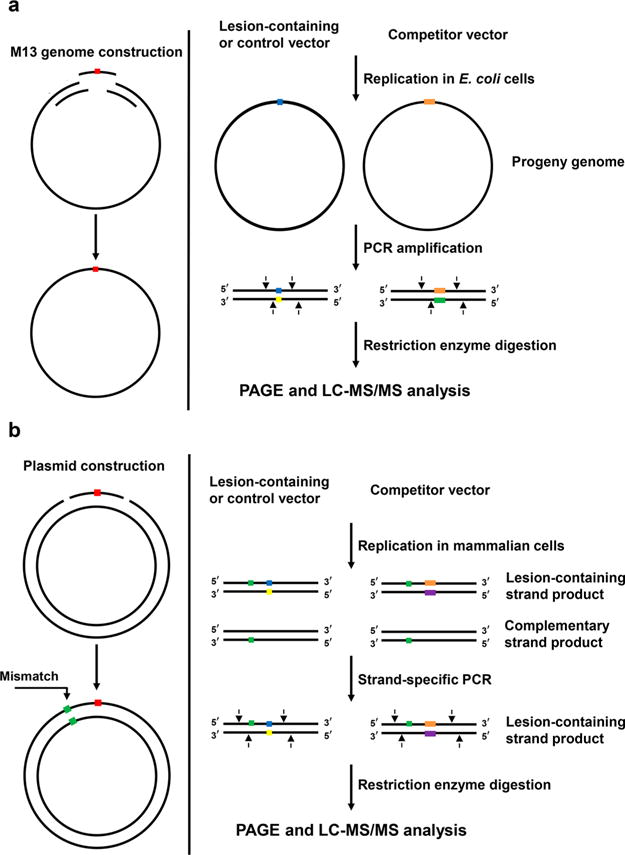
Schematic diagrams showing the experimental procedures for the competitive replication and adduct bypass assay in E. coli cells (a) and mammalian cells (b).
The experimental system begins with the construction of lesion-containing and lesion-free M13 genomes as well as a competitor M13 genome, which has three additional nucleotides. Because transfection efficiencies can vary, premixing the lesion-bearing or the respective lesion-free genome with a non-lesion competitor genome prior to transfection allows the blockage to replication to be accurately quantified from the attenuation in ratio of output signal for the lesion/competitor genome relative to that for the corresponding control/competitor genome. In this vein, the competitor genome acts as an internal standard and the bypass efficiency of the corresponding control lesion-free M13 genome is considered 100%. After replication in E. coli cells, the region of the isolated progeny M13 genomes harboring the initial lesion or corresponding control site is amplified by PCR. The PCR products are digested with appropriate restriction enzymes, and the resulting digestion products are subjected to LC-MS/MS and PAGE analyses.
This assay allows for the determination of bypass efficiency and mutation frequency for structurally defined DNA lesions. Additionally, by conducting the replication studies in E. coli cells that are proficient or deficient in specific repair proteins or TLS DNA polymerases, their respective roles in repair and translesion synthesis of the lesion under consideration could be revealed.
In the past few years, this MS-based CRAB assay has been applied to investigate the impact of ethylated thymidine lesions on DNA replication in E. coli cells.139–141 Among the regioisomeric O2-, N3-, and O4-Et-dT, the former two were found to be strong impediments to DNA replication and direct promiscuous nucleotide misincorporation, whereas O4-Et-dT was bypassed efficiently and exclusively resulted in the T → C mutation. Moreover, Pol V is involved in the replicative bypass of O2- and N3-Et-dT, while the three SOS-induced polymerases, i.e., Pol II, Pol IV, and Pol V, play a somewhat redundant role in bypassing O4-Et-dT.139
These findings still hold true when other alkyl groups are considered, from a simple methyl to more complex sec-butyl group, conjugated with the O2 and O4 positions of thymidine. It was demonstrated that alkylation at the O2 position of thymidine led to promiscuous miscoding and strong blockage to DNA replication, with the replicative bypass efficiency decreasing with the size of the alkyl group,140 which is consistent with the mutagenic properties observed for O2-POB-dT in E. coli cells.144 However, alkylation at the O4 position only directed dGMP misincorporation opposite the lesion site and was not a strong impediment to DNA replication in E. coli cells.141 Similar as what was observed for O2-Et-dT, Pol V was involved in the replicative bypass of the O2-alkyl-dT lesions and the three TLS polymerases played somewhat redundant roles in bypassing the O4-alkyl-dT lesions. Furthermore, repair of the O4-alkyl-dT lesions was also investigated, and the results showed that Ogt, but not Ada, was more efficient in removing alkyl groups from the O4 position.140,141
DNA epimeric lesions were also investigated, and it was found that all α-dN lesions except α-dA strongly blocked DNA replication and, while replication across α-dA was error-free, replicative bypass of α-dC and α-dG yielded mainly C → A and G → A mutations, respectively.142 Unlike the α-dN lesions, the C3′-epimeric lesions are not strong impediments to DNA replication, where only the C3′-epimer of dA induced a moderate frequency of A → G mutation.143
LC-MS-Based in Vivo Replication Studies in Mammalian Cells
The CRAB assay can also be extended to mammalian cells by replacing the single-stranded bacteriophage plasmid with a double-stranded shuttle vector that is able to propagate in mammalian cells (Figure 8b).145–147 Similar to the assay performed in bacteria, lesion-containing or lesion-free double-stranded plasmid and competitor plasmid are first constructed using a gapped vector-based strategy, which are then allowed to replicate in mammalian cells. In this regard, the undamaged strand in the double-stranded plasmid may be preferentially replicated over the complementary lesion-carrying strand, rendering it difficult to accurately determine the mutation frequencies and bypass efficiencies. Hence, a mismatch is incorporated into the lesion-containing double-stranded plasmid. The progenies of the plasmid are isolated from the host cells, and the residual unreplicated plasmid DNA is removed by DpnI digestion.
The progeny plasmids are subsequently amplified by PCR with a pair of primers flanking the site initially housing the lesion. In this respect, strand-specific PCR (SSPCR) is performed by introducing a mismatch in one of the primers to amplify the region of interest in the progeny genomes arising from the replication of the lesion-carrying strand or the corresponding control strand. The resulting PCR products are restriction digested and subjected to PAGE and LC-MS/MS analyses.
This assay has been employed to investigate alkylated and carboxyalkylated DNA lesions. Consistent with the replication studies in E. coli, O4-alkyl-dT lesions were found to be moderate blockades to DNA replication and only directed T → C transition mutation.145 In addition, it was revealed that Pol η and Pol ζ were important TLS polymerases in bypassing this type of lesions.145 Alkylation at the O6 position of dG, e.g., O6-Me-dG and O6-POB-dG, mispaired with dTMP during replication, leading to G → A mutation,148 which also holds true for O6-CM-dG.146 For other carboxyalkylated lesions, it turned out that neither N6-CM-dA nor N4-CM-dC substantially blocked DNA replication in cells, and replication past these two lesions was also highly accurate. O4-CMdT and N3-CMdT, on the other hand, moderately blocked DNA replication in HEK293T cells, and induced T → C and T → A mutations, respectively.146 In addition, significant decreases in bypass efficiencies were observed for N4-CM-dC and N3-CM-dT in cells depleted of Pol η, for N6-CM-dA and O6-CM-dG in cells lacking Pol κ, and for O6-CM-dG in cells deficient in Pol ζ.146 Intriguingly, carboxyalkylation at the N2 position of dG, i.e., (S)- and (R)-N2-CEdG and N2-CMdG, does not impede replication bypass, and these lesions direct G → A and G → T mutations. Moreover, Pol κ and Pol ι were responsible for error-free bypass of these lesions.149
Apart from single modified nucleobases, the effects of ICLs on DNA replication can also be evaluated with a modified strategy for constructing double-strand shuttle vector.147 It was recently reported that a reduced dG-AP cross-link constituted a strong blockage to DNA replication and multiple TLS polymerases were involved in bypassing the lesion.147 In addition, replication past the cross-linked AP residue in cells was moderately error-prone, whereas replication across the cross-linked dG component occurred at a low mutation frequency.147
LC-MS-Based Transcription Studies
A novel competitive transcription and adduct bypass (CTAB) assay was developed to investigate transcriptional alterations conferred by DNA lesions in vitro and in cells. This assay employs a double-stranded shuttle vector incapable of replicating in mammalian cells, which avoids the complication from the transcriptional bypass of progenies of replication products of damage-containing plasmids (Figure 9).150,151
Figure 9.
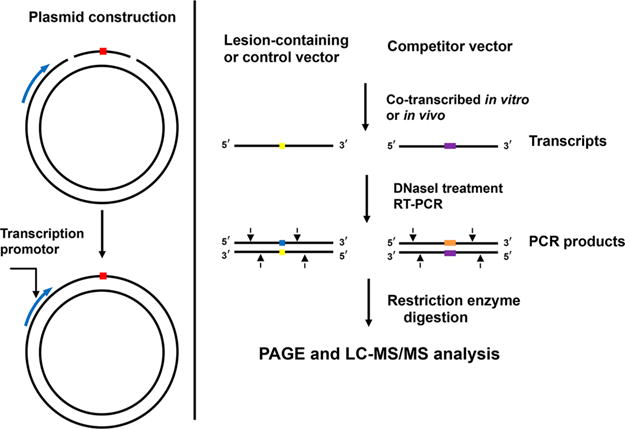
A scheme outlining the experimental procedures of the competitive transcription and adduct bypass assay.
The constructed lesion-containing or lesion-free plasmid is premixed with a competitor plasmid and is used as DNA template for transcription both in vitro and in cells. The resulting transcription products are isolated, and the residual contaminated DNA is removed by DNase treatment. The resulting runoff transcripts of interest are then amplified by RT-PCR, and the RT-PCR products are subsequently digested with appropriate restriction enzymes and subjected to PAGE and LC-MS/MS analyses. The impact of lesions on transcription is characterized by relative bypass efficiency (RBE) and base substitution frequency. It should be noted that, by placing DNA lesions downstream of specific promoter sequences of RNA polymerases, the DNA lesion-induced perturbation of the corresponding RNA polymerase-mediated transcription can be investigated. In addition, by conducting the assay in mammalian cells deficient in repair proteins, the repair mechanism, e.g., global-genome nucleotide excision repair and transcription-coupled nucleotide excision repair, of specific DNA lesion can be revealed.152–154
This method has been recently applied to investigate the transcriptional perturbations elicited by ethylated thymidines and carboxymethylated lesions. Consistent with the replication study in E. coli cells, it was found that O4-Et-dT was highly mutagenic and exclusively induced the misincorporation of guanosine opposite the lesion, whereas N3-Et-dT and O2-Et-dT displayed promiscuous miscoding properties during transcription, and N3-Et-dT and O2-Et-dT were found to strongly inhibit DNA transcription both in vitro and in cells.152 In addition, N3-Et-dT, but not O2-Et-dT or O4-Et-dT, was an efficient substrate for ALKBH2, ALKBH3, and transcription-coupled NER (TC-NER).152,154 For carboxymethylated lesions, N3-CMdT and O4-CMdT were found to substantially inhibit DNA transcription, where they predominantly led to misinsertions of uridine and guanosine, respectively.153 Moreover, these two lesions were found to be substrates for TC-NER but cannot be repaired by ALKBH2 or ALKBH3.154
Application of Next-Generation Sequencing in Replication Studies
NGS technology has become an affordable and reliable method to perform massively parallel sequencing.155,156 When coupled with the aforementioned CRAB assay, NGS enables a multiplexed site-specific mutagenesis assay and facilitates the quantification of insertion and deletion mutations readily (Figure 10).149,157–159 Briefly, individual lesion-containing or lesion-free control shuttle vector, i.e., M13 bacteriophage, is specifically barcoded. These barcoded genomes are all mixed together and transfected into cells with specific repair/replication background. After in vivo replication, the progeny genomes are isolated and the region in the progeny genomes of interest is amplified by PCR. The resulting PCR products are ligated to two paired-end adaptors, and a second PCR is performed to introduce another set of barcodes or tagged using a DNA library preparation kit. Thus, the repair/replication backgrounds and biological replicates can be distinguished by the second sets of barcodes. This prepared DNA library is then subjected to NGS. Mutations induced by lesions can be identified by aligning sequencing reads from each lesion to the expected M13 sequence, thereby facilitating the determination of mutation frequency. The bypass efficiency is determined on the basis of the change in number of sequencing reads from the lesion to that of the lesion-free control.
Figure 10.
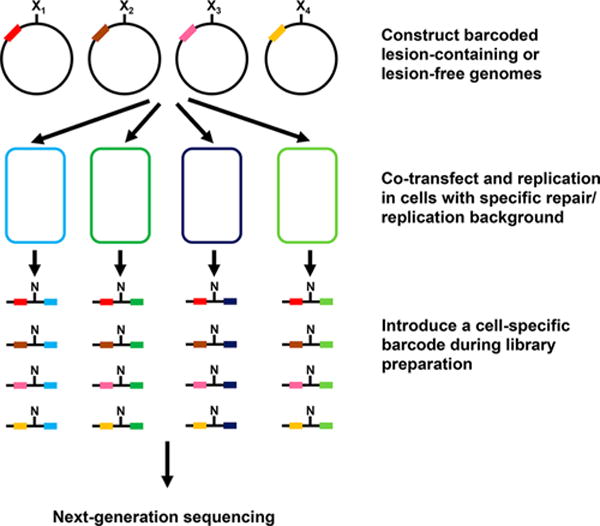
Overall procedures of the in vivo replication assay using next-generation sequencing.
The NGS has been employed for assessing the genotoxicity and cytotoxicity of various DNA adducts or modifications in the past few years. It was found that N4-CM-dC and N6-CM-dA did not block DNA replication in wild-type E. coli cells, whose bypass efficiencies were not affected considerably by TLS polymerases. O4-CM-dT and N3-CM-dT, on the other hand, block appreciably DNA replication in wild-type E. coli cells, and Pol V was involved in the bypass of O4-CM-dT.149 Moreover, cPus were found to be strong impediments to DNA replication, and the bypass of these two lesions also required Pol V.149 The impact of epigenetic marks 5mC and its oxidative derivatives, 5hmC, 5fC, and 5-carboxylcytosine (5caC) on DNA replication were also investigated, and they were found not to block replication and only direct minimal C → T mutation.157
DNA adducts formed from carcinogen exposure have also been studied using NGS. The etheno dG adducts, N2,3-ethenoguanine (N2,3-εG) and 1,N2-ethenoguanine (1,N2-εG), formed from vinyl chloride exposure, were strong replication blocks, and DinB facilitated the mutagenic bypass. In addition, N2,3-εG was found to direct mainly G → A transition mutation and cannot be repaired by AlkB, while mutations induced by 1,N2-εG were promiscuous.158 1,3-Butadiene-induced adenine DNA adducts, reported recently, were found to direct a low level of mutation (<5%) in repair-proficient or deficient cells, though they conferred some inhibitory effect on DNA replication in E. coli cells.159
PERSPECTIVES
In this Review, we summarized the occurrence of a series of DNA lesions, as well as recent studies about the quantification of DNA lesions in tissues and other biological matrices such as blood and urine. The results of these studies suggest that DNA lesions can serve as potential biomarkers for evaluating the exposure toward exogenous and endogenous DNA damaging agents. Moreover, the application of shuttle vector-based replication and transcription assays provided essential information about the repair and biological consequences of DNA adducts in vivo. Comprehensive understanding of the formation, removal, and biological implications of DNA adducts in vivo will facilitate DNA adduct analysis in clinical research, diagnostic applications, and risk assessment.
During the past two decades, the LC-MS/MS coupled with the stable isotope dilution method has evolved to be the gold standard for unequivocal identification and accurate quantification of DNA lesions in biological samples. Further development of the LC-MS-based method for high-throughput, accurate, and efficient quantifications of DNA lesions will still be the top priority of future studies. In this vein, we expect to observe further improvements in sample preparation techniques and increasing application of high-resolution Orbitrap and time-of-flight mass analyzers in the detection of low levels of DNA lesions in cellular and tissue samples.
Future efforts should also be made for the systematic analysis of DNA adductome since some DNA damaging agents can induce a series of DNA lesions rather than limited types of lesions. For example, tobacco smoke contains a variety of reactive chemical species, such as NNN, NNK, aldehydes, PAH, and ROS. The same situation applies to diet-related DNA adducts. Such complex exposure to DNA damaging agents implies the intricate involvement of DNA damage in the etiology of human diseases, where multiple types of lesions may act together to contribute to the disease development. Hence, DNA adductomics studies will expedite the discovery of novel cancer-relevant DNA adducts. In addition, the investigation of tissue- and tumor type-specific adductome will also provide important insights into the molecular mechanisms through which DNA damage induces carcinogenesis and other human diseases. We envision that a standard, comprehensive, and publicly available DNA adductome database based on high resolution MS analysis will significantly facilitate the use of DNA adductome in clinical applications.
While the LC-MS/MS-based DNA adduct analysis provides important quantitative information about the levels of DNA damage, sequencing-based detection methods can reveal the genome-wide distribution of DNA adducts. Currently, these approaches are limited to certain lesions, such as UV light- and cisplatin-induced DNA damage and PAH–DNA adducts. Further development of the DNA adduct sequencing method may enable the future assessment about the genome-wide distributions of other types of DNA lesions. Such information may allow for the revelation of direct correlation, at genome-wide scale, between DNA adduct formation and alterations in genomic DNA and the transcriptome.
Acknowledgments
The authors thank the National Institutes of Health for supporting this research (R01 CA210072 and P01 AG043376).
Biographies
Yang Yu received his B.S. (2009) and M.S. (2012) degrees from Nankai University, China. He obtained his Ph.D. degree in Environmental Toxicology from the University of California Riverside in 2017 under the supervision of Professor Yinsheng Wang. He is currently an Assistant Specialist in Chemistry at the University of California Riverside. His research mainly involves the development of mass spectrometric methods for the quantification of different types of structurally modified DNA and the application of these methods in assessing the biological implications of modified nucleosides in living cells and organisms.
Pengcheng Wang received his B.S. in Pharmacy and M.S. in Microbiological and Biochemical Pharmacy from Wuhan University, China. He earned his Ph.D. in Environmental Toxicology from University of California Riverside in 2016. Currently, he is a postdoctoral fellow at the University of California Riverside, and his research involves the application of LC-MS methods for examining how DNA adducts perturb the efficiency and fidelity of DNA replication in cells.
Yuxiang Cui is a Ph.D. student in the Environmental Toxicology Graduate Program at the University of California, Riverside. She received her B.S. degree in Chemistry at Nankai University and M.S. degree in Food Safety and Toxicology at the University of Hong Kong. Presently, she is working under the guidance of Prof. Yinsheng Wang, focusing on the development and applications of LC-MS methods for the quantitative measurement of DNA lesions.
Yinsheng Wang is a Professor of Chemistry at the University of California Riverside. He obtained his B.S., M.S., and Ph.D. degrees from Shandong University (1993), Dalian Institute of Chemical Physics (1996), and Washington University in St. Louis (2001), respectively. One major emphasis of his research program is placed on the investigation of the occurrence, repair, and biological consequences of DNA damage. The interests of his research group also include epigenetics and proteomics.
Footnotes
Special Issue: Fundamental and Applied Reviews in Analytical Chemistry 2018
ORCID
Yinsheng Wang: 0000-0001-5565-283X
Notes
The authors declare no competing financial interest.
References
- 1.Bauer NC, Corbett AH, Doetsch PW. Nucleic Acids Res. 2015;43:10083–10101. doi: 10.1093/nar/gkv1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee N, Walker GC. Environ Mol Mutag. 2017;58:235–263. doi: 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delaney JC, Essigmann JM. Chem Res Toxicol. 2008;21:232–252. doi: 10.1021/tx700292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.You C, Wang Y. Acc Chem Res. 2016;49:205–213. doi: 10.1021/acs.accounts.5b00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finkel T, Holbrook NJ. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 6.Yu Y, Cui Y, Niedernhofer LJ, Wang Y. Chem Res Toxicol. 2016;29:2008–2039. doi: 10.1021/acs.chemrestox.6b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cadet J, Davies KJA. Free Radical Biol Med. 2017;107:2–12. doi: 10.1016/j.freeradbiomed.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balaban RS, Nemoto S, Finkel T. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Stohs SJ, Bagchi D. Free Radical Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 10.Marnett LJ. Mutat Res, Fundam Mol Mech Mutagen. 1999;424:83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- 11.Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chem Res Toxicol. 2009;22:759–778. doi: 10.1021/tx9000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minko IG, Kozekov ID, Kozekova A, Harris TM, Rizzo CJ, Lloyd RS. Mutat Res, Fundam Mol Mech Mutagen. 2008;637:161–172. doi: 10.1016/j.mrfmmm.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.el Ghissassi F, Barbin A, Nair J, Bartsch H. Chem Res Toxicol. 1995;8:278–283. doi: 10.1021/tx00044a013. [DOI] [PubMed] [Google Scholar]
- 14.Burcham PC. Mutagenesis. 1998;13:287–305. doi: 10.1093/mutage/13.3.287. [DOI] [PubMed] [Google Scholar]
- 15.Chung F-L, Chen H-JC, Nath RG. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. [DOI] [PubMed] [Google Scholar]
- 16.Bartsch H, Nair J, Owen RW. Biol Chem. 2002;383:915–921. doi: 10.1515/BC.2002.098. [DOI] [PubMed] [Google Scholar]
- 17.Drabløs F, Feyzi E, Aas PA, Vaagbø CB, Kavli B, Bratlie MS, Peña-Diaz J, Otterlei M, Slupphaug G, Krokan HE. DNA Repair. 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Fu D, Calvo JA, Samson LD. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Wang Y. Chem Soc Rev. 2015;44:7829–7854. doi: 10.1039/c5cs00316d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tretyakova N, Goggin M, Sangaraju D, Janis G. Chem Res Toxicol. 2012;25:2007–2035. doi: 10.1021/tx3002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tretyakova N, Villalta PW, Kotapati S. Chem Rev. 2013;113:2395–2436. doi: 10.1021/cr300391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shrivastav N, Li D, Essigmann JM. Carcinogenesis. 2010;31:59–70. doi: 10.1093/carcin/bgp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttenplan JB. Mutat Res, Fundam Mol Mech Mutagen. 1990;233:177–187. doi: 10.1016/0027-5107(90)90161-v. [DOI] [PubMed] [Google Scholar]
- 24.Montesano R, Pegg AE, Margison GP. J Toxicol Environ Health. 1980;6:1001–1008. doi: 10.1080/15287398009529922. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Wang Y. Adv Mol Toxicol. 2011;5:219–243. [Google Scholar]
- 26.Shuker DE, Margison GP. Cancer Res. 1997;57:366–369. [PubMed] [Google Scholar]
- 27.Harrison KL, Jukes R, Cooper DP, Shuker DEG. Chem Res Toxicol. 1999;12:106–111. doi: 10.1021/tx980057n. [DOI] [PubMed] [Google Scholar]
- 28.Gottschalg E, Scott GB, Burns PA, Shuker DEG. Carcinogenesis. 2007;28:356–362. doi: 10.1093/carcin/bgl150. [DOI] [PubMed] [Google Scholar]
- 29.Ewa B, Danuta M-Š. J Appl Genet. 2017;58:321. doi: 10.1007/s13353-016-0380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henkler F, Stolpmann K, Luch A. In: Molecular, Clinical and Environmental Toxicology: Vol– 3: Environmental Toxicology. Luch A, editor. Springer; Basel: 2012. pp. 107–131. [Google Scholar]
- 31.Shimada T, Fujii-Kuriyama Y. Cancer Sci. 2004;95:1–6. doi: 10.1111/j.1349-7006.2004.tb03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Pla RC, Bowman KJ, Farmer PB, Jones GDD. Chem Res Toxicol. 2006;19:407–413. doi: 10.1021/tx050298c. [DOI] [PubMed] [Google Scholar]
- 33.Jones GDD, Le Pla RC, Farmer PB. Mutagenesis. 2010;25:3–16. doi: 10.1093/mutage/gep038. [DOI] [PubMed] [Google Scholar]
- 34.Ma B, Villalta PW, Zarth AT, Kotandeniya D, Upadhyaya P, Stepanov I, Hecht SS. Chem Res Toxicol. 2015;28:2151–2159. doi: 10.1021/acs.chemrestox.5b00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawley PD, Phillips DH. Mutat Res, Fundam Mol Mech Mutagen. 1996;355:13–40. doi: 10.1016/0027-5107(96)00020-6. [DOI] [PubMed] [Google Scholar]
- 36.Sanderson BJS, Shield AJ. Mutat Res, Fundam Mol Mech Mutagen. 1996;355:41–57. doi: 10.1016/0027-5107(96)00021-8. [DOI] [PubMed] [Google Scholar]
- 37.Noll DM, Mason TM, Miller PS. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turesky RJ. Drug Metab Rev. 2002;34:625–650. doi: 10.1081/dmr-120005665. [DOI] [PubMed] [Google Scholar]
- 39.Turesky RJ, Le Marchand L. Chem Res Toxicol. 2011;24:1169–1214. doi: 10.1021/tx200135s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng K-W, Chen F, Wang M. Mol Nutr Food Res. 2006;50:1150–1170. doi: 10.1002/mnfr.200600086. [DOI] [PubMed] [Google Scholar]
- 41.Turesky RJ, Vouros P. J Chromatogr B: Anal Technol Biomed Life Sci. 2004;802:155–166. doi: 10.1016/j.jchromb.2003.10.053. [DOI] [PubMed] [Google Scholar]
- 42.Turesky RJ. Mol Nutr Food Res. 2005;49:101–117. doi: 10.1002/mnfr.200400076. [DOI] [PubMed] [Google Scholar]
- 43.Turesky RJ. Toxicol Lett. 2007;168:219–227. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 44.Dudley E, Bond L. Mass Spectrom Rev. 2014;33:302–331. doi: 10.1002/mas.21388. [DOI] [PubMed] [Google Scholar]
- 45.Smith RD, Shen Y, Tang K. Acc Chem Res. 2004;37:269–278. doi: 10.1021/ar0301330. [DOI] [PubMed] [Google Scholar]
- 46.Blue LE, Franklin EG, Godinho JM, Grinias JP, Grinias KM, Lunn DB, Moore SM. J Chromatogr. 2017;1523:17. doi: 10.1016/j.chroma.2017.05.039. [DOI] [PubMed] [Google Scholar]
- 47.Denoroy L, Zimmer L, Renaud B, Parrot S. J Chromatogr B: Anal Technol Biomed Life Sci. 2013;927:37–53. doi: 10.1016/j.jchromb.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 48.Núñez O, Gallart-Ayala H, Martins CPB, Lucci P, Busquets R. J Chromatogr B: Anal Technol Biomed Life Sci. 2013;927:3–21. doi: 10.1016/j.jchromb.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Hou H, Chen H, Liu Y, Wang A, Hu Q. J Chromatogr B: Anal Technol Biomed Life Sci. 2015;1002:123–129. doi: 10.1016/j.jchromb.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 50.Cho S-H, Jung BH, Lee SH, Lee W-Y, Kong G, Chung BC. Biomed Chromatogr. 2006;20:1229–1236. doi: 10.1002/bmc.689. [DOI] [PubMed] [Google Scholar]
- 51.Ma B, Jing M, Villalta PW, Kapphahn RJ, Montezuma SR, Ferrington DA, Stepanov I. Sci Rep. 2016;6:22375. doi: 10.1038/srep22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torres-Cuevas I, Aupi M, Asensi MA, Vento M, Ortega Á, Escobar J. Talanta. 2017;170:97–102. doi: 10.1016/j.talanta.2017.03.072. [DOI] [PubMed] [Google Scholar]
- 53.Guo C, Li X, Ye M, Xu F, Yu J, Xie C, Cao X, Guo M, Yuan Y, Zheng S. Oncotarget. 2017;8:53100–53109. doi: 10.18632/oncotarget.17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yin J, Xu T, Zhang N, Wang H. Anal Chem. 2016;88:7730–7737. doi: 10.1021/acs.analchem.6b01682. [DOI] [PubMed] [Google Scholar]
- 55.Yu Y, Guerrero CR, Liu S, Amato NJ, Sharma Y, Gupta S, Wang Y. Mol Cell Proteomics. 2016;15:810–817. doi: 10.1074/mcp.M115.052696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krokidis MG, Terzidis MA, Efthimiadou E, Zervou S-K, Kordas G, Papadopoulos K, Hiskia A, Kletsas D, Chatgilialoglu C. Free Radical Res. 2017;51:470–482. doi: 10.1080/10715762.2017.1325485. [DOI] [PubMed] [Google Scholar]
- 57.Hemeryck LY, Decloedt AI, Vanden Bussche J, Geboes KP, Vanhaecke L. Anal Chim Acta. 2015;892:123–131. doi: 10.1016/j.aca.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 58.Zhang F, Bartels MJ, LeBaron MJ, Schisler MR, Jeong Y-C, Gollapudi BB, Moore NP. J Chromatogr B: Anal Technol Biomed Life Sci. 2015;976-977:33–48. doi: 10.1016/j.jchromb.2014.10.042. [DOI] [PubMed] [Google Scholar]
- 59.Li X, Liu L, Wang H, Chen J, Zhu B, Chen H, Hou H, Hu Q. J Chromatogr B: Anal Technol Biomed Life Sci. 2017;1060:451–459. doi: 10.1016/j.jchromb.2017.06.031. [DOI] [PubMed] [Google Scholar]
- 60.Mutlu E, Gao L, Collins LB, Walker NJ, Hartwell HJ, Olson JR, Sun W, Gold A, Ball LM, Swenberg JA. Chem Res Toxicol. 2016;29:1335–1344. doi: 10.1021/acs.chemrestox.6b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Cui S, Wang S, Jiang X, Zhang S, Zhang R, Fu PP, Sun X. Free Radical Res. 2015;49:1049–1054. doi: 10.3109/10715762.2015.1006213. [DOI] [PubMed] [Google Scholar]
- 62.Zhang N, Song Y, Wu D, Xu T, Lu M, Zhang W, Wang H. J Chromatogr. 2016;1450:38–44. doi: 10.1016/j.chroma.2016.04.067. [DOI] [PubMed] [Google Scholar]
- 63.Zhang N, Song Y, Zhang W, Wang H. J Chromatogr B: Anal Technol Biomed Life Sci. 2016;1023-1024:68–71. doi: 10.1016/j.jchromb.2016.04.029. [DOI] [PubMed] [Google Scholar]
- 64.Ma F, Zhang Z, Jiang J, Hu J. Chemosphere. 2015;139:256–259. doi: 10.1016/j.chemosphere.2015.06.077. [DOI] [PubMed] [Google Scholar]
- 65.Limón-Pacheco J, Gonsebatt ME. Mutat Res, Genet Toxicol Environ Mutagen. 2009;674:137–147. doi: 10.1016/j.mrgentox.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 66.Yu Y, Wang J, Wang P, Wang Y. Anal Chem. 2016;88:8036–8042. doi: 10.1021/acs.analchem.6b01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sangaraju D, Boldry EJ, Patel YM, Walker V, Stepanov I, Stram D, Hatsukami D, Tretyakova N. Chem Res Toxicol. 2017;30:678–688. doi: 10.1021/acs.chemrestox.6b00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaramillo R, Shuck SC, Chan YS, Liu X, Bates SE, Lim PP, Tamae D, Lacoste S, O’Connor TR, Termini J. Chem Res Toxicol. 2017;30:689–698. doi: 10.1021/acs.chemrestox.6b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan B, Cao H, Jiang Y, Hong H, Wang Y. Proc Natl Acad Sci U S A. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hecht SS. Chem Res Toxicol. 2017;30:367–375. doi: 10.1021/acs.chemrestox.6b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang J, Villalta PW, Upadhyaya P, Hecht SS. Chem Res Toxicol. 2016;29:87–95. doi: 10.1021/acs.chemrestox.5b00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zarth AT, Upadhyaya P, Yang J, Hecht SS. Chem Res Toxicol. 2016;29:380–389. doi: 10.1021/acs.chemrestox.5b00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma B, Ruszczak C, Jain V, Khariwala SS, Lindgren B, Hatsukami DK, Stepanov I. Chem Res Toxicol. 2016;29:1849–1856. doi: 10.1021/acs.chemrestox.6b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michel AK, Zarth AT, Upadhyaya P, Hecht SS. ACS Omega. 2017;2:1180–1190. doi: 10.1021/acsomega.7b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P, Wang M. Chem Res Toxicol. 2004;17:588–597. doi: 10.1021/tx034263t. [DOI] [PubMed] [Google Scholar]
- 76.Leng J, Wang Y. Anal Chem. 2017;89:9124–9130. doi: 10.1021/acs.analchem.7b01857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun Y-W, El-Bayoumy K, Aliaga C, Awad AS, Gowda K, Amin S, Chen K-M. Chem Res Toxicol. 2015;28:1427–1433. doi: 10.1021/acs.chemrestox.5b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klaene JJ, Flarakos C, Glick J, Barret JT, Zarbl H, Vouros P. J Chromatogr. 2016;1439:112–123. doi: 10.1016/j.chroma.2015.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo J, Yun BH, Upadhyaya P, Yao L, Krishnamachari S, Rosenquist TA, Grollman AP, Turesky RJ. Anal Chem. 2016;88:4780–4787. doi: 10.1021/acs.analchem.6b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yao C, Foster WG, Sadeu JC, Siddique S, Zhu J, Feng Y-L. Sci Total Environ. 2017;575:742–749. doi: 10.1016/j.scitotenv.2016.09.122. [DOI] [PubMed] [Google Scholar]
- 81.Corte-Rodríguez M, Espina M, Sierra LM, Blanco E, Ames T, Montes-Bayón M, Sanz-Medel A. Biochem Pharmacol. 2015;98:69–77. doi: 10.1016/j.bcp.2015.08.112. [DOI] [PubMed] [Google Scholar]
- 82.Ming X, Groehler A, Michaelson-Richie ED, Villalta PW, Campbell C, Tretyakova NY. Chem Res Toxicol. 2017;30:980–995. doi: 10.1021/acs.chemrestox.6b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stornetta A, Villalta PW, Hecht SS, Sturla SJ, Balbo S. Anal Chem. 2015;87:11706–11713. doi: 10.1021/acs.analchem.5b02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gruppi F, Hejazi L, Christov PP, Krishnamachari S, Turesky RJ, Rizzo CJ. Chem Res Toxicol. 2015;28:1850–1860. doi: 10.1021/acs.chemrestox.5b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang P, Zhang Y, Chen J, Guo L, Xu B, Wang L, Xu H, Xie J. Chem Res Toxicol. 2015;28:1224–1233. doi: 10.1021/acs.chemrestox.5b00055. [DOI] [PubMed] [Google Scholar]
- 86.Groehler A, Villalta PW, Campbell C, Tretyakova N. Chem Res Toxicol. 2016;29:190–202. doi: 10.1021/acs.chemrestox.5b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Høie AH, Monien BH, Sakhi AK, Glatt H, Hjertholm H, Husøy T. Mutagenesis. 2015;30:643–649. doi: 10.1093/mutage/gev023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turesky RJ, Yun BH, Brennan P, Mates D, Jinga V, Harnden P, Banks RE, Blanche H, Bihoreau M-T, Chopard P, Letourneau L, Lathrop GM, Scelo G. Br J Cancer. 2016;114:76–80. doi: 10.1038/bjc.2015.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu L, Xue J, Xia Q, Fu PP, Lin G. Arch Toxicol. 2017;91:949–965. doi: 10.1007/s00204-016-1713-z. [DOI] [PubMed] [Google Scholar]
- 90.Fu PP, Xia Q, He X, Barel S, Edery N, Beland FA, Shimshoni JA. Chem Res Toxicol. 2017;30:851–858. doi: 10.1021/acs.chemrestox.6b00456. [DOI] [PubMed] [Google Scholar]
- 91.Turesky RJ, Konorev D, Fan X, Tang Y, Yao L, Ding X, Xie F, Zhu Y, Zhang Q-Y. Chem Res Toxicol. 2015;28:2400–2410. doi: 10.1021/acs.chemrestox.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim S, Guo J, O’Sullivan MG, Gallaher DD, Turesky RJ. Environ Mol Mutag. 2016;57:125–136. doi: 10.1002/em.21993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pathak KV, Bellamri M, Wang Y, Langouët S, Turesky RJ. J Biol Chem. 2015;290:16304. doi: 10.1074/jbc.M115.646539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cai T, Bellamri M, Ming X, Koh W-P, Yu MC, Turesky RJ. Chem Res Toxicol. 2017;30:1333–1343. doi: 10.1021/acs.chemrestox.7b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xiao S, Guo J, Yun BH, Villalta PW, Krishna S, Tejpaul R, Murugan P, Weight CJ, Turesky RJ. Anal Chem. 2016;88:12508–12515. doi: 10.1021/acs.analchem.6b04157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Singh R, Arlt VM, Henderson CJ, Phillips DH, Farmer PB, Costa GGd. J Chromatogr B: Anal Technol Biomed Life Sci. 2010;878:2155–2162. doi: 10.1016/j.jchromb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Krais AM, Speksnijder EN, Melis JPM, Singh R, Caldwell A, Gamboa da Costa G, Luijten M, Phillips DH, Arlt VM. Int J Cancer. 2016;138:976–982. doi: 10.1002/ijc.29836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Høie AH, Monien BH, Glatt H, Hjertholm H, Husøy T. Toxicol Lett. 2016;248:34–38. doi: 10.1016/j.toxlet.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 99.Kim JK, Gallaher DD, Chen C, Gallaher CM, Yao D, Trudo SP. Mol Nutr Food Res. 2016;60:1956–1966. doi: 10.1002/mnfr.201500790. [DOI] [PubMed] [Google Scholar]
- 100.Kim JK, Gallaher DD, Chen C, Yao D, Trudo SP. J Nutr. 2015;145:442–451. doi: 10.3945/jn.114.202622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bessette EE, Spivack SD, Goodenough AK, Wang T, Pinto S, Kadlubar FF, Turesky RJ. Chem Res Toxicol. 2010;23:1234–1244. doi: 10.1021/tx100098f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gu D, Turesky RJ, Tao Y, Langouët SA, Nauwelaërs GC, Yuan J-M, Yee D, Yu MC. Carcinogenesis. 2012;33:124–130. doi: 10.1093/carcin/bgr252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Balbo S, Turesky RJ, Villalta PW. Chem Res Toxicol. 2014;27:356–366. doi: 10.1021/tx4004352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hemeryck LY, Moore SA, Vanhaecke L. Anal Chem. 2016;88:7436–7446. doi: 10.1021/acs.analchem.6b00863. [DOI] [PubMed] [Google Scholar]
- 105.Kanaly RA, Micheletto R, Matsuda T, Utsuno Y, Ozeki Y, Hamamura N. MicrobiologyOpen. 2015;4:841–856. doi: 10.1002/mbo3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yao C, Feng Y-L. Talanta. 2016;159:93–102. doi: 10.1016/j.talanta.2016.05.074. [DOI] [PubMed] [Google Scholar]
- 107.Hemeryck LY, Van Hecke T, Vossen E, De Smet S, Vanhaecke L. Food Chem. 2017;230:378–387. doi: 10.1016/j.foodchem.2017.02.129. [DOI] [PubMed] [Google Scholar]
- 108.Fleming AM, Ding Y, Burrows CJ. Proc Natl Acad Sci U S A. 2017;114:2604–2609. doi: 10.1073/pnas.1619809114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ding Y, Fleming AM, Burrows CJ. J Am Chem Soc. 2017;139:2569–2572. doi: 10.1021/jacs.6b12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu JC, Adar S, Selby CP, Lieb JD, Sancar A. Genes Dev. 2015;29:948–960. doi: 10.1101/gad.261271.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Adar S, Hu JC, Lieb JD, Sancar A. Proc Natl Acad Sci U S A. 2016;113:E2124–E2133. doi: 10.1073/pnas.1603388113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li WT, Hu JC, Adebali O, Adar S, Yang YY, Chiou YY, Sancar A. Proc Natl Acad Sci U S A. 2017;114:6752–6757. doi: 10.1073/pnas.1706021114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hu JC, Adebali O, Adar S, Sancar A. Proc Natl Acad Sci U S A. 2017;114:6758–6763. doi: 10.1073/pnas.1706522114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu JC, Lieb JD, Sancar A, Adar S. Proc Natl Acad Sci U S A. 2016;113:11507–11512. doi: 10.1073/pnas.1614430113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mao P, Smerdon MJ, Roberts SA, Wyrick JJ. Proc Natl Acad Sci U S A. 2016;113:9057–9062. doi: 10.1073/pnas.1606667113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li MY, Ko TY, Li SS. J Biol Chem. 2015;290:23148–23161. doi: 10.1074/jbc.M115.676296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shu XT, Xiong XS, Song JH, He C, Yi CQ. Angew Chem, Int Ed. 2016;55:14246–14249. doi: 10.1002/anie.201607380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu L, Li YH, Li SL, Hu N, He YM, Pong R, Lin DN, Lu LH, Law M. J Biomed Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Riedl J, Ding Y, Fleming AM, Burrows CJ. Nat Commun. 2015;6:8807. doi: 10.1038/ncomms9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Riedl J, Fleming AM, Burrows CJ. J Am Chem Soc. 2016;138:491–494. doi: 10.1021/jacs.5b11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu L, Li YR, Li T, Xie JI, Chen CF, Liu QS, Zhang SW, Wu HC. Anal Chem. 2016;88:1073–1077. doi: 10.1021/acs.analchem.5b04102. [DOI] [PubMed] [Google Scholar]
- 122.Li CP, Wang HL. J Chromatogr. 2015;1406:324–330. doi: 10.1016/j.chroma.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 123.Lee J, Kim Y, Lim S, Jo K. Analyst. 2016;141:847–852. doi: 10.1039/c5an01875g. [DOI] [PubMed] [Google Scholar]
- 124.Guthrie JW, Limmer RT, Brooks EA, Wisnewski CC, Loggins-Davis ND, Bouzid A. Anal Chim Acta. 2015;853:676–681. doi: 10.1016/j.aca.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 125.Kudr J, Richtera L, Xhaxhiu K, Hynek D, Heger Z, Zitka O, Adam V. Biosens Bioelectron. 2017;92:133–139. doi: 10.1016/j.bios.2017.01.067. [DOI] [PubMed] [Google Scholar]
- 126.Trantakis IA, Nilforoushan A, Dahlmann HA, Stauble CK, Sturla SJ. J Am Chem Soc. 2016;138:8497–8504. doi: 10.1021/jacs.6b03599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhong SW, Li Z, Jiang T, Li XJ, Wang HL. Anal Chem. 2017;89:5702–5707. doi: 10.1021/acs.analchem.7b01428. [DOI] [PubMed] [Google Scholar]
- 128.Ma SX, Sun HP, Li Y, Qi HL, Zheng JB. Anal Chem. 2016;88:9934–9940. doi: 10.1021/acs.analchem.6b01265. [DOI] [PubMed] [Google Scholar]
- 129.Evison BJ, Actis ML, Fujii N. Bioorg Med Chem. 2016;24:1071–1078. doi: 10.1016/j.bmc.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Condie AG, Yan Y, Gerson SL, Wang YM. PLoS One. 2015;10:e0131330. doi: 10.1371/journal.pone.0131330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pan D, Zhou Q, Rong SZ, Zhang GT, Zhang YN, Liu FH, Li MJ, Chang D, Pan HZ. Microchim Acta. 2016;183:361–368. [Google Scholar]
- 132.Martins GV, Marques AC, Fortunato E, Sales MGF. Biosens Bioelectron. 2016;86:225–234. doi: 10.1016/j.bios.2016.06.052. [DOI] [PubMed] [Google Scholar]
- 133.Fan JH, Liu YJ, Xu ES, Zhang YJ, Wei W, Yin LH, Pu YP, Liu SQ. Anal Chim Acta. 2016;946:48–55. doi: 10.1016/j.aca.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 134.Song BY, Shen M, Jiang D, Malla S, Mosa IM, Choudhary D, Rusling JF. Analyst. 2016;141:5722–5729. doi: 10.1039/c6an01237j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bregeon D, Doetsch PW. Nat Rev Cancer. 2011;11:218–227. doi: 10.1038/nrc3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ziv O, Diamant N, Shachar S, Hendel A, Livneh Z. In: DNA Repair Protocols. Bjergbæk L, editor. Humana Press; Totowa, NJ: 2012. pp. 529–542. [Google Scholar]
- 137.Livingston AL, O’Shea VL, Kim T, Kool ET, David SS. Nat Chem Biol. 2008;4:51–58. doi: 10.1038/nchembio.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Delaney JC, Essigmann JM. Methods Enzymol. 2006;408:1–15. doi: 10.1016/S0076-6879(06)08001-3. [DOI] [PubMed] [Google Scholar]
- 139.Zhai Q, Wang P, Wang Y. Carcinogenesis. 2014;35:2002–2006. doi: 10.1093/carcin/bgu085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhai Q, Wang P, Cai Q, Wang Y. Nucleic Acids Res. 2014;42:10529–10537. doi: 10.1093/nar/gku748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang PC, Amato NJ, Zhai QQ, Wang YS. Nucleic Acids Res. 2015;43:10795–10803. doi: 10.1093/nar/gkv941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Amato NJ, Zhai QQ, Navarro DC, Niedernhofer LJ, Wang YS. Nucleic Acids Res. 2015;43:8314–8324. doi: 10.1093/nar/gkv725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang PC, Amato NJ, Wang YS. Biochemistry. 2017;56:3725–3732. doi: 10.1021/acs.biochem.7b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jasti VP, Spratt TE, Basu AK. Chem Res Toxicol. 2011;24:1833–1835. doi: 10.1021/tx200435d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu J, Li L, Wang P, You C, Williams NL, Wang Y. Nucleic Acids Res. 2016;44:9256–9265. doi: 10.1093/nar/gkw662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wu J, Wang P, Li L, Williams NL, Ji D, Zahurancik WJ, You C, Wang J, Suo Z, Wang Y. Nucleic Acids Res. 2017;45:7276–7284. doi: 10.1093/nar/gkx442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Price NE, Li L, Gates KS, Wang YS. Nucleic Acids Res. 2017;45:6486–6493. doi: 10.1093/nar/gkx266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pauly GT, Peterson LA, Moschel RC. Chem Res Toxicol. 2002;15:165–169. doi: 10.1021/tx0101245. [DOI] [PubMed] [Google Scholar]
- 149.Yuan BF, Wang JS, Cao HC, Sun RB, Wang YS. Nucleic Acids Res. 2011;39:5945–5954. doi: 10.1093/nar/gkr159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.You CJ, Wang YS. Nat Protoc. 2015;10:1389–1406. doi: 10.1038/nprot.2015.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.You C, Dai X, Yuan B, Wang J, Wang J, Brooks PJ, Niedernhofer LJ, Wang Y. Nat Chem Biol. 2012;8:817–822. doi: 10.1038/nchembio.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.You CJ, Wang PC, Dai XX, Wang YS. Nucleic Acids Res. 2014;42:13706–13713. doi: 10.1093/nar/gku1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.You CJ, Wang JS, Dai XX, Wang YS. Nucleic Acids Res. 2015;43:1012–1018. doi: 10.1093/nar/gku1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.You C, Wang P, Nay SL, Wang J, Dai X, O’Connor TR, Wang Y. ACS Chem Biol. 2016;11:1332–1338. doi: 10.1021/acschembio.6b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Herzyk P. Handbook of Pharmacogenomics and Stratified Medicine. Academic Press; San Diego: 2014. pp. 125–145. [Google Scholar]
- 156.Pareek CS, Smoczynski R, Tretyn A. J Appl Genet. 2011;52:413–435. doi: 10.1007/s13353-011-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xing XW, Liu YL, Vargas M, Wang YS, Feng YQ, Zhou X, Yuan BF. PLoS One. 2013;8:e72993. doi: 10.1371/journal.pone.0072993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chang SC, Fedeles BI, Wu J, Delaney JC, Li DY, Zhao LL, Christov PP, Yau E, Singh V, Jost M, Drennan CL, Marnett LJ, Rizzo CJ, Levine SS, Guengerich FP, Essigmann JM. Nucleic Acids Res. 2015;43:5489–5500. doi: 10.1093/nar/gkv243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chang SC, Seneviratne UI, Wu J, Tretyakova N, Essigmann JM. Chem Res Toxicol. 2017;30:1230–1239. doi: 10.1021/acs.chemrestox.7b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]