

Abstract
Diabetes mellitus contributes greatly to morbidity, mortality, and overall health care costs. In major part, these outcomes derive from the high incidence of progressive kidney dysfunction in patients with diabetes making diabetic nephropathy a leading cause of end-stage renal disease. A better understanding of the molecular mechanism involved and of the early dysfunctions observed in the diabetic kidney may permit the development of new strategies to prevent diabetic nephropathy. Here we review the pathophysiological changes that occur in the kidney in response to hyperglycemia, including the cellular responses to high glucose and the responses in vascular, glomerular, podocyte, and tubular function. The molecular basis, characteristics, and consequences of the unique growth phenotypes observed in the diabetic kidney, including glomerular structures and tubular segments, are outlined. We delineate mechanisms of early diabetic glomerular hyperfiltration including primary vascular events as well as the primary role of tubular growth, hyperreabsorption, and tubuloglomerular communication as part of a “tubulocentric” concept of early diabetic kidney function. The latter also explains the “salt paradox” of the diabetic kidney, that is, a unique and inverse relationship between glomerular filtration rate and dietary salt intake. The mechanisms and consequences of the intrarenal activation of the renin-angiotensin system and of diabetes-induced tubular glycogen accumulation are discussed. Moreover, we aim to link the changes that occur early in the diabetic kidney including the growth phenotype, oxidative stress, hypoxia, and formation of advanced glycation end products to mechanisms involved in progressive kidney disease.
Introduction
Diabetes is the major cause of end-stage renal disease in the United States and elsewhere, and its incidence has increased by about 50% in the past 10 years (644). Notably, only about 20% of individuals with either type 1 or type 2 diabetes (T1DM; T2DM) actually develop nephropathy, indicating that specific genetic and/or environmental factors contribute to its initiation and progression. In fact, family-based studies including genome-wide scans suggest that a significant genetic component confers risk for diabetic nephropathy (73, 265, 266, 336, 553, 567). Moreover, diabetes-induced end-stage renal disease appears to be more likely to be present in African Americans (adjusted odds ratio 1.9), Hispanics (1.4), Asians (1.8), and Native Americans (1.9) than Caucasians (733). The pathogenesis of diabetic nephropathy is still incompletely understood and we do not know which genes are critically involved and also can not predict which individual diabetic patient will eventually develop end-stage renal disease. It is urgent to better understand the genes and events that lead from the onset of diabetes to impairment of renal function with the goal of identifying the patients at risk and achieving effective prevention. To prevent diabetic nephropathy, it may prove more reasonable and effective to identify and understand more completely the very early molecular events that initiate the progressive disease.
High-glucose concentrations induce specific cellular effects, which in the kidney affect many types of cells including endothelial cells, smooth muscle cells, mesangial cells, podocytes, cells of the tubular and collecting duct system and inflammatory cells and myofibroblasts. A better understanding of the molecular mechanisms underlying these effects is expected to be crucial to better understand the disease. In this regard, it will be important to identify effects that are not only observed in vitro in cultured cells but that are observed in the intact organism and are functionally relevant.
The hemodynamic phenotype in early diabetes is characterized by glomerular hyperfiltration which has been associated with progressive diabetic nephropathy (415), although this is still a matter of debate (see below for further discussion). Glomerular hyperfiltration has been attributed to abnormalities of the glomerulus and preglomerular vessels (447), which are related to changes in the metabolic milieu, vasoactive factors, alterations in signal transduction, as well as intrinsic defects in glomerular arterioles including electromechanical coupling. Albuminuria and proteinuria indicate relevant tissue damage in the diabetic kidney and, besides changes in renal hemodynamics, have been linked to specific alterations in podocyte function. Another notable phenotype of the early diabetic kidney is that it grows. This “growth” phenotype is characterized by enlargement of the kidney through both hyperplasia and hypertrophy which begin at the very onset of diabetes (512). The proximal tubule accounts for most of the cortical mass to begin with, and the proximal tubule also accounts for the greatest share of growth in diabetes (147, 571). As the tubule grows, more of the glomerular filtrate is reabsorbed and less reaches the macula densa (MD) at the end of Henle’s loop. This causes the glomerular filtration rate (GFR) to increase through the normal physiologic action of the tubuloglomerular feedback (TGF) system (648). As a consequence of hyperfiltration and the diabetic milieu, the glomeruli filter increased amounts of proteins, growth factors, and advanced glycation end products (AGEs). The diabetic milieu and the prolonged interaction of these proteins and factors with the tubular system trigger renal oxidative stress and cortical interstitial inflammation (2, 3), with the resulting hypoxia and tubulointerstitial fibrosis determining to a great extent the progression of renal disease (33, 65, 126, 154, 198, 386, 396, 581, 606). Moreover, the unique molecular mechanisms involved in the early growth phenotype of the diabetic kidney may contribute to set the stage for long-term kidney failure.
This review addresses these aspects in detail and aims to link changes that occur early in the diabetic kidney to progressive kidney disease. Most of the evidence currently available on these issues has been derived from patients and experimental models with T1DM. Fewer data have been acquired on the early renal pathophysiology in T2DM. While for many aspects similar principles may apply, differences in circulating insulin levels (and C-peptide) and comorbidities are expected to modify the phenotypes in T1DM versus T2DM.
General Mechanisms of Glucose-Induced Cell Injury
The diabetic milieu affects most renal cell types and compartments, although the effects and consequences of high glucose and other components of the diabetic milieu may be specific in individual renal cells. Some cells may be more susceptible to high glucose-induced injury than others. As suggested by Brownlee (86), the cell susceptibility to glucose-induced toxicity is determined by its expression of glucose transporters that mediate cellular uptake of glucose from the extracellular compartment, allowing deleterious increases in intracellular glucose concentrations. Thus, the cell populations most susceptible to the changing milieu of diabetes are those that are unable to sufficiently downregulate glucose uptake and prevent high intracellular glucose levels in the setting of hyperglycemia. In this regard, both mesangial cells (228) and proximal tubular cells (418) are unable to decrease glucose transport rates adequately to prevent excessive changes in intracellular glucose when exposed to high glucose concentrations.
The following sections describe general mechanisms of glucose-induced cell injury that have been implicated in functional and structural changes observed in the diabetic kidney. These mechanisms often act upstream from a variety of processes discussed in the later sections dedicated to specific renal cells, compartments, and functions.
Generation and effects of advanced glycation end products
Prolonged hyperglycemia, but also dyslipidemia and oxidative stress in diabetes result in the production and accumulation of AGEs in the kidney, and at other sites of diabetic complications (86, 92, 591, 612). AGEs are formed by nonenzymatic Maillard or “browning” reaction between carbonyl groups of reducing sugars, like glucose, and amino groups on proteins, lipids, or nucleic acids at a rate determined by a number of factors including intracellular glucose concentrations, pH, and time. The first stable adduct between glucose and protein is fructose-lysine or the Amadori product (see Fig. 1). In addition, the glycation of intracellular proteins is initiated by the elevation of intracellular glucose degradation products (such as glyoxal resulting from glycolysis and the tricarboxylic acid cycle), which occurs more rapidly than with glucose itself. These AGEs can be generated from intra-cellular auto-oxidation of glucose to glyoxal, decomposition of early glycation (Amadori) products to 3-deoxyglucosone, and fragmentation of metabolites of the pentose phosphate pathway such as glyceraldehyde-3-phosphate and dihydroxyacetone phosphate to the reactive carbonyl methylglyoxal (13). Subsequent rearrangement and fragmentation reactions lead to the formation of AGEs. Under physiological conditions, these reactions are slow. However, in diabetes, persistent hyperglycemia, dyslipidemia, and oxidative stress all act to hasten the formation of AGEs (86, 87) and modification of both long-lived as well as short-lived proteins (86).
Figure 1.

Simplified biochemistry of advanced glycation end-product formation. Prolonged hyperglycemia, but also dyslipidemia and oxidative stress in diabetes result in the production and accumulation of advanced glycation end products (AGEs) in the kidney, and at other sites of diabetic complications. AGEs are formed by nonenzymatic Maillard or “browning” reaction between carbonyl groups of reducing sugars, like glucose, and amino groups on proteins, lipids, or nucleic acids. The first stable adduct between glucose and protein is the Amadori product. In addition, the glycation of intracellular proteins is initiated by the elevation of intracellular glucose degradation products or decomposition of early glycation products leading to formation of intermediates such as glyoxal or methylglyoxal. Subsequent rearrangement and fragmentation reactions lead to the formation of AGEs. Under physiological conditions these reactions are slow. However, in diabetes, persistent hyperglycemia, dyslipidemia, and oxidative stress all act to hasten the formation of AGEs.
AGEs are a chemically heterogeneous group of compounds. The biochemistry of AGEs is remarkably complex and beyond the scope of this article [see (13, 614) for further details]. In brief, Ne-(carboxymethyl)lysine is the simplest and best characterized AGE, derived predominantly from the carbonyl modification of lysine. Tissue Ne-(carboxymethyl)lysine concentrations are increased in diabetes, and elevated levels are associated with the presence of vascular complications in patients with diabetes (47). Other more complex AGEs form “cross-links” both between and within modified proteins, such as pentosidine, MOLD (methylglyoxal lysine dimer) and GOLD (glyoxal lysine dimer). Once AGEs are formed, they are nearly irreversible, although enzymes, such as glyoxalase-1, have the ability to detoxify AGE precursors and inhibit AGE production (579). AGEs induce cell injury and contribute to diabetic complications by changing protein structure and function, such as the formation of cross-links between key molecules of basement membranes and the extracellular matrix (ECM) proteins (see Fig. 2). Importantly, AGEs also interact with a receptor (RAGE) on the cell surface thereby altering cellular function (see below).
Figure 2.

Consequences of accelerated formation of advanced glycation end products (AGEs) in the diabetic kidney. The schematic shows major mechanisms whereby AGEs exert their harmful effects in the diabetic kidney. AGEs typically crosslink and alter protein structure and function in extracellular compartment, modify cytosolic molecules and exert receptor-mediated effects that lead to activation of multiple signaling pathways and genes implicated in a variety of pathophysiological mechanisms in the diabetic kidney. RAGE, receptor for AGEs; ROS, reactive oxygen species; MAPKs, mitogen-activated protein kinases; ROCKs, Rho kinases; NO, nitric oxide; BM, basement membrane; ECM, extracellular matrix; NF-κB, nuclear factor kappa B.
Formation of AGEs in the ECM occurs on proteins with a slow turnover rate. Accumulation of AGEs on large matrix proteins such as collagens, fibronectin, and laminin through AGE-AGE intermolecular covalent bonds or cross linking increases collagen stiffness, reduces thermal stability, and causes resistance to digestion by metalloproteinases (MMPs) [reviewed in (614)]. In the kidney, this phenomenon is manifested by expansion of mesangial ECM (580), one of the hallmarks of diabetic nephropathy. In the vascular system, the process results in increased stiffness of the vasculature (300). The physiological relevance of these mechanisms has been validated in experimental studies showing nephroprotective effects of inhibitors of AGE formation (155, 306, 592) or by applying “cross-link breakers,” compounds that dissolve the above-mentioned cross-links between the ECM molecules in experimental diabetic nephropathy (178).
Glycation of extracellular lipids, as evidenced by the increased lipid-linked AGEs in low-density lipoproteins (LDL) samples from persons with diabetes, may also play a role in pathophysiology. For example, glycated LDL can contribute to endothelial dysfunction by reducing nitric oxide (NO) production and suppressing uptake and clearance of LDL through its receptor on endothelial cells (502).
Receptor-mediated effects of advanced glycation end products and their consequences
Several different receptors for AGEs have been discovered. The best studied AGE receptor, termed RAGE, initiates the intracellular signaling mechanisms that impair cellular function following the binding of AGEs (see Fig. 2). RAGE is a member of the immunoglobulin superfamily of receptors involved in inflammatory response (556). Upregulation of RAGE in diabetes occurs in the kidney, vasculature, and mononuclear phagocytes (557, 591). RAGE ligands include AGEs of at least two varieties: Ne-(carboxymethyl)lysine adducts (313), and hydroimidazolones derived from methylglyoxal and 3-deoxyglucosone. AGE-RAGE interaction stimulates multiple signals and activates important players in the pathophysiology of nephropathy, such as NAD(P)H oxidase, p21 ras, the mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase ERK 1/2 and p38, Src, and the GTPases Cdc42 and Rac, resulting in activation and translocation of nuclear transcription factors, including NF-κB (57,58, 515). Target genes of NF-κB include genes for endothelin-1 (ET-1), vascular cell adhesion molecule-1 (VCAM-1), inter-cellular adhesion molecule-1 (ICAM-1), tissue factor, thrombomodulin, vascular endothelial growth factor (VEGF), and likely, proinflammatory cytokines, including interleukins 1 and 6 (IL-1, IL-6), and tumor necrosis factor-α (TNF-α), as well as RAGE itself (57, 313, 515). Moreover, RAGE has been implicated in diabetes-induced mitochondrial superoxide formation in renal cells (123). Blockade of RAGE with anti-RAGE IgG, soluble RAGE (sRAGE), the extracellular ligand that mediates the clearance of AGEs and RAGE knockout all inhibit NF-κB activation, the production of mitochondrial superoxide, and protect the kidney against deleterious effects of the diabetic milieu in experimental settings (57,58, 123, 515). More recent studies in mice indicated that blockade of RAGE may exert its renoprotective effects in the diabetic kidney in part via induction of the angiotensin (Ang) II type 2 (AT2) receptor (593).
Activation of the hexosamine pathway and consequences
Under normoglycemic conditions, only 3% of total glucose is utilized via the hexosamine pathway. However, when the intracellular concentrations of glucose are high, fructose-6-phosphate, an intermediate in the glycolytic pathway, is partially diverted into the hexosamine pathway (see Fig. 3). Entry into the hexosamine pathway is catalyzed by the first and rate-limiting enzyme glutamine:fructose 6-phosphate (F-6-P) amidotransferase (GFAT), which converts F-6-P and glutamine to glucosamine 6-phosphate (GlcN-6-P) and glutamate. Subsequent steps metabolize GlcN-6-P to UDP-N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is a molecule of particular interest with respect to the development of diabetic complications. It serves as a substrate of O-GlcNAc transferase (OGT). This is a cytosolic and nuclear enzyme that catalyzes a reversible posttranslational protein modification, whereby GlcNAc is transferred in O-linkage to specific serine/threonine residues of numerous proteins (86, 93, 555). The sites of O-GlcNAc modification (O-GlcNAcylation) are often identical or adjacent to known phosphorylation sites, suggesting a regulatory function. Functional significance of O-GlcNAcylation has been reported for several proteins, including the transcription factors Sp1 and c-myc, as well as cytosolic and nuclear enzymes, such as glycogen synthase (GS) and RNA polymerase II [reviewed in (93)]. Sp1 was the first transcription factor identified as an O-GlcNAc-modified protein; it has multiple O-GlcNAc modification sites, and its phosphorylation on serine and threonine residues is inversely proportional to its O-GlcNAc modification (93).
Figure 3.
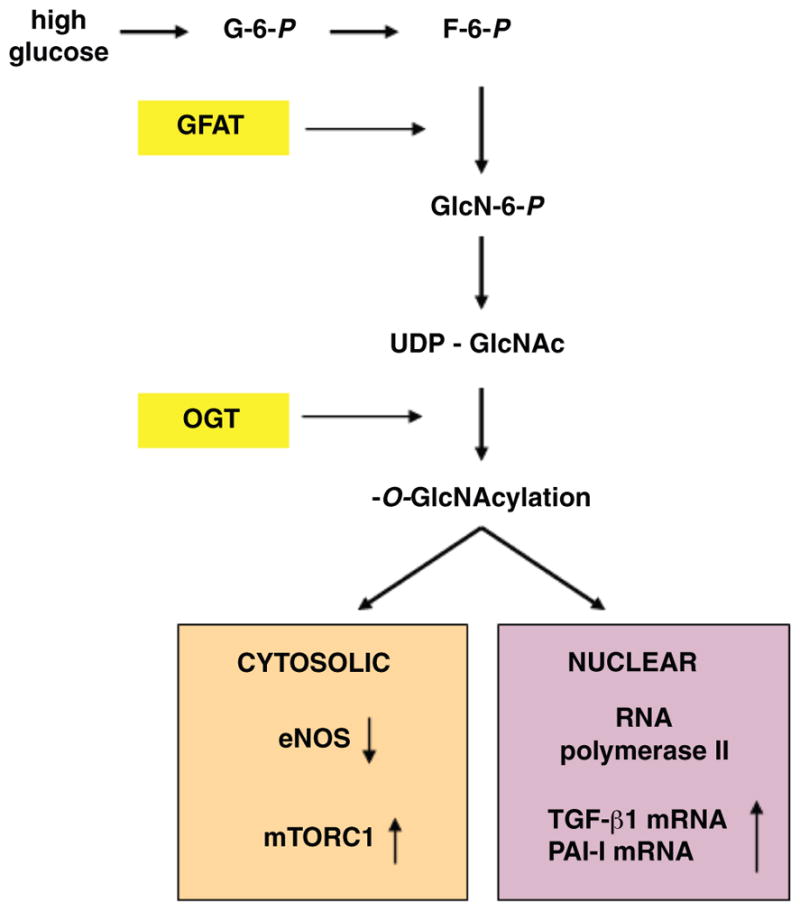
Activation and downstream targets of the hexosamine pathway in diabetes. When the intracellular concentrations of glucose are high, fructose-6-phosphate, an intermediate in the glycolytic pathway, is partially diverted into the hexosamine pathway. After a series of enzymatic steps glucosamine (GlcNAc) is transferred in O-linkage to specific serine/threonine residues of numerous proteins, including transcription factors as well as cytosolic and nuclear enzymes, with patho-physiological consequences in affected cells. Key enzymes in the pathway are highlighted in yellow. More detailed description is provided in the text. G-6-P, fructose-6-phosphate; F-6-P, fructose-6-phosphate; GFAT, glutamine:fructose 6-phosphate amidotransferase; GlcN-6-P, glucosamine 6-phosphate; UDP-GlcNAc, UDP-N-acetylglucosamine; OGT, O-GlcNAc transferase; eNOS, endothelial NO synthase; mTOR, mammalian target of rapamycin; TGF-β, transforming growth factor-β; PAI-I, plasminogen activator inhibitor-I.
The hexosamine pathway has been causally linked to up-regulation of several molecules implicated in the pathophysiology of nephropathy. Glucosamine reproduced the effect of glucose on induction of transforming growth factor β1 (TGF-β1), and blocking GFAT activity with antisense GFAT oligonucleotide or with a chemical inhibitor ameliorated high glucose/GlcN stimulation of TGF-β1 synthesis; vice versa overexpressing GFAT-induced TGF-β1 and fibronectin expression in mesangial cells incubated in 5 mM glucose (696). A more recent study (695) has suggested an ability of the hexosamine pathway to increase TGF-β1 promoter activity. Thus TGF-β1 expression seems to be dependent, at least in part, on the activity of the hexosamine pathway (see Fig. 3). The hexosamine pathway has been also implicated in diabetes-induced expression of plasminogen activator inhibitor 1 (PAI-1) in mesangial cells (271) and in high glucose-induced upregulation of angiotensinogen in renal tubular cells (247). Another line of evidence has shown the capability of hexosamines to activate Rho, a small GTPase protein, and its immediate downstream target, Rho kinase (ROCK), in vascular smooth muscle cells (303), a pathway recently implicated in the pathophysiology of nephropathy (316, 323, 484).
The O-linked post-translational modification has been described for endothelial NO synthase (eNOS), an enzyme responsible for endothelial NO production and essential for normal endothelial function. Du et al. (150) and Federici et al. (165) reported that hexosamine pathway-induced modification of eNOS protein interferes with its activation by phosphorylation thereby resulting in a reduced ability of eNOS to produce NO. More recent studies have also implicated the pathway in mesangial proliferation mediated by rapamycin-sensitive mTOR complex 1 (mTORC1) as well as in mesangial apoptosis (272). Further studies are necessary to define the relevance of the hexosamine pathway in the diabetic kidney in vivo.
The polyol pathway
The key enzyme in this pathway, aldose reductase, catalyses reduction of toxic aldehydes in the cell to inactive alcohols. When the glucose concentration in the cell becomes too high, aldose reductase reduces glucose to sorbitol, which is later oxidized to fructose. In the process of reducing high amounts of intracellular glucose to sorbitol, the aldose reductase consumes the cofactor NADPH, which is the essential cofactor for regenerating the intracellular antioxidant, reduced glutathione. By reducing the amount of reduced glutathione, the polyol pathway increases susceptibility to intracellular oxidative stress (86). In addition, since sorbitol does not cross cell membranes, its intracellular accumulation results in osmotic stress. A small number of studies conducted in the 1990s suggested that aldose reductase inhibition could attenuate glucose-induced activation of protein kinase C (PKC) and TGF-β production in mesangial cells in vitro (261), as well as inhibit some other markers of experimental nephropathy (42, 394). Moreover, the polyol pathway has been implicated in glucose-mediated alterations in proximal tubular cell matrix generation (62, 418, 749), whereas it does not seem to contribute to tubular hypertrophy (749). Further studies are necessary to define the significance of the polyol pathway in renal pathophysiology and outcome.
Activation of protein kinase C and downstream targets
The activation of the diacylglycerol (DAG)-PKC pathway is associated with many abnormalities in retinal, cardiovascular, and renal tissues in diabetic- and insulin-resistant states (332). PKC is a family of serine/threonine kinases that consist of 12 isoforms. PKC isoforms are classified according to whether they contain domains that bind calcium or DAG, both of which positively regulate the kinase activity. Conventional PKC isoforms (α, β 1/2, and γ) bind both calcium and DAG, novel PKC isoforms (δ, ε, η, θ, and μ) bind DAG, but not calcium, and atypical PKC isoforms (ς, λ) bind neither. The activation of conventional and novel PKC isoforms require the phosphorylation of the isoforms and the presence of cofactors such as calcium and/or DAG. When phosphorylated, increases in calcium and/or DAG induce PKC translocation to the membranous compartments of the cells to elicit biological actions. Rapid and short-term increases of DAG and calcium levels are usually induced via the activation of phospholipase C. Chronic activation of conventional and novel PKC isoforms require sustained elevations of DAG, which involves the activation of phospholipase D/C or the de novo synthesis of DAG (131, 332).
In diabetes, PKC is activated via multiple mechanisms. PKC activation in diabetic tissues is linked to enhanced formation of DAG. The latter can be formed by de novo synthesis from glycolytic intermediates, dihydroxyacetone phosphate and glycerol-3-phosphate, a process stimulated by high glucose levels (86, 131). DAG can be also derived from the hydrolysis of phosphatidylinositides, which are derived from the metabolism of phosphatidylcholine by phospholipase C (131). Another mechanism leading to activation of the DAG–PKC pathway involves hyperglycemia-induced increases in oxidants such as H2O2 which are known to activate PKC either directly or by increasing DAG production (442). In addition, PKC is activated as part of post-receptor signaling of vasoactive peptides known to be involved in the pathophysiology of diabetic complications, such as angiotensin (Ang) II (695) or ET-1 (141). AGEs, as another important component of the diabetic milieu, have been also shown to activate PKC, via ROS-mediated mechanisms (177). Thus, PKC activation is part of several signaling pathways implicated in the pathophysiology of the diabetic kidney.
Studies have suggested the importance of the activation of specific PKC isoforms, mainly the α, β 1/2, and δ isoforms, in causing cardiac or microvascular pathologies in diabetic animals (295, 332, 404, 489). In the kidney, activation of PKC has multiple consequences such as altered regulation of endothelial permeability, vasoconstriction, tubular transport, ECM synthesis/turnover, cell growth, angiogenesis, cytokine activation and leukocyte adhesion (156, 445). Selective inhibition of PKCβ has been shown to be nephroprotective in several models of diabetic kidney disease, which may involve effects on multiple target cells including mesangial cells, tubular cells, and antigen-presenting interstitial cells (260, 305, 308, 330, 489). First, clinical trials with a selective inhibitor of PKCβ showed beneficial short-term effects, whereas long-term kidney outcomes, assessed in patients with diabetic eye disease, were similar to placebo (638, 641). Notably, genetic variants of the PKCβ1 gene have been linked to the development of end-stage renal disease in patients with T2DM (378).
While the activation of PKC has been mostly studied as mediators of diabetes-induced cell injury, it should be noted that some isoforms, such as PKCε, may exert protective actions in the diabetic kidney (400).
Oxidative stress
Reactive oxygen species (ROS) include free radicals such as superoxide anion (•O2−), hydroxyl anion (•OH−), and peroxyl (•RO2) and nonradical species such as hydrogen peroxide (H2O2) and hydrochlorous acid (HOCl). It is important to note that there are also reactive nitrogen species produced from similar pathways, which include the radicals NO (•NO) and nitrogen dioxide (•NO2), as well as the nonradical peroxynitrite (ONOO−), nitrous oxide (HNO2), and alkyl peroxynitrates (RONOO). Of these, •O2−, •NO, H2O2, and ONOO− have been the most widely investigated in the diabetic kidney (177). Formation of ROS has been intensively studied as one of the principal mechanisms of glucose-induced cell toxicity, resulting in oxidation of important macromolecules including proteins, lipids, carbohydrates, and DNA. ROS can also act as signaling molecules for growth and vasoactive factors, such as Ang II (209).
The sources of ROS in cells exposed to the diabetic milieu can be divided into mitochondrial and cytosolic, as discussed in the following and outlined in Figure 4.
Figure 4.
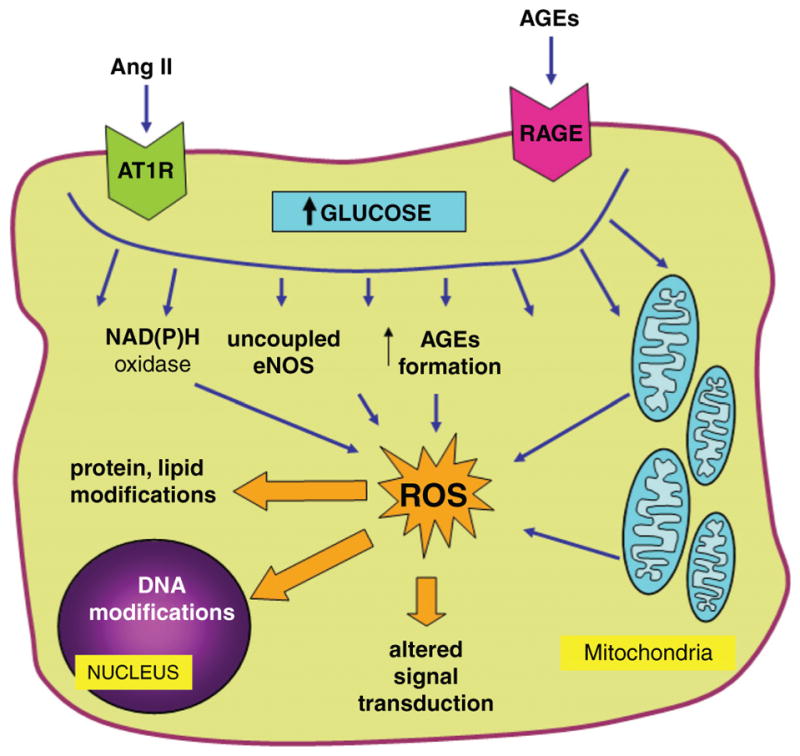
Mechanisms of enhanced formation of reactive oxygen species (ROS) in renal cells in diabetes. High intracellular glucose, stimulation of RAGE, as well as increased activity of vasoactive and pro-growth factors, such as the renin-angiotensin system, lead to formation of cytosolic and mitochondrial ROS via multiple mechanisms. ROS alter protein, lipid and DNA structure, and act as signaling molecules in pathways implicated in the pathophysiology of diabetic complications. See text for further details. Ang II, angiotensin II; AT1R, angiotensin II type 1 receptor; AGEs, advanced glycation end products; RAGE, receptor for AGEs.
Mitochondrial source of reactive oxygen species in diabetes
Mitochondria provide energy to the cell as ATP through oxidative phosphorylation. Oxidative phosphorylation is the process by which ATP is formed as electrons are transferred from NADH or FADH2 (generated through the Krebs cycle or indirectly via glycolysis) to molecular oxygen (O2). The generation of ROS, specifically •O2−, by damaged or dysfunctional mitochondria, has been postulated as the primary initiating event in the development of diabetes complications (86, 177, 442). In diabetes, where there is an excess of fuels supplied as a result of chronic hyperglycemia feeding into the respiratory chain, it has been hypothesized that excess production of •O2− occurs via the premature collapse of the mitochondrial membrane potential, which, rather than driving ATP production, leaks electrons to oxygen to form •O2−. A number of functional enzymes within the mitochondria are particularly susceptible to ROS-mediated damage, leading to altered ATP synthesis, cellular calcium dysregulation, and induction of mitochondrial permeability transition, all of which predispose the cell to necrosis or apoptosis (86, 177). However, it should be noted that these predominantly tissue culture studies (312) remain to be fully substantiated in vivo, particularly with respect to their role in nephropathy. More recent studies showed that RAGE induce cytosolic ROS and promote mitochondrial •O2− generation in diabetes. Notably, no increase in the production of mitochondrial •O2− was seen in renal cortices from gene-targeted diabetic mice lacking RAGE (123).
Cytosolic sources of reactive oxygen species in diabetes
The system of NAD(P)H oxidases seems to be, at this stage, the most important source of cytosolic ROS in the diabetic kidney. The phagocyte NADPH oxidase catalyzes the NADPH-dependent reduction of molecular oxygen to generate superoxide anion (•O2−) which is dismuted to form H2O2 (177). The enzyme consists of two plasma membrane-associated proteins, gp91phox (the catalytic subunit) and p22phox which comprise flavocytochrome b558, and cytosolic factors, p47phox, p67phox, and the small GTPase Rac (177). Studies over the last decade have documented significant NAD(P)H-dependent ROS-producing activities, not only in phagocytes, but also in nonphagocytic cells, including kidney cells (mesangial cells, podocytes, tubular cells) (177, 604).
The family of gp91phox homologues termed Nox (for NAD(P)H oxidase) proteins consists of five members (Nox1–5), of which Nox2 is gp91phox or the neutrophil isoform (347). These oxidases are proposed to play a role in a variety of signaling events, such as cell growth, cell death or survival, oxygen sensing, and inflammatory processes. The kidney seems to be particularly susceptible to NADPH oxidase-induced oxidative damage. The isoform Nox4/Renox was cloned from the kidney and found to be highly expressed in renal tubular epithelial and mesangial cells (205). Enzyme activity and subunit expression are upregulated in response to high glucose in vascular and renal cells as well as in experimental models of diabetes and mediate diabetes-induced oxidative stress and downstream signaling, as well as renal cell structural and functional abnormalities characteristic for the diabetic state (177, 205, 235, 547, 604).
Cytosolic ROS are also formed during the formation of intracellular AGEs (177) and as a result of activation of the polyol pathway, processes discussed above in more detail. Other sources of cytosolic ROS include uncoupled eNOS (235) and xanthine oxidase (177). Further information about the individual sources of ROS and their pathophysiological roles in specific kidney compartments and cells is provided in the following sections.
In parallel with enhanced ROS generation in the diabetic environment, the cells exposed to high glucose and other components of the diabetic milieu lose their ability to fight oxidative stress. Several mechanisms of endogenous antioxidant protection have been shown to be altered in diabetic kidney as discussed in the following.
Superoxide dismutase
Superoxide dismutase (SOD) is the major antioxidant enzyme for superoxide removal, which converts superoxide into H2O2 and molecular oxygen (184). The hydrogen peroxide is further detoxified to H2O by other important components of the cellular antioxidant system, catalase or glutathione peroxidase. In mammals, three SOD isoforms exist: cytoplasmic CuZnSOD (SOD1), mitochondrial MnSOD (SOD2), and extracellular CuZnSOD (SOD3, ecSOD) (177). In addition to experimental evidence, lower SOD activity has also been observed in the diabetic patients with nephropathy as compared with those without diabetic complications (177).
Overexpression of cytoplasmic Cu/ZnSOD1 in mice attenuated diabetes-induced albuminuria, glomerular hypertrophy, and ECM accumulation (142). Recent studies in mice with different susceptibility to the development of nephropathy have shown that the differences in strains may be attributable to differences in activities of SOD isoforms (189). AGE modifications could be responsible for altered activity of these enzymes in diabetic nephropathy (177).
Glutathione
The tripeptide glutathione (GSH, glutamylcysteinylglycine) is the abundant intracellular nonprotein thiol. Some 20% of intracellular glutathione is located in the mitochondria where it helps protect against mitochondrial ROS production. There are several mechanisms whereby hyperglycemia may bring about oxidative stress via changes in glutathione metabolism (86, 142, 177). Excessive glucose resulting in mitochondrial or cytoplasmic ROS generation (described above) may exhaust glutathione pool. Furthermore, as mentioned, hyperglycemia results in increased flux through the polyol pathway causing glutathione depletion.
Thioredoxin
The major thioredoxins are thioredoxin-1, a cytosolic and nuclear form, and thioredoxin-2, a mitochondrial form. Thioredoxin-1 efficiently reduces cellular ROS through thioredoxin peroxidases, which belong to a conserved family of antioxidant proteins, the peroxiredoxins. The reduced form of thioredoxin peroxidase scavenges oxidant species such as H2O2 and in the process homo- or heterodimerizes with other family members. Thioredoxin interacting protein (Txnip), also known as vitamin D3 upregulated protein-1 or thioredoxin binding protein-2, is the endogenous inhibitor of cellular thioredoxin, inactivating its antioxidative function. Hyperglycemia promotes oxidative stress, in part, through inhibition of thioredoxin activity by increased expression and activity of Txnip (566), mechanism recently demonstrated to be operating also in the diabetic kidney (12).
Detailed discussion of these exceedingly complex biochemical reactions is beyond the scope of this articles and the readers are referred to excellent reviews addressing this topic (86, 177, 475).
Glomerular Pathophysiology in Diabetes
The diabetic milieu has profound effects on glomerular function and morphology, affecting all types of glomerular cells and structures. In the following section, we will discuss diabetes-induced mechanisms of glomerular cell injury in specific cell types and their consequences for the development of proteinuria and structural changes.
Mesangial cell pathophysiology in the diabetic kidney
Although there are some exceptions to the rule, the hypertrophic phenotype prevails over the hyperplastic phenotype in diabetic glomeruli. Mesangial expansion is considered to be one of the most important structural lesions during the development of nephropathy in T1DM. The importance of mesangial expansion was proposed in seminal human biopsy studies by Mauer et al. (396) and Osterby et al. (464, 466). These studies formulated the so-called structural-functional relationship in diabetic nephropathy based on tight correlations between the degree of mesangial expansion and clinical characteristics of nephropathy, such as proteinuria, blood pressure, and the rate of decline in GFR. Mesangial expansion is determined both by mesangial cell hypertrophy and accumulation of mesangial matrix. These processes are closely related and are discussed together. Many of the following pathways are further discussed and illustrated in the section on tubular growth.
Factors driving mesangial expansion
Factors that are driving mesangial expansion are discussed in the following. We begin with growth factors and cytokines, which are stimulated by hyperglycemia, AGEs, oxidative stress, and physical forces, such as mechanical stress (211), and then discuss the role of cell-cycle regulation.
Transforming growth factor-β system
This system includes TGF-β peptides and receptors as well as Smad signaling molecules. TGF-β signals via TGF-β types I and II receptors. Upon TGF-β binding to the type II receptor on the cell surface, this complex activates the type I receptor, which then phosphorylates and activates members of the Smad family (Smad2 and Smad3). The activated Smads form oligomers with the unique co-Smad, Smad4, and rapidly translocate to the nucleus to regulate expression of target genes. Smad7 is an inhibitory Smad that regulates TGF-β signaling, preventing recruitment and phosphorylation of Smad2 and Smad3 (364). In addition, recruitment of a complex of Smad7 with Smurf1 or Smurf2 to the type I TGF-β receptor results in receptor ubiquitination by the Smurf proteins and targets the receptor for degradation.
TGF-β is a prosclerotic factor considered to be crucial for the development of mesangial cell hypertrophy and production of ECM proteins. This factor not only stimulates genes for the components of ECM (68), but also inhibits the synthesis of MMPs, enzymes responsible for ECM degradation, and stimulates production of inhibitors of metalloproteinases (TIMP). Thus, TGF-β is one of the major determinants of the dysbalance between production and degradation of ECM in the diabetic kidney (68). In addition, TGF-β influences the cell cycle as discussed below (706).
Numerous studies have shown that TGF-β plays a key role in the process of mesangial expansion and the development of glomerulosclerosis. Renal expression of TGF-β and other components of this system are increased in the kidney both in diabetic patients and in a variety of experimental models of diabetes (71, 191, 220, 263, 331, 575, 578, 723, 748). In addition to the components of the diabetic milieu discussed in the introductory part of this article (103), it is important to note that TGF-β is also activated by factors more typical for the metabolic environment in T2DM, such as leptin (220, 709).
TGF-β is also activated by Ang II and acts as one of the mediators of growth and prosclerotic effects of the renin-angiotensin-aldosterone-system (RAS) (69, 119, 286, 707). Indeed, inhibitors of the RAS such as ACE inhibitors and Ang AT1 receptor blockers (ARB), or more recently with renin inhibitors (251) reduce TGF-β expression in the diabetic kidney, which is considered to be important for antifibrotic effects of these agents (70, 191). This phenomenon is discussed in more detail in a section devoted to the intrarenal activation of the RAS in the diabetic kidney. In addition, TGF-β has been show to operate downstream of other vasoactive and growth factors acting in the diabetic kidney such as ET-1 (231) or vasoconstrictor prostanoids (107, 430, 712).
The role of TGF-β as a mediator of diabetic glomerulosclerosis, as well as interstitial fibrosis (discussed in the following sections) has been documented in a number of studies using a variety of experimental approaches. A seminal study by Ziyadeh et al. (748) utilized long-term treatment with a neutralizing antibody to demonstrate that TGF-β inhibition ameliorated mesangial expansion and glomerulosclerosis in db/db mice, a model of T2DM. Interestingly, these protective effects were not associated with a significant reduction of proteinuria, suggesting that the glomerular pathology is driven by multiple factors acting on various cell types. More recently, the results by Ziyadeh et al. (748) were reproduced in studies with Smad3 knockout (KO) mice (686).
Several proteins from the TGF-β family act as endogenous antagonists of TGF-β and have also been implicated in the pathophysiology of nephropathy. For example, bone morphogenic protein-7 (BMP-7) prevents the development of nephropathy in streptozotocin (STZ)-diabetic rats (314). The small proteoglycan decorin is another endogenous inhibitor of TGF-β, acting at least in part downstream of the protective hepatocyte growth factor (HGF) (315).
Connective tissue growth factor
Connective tissue growth factor (CTGF), a 36 to 38 kDa cysteine-rich secreted protein, has been identified as a prosclerotic cytokine involved in the regulation of ECM accumulation by acting as a downstream mediator of TGF-β (91, 521). However, there is also evidence for TGF-β-independent regulation of CTGF by other components of the diabetic milieu (684). Exposure of mesangial cells to high glucose concentrations, mechanical strain, AGEs, or TGF-β leads to increased levels of CTGF gene expression and protein secretion (91, 525). In their seminal study, Riser et al. (525) examined cultured rat mesangial cells, kidney cortex, and microdissected glomeruli from diabetic db/db mice, a model of T2DM, and their control counterparts. Their results suggested that diabetes or hyperglycemia-induced CTGF upregulation is important for mesangial matrix accumulation and progressive glomerulosclerosis, acting downstream of TGF-β. Similarly, in STZ-diabetic mice, a model of T1DM, Roestenberg et al. (529) reported that CTGF expression coincided with the initiation of mesangial expansion and preceded increases in renal fibronectin and collagen IV expression.
Importantly, several observations have suggested that urinary CTGF may serve as an early biomarker and predictor of progression of nephropathy in diabetic patients. In patients with diabetes and microalbuminuria or overt diabetic nephropathy, urinary CTGF mRNA (10) or protein excretion are increased (197, 436, 522) and correlate with urinary albumin excretion and GFR. Moreover, a prospective study by Nguyen et al. has identified plasma CTGF as an independent predictor of end-stage renal disease and mortality in patients with diabetic nephropathy (437). Similarly to TGF-β, CTGF is present in the renal tubular fluid where it may act directly on tubular epithelial cells and mediate epithelial-mesenchymal transition (91), a process discussed in more detail in the following sections.
Further support for a pathogenetic role of CTGF in nephropathy provided studies with antisense oligodeoxynucleotides. This intervention was successful in reducing pro-teinuria and expression of genes involved in mesangial matrix expansion in mouse models of diabetes (212). In a first small and open-label phase 1 safety trial in microalbuminuric diabetic patients, the human monoclonal antibody to CTGF, FG-3019, rapidly and significantly lowered urine albumin to creatinine ratios, which warrant further prospective, randomized and blinded clinical trials (11).
Growth hormone and insulin-like growth factor axis
Diabetes is associated with complex alterations of growth hormone (GH) and insulin-like growth factor I (IGF-I) secretion, activity and actions. Although a very detailed analysis is beyond the scope of this article, it is important to note that relatively abundant evidence has been amassed with respect to renal actions of these hormones and their possible contribution to the development of nephropathy. The kidneys are an important source for the synthesis of IGF-I, some of which is released into the circulation (670). GH and IGF-I have profound effects on renal growth, glomerular hemodynamics, and tubular function. Administration of IGF-I to animals promotes renal growth through a process of cellular hypertrophy and hyperplasia, and induces a rapid increase in renal blood flow and GFR together with a reduction in renal vascular resistance (163, 670). Subjects with diabetes and microalbuminuria present several alterations in the GH-IGF-IGF-binding proteins (IGFBP) system, with increased IGF-I renal levels and IGFBP-3 protease activity, increased excretion of bioactive GH, IGF-I, and IGFBP-3, but decreased circulating IGFBP-3 levels. Circulating levels of IGF-I are normal or reduced in patients with diabetes when compared with normal controls, but renal IGF-I production can be increased and contributes to the pathogenesis of diabetic nephropathy (670).
The most relevant evidence about the roles of the GH-IGF-I pathway in diabetic renal pathophysiology has been provided by studies with inhibitors of the system. Antagonists of the GH-IGF-I pathway, including somatostatin analogues or GH and IGF-I receptors antagonists, have been found to have beneficial effects on the diabetic kidney in animal models (340, 568). Landau et al. (340) investigated the effects of PTR-3173, a novel somatostatin analogue which exerts a prolonged inhibition of the GH-IGF axis without affecting insulin secretion, on markers of diabetic nephropathy in the nonobese diabetic (NOD) mouse. The treatment with PTR-3173 blunted renal and glomerular hypertrophy, albuminuria, and glomerular hyperfiltration in diabetic NOD mice. These effects were associated with the prevention of renal IGF-I accumulation.
Specific inhibitors of GH action [i.e., specific GH receptor antagonists (GHRAs)] represent another group of agents with nephroprotective potential in diabetes. Diabetic mice treated from the onset of diabetes with the GHRA, G120K-PEG, showed normalization of diabetes-induced renal hypertrophy, glomerular enlargement, and slower development of albuminuria (568). In addition, beneficial renal effects were reported even after late intervention with GHRA in NOD mice with manifest renal changes (568).
Platelet-derived growth factor
Platelet-derived growth factor (PDGF) is expressed mainly in glomerular mesangial and epithelial cells and it plays an important role in the production of ECM proteins in renal diseases. Several studies have suggested that PDGF may be involved in the pathogenesis of DN, and increased glomerular expression of this factor has been documented in models of diabetic nephropathy in vitro (633) and in vivo (306), as well as in renal biopsies from patients with nephropathy (345). However, the pathogenetic role of PDGF in diabetic nephropathy still remains to be tested in studies with selective inhibitors.
The role of cell-cycle regulation
Although diabetes-induced changes in cell-cycle regulation occurs in a variety of cell types, this topic is introduced in this section and further discussed in the section on tubular growth. Following activation, for example by growth factors, quiescent cells in G0 phase, progress through G1 phase and initiate DNA synthesis in S phase. During the G2 phase, cell growth continues and proteins are synthesized in preparation for entry into the M phase, where mitosis occurs. The process requires orchestrated activation of cyclin-dependent kinases (CDK), which, after binding specific proteins (cyclins) form active complexes, and act as positive regulators of the cell cycle. The cyclin-CDK complexes are regulated by multiple phosphorylation and dephosphorylation events and by several proteins called CDK-inhibitors. Two classes of CDK-inhibitors have been identified: the INK4 family and the Cip/Kip family.
The transition from late G1 into the S phase determines the cell’s growth characteristics. G1 arrest results in hypertrophy, whereas G1 exit is associated with apoptosis. Transition from G1 to S phase results in DNA synthesis and proliferation. On entry into G1, cells normally undergo a physiologic increase in protein synthesis before S phase DNA synthesis. In hypertrophic cells, as in the kidney cells exposed to the diabetic milieu, this increase in protein content is not matched by a concurrent increase in DNA. Consequently, one mechanism underlying cellular hypertrophy is cell-cycle arrest at the G1/S checkpoint, leading to increased protein to DNA ratio (388, 573).
Mesangial cells respond to high glucose with an initial proliferation phase, which is followed by G1 arrest and hypertrophy (732). Hyperglycemia increases the expression of cyclin D1 and the activation of CDK4, evidence of cell-cycle entry (167). Importantly, exposure to high glucose increases the levels of both CDK-inhibitors p21 Cip1 and p27 Kip1 in cultured mesangial cells (709), and antisense oligonucleotides (ASOs) to p21 Cip1 or p27 Kip1 reduce the hypertrophic effects of hyperglycemia (163). Further support for the roles of CDK inhibitors in diabetes-induced mesangial hypertrophy has been provided in studies with p21 or p27 KO mice. High glucose-induced mesangial hypertrophy was not observed in cells harvested from p21 KO or p27 KO mice. Moreover, the reconstitution of p27 Kip1 by transfection in p27 KO mesangial cells restored the hypertrophic phenotype (417, 710). In addition, both p21 Cip1 and p27 Kip1 are required for maximal mesangial cell hypertrophy induced by TGF-β (163) and hypertrophic effects of CTGF are at least in part mediated by increased expression p15 INK4, p21 Cip1, and p27 Kip1 (4).
The in vitro evidence discussed above has been reproduced in in vivo studies in experimental models of diabetic nephropathy. In STZ-diabetic mice, glomerular hypertrophy was associated with a selective increase in p21 Cip1 (338). Diabetic p21 KO mice are protected from glomerular hypertrophy and development of progressive renal failure (16). Diabetic p27 KO mice exhibit only mild mesangial expansion without glomerular hypertrophy, despite increases in glomerular TGF-β (32, 708). In summary, these cell culture and experimental models show that p21 Cip1 and p27 Kip1 play a critical role in mediating diabetes-associated glomerular hypertrophy and the consequential increase in matrix proteins, associated with the deterioration in renal function. These pathways are further discussed and illustrated in the section on tubular growth.
Notably, an influence of the presence of ECM proteins on the response of mesangial cells to diabetes has been indicated in a recent study in STZ-diabetic mice. Using KO mice for type VIII collagen, Hopfer et al. linked mesangial expansion and cellularity, ECM expansion and albuminuria to the expression of type VIII collagen which is enhanced in this model (243).
Changes in the Glomerular Filtration Barrier in the Diabetic Kidney
The glomerular filtration barrier (GFB) is composed of glomerular endothelial cells (GEC), the glomerular basement membrane, and podocytes (glomerular epithelial cells). All three components of the barrier undergo changes in diabetes and contribute both to the structural glomerular alterations and changes in the permeability to macromolecules. In addition to other functions, the integrity of the GFB determines the extent to which albumin and other proteins will be filtered into the urinary space and, thereby, contributes to the development of albuminuria/proteinuria, a hallmark of diabetic nephropathy. Figure 5 illustrates and summarizes diabetes-induced changes in the GFB.
Figure 5.

Diabetes-induced alterations of glomerular filtration barrier. The schematic presentation describes major consequences of diabetes-induced changes in glomerular endothelial cells, glomerular basement membrane, and podocytes, as discussed in more detail in the text.
The GFB contributes to the impermeability to macromolecules by size and charge selectivity. Under physiological conditions, low molecular weight proteins (≤40 kDa) are essentially freely filtered, whereas, high molecular weight proteins (≥100 kDa) are almost completely restricted (125, 390). The molecular weight of albumin is 69 kDa, that is, of borderline size. In patients with diabetes and low-grade albumin excretion (microalbuminuria), no changes in size selectivity, determined mainly by the radius of glomerular endothelial pores, have been reported (550), suggesting a possible role for alterations in the charge barrier at the early stages of microalbuminuria. The charge barrier is formed by anionic sites within the GBM, on podocytes and the endothelial glycocalyx. As the disease progresses, anatomical derangements of the GFB result in gradual loss of selectivity.
Changes in the glomerular endothelium in the diabetic kidney
The changes in the glomerular endothelium are similar to the well-described generalized endothelial dysfunction in diabetes, including reduced bioavailability of NO, impaired endothelium-dependent regulation of vascular tone, upregulation of adhesion molecules, development of a pro-thrombotic phenotype and increased permeability (203, 318, 500, 551). These changes are not specific for GEC, but are applicable to the entire renal vascular tree.
Unlike other capillaries, glomerular capillaries have a high permeability to water (hydraulic conductivity), yet, like other capillaries, they are relatively impermeable to macromolecules. GEC are highly specialized cells with regions of attenuated cytoplasm punctuated by numerous fenestrae, circular transcellular pores 60 to 80 nm in diameter (36). The glycocalyx is a layer on the luminal surface of GEC, largely composed of glycoproteins and proteoglycans with adsorbed plasma proteins. Heparan sulphate proteoglycans (HSPGs) may be responsible for the negative charge characteristics of the glycocalyx (36). Relevant for the pathophysiology of diabetic nephropathy, ROS have been shown to disrupt the glycocalyx (230). In diabetes, a specific assessment of structural changes in GEC and the associated glycocalyx has not been performed. However, indirect evidence supports the notion that abnormalities of the glycocalyx exist and may be relevant. For example, total systemic glycocalyx volume is reduced by acute hyperglycemia in humans (440). Furthermore, patients with T1DM have decreased systemic glycocalyx volume, which correlates with the presence of microalbuminuria (439).
Impaired activity of endothelial nitric oxide synthase and of nitric oxide
eNOS and eNOS-derived NO are crucial molecules for endothelial function and integrity. NO acts as a potent vasodilator and as a molecule with potent anti-thrombotic and anti-growth properties in the vasculature (318). Earlier studies suggested several direct mechanisms whereby the diabetic milieu antagonizes NO bioavailability. These mechanisms include NO quenching by AGEs (89, 673) or ROS (235), and NO capture by glucose (82).
More recent studies revealed a spectrum of mechanisms related to posttranslational alterations of eNOS molecules in diabetes. To function as an endothelial NO-producing enzyme, eNOS requires a battery of co-factors, posttranslational modifications such as phosphorylation and dimerization, protein-protein interactions, and subcellular targeting (207). Phosphorylation of eNOS on Ser1177 by several serine/threonine kinases, such as Akt (protein kinase B) (143, 192) or protein kinase A (PKA) (94), in response to a variety of physiological stimuli, is a critical control step for NO production by the enzyme. This process enhances the rate of electron flux from the reductase to the oxygenase domain of eNOS, and reduces the calcium requirements for the enzyme, thus increasing NO synthesis (398). Also the formation of a homodimer is crucial for NO production by eNOS (229, 350), creating high affinity binding sites for the NOS substrate L-arginine, and enabling electron transfer from the reductase domain of one NOS monomer to the oxygenase domain of the other (541). Finally, activation of eNOS is not only dependent on phosphorylation by upstream kinases and conformational changes, but is also determined by its specific subcellular localization with the activatable enzyme being localized within the plasma membrane.
As mentioned in the “General mechanisms of glucose-induced cell injury” section, hexosamine pathway-induced modification of eNOS molecule in vitro interferes with its stimulatory phosphorylation resulting in a reduced ability of eNOS to produce NO (150, 165). To address these issues in in vivo settings, Komers et al. (326) analyzed renal cortical samples from control and STZ-diabetic rats, and described impaired eNOS dimerization, membrane targeting, and stimulatory phosphorylation at Ser1177 in diabetic samples despite of similar whole cell eNOS expression in diabetic and control animals. These findings further suggest that a process called “eNOS uncoupling” operates also in the diabetic kidney. During this process, electron transfer within the active site of eNOS becomes “uncoupled” from L-arginine oxidation resulting in reduction of molecular oxygen to superoxide (669, 720). Thus, eNOS can become a ROS producing enzyme in sharp contrast to its protective role as a machinery for production of protective NO. Indeed, renal production of ROS has been shown to be attenuated by L-arginine-analogues (547) that act as NOS inhibitors. The mechanisms of uncoupling are currently being investigated, but well-described diabetes-induced deficiency in co-factors, such as tetrahydrobiopterin (402, 503), necessary for conformational changes of eNOS allowing NO production, are likely to play a role.
Moreover, as postulated by Nakagawa et al. (423) high glucose-induced posttranslational changes in eNOS may be responsible for uncoupling of VEGF actions from eNOS, allowing unhindered growth and vasoactive signaling of VEGF (discussed in detail below) without parallel activation of protective eNOS-derived NO production. VEGF normally stimulates endothelial NO release and acts in concert with elevated NO levels as a trophic factor for vascular endothelium. The increased NO derived from the endothelial cell acts as an inhibitory factor that prevents excess endothelial cell proliferation, mesangial or vascular smooth muscle cell proliferation, and macrophage infiltration. In the setting of reduced NO bioavailability, high levels of VEGF lead to excessive endothelial cell proliferation, stimulation of macrophage chemotaxis, and mesangial or vascular smooth muscle cell activation.
Role of vascular endothelial growth factor in the early and later stages of the diabetic kidney
Several kidney biopsy studies have confirmed increased glomerular vascularity and endothelial cell proliferation at early stages of diabetic nephropathy (465). VEGF, a pro-angiogenic factor, is prominently produced by podocytes (122). VEGF may affect podocyte function in an autocrine fashion (via VEGFR1) (106). VEGF protein also crosses the GBM and acts on GEC (via VEGFR1 and VEGFR2) to promote endothelial cell survival and to induce the formation of fenestrae, which enhance glomerular endothelial permeability. VEGF expression is increased at early stages of nephropathy, and has been implicated in diabetic renal patho-physiology, and in accord with its actions, as a mediator of albuminuria or even structural changes (122, 134). Indeed, inhibition of VEGF by a variety of means reduces albuminuria in diabetic rodents (134, 337, 603).
However, the role VEGF and angiogenesis in the development of diabetic kidney disease, even albuminuria is more complex. VEGF may be elevated in the early phases of the diabetic kidney, but it may not be maintained as more chronic fibrotic changes occur in the kidney, and VEGF may decrease in the advanced stages of the disease. In a remnant kidney model, VEGF levels are reduced, correlating with the progression of renal damage (294). In human as well as experimental diabetic nephropathy, more advanced stages are also associated with lower VEGF expression (34, 195, 238, 372). These observations suggest that the upregulation of VEGF in early stages of diabetic nephropathy may provide a mechanism for the initial progression of the disease, leading to excessive blood vessel formation and albuminuria. The decline of VEGF in the later phase of diabetic nephropathy may reflect a loss of endogenous VEGF due to the disruption of podocytes and tubular cells in chronic kidney damage. In fact, at later stages of chronic kidney disease, VEGF is currently considered to be a protective factor with beneficial effects on glomerular endothelium, podocyte viability, and, as described in the remnant kidney models and in diabetic patients, on the peritubular network of capillaries (372), preventing the progression of interstitial fibrosis.
Role of inhibitors of angiogenesis and angiopoietins in the diabetic kidney
The pathophysiological roles of angiogenic factors in the diabetic kidney are further supported by studies focusing on endogenous inhibitors of angiogenesis. These molecules originate as cleavage products of ECM proteins (tumstatin and endostatin) or plasminogen (angiostatin) and specifically target endothelial cells and inhibit their proliferation, survival, migration, and sprouting (218, 459, 460). The levels of these proteins are dramatically decreased in the kidney of STZ-diabetic rats, most likely due to decreased activity of MMPs [see section “Glomerular pathophysiology in the diabetic kidney” and (740)]. Administration of tumstatin (a cleavage product of collagen IV), endostatin (a cleavage product of collagen XVIII), or angiostatin has been shown to reduce glomerular hypertrophy, hyperfiltration, albuminuria, mesangial matrix expansion, ECM accumulation, endothelial cell proliferation, and monocyte/macrophage infiltration in STZ-induced diabetic mice (255, 724, 740) in parallel with the suppression of multiple molecular markers of nephropathy. The treatments also increased nephrin expression (255, 724) and enhanced the levels of pigment epithelium-derived factor, another endogenous antiangiogenic factor (740). Similar renoprotective effects have been observed in STZ-diabetic mice treated with vasohibin-1, an endogenous angiogenesis inhibitor that is, unlike the angiostatins discussed above, induced in endothelial cells by proangiogenic factors (428).
The studies with endogenous angiogenesis inhibitors should be also interpreted with caution. Despite impressive effects of these molecules on a variety of functional, structural and molecular markers of nephropathy, these studies were conducted only for several weeks in STZ-diabetic mice known to be relatively resistant to the development of structural changes characteristic of diabetic nephropathy. Although those studies suggest beneficial effects of angiogenesis inhibition at early stages of nephropathy, their long-term nephroprotective effects, and consequently the role of angiogenesis later in the course of the disease, remain to be established. Moreover, observed beneficial effects of angiogenesis inhibitors in mesangial cells and podocytes suggest involvement of additional, less understood, effects unrelated to modulation of angiogenesis.
Angiopoietins are another family of vascular growth factors that have been implicated in the pathophysiology of nephropathy. The best-studied are angiopoietin 1 (Ang-1) and Ang-2. During normal development, they are considered critical for vascular differentiation through angiogenesis, the process of growth and remodeling of existing vessels. In mature organisms, angiopoietins are involved in maintenance and turnover of blood vessels (715). Ang-1 causes enhanced endothelial survival and endothelial cell stabilization, whereas Ang-2 acts as a natural antagonist of Ang-1 (310). The in vivo biologic effects of the angiopoietins also depend on ambient levels of VEGF-A (715). Angiopoietins seem to be linked to nephrin expression, since Ang-2 overexpression in non-diabetic mice leads to downregulation of nephrin (132). Experimental models of T1DM are associated with altered renal expression of angiopoietins. Rizkalla et al. (527) reported that in STZ-diabetic rats whole-kidney Ang-1 and Ang-2 mRNA and protein levels rose at 4 weeks, but at 8 weeks, Ang-1 levels were lower than those in nondiabetic controls, whereas Ang-2 remained elevated. Ang-1 was immunolocalized in diabetic kidney tubules, whereas Ang-2 was prominent in glomerular endothelia and podocytes. Yamamoto et al. (724) also reported renal upregulation of Ang-2 in STZ-diabetic mice. In accordance with this in vivo evidence in models of diabetes, more recent in vitro studies in endothelial cells identified mechanisms of high glucose-induced Ang-2 expression (727). These observations suggest that a decreased ratio of Ang-1/Ang-2 might play a role in the pathobiology of glomerular disease in diabetic nephropathy. However, considering the above discussed effects of angiogenesis inhibitors in the diabetic kidney, the pathophysiological roles of Ang-1/Ang-2 remain rather murky. It is also possible, that the primary target of these molecules is not the GEC but the podocytes.
Other factors influencing glomerular endothelial permeability in diabetes
Glomerular endothelial permeability can also be increased by proinflammatory cytokines and adipokines, such as TNF-α, IL-6 and leptin, that is, factors more typical for T2DM [reviewed in (546)]. In contrast, adiponectin, unlike other adipokines, appears to have protective effects in the vasculature by reducing endothelial cell activation and inflammation, in addition to protective and antiproteinuric effects on podocytes (577).
The glomerular basement membrane in the diabetic kidney
The normal GBM is a 300- to 400-nm thick structure consisting predominantly of type IV collagen, laminin, nidogen, and heparan sulfate proteoglycans (HSPGs). In T1DM, GBM thickening occurs early in the course of the disease, long before the development of microalbuminuria. Thickening of the capillary basement membrane has been observed in other vascular beds in patients with T1DM and does not seem to be specific for the kidney (149, 392). The thickening of the GBM is due to the accumulation of ECM, which originates most likely in the podocyte (see Fig. 6).
Figure 6.

Podocyte foot process effacement and glomerular basement membrane (GBM) thickening in experimental diabetes. Transmission electron microscopy was used to investigate the glomerular filtration barrier (GFB) in normal (C) and STZ-diabetic DBA/2J mice (V, L, DL, L + DL, and L + DH). Compare the appearance of the GFB in normal (C) and diabetic mice (V), and the effects of nephroprotective treatments [L, DL, L + DL, and L + DH; for details on treatments see (741)]. Stars indicate the areas of podocyte foot process effacement; arrow pairs point to both sides of the glomerular basement membrane (GBM). Adapted with permission from (741).
The GBM may constitute a charge barrier, due to its HSPG content. The evidence for a role of HSPG in the diabetic kidney remains controversial with equally persuasive reports supporting a role of loss of HSPG in diabetes (136, 404, 508, 586, 610) or arguing against it (666, 700, 701).
Paradoxically, diabetes-induced increases in GBM thickness might be expected to reduce protein transit, but it is associated with proteinuria. However, as postulated by Jefferson and coworkers (273), the GBM thickening occurs in an irregular manner with areas of thinned GBM with presumably enhanced permeability for proteins. Moreover, altered GBM may affect the adjacent cellular elements, reducing cell binding and promoting podocyte detachment (see below).
Podocyte pathophysiology in diabetes
Podocytes are terminally differentiated and highly specialized cells. They line the urinary side of the GBM and function as a fine filter contributing to ultimate size-selectivity and permitting permeability to molecules smaller than albumin. They also synthesize components of the GBM and counteract capillary hydrostatic pressure. The podocyte is composed of the cell body and interdigitating foot processes. The narrow gaps (30–40 nm) between neighboring processes are bridged by the glomerular slit diaphragm. This crucial structure is a highly specialized gap junction with small pores, permeable to water and solutes but relatively impermeable to plasma proteins. The shape of podocytes is supported by an actin cytoskeleton, which allows the podocyte to dynamically change its shape. Podocytes are anchored to the GBM by integrins and α- and β-dystroglycans. Similar to GEC and GBM, podocytes are also negatively charged on their apical membrane domain owing to surface anionic proteins such as podocalyxin, podoplanin, and podoendin. The negative charge helps limit passage of negatively charged molecules (like albumin). However, podocytes also serve as a size barrier to proteins, predominantly due to the properties of the slit diaphragm.
When exposed to the diabetic milieu, podocytes undergo a spectrum of changes that ultimately contribute to the development of proteinuria and glomerulosclerosis. These include a reduction in podocyte number (podocytopenia), foot process widening and effacement, and altered secretory functions. These processes are closely linked, but will be, for better clarity, discussed separately and are summarized in Figure 5. Importantly, kidney biopsies from Pima Indians with type II diabetes suggested that podocyte loss contributes to the progression of diabetic nephropathy (354, 355, 468).
Reduction in podocyte number (podocytopenia)
Several mechanisms have been implicated in podocyte loss in the diabetic kidney as outlined in the following.
Podocyte detachment
The appearance of podocytes in the urine (podocyturia), which has been described in 53% of microalbuminuric and 80% of macroalbuminuric patients with T2DM, while being absent in subjects with normal albumin excretion (424, 488), has been considered to be a persuasive in vivo indicator of podocyte detachment in the diabetic kidney. It has been proposed that this abnormality occurs due to reduced expression of the α3β1 integrin, the predominant integrin tethering the podocyte to the GBM (102).
Podocyte apoptosis
There is increasing and relatively abundant evidence implicating apoptosis of podocytes as another cause of podocytopenia in diabetes. Several mechanisms might underlie podocyte apoptosis in diabetes. Studies by Susztak et al. (604) have implicated high glucose-induced oxidative stress and proapoptotic p38 MAPK signaling in this process. Importantly, this study demonstrated that apoptosis preceded podocyte depletion and coincided with the onset of albuminuria. It should be noted, however, that early activation of p38 MAPK has also been associated with maintenance of the actin cytoskeleton in diabetic podocytes, and its loss correlated with progression of proteinuria (130). Other factors operating in the diabetic kidney, such as TGF-β (554, 683) or activation of RAS (151), have also been shown to induce podocyte apoptosis. Other studies have implicated a stimulation of RAGE, which is overexpressed on podocytes in experimental T2DM, in proapoptotic signaling (699).
More recently, Isermann et al. (258) reported interesting findings implicating activated protein C (APC) formation in several alterations in the diabetic kidney. Protein C is an antithrombotic system, regulated by endothelial thrombomodulin. Plasma protein C activity is reduced in diabetic patients (682), but with no clear link to the pathophysiology of diabetic nephropathy. Isermann and colleagues described a lower activity of a new pathway involving thrombomodulin-dependent APC formation in the diabetic kidney, resulting in attenuated cytoprotection in renal cells and enhanced glomerular apoptosis including podocytes. APC prevented the mitochondrial apoptosis pathway via the protease-activated receptor PAR-1 and the endothelial protein C receptor EPCR in glucose-stressed cells. In vivo, maintaining high APC levels during long-term diabetes protected against diabetic nephropathy in mice (258).
Activation of the Notch signaling pathway has been the most recent addition to a spectrum of mechanisms underlying podocyte apoptosis in diabetes. Notch proteins and numerous components of their down-stream signaling pathway are known to play a crucial role in mammalian kidney development. However, Notch1 is practically undetectable in the mature kidney. Niranjan et al. (441) have reported the expression of genes in the Notch pathway in mature podocytes in humans with diabetic nephropathy and in mice with STZ-induced diabetes. Moreover, expression of ICN1, a Notch intracellular domain, in podocytes resulted in apoptosis in vitro and led to podocyte loss, albuminuria, and glomerulosclerosis in vivo. Conversely, genetic or pharmacological inhibition of Notch signaling prevented podocyte apoptosis and albuminuria. Most recently, Lin et al. (370) extended these observations and showed that, in addition to the antiapoptotic effect, inhibition of Notch1 abrogated high glucose-induced VEGF synthesis and nephrin loss in podocytes, with a beneficial effect on proteinuria in diabetic animals.
Inability to proliferate and restore podocyte number
In contrast to mesangial cells, mature podocytes do not actively synthesize DNA nor proliferate under normal conditions (480). In the diabetic kidney, where podocytes undergo detachment or apoptosis, the relative inability of these cells to proliferate may lead to an eventual decline in number and become a contributing factor for the reduced podocyte number. As shown in several studies, high expression of CDK inhibitors may be responsible for this phenomenon (573).
High expression of CDK inhibitors has also been linked to another diabetes-induced podocytic change, namely hypertrophy (708). Under diabetic conditions, podocytes undergo hypertrophic changes like mesangial cells, resulting in increased cell size (517). The high glucose-induced podocyte hypertrophy can be ameliorated by Ang II AT1 receptor blockade, suggesting the involvement of RAS activation (365). Downstream effectors may include parathyroid hormone-related protein, which induces hypertrophy in podocytes via TGF-β1 and p27 Kip1 (530). In addition to high glucose, cyclic mechanical stretch, as a result of elevated intraglomerular pressure and impaired autoregulation (see below), can induce podocyte hypertrophy (487). The pathophysiological consequences of the increased size of podocytes are not clear. However, as shown by Wolf et al., the protective effect against the development of nephropathy in STZ-diabetic mice by deletion of the CDK inhibitor p27 Kip1 was associated with reduced podocyte hypertrophy and collagen type IV and laminin expression (708). Additional studies indicated that exposure of podocytes in culture to AGE-modified bovine serum albumin (AGE-BSA) induces cell-cycle arrest and cell hypertrophy by a mechanism that involves upregulation of p27 Kip1 (535). More recently, Bondeva et al. exposed differentiated mouse podocytes in culture to AGE-BSA. Using differential display and real-time PCR analyses they observed a downregulation of neuropilin-1 and confirmed a reduction in neuropilin-1 expression in glomeruli of diabetic db/db mice and in renal biopsies from patients with diabetic nephropathy compared to transplant donors (66). Additional in vitro studies indicated that AGE-BSA inhibited podocyte migration by down-regulating neuropilin-1. The authors proposed that decreased podocyte migration could lead to adherence of uncovered areas of the glomerular basement membrane to Bowman’s capsule thereby contributing to focal glomerulosclerosis.
Podocyte foot process widening and effacement
Foot process effacement is a result of retraction, widening, and shortening of the podocyte foot processes (see Fig. 6). The retraction results in large areas of flattened epithelium covering the capillary loop. This leads to a decrease in slit diaphragm pores per unit of length of GBM, causing a reduction in filtration surface. However, as postulated by Jefferson et al. (273), focal epithelial defects may allow increased protein flux across these denuded areas. This may be further facilitated by defects in slit diaphragm protein composition. This phenomenon is not specific for the diabetic kidney, and proteinuria may occur without effacement (730). Conversely, effacement is not always associated with proteinuria (273). Mundel and Shankland (419) proposed that the underlying mechanisms leading to foot process effacement include (1) changes in slit diaphragm-associated proteins, (2) actin cytoskeleton abnormalities, (3) alterations in the negative apical membrane domain of podocytes, and (4) interference with podocyte-GBM interaction.
Slit diaphragm abnormalities
The slit diaphragm, which bridges adjacent foot processes derived from different podocytes, functions as the ultimate molecular size filter. Several podocyte-specific proteins, such as nephrin, NEPH1, P-cadherin, and FAT-1 have been located at this region, whereas CD2AP, podocin, and zonula occludens-1 (ZO-1) are found in the slit diaphragm inserted region of the foot process [reviewed in (365)]. The relevance of these proteins has been documented by detection of foot process effacements, often associated with proteinuria, in patients with mutations of these genes or experimental models with targeted deletion or administration of antibodies to some of these molecules (365).
Proteinuria is a hallmark of diabetic nephropathy. Consequently, studies have focused on the changes in slit diaphragm-associated molecules in diabetic nephropathy. Thus far, nephrin is the best studied slit diaphragm protein in the diabetic context. Bonnet et al. (67) demonstrated a reduction in nephrin mRNA and protein expression in STZ-induced diabetic spontaneously hypertensive rats at 32 weeks after diabetes induction. These experimental findings are in accord with observations in kidney biopsies derived from patients with nephropathy and T1DM or T2DM (148). These changes in nephrin expression have been linked to activation of PKCα (403) and/or the RAS (67, 323, 344). Similar changes in nephrin expression were observed in cultured podocytes exposed to glycated albumin and Ang II (148). In contrast, Aaltonen et al. (1) showed an increase in nephrin mRNA levels in STZ-diabetic rats and in nonobese diabetic mice even before the development of significant albuminuria. In another study, nephrin protein expression was increased in renal cortex of db/db mice (603), whereas others found no changes in renal cortical nephrin expression in diabetes (337). The reasons for the disparate findings may be due to differences in duration of diabetes, the possibility of a biphasic response in nephrin expression and the influence of confounding factors, like hypertension.
Compared to nephrin, there have been relatively fewer investigations on the changes of other slit diaphragm proteins. P-cadherin mRNA and protein expression were reduced in diabetic glomeruli and in podocytes exposed to high glucose, a process mediated by PKC (722). Another study revealed decreases in ZO-1 protein expression in glomeruli of animal models of T1DM and T2DM and in high glucose-stimulated podocytes (520). In comparison, the expression of CD2AP and podocin has been reported to be unchanged under diabetic conditions (51, 329).
Actin cytoskeleton abnormalities
The actin cytoskeleton and several actin-binding proteins such as α-actinin-4 and synaptopodin are important in maintaining podocyte shape. The main cytoskeleton components of the foot process are microfilaments, containing a dense network of F-actin and myosin. Dai et al. (130) studied the podocytic cytoskeletal response to diabetes both in vitro and in vivo. They reported early high glucose- or diabetes-induced activation of the p38 module of MAPK and its downstream effector small heat-shock protein 25 (HSP25), resulting in a stabilization of fibrillar actin (FA) and prevention of its cleavage to G-actin, and, thus, maintenance of the actin cytoskeleton. This response was gradually lost with increasing duration of diabetes, correlating with slit diaphragm pore density and albuminuria. The authors concluded that p38 MAPK and HSP25 activation comprise an acute adaptation to glycemic stress with beneficial effects on actin cytoskeleton, which is being lost with increasing duration of the disease with structural and functional consequences.
α-Actinin-4 is an actin cross-linking protein, and its mutation in human and animal leads to proteinuria and foot process fusion. A recent study revealed that glomerular α-actinin-4 mRNA expression is increased in glomeruli from db/db mice with T2DM, which might reflect a repairing process (383).
Small GTPase RhoA and its downstream effectors Rho-associated kinases (ROCK) are the major signaling pathway involved in regulation of actin cytoskeleton (162). Therefore, the Rho-ROCK pathway has attracted considerable interest as a potential mediator of podocytic cytoskeletal changes in pro-teinuric kidney disease. The pathway mediates the podocyte cytoskeletal response (F-actin reorganization) to mechanical stress (160). More recently, RhoA has been linked to synaptopodin, another important actin-associated protein. Asanuma et al. reported that synaptopodin induced stress fibers in podocytes by competitive blocking of Smurf1-mediated ubiquitination of RhoA and promoted contractile stress fiber formation in the cell body (30). Moreover, gene silencing of synaptopodin in podocytes causes loss of stress fibers, formation of aberrant nonpolarized filopodia, and impairment of cell migration (30). This suggests that synaptopodin is essential for the integrity of the podocyte actin cytoskeleton and for the regulation of podocyte cell migration. Despite the functional role of synaptopodin in maintaining podocyte shape, only one study by Blanco et al. (60) investigated the changes in synaptopodin expression in diabetic nephropathy. They observed a trend toward a decrease in glomerular synaptopodin expression in obese Zucker rats with T2DM.
The above-mentioned evidence suggests that the Rho-ROCK pathway is important for maintaining podocyte cytoskeletal integrity and associated cell functions. However, as indicated by studies with ROCK inhibitors, the pathway plays a role in progressive kidney disease (262, 291, 443, 444), including diabetic nephropathy (202, 316, 323, 484). Interestingly, long-term inhibition of ROCK led to amelioration of foot process effacement in STZ-diabetic rats (484), and to nephrin upregulation in uninephrectomized diabetic rats with accelerated nephropathy (323). These effects were associated with blood pressure-independent modest antiprotein-uric effects (323, 484). Further studies are needed to establish whether the nephroprotection of ROCK inhibition is mediated via its effects on podocytic cytoskeleton.
Alterations in the negatively charged apical membrane domain of podocytes
Similar to GEC or GBM, the apical membrane domain of podocytes is negatively charged due to the surface anionic proteins, such as podocalyxin and podoplanin. As the cytoplasmic domain of podocalyxin is linked to the actin cytoskeleton, podocalyxin is also considered important in maintaining podocyte shape. mRNA and protein expression of podocalyxin have been shown to be suppressed in podocytes cultured in high glucose conditions, and glomerular podocalyxin protein expression was decreased in STZ-induced diabetic rats (152). Podoplanin is another apical membrane protein. In contrast to animals with puromycin aminonucleoside nephrosis (77), podoplanin mRNA expression was increased in human diabetic glomeruli and was positively correlated with the mean foot process width (329), suggesting a presumable compensatory reaction to the damage inflicted on the podocyte.
Interference with podocyte-GBM interaction
This phenomenon is discussed above as a possible cause of podocyte detachment. In addition to its role in podocyte detachment, decreased expression of α3β1 integrin is associated with foot process effacement, suggesting a role for α3β1 integrin in maintaining cell shape (9). However, the evidence indicating a role for this mechanism specifically in the patho-physiology of diabetic nephropathy is still scarce. In addition to α3β1 integrin, the DG protein complex, another podocyte anchoring molecule, is also important in keeping foot process shape. DG1, a heavily glycosylated peripheral membrane protein located in podocytes, is known to be decreased in various glomerular diseases with foot process effacement (517). However, this has not been observed in experimental diabetic nephropathy. In fact, an increase in DG1 expression was reported in isolated glomeruli from db/db mice (383) suggesting involvement of this protein in a glomerular repair process.
Altered secretory functions of podocytes
In addition to the functions described above, podocytes act as secretory cells. These actions and consequences on the surrounding cells and structures are described in detail in other sections describing alterations of the GFB in diabetes. In brief, podocytes exposed to the diabetic milieu could be an important source of the components of the RAS including Ang II, angiotensinogen and renin as indicated in studies in immortalized cultured podocytes (151). Podocytes also secrete GBM proteins (296) and act as an important source of glomerular VEGF (122) and lipid mediators, such as 12-lipoxygenase (296) and cyclooxygenase (COX) products (390).
The Unique Growth Phenotype and Morphological Changes of the Tubular System in the Diabetic Kidney
Growth of the kidney occurs with the onset of diabetes (116, 413). Understanding the involved mechanisms is crucial because diabetic kidney growth has been associated with the development of nephropathy (44, 64, 349, 738). Zerbini et al. (738) observed patients with T1DM and normal renal function for a mean duration of 9.5 years and found that increased kidney volume enhanced the risk of progression to microalbuminuria along with a faster decline in GFR. Diabetes-induced kidney growth in early diabetes may affect not only mesangial and podocyte function (see above), but also tubular hyperreabsorption, and as a consequence, glomerular hyperfiltration. In addition, the unique molecular mechanisms involved in early tubular growth in diabetes may set the stage for progressive tubulointerstitial damage. In the following we discuss the unique mechanisms involved in the growth of the tubular system and other morphological changes in the diabetic kidney.
Tubular growth in diabetes
The kidney in general and the proximal and distal tubules in particular, grow large from the onset of diabetes with the proximal tubule accounting for the greatest share of growth (116, 510, 511, 571). Rasch described in 50-day STZ-diabetic rats that the length of the proximal and distal tubules increased by 22% and 20%, respectively. Whereas no structural abnormalities were found in the proximal tubules, the total increase of the distal tubule was due to abnormal tubules confined to the cortex and the outer stripe of the outer medulla, which were filled with glycogen deposits, had strikingly few organelles and the basal infoldings were greatly reduced (see below for further details). In comparison, the total length of the collecting ducts was not affected in diabetes and most cells appeared normal (510). As described above for mesangial cells, both hyperplasia and hypertrophy are involved in diabetes-induced tubular growth (512), as illustrated in Figure 7.
Figure 7.

Regulation of tubular growth in the diabetic kidney. Illustrated is a conceptional frame work that links hyperglycemia to tubular growth including an early phase of hyperplasia that is followed by G1 cell-cycle arrest and development of hypertrophy and a senescence-like phenotype. Potential links to enhanced formation of extracellular matrix, inflammation, and tubulointerstitial injury are shown. See text for further explanations. ECM, extracellular matrix; EMT, epithelial mesenchymal transition; TSC, tuberous sclerosis complex.
The role of ornithine decarboxylase and polyamine synthesis in the growing diabetic tubule
The growth of the early diabetic kidney depends, at least in part, on overexpression of the enzyme, ornithine decarboxylase (ODC) (482). ODC converts ornithine to putrescine. This is the first and rate-limiting step in polyamine synthesis and appears to be a necessary early step for all cell division and for some forms of hypertrophy. In the early diabetic kidney, polyamines are required components for hyperplasia and most likely also for hypertrophy of the proximal tubule (140, 481, 482, 617). Renal ODC mRNA and protein expression are increased as a very early response to the onset of diabetes (358, 481, 617). Increased ODC activity can persist in the STZ-diabetic kidney for several weeks (482, 617). The specific mechanisms leading to sustained ODC expression in diabetic kidneys are not known, but the finding is characteristic of this form of kidney growth. The earliest change in the local environment of the proximal tubular cell in diabetes is the increase in filtered glucose leading to increased apical entry of sodium and glucose via sodium glucose co-transport (SGLT). One might imagine glucose and/or sodium in the cell providing some stimulus for the cell to grow (339, 595). However, based on immunostaining, Deng et al. proposed that the increase in ODC expression in early diabetes mainly occurs in the distal nephron and suggested that polyamines may pass from the distal to proximal tubule in a paracrine fashion to trigger proximal tubular growth (140). More studies are necessary to clarify this issue and what may trigger ODC expression in the distal tubule.
Growth of the proximal tubule in diabetes — early hyperplasia
For the early growth of the proximal tubule in the diabetic kidney, a period of hyperplasia precedes hypertrophy (250). DNA synthesis is increased in the proximal tubules of STZ-induced diabetes, peaking at day 2 (512). Numerous growth factors are associated with the early onset of diabetes and diabetic kidney growth, including IGF-I, HGF, PDGF, fibroblast growth factor (FGF), VEGF, epidermal growth factor (EGF), and DAG (134, 176, 199, 538, 712). All of these factors can induce ODC activity (549). IGF-I may be the most studied mediator of the very early growth phase in diabetes, and its effects are attributed principally to activation of the phosphoinositide 3-kinase (PI3K)/Akt pathways (see Fig. 7). At the onset of STZ-diabetes, the rapid yet transient renal induction of IGF-I (171, 506) temporally correlates with the upregulation of renal ODC expression and activity (140, 481, 617), induction of intracellular polyamines in the kidney cortex (140), and the proliferative phase. Also VEGF, which is induced in both the STZ-model (T1DM) and the db/db mouse model (T2DM), increases protein synthesis in renal epithelial cells utilizing the PI3K/Akt pathway (569). Diabetes also enhances proximal tubular activity of PKCβ1 (489). PKC activation can produce a myriad of consequences in diabetes including a mitogen-induced early proliferation phase (85). PKCβ has been implicated in Akt activation in the renal cortex of diabetic rats (719). Activation of PI3K/Akt (574) and PKC pathways (245, 277) can both increase ODC activity. PKC activation of ODC is likely by induction of immediate early genes c-myc, c-jun, and c-fos (232, 698). In accordance with a role of PKCβ in kidney growth, the early diabetes-induced increase in kidney weight was blunted in mice lacking this PKC isoform (401).
In rats with T1DM, renal growth is associated with reduced phosphorylation of AMP-activated protein kinase (AMPK) (351). Phosphorylated AMPK inhibits the mammalian target of rapamycin (mTOR) activity by phosphorylating and activating tuberous sclerosis complex (TSC). TSC negatively regulates mTOR complex 1 (mTORC1) by inhibiting Rheb, a small cytoplasmic GTPase and activator of mTORC1 [for review see (369)]. In accordance, mTOR activity is enhanced in the diabetic kidney and increasing AMPK phosphorylation by pharmacological means reversed mTOR activation and inhibited renal growth without affecting hyperglycemia (351). Together these studies propose a link between diabetes-induced growth factor filtration and expression, activation of PKC, inhibition of AMPK, and activation of both mTORC1 and ODC in the early tubular proliferation in diabetes (see Fig. 7).
Growth of the proximal tubule in diabetes — switch from hyperplasia to hypertrophy
The diabetic kidney switches from hyperplastic to hypertrophic growth very early in the course of hyperglycemia, for example, at around day 4 in the model of STZ-diabetes (250), which matches the time frame of hyperplasia observed using 5-bromodeoxyuridine (BrdU) incorporation (140), an indicator of cell-cycle progression. TGF-β1 is a critical mediator of this growth switch in diabetes and critical to diabetic kidney growth in general (219) (see Fig. 7). PKCβ has been implicated in the induction of TGF-β in the diabetic kidney (260, 308, 330, 331). PKCβ1 has been localized to the proximal tubule of the rat (490) and its membrane-associated expression (activation) was further upregulated in response to STZ-diabetes (489). Other studies indicated that ERK and p38, which are activated in renal tubular cells of STZ-diabetic rats, may mediate high glucose-induced TGF-β expression and cellular hypertrophy (190). High glucose administered to primary tubule cells from TGF-β KO mice induced an increased rate of proliferation relative to isolated cells from wild type littermates, and no hypertrophy (105). Expression of the CDK inhibitor p27KIP1 (p27) increases in response to hyperglycemia or diabetes, which, based on studies in non-tubular cells, can be attributed to induction by PKC (711) and TGF-β (288). TGF-β can arrest cells in the G1 phase of the cell cycle by induction of p27 (288). In mesangial cells, it is this G1 arrest that likely switches the hyperplastic growth to the hypertrophic phenotype observed in diabetes (32, 417, 708, 710). In accordance, targeted disruption of the p27 gene does not affect hyperglycemia in the STZ diabetic model, but decreases glomerular hypertrophy (32, 708) (also see sections on glomerular pathophysiology above). Experimental diabetes also increases the renal expression of p21 (16, 548). Moreover, the loss of p21 increases tubular cell proliferation, consistent with a role of p21 in the switch from hyperplasia to hypertrophy in the diabetic kidney (16) (see Fig. 7).
By 7 days after STZ administration, renal ODC activity is still elevated (482, 617), and may drop to below control by 3 or 4 weeks (358, 587). A temporal activation of ODC activity may be attributed to temporal changes in the local growth factor/mitogen content. For example, IGF-I induction is transient, with levels peaking at day 3 and normalizing by day 7 (171). Therefore, the sustained hypertrophy observed later in diabetes may be more a consequence of decreased proteolysis than cell-cycle arrest (179), and may also not be dependent upon growth factor/PI3K/Akt induction (179).
Tubular senescence in the early diabetic kidney
Satriano et al. recently demonstrated that tubular hypertrophy in the STZ-diabetic kidney is associated with a phenotypic transition to senescence (548) (see Fig. 7). Senescence is a tumor suppressor mechanism to halt cells from replicating and passing on a potentially damaged genome. Like early diabetic hypertrophy, senescent arrest requires upregulation of CDK inhibitor (518). Although the prototypical senescent arrest involves the temporal induction of p21WAF1 (p21) and p16INK4A (p16), studies demonstrate that induction of p27 can impose a senescent-like growth arrest (17, 729). Satriano et al. further described that proximal tubule cells in culture transition to senescence in response to oxidative stress. Moreover, STZ-diabetic kidneys exhibited an early temporal induction of growth phase components followed by their suppression at day 10 after STZ application concurrent with the induction of CDK inhibitors p16, p21, and p27 and markers of senescence including expression of senescence associated beta-galactosidase activity in cortical tubules (548). Notably, kidneys of patients with T2DM and nephropathy display an accelerated senescent phenotype in tubule cells and, to a lesser extent, in podocytes (675). Senescent cells exhibit several aspects of a fairly well differentiated phenotype; however, this phenotype is skewed in several parameters (518), including the release of inflammatory cytokines, production of growth factors and ECM, and a resistance to apoptotic remodeling (18, 583). Thus, one may speculate that the senescent arrest of tubular cells may not only alter the early transport phenotype [e.g., the “salt paradox” of the diabetic kidney (651)] but also contribute to the development of diabetic nephropathy (see Fig. 7).
Evidence for epithelial-mesenchymal transition in the diabetic kidney
Tubulointerstitial fibrosis represents the final common pathway of many forms of kidney disease including diabetic nephropathy (426). The accumulation of fibrotic material in the tubular interstitium tends to accompany disease progression, correlating with a gradual decline in renal function in patients with T1DM (302). Much of the accumulated interstitial matrix in fibrosis is synthesized by interstitial myofibroblasts. In addition to interstitial fibroblasts and circulating precursor cells, a proportion of interstitial myofibroblasts in fibrotic kidneys can originate from tubuloepithelial cells through epithelial to mesenchymal transition (EMT) (264, 374, 433, 599, 600, 725). In the model of unilateral ureter occlusion, very elegant studies using bone marrow chimeras and transgenic reporter mice indicated that EMT may account for at least a third of the fibroblasts (264). EMT is associated with an early decrease in the expression of adhesion proteins that keep cell polarity and intercellular junctions, including E-cadherin and zonula occludens protein ZO-1 (234, 287). Upregulated EMT markers include metalloproteinases MMP-2 (108, 109, 601) and MMP-9 (601, 725) involved in the degradation of the tubular basement membrane, as well as alpha-SMA (236, 433) and integrin linked kinase (ILK) (367, 368), which increase cell contractility/motility and movement, respectively. EMT leads to enhanced formation of ECM proteins (e.g., fibronectin) (287, 599).
The extent to which EMT contributes to renal fibrosis in vivo, especially in human, remains a matter of intense debate (371, 498, 679) and may depend on the experimental and clinical context [for review see (375)]. Features of EMT have been observed in human renal biopsies from diseased kidney, in which the number of tubuloepithelial cells with EMT features were associated with serum creatinine and the degree of interstitial damage (513). Markers of EMT have also been observed in kidneys of STZ-diabetic uninephrectomized mice (368), STZ-diabetic Wistar Kyoto (457), Sprague Dawley (91, 457), uninephrectomized Sprague Dawley (323), and Ren-2 rats (240), and in a renal biopsy from a patient with T1DM (457). Recent studies using cell lineage-tracing techniques indicated that a substantial number of interstitial fibroblasts, including in the diabetic kidney, also come from capillary endothelia by endothelial-to-mesenchymal transition (363, 736). Endothelial cells are a specialized type of epithelia and, thus, the latter process represents another form of EMT occurring in the injured kidney.
TGF-β1 is upregulated in the kidney in experimental diabetes and in patients, plays a critical role for tubulointerstitial fibrosis (493), and is the most potent inducer that is capable of initiating and completing the entire EMT course (90, 234, 374) (see Fig. 7). Other endogenous factors of EMT include EGF, FGF-2, IL-1, Ang II, AGEs, Kruppel-like factor 6 (KLF6), CTGF, and MMP-2, part of which act through TGF-β1 [KLF6 (240), IL-1, MMP-2] or exhibit a dramatic additive or synergistic effects with TGF-β1 to promote EMT (EGF, FGF-2, Ang II) [for review see (374)]. Signaling events in EMT downstream of TGF-β receptor activation include activation of MAPK, Smad/ILK, and CTGF (90, 91, 234, 287, 374). Recent results suggested that parathyroid hormone (PTH)-related protein (PTHrP), TGF-β1, EGF, and VEGF might cooperate through activation of ERK1/2 to induce EMT in renal tubuloepithelial cells (27). In this regard, PTHrP and its receptor are upregulated in tubules and glomeruli of STZ-induced diabetic mice (267) and a preliminary report has shown that PTHrP upregulation also occurs in both tubules and glomeruli in patients with diabetic nephropathy (72). In primary cultured renal proximal tubular epithelial cells, hyperglycemia induces EMT through ROS, PI3K/Akt, GSK-3beta, snail, and beta-catenin (352) (see Fig. 7).
Conversely, application of BMP-7 (737) and HGF (129) has been shown to inhibit fibrosis and markers of EMT in the kidney in vivo, the latter in the diabetic kidney of mice. HGF appears to specifically induce Smad transcriptional co-repressor SnoN expression, which in turn physically interacts with activated Smad-2 by forming a transcriptionally inactive complex and blocks the trans-activation of Smad-mediated genes, including EMT mediator ILK (368). Thus, there may be intrinsic renoprotective factors that can safeguard tubular epithelial phenotypes by preventing EMT, which may have therapeutic potential.
A partial EMT phenotype has been recently described (356) that is not necessarily associated with E-cadherin down-regulation, and there is evidence that this process might operate in the diabetic kidney of mice (687). It has been postulated that the entire classical EMT program observed primarily in vitro, with the associated decrease in E-cadherin, may not be critical for the development and progression of tubulointerstitial fibrosis (687). Both TGF-β and CTGF increase ECM expression independently of E-cadherin. While TGF-β lowers E-cadherin, CTGF (a driver of partial EMT) increases E-cadherin expression. In the nondiabetic context, an increase in E-cadherin has been recently reported in rats with ureteral obstruction, a classical model of EMT-mediated kidney fibrosis (144). Further studies are needed to better understand the relevance, the molecular signature, and the therapeutic potential of EMT in the diabetic kidney, especially in human.
Hyperglycemia Induces Glycogen Accumulation in Thick Ascending Limb and Further Distal Cortical Nephron Segments
In hyperglycemic conditions, cells of the thick ascending limb (TAL) and, to a lesser extent, of the distal convoluted tubule and the collecting duct show glycogen deposits, also know as glycogen nephrosis or Armanni-Ebstein lesions (54, 239, 510). Holck and Rasch reported that on a continuous basis, glycogen is present only in the cortical TAL, whereas scattered dot-shaped and diffuse glycogen accumulations are discretely distributed in the MD, the distal convoluted tubule and the cortical collecting duct system (239). The cells appear either empty (“clear cells”) or full of a PAS-positive material, digestible with alpha-amylase, and at the electron microscope level the cytoplasm contains glycogen-like granules, strikingly few organelles and the basal infoldings are greatly reduced (510). Notably, these abnormalities are confined to the cortex and the outer stripe of the outer medulla (510). The fate of glycogen-accumulating “clear cells” and the role in diabetic kidney disease is still unclear. Under severe hyperglycemic conditions and ketoacidosis, apoptosis of these cells may occur (39).
Holck and Rasch proposed that the glycogen accumulation in the distal nephron of diabetic rats correlates with the delivery of glucose via the tubular fluid (239) (see Fig. 8). Alternatively, glycogen accumulation in the TAL in diabetes may reflect activation of GS. Studies by Cammisotto et al. in normal rats indicated that globular adiponectin (derived from white adipocyte secretion and being filtered by the glomeruli) through binding to its receptor, ADIPOR1, in the luminal membrane of the TAL activates AMPK leading to inhibition of GS. Moreover, they showed that this regulation is impaired in STZ-diabetic rats, which might contribute to glycogen nephrosis (95) and is similar to adiponectin resistance in other tissues (88). This may be due to high glucose-induced dephosphorylation of AMPK (351), inasmuch as high glucose-activation of the phosphatase PP2A can dephosphorylate AMPK and prevent the association of AMPK subunits (127, 201, 278). Moreover, TALs from diabetic animals have higher concentrations of glucose-6-phosphate (95), an allosteric activator of GS. Further studies are necessary to better understand the tubular formation of glycogen deposits and their relevance in the diabetic kidney.
Figure 8.

Hyperglycemia induces glycogen accumulation in thick ascending limb and further distal cortical nephron segments. Glycogen accumulation, also know as glycogen nephrosis or Armanni-Ebstein lesions, may relate to increased delivery of glucose via the tubular fluid or peritubular capillaries. Glycogen accumulation may also reflect activation of glycogen synthase, due to accumulation of the allosteric activator, glucose-6-phosphate. Adiponectin is secreted from white adipocytes and signals through its receptor, ADIPOR1, to activate AMP-activated protein kinase (AMPK) thereby inhibiting glycogen synthase. Thus, glycogen synthase can be activated as a consequence of reduced adiponectin levels or adiponectin resistance with impaired activation of AMPK due to activation of phosphatase PP2A. See text for further details.
Mechanisms of Tubulointerstitial Injury in the Diabetic Kidney
Glomerular mesangial expansion and podocyte loss are important early features of diabetic nephropathy, whereas tubulointerstitial injury and fibrosis are critical for progression of diabetic nephropathy to kidney failure. Increased proximal tubular basement membrane width is an integral component of early nephropathology in patients with T1DM (80). Tubulointerstitial injury is a final common pathway to end-stage renal failure in many forms of kidney disease (425,426) and the focus of this section. Important primary factors contributing to tubulointerstitial injury include hyperglycemia, proteinuria, AGEs and chronic hypoxia. For recent reviews see (112, 297, 382, 581, 751). Figure 9 aims to integrate and illustrate some of the aspects that are discussed in the following.
Figure 9.

Mechanisms of tubulointerstitial injury in the diabetic kidney. Illustrated is the interplay of hyperglycemia, luminal factors (derived from glomerular filtration and tubular release), reabsorption, and blood flow in the interaction of tubular cells with fibroblasts and inflammatory cells. TGF-β, chemokines, and the complex interactions between advanced glycation end products (AGE), hypoxia and oxidative stress play key roles in the development of diabetic tubulointerstitial injury. ECM, extracellular matrix. See text for further details.
Enhanced glomerular filtration of proteins, growth factors, and other molecules in diabetes
The mechanisms by which proteinuria results in tubular and interstitial damage may include lysosomal injury, energy depletion and direct tubular injury by various components in the proteinuric urine, such as AGEs, transferrin, albumin, albumin-bound fatty acids, and complement components. Urinary albumin induces pro-inflammatory chemokines in proximal tubular epithelial cells such as IL-8 (382, 611) and monocyte chemotactic protein 1 (MCP-1), the latter is mediated by NFκB (694) (see Fig. 9). In addition, ultrafiltered growth factors, such as IGF-I, HGF, and TGF-β have been linked to proteinuria-associated interstitial fibrosis in the diabetic kidney (688, 690, 691). The glomerular filtration of fatty acids bound to albumin has also been implicated in tubular damage. Fatty acids can activate peroxisome proliferator-activated receptor-gamma (PPAR-γ) in human proximal tubular cells and induce apoptotic cell death (28). This, however, is in contrast with the tubuloprotective effects of PPAR-γ agonists in animal models of diabetes against oxidative injury (588), indicating the need for further clarification. Proteinuric urine contains components of the complement system and, in addition, both transferrin and IL-2 can stimulate C3 synthesis (83, 237). In this regard, the degree of nonselective proteinuria correlates with both the level of intratubular C3 activation as well as tubulointerstitial injury (425).
Linking high glucose to tubulointerstitial fibrosis
It has been proposed that it is the tubular injury which initiates the deleterious cascade of events in chronic kidney disease (173). High glucose concentrations induce collagen gene transcription and secretion in a murine cortical tubular cell line (750). Similarly, exposure of primary cultures of human renal proximal tubular cells to high glucose increased the amount of type IV collagen and fibronectin (494). This was associated with a net decrease in gelatinolytic activity which accounted for the accumulation of fibronectin and type IV collagen (492). Exposure of human proximal tubular cells and cortical fibroblasts to high extracellular glucose concentrations can directly stimulate cell growth and collagen synthesis, independent of hemodynamic, glomerular or vascular pathology (283). The polyol pathway has been implicated in high glucose induced alterations in proximal tubular cell matrix generation (62, 418, 749).
In addition to alterations in the turnover of “normal” matrix constituents, the diabetic state may also lead to the de novo induction of “abnormal” structural elements, such as hyaluronic acid. Both elevated glucose and IL-1β can increase hyaluronic acid synthase 2 and HA generation by proximal tubular cells via activation of NF-kB, whereas the profibrotic cytokines TGF-β1, PDGF, and FGF2 did not increase hyaluronic acid synthesis (282). The functional role of hyaluronic acid in the diabetic kidney remains to be established.
A role for transforming growth factor-β
Several growth factors contribute to the pathogenesis of diabetic nephropathy, including TGF-β, CTGF, VEGF, GH, and IGF-I with some evidence being available also for EGF and PDGF (see Fig. 9). They are generally expressed in the normal kidney but their levels further increase in diabetes, where they have been implicated in the control of cell hypertrophy, proliferation and survival, as well as renal matrix composition [for review see (112)]. The expression of TGF-β is elevated in human and experimental diabetic nephropathy (723) and its upregulation is one of the key factors responsible for the fibrotic changes and scarring observed in the diabetic kidney (493, 747). Importantly, TGF-β is relevant to progression of renal disease as indicated by studies in db/db mice, a model of T2DM, in which treatment with a monoclonal anti-TGF-β antibody prevented renal insufficiency (748). In the diabetic kidney, the synergism of high glucose concentrations with cytokines such as PDGF or the pro-inflammatory macrophage-derived cytokine, IL-1β, can stimulate TGF-1 synthesis by proximal tubular cells (495, 496) (see Fig. 9). The latter studies with IL-1β further indicated that glucose stimulated leukocyte recruitment contributes to tubular TGF-β1 protein synthesis indicating a close link between inflammation and the development of fibrosis in the diabetic kidney.
Reciprocal paracrine activation of proximal tubular cells and fibroblasts
Notably, in chronic kidney disease, the regions of active interstitial fibrosis predominantly exhibit a peritubular rather than perivascular distribution (21), indicating that injured proximal tubular cells may release fibrogenic signals to cortical fibroblasts. In fact, TGF-β1 can stimulate the release of pre-formed basic fibroblast growth factor (bFGF) from renal proximal tubular cells (284). Glucose can also stimulate the interstitial fibroblasts to increase their synthesis of ECM and fibrillar collagens via an autocrine TGF-β1 system (221) (see Fig. 9). Importantly, there is evidence for reciprocal paracrine activation of proximal tubular cells and fibroblasts. Proximal tubular cells in the human kidney modulate the biological behavior of neighboring cortical fibroblasts through paracrine mechanisms, which include the production and release of the AB heterodimer of PDGF and TGF-β1 (280). Vice versa, studies in human renal fibroblasts indicated that they can modulate proximal tubule cell growth and transport via the secretion of IGF-I and IGF binding protein-3 (281) (see Fig. 9). These interactions in the tubulointerstitium appear to be modified by the tubular basement membrane components laminin and collagen type IV: conditioned medium from rat proximal tubular cells cultured on collagen-type IV stimulated proliferation of cortical fibroblast. Moreover, co-culture of proximal tubular cells on laminin with cortical fibroblasts suppressed the activity of MMPs collagenase and gelatinase in both cell types (361). Thus, a complex cross-talk between proximal tubular cells, ECM proteins and fibroblasts appears to contribute to the regulation of ECM and one may speculate that tubular injury in diabetes alters these interactions and contributes to tubulointerstitial fibrosis.
A role for connective tissue growth factor
CTGF is a prosclerotic cytokine that is mainly induced by TGF-β and involved in the regulation of matrix accumulation (542). Vice versa, CTGF may enhance the profibrotic effects of TGF-β by enhancing TGF-β expression (212) as well as the ability of TGF-β to bind to its receptor at low TGF-β concentrations (6). Urinary CTGF is detectable in diabetic patients (see Fig. 9) and the levels correlate with the degree of microalbuminuria (197, 522). Wang et al. found that CTGF mRNA levels were increased in the renal cortex of rats with STZ-induced diabetes and immunohistology localized the CTGF protein expression particularly to dilated-appearing proximal tubules, where it appeared to colocalize with IGF-I (688). These studies also provided evidence that glomerular ultrafiltrate from diabetic rats, which contained bioactive TGF-β and HGF, can induce CTGF expression in proximal tubular cells (see Fig. 9). CTGF has an IGF-binding domain and binds to IGF-I, and studies in NRK-49F renal fibroblast cells showed that IGF-I increased the activity of CTGF toward the expression of collagen α1III (688). Guha et al. evaluated the specific role of CTGF in the diabetic kidney by using a CTGF ASO (212). In STZ-diabetic mice, CTGF-ASO reduced renal CTGF expression as well as proteinuria, albuminuria, and kidney growth. In db/db mice, CTGF-ASO reduced serum creatinine and attenuated urinary albuminuria and proteinuria. CTGF-ASO also reduced renocortical expression of fibronectin, collagen (I and IV) and PAI-1, an inhibitor of matrix degradation. Further analyses implicated a role for CTGF in diabetes-induced phosphorylation of p38 MAPK and its downstream target CREB that have been implicated in regulating matrix proteins (212).
A potential role for serum- and glucocorticoid-inducible kinase 1
The serum- and glucocorticoid-inducible kinase 1 (SGK1) is a serine-threonine kinase that has been implicated in many forms of fibrosis and is stimulated by multiple diabetes-associated factors and conditions including high glucose, hyperosmolality, TGF-β, FGF, PDGF, AGEs, and insulin (341). In primary culture of human proximal tubular cells, high glucose increased SGK-1 mRNA and protein expression (538). Moreover, high glucose and overexpression of SGK-1 increased Na/H-exchanger NHE3 mRNA and EGFR phosphorylation. SGK-1 overexpression increased proximal tubular cell growth, progression through the cell cycle, and inhibited apoptosis. Upregulation of tubular SGK-1 mRNA was confirmed in STZ-diabetic animals in vivo. These in vitro and in vivo effects of high glucose were sensitive to the EGFR blocker PKI166, indicating that the effects of high glucose on proximal tubular cell proliferation, reduced apoptosis and increased NHE3 mRNA levels are mediated by EGFR-dependent upregulation of SGK-1 (538). Importantly, SGK transcription was markedly enhanced in patients with diabetic nephropathy, with particularly high expression in mesangial cells, interstitial cells, and cells in TAL of Henle’s loop and distal tubules (343). SGK1 message and protein expression is also upregulated in the kidney of STZ-diabetic mice, where it has been implicated in enhanced proximal tubular glucose reabsorption (7) and fibronectin formation (168). In fibroblasts, SGK1 can induce CTGF promoter activity and CTGF expression through activation of NFκB (665). Thus, SGK-1 has been linked to TGF-β and CTGF and the outlined studies implicate potential roles of the kinase in the early and late stages of the diabetic kidney including potential effects on kidney growth, transport, and fibrosis. Further studies are necessary to better define the role of SGK1 in the diabetic kidney in vivo and determine the relevance of SGK-1 for the long-term renal outcome of the diabetic kidney.
Interrelation between hypoxia and oxidative stress in the diabetic kidney
Abnormalities in oxygen metabolism have been implicated in the development and progression of diabetic nephropathy and include hypoxia, oxidative stress, nitrosative stress, and advanced glycation and/or carbonyl stress [for review see (183, 187, 297, 409)]. The primary cause for local oxidative stress in the diabetic kidney remains a matter of debate (see earlier section on ROS). Most of the ROS are generated during mitochondrial oxidative phosphorylation and smaller amounts via the NADPH-oxidase system (200, 293, 366) (see Fig. 9). Hypoxia has also been implicated as a cause of oxidative stress in the diabetic kidney and in the pathophysiology of diabetic nephropathy (581).
Renal hypoxia has been demonstrated in animal models of T1DM and T2DM (200, 268, 469, 519, 531), particularly in the outer medullary region including the medullary TAL (519, 531). A role of hypoxia in chronic renal disease was propeposed by (174) and has been confirmed in human and animal models including the diabetic kidney (426, 581). Hypoxia can be due to enhanced oxygen consumption, as shown ex vivo in cortical and medullary tubular cells of STZ diabetic rats (469). Changes in vasoactive factors such as Ang II and/or NO can lower postglomerular blood flow in diabetes thereby contributing to hypoxia (471–473, 582). In later stages of the disease, glomerular lesions and interstitial vascular rarefaction, which is associated with reduced VEGF-A expression in human diabetic nephropathy (372), further impair peritubular blood flow and oxygen delivery to the tubules (426). Hypoxia can be further enhanced by concurrent anemia (35) and enhanced sodium transport load and oxygen consumption in remnant nephrons (429) (see Fig. 9).
In general, the cells in hypoxia depend on anaerobic glycolysis to generate ATP. However, due to the residual low oxygen supply, some level of oxidative ATP production remains via the Krebs cycle and electron transport chain. Electron leakage from the mitochondrial electron transport chain under hypoxic conditions results in excessive ROS generation. Studies in diabetic rats indicated increased mitochondrial expression of uncoupling protein 2 (UCP-2) in renal proximal tubular cells, which mediates mitochondrial uncoupling and increased O2 consumption (186). Whereas this may protect against diabetes-induced oxidative stress, it may aggravate hypoxia (see Fig. 9).
Hypoxia can enhance oxidative stress as outlined. Vice versa, oxidative stress can enhance hypoxia. Overproduction of ROS, in part due to activation of NADPH oxidase with translocation of p47phox to the membrane, limits NO generation in the diabetic kidney (29), which enhances hypoxia by affecting the use and supply of oxygen (411, 471, 473). The latter is enhanced through NO quenching by AGEs (89, 673) or ROS (235), and NO capture by glucose (82). Superoxide can induce renal vascular constriction (752) and enhance tubular salt reabsorption (285), which can enhance renal hypoxia., treatment of STZ-diabetic rats with the antioxidant alpha-tocopherol prevented diabetes-induced disturbances in oxidative stress, oxygen tension, and oxygen consumption. Notably, diabetic hypertrophy and glomerular hyperfiltration were unaffected by alpha-tocopherol (469).
Hypoxia and oxidative stress also affect the function of the endoplasmatic reticulum (ER) which is critical for the processing and function of newly synthesized proteins (721). Impaired ER function—ER stress—has been documented in the diabetic kidney (373). ER stress but also oxidative stress and nitrosative stress (50, 458), carbonyl stress and AGE formation (410, 427) are sensitive to Ang II AT1 receptor blockade (or ACE inhibition). In addition, these maneuvers can improve tissue hypoxia by increasing postglomerular peritubular blood flow (384) and/or through preferred activation of Ang II AT2 receptor activation and NO production (470) as shown in other models of kidney injury. A protective role of these effects has been implicated in experimental models of T2DM (268), which support a prominent role of the RAS in the diabetic kidney beyond blood pressure regulation.
Stimulation and inhibition of hypoxia inducible factor in the diabetic kidney
Defense against hypoxia involves the hypoxia-inducible factor (HIF) (561). Its activation induces a wide variety of genes [e.g., erythropoietin, VEGF, hemoxygenase 1 (HO-1)], which can help to protect hypoxic tissues. The cell-protective effect of HO-1 relies, at least in part, on its ROS scavenging ability (5). The stability of HIF is reduced through hydroxylation by the oxygen sensor prolyl hydroxylase (PHD). Rosenberger et al. showed that STZ-diabetic and Cohen diabetes-sensitive rats transiently upregulate the hypoxia marker pimonidazole (PIM) and HIF-1a, HIF-2a, as well as HO-1 in the renal outer medulla, with increases at 14 and 30 days and normalization after 90 days (531). PIM was detected in TALs and CDs, increasing with distance from vascular bundels. Whereas HIF-1a appeared in the same cells as PIM, HIF-2a was localized to capillary endothelial cells and in the interstitial cells of the inter-bundle zone. The HIF target gene HO-1 was also detected in interstitial cells (531). Studies by Katavetin et al. showed that the activation of the HIF hypoxia-responsible element pathway is blunted in STZ-diabetic rats (301). Superoxide has been proposed to reduce HIF activity (726). In accordance treatment of diabetic rats with the SOD mimetic tempol increased HIF-1a, HIF-2a, and HO-1 in the renal outer medulla (531) indicating that oxidative stress in the diabetic kidney may suppress HIF activation (see Fig. 9). Similarly, high glucose levels inhibit the hypoxia-induced activation of HIF and VEGF expression in rat proximal tubular cells in vitro; this effect was reversed by the antioxidant α-tocopherol (301).
The role of HIF was further tested in spontaneous hypertensive rats (SHR)/NDmcr-cp rats, a hypertensive model of T2DM, by application of cobalt (for 20 weeks), which inhibits HIF degradation by PHD (454). Cobalt did not affect hypertension and metabolic abnormalities but reduced proteinuria and histological kidney damage. This was associated with increased expressions of HIF-regulated genes, including erythropoietin, VEGF, and HO-1. Moreover, cobalt reduced the renal expressions of TGF-β and AGE formation (454). The toxicity of cobalt prohibits its use in humans. Small molecular activators of HIF that are currently developed include inhibitors of PHD1, which can induce hypoxia tolerance by reprogramming basal oxygen metabolism (25, 559), without impairing the regulation of angiogenesis and erythropoiesis mediated by PHD2 (608, 609).
Linking hypoxia and oxidative stress to the accumulation of extracellular matrix
in vitro studies linked renal hypoxia to enhanced ECM: studies in human proximal tubular epithelial cells showed that hypoxia (1% O2, 24 h) increased total collagen production, which was associated with decreased MMP-2 activity and increased tissue inhibitor of metalloproteinase-1 (TIMP-1) protein (461). Collagen IV mRNA levels decreased while collagen I mRNA increased, suggesting induction of interstitial collagen. Although hypoxia stimulated TGF-β production, this did not appear to mediate the pro-fibrogenic stimulus of hypoxia (461). Similarly, hypoxia (1% O2) applied to human renal tubular endothelia, interstitial fibroblasts and microvascular endothelial cells in vitro increases ECM production, which was associated with decreased turnover via effects on matrix-degrading enzymes and their inhibitors (446). These effects were independent of HIF but involved activation of both PKC- and tyrosine kinase-mediated signal transduction pathways (446). Superoxide activated ERK-dependent fibrosis-stimulatory factor and ECM gene transcription have been implicated in STZ-diabetic rats. These findings link both hypoxia and oxidative stress to the accumulation of ECM and fibrosis.
Inflammatory response in the diabetic kidney
Renal inflammation is an important pathway that is common to diverse chronic kidney diseases, preluding renal fibro-sis and driving progression to end-stage renal failure (153). Studies in STZ-diabetic rats have demonstrated prominent macrophage infiltration (545, 732). Moreover, studies of renal biopsies taken from patients with T2DM have suggested a role for macrophages and their products in the initiation of the pathological changes of human diabetic nephropathy (193). Chemokines are important for the recruitment of specific inflammatory cells into the kidney including MCP-1, which has a key role in the recruitment of macrophages as indicated in diabetic animal models and in renal biopsies from patients with T1DM and T2DM [for review see (537)]. As mentioned above, urinary albumin can induce pro-inflammatory chemokines in proximal tubular epithelial cells including IL-8 (611) and MCP-1 (694) (see Fig. 9).
Interaction between growth factors, such as IGF-I, HGF, and TGF-β, and their respective receptors in the apical membranes in proximal and distal tubules and collecting ducts enhances the levels of MCP-1, RANTES, and PDGF-β, which activate the proliferation of fibroblasts but also of macrophages (112, 237, 689). Many proinflammatory mediators, including chemokines MCP-1 and RANTES, are under the tight control of transcription factor NFκB (213). ROS are thought to be critical inducers of renal inflammation and destruction (455, 572). In accordance, recent studies in renal epithelial cells and in animal studies showed that candesartan, by a direct antioxidant effect, independent of Ang II AT1 receptor blockade, inhibits NFκB activation and chemokine production, including MCP-1 and RANTES, thereby ameliorating renal inflammation and injury (104) (see Fig. 9).
Stimulation of proximal tubular TGF-β1 synthesis by IL-1β derived from pro-inflammatory macrophages has been described in the diabetic kidney (496). High glucose has also been shown to induce macrophage inflammatory protein-3 alpha (MIP-3α) in human proximal tubular cells in a TGF-β1-dependent way (507) (see Fig. 9). Moreover, MIP-3α was upregulated in the dilated tubules of diabetic rats, which were surrounded by CD3-positive cells, a marker of T lymphocytes. Notably, this upregulation was attenuated in the presence of an ACE inhibitor (507). Blockade of the renin-angiotensin-aldosterone system is currently the only clinical used anti-inflammatory strategy to treat diabetic nephropathy. Studies in STZ-diabetic rats revealed that ACE inhibition by lisinopril reduced the renocortical accumulation of interstitial cells that have activated PKCβ2 and express markers of antigen-presenting cells and of macrophages that are infiltrating affected tissue (489). PKCβ2 has been implicated in glucose-dependent monocyte transformation to macrophages (101). This would indicate that pharmacological inhibition of ACE or PKCβ2 could have nephroprotective effects in diabetes in part by inhibiting this transformation and renal inflammation. The clinical relevance of new experimental approaches like chemokine receptor antagonists or even immunosuppressive therapy to prevent/treat diabetic nephropathy remains to be established (537).
Nuclear hormone receptors in the diabetic kidney
Many nuclear hormone receptors have been suggested to provide protection against metabolic, inflammatory and cardiovascular diseases. A number of studies indicated a potential role for PPARα (476, 477), PPARγ (532), PPARδ (532), estrogen receptors (ER) (100, 387), hepatic nuclear factor 4 alpha (HNF4α) (438) and, especially, the vitamin D receptor (VDR) (135, 741, 743, 744) and the farnesoid X receptor (FXR) (279, 693) in the pathogenesis of diabetic nephropathy. Notably, activation of all of these nuclear hormone receptors has been linked to lowering proteinuria, while some have, in addition, been implicated in attenuating mesangial expansion (PPARs, ER, VDR, FXR), renal fibrosis (PPARs, ER, FXR), and macrophage infiltration (PPARs, VDR, FXR) [for review see (692)]. Although there is first evidence that at least some of these receptors play a direct role in the diabetic kidney, the interpretation of renal outcomes in the setting of diabetes has been confounded by extrarenal effects on metabolism and other functions with potential secondary effects on the kidney. Studies using animal models with cell-specific knockdown of these genes in the kidney will greatly facilitate to separate the systemic versus kidney-specific effects of these nuclear hormone receptors in the diabetic kidney.
Glomerular Function in Diabetes
Early glomerular hyperfiltration in T1DM, which is often associated with nephromegaly, has been recognized for many years (413). Similar studies have more recently been performed in the much larger patient population with T2DM. These studies revealed a significant variation in renal hemo-dynamics, but provided clear evidence for elevations of GFR and renal plasma flow (RPF) in significant proportions of patients with T2DM of Caucasian, Native and African American origin (420, 474, 680). Compelling evidence for the presence of renal hemodynamic abnormalities in T2DM has also been reported in Pima Indians (432), in which transition from impaired glucose tolerance to T2DM was accompanied by a 30% increase in GFR. An increase in GFR has been also reported in obesity (215), a condition which often accompanies T2DM.
Alterations in renal hemodynamics in diabetes can also be associated with loss of renal functional reserve, that is, the ability to increase GFR in response to amino acid infusion or to ingestion of a meal rich in protein (540). These maneuvers may identify altered renal hemodynamics in a subset of patients with GFR within the normal range. Notably, another study found in fasted diabetic patients who have normal renal function that the renal hemodynamic responses to increased plasma amino acid concentrations were augmented (640).
It has been proposed that early glomerular hyperfiltration may predispose the subsequent development of diabetic glomerulopathy (22, 128, 412, 414, 533, 596). Early support for this hypothesis emanated from the finding that diabetic glomerulopathy only occurred in the nonstenosed kidney in the setting of unilateral renal artery stenosis (52). However, the clinical relevance of early hyperfiltration as a risk and/or pathogenic factor for the later development of overt nephropathy is debated since other studies have failed to document such a relationship (357, 728), including a recent analysis by Ficociello et al. (172). The ongoing debate is well documented by two recent publications: a meta-analysis of 10 studies in T1DM conducted by Magee et al. (380) concluded that the presence of hyperfiltration at baseline more than doubled the risk of developing micro- or macroalbuminuria at follow-up. In contrast, a thorough analysis of available data led Jerums et al. (276) to the notion that the available data in support of a pathogenetic role for hyperfiltration in diabetic nephropathy are at present too weak to allow reliable conclusions. The available studies suffer from one or more of the following defects: they are too small, far too short and have failed to control for confounders. In addition, the putative link between hyperfiltration and onset of nephropathy has focused entirely on albuminuria and the definition of progression of albuminuria has been variable. By contrast, no study has shown prospectively that hyperfiltration leads to subnormal GFR independently of other variables.
Renal hemodynamics in experimental diabetes mellitus
Despite the controversy in human diabetes concerning the significance of hyperfiltration in the subsequent development of overt nephropathy, extensive experimental data discussed in this section provides considerable insight into the importance of hemodynamic factors in the initiation and progression of diabetic glomerulopathy. As in diabetic patients, diabetic rats tend to exhibit reduced values for whole kidney GFR during periods of severe uncontrolled hyperglycemia; single nephron GFR (SNGFR) and plasma flow rates are also normal or reduced in animals in such catabolic states (244). In the more clinically applicable model of T1DM with moderate hyperglycemia, whole kidney GFR and SNGFR increase as compared to normal rats (244, 274, 734, 735). Reductions in intrarenal vascular resistances result in elevation of glomerular plasma flow rate, QA. Despite normal blood pressure levels, transmission of systemic pressures to the glomerular capillaries can be facilitated by proportionally greater reductions in afferent compared to efferent arteriolar resistances (see Fig. 10). This phenomenon was documented not only by micropuncture studies, but also by direct measurements of afferent and efferent arteriolar diameters using videomicroscopic techniques (99). Consequently, the glomerular capillary hydraulic pressure (PGC) can rise, although this is not a prerequisite for diabetic hyperfiltration (274, 735). Thus, the observed single nephron hyperfiltration can result from both glomerular capillary hyperperfusion and hypertension (244, 734, 735). The potential minor contribution of a lower hydrostatic pressure in Bowman space (274, 654, 657, 734) as a cause of a greater effective filtration pressure in diabetes is further outlined in the tubular section below.
Figure 10.

Renal hemodynamic changes in diabetes at the whole kidney and single nephron level. Early stages of diabetes are associated with specific renal hemodynamic changes schematically presented in this figure. The schematic shows regulators of afferent and efferent arteriolar tone, which have been implicated in these changes. GFR, glomerular filtration rate; RPF, renal plasma flow; SNGFR, single nephron glomerular filtration rate; QA, glomerular plasma flow rate; RA, afferent arteriolar resistance; RE, efferent arteriolar resistance; PGC, glomerular capillary pressure; Kf, ultrafiltration coefficient.
In long-term studies, diabetic rats develop morphologic changes reminiscent of those in the diabetic human, including glomerular basement membrane thickening, renal and glomerular hypertrophy, mesangial matrix thickening and hyaline deposition, and ultimately glomerular sclerosis (734, 735). In comparison, mice in general are more resistant to the development of diabetic nephropathy (79, 84) (see animal models below for further discussion). Evidence that glomerular hemodynamic maladaptations contribute to the development and progression of diabetic glomerulopathy has been shown by studies of maneuvers which aggravate or ameliorate glomerular hyperperfusion and hyperfiltration, without affecting metabolic control. Uninephrectomy, which increases SNGFR, QA and PGC in normal rats, accelerates the development of albuminuria and glomerular sclerosis in diabetic rats (448). Intensification of glomerular lesions is observed in the unclipped kidney of diabetic rats with two-kidney Goldblatt hypertension, while the clipped kidney is substantially protected from glomerular injury (395). Diabetic renal injury is similarly amplified by augmentation of dietary protein content, which increases glomerular perfusion and filtration (735): diabetic rats fed a high protein diet exhibited glomerular capillary hyperfiltration, hyperperfusion, and hypertension, and marked increases in albuminuria and glomerular morphologic injury. By contrast, dietary protein restriction, which reduces SNGFR, QA and PGC in other models, limited SNGFR in long-term diabetes by reducing the elevated PGC and QA, and virtually prevented albuminuria and glomerular injury (735). As there were no differences in metabolic control between the various groups, this study provided strong evidence that amelioration of the maladaptive glomerular hemodynamic pattern can dramatically lower the risk of diabetic glomerulopathy and renal disease.
Mechanisms of hyperfiltration in diabetes
The pathogenesis of diabetic hyperfiltration is multifactorial. Numerous mechanisms and mediators for this effect have been proposed, and are briefly reviewed here. For better clarity, these mechanisms are divided in five areas separately discussed below. However, the reader should be aware of overlap between these processes, and the fact that they act in concert to promote renal hemodynamic alterations in diabetes.
Metabolic milieu
Considering the simple fact that without hyperglycemia and other factors characteristic for the diabetic milieu there would be no hemodynamic changes or nephropathy, the diabetic metabolic milieu must contribute. Hyperglycemia and/or insulinopenia per se (118), together with augmented GH and glucagon levels (117, 478) have been invoked in this process. Reduction of plasma glucose with initial institution of therapy reduces GFR in patients with both T1DM and T2DM (118, 680). In moderately hyperglycemic diabetic rats, normalization of blood glucose levels reverses hyperfiltration (594), and insulin infusion reduces PGC (562). By contrast, insulin infusion sufficient to produce hyperinsulinemia, with euglycemia, increases PGC and hyperfiltration in normal rats (635). Further, infusion of blood containing early glycosylation products reproduces glomerular hyperfiltration in normal rats (539). However, it should be appreciated that the most pronounced changes in renal hemodynamics as compared to nondiabetic animals occur in moderately hyperglycemic rats treated with suboptimal doses of exogenous insulin. Hyperfiltration may not always develop in rats without exogenous insulin treatment, despite moderate hyperglycemia. This finding suggests that some insulin levels, in concert with hyperglycemia, may facilitate the development of renal hemodynamic changes in diabetes. Whether these effects are linked to vasodilator actions of insulin remains to be established. Furthermore, C-peptide (685), which is a part of the endogenous insulin molecule but that is not contained in exogenous insulin preparations, has its own separate physiological and pathophysiological effects. In addition to alterations in plasma insulin levels and actions, it has been suggested that the lack of C-peptide also contributes to diabetic hyperfiltration (233, 249, 514).
Vasoactive factors
Apart from the above-mentioned mechanisms, which are closely related to the diabetic metabolic milieu, there is substantial evidence suggesting that renal hemodynamic alterations are a consequence of an imbalance between the vasoactive humoral systems controlling the glomerular circulation. There is evidence that the balance between factors influencing the afferent arteriolar tone is shifted toward vasodilators, whereas opposite changes may occur on the efferent arteriole.
Atrial natriuretic peptide
Atrial natriuretic peptide (ANP) which induces afferent dilation and efferent constriction represents one such promising candidate for mediating diabetic hyperfiltration. Plasma ANP levels are elevated in diabetes (462), and blockade of the ANP action with an antibody (462) or a specific receptor antagonist (739) blunts hyperfiltration in diabetic rats. It is likely that altered levels of ANP in diabetes are a consequence of an increase in total exchangeable body sodium and the hypervolemic state (166, 450), although resistance to ANP may be also involved (175). Although these observations may suggest that sodium homeostasis is an important factor in the pathogenesis of hyperfiltration, it is clear that the latter does not absolutely depend on accumulation of NaCl in the body, since GFR can increase relentlessly in early diabetes, notwithstanding a decline in extracellular volume (283).
Nitric oxide
NO is a potent vasodilator acting on both afferent and efferent arterioles, presumably with predominant afferent actions in vivo (139). In mammalian tissues, NO is synthesized by a family of isoenzymes known as NO synthases (NOS). Considering its renal actions, an enhanced NO activity appears to be a good candidate for mediating diabetic hyperfiltration. Paradoxically, diabetes is considered to be a state with reduced NO bioavailability (203, 499). However, the situation in the diabetic kidney, in particular at the early stages of diabetes, is more complex (318).
Most of the renal hemodynamic studies conducted in hyperfiltering diabetic rats demonstrated increased renal hemo-dynamic responses and near-normalization of renal hemodynamics in response to inhibition of NO synthesis with nonspecific NOS inhibitors (i.e., affecting all NOS isoforms) (317, 391, 626). Subsequent studies attempted to identify the contribution of individual NOS isoforms in the process and focused on renal hemodynamic roles of neuronal NOS (nNOS, NOS1). Under physiological conditions, NOS1-derived NO from the MD counteracts afferent vasoconstrictor signals mediated by the TGF mechanism, thus inducing a rightward shift and attenuation of the slope of the TGF function and contributing to the control of QA and PGC (662, 663, 702). The renal vascular tree is more sensitive to systemic NOS1 inhibition in diabetic rats as compared to controls (320, 324). Furthermore, we observed complete amelioration of hyperfiltration in response to acute intrarenal selective NOS1 inhibition in conjunction with increased number of nNOS-positive cells in MD regions of diabetic kidneys (320). Thomson et al. confirmed the GFR-lowering effect of acute selective NOS1 inhibition in STZ-diabetic rats, when the drug was given intravenously, and reported an ongoing overactivity of MD NOS1 that is not regulated by MD salt (585). These observations implicated NOS1-derived NO as an important player in the pathogenesis of diabetic hyperfiltration, and are in accordance with previous reports showing a modest attenuation of the slope of the TGF function in diabetic rats (61, 647), as further discussed below. Moreover, unlike the chronic treatment with nonspecific NOS inhibitors, which aggravates the course of experimental nephropathy, selective NOS1 inhibition slowed down the development of nephropathy in uninephrectomized diabetic rats most likely via a hemodynamic mechanism (324).
There is also evidence suggesting that the activity of the endothelial NOS isoform (eNOS, NOS3) may be increased, and responsible for enhanced renal NO production in diabetes (602, 671). However, the relevance of such findings could not be further validated due to the lack of specific eNOS inhibitor. The contribution of eNOS to the development of renal hemo-dynamic changes in diabetes is in our opinion less likely, due to the complex pathobiochemistry of this enzyme as discussed in more detail in the sections devoted to endothelial dysfunction. Notably, genetic knockout of Nos3 has been used to induce nephropathic changes in mouse models of both T1DM and T2DM that mimic many aspects of human disease (278, 392, 398, 707) (see discussion of animal models below). A possible role for NO-ROS interactions in the regulation of efferent arteriolar tone is outlined below.
In addition to possible direct effects of diabetes on NOS activities, the NO-mediated alterations in renal hemodynamics may be related to increased activity of factors that act as NO-dependent vasodilators or activate the NO-cGMP pathway as a part of their signal transduction. De Vriese et al. (134) reported that neutralization of VEGF with an antibody ameliorated diabetic hyperfiltration. VEGF has been implicated in nonhemodynamic pathways in the pathogenesis of diabetic complications, but also possesses vasomotor effects mediated by NO. However, as discussed above and below, the more recently described phenomenon of uncoupling of VEGF with the eNOS pathway as a mechanism for diabetic vasculopathy (423) does not support the role of this mechanism in the pathogenesis of hyperfiltration.
Cyclooxygenase
A role for COX metabolites of arachidonic acid in the pathogenesis of diabetic nephropathy has been suggested in a number of clinical and experimental studies. Schambelan et al. (552) demonstrated an increase in conversion of exogenous arachidonate to prostaglandin E2 (PGE2), prostaglandin F2, prostaglandin D2, and thromboxane B2 (TxB2) in glomeruli from diabetic rats. In the early stages of nephropathy, vasodilator prostaglandins, such as PGE2 and prostacyclin, have been implicated in mediating alterations in renal hemodynamics in humans with T1DM (161, 194, 242, 677), as well as in experimental models of diabetes (124, 275, 299, 486). As demonstrated by Jensen et al. (275), inhibition of PG synthesis results in significant reductions in SNGFR, QA and PGC in diabetic rats.
The above-mentioned evidence about the role of prostaglandins in the renal hemodynamics in diabetes relied on measurements of renal function in response to nonspecific inhibitors that inhibit COX-1 and COX-2 isoenzymes. Follow-up studies demonstrated an increased expression of the COX-2 isoform in the diabetic kidney and a modest GFR lowering effect of selective COX-2 inhibition in these diabetic rats; in comparison, COX-1 expression was unchanged in diabetic rats and acute COX-1 inhibition ineffective with regard to renal hemodynamics (321). These experimental observations, indicating a role for COX-2 in the regulation of renal hemodynamics in the diabetic kidney, were more recently reproduced in the clinical setting (111).
Other mediators
Diabetes-related abnormalities of other vasodilator mechanisms have also been suggested, including activation of the kallikrein-kinin system (96, 269). However, studies with kinin receptor antagonists have been inconsistent (270, 319, 681).
Efferent arteriolar vasoconstrictors
Reduced afferent arteriolar or mesangial constriction may occur in the diabetic kidney due to reduced glomerular receptor sites for the vasoconstrictors Ang II and thromboxane (38, 703, 704) and altered vascular responsiveness (452). Notably, inhibition of vasoconstrictor systems such as the RAS (734), ET (49), or thromboxane A2 (328, 643) has beneficial effects on the development of nephropathy including amelioration of diabetic hyperfiltration. The most plausible explanation is, at least in part, inhibition of their efferent arteriolar vasoconstrictor actions. This suggests that diabetes is associated with normal or increased efferent actions of vasoconstrictors. Supporting this view are our whole kidney data showing enhanced renal vasodilation to Ang II inhibition in diabetic rats (309, 324) in conjunction with reduced filtration fraction. Also Hollenberg’s group has documented enhanced activity of the renal RAS in both types of diabetes by demonstrating enhanced hemodynamic responses in these patients as compared to nondiabetic subjects (346, 505).
Small GTPase RhoA and its down-stream effectors ROCK mediate vasoconstrictor signals and recent studies showed altered renal hemodynamic responses in the diabetic kidney. Unlike the control rats, diabetic animals responded to acute inhibition of ROCK (with the selective inhibitors Y27632 or fasudil) with renal vasodilation and lower filtration fraction (322). Considering the hemodynamic patterns induced by inhibitors of these systems, which include reductions in filtration fraction and thus potentially in PGC, it is possible that these interventions have important efferent actions and target diabetes-induced dysbalances in afferent and efferent vasoconstrictor signals.
It should be noted that similar to the glomerular micro-circulatory pattern, renal hemodynamic responses to various mediators in diabetes are largely dependent upon the state of metabolic control and insulin treatment. These differences may explain some disparate findings of studies exploring the activities of vasoactive systems in diabetes.
Reactive oxygen species
Enhanced production of ROS has been also implicated in the renal hemodynamic changes of the diabetic kidney. ROS can induce renal vasoconstriction by decreasing the bioavailability of NO or limiting the buffering capacity of NO against vasoconstrictors (451, 558, 565). Therefore, one would expect that enhanced ROS may induce renal vasoconstriction in diabetes. Importantly, there is no evidence demonstrating the effect of ROS scavenging on basal renal arteriolar tone in diabetic rats (558). Furthermore, considering the described NO-ROS interaction and a dominant role of NO on the afferent arteriole, one would expect enhanced afferent vasodilator responses to antioxidant treatment in diabetes. In contrast, in vivo studies demonstrated that treatment with the antioxidant α-tocopherol normalizes glomerular hyperfiltration in diabetic rats (333). This reduction in GFR was associated with a decrease in filtration fraction, which may implicate an effect of the antioxidant treatment on the efferent arteriole, which may include an NO effect. The observed renal microvascular effects of antioxidant treatment may also include other mechanisms, such as attenuation of PKC activity (333) or Ang II signaling via ROS (208).
Alterations in signal transduction
In addition to a myriad of other effects in diabetic cells and tissues, PKC transduces vasoactive signals. Importantly, some PKC isoenzymes are not only activated by hyperglycemia via de novo synthesis of DAG, but also operate in signaling cascades of some vasoactive peptides, such as Ang II (564). The role for PKCβ in altered hemodynamics in the diabetic kidney is best documented by amelioration of hyperfiltration, associated with an overall renoprotective effect in the diabetic kidney, in diabetic rats and, as shown more recently, in patients with T1DM treated with the PKCβ inhibitor ruboxistaurin (110, 260). PKCβ is also expressed in the brush border of the proximal tubule (489, 490) and thus may affect GFR in diabetes indirectly via its tubular effects as discussed below. In addition to PKC, other signaling pathways have been studied as potential mediators of diabetic hyperfiltration, in particular those that are involved in the control of vascular tone. For example, selective inhibition of p38 MAPK near-normalized hyperfiltration in diabetic rats, however, this effect was not specific for the diabetic state and similar effects of this intervention were observed in control animals (325).
Renal sodium handling and tubuloglomerular feedback
In the normal kidney, cells of the MD sense early distal intratubular concentrations of Na+ and Cl− and in response to increasing ion concentrations, they send vasoconstrictor signals to the afferent arteriole. This autoregulatory mechanism, known as TGF, prevents excessive NaCl losses from the organism and contributes to renal autoregulation. The contribution of TGF to the pathogenesis of glomerular hyperfiltration is discussed below in detail as part of the “tubulocentric concept” of renal hemodynamics in the early diabetic kidney.
Intrinsic defects in glomerular arterioles, electromechanical coupling
Data originating mainly from Carmines’ laboratory have indicated primary changes in preglomerular microvascular smooth muscle function as an underlying mechanism of afferent arteriolar dilation during the hyperfiltration state of diabetes. Using videomicroscopy in isolated blood-perfused juxtamedullary nephrons, they described several diabetes-induced alterations in afferent arteriolar ion channels that can result in increased baseline diameter and impaired responses to vasoconstrictors. This impaired electromechanical coupling in afferent arteriolar smooth muscle could impair vasoactive responses to multiple humoral agents as well as the myogenic response, TGF, and, ultimately, autoregulation of renal blood flow and GFR.
They showed that STZ-diabetic rats have suppressed vasoconstrictor-induced increases in intracellular Ca2+ concentrations due to functional defects in afferent arteriolar L-type calcium channels (99). Brosius and colleagues had earlier proposed that diabetes attenuates glucose uptake via GLUT4 in afferent microvessels and renal glomeruli, and they proposed that this may induce vasodilation by lowering intracellular glucose-induced ATP formation and thus the inhibition of ATP-sensitive K+ channels (K-ATP) (376, 385). Subsequent studies (256) implicated increased expression and function of K-ATP channels in the renal afferent arteriolar dilation in experimental diabetes. Pharmacological blockade of K-ATP channels contracts afferent arterioles from rats with STZ-induced diabetes mellitus while having minimal impact on afferent arterioles from normal rats (256). More recent studies evaluated the contributions of other K+ channels to the regulation of afferent arteriolar tone in normal and diabetic rat kidney using pharmacological inhibitors of voltage-gated K+ channels (KV), inward rectifier K+ channels (KIR), Kir1.1 and Kir3.x subfamilies of KIR channels, large-conductance Ca2+-activated K+ channels (BKCa), and small-conductance Ca2+-activated K+ channels (SKCa) (631). While Blockers of SKCa and BKCa had no discernible impact on afferent arteriolar tone in normal rat kidney, STZ-diabetic rats displayed altered relative contributions of specific K+ channels to afferent arteriolar tone, such that there was an increased tonic dilator impact of KIR channels (including Kir1.1 and/or Kir3.x). Thus, exposure to blockers of K-ATP or KIR channels reversed the afferent arteriolar dilation in diabetic rats. This observation suggests that K+ channel activation is important for sustaining tonic afferent arteriolar dilation during the hyperfiltration stage of diabetes mellitus in the rat. These K+ channels can contribute to afferent arteriolar dilation in diabetes mellitus by promoting vascular smooth muscle membrane hyperpolarization, thereby reducing Ca2+ influx through voltage-gated channels and, ultimately, decreasing cytosolic Ca2+ concentration. This situation can be exacerbated by the mentioned suppression of afferent arteriolar Ca2+ influx responsiveness to membrane potential in kidneys from diabetic rats (99). In the more complex in vivo setting, systemic application of the K-ATP channel blocker, U37883A, did not alter renal vascular resistance, renal blood flow, or GFR in both control and STZ-diabetic rats (645).
Role of glomerular capillary hypertension in renal injury
Of the glomerular hemodynamic determinants of hyperfiltration, the available evidence suggests that glomerular capillary hypertension plays the key role in progression of renal injury. Studies in a variety of experimental models, including diabetes, have consistently shown that interventions which control glomerular capillary hypertension are associated with marked slowing of the development of structural injury (23).
Glomerular capillary hypertension can modify the growth and activity of glomerular cells, inducing the elaboration or altered expression of growth factors and cytokines, enzymes and a variety of signaling molecules (37, 334, 453, 467, 485, 523, 742). As outlined above, podocytes may be particularly sensitive to mechanical stress and capillary stretch associated with increases in PGC. The enhanced expression and/or release of these mediators then stimulate mesangial matrix production and promote structural injury as discussed in various contexts in this article.
Tubular Function in Diabetes
Hyperglycemia induces specific primary effects on the tubular system of the kidney. This includes changes in tubular growth (discussed above) and transport with secondary consequences on renal hemodynamics via the physiology of tubuloglomerular communication. In addition to primary defects in the GFB (see above), an inhibition of proximal tubular reabsorption of albumin has been proposed to contribute to the manifestation of albuminuria in early diabetic nephropathy (534, 637). An increased sensitivity of proximal tubular NaCl reabsorption to changes in dietary NaCl intake explains the unique phenomenon of the “salt paradox” of the early diabetic kidney. Moreover, kidney growth via its role in the development of tubulointerstitial injury is though to play a crucial role in the long-term progression to kidney failure in diabetes. This article summarizes the effects of hyperglycemia on tubular transport of glucose, NaCl, and fluid [for previous reviews see (382, 493, 613, 646, 648)]. We also include a brief discussion on hyperinsulinemia-induced renal sodium retention in T2DM.
Evidence for a primary increase in reabsorption by the proximal tubule
Hyperfiltering adult patients with T1DM (81, 223) or T2DM (397) have increased absolute and fractional proximal reabsorption as determined by lithium clearances. Enhanced fractional proximal reabsorption was confirmed in children with T1DM (449). Similarly, hyperfiltering rats with STZ-diabetes (40, 501, 647, 657) have increased absolute and fractional reabsorption in the nephron segments upstream from the MD associated with lower early distal delivery of Na+, Cl−, and K+ (501, 647, 657). Operating solely under the influence of glomerulo-tubular balance (GTB), that is, load-dependence of tubular reabsorption, an increase in GFR should lead to an increase in absolute reabsorption. Due to the imperfect nature of GTB, however, an increase in GFR should also decrease fractional reabsorption and increase the distal delivery. Therefore, if fractional reabsorption and GFR happen to change in the same direction, this cannot be explained by GTB alone. There must be a change in the number of transporters or in their avidity, that is, a primary change in tubular reabsorption. To follow up on this issue, we performed micropuncture experiments in STZ-diabetic rats. In these experiments, SNGFR was manipulated by perfusing Henle’s loop to activate TGF. This enabled proximal reabsorption to be established as a function of SNGFR. Using this method to control for the effects of SNGFR, a major primary increase in proximal reabsorption was documented for rats with early STZ-diabetes (617, 651) (see Fig. 11). The following paragraphs discuss mechanisms that contribute to this primary increase in proximal tubular reabsorption in the early diabetic kidney.
Figure 11.
Primary increase in proximal tubular reabsorption and inhibition by high NaCl diet in the early diabetic kidney. Absolute proximal fluid reabsorption (Jprox) shown as a function of SNGFR. SNGFR was manipulated by perfusing Henle’s loop to activate TGF to characterize proximal reabsorption as a function of SNGFR in control rats (CON) and diabetic rats (STZ) on normal versus high NaCl diet. *P < 0.05 for a GFR-independent effect of STZ (left panel) or high NaCl diet (right panel) on proximal tubular reabsorption. Adapted with permission from (651).
Tubular growth as a cause of primary tubular hyperreabsorption in diabetes
The kidney in general and the proximal tubule in particular, grow large from the onset of diabetes (116, 511, 571) (see above). On the basis of the principle of mass action, an increase in the size of the tubule results in increased proximal reabsorption (648). To test whether tubular growth per se contributes to the primary increase in proximal reabsorption in early diabetes mellitus, we used difluoromethylornithine, an inhibitor of ODC, which had been shown previously to attenuate kidney growth in early STZ-diabetic rats (482). We observed that difluoromethylornithine not only attenuated kidney growth but also eliminated the primary increase in proximal reabsorption in STZ-diabetic rats (617).
Tubular growth in diabetes involves PKCβ activation (see above). Moreover, the PKCβ1 isoenzyme is expressed in the brush border of proximal tubule (490) and stimulation of sodium transport in proximal tubule by Ang II is mediated by PKCβ activation (157). STZ-diabetes activates PKCβ1 in the proximal tubule, which was inhibited by ACE inhibition (489). Further studies are needed to more directly test the role of PKCβ1 for proximal tubular hyperreabsorption, which could contribute to the beneficial effects of specific inhibition of PKCβ isoenzymes on the diabetic kidney (260). Notably, genetic variants of the PKCβ1 gene have been linked to the development of end-stage renal disease in patients with T2DM (378).
Sodium-glucose co-transport as a cause of primary tubular hyperreabsorption in diabetes
The kidneys reabsorb large amounts of glucose, thereby contributing to the overall metabolic balance of the body. In healthy humans, about 180 g/day of glucose are filtered through the renal glomerulus, more than 99% of which is reabsorbed along the tubular system. Glucose entry into proximal tubular cells is insulin-independent, which makes proximal tubular cells particularly sensitive to the deleterious effects of chronic hyperglycemia in diabetic conditions. Transport of glucose across cell membranes is accomplished by two gene families: the facilitative glucose transporters—the GLUTs—and by an active sodium-dependent transport process mediated by the SGLT. At least two sodium-coupled glucose transporters, SGLT1 and SGLT2, play an important role in the apical membrane of proximal tubular cells in the kidney (718). The high-capacity, low-affinity SGLT2 is found primarily in the apical membrane of the S1 segment of the proximal tubule, whereas the low-capacity, high-affinity SGLT1 is located more distally in the S3 segment of the proximal tubule (290, 716, 718, 731). Two basolateral membrane glucose transporters facilitate transcellular glucose transport—the low-affinity GLUT2, which works in concert with SGLT2 in the S1 segment, and the high-affinity GLUT1, which works with SGLT1 in the S3 segment (114, 713).
The phenotype of humans carrying gene mutations in the genes for SGLT1 (SLC5A1) and SGLT2 (SLC5A2) provided much of the evidence for the quantitative contribution of these proteins to renal glucose reabsorption. Gene mutation of SGLT1 has serious intestinal consequences for afflicted individuals, who experience glucose-galactose malabsorption, frequent watery diarrhea, and dehydration, demonstrating the role of SGLT1 in intestinal glucose uptake (717). However, these individuals have little or no glucosuria. In contrast, those who have SGLT2 gene mutations have persistent renal glucosuria, with glucose excretion of up to 160 g/day in severe cases (180, 381, 543, 563, 667). Recently we directly localized the expression of SGLT2 protein to the apical brush border of the early proximal tubule and demonstrated by free-flow micropuncture that SGLT2 KO mice lack glucose reabsorption in the early proximal tubule and that fractional renal reabsorption of glucose is reduced compared with wild-type mice (36 ± 8 vs. 99.7 ± 0.1%) and varied inversely with the amount of filtered glucose in the KO mice (between 60% and 10%) (656). Thus, early proximal SGLT2 reabsorbs the bulk of glucose filtered by the kidneys.
The capacity of glucose transport through SGLTs depends on the level of SGLT protein expression. Moreover, modeling the effects of sodium-linked glucose transport on the active and passive components of proximal reabsorption predicts a positive effect of filtered glucose on net proximal sodium reabsorption up to the point where the filtered load approximately doubles the transport maximum for glucose (697). Glucosuria starts to develop when blood glucose levels reach the renal threshold for reabsorption, which is about 8 to 10 mmol/l (180 mg/dl). Thus, modest hyperglycemia enhances sodium reabsorption in the proximal tubule, and the extent of sodium reabsorption depends on the expression of SGLT. In this regard, STZ-diabetic rat renal cortex was found to contain increased mRNA expression for SGLT 1 and SGLT 2 (676) and greater renal SGLT1 protein expression (678). Likewise, renal SGLT1 and SGLT2 mRNA levels in diabetic obese Zucker rats were higher than in age-matched leans (605). Primary cultures of human exfoliated proximal tubular epithelial cells from fresh urine of patients with T2DM indicated an increased renal glucose uptake associated with increased mRNA and protein expression of SGLT2 and GLUT2 compared with healthy control subjects (509). Upregulation of SGLT2 expression in diabetes may involve Ang II AT1 receptors (463). In accordance, treatment of rats with either ramipril or losartan showed significant reduction in the intensity of immunostaining and levels of SGLT2 protein and mRNA (45). Upregulation of SGLT2 expression in diabetes may also involve the transcription factor, hepatocyte nuclear factor HNF-1α (181). There is also evidence for upregulation of GLUT2 mRNA and protein expression in renal proximal tubules in diabetic rat models (113, 145, 289), which may be due to transcriptional activity of both HNF-1α and HNF-3β (182). Whereas multiple studies found evidence for an upregulation of SGLT2 expression in the diabetic kidney, other studies showed unchanged (8) or reduced (15) renal expression in diabetic rodent models, and in vitro studies indicated that high glucose-induced oxidative stress can inhibit Na+/glucose co-transporter activity in renal proximal tubule cells (222). Downregulation of SGLT could be an approach to limit glucose uptake and glucose toxicity. Notably and possibly relevant in this regard, gene-knockout of SGLT2 lowers renal SGLT1 mRNA and protein expression by 40% (656).
The serum and glucocorticoid-inducible kinase SGK1 is upregulated in proximal tubules in STZ-diabetic rats (538) and in patients with diabetic nephropathy (343). Excessive SGK1 expression in the diabetic kidney contributes to stimulation of SGLT1 activity in proximal renal tubules, which attenuates glucosuria (7). SGK1 could also increase proximal renal tubular glucose transport by stimulation of the K+ channel KCNQ1/KCNE1 (159), which establishes the electrical driving force for electrogenic glucose transport in the proximal tubule (649, 650). Moreover, SGK1 has been proposed to mediate in STZ-diabetic rats the upregulation of mRNA levels of the Na-H-exchanger NHE3 (538), which is of critical importance for proximal tubular sodium reabsorption (660). SGK1 effects on proximal and distal tubular transport (see below) as well as fibrosis (see above) make this kinase an interesting targeting in diabetes (342). NHE3 is thus also a candidate to be involved in proximal tubular hyperreabsorption in the diabetic kidney. Whereas renal NHE3 expression was found to be increased, unchanged or decreased in STZ-diabetes (431, 590, 678), the expression was found to be reduced in Zucker diabetic fatty rats (55, 63).
Based on microperfusion studies in normal and STZ-diabetic rats, Bank and Aynedjian proposed that high glucose in the proximal tubular fluid stimulates sodium absorption through SGLT (40). They found that increasing luminal glucose (from 100 to 500 mg/dl) induced significantly greater increases in sodium versus glucose absorption on a molar basis, which may reflect, in part, the well-documented sodium:glucose coupling ratio for SGLT1 of 2:1 [in comparison, the ratio is 1:1 for SGLT2 (718)]. The contribution of SGLT1 to renal glucose absorption is expected to increase in diabetes when the luminal glucose delivery overwhelms the capacity of SGLT2. Confirmation of increased SGLT-mediated sodium transport was demonstrated with micropuncture in STZ-diabetic rats with moderate hyperglycemia, by delivering the SGLT inhibitor, phlorizin, directly into the free-flowing early proximal tubule of nephrons with superficial glomeruli: in diabetic rats, phlorizin elicited a greater decline in absolute and fractional sodium reabsorption up to the early distal tubule and abolished the hyperreabsorption (657). These studies support the concept that moderate hyperglycemia enhances SGLT-mediated glucose and sodium reabsorption in the proximal tubule.
Earlier, Seyer-Hansen had reported that in early STZ-diabetic rats the glucose reabsorptive rate increased to the same extent as kidney weight (570). Hence, the primary increase in proximal reabsorption in early diabetes is the combined result of growth of the tubule and increased SGLT. This could be relevant for the regulation of GFR by TGF in early diabetes (see below).
Inhibition of SGLT2 as a new therapeutic approach in diabetes
Good control of blood glucose levels is important in diabetic patients but difficult to achieve without the potential for hypoglycemia and weight gain. Moreover, existing therapies may not always reduce cardiovascular complications, indicating the need for new therapies with novel mechanisms of action. SGLT inhibition offers the potential to decrease the deleterious effects of hyperglycemia while reducing blood glucose levels by inhibiting renal glucose reabsorption. Promising results have been achieved in diabetic animal with the nonselective SGLT inhibitor, phlorizin (158), its derivative T-1095 and its metabolite T-1095A (26, 642), including beneficial effects on blood glucose levels and HbA1c levels, partial improvement of glucose tolerance and insulin resistance, and suppression of albuminuria and the expansion of the glomerular mesangial area in diabetic rodents (456, 642). Due to their effect on intestinal SGLT1, however, nonselective inhibitors can have unacceptable gastrointestinal side effects and did not proceed to clinical development. Selective SGLT2 inhibitors offer the potential to primarily increase renal excretion of glucose and create a negative energy balance without affecting intestinal function. Therefore, various selective SGLT2 inhibitors are currently in development for the treatment of diabetes mellitus [for review see (421, 661)]. It remains to be determined whether these drugs lower the toxicity of glucose directly on renal cells, independent of hyperglycemia, which may slow or prevent the progressive nature of diabetic nephropathy.
Is there relevant hyperinsulinemia-induced renal NaCl retention in type 2 diabetes mellitus?
Hyperinsulinemia-mediated sodium retention has been hypothesized to contribute to hypertension, particularly in obese subjects with T2DM (137, 170, 516, 526, 544, 624). An acute increase in insulin levels induces an antinatriuretic response, which is at least in part due to enhanced renal reabsorption, as indicated by studies in human (31, 138, 185, 584, 597) and animals (169, 196, 311, 407). These antinatriuretic effects of insulin have been ascribed to its actions on the proximal tubule (169, 196), loop of Henle (311), and especially on the distal nephron (138, 169, 185, 589, 597). Studies by Tiwari et al. in mice showed that the acute antinatriuretic response to insulin is associated with enhanced expression of total and phosphorylated SGK1 in the membrane fraction as well as trafficking of the epithelial sodium channel, ENaC, to the apical membrane compartment of the distal nephron (623). In accordance, Huang et al. showed that the acute insulin-induced antinatriuresis observed in wild-type mice (during glucose clamp) is significantly blunted in mice lacking SGK1, and these mice lack the salt-sensitizing hypertensive effect of high fructose intake, which is associated with hyperinsulinemia (248). SGK1 may also contribute to the enhanced expression and activation of the thiazide-sensitive Na-Cl co-transporter (NCC or TSC) observed in a rat model of hyperinsulinemia (56, 658).
One study in patients with T1DM reported that the acute sodium-retaining effect of insulin was blunted (483). However, others concluded an intact distal antinatriuretic effect of insulin in T1DM (598), and studies in hyperinsulinemic patients with T2DM and peripheral insulin resistance also showed a preserved acute antinatriuretic effect of insulin (585). Yet, studies in rats showed that insulin receptors are downregulated in the kidneys of insulin resistant animals as well as in STZ-diabetes, possibly mediated by hyperglycemia and Ang II (622). Furthermore, studies with partial knockdown of insulin receptors in renal tubules implicated that activation of renal insulin receptors can actually facilitate renal sodium and water excretion and reduce blood pressure, possibly via an effect on NO production (625). These findings are consistent with the notion that the antinatriuretic and ENaC-mediated effects of insulin are actually mediated by its binding to the IGF-I receptor and downstream activation of SGK1 (204).
Notably, all the currently available data that support a sodium-retaining or blood pressure increasing effect of chronic hyperinsulinemia have been generated in rodent studies (75,76, 399, 589, 628). In comparison, chronic increases in plasma insulin levels in humans and dogs have not been shown to cause hypertension (74, 217), and sodium retention was not apparent during chronic intra-renal insulin infusion in dogs (216). Therefore, further studies are necessary to translate the results from acute insulin infusion studies in humans and chronic rat and mice studies that support a sodium-retaining effect of hyperinsulinemia to the situation of chronic hyperinsulinemia in humans with T2DM.
Role of primary tubular hyperreabsorption for glomerular hyperfiltration in the diabetic kidney
Common sense may suggest that glomerular hyperfiltration must be due to some defect in microvascular function or an imbalance of hormones impinging directly on the glomerulus, as outlined above. However, in early diabetes glomerular filtration is also inordinately influenced by feedback from the proximal tubule and we have proposed a general tubulocentric principle for the control of GFR in early diabetes [reviewed in (648)]. In this principle, primary changes in tubular transport induce secondary changes in GFR through TGF (see Fig. 12). Manifestations of this tubulocentric model in the early diabetic kidney include glomerular hyperfiltration discussed here as well as the salt paradox, which is outlined below.
Figure 12.
Tubular basis of glomerular hyperfiltration in the early diabetic kidney. Hyperglycemia causes a primary increase in proximal tubular reabsorption through enhanced tubular growth and Na+-glucose (Gluc) cotransport (1). The enhanced reabsorption reduces the signal of the tubuloglomerular feedback (TGF) at the macula densa ([Na-Cl-K]MD) (2) and via TGF increases SNGFR (4). Enhanced growth and tubular reabsorption also reduce the hydrostatic pressure in Bowman space (PBOW) (3), which by increasing effective filtration pressure can also increase SNGFR (4). The resulting increase in SNGFR serves to partly restore the fluid and electrolyte load to the distal nephron (5). SNGFR0 is the input to SNGFR independent of TGF. Adapted with permission from (648).
The TGF system senses changes in the concentration of Na+, Cl−, and K+ at the luminal MD and induces reciprocal changes in SNGFR (560, 655). TGF, thereby, stabilizes electrolyte delivery to the distal tubule which in these nephron segments allows fine adjustment of reabsorption and excretion according to body needs. As outlined above, there is a primary increase in proximal reabsorption in early diabetes, which reduces delivery of NaCl and K+ to the MD. Pollock et al. reported that the Na+ concentration in early distal tubules of hyperfiltering STZ-diabetic Sprague Dawley rats is remarkably low (about 30 vs. 76 mM) (501). We performed similar experiments in diabetic rats of the Hannover-Wistar-Froemter strain with superficial glomeruli from which it is possible to sample tubular fluid closer to the MD. In these studies, respective ambient early distal tubular concentrations of Na+, Cl−, and K+ in nondiabetic rats were 21, 20, and 1.2 mM, respectively. In hyperfiltering STZ-diabetic rats of this strain, early distal concentrations of Na+, Cl−, and K+ were reduced by 20% to 28%, consistent with a primary increase in upstream reabsorption (657). The MD senses the decline in salt delivery as an error signal and elicits an increase in GFR, which offsets a portion of the original error signal through negative TGF.
The role of tubular reabsorption and a reduced TGF stimulus as antecedents to diabetic hyperfiltration is further supported by the observation that adding phlorizin to the early proximal tubule of diabetic rats caused a significant increase in early distal electrolyte concentration along with a decisive reduction in SNGFR in diabetic rats (657). Tubular control of GFR has also been demonstrated in dogs where acute hyperglycemia caused GFR to increase, but only if TGF was intact (714). Evidence for a primary hyperreabsorption upstream of the MD and a potential role in glomerular hyperfiltration was also proposed in diabetic patients (81, 223, 674), including studies which showed that fractional proximal reabsorption was elevated and positively correlated with GFR (223). Again, based on the principle that GTB cannot explain a positive correlation between GFR and fractional proximal reabsorption, these findings imply a primary increase in proximal reabsorption leading to a TGF-mediated increase in GFR.
Increasing by pharmacological means the cortical interstitial concentrations of adenosine, which acts as the mediator of TGF (615), normalizes GFR in STZ-diabetic rats (654). Excessive hyperglycemia may inhibit proximal reabsorption (697) and, thus, TGF may limit hyperfiltration under these conditions (164). However, moderate hyperglycemia increases proximal reabsorption and, in accordance, glomerular hyperfiltration is blunted in moderately hyperglycemic STZ-diabetic mice that lack TGF (due to gene-targeting of the adenosine A1 receptor) (653).
Intervening with difluoromethylornithine, an inhibitor of ODC, to reduce early diabetic tubular hypertrophy and hyperreabsorption also diminished glomerular hyperfiltration in direct proportion to the effect on kidney size (617). Along these lines, each maneuver that succeeds at attenuating kidney growth in diabetes (which may occur with normalization of blood glucose levels by insulin treatment (115, 511, 705), manipulations of GH signaling, etc.) is expected to also reduce GFR by preventing hyperreabsorption. Furthermore, in some diabetic patients, glomerular hyperfiltration persists even after euglycemia is achieved through aggressive insulin therapy. Since established tubular growth reverses slowly, and complete normalization of kidney size may not be achieved in patients even after intensive treatment with insulin supplementation (115, 639, 640, 705), glomerular hyperfiltration may endure in these patients due to persistent tubular enlargement and hyperreabsorption independent of the average blood glucose level. Moreover, patients may be heterogeneous in their response to hyperglycemia with regard to kidney growth, and therefore in the resulting tubular hyperreabsorption and glomerular hyperfiltration.
The primary increase in tubular reabsorption in diabetes, in addition to reducing the TGF signal, can lower the hydrostatic pressure in Bowman space (PBOW) (274, 654, 657, 734). Enhanced reabsorption is expected to reduce PBOW by lowering the flow rate through distal nephron segments where flow resistance is high (362). Furthermore, in early STZ-diabetic rats, tubular growth increases luminal diameter of high flow resistance distal nephron segments (511). Hence, tubular growth might contribute to the decrease in PBOW in diabetes through its effects on tubular reabsorption and tubular diameter. All else remaining equal, this reduction in PBOW could make a small, but significant, contribution to glomerular hyperfiltration by increasing the effective glomerular filtration pressure in diabetes (657). Figure 12 integrates the outlined concept of a primary role of the proximal tubule and tubuloglomerular communication for glomerular hyperfiltration in early diabetes mellitus.
Reduced efficiency of tubuloglomerular communication and potential consequences in diabetes
The incremental slope of the TGF response to any given positive or negative change in MD salt around the operating point is modestly reduced in STZ-diabetes as determined by free-flow perturbation analysis (647) (see Fig. 13). This is associated with ongoing overactivity of NOS I that is not regulated by MD salt (618). A change in TGF efficiency is irrelevant to an undisturbed nephron in steady state in which SNGFR and PGC would remain constant. However, a consequence is that TGF buffers any given disturbance less efficiently in the diabetic kidney, and a greater variability in glomerular capillary pressure will result, which could contribute to the development of diabetic glomerular sclerosis by exposing the glomerular mesangium and podocytes to greater fluctuating physical stress (14, 335, 524). In addition to TGF, glomerular capillary pressure is stabilized by a myogenic mechanism when blood pressure fluctuates or increases. Whereas micropuncture is restricted to superficial nephrons, diabetes has been found to impair renal autoregulation also at the whole kidney level (225, 226, 259), although improved autoregulation at low perfusion pressures has also been reported (348, 393). Bell et al. measured mean arterial pressure and renal blood flow continuously (18 h/day) in uninephrectomized control and STZ-diabetic rats. Transfer function analysis of blood pressure and renal blood flow power spectra revealed rapid and sustained increases in gain over the TGF and myogenic frequency ranges, suggesting that renal autoregulation is impaired at the very onset of hyperglycemia (48). Using a similar approach in the ZSF1 (fa/fa cp) model of type 2 diabetes, Griffin et al. could not detect a significant difference in renal transmission of blood pressure (210) further indicating that the finding of impaired autoregulation at the whole kidney level is not uniform in diabetes.
Figure 13.

Lower efficiency of tubuloglomerular feedback (TGF) in the diabetic kidney. (A) In vivo free-flow perturbation analysis of tubular flow rate in control and STZ-diabetic rats. The fractional compensation profiles illustrate the ability of the TGF system to stabilize tubular flow in response to a perturbation in late proximal tubular flow (by adding or subtracting tubular fluid). The data show that the efficiency of the TGF system to stabilize tubular flow rate (as an indirect measure of stabilizing SNGFR or PGC) is reduced in diabetic rats with a compensation of small perturbations around the operating point of 40% versus 70% in control rats. (B) Data from perturbation analysis of late proximal tubular flow rate were combined with data for fractional proximal reabsorption to synthesize “traditional” TGF functions describing the dependence of SNGFR on late proximal tubular flow rate. The triangles indicate the operating points. The gain of the curve at the operating point is modestly reduced in diabetic rats (red lines). Adapted with permission from (647).
The salt paradox in the diabetic kidney
In addition to causing a high baseline GFR, diabetes also alters the renal hemodynamic response to a variety of stimuli. For example, acute insulin application, which increases GFR in the nondiabetic rat, lowers RPF and GFR in STZ-diabetic rats while being glucose clamped at euglycemic levels (636). Changes in dietary salt constitute another such stimulus, the renal response to which is abnormal in diabetes. In 1995, we reported studies in which we placed male STZ-diabetic rats on a low salt diet for 7 to 8 days after 6 weeks of diabetes. Surprisingly, the maneuver increased renal blood flow, GFR and kidney weight in diabetic rats whereas low NaCl diet did not significantly alter these parameters in nondiabetic controls (664). This observation was extended by demonstrating renal vasoconstriction in response to a high NaCl diet among female rats with early (one week) or established (4–5 weeks) STZ-diabetes (652). Since the negative impact of dietary NaCl on GFR in diabetes is counterintuitive with regard to salt balance, we refer to it as the salt paradox. We have subsequently confirmed the paradoxical effect of dietary salt by micropuncture on the level of the single nephron in male (651) and female (unpublished observation) rats. More recently, the salt paradox was confirmed in STZ-diabetic mice (659) and STZ-diabetic Long-Evans rats (348).
Most importantly, in 1997 Miller reported the same phenomenon in young patients with T1DM: restriction of dietary sodium to 20 mmol/day was found to induce a fall in renal vascular resistance, accompanied by a rise in effective RPF as well as GFR, in spite of a significant rise in plasma renin activity (406). In 2002 Luik et al. similarly showed that short-term moderate sodium restriction induces relative hyperfiltration in uncomplicated T1DM (377). Almost all evidence currently available on kidney function in early diabetes has been derived from patients and experimental models with T1DM. Fewer data have been acquired on the early renal pathophysiology in T2DM. With regard to the influence of NaCl diet and consistent with the salt paradox, one study reported that T2DM with hypertension is associated with reduced RPF when dietary NaCl intake is high for 5 to 7 days (133). Another study, however, found no significant effect on RPF or GFR by variation in NaCl intake for 9 to 14 days in this kind of patients (97). Future studies are needed to further elucidate this issue and to answer the question whether the salt paradox is present in patients with T2DM in the early phase of the disease and in the absence of confounding complications.
The salt paradox in the diabetic kidney is due to hypersensitivity of proximal reabsorption to dietary salt and linked to tubular growth
In normal subjects, GFR is either insensitive to dietary NaCl or changes in the same direction as dietary NaCl (616, 619, 651). The negative impact of dietary NaCl on GFR in diabetes is counterintuitive, but has been observed in diabetic human and animal subjects, and may reveal something basic about the main controller of renal function in diabetes. The paradoxical effect of dietary NaCl on the diabetic kidney cannot be explained by primary vascular effects of the neurohumoral or pressure natriuresis system (648) (see Fig. 14). We propose that the salt paradox is another manifestation of the tubulocentric nature of glomerular function in diabetes.
Figure 14.
Tubular basis of the salt paradox in the early diabetic kidney. (A) In classical physiology, renal function and total body NaCl are linked by several parallel feedback loops. Paths from dietary NaCl to GFR and renal blood flow (RBF) are highlighted (black arrows). Each highlighted path contains only “+” signs or an even number of “−” signs, indicating a positive influence of dietary NaCl on GFR and RBF. Interfering with these processes can alter the strength of this influence, but cannot make it paradoxical. (B) Incorporating tubuloglomerular feedback (TGF) provides a pathway whereby dietary NaCl can inversely impact GFR and RBF via a primary change in proximal (i.e., upstream to macula densa) reabsorption (note the odd number of “−” signs along the dotted path). Thus, GFR and RBF are subject to competing influences in response to changes in dietary NaCl. The NaCl paradox arises when TGF prevails. Adapted with permission from (648).
Micropuncture studies revealed that nondiabetic rats on various NaCl intakes were able to manage salt balance with no significant primary effect on reabsorption in the proximal tubule (see Fig. 11) and upstream to the MD. Thus, an inverse effect of dietary NaCl on GFR mediated by TGF did not occur (651). From a teleological standpoint, this is appealing. In comparison, a prominent negative impact of dietary NaCl on reabsorption upstream from the MD was observed in STZ-diabetic rats (651). Feeding a high NaCl diet to diabetic rats led to a major primary decrease in proximal reabsorption (i.e., a change in reabsorption that is not attributable to GTB) (see Fig. 11). Furthermore, by measuring concentrations of Na+, Cl−, and K+ in early distal tubular fluid in rats on high and low NaCl diet, it was confirmed that this primary effect of dietary NaCl on tubular reabsorption strongly links the TGF signal and the consequent reduction in GFR to dietary NaCl in diabetes. Thus, the salt paradox arises in diabetes because the proximal tubule is strikingly sensitive to NaCl intake, making GFR a “slave” to tubular function via tubuloglomerular communication (see Fig. 14). In accordance, the salt paradox is absent in STZ-diabetic mice lacking an intact TGF response (659). This dovetails with the “tubular hypothesis” of basal diabetic hyperfiltration according to which diabetic hyperfiltration, per se, results from a primary increase in proximal reabsorption as outlined above.
Given the limits of TGF and the need to maintain effective circulating volume, the capacity to increase GFR by reducing distal salt delivery must be less than the capacity to reduce GFR through the systemic influences of salt depletion which, in the extreme case, will result in zero GFR. Hence, if dietary salt restriction progresses to actual salt depletion, the salt paradox will become unapparent (20, 41, 630) (see Fig. 14).
The mechanism which makes the diabetic proximal tubule more sensitive to dietary NaCl is little understood. Ang II and renal nerves are the prominent effectors that link proximal reabsorption to total body salt. Clearance experiments, however, indicate that the enhanced salt-sensitivity of proximal tubular reabsorption is not mediated by renal nerves or Ang II acting through Ang II AT1 receptors, inasmuch as chronic renal denervation (59) or chronic treatment with losartan (664) did not prevent the rise in GFR in response to low NaCl diet in STZ-diabetic rats.
Growth of the proximal tubule in the early diabetic kidney is characterized by a combination of hyperplasia and hypertrophy, and it may be that several peculiar aspects of kidney function in early diabetes, including the salt paradox, are consequences of the mechanism whereby the kidney has grown. In early diabetes hypertrophic proximal tubular cells that are continuously pushed by mitogens and at the same time are prevented from entering the cell cycle (see above) could exhibit a less differentiated transport phenotype than normal differentiated proximal tubular cells: a normal proximal tubule cell not to respond to every change in local hormones that contribute to salt balance as this balance is normally taken care of in nephron segments downstream of the MD. The diabetic proximal tubule, however, may have lost this feature of a differentiated nephron segment, and possibly as a consequence, responds significantly to dietary NaCl forming the basis for the salt paradox. Indeed, more recent studies linked the salt paradox to kidney growth of the early diabetic kidney: pharmacological inhibition of ODC and tubular growth prevented the salt paradox (408).
Renin Release and the Intrarenal Renin-Angiotensin System in the Diabetic Kidney
The renin-angiotensin system (RAS) is of primary importance for the pathophysiology of the diabetic kidney (214). Human trials show that RAS inhibition significantly slows the progression of renal damage in patients with T1DM and T2DM (78, 359, 360), including early stages of the disease (479). The mechanisms involved in renal protection in diabetes by RAS inhibition are less clear but may include effects on blood pressure and glomerular hemodynamics (214). However, clear evidence for systemic activation of the RAS is difficult to detect in diabetic patients, and plasma renin activity is often normal or even suppressed under regular diet (406, 505, 632, 634). Likewise, supine serum Ang II is suppressed in patients with T1DM (166). It has been proposed that this may be misleading and perhaps reflecting an activated intrarenal RAS system (505). Whereas plasma renin concentration and serum ACE activity were normal in modestly hyperglycemic STZ-diabetic rats, intrarenal renin protein content and renin and angiotensinogen mRNAs were increased, consistent with disproportionate activation of the intrarenal RAS (24). Acute increases in RPF and GFR in response to ACE inhibition in patients with T1DM are consistent with intrarenal activation of the RAS (241). Immunohistochemical studies indicated a glomerular redistribution of ACE activity in the diabetic kidney inasmuch as proximal tubule ACE activity was reduced and ACE immunostaining intensity enhanced in glomeruli and renal vasculature of diabetic rats (24).
Whereas ACE converts Ang I to Ang II, ACE2 is a homologue of ACE that both degrades the vasoconstrictor Ang II and generates the vasodilator peptide Ang 1–7, thereby counterbalancing effects of ACE. ACE2 is highly expressed in the kidney, particularly in the proximal tubules. STZ-diabetes reduced ACE2 protein expression in the kidney, an effect that was prevented by ACE inhibitor therapy (621). Moreover, immunostaining revealed that in the diabetic kidney, ACE2 protein was localized predominantly to renal tubules. Also in mice, induction of STZ-diabetes was associated with a reduction in renal ACE2 expression and Ang 1–7 was decreased (620). The ACE2 inhibitor, MLN-4760, and gene-knockout of ACE2 enhanced diabetes-associated albuminuria and increased blood pressure. However, renal hypertrophy and fibrogenesis were reduced in diabetic mice with ACE2 deficiency, and hyperfiltration was attenuated (620). These data indicate a complex role of ACE2 in the diabetic kidney. In Finnish patients with T1DM, polymorphisms were not associated with diabetic nephropathy or any studied risk factor for this complication (188).
As mentioned before, high glucose can induce podocyte hypertrophy, which can be ameliorated by Ang II AT1 receptor blockade, suggesting the involvement of RAS activation (365). Moreover, podocytes express RAGE and studies in cultured podocytes, which express Ang II AT1 and AT2 receptors, surprisingly, revealed that Ang II induced RAGE mRNA and protein expression through activation of the Ang II AT2 receptor (536). This involves a NF-kappaB binding site on the RAGE promoter region and has functional consequences like AGE-induced increases in the expression of TNF-α, but did not affect podocyte proliferation or apoptosis. Further studies are needed to determine the relevance and consequences for the diabetic kidney in vivo.
More recent studies provided strong evidence for a role of the metabolite, succinate, and its receptor in the juxtaglomerular apparatus for an upregulation of intrarenal renin in diabetes and also pointed to a specific role of prorenin in the diabetic kidney, as discussed in the following and illustrated in Figure 15.
Figure 15.

Diabetes-induced upregulation of the renin-angiotensin-system in glomeruli. Illustrated is a proposed role of succinate acting on its receptor (SUCNR1) in glomerular endothelial cells and macula densa cells in the activation of renin. Renin and prorenin can induce angiotensin II-dependent and independent effects by activation of renin receptors on glomerular structures including mesangial cells and podocytes. See text for further details. ADO, adenosine; Ado A1, adenosine A1 receptor; Agten, angiotensinogen; RR, renin receptor.
A role for succinate and the succinate receptor 1 (SUCNR1) in the regulation of renin in diabetes
Baumbach et al. had shown in 1976 that succinate increases renin release from viable juxtaglomerular cells during superfusion of isolated rat glomeruli (43). More recently, the G protein-coupled metabolic receptor SUCNR1 (previously GPR91) has been identified as a succinate receptor that is highly expressed in the kidney, where it can contribute to renin-dependent activation of the RAS and increases in blood pressure (227). SUCNR1 has been located in the luminal membrane of MD cells of the juxtaglomerular apparatus in close proximity to renin-producing granular cells, as well as in cortical TAL, and cortical and inner medullary collecting duct cells (528). Toma et al. proposed that high levels of glucose enhance the local accumulation of succinate in the juxtaglomerular apparatus and that activation of SUCNR1 in the glomerular endothelium triggers the release of renin (627). In addition, renin may be stimulated in the diabetic kidney by luminal succinate that activates the SUCNR1 in the luminal membrane of MD cells (668).
In patients with T2DM plasma, renin activity can be less suppressible in response to high NaCl intake (504). Further studies are needed to elucidate whether this may be due to tonic activation of the succinate system. Since an increase in systemic renin activity is not a general phenomenon in diabetes, it may be that the succinate system contributes to the activation of a local intraglomerular RAS. Vargas and colleagues showed that STZ-diabetes caused SUCNR1-dependent upregulation of renal renin content (668); the signal transduction pathway activated by succinate included the production of NO and PGE2. Further studies are needed to test whether the succinate-induced NO and PGE2 contribute to the development of diabetic glomerular hyperfiltration. This could constitute an extension of the tubular hypothesis (see above) with luminal succinate delivered from upstream tubular segments affecting glomerular function via SUCNR1 in the MD. Inhibition of SUCNR1 and a better understanding of the determinants of succinate accumulation in tubular fluid and the juxtaglomerular apparatus and its role in the long-term outcome of the diabetic kidney may provide further therapeutic approaches.
Activation of the intraglomerular renin/prorenin system in diabetes
Renin was long suspected to have cellular receptors and in 2002 Nguyen and colleagues reported the cloning of the human renin receptor, a 350-amino acid protein with a single transmembrane domain (435). The receptor specifically binds renin and prorenin. Binding of renin increases the catalytic efficiency of angiotensinogen conversion to Ang I and induces an intracellular signal with phosphorylation of serine and tyrosine residues associated to an activation of MAP kinases ERK1/2. Within the kidney the receptor is localized in the mesangium of glomeruli and in the subendothelium of kidney arteries, associated to smooth muscle cells and co-localized with renin (435). These results are consistent with a prominent intraglomerular RAS and propose renin effects independent of Ang II. In accordance, renin may exert receptor-mediated, Ang II-independent, tissue-damaging effects by increasing the expression of profibrotic pathways and molecules such as TGF-β1 (251, 434).
Studies in cultured immortalized mouse podocytes revealed that exposure to high glucose induced a 2-fold increase in Ang II levels mediated through increased renin activity and also increased Ang II AT1 receptor levels (151). The same study proposed a glomerular upregulation of the prorenin receptor in a podocyte distribution early in the course of STZ diabetes in rats. The prorenin-to-active renin ratio significantly increases in the diabetic kidney. Ichihara et al. reported that when a site-specific binding protein interacts with the “handle” region of the prorenin prosegment, the prorenin molecule undergoes a conformational change to its enzymatically active state. Moreover, they provided evidence that the nonproteolytic activation of prorenin may be a significant mechanism in the STZ-diabetic kidney (252). Subsequent studies provided evidence that prorenin binding to its receptor in the diabetic kidney induces kidney injury without its conventional proteolytic activation. Additional studies, using Ang II AT1a receptor gene-deficient mice and (pro)renin receptor blockade with a short peptide of prorenin, indicated that not only Ang II but also intraglomerular activation of MAPK by the receptor-associated prorenin plays a pivotal role in diabetes-induced kidney injury (254). In additional studies, heminephrectomized STZ-diabetic rats with an increased urinary protein excretion and a significant glomerulosclerosis were treated for 12 weeks with a (pro)renin receptor blocker (PRRB), an ACE inhibitor, or vehicle peptide. Whereas treatment with PRRB prevented any further increase in urinary protein excretion or glomerulosclerosis, treatment with an ACE inhibitor only attenuated the further increases (607). The data indicate a prominent role of a local intraglomerular renin system in the diabetic kidney and a potential therapeutic target (253).
The outlined studies in rats and mice were performed in the absence of any insulin treatment and under conditions of severe hyperglycemia (~400–550 mg/dl). Notably, whole kidney Ang II content is lower in STZ-diabetic rats compared with nondiabetic controls when diabetic rats were daily treated with insulin to adjust blood glucose levels around 350 mg/dl (664). In contrast, in the absence of insulin treatment and blood glucose levels of about 500 mg/dl, kidney content of both Ang I and II were reported to be increased in STZ-diabetic rats (252), indicating a potential influence of insulin treatment and/or blood glucose control. Further studies are needed to elucidate the influence of the degree of hyperglycemia and of insulin and other antidiabetic treatments on the activation of the intrarenal prorenin/renin system and, most importantly, its role in long-term diabetic renal failure.
Animal Models of Diabetic Nephropathy
Reliable animal models that mimic the human diabetic kidney disease are very important to better understand the molecular pathways involved and to identify key targets for better prevention and therapy. Diabetic animal models have mainly been developed in the rat and mouse and a brief overview on current models, problems and perspectives is presented. See also (19, 79, 84, 576, 672) for recent reviews on the topic.
Rat models of diabetic nephropathy
Models of nephropathy in type 1 diabetes mellitus
The streptozotocin (STZ)-diabetic rat is the most frequently used model of T1DM. Administration of STZ, a beta cell toxin, leads to beta cell destruction and insulinopenic diabetes within 24 to 36 h. Residual insulin secretion may be detectable in plasma for several weeks. Spontaneous recovery after several weeks of hyperglycemia is also possible. Without insulin treatment, STZ-diabetic rats develop severe hyperglycemia, dehydration, salt wasting, slow weight gain, and corresponding polydipsia and hyperphagia. To improve long-term survival and to create a more clinically relevant model of T1DM and its complications, studies are frequently conducted with STZ-diabetic rats treated with suboptimal doses of insulin to achieve moderate hyperglycemia (~20–25 mmol/l).
STZ-diabetic rats develop early renal and glomerular hypertrophy, initially without any structural changes. Within weeks, kidneys from STZ-diabetic rats display molecular markers of nephropathy including prosclerotic cytokines, growth factors, and components of ECM and a gradual increase in albuminuria as compared to age-matched nondiabetic controls. Although some degree of mesangial expansion or diffuse glomerulosclerosis could be detected earlier, measurable glomerulosclerosis, and in particular tubulointerstitial fibrosis, develop 6 to 12 months after induction of diabetes. Electron microscopy may reveal foot process effacement.
There are differences in rat strains in propensity to diabetes-induced renal pathology. The Sprague-Dawley strain is in our experience more susceptible than Wistar rats. Moreover, male rats are more prone to the development of albuminuria and structural changes than females. Without additional interventions STZ-diabetic rats do not develop decreases in GFR.
The relatively long period of time required for the development of renal structural changes and the lack of progressive decrease in GFR in STZ-diabetic rats motivated additional approaches to accelerate the course of nephropathy. These approaches include induction of STZ-diabetes in rat models of hypertension both spontaneous, such as in spontaneously hypertensive rats (121), and acquired, such as in DOCA-salt hypertensive rats (107). An aggressively progressive course of nephropathy with the development of severe hypertension, albuminuria, progressive decrease in GFR, and glomerular and interstitial lesions is characteristic for STZ-diabetic mRen-2 transgenic rats (307), harboring mouse renin gene. However, this model has been recently increasingly criticized. Massive activation of the RAS and hypertension lead to end-organ damage without concomitant diabetes, and, as demonstrated by Hartner et al. (224) these factors are the major driving force of nephropathy in this model.
The accelerated course of nephropathy can be also achieved by combining STZ-induced diabetes with unilateral nephrectomy, usually performed prior to induction of diabetes. These rats develop modest increases in blood pressure, massive proteinuria, reductions in GFR, and significantly more severe glomerulosclerosis and interstitial fibrosis as compared to uninephrectomized nondiabetic counterparts within 16 to 20 weeks after onset of diabetes (322, 323).
Rat models of nephropathy in type 2 diabetes mellitus
T2DM is part of the “metabolic syndrome”, which is, in addition to T2DM, defined by insulin resistance, truncal obesity, lipid abnormalities, and hypertension. The patients with metabolic syndrome also develop hyperuricemia, hypercoagulation state, and microalbuminuria. Therefore, the models of T2DM and associated vascular complications should be viewed from this broader perspective. Indeed, some of these models display clinical course resembling the evolution of metabolic syndrome and T2DM in humans. Moreover, these animals demonstrate a variety of renal abnormalities prior to the development of hyperglycemia (55, 327, 379), but in parallel with rising plasma insulin, FFA and triglyceride levels.
The best studied model of metabolic syndrome and T2DM with kidney involvement is the Zucker obese (fatty, fa/fa) rat. These rats are metabolically well characterized—an autosomal recessive mutation of the fagene, encoding the leptin receptor, results in hyperphagia, obesity, and hyperlipidemia (497). Similar to patients with T2DM, ZDF rats display insulin resistance, hyperinsulinemia, and impaired glucose tolerance, and slowly progressive increases in blood glucose levels (257). During the first 3 to 4 months of age, these rats do not display frank hyperglycemia, and are therefore suitable for investigations of pathophysiological roles of other factors associated with metabolic syndrome insulin resistance and T2DM, for example, increased levels of FFA, hyperinsulinemia, and adipokines. Zucker lean rats without leptin receptor abnormality do not display any abnormalities and serve as controls.
Extensive studies by Kasiske et al. (298) and Coimbra et al. (120) have shown that the first detectable changes in glomerular morphology suggestive of glomerulosclerosis and significant proteinuria occur at about 4 to 5 months of age, whereas tubulointerstitial damage after at least 40 weeks. More subtle changes characterized by glomerular hypertrophy and mesangial matrix expansion and albuminuria occur by 10 to 14 weeks of age. Mild elevations of blood pressure have been also reported (298). Similar to a number of other models of kidney disease, the kidney involvement is more severe in males than in females (146).
Otsuka Long-Evans Tokushima Fatty (OLETF) Strain and Goto Kakizaki (GK) rats are additional models utilized in studies exploring the pathophysiology and management of nephropathy in metabolic syndrome and T2DM. The OLETF Strain, a spontaneously diabetic rat with polyuria, polydipsia, and mild obesity, was discovered in 1984 in an outbred colony of Long-Evans rats. The characteristic features of OLETF rats include late onset of hyperglycemia (after 18 weeks of age), mild obesity, inheritance by males, hyperplastic foci of pancreatic islets; and renal complications including nodular lesions. After 40 weeks of age, the beta cell function is exhausted leading to low plasma insulin levels with gradual development of polyphagia, polyuria, decrease of body weight, and hyperglycemia, that is, symptoms of insulinopenic diabetes. Since this phenomenon occurs in a proportion of patients with T2DM, OLETF rats might be a valuable model for studies focusing on the impact of this transition on progression of microvascular complications. Interestingly, mild glomerular involvement, such as mesangial proliferation and thickening of the basement membranes were observed from week 40, associated with the appearance of glycosuria. The nodular lesions, have been described in 70-week old animals (304).
The GK rat, which was developed by Goto and collaborators (206) is characterized by glucose intolerance, which appears after 2 weeks of age in all animals. The etiology of diabetes in GK rats is thought to be exclusively genetic, because they were obtained by repetitive selective breeding of Wistar rats with abnormal glucose tolerance and became stably diabetic after several generations (206).
Unlike Zucker obese rats, GK rat do not display most of features of metabolic syndrome, such as hyperlipidemia, obesity, and remain normotensive until advanced age. Phillips et al. (491) described the course of renal changes in this model. They showed that GK rats did not develop significant proteinuria, but displayed age-dependent renal structural changes. These changes consisted of thickening of glomerular and tubular basement membranes at week 35. This thickening was accompanied by marked glomerular hypertrophy, although fractional capillary and mesangial volumes remained unchanged. There was evidence of podocyte injury, assessed by de novo expression of desmin. Interstitial monocyte/macrophage influx increased significantly in GK rats at 12 weeks compared with Wistar controls. Glomerular macrophage infiltration was elevated significantly in 35-week old GK rats. The authors concluded that structural changes in the GK rats resembled those described in prolonged non-insulin-dependent diabetes mellitus patients without overt renal disease. The model might be suitable to study the effects of hyperglycemia in T2DM on the development of organ complications without other confounding factors such as lipid abnormalities or obesity.
Zucker obese rats can be also crossed with nondiabetic rat models of hypertension to generate models with an accelerated course of nephropathy. For example, hybrid rats derived from the Zucker obese rats and the spontaneous hypertensive heart failure rat (SHHF/Gmi-fa) have been shown to develop severe renal injury and renal failure (146). A similar approach has been used to create spontaneously hypertensive/NIH-corpulent (SHR/NDmc-cp (fat/fat)) (405, 427).
Mouse models of diabetic nephropathy and the Animal Models of Diabetic Complications Consortium (AMDCC)
In 2001, the AMDCC (http://www.amdcc.org) was initiated by the National Institutes of Health to develop and characterize mouse models that mimic important aspects of human diabetic disease. The availability of >450 inbred strains (46, 389), the ongoing assembly of mice that bear multiple mutations that alter function in each known gene (246, 353, 629), and the application of the available unique strategies for genetic manipulations to such a mouse disease model may facilitate the understanding of the underlying genetic mechanisms that contribute to the development of diabetic nephropathy in human (79). However, no current murine strain of T1DM or T2DM reliably develops all of the features of human diabetic nephropathy (84). The FVB-OVE26 mouse (JAX # 5564) is a transgenic model of early-onset T1DM, generated directly onto the FVB/N background in the Epstein laboratory (746), which has been described to come relatively close. These mice showed progressively enlarged glomeruli and expanded mesangium with diffuse and nodular expansion of mesangial matrix and thickening of the glomerular basement membrane. Moreover, tubulointerstitial fibrosis, and some decrease in GFR in the later stages have been observed in these mice (746). More extensive studies are necessary to establish the value of this mouse model.
The AMDCC recommends the following criteria for validating a progressive mouse model of diabetic nephropathy: greater than 50% decline in GFR over the lifetime of the animal; greater than 10-fold increase in albuminuria compared with controls for that strain at the same age and gender; kidney pathology including advanced mesangial matrix expansion ± nodular sclerosis and mesangiolysis, any degree of arteriolar hyalinosis, glomerular basement membrane thickening by >50% over baseline, and tubulointerstitial fibrosis (84). The explicit focus on renal insufficiency is important, yet no current murine strain of T1DM or T2DM progresses to renal failure. An additional useful source for comparing basal metabolic parameters in the more commonly used inbred strains, including urinary albumin/creatinine ratios, is The Mouse Phenome Database (http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=docs/home).
Very helpful are the strategies and insights provided by the AMDCC on phenotyping standards (e.g., analysis of hyperglycemia, HgbA1C, albuminuria, creatinine, and GFR; staining methods for histology), useful T1DM and T2DM mouse models (including use of low-dose STZ protocol), validation criteria, the role of animal age, and the importance of genetic background (79). With regard to the latter, it appears that strains like BKS, 129S6/SvEvTac, KK/HIJ and, in particular, the DBA/2 mouse are more susceptible to diabetic glomerulopathy than the B6 strain, which seems to be the most resistant. However, to which extend these differences are associated with or predictive of renal insufficiency (i.e., a reduction in GFR), is unclear. Considering the pathophysiological importance of podocyte loss and that tubulointerstitial fibrosis determines to a major degree the outcome of the diabetic kidney, these aspects may deserve stronger attention in the model criteria. Furthermore, it seems important to also characterize these experimental models for important early aspects of the diabetic kidney that have been associated with later injury (98) including tubular growth.
The recent AMDCC review (84) concluded that targeting of Nos3, the gene encoding eNOS induces nephropathic changes in mouse models of both T1DM and T2DM that mimic many aspects of human disease (292, 416, 422, 745). Notably, this model is one of the very few to develop appreciable degrees of tubulointerstitial fibrosis and available on different backgrounds from The Jackson Laboratory [B6 (JAX# 2684), BKS-db/db (JAX# 8340), and BALB/cBy (JAX# 7073)]. Likewise important are new information on negative diabetic models that unexpectedly did not show disease progression (84). Details on these models including possible explanations are outlined on the AMDCC web site. Examples include diabetes induction in C57B6 mice with ACE2 gene deficiency, B6-Sod2 heterozygous mice, and endothelial-specific receptors of advances glycation end-product monotransgenic mice on a mixed B6 and CD-1 background.
Considering the relative resistance of the mouse to diabetic nephropathy, important clues may be derived by comparing gene expression profiles in diabetic human and mice. In this regard, Berthier et al. analyzed the expression of genes in glomeruli and tubulointerstitium in kidney biopsies from diabetic nephropathy patients to identify pathways that may be activated in humans but not in murine models that are resistant to diabetic nephropathy and kidney failure, using a mRNA microarray approach, which was followed up by real-time RT-PCR and immunohistological staining. db/db C57BLKS and STZ-induced diabetic DBA/2J mice were used as commonly studied murine models of diabetic nephropathy. The study found a direct relationship between tubulointerstitial expression of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway and progression of kidney failure in patients with T2DM and nephropathy. In contrast, there was little or no increase in expression of these pathways in the nonprogressive murine diabetic mouse models (53).
Conclusion
Much has been learned about the (patho)physiology of high glucose concentrations in the context of a single cell as well as the intact kidney. This includes mechanisms of AGEs formation and oxidative stress and their central role in the diabetic kidney. A better understanding of the molecular determinants and the complex interplay between the diabetic milieu and the structures involved in the GFB, including the podocytes, is emerging. A role for primary vascular as well as tubular events in early diabetic glomerular hyperfiltration has been established, the latter being related to tubular growth and hyperreabsorption and the physiology of tubuloglomerular communication. We are beginning to understand the molecular basis of the complex interactions between the diabetic milieu and the tubulointerstitium as well as of the unique growth phenotype of the diabetic kidney, and their potentially critical role in linking early changes of the diabetic kidney to tubulointerstitial fibrosis, hypoxia, and renal failure. A better understanding of a proposed senescent phenotype of diabetic tubular cells is necessary as well as the molecular signature and the relevance of epithelial mesenchymal transition in the diabetic kidney. More insight in the activation of the intraglomerular renin/prorenin system, including the role of succinate and its receptor, and of their relevance to the long-term kidney outcome is highly desirable. Further insights are also expected from the rapidly expanding field of epigenetics and transcriptional control, which may identify and explain the phenomenon of “metabolic memory” in the pathogenesis and perpetuation of microvascular complications of diabetes including nephropathy. The identification and better understanding of the molecular pathophysiology of key mechanisms may help one day to predict which diabetic patient will eventually develop diabetic nephropathy and renal failure, and to develop new therapeutic strategies that target these mechanisms. Our progress in this regard will also depend on the further development of reliable animal models that mimic the phenotypic and molecular signature of human diabetic kidney disease. To identify and further explore these models, experimental studies are needed that follow up into the later stages of diabetic nephropathy including the decline in GFR and renal failure.
Acknowledgments
The authors were supported by the American Heart Association (GRNT3440038 to VV), the National Institutes of Health (R01DK56248, R01DK28602, R01GM66232, R01HL094728, P30DK079337 to VV), the Department of Veterans Affairs (to VV), and the Juvenile Diabetes Research Foundation (1-2008-314, 42-2009-779 to RK).
References
- 1.Aaltonen P, Luimula P, Astrom E, Palmen T, Gronholm T, Palojoki E, Jaakkola I, Ahola H, Tikkanen I, Holthofer H. Changes in the expression of nephrin gene and protein in experimental diabetic nephropathy. Lab Invest. 2001;81:1185–1190. doi: 10.1038/labinvest.3780332. [DOI] [PubMed] [Google Scholar]
- 2.Abbate M, Remuzzi G. Proteinuria as a mediator of tubulointerstitial injury. Kidney Blood Press Res. 1999;22:37–46. doi: 10.1159/000025907. [DOI] [PubMed] [Google Scholar]
- 3.Abbate M, Zoja C, Corna D, Capitanio M, Bertani T, Remuzzi G. In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol. 1998;9:1213–1224. doi: 10.1681/ASN.V971213. [DOI] [PubMed] [Google Scholar]
- 4.Abdel-Wahab N, Weston BS, Roberts T, Mason RM. Connective tissue growth factor and regulation of the mesangial cell cycle: Role in cellular hypertrophy. J Am Soc Nephrol. 2002;13:2437–2445. doi: 10.1097/01.asn.0000031828.58276.02. [DOI] [PubMed] [Google Scholar]
- 5.Abraham NG, Kappas A. Heme oxygenase and the cardiovascular-renal system. Free Radic Biol Med. 2005;39:1–25. doi: 10.1016/j.freeradbiomed.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 6.Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackermann TF, Boini KM, Volkl H, Bhandaru M, Bareiss PM, Just L, Vallon V, Amann K, Kuhl D, Feng Y, Hammes HP, Lang F. SGK1-sensitive renal tubular glucose reabsorption in diabetes. Am J Physiol Renal Physiol. 2009;296:F859–F866. doi: 10.1152/ajprenal.90238.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adachi T, Yasuda K, Okamoto Y, Shihara N, Oku A, Ueta K, Kitamura K, Saito A, Iwakura I, Yamada Y, Yano H, Seino Y, Tsuda K. T-1095, a renal Na+-glucose transporter inhibitor, improves hyperglycemia in streptozotocin-induced diabetic rats. Metabolism. 2000;49:990–995. doi: 10.1053/meta.2000.7729. [DOI] [PubMed] [Google Scholar]
- 9.Adler S, Chen X. Anti-Fx1A antibody recognizes a beta 1-integrin on glomerular epithelial cells and inhibits adhesion and growth. Am J Physiol. 1992;262:F770–F776. doi: 10.1152/ajprenal.1992.262.5.F770. [DOI] [PubMed] [Google Scholar]
- 10.Adler SG, Kang SW, Feld S, Cha DR, Barba L, Striker L, Striker G, Riser BL, Lapage J, Nast CC. Glomerular mRNAs in human type 1 diabetes: Biochemical evidence for microalbuminuria as a manifestation of diabetic nephropathy. Kidney Int. 2001;60:2330–2336. doi: 10.1046/j.1523-1755.2001.00073.x. [DOI] [PubMed] [Google Scholar]
- 11.Adler SG, Schwartz S, Williams ME, Arauz-Pacheco C, Bolton WK, Lee T, Li D, Neff TB, Urquilla PR, Sewell KL. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol. 2010;5:1420–1428. doi: 10.2215/CJN.09321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Advani A, Gilbert RE, Thai K, Gow RM, Langham RG, Cox AJ, Connelly KA, Zhang Y, Herzenberg AM, Christensen PK, Pollock CA, Qi W, Tan SM, Parving HH, Kelly DJ. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J Am Soc Nephrol. 2009;20:730–741. doi: 10.1681/ASN.2008020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed N, Thornalley PJ. Advanced glycation endproducts: What is their relevance to diabetic complications? Diabetes Obes Metab. 2007;9:233–245. doi: 10.1111/j.1463-1326.2006.00595.x. [DOI] [PubMed] [Google Scholar]
- 14.Akai Y, Homma T, Burns KD, Yasuda T, Badr KF, Harris RC. Mechanical stretch/relaxation of cultured rat mesangial cells induces protooncogenes and cyclooxygenase. Am J Physiol. 1994;267:C482–C490. doi: 10.1152/ajpcell.1994.267.2.C482. [DOI] [PubMed] [Google Scholar]
- 15.Albertoni Borghese MF, Majowicz MP, Ortiz MC, Passalacqua MR, Sterin Speziale NB, Vidal NA. Expression and activity of SGLT2 in diabetes induced by streptozotocin: Relationship with the lipid environment. Nephron Physiol. 2009;112:45–52. doi: 10.1159/000214214. [DOI] [PubMed] [Google Scholar]
- 16.Al-Douahji M, Brugarolas J, Brown PA, Stehman-Breen CO, Alpers CE, Shankland SJ. The cyclin kinase inhibitor p21WAF1/CIP1 is required for glomerular hypertrophy in experimental diabetic nephropathy. Kidney Int. 1999;56:1691–1699. doi: 10.1046/j.1523-1755.1999.00728.x. [DOI] [PubMed] [Google Scholar]
- 17.Alexander K, Hinds PW. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol Cell Biol. 2001;21:3616–3631. doi: 10.1128/MCB.21.11.3616-3631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen RG, Tresini M, Keogh BP, Doggett DL, Cristofalo VJ. Differences in electron transport potential, antioxidant defenses, and oxidant generation in young and senescent fetal lung fibroblasts (WI-38) J Cell Physiol. 1999;180:114–122. doi: 10.1002/(SICI)1097-4652(199907)180:1<114::AID-JCP13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 19.Allen TJ, Cooper ME, Lan HY. Use of genetic mouse models in the study of diabetic nephropathy. Curr Atheroscler Rep. 2004;6:197–202. doi: 10.1007/s11883-004-0032-7. [DOI] [PubMed] [Google Scholar]
- 20.Allen TJ, Waldron MJ, Casley D, Jerums G, Cooper ME. Salt restriction reduces hyperfiltration, renal enlargement, and albuminuria in experimental diabetes. Diabetes. 1997;46:19–24. doi: 10.2337/diabetes.46.1.19. [DOI] [PubMed] [Google Scholar]
- 21.Alpers CE, Hudkins KL, Floege J, Johnson RJ. Human renal cortical interstitial cells with some features of smooth muscle cells participate in tubulointerstitial and crescentic glomerular injury. J Am Soc Nephrol. 1994;5:201–209. doi: 10.1681/ASN.V52201. [DOI] [PubMed] [Google Scholar]
- 22.Amin R, Turner C, van AS, Bahu TK, Watts A, Lindsell DR, Dalton RN, Dunger DB. The relationship between microalbuminuria and glomerular filtration rate in young type 1 diabetic subjects: The Oxford Regional Prospective Study. Kidney Int. 2005;68:1740–1749. doi: 10.1111/j.1523-1755.2005.00590.x. [DOI] [PubMed] [Google Scholar]
- 23.Anderson S. Progression of chronic renal disease: Role of systemic and glomerular hypertension. Am J Kidney Dis. 1989;13:8–12. [PubMed] [Google Scholar]
- 24.Anderson S, Jung FF, Ingelfinger JR. Renal renin-angiotensin system in diabetes: Functional, immunohistochemical, and molecular biological correlations. Am J Physiol. 1993;265:F477–F486. doi: 10.1152/ajprenal.1993.265.4.F477. [DOI] [PubMed] [Google Scholar]
- 25.Aragones J, Schneider M, Van GK, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van NP, De BK, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van HP, Schuit F, Van VP, Ratcliffe P, Baes M, Maxwell P, Carmeliet P. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 26.Arakawa K, Ishihara T, Oku A, Nawano M, Ueta K, Kitamura K, Matsumoto M, Saito A. Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na(+)-glucose cotransporter inhibitor T-1095. Br J Pharmacol. 2001;132:578–586. doi: 10.1038/sj.bjp.0703829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ardura JA, Rayego-Mateos S, Ramila D, Ruiz-Ortega M, Esbrit P. Parathyroid hormone-related protein promotes epithelial-mesenchymal transition. J Am Soc Nephrol. 2010;21:237–248. doi: 10.1681/ASN.2009050462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arici M, Chana R, Lewington A, Brown J, Brunskill NJ. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-gamma. J Am Soc Nephrol. 2003;14:17–27. doi: 10.1097/01.asn.0000042167.66685.ea. [DOI] [PubMed] [Google Scholar]
- 29.Asaba K, Tojo A, Onozato ML, Goto A, Quinn MT, Fujita T, Wilcox CS. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005;67:1890–1898. doi: 10.1111/j.1523-1755.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 30.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 31.Atchley DW, Loeb RF, Richards DW, Benedict EM, Driscoll ME. On diabetic acidosis: A detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest. 1933;12:297–326. doi: 10.1172/JCI100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awazu M, Omori S, Ishikura K, Hida M, Fujita H. The lack of cyclin kinase inhibitor p27(Kip1) ameliorates progression of diabetic nephropathy. J Am Soc Nephrol. 2003;14:699–708. doi: 10.1097/01.asn.0000051726.41601.c0. [DOI] [PubMed] [Google Scholar]
- 33.Bader R, Bader H, Grund KE, Mackensen-Haen S, Christ H, Bohle A. Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathol Res Pract. 1980;167:204–216. doi: 10.1016/S0344-0338(80)80051-3. [DOI] [PubMed] [Google Scholar]
- 34.Baelde HJ, Eikmans M, Lappin DW, Doran PP, Hohenadel D, Brinkkoetter PT, Van Der Woude FJ, Waldherr R, Rabelink TJ, de HE, Bruijn JA. Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int. 2007;71:637–645. doi: 10.1038/sj.ki.5002101. [DOI] [PubMed] [Google Scholar]
- 35.Bahlmann FH, Fliser D. Erythropoietin and renoprotection. Curr Opin Nephrol Hypertens. 2009;18:15–20. doi: 10.1097/MNH.0b013e32831a9dde. [DOI] [PubMed] [Google Scholar]
- 36.Ballermann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007;106:19–25. doi: 10.1159/000101796. [DOI] [PubMed] [Google Scholar]
- 37.Ballermann BJ, Dardik A, Eng E, Liu A. Shear stress and the endothelium. Kidney Int Suppl. 1998;67:S100–S108. doi: 10.1046/j.1523-1755.1998.06720.x. [DOI] [PubMed] [Google Scholar]
- 38.Ballermann BJ, Skorecki KL, Brenner BM. Reduced glomerular angiotensin II receptor density in early untreated diabetes mellitus in the rat. Am J Physiol. 1984;247:F110–F116. doi: 10.1152/ajprenal.1984.247.1.F110. [DOI] [PubMed] [Google Scholar]
- 39.Bamri-Ezzine S, Ao ZJ, Londono I, Gingras D, Bendayan M. Apoptosis of tubular epithelial cells in glycogen nephrosis during diabetes. Lab Invest. 2003;83:1069–1080. doi: 10.1097/01.lab.0000078687.21634.69. [DOI] [PubMed] [Google Scholar]
- 40.Bank N, Aynedjian HS. Progressive increases in luminal glucose stimulate proximal sodium absorption in normal and diabetic rats. J Clin Invest. 1990;86:309–316. doi: 10.1172/JCI114700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bank N, Lahorra G, Aynedjian HS, Wilkes BM. Sodium restriction corrects hyperfiltration of diabetes. Am J Physiol. 1988;254:F668–F676. doi: 10.1152/ajprenal.1988.254.5.F668. [DOI] [PubMed] [Google Scholar]
- 42.Bank N, Mower P, Aynedjian HS, Wilkes BM, Silverman S. Sorbinil prevents glomerular hyperperfusion in diabetic rats. Am J Physiol. 1989;256:F1000–F1006. doi: 10.1152/ajprenal.1989.256.6.F1000. [DOI] [PubMed] [Google Scholar]
- 43.Baumbach L, Leyssac PP, Skinner SL. Studies on renin release from isolated superfused glomeruli: Effects of temperature, urea, ouabain and ethacrynic acid. J Physiol. 1976;258:243–256. doi: 10.1113/jphysiol.1976.sp011417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumgartl HJ, Sigl G, Banholzer P, Haslbeck M, Standl E. On the prognosis of IDDM patients with large kidneys. Nephrol Dial Transplant. 1998;13:630–634. doi: 10.1093/ndt/13.3.630. [DOI] [PubMed] [Google Scholar]
- 45.Bautista R, Manning R, Martinez F, Avila-Casado MC, Soto V, Medina A, Escalante B. Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am J Physiol Renal Physiol. 2004;286:F127–F133. doi: 10.1152/ajprenal.00113.2003. [DOI] [PubMed] [Google Scholar]
- 46.Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM. Genealogies of mouse inbred strains. Nat Genet. 2000;24:23–25. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- 47.Beisswenger PJ, Makita Z, Curphey TJ, Moore LL, Jean S, Brinck-Johnsen T, Bucala R, Vlassara H. Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes. 1995;44:824–829. doi: 10.2337/diab.44.7.824. [DOI] [PubMed] [Google Scholar]
- 48.Bell TD, DiBona GF, Wang Y, Brands MW. Mechanisms for renal blood flow control early in diabetes as revealed by chronic flow measurement and transfer function analysis. J Am Soc Nephrol. 2006;17:2184–2192. doi: 10.1681/ASN.2006030216. [DOI] [PubMed] [Google Scholar]
- 49.Benigni A, Colosio V, Brena C, Bruzzi I, Bertani T, Remuzzi G. Unselective inhibition of endothelin receptors reduces renal dysfunction in experimental diabetes. Diabetes. 1998;47:450–456. doi: 10.2337/diabetes.47.3.450. [DOI] [PubMed] [Google Scholar]
- 50.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benigni A, Gagliardini E, Tomasoni S, Abbate M, Ruggenenti P, Kalluri R, Remuzzi G. Selective impairment of gene expression and assembly of nephrin in human diabetic nephropathy. Kidney Int. 2004;65:2193–2200. doi: 10.1111/j.1523-1755.2004.00636.x. [DOI] [PubMed] [Google Scholar]
- 52.Berkman J, Rifkin H. Unilateral nodular diabetic glomerulosclerosis (Kimmelstiel-Wilson): Report of a case. Metabolism. 1973;22:715–722. doi: 10.1016/0026-0495(73)90243-6. [DOI] [PubMed] [Google Scholar]
- 53.Berthier CC, Zhang H, Schin M, Henger A, Nelson RG, Yee B, Boucherot A, Neusser MA, Cohen CD, Carter-Su C, Argetsinger LS, Rastaldi MP, Brosius FC, Kretzler M. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58:469–477. doi: 10.2337/db08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biava C, Grossman A, West M. Ultrastructural observations on renal glycogen in normal and pathologic human kidneys. Lab Invest. 1966;15:330–356. [PubMed] [Google Scholar]
- 55.Bickel CA, Knepper MA, Verbalis JG, Ecelbarger CA. Dysregulation of renal salt and water transport proteins in diabetic Zucker rats. Kidney Int. 2002;61:2099–2110. doi: 10.1046/j.1523-1755.2002.00353.x. [DOI] [PubMed] [Google Scholar]
- 56.Bickel CA, Verbalis JG, Knepper MA, Ecelbarger CA. Increased renal Na-K-ATPase, NCC, and beta-ENaC abundance in obese Zucker rats. Am J Physiol Renal Physiol. 2001;281:F639–F648. doi: 10.1152/ajprenal.2001.281.4.F639. [DOI] [PubMed] [Google Scholar]
- 57.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 58.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 59.Birk C, Richter K, Huang DY, Piesch C, Luippold G, Vallon V. The salt paradox of the early diabetic kidney is independent of renal innervation. Kidney Blood Press Res. 2003;26:344–350. doi: 10.1159/000073941. [DOI] [PubMed] [Google Scholar]
- 60.Blanco S, Bonet J, Lopez D, Casas I, Romero R. ACE inhibitors improve nephrin expression in Zucker rats with glomerulosclerosis. Kidney Int Suppl. 2005:S10–S14. doi: 10.1111/j.1523-1755.2005.09303.x. [DOI] [PubMed] [Google Scholar]
- 61.Blantz RC, Peterson OW, Gushwa L, Tucker BJ. Effect of modest hyperglycemia on tubuloglomerular feedback activity. Kidney Int Suppl. 1982;12:S206–S212. [PubMed] [Google Scholar]
- 62.Bleyer AJ, Fumo P, Snipes ER, Goldfarb S, Simmons DA, Ziyadeh FN. Polyol pathway mediates high glucose-induced collagen synthesis in proximal tubule. Kidney Int. 1994;45:659–666. doi: 10.1038/ki.1994.88. [DOI] [PubMed] [Google Scholar]
- 63.Bobulescu IA, Dubree M, Zhang J, McLeroy P, Moe OW. Effect of renal lipid accumulation on proximal tubule Na+/H+ exchange and ammonium secretion. Am J Physiol Renal Physiol. 2008;294:F1315–F1322. doi: 10.1152/ajprenal.00550.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bognetti E, Zoja A, Meschi F, Paesano PL, Chiumello G. Relationship between kidney volume, microalbuminuria and duration of diabetes mellitus. Diabetologia. 1996;39:1409. [PubMed] [Google Scholar]
- 65.Bohle A, Wehrmann M, Bogenschutz O, Batz C, Muller CA, Muller GA. The pathogenesis of chronic renal failure in diabetic nephropathy. Investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187:251–259. doi: 10.1016/s0344-0338(11)80780-6. [DOI] [PubMed] [Google Scholar]
- 66.Bondeva T, Ruster C, Franke S, Hammerschmid E, Klagsbrun M, Cohen CD, Wolf G. Advanced glycation end-products suppress neuropilin-1 expression in podocytes. Kidney Int. 2009;75:605–616. doi: 10.1038/ki.2008.603. [DOI] [PubMed] [Google Scholar]
- 67.Bonnet F, Cooper ME, Kawachi H, Allen TJ, Boner G, Cao Z. Irbesartan normalises the deficiency in glomerular nephrin expression in a model of diabetes and hypertension. Diabetologia. 2001;44:874–877. doi: 10.1007/s001250100546. [DOI] [PubMed] [Google Scholar]
- 68.Border WA, Brees D, Noble NA. Transforming growth factor-beta and extracellular matrix deposition in the kidney. Contrib Nephrol. 1994;107:140–145. doi: 10.1159/000422972. [DOI] [PubMed] [Google Scholar]
- 69.Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension. 1998;31:181–188. doi: 10.1161/01.hyp.31.1.181. [DOI] [PubMed] [Google Scholar]
- 70.Border WA, Noble N. Maximizing hemodynamic-independent effects of angiotensin II antagonists in fibrotic diseases. Semin Nephrol. 2001;21:563–572. doi: 10.1053/snep.2001.26795. [DOI] [PubMed] [Google Scholar]
- 71.Border WA, Noble NA, Ketteler M. TGF-beta: A cytokine mediator of glomerulosclerosis and a target for therapeutic intervention. Kidney Int Suppl. 1995;49:S59–S61. [PubMed] [Google Scholar]
- 72.Bover J, Izquierdo A, Arce Y, Ortega A, Fernández S, Romero M, Algaba F, Calero F, Ballarin J, Esbrit P, Bosch RJ. Parathyroid hormone-related protein (PTHrP) up-regulation in glomeruli and tubuli of human glomerular nephropathies. Nephrol Dial Transplant. 2006;21:iv25. (Abstract) [Google Scholar]
- 73.Bowden DW, Sale M, Howard TD, Qadri A, Spray BJ, Rothschild CB, Akots G, Rich SS, Freedman BI. Linkage of genetic markers on human chromosomes 20 and 12 to NIDDM in Caucasian sib pairs with a history of diabetic nephropathy. Diabetes. 1997;46:882–886. doi: 10.2337/diab.46.5.882. [DOI] [PubMed] [Google Scholar]
- 74.Brands MW, Hall JE, Keen HL. Is insulin resistance linked to hypertension? Clin Exp Pharmacol Physiol. 1998;25:70–76. doi: 10.1111/j.1440-1681.1998.tb02148.x. [DOI] [PubMed] [Google Scholar]
- 75.Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Sustained hyperinsulinemia increases arterial pressure in conscious rats. Am J Physiol. 1991;260:R764–R768. doi: 10.1152/ajpregu.1991.260.4.R764. [DOI] [PubMed] [Google Scholar]
- 76.Brands MW, Lee WF, Keen HL, Alonso-Galicia M, Zappe DH, Hall JE. Cardiac output and renal function during insulin hypertension in Sprague-Dawley rats. Am J Physiol. 1996;271:R276–R281. doi: 10.1152/ajpregu.1996.271.1.R276. [DOI] [PubMed] [Google Scholar]
- 77.Breiteneder-Geleff S, Matsui K, Soleiman A, Meraner P, Poczewski H, Kalt R, Schaffner G, Kerjaschki D. Podoplanin, novel 43-kd membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am J Pathol. 1997;151:1141–1152. [PMC free article] [PubMed] [Google Scholar]
- 78.Brenner BM, Cooper ME, de ZD, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 79.Breyer MD, Bottinger E, Brosius FC, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 80.Brito PL, Fioretto P, Drummond K, Kim Y, Steffes MW, Basgen JM, Sisson-Ross S, Mauer M. Proximal tubular basement membrane width in insulin-dependent diabetes mellitus. Kidney Int. 1998;53:754–761. doi: 10.1046/j.1523-1755.1998.00809.x. [DOI] [PubMed] [Google Scholar]
- 81.Brochner-Mortensen J, Stockel M, Sorensen PJ, Nielsen AH, Ditzel J. Proximal glomerulotubular balance in patients with type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1984;27:189–192. doi: 10.1007/BF00273804. [DOI] [PubMed] [Google Scholar]
- 82.Brodsky SV, Morrishow AM, Dharia N, Gross SS, Goligorsky MS. Glucose scavenging of nitric oxide. Am J Physiol Renal Physiol. 2001;280:F480–F486. doi: 10.1152/ajprenal.2001.280.3.F480. [DOI] [PubMed] [Google Scholar]
- 83.Brooimans RA, Stegmann AP, van Dorp WT, Van Der Ark AA, Van Der Woude FJ, van Es LA, Daha MR. Interleukin 2 mediates stimulation of complement C3 biosynthesis in human proximal tubular epithelial cells. J Clin Invest. 1991;88:379–384. doi: 10.1172/JCI115314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brosius FC, III, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 86.Brownlee M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 87.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 88.Bruce CR, Mertz VA, Heigenhauser GJ, Dyck DJ. The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes. 2005;54:3154–3160. doi: 10.2337/diabetes.54.11.3154. [DOI] [PubMed] [Google Scholar]
- 89.Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest. 1991;87:432–438. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burns WC, Kantharidis P, Thomas MC. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs. 2007;185:222–231. doi: 10.1159/000101323. [DOI] [PubMed] [Google Scholar]
- 91.Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, Thomas MC, Cooper ME, Kantharidis P. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: Implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- 92.Busch M, Franke S, Ruster C, Wolf G. Advanced glycation end-products and the kidney. Eur J Clin Invest. 2010;40:742–755. doi: 10.1111/j.1365-2362.2010.02317.x. [DOI] [PubMed] [Google Scholar]
- 93.Buse MG. Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am J Physiol Endocrinol Metab. 2006;290:E1–E8. doi: 10.1152/ajpendo.00329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem. 2000;275:5179–5187. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- 95.Cammisotto PG, Londono I, Gingras D, Bendayan M. Control of glycogen synthase through ADIPOR1-AMPK pathway in renal distal tubules of normal and diabetic rats. Am J Physiol Renal Physiol. 2008;294:F881–F889. doi: 10.1152/ajprenal.00373.2007. [DOI] [PubMed] [Google Scholar]
- 96.Campbell DJ, Kelly DJ, Wilkinson-Berka JL, Cooper ME, Skinner SL. Increased bradykinin and “normal” angiotensin peptide levels in diabetic Sprague-Dawley and transgenic (mRen-2)27 rats. Kidney Int. 1999;56:211–221. doi: 10.1046/j.1523-1755.1999.00519.x. [DOI] [PubMed] [Google Scholar]
- 97.Campese VM, Wurgaft A, Safa M, Bianchi S. Dietary salt intake, blood pressure and the kidney in hypertensive patients with non-insulin dependent diabetes mellitus. J Nephrol. 1998;11:289–295. [PubMed] [Google Scholar]
- 98.Caramori ML, Fioretto P, Mauer M. The need for early predictors of diabetic nephropathy risk: Is albumin excretion rate sufficient? Diabetes. 2000;49:1399–1408. doi: 10.2337/diabetes.49.9.1399. [DOI] [PubMed] [Google Scholar]
- 99.Carmines PK, Ohishi K, Ikenaga H. Functional impairment of renal afferent arteriolar voltage-gated calcium channels in rats with diabetes mellitus. J Clin Invest. 1996;98:2564–2571. doi: 10.1172/JCI119075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Catanuto P, Doublier S, Lupia E, Fornoni A, Berho M, Karl M, Striker GE, Xia X, Elliot S. 17 beta-estradiol and tamoxifen upregulate estrogen receptor beta expression and control podocyte signaling pathways in a model of type 2 diabetes. Kidney Int. 2009;75:1194–1201. doi: 10.1038/ki.2009.69. [DOI] [PubMed] [Google Scholar]
- 101.Ceolotto G, Gallo A, Miola M, Sartori M, Trevisan R, Del PS, Semplicini A, Avogaro A. Protein kinase C activity is acutely regulated by plasma glucose concentration in human monocytes in vivo. Diabetes. 1999;48:1316–1322. doi: 10.2337/diabetes.48.6.1316. [DOI] [PubMed] [Google Scholar]
- 102.Chen HC, Chen CA, Guh JY, Chang JM, Shin SJ, Lai YH. Altering expression of alpha3beta1 integrin on podocytes of human and rats with diabetes. Life Sci. 2000;67:2345–2353. doi: 10.1016/s0024-3205(00)00815-8. [DOI] [PubMed] [Google Scholar]
- 103.Chen S, Cohen MP, Lautenslager GT, Shearman CW, Ziyadeh FN. Glycated albumin stimulates TGF-beta 1 production and protein kinase C activity in glomerular endothelial cells. Kidney Int. 2001;59:673–681. doi: 10.1046/j.1523-1755.2001.059002673.x. [DOI] [PubMed] [Google Scholar]
- 104.Chen S, Ge Y, Si J, Rifai A, Dworkin LD, Gong R. Candesartan suppresses chronic renal inflammation by a novel antioxidant action independent of AT1R blockade. Kidney Int. 2008;74:1128–1138. doi: 10.1038/ki.2008.380. [DOI] [PubMed] [Google Scholar]
- 105.Chen S, Hoffman BB, Lee JS, Kasama Y, Jim B, Kopp JB, Ziyadeh FN. Cultured tubule cells from TGF-beta1 null mice exhibit impaired hypertrophy and fibronectin expression in high glucose. Kidney Int. 2004;65:1191–1204. doi: 10.1111/j.1523-1755.2004.00492.x. [DOI] [PubMed] [Google Scholar]
- 106.Chen S, Kasama Y, Lee JS, Jim B, Marin M, Ziyadeh FN. Podocyte-derived vascular endothelial growth factor mediates the stimulation of alpha3(IV) collagen production by transforming growth factor-beta1 in mouse podocytes. Diabetes. 2004;53:2939–2949. doi: 10.2337/diabetes.53.11.2939. [DOI] [PubMed] [Google Scholar]
- 107.Cheng HF, Wang CJ, Moeckel GW, Zhang MZ, McKanna JA, Harris RC. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Int. 2002;62:929–939. doi: 10.1046/j.1523-1755.2002.00520.x. [DOI] [PubMed] [Google Scholar]
- 108.Cheng S, Lovett DH. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am J Pathol. 2003;162:1937–1949. doi: 10.1016/S0002-9440(10)64327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng S, Pollock AS, Mahimkar R, Olson JL, Lovett DH. Matrix met-alloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006;20:1898–1900. doi: 10.1096/fj.06-5898fje. [DOI] [PubMed] [Google Scholar]
- 110.Cherney DZ, Konvalinka A, Zinman B, Diamandis EP, Soosaipillai A, Reich H, Lorraine J, Lai V, Scholey JW, Miller JA. Effect of protein kinase Cbeta inhibition on renal hemodynamic function and urinary biomarkers in humans with type 1 diabetes: A pilot study. Diabetes Care. 2009;32:91–93. doi: 10.2337/dc08-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cherney DZ, Miller JA, Scholey JW, Bradley TJ, Slorach C, Curtis JR, Dekker MG, Nasrallah R, Hebert RL, Sochett EB. The effect of cyclooxygenase-2 inhibition on renal hemodynamic function in humans with type 1 diabetes. Diabetes. 2008;57:688–695. doi: 10.2337/db07-1230. [DOI] [PubMed] [Google Scholar]
- 112.Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Horm Metab Res. 2009;41:585–593. doi: 10.1055/s-0029-1220752. [DOI] [PubMed] [Google Scholar]
- 113.Chin E, Zamah AM, Landau D, Gronbcek H, Flyvbjerg A, LeRoith D, Bondy CA. Changes in facilitative glucose transporter messenger ribonucleic acid levels in the diabetic rat kidney. Endocrinology. 1997;138:1267–1275. doi: 10.1210/endo.138.3.5015. [DOI] [PubMed] [Google Scholar]
- 114.Chin E, Zhou J, Bondy C. Anatomical and developmental patterns of facilitative glucose transporter gene expression in the rat kidney. J Clin Invest. 1993;91:1810–1815. doi: 10.1172/JCI116392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Christensen CK, Christiansen JS, Christensen T, Hermansen K, Mogensen CE. The effect of six months continuous subcutaneous insulin infusion on kidney function and size in insulin-dependent diabetics. Diabet Med. 1986;3:29–32. doi: 10.1111/j.1464-5491.1986.tb00701.x. [DOI] [PubMed] [Google Scholar]
- 116.Christiansen JS, Gammelgaard J, Frandsen M, Parving HH. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetologia. 1981;20:451–456. doi: 10.1007/BF00253406. [DOI] [PubMed] [Google Scholar]
- 117.Christiansen JS, Gammelgaard J, Orskov H, Andersen AR, Telmer S, Parving HH. Kidney function and size in normal subjects before and during growth hormone administration for one week. Eur J Clin Invest. 1981;11:487–490. doi: 10.1111/j.1365-2362.1981.tb02018.x. [DOI] [PubMed] [Google Scholar]
- 118.Christiansen JS, Gammelgaard J, Tronier B, Svendsen PA, Parving HH. Kidney function and size in diabetics before and during initial insulin treatment. Kidney Int. 1982;21:683–688. doi: 10.1038/ki.1982.81. [DOI] [PubMed] [Google Scholar]
- 119.Cohen MP, Sharma K, Guo J, Eltayeb BO, Ziyadeh FN. The renal TGF-beta system in the db/db mouse model of diabetic nephropathy. Exp Nephrol. 1998;6:226–233. doi: 10.1159/000020527. [DOI] [PubMed] [Google Scholar]
- 120.Coimbra TM, Janssen U, Grone HJ, Ostendorf T, Kunter U, Schmidt H, Brabant G, Floege J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000;57:167–182. doi: 10.1046/j.1523-1755.2000.00836.x. [DOI] [PubMed] [Google Scholar]
- 121.Cooper ME, Rumble JR, Allen TJ, O’Brien RC, Jerums G, Doyle AE. Antihypertensive therapy in a model combining spontaneous hypertension with diabetes. Kidney Int. 1992;41:898–903. doi: 10.1038/ki.1992.137. [DOI] [PubMed] [Google Scholar]
- 122.Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, Casley DJ, Bach LA, Kelly DJ, Gilbert RE. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–2239. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- 123.Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, Tan AL, Fukami K, Thallas-Bonke V, Nawroth PP, Brownlee M, Bierhaus A, Cooper ME, Forbes JM. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. 2009;20:742–752. doi: 10.1681/ASN.2008050514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Craven PA, Caines MA, DeRubertis FR. Sequential alterations in glomerular prostaglandin and thromboxane synthesis in diabetic rats: Relationship to the hyperfiltration of early diabetes. Metabolism. 1987;36:95–103. doi: 10.1016/0026-0495(87)90070-9. [DOI] [PubMed] [Google Scholar]
- 125.D’Amico G, Bazzi C. Pathophysiology of proteinuria. Kidney Int. 2003;63:809–825. doi: 10.1046/j.1523-1755.2003.00840.x. [DOI] [PubMed] [Google Scholar]
- 126.D’Amico G, Ferrario F, Rastaldi MP. Tubulointerstitial damage in glomerular diseases: Its role in the progression of renal damage. Am J Kidney Dis. 1995;26:124–132. doi: 10.1016/0272-6386(95)90165-5. [DOI] [PubMed] [Google Scholar]
- 127.da Gabriela SX, Leclerc I, Salt IP, Doiron B, Hardie DG, Kahn A, Rutter GA. Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc Natl Acad Sci U S A. 2000;97:4023–4028. doi: 10.1073/pnas.97.8.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dahlquist G, Stattin EL, Rudberg S. Urinary albumin excretion rate and glomerular filtration rate in the prediction of diabetic nephropathy; a long-term follow-up study of childhood onset type-1 diabetic patients. Nephrol Dial Transplant. 2001;16:1382–1386. doi: 10.1093/ndt/16.7.1382. [DOI] [PubMed] [Google Scholar]
- 129.Dai C, Yang J, Bastacky S, Xia J, Li Y, Liu Y. Intravenous administration of hepatocyte growth factor gene ameliorates diabetic nephropathy in mice. J Am Soc Nephrol. 2004;15:2637–2647. doi: 10.1097/01.ASN.0000139479.09658.EE. [DOI] [PubMed] [Google Scholar]
- 130.Dai T, Natarajan R, Nast CC, Lapage J, Chuang P, Sim J, Tong L, Chamberlin M, Wang S, Adler SG. Glucose and diabetes: Effects on podocyte and glomerular p38MAPK, heat shock protein 25, and actin cytoskeleton. Kidney Int. 2006;69:806–814. doi: 10.1038/sj.ki.5000033. [DOI] [PubMed] [Google Scholar]
- 131.Das EN, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55:498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 132.Davis B, Dei CA, Long DA, White KE, Hayward A, Ku CH, Woolf AS, Bilous R, Viberti G, Gnudi L. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. doi: 10.1681/ASN.2006101093. [DOI] [PubMed] [Google Scholar]
- 133.De’Oliveira JM, Price DA, Fisher ND, Allan DR, McKnight JA, Williams GH, Hollenberg NK. Autonomy of the renin system in type II diabetes mellitus: Dietary sodium and renal hemodynamic responses to ACE inhibition. Kidney Int. 1997;52:771–777. doi: 10.1038/ki.1997.394. [DOI] [PubMed] [Google Scholar]
- 134.de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12:993–1000. doi: 10.1681/ASN.V125993. [DOI] [PubMed] [Google Scholar]
- 135.Deb DK, Chen Y, Zhang Z, Zhang Y, Szeto FL, Wong KE, Kong J, Li YC. 1,25-Dihydroxyvitamin D3 suppresses high glucose-induced an-giotensinogen expression in kidney cells by blocking the NF-{kappa}B pathway. Am J Physiol Renal Physiol. 2009;296:F1212–F1218. doi: 10.1152/ajprenal.00002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia. 1989;32:219–226. doi: 10.1007/BF00285287. [DOI] [PubMed] [Google Scholar]
- 137.DeFronzo RA. The effect of insulin on renal sodium metabolism. A review with clinical implications. Diabetologia. 1981;21:165–171. doi: 10.1007/BF00252649. [DOI] [PubMed] [Google Scholar]
- 138.DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55:845–855. doi: 10.1172/JCI107996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deng A, Baylis C. Locally produced EDRF controls preglomerular resistance and ultrafiltration coefficient. Am J Physiol. 1993;264:F212–F215. doi: 10.1152/ajprenal.1993.264.2.F212. [DOI] [PubMed] [Google Scholar]
- 140.Deng A, Munger KA, Valdivielso JM, Satriano J, Lortie M, Blantz RC, Thomson SC. Increased expression of ornithine decarboxylase in distal tubules of early diabetic rat kidneys: Are polyamines paracrine hypertrophic factors? Diabetes. 2003;52:1235–1239. doi: 10.2337/diabetes.52.5.1235. [DOI] [PubMed] [Google Scholar]
- 141.Deng D, Evans T, Mukherjee K, Downey D, Chakrabarti S. Diabetes-induced vascular dysfunction in the retina: Role of endothelins. Diabetologia. 1999;42:1228–1234. doi: 10.1007/s001250051296. [DOI] [PubMed] [Google Scholar]
- 142.DeRubertis FR, Craven PA, Melhem MF, Salah EM. Attenuation of renal injury in db/db mice overexpressing superoxide dismutase: Evidence for reduced superoxide-nitric oxide interaction. Diabetes. 2004;53:762–768. doi: 10.2337/diabetes.53.3.762. [DOI] [PubMed] [Google Scholar]
- 143.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 144.Docherty NG, Calvo IF, Quinlan MR, Perez-Barriocanal F, McGuire BB, Fitzpatrick JM, Watson RW. Increased E-cadherin expression in the ligated kidney following unilateral ureteric obstruction. Kidney Int. 2009;75:205–213. doi: 10.1038/ki.2008.482. [DOI] [PubMed] [Google Scholar]
- 145.Dominguez JH, Camp K, Maianu L, Feister H, Garvey WT. Molecular adaptations of GLUT1 and GLUT2 in renal proximal tubules of diabetic rats. Am J Physiol. 1994;266:F283–F290. doi: 10.1152/ajprenal.1994.266.2.F283. [DOI] [PubMed] [Google Scholar]
- 146.Dominguez JH, Wu P, Hawes JW, Deeg M, Walsh J, Packer SC, Nagase M, Temm C, Goss E, Peterson R. Renal injury: Similarities and differences in male and female rats with the metabolic syndrome. Kidney Int. 2006;69:1969–1976. doi: 10.1038/sj.ki.5000406. [DOI] [PubMed] [Google Scholar]
- 147.Dorup J, Morsing P, Rasch R. Tubule-tubule and tubule-arteriole contacts in rat kidney distal nephrons. A morphologic study based on computer-assisted three-dimensional reconstructions. Lab Invest. 1992;67:761–769. [PubMed] [Google Scholar]
- 148.Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 149.Drummond K, Mauer M. The early natural history of nephropathy in type 1 diabetes: II. Early renal structural changes in type 1 diabetes. Diabetes. 2002;51:1580–1587. doi: 10.2337/diabetes.51.5.1580. [DOI] [PubMed] [Google Scholar]
- 150.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by post-translational modification at the Akt site. J Clin Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294:F830–F839. doi: 10.1152/ajprenal.00266.2007. [DOI] [PubMed] [Google Scholar]
- 152.Economou CG, Kitsiou PV, Tzinia AK, Panagopoulou E, Marinos E, Kershaw DB, Kerjaschki D, Tsilibary EC. Enhanced podocalyxin expression alters the structure of podocyte basal surface. J Cell Sci. 2004;117:3281–3294. doi: 10.1242/jcs.01163. [DOI] [PubMed] [Google Scholar]
- 153.Eddy AA, Neilson EG. Chronic kidney disease progression. J Am Soc Nephrol. 2006;17:2964–2966. doi: 10.1681/ASN.2006070704. [DOI] [PubMed] [Google Scholar]
- 154.Eddy AA, Schnaper HW. The nephrotic syndrome: From the simple to the complex. Semin Nephrol. 1998;18:304–316. [PubMed] [Google Scholar]
- 155.Edelstein D, Brownlee M. Aminoguanidine ameliorates albuminuria in diabetic hypertensive rats. Diabetologia. 1992;35:96–97. doi: 10.1007/BF00400859. [DOI] [PubMed] [Google Scholar]
- 156.Efendiev R, Bertorello AM, Pedemonte CH. PKC-beta and PKC-zeta mediate opposing effects on proximal tubule Na+, K+-ATPase activity. FEBS Lett. 1999;456:45–48. doi: 10.1016/s0014-5793(99)00925-4. [DOI] [PubMed] [Google Scholar]
- 157.Efendiev R, Budu CE, Cinelli AR, Bertorello AM, Pedemonte CH. Intracellular Na+ regulates dopamine and angiotensin II receptors availability at the plasma membrane and their cellular responses in renal epithelia. J Biol Chem. 2003;278:28719–28726. doi: 10.1074/jbc.M303741200. [DOI] [PubMed] [Google Scholar]
- 158.Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: A review. Diabetes Metab Res Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- 159.Embark HM, Bohmer C, Vallon V, Luft F, Lang F. Regulation of KCNE1-dependent K(+) current by the serum and glucocorticoid-inducible kinase (SGK) isoforms. Pflugers Arch. 2003;445:601–606. doi: 10.1007/s00424-002-0982-y. [DOI] [PubMed] [Google Scholar]
- 160.Endlich N, Kress KR, Reiser J, Uttenweiler D, Kriz W, Mundel P, Endlich K. Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol. 2001;12:413–422. doi: 10.1681/ASN.V123413. [DOI] [PubMed] [Google Scholar]
- 161.Esmatjes E, Fernandez MR, Halperin I, Camps J, Gaya J, Arroyo V, Rivera F, Figuerola D. Renal hemodynamic abnormalities in patients with short term insulin-dependent diabetes mellitus: Role of renal prostaglandins. J Clin Endocrinol Metab. 1985;60:1231–1236. doi: 10.1210/jcem-60-6-1231. [DOI] [PubMed] [Google Scholar]
- 162.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 163.Fan YP, Weiss RH. Exogenous attenuation of p21(Waf1/Cip1) decreases mesangial cell hypertrophy as a result of hyperglycemia and IGF-1. J Am Soc Nephrol. 2004;15:575–584. doi: 10.1097/01.asn.0000114557.75244.5f. [DOI] [PubMed] [Google Scholar]
- 164.Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, Mizel D, Kajiyama H, Zerfas P, Briggs JP, Kopp JB, Schnermann J. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J Am Soc Nephrol. 2008;19:722–730. doi: 10.1681/ASN.2007060721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106:466–472. doi: 10.1161/01.cir.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 166.Feldt-Rasmussen B, Mathiesen ER, Deckert T, Giese J, Christensen NJ, Bent-Hansen L, Nielsen MD. Central role for sodium in the pathogenesis of blood pressure changes independent of angiotensin, aldosterone and catecholamines in type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1987;30:610–617. doi: 10.1007/BF00277316. [DOI] [PubMed] [Google Scholar]
- 167.Feliers D, Frank MA, Riley DJ. Activation of cyclin D1-Cdk4 and Cdk4-directed phosphorylation of RB protein in diabetic mesangial hypertrophy. Diabetes. 2002;51:3290–3299. doi: 10.2337/diabetes.51.11.3290. [DOI] [PubMed] [Google Scholar]
- 168.Feng Y, Wang Q, Wang Y, Yard B, Lang F. SGK1-mediated fibronectin formation in diabetic nephropathy. Cell Physiol Biochem. 2005;16:237–244. doi: 10.1159/000089849. [DOI] [PubMed] [Google Scholar]
- 169.Feraille E, Marsy S, Cheval L, Barlet-Bas C, Khadouri C, Favre H, Doucet A. Sites of antinatriuretic action of insulin along rat nephron. Am J Physiol. 1992;263:F175–F179. doi: 10.1152/ajprenal.1992.263.1.F175. [DOI] [PubMed] [Google Scholar]
- 170.Ferrannini E, Natali A. Insulin resistance and hypertension: Connections with sodium metabolism. Am J Kidney Dis. 1993;21:37–42. doi: 10.1016/s0272-6386(12)70253-6. [DOI] [PubMed] [Google Scholar]
- 171.Fervenza FC, Tsao T, Hoffman AR, Rabkin R. Regional changes in the intrarenal insulin-like growth factor-I axis in diabetes. Kidney Int. 1997;51:811–818. doi: 10.1038/ki.1997.114. [DOI] [PubMed] [Google Scholar]
- 172.Ficociello LH, Perkins BA, Roshan B, Weinberg JM, Aschengrau A, Warram JH, Krolewski AS. Renal hyperfiltration and the development of microalbuminuria in type 1 diabetes. Diabetes Care. 2009;32:889–893. doi: 10.2337/dc08-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fine LG, Ong AC, Norman JT. Mechanisms of tubulo-interstitial injury in progressive renal diseases. Eur J Clin Invest. 1993;23:259–265. doi: 10.1111/j.1365-2362.1993.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 174.Fine LG, Orphanides C, Norman JT. Progressive renal disease: The chronic hypoxia hypothesis. Kidney Int Suppl. 1998;65:S74–S78. [PubMed] [Google Scholar]
- 175.Fioretto P, Sambataro M, Cipollina MR, Giorato C, Carraro A, Opocher G, Sacerdoti D, Brocco E, Morocutti A, Mantero F. Role of atrial natriuretic peptide in the pathogenesis of sodium retention in IDDM with and without glomerular hyperfiltration. Diabetes. 1992;41:936–945. doi: 10.2337/diab.41.8.936. [DOI] [PubMed] [Google Scholar]
- 176.Flyvbjerg A, Dagnaes-Hansen F, de Vriese AS, Schrijvers BF, Tilton RG, Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–3094. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- 177.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 178.Forbes JM, Soulis T, Thallas V, Panagiotopoulos S, Long DM, Vasan S, Wagle D, Jerums G, Cooper ME. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia. 2001;44:108–114. doi: 10.1007/s001250051587. [DOI] [PubMed] [Google Scholar]
- 179.Franch HA. Pathways of proteolysis affecting renal cell growth. Curr Opin Nephrol Hypertens. 2002;11:445–450. doi: 10.1097/00041552-200207000-00012. [DOI] [PubMed] [Google Scholar]
- 180.Francis J, Zhang J, Farhi A, Carey H, Geller DS. A novel SGLT2 mutation in a patient with autosomal recessive renal glucosuria. Nephrol Dial Transplant. 2004;19:2893–2895. doi: 10.1093/ndt/gfh426. [DOI] [PubMed] [Google Scholar]
- 181.Freitas HS, Anhe GF, Melo KF, Okamoto MM, Oliveira-Souza M, Bordin S, Machado UF. Na(+)-glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: Involvement of hepatocyte nuclear factor-1alpha expression and activity. Endocrinology. 2008;149:717–724. doi: 10.1210/en.2007-1088. [DOI] [PubMed] [Google Scholar]
- 182.Freitas HS, Schaan BD, David-Silva A, Sabino-Silva R, Okamoto MM, Alves-Wagner AB, Mori RC, Machado UF. SLC2A2 gene expression in kidney of diabetic rats is regulated by HNF-1alpha and HNF-3beta. Mol Cell Endocrinol. 2009;305:63–70. doi: 10.1016/j.mce.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 183.Fridlyand LE, Philipson LH. Oxidative reactive species in cell injury: Mechanisms in diabetes mellitus and therapeutic approaches. Ann N Y Acad Sci. 2005;1066:136–151. doi: 10.1196/annals.1363.019. [DOI] [PubMed] [Google Scholar]
- 184.Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 185.Friedberg CE, van BM, Bijlsma JA, Koomans HA. Insulin increases sodium reabsorption in diluting segment in humans: Evidence for indirect mediation through hypokalemia. Kidney Int. 1991;40:251–256. doi: 10.1038/ki.1991.207. [DOI] [PubMed] [Google Scholar]
- 186.Friederich M, Fasching A, Hansell P, Nordquist L, Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. 2008;1777:935–940. doi: 10.1016/j.bbabio.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 187.Friederich M, Hansell P, Palm F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Curr Diabetes Rev. 2009;5:120–144. doi: 10.2174/157339909788166800. [DOI] [PubMed] [Google Scholar]
- 188.Frojdo S, Sjolind L, Parkkonen M, Makinen VP, Kilpikari R, Pettersson-Fernholm K, Forsblom C, Fagerudd J, Tikellis C, Cooper ME, Wessman M, Groop PH. Polymorphisms in the gene encoding angiotensin I converting enzyme 2 and diabetic nephropathy. Diabetologia. 2005;48:2278–2281. doi: 10.1007/s00125-005-1955-4. [DOI] [PubMed] [Google Scholar]
- 189.Fujita H, Fujishima H, Chida S, Takahashi K, Qi Z, Kanetsuna Y, Breyer MD, Harris RC, Yamada Y, Takahashi T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol. 2009;20:1303–1313. doi: 10.1681/ASN.2008080844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fujita H, Omori S, Ishikura K, Hida M, Awazu M. ERK and p38 mediate high-glucose-induced hypertrophy and TGF-beta expression in renal tubular cells. Am J Physiol Renal Physiol. 2004;286:F120–F126. doi: 10.1152/ajprenal.00351.2002. [DOI] [PubMed] [Google Scholar]
- 191.Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, Motomiya Y, Bucala R, Iida S, Tamaki K, Imaizumi T, Cooper ME, Okuda S. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004;66:2137–2147. doi: 10.1111/j.1523-1755.2004.66004.x. [DOI] [PubMed] [Google Scholar]
- 192.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Furuta T, Saito T, Ootaka T, Soma J, Obara K, Abe K, Yoshinaga K. The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis. 1993;21:480–485. doi: 10.1016/s0272-6386(12)80393-3. [DOI] [PubMed] [Google Scholar]
- 194.Gambardella S, Andreani D, Cancelli A, Di MU, Cardamone I, Stirati G, Cinotti GA, Pugliese F. Renal hemodynamics and urinary excretion of 6-keto-prostaglandin F1 alpha and thromboxane B2 in newly diagnosed type I diabetic patients. Diabetes. 1988;37:1044–1048. doi: 10.2337/diab.37.8.1044. [DOI] [PubMed] [Google Scholar]
- 195.Gealekman O, Brodsky SV, Zhang F, Chander PN, Friedli C, Nasjletti A, Goligorsky MS. Endothelial dysfunction as a modifier of angiogenic response in Zucker diabetic fat rat: Amelioration with Ebselen. Kidney Int. 2004;66:2337–2347. doi: 10.1111/j.1523-1755.2004.66035.x. [DOI] [PubMed] [Google Scholar]
- 196.Gesek FA, Schoolwerth AC. Insulin increases Na(+)-H+ exchange activity in proximal tubules from normotensive and hypertensive rats. Am J Physiol. 1991;260:F695–F703. doi: 10.1152/ajprenal.1991.260.5.F695. [DOI] [PubMed] [Google Scholar]
- 197.Gilbert RE, Akdeniz A, Weitz S, Usinger WR, Molineaux C, Jones SE, Langham RG, Jerums G. Urinary connective tissue growth factor excretion in patients with type 1 diabetes and nephropathy. Diabetes Care. 2003;26:2632–2636. doi: 10.2337/diacare.26.9.2632. [DOI] [PubMed] [Google Scholar]
- 198.Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: More than an aftermath of glomerular injury? Kidney Int. 1999;56:1627–1637. doi: 10.1046/j.1523-1755.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- 199.Gilbert RE, Cox A, McNally PG, Wu LL, Dziadek M, Cooper ME, Jerums G. Increased epidermal growth factor in experimental diabetes related kidney growth in rats. Diabetologia. 1997;40:778–785. doi: 10.1007/s001250050749. [DOI] [PubMed] [Google Scholar]
- 200.Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 201.Gimeno-Alcaniz JV, Sanz P. Glucose and type 2A protein phosphatase regulate the interaction between catalytic and regulatory subunits of AMP-activated protein kinase. J Mol Biol. 2003;333:201–209. doi: 10.1016/j.jmb.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 202.Gojo A, Utsunomiya K, Taniguchi K, Yokota T, Ishizawa S, Kanazawa Y, Kurata H, Tajima N. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2007;568:242–247. doi: 10.1016/j.ejphar.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 203.Goligorsky MS, Chen J, Brodsky S. Workshop: Endothelial cell dysfunction leading to diabetic nephropathy: Focus on nitric oxide. Hypertension. 2001;37:744–748. doi: 10.1161/01.hyp.37.2.744. [DOI] [PubMed] [Google Scholar]
- 204.Gonzalez-Rodriguez E, Gaeggeler HP, Rossier BC. IGF-1 vs insulin: Respective roles in modulating sodium transport via the PI-3 kinase/Sgk1 pathway in a cortical collecting duct cell line. Kidney Int. 2007;71:116–125. doi: 10.1038/sj.ki.5002018. [DOI] [PubMed] [Google Scholar]
- 205.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. 2005;280:39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 206.Goto Y, Kakizaki M. The spontaneous diabetes rat: A model of non-insulin-dependent diabetes mellitus. Proc Jpn Acad. 1981;57:381–384. [Google Scholar]
- 207.Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol. 2001;280:F193–F206. doi: 10.1152/ajprenal.2001.280.2.F193. [DOI] [PubMed] [Google Scholar]
- 208.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 209.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 210.Griffin KA, Abu-Naser M, Abu-Amarah I, Picken M, Williamson GA, Bidani AK. Dynamic blood pressure load and nephropathy in the ZSF1 (fa/fa cp) model of type 2 diabetes. Am J Physiol Renal Physiol. 2007;293:F1605–F1613. doi: 10.1152/ajprenal.00511.2006. [DOI] [PubMed] [Google Scholar]
- 211.Gruden G, Zonca S, Hayward A, Thomas S, Maestrini S, Gnudi L, Viberti GC. Mechanical stretch-induced fibronectin and transforming growth factor-beta1 production in human mesangial cells is p38 mitogen-activated protein kinase-dependent. Diabetes. 2000;49:655–661. doi: 10.2337/diabetes.49.4.655. [DOI] [PubMed] [Google Scholar]
- 212.Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355–3368. doi: 10.1096/fj.06-6713com. [DOI] [PubMed] [Google Scholar]
- 213.Guijarro C, Egido J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int. 2001;59:415–424. doi: 10.1046/j.1523-1755.2001.059002415.x. [DOI] [PubMed] [Google Scholar]
- 214.Gurley SB, Coffman TM. The renin-angiotensin system and diabetic nephropathy. Semin Nephrol. 2007;27:144–152. doi: 10.1016/j.semnephrol.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 215.Hall JE. Hyperinsulinemia: A link between obesity and hypertension? Kidney Int. 1993;43:1402–1417. doi: 10.1038/ki.1993.197. [DOI] [PubMed] [Google Scholar]
- 216.Hall JE, Brands MW, Mizelle HL, Gaillard CA, Hildebrandt DA. Chronic intrarenal hyperinsulinemia does not cause hypertension. Am J Physiol. 1991;260:F663–F669. doi: 10.1152/ajprenal.1991.260.5.F663. [DOI] [PubMed] [Google Scholar]
- 217.Hall JE, Brands MW, Zappe DH, Alonso-Galicia M. Cardiovascular actions of insulin: Are they important in long-term blood pressure regulation? Clin Exp Pharmacol Physiol. 1995;22:689–700. doi: 10.1111/j.1440-1681.1995.tb01922.x. [DOI] [PubMed] [Google Scholar]
- 218.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Han DC, Hoffman BB, Hong SW, Guo J, Ziyadeh FN. Therapy with antisense TGF-beta1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am J Physiol Renal Physiol. 2000;278:F628–F634. doi: 10.1152/ajprenal.2000.278.4.F628. [DOI] [PubMed] [Google Scholar]
- 220.Han DC, Isono M, Chen S, Casaretto A, Hong SW, Wolf G, Ziyadeh FN. Leptin stimulates type I collagen production in db/db mesangial cells: Glucose uptake and TGF-beta type II receptor expression. Kidney Int. 2001;59:1315–1323. doi: 10.1046/j.1523-1755.2001.0590041315.x. [DOI] [PubMed] [Google Scholar]
- 221.Han DC, Isono M, Hoffman BB, Ziyadeh FN. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: Mediation by autocrine activation of TGF-beta. J Am Soc Nephrol. 1999;10:1891–1899. doi: 10.1681/ASN.V1091891. [DOI] [PubMed] [Google Scholar]
- 222.Han HJ, Lee YJ, Park SH, Lee JH, Taub M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am J Physiol Renal Physiol. 2005;288:F988–F996. doi: 10.1152/ajprenal.00327.2004. [DOI] [PubMed] [Google Scholar]
- 223.Hannedouche TP, Delgado AG, Gnionsahe DA, Boitard C, Lacour B, Grunfeld JP. Renal hemodynamics and segmental tubular reabsorption in early type 1 diabetes. Kidney Int. 1990;37:1126–1133. doi: 10.1038/ki.1990.95. [DOI] [PubMed] [Google Scholar]
- 224.Hartner A, Cordasic N, Klanke B, Wittmann M, Veelken R, Hilgers KF. Renal injury in streptozotocin-diabetic Ren2-transgenic rats is mainly dependent on hypertension, not on diabetes. Am J Physiol Renal Physiol. 2007;292:F820–F827. doi: 10.1152/ajprenal.00088.2006. [DOI] [PubMed] [Google Scholar]
- 225.Hashimoto Y, Ideura T, Yoshimura A, Koshikawa S. Autoregulation of renal blood flow in streptozocin-induced diabetic rats. Diabetes. 1989;38:1109–1113. doi: 10.2337/diab.38.9.1109. [DOI] [PubMed] [Google Scholar]
- 226.Hayashi K, Epstein M, Loutzenhiser R, Forster H. Impaired myogenic responsiveness of the afferent arteriole in streptozotocin-induced diabetic rats: Role of eicosanoid derangements. J Am Soc Nephrol. 1992;2:1578–1586. doi: 10.1681/ASN.V2111578. [DOI] [PubMed] [Google Scholar]
- 227.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 228.Heilig CW, Kreisberg JI, Freytag S, Murakami T, Ebina Y, Guo L, Heilig K, Loberg R, Qu X, Jin Y, Henry D, Brosius FC., III Antisense GLUT-1 protects mesangial cells from glucose induction of GLUT-1 and fibronectin expression. Am J Physiol Renal Physiol. 2001;280:F657–F666. doi: 10.1152/ajprenal.2001.280.4.F657. [DOI] [PubMed] [Google Scholar]
- 229.Hellermann GR, Solomonson LP. Calmodulin promotes dimerization of the oxygenase domain of human endothelial nitric-oxide synthase. J Biol Chem. 1997;272:12030–12034. doi: 10.1074/jbc.272.18.12030. [DOI] [PubMed] [Google Scholar]
- 230.Henry CB, Duling BR. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. 2000;279:H2815–H2823. doi: 10.1152/ajpheart.2000.279.6.H2815. [DOI] [PubMed] [Google Scholar]
- 231.Herman WH, Emancipator SN, Rhoten RL, Simonson MS. Vascular and glomerular expression of endothelin-1 in normal human kidney. Am J Physiol. 1998;275:F8–17. doi: 10.1152/ajprenal.1998.275.1.F8. [DOI] [PubMed] [Google Scholar]
- 232.Hickok NJ, Wahlfors J, Crozat A, Halmekyto M, Alhonen L, Janne J, Janne OA. Human ornithine decarboxylase-encoding loci: Nucleotide sequence of the expressed gene and characterization of a pseudogene. Gene. 1990;93:257–263. doi: 10.1016/0378-1119(90)90233-h. [DOI] [PubMed] [Google Scholar]
- 233.Hills CE, Brunskill NJ, Squires PE. C-Peptide as a Therapeutic Tool in Diabetic Nephropathy. Am J Nephrol. 2010;31:389–397. doi: 10.1159/000289864. [DOI] [PubMed] [Google Scholar]
- 234.Hills CE, Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol. 2010;31:68–74. doi: 10.1159/000256659. [DOI] [PubMed] [Google Scholar]
- 235.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 236.Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hirschberg R, Wang S. Proteinuria and growth factors in the development of tubulointerstitial injury and scarring in kidney disease. Curr Opin Nephrol Hypertens. 2005;14:43–52. doi: 10.1097/00041552-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 238.Hohenstein B, Hausknecht B, Boehmer K, Riess R, Brekken RA, Hugo CP. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006;69:1654–1661. doi: 10.1038/sj.ki.5000294. [DOI] [PubMed] [Google Scholar]
- 239.Holck P, Rasch R. Structure and segmental localization of glycogen in the diabetic rat kidney. Diabetes. 1993;42:891–900. doi: 10.2337/diab.42.6.891. [DOI] [PubMed] [Google Scholar]
- 240.Holian J, Qi W, Kelly DJ, Zhang Y, Mreich E, Pollock CA, Chen XM. Role of Kruppel-like factor 6 in transforming growth factor-beta1-induced epithelial-mesenchymal transition of proximal tubule cells. Am J Physiol Renal Physiol. 2008;295:F1388–F1396. doi: 10.1152/ajprenal.00055.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Hollenberg NK, Price DA, Fisher ND, Lansang MC, Perkins B, Gordon MS, Williams GH, Laffel LM. Glomerular hemodynamics and the renin-angiotensin system in patients with type 1 diabetes mellitus. Kidney Int. 2003;63:172–178. doi: 10.1046/j.1523-1755.2003.00701.x. [DOI] [PubMed] [Google Scholar]
- 242.Hommel E, Mathiesen E, Arnold-Larsen S, Edsberg B, Olsen UB, Parving HH. Effects of indomethacin on kidney function in type 1 (insulin-dependent) diabetic patients with nephropathy. Diabetologia. 1987;30:78–81. doi: 10.1007/BF00274575. [DOI] [PubMed] [Google Scholar]
- 243.Hopfer U, Hopfer H, Meyer-Schwesinger C, Loeffler I, Fukai N, Olsen BR, Stahl RA, Wolf G. Lack of type VIII collagen in mice ameliorates diabetic nephropathy. Diabetes. 2009;58:1672–1681. doi: 10.2337/db08-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hostetter TH, Troy JL, Brenner BM. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int. 1981;19:410–415. doi: 10.1038/ki.1981.33. [DOI] [PubMed] [Google Scholar]
- 245.Hovis JG, Stumpo DJ, Halsey DL, Blackshear PJ. Effects of mitogens on ornithine decarboxylase activity and messenger RNA levels in normal and protein kinase C-deficient NIH-3T3 fibroblasts. J Biol Chem. 1986;261:10380–10386. [PubMed] [Google Scholar]
- 246.Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A, Peters C, Meitinger T, Strom T, Steckler T, Holsboer F, Klopstock T, Gekeler F, Schindewolf C, Jung T, Avraham K, Behrendt H, Ring J, Zimmer A, Schughart K, Pfeffer K, Wolf E, Balling R. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25:444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- 247.Hsieh TJ, Fustier P, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Fantus IG, Hamet P, Chan JS. High glucose stimulates angiotensinogen gene expression and cell hypertrophy via activation of the hexosamine biosynthesis pathway in rat kidney proximal tubular cells. Endocrinology. 2003;144:4338–4349. doi: 10.1210/en.2003-0220. [DOI] [PubMed] [Google Scholar]
- 248.Huang DY, Boini KM, Friedrich B, Metzger M, Just L, Osswald H, Wulff P, Kuhl D, Vallon V, Lang F. Blunted hypertensive effect of combined fructose and high-salt diet in gene-targeted mice lacking functional serum- and glucocorticoid-inducible kinase SGK1. Am J Physiol Regul Integr Comp Physiol. 2006;290:R935–R944. doi: 10.1152/ajpregu.00382.2005. [DOI] [PubMed] [Google Scholar]
- 249.Huang DY, Richter K, Breidenbach A, Vallon V. Human C-peptide acutely lowers glomerular hyperfiltration and proteinuria in diabetic rats: A dose-response study. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:67–73. doi: 10.1007/s00210-001-0502-1. [DOI] [PubMed] [Google Scholar]
- 250.Huang HC, Preisig PA. G1 kinases and transforming growth factor-beta signaling are associated with a growth pattern switch in diabetes-induced renal growth. Kidney Int. 2000;58:162–172. doi: 10.1046/j.1523-1755.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- 251.Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L, Noble NA, Border W. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006;69:105–113. doi: 10.1038/sj.ki.5000011. [DOI] [PubMed] [Google Scholar]
- 252.Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, Koura Y, Nishiyama A, Okada H, Uddin MN, Nabi AH, Ishida Y, Inagami T, Saruta T. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest. 2004;114:1128–1135. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ichihara A, Sakoda M, Kurauchi-Mito A, Narita T, Kinouchi K, Murohashi-Bokuda K, Itoh H. Possible roles of human (pro)renin receptor suggested by recent clinical and experimental findings. Hypertens Res. 2010;33:177–180. doi: 10.1038/hr.2009.214. [DOI] [PubMed] [Google Scholar]
- 254.Ichihara A, Suzuki F, Nakagawa T, Kaneshiro Y, Takemitsu T, Sakoda M, Nabi AH, Nishiyama A, Sugaya T, Hayashi M, Inagami T. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2006;17:1950–1961. doi: 10.1681/ASN.2006010029. [DOI] [PubMed] [Google Scholar]
- 255.Ichinose K, Maeshima Y, Yamamoto Y, Kitayama H, Takazawa Y, Hirokoshi K, Sugiyama H, Yamasaki Y, Eguchi K, Makino H. Antian-giogenic endostatin peptide ameliorates renal alterations in the early stage of a type 1 diabetic nephropathy model. Diabetes. 2005;54:2891–2903. doi: 10.2337/diabetes.54.10.2891. [DOI] [PubMed] [Google Scholar]
- 256.Ikenaga H, Bast JP, Fallet RW, Carmines PK. Exaggerated impact of ATP-sensitive K(+) channels on afferent arteriolar diameter in diabetes mellitus. J Am Soc Nephrol. 2000;11:1199–1207. doi: 10.1681/asn.v1171199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ionescu E, Sauter JF, Jeanrenaud B. Abnormal oral glucose tolerance in genetically obese (fa/fa) rats. Am J Physiol. 1985;248:E500–E506. doi: 10.1152/ajpendo.1985.248.5.E500. [DOI] [PubMed] [Google Scholar]
- 258.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh J, Gerlitz B, Berg DT, Grinnell BW, Chavakis T, Esmon CT, Weiler H, Bierhaus A, Nawroth PP. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 259.Ishida K, Ishibashi F, Takashina S. Comparison of renal hemodynamics in early non-insulin-dependent and insulin-dependent diabetes mellitus. J Diabet Complications. 1991;5:143–145. doi: 10.1016/0891-6632(91)90049-u. [DOI] [PubMed] [Google Scholar]
- 260.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 261.Ishii H, Tada H, Isogai S. An aldose reductase inhibitor prevents glucose-induced increase in transforming growth factor-beta and protein kinase C activity in cultured mesangial cells. Diabetologia. 1998;41:362–364. doi: 10.1007/s001250050916. [DOI] [PubMed] [Google Scholar]
- 262.Ishikawa Y, Nishikimi T, Akimoto K, Ishimura K, Ono H, Matsuoka H. Long-term administration of rhokinase inhibitor ameliorates renal damage in malignant hypertensive rats. Hypertension. 2006;47:1075–1083. doi: 10.1161/01.HYP.0000221605.94532.71. [DOI] [PubMed] [Google Scholar]
- 263.Isono M, Mogyorosi A, Han DC, Hoffman BB, Ziyadeh FN. Stimulation of TGF-beta type II receptor by high glucose in mouse mesangial cells and in diabetic kidney. Am J Physiol Renal Physiol. 2000;278:F830–F838. doi: 10.1152/ajprenal.2000.278.5.F830. [DOI] [PubMed] [Google Scholar]
- 264.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Iyengar SK, Abboud HE, Goddard KA, Saad MF, Adler SG, Arar NH, Bowden DW, Duggirala R, Elston RC, Hanson RL, Ipp E, Kao WH, Kimmel PL, Klag MJ, Knowler WC, Meoni LA, Nelson RG, Nicholas SB, Pahl MV, Parekh RS, Quade SR, Rich SS, Rotter JI, Scavini M, Schelling JR, Sedor JR, Sehgal AR, Shah VO, Smith MW, Taylor KD, Winkler CA, Zager PG, Freedman BI. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: The family investigation of nephropathy and diabetes (FIND) Diabetes. 2007;56:1577–1585. doi: 10.2337/db06-1154. [DOI] [PubMed] [Google Scholar]
- 266.Iyengar SK, Fox KA, Schachere M, Manzoor F, Slaughter ME, Covic AM, Orloff SM, Hayden PS, Olson JM, Schelling JR, Sedor JR. Linkage analysis of candidate loci for end-stage renal disease due to diabetic nephropathy. J Am Soc Nephrol. 2003;14:S195–S201. doi: 10.1097/01.asn.0000070078.66465.55. [DOI] [PubMed] [Google Scholar]
- 267.Izquierdo A, Lopez-Luna P, Ortega A, Romero M, Guitierrez-Tarres MA, Arribas I, Alvarez MJ, Esbrit P, Bosch RJ. The parathyroid hormone-related protein system and diabetic nephropathy outcome in streptozotocin-induced diabetes. Kidney Int. 2006;69:2171–2177. doi: 10.1038/sj.ki.5000195. [DOI] [PubMed] [Google Scholar]
- 268.Izuhara Y, Nangaku M, Inagi R, Tominaga N, Aizawa T, Kurokawa K, van Ypersele de SC, Miyata T. Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J Am Soc Nephrol. 2005;16:3631–3641. doi: 10.1681/ASN.2005050522. [DOI] [PubMed] [Google Scholar]
- 269.Jaffa AA, Miller DH, Bailey GS, Chao J, Margolius HS, Mayfield RK. Abnormal regulation of renal kallikrein in experimental diabetes. Effects of insulin on prokallikrein synthesis and activation. J Clin Invest. 1987;80:1651–1659. doi: 10.1172/JCI113254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Jaffa AA, Rust PF, Mayfield RK. Kinin, a mediator of diabetes-induced glomerular hyperfiltration. Diabetes. 1995;44:156–160. doi: 10.2337/diab.44.2.156. [DOI] [PubMed] [Google Scholar]
- 271.James LR, Fantus IG, Goldberg H, Ly H, Scholey JW. Overexpression of GFAT activates PAI-1 promoter in mesangial cells. Am J Physiol Renal Physiol. 2000;279:F718–F727. doi: 10.1152/ajprenal.2000.279.4.F718. [DOI] [PubMed] [Google Scholar]
- 272.James LR, Le C, Scholey JW. Influence of glucosamine on glomerular meseangial cell turnover: Implications for hyperglycemia and hexosamine pathway flux. Am J Physiol Endocrinol Metab. 2010;298:E210–E221. doi: 10.1152/ajpendo.00232.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: A mechanistic viewpoint. Kidney Int. 2008;74:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 274.Jensen PK, Christiansen JS, Steven K, Parving HH. Renal function in streptozotocin-diabetic rats. Diabetologia. 1981;21:409–414. [PubMed] [Google Scholar]
- 275.Jensen PK, Steven K, Blaehr H, Christiansen JS, Parving HH. Effects of indomethacin on glomerular hemodynamics in experimental diabetes. Kidney Int. 1986;29:490–495. doi: 10.1038/ki.1986.26. [DOI] [PubMed] [Google Scholar]
- 276.Jerums G, Premaratne E, Panagiotopoulos S, Macisaac RJ. The clinical significance of hyperfiltration in diabetes. Diabetologia. 2010;53:2093–2104. doi: 10.1007/s00125-010-1794-9. [DOI] [PubMed] [Google Scholar]
- 277.Jetten AM, Ganong BR, Vandenbark GR, Shirley JE, Bell RM. Role of protein kinase C in diacylglycerol-mediated induction of ornithine decarboxylase and reduction of epidermal growth factor binding. Proc Natl Acad Sci U S A. 1985;82:1941–1945. doi: 10.1073/pnas.82.7.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Jiang R, Carlson M. Glucose regulates protein interactions within the yeast SNF1 protein kinase complex. Genes Dev. 1996;10:3105–3115. doi: 10.1101/gad.10.24.3105. [DOI] [PubMed] [Google Scholar]
- 279.Jiang T, Wang XX, Scherzer P, Wilson P, Tallman J, Takahashi H, Li J, Iwahashi M, Sutherland E, Arend L, Levi M. Farnesoid X receptor modulates renal lipid metabolism, fibrosis, and diabetic nephropathy. Diabetes. 2007;56:2485–2493. doi: 10.2337/db06-1642. [DOI] [PubMed] [Google Scholar]
- 280.Johnson DW, Saunders HJ, Baxter RC, Field MJ, Pollock CA. Paracrine stimulation of human renal fibroblasts by proximal tubule cells. Kidney Int. 1998;54:747–757. doi: 10.1046/j.1523-1755.1998.00048.x. [DOI] [PubMed] [Google Scholar]
- 281.Johnson DW, Saunders HJ, Brew BK, Ganesan A, Baxter RC, Poronnik P, Cook DI, Gyory AZ, Field MJ, Pollock CA. Human renal fibroblasts modulate proximal tubule cell growth and transport via the IGF-I axis. Kidney Int. 1997;52:1486–1496. doi: 10.1038/ki.1997.479. [DOI] [PubMed] [Google Scholar]
- 282.Jones S, Jones S, Phillips AO. Regulation of renal proximal tubular epithelial cell hyaluronan generation: Implications for diabetic nephropathy. Kidney Int. 2001;59:1739–1749. doi: 10.1046/j.1523-1755.2001.0590051739.x. [DOI] [PubMed] [Google Scholar]
- 283.Jones SC, Saunders HJ, Pollock CA. High glucose increases growth and collagen synthesis in cultured human tubulointerstitial cells. Diabet Med. 1999;16:932–938. doi: 10.1046/j.1464-5491.1999.00174.x. [DOI] [PubMed] [Google Scholar]
- 284.Jones SG, Morrisey K, Williams JD, Phillips AO. TGF-beta1 stimulates the release of pre-formed bFGF from renal proximal tubular cells. Kidney Int. 1999;56:83–91. doi: 10.1046/j.1523-1755.1999.00517.x. [DOI] [PubMed] [Google Scholar]
- 285.Juncos R, Garvin JL. Superoxide enhances Na-K-2Cl cotransporter activity in the thick ascending limb. Am J Physiol Renal Physiol. 2005;288:F982–F987. doi: 10.1152/ajprenal.00348.2004. [DOI] [PubMed] [Google Scholar]
- 286.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J Clin Invest. 1994;93:2431–2437. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kamesaki H, Nishizawa K, Michaud GY, Cossman J, Kiyono T. TGF-beta 1 induces the cyclin-dependent kinase inhibitor p27Kip1 mRNA and protein in murine B cells. J Immunol. 1998;160:770–777. [PubMed] [Google Scholar]
- 289.Kamran M, Peterson RG, Dominguez JH. Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J Am Soc Nephrol. 1997;8:943–948. doi: 10.1681/ASN.V86943. [DOI] [PubMed] [Google Scholar]
- 290.Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest. 1994;93:397–404. doi: 10.1172/JCI116972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kanda T, Wakino S, Hayashi K, Homma K, Ozawa Y, Saruta T. Effect of fasudil on Rhokinase and nephropathy in subtotally nephrectomized spontaneously hypertensive rats. Kidney Int. 2003;64:2009–2019. doi: 10.1046/j.1523-1755.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 292.Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, Takahashi T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol. 2007;170:1473–1484. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Kang D, Hamasaki N. Mitochondrial oxidative stress and mitochondrial DNA. Clin Chem Lab Med. 2003;41:1281–1288. doi: 10.1515/CCLM.2003.195. [DOI] [PubMed] [Google Scholar]
- 294.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol. 2001;12:1448–1457. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 295.Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, Haller H. Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int. 1999;56:1737–1750. doi: 10.1046/j.1523-1755.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 296.Kang SW, Natarajan R, Shahed A, Nast CC, Lapage J, Mundel P, Kashtan C, Adler SG. Role of 12-lipoxygenase in the stimulation of p38 mitogen-activated protein kinase and collagen alpha5(IV) in experimental diabetic nephropathy and in glucose-stimulated podocytes. J Am Soc Nephrol. 2003;14:3178–3187. doi: 10.1097/01.asn.0000099702.16315.de. [DOI] [PubMed] [Google Scholar]
- 297.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: Mechanisms of renal disease progression. Exp Biol Med (Maywood) 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 298.Kasiske BL, Cleary MP, O’Donnell MP, Keane WF. Effects of genetic obesity on renal structure and function in the Zucker rat. J Lab Clin Med. 1985;106:598–604. [PubMed] [Google Scholar]
- 299.Kasiske BL, O’Donnell MP, Keane WF. Glucose-induced increases in renal hemodynamic function. Possible modulation by renal prostaglandins. Diabetes. 1985;34:360–364. doi: 10.2337/diab.34.4.360. [DOI] [PubMed] [Google Scholar]
- 300.Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, deGroof RC, Lakatta EG. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104:1464–1470. doi: 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- 301.Katavetin P, Miyata T, Inagi R, Tanaka T, Sassa R, Ingelfinger JR, Fujita T, Nangaku M. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. J Am Soc Nephrol. 2006;17:1405–1413. doi: 10.1681/ASN.2005090918. [DOI] [PubMed] [Google Scholar]
- 302.Katz A, Caramori ML, Sisson-Ross S, Groppoli T, Basgen JM, Mauer M. An increase in the cell component of the cortical interstitium antedates interstitial fibrosis in type 1 diabetic patients. Kidney Int. 2002;61:2058–2066. doi: 10.1046/j.1523-1755.2002.00370.x. [DOI] [PubMed] [Google Scholar]
- 303.Kawamura H, Yokote K, Asaumi S, Kobayashi K, Fujimoto M, Maezawa Y, Saito Y, Mori S. High glucose-induced upregulation of osteopontin is mediated via Rho/Rho kinase pathway in cultured rat aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:276–281. doi: 10.1161/01.ATV.0000112012.33770.2a. [DOI] [PubMed] [Google Scholar]
- 304.Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41:1422–1428. doi: 10.2337/diab.41.11.1422. [DOI] [PubMed] [Google Scholar]
- 305.Kelly DJ, Chanty A, Gow RM, Zhang Y, Gilbert RE. Protein kinase Cbeta inhibition attenuates osteopontin expression, macrophage recruitment, and tubulointerstitial injury in advanced experimental diabetic nephropathy. J Am Soc Nephrol. 2005;16:1654–1660. doi: 10.1681/ASN.2004070578. [DOI] [PubMed] [Google Scholar]
- 306.Kelly DJ, Gilbert RE, Cox AJ, Soulis T, Jerums G, Cooper ME. Aminoguanidine ameliorates overexpression of prosclerotic growth factors and collagen deposition in experimental diabetic nephropathy. J Am Soc Nephrol. 2001;12:2098–2107. doi: 10.1681/ASN.V12102098. [DOI] [PubMed] [Google Scholar]
- 307.Kelly DJ, Skinner SL, Gilbert RE, Cox AJ, Cooper ME, Wilkinson-Berka JL. Effects of endothelin or angiotensin II receptor blockade on diabetes in the transgenic (mRen-2)27 rat. Kidney Int. 2000;57:1882–1894. doi: 10.1046/j.1523-1755.2000.00038.x. [DOI] [PubMed] [Google Scholar]
- 308.Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, Wilkinson-Berka JL, Gilbert RE. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes. 2003;52:512–518. doi: 10.2337/diabetes.52.2.512. [DOI] [PubMed] [Google Scholar]
- 309.Kennefick TM, Oyama TT, Thompson MM, Vora JP, Anderson S. Enhanced renal sensitivity to angiotensin actions in diabetes mellitus in the rat. Am J Physiol. 1996;271:F595–F602. doi: 10.1152/ajprenal.1996.271.3.F595. [DOI] [PubMed] [Google Scholar]
- 310.Kim KT, Choi HH, Steinmetz MO, Maco B, Kammerer RA, Ahn SY, Kim HZ, Lee GM, Koh GY. Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate Tie2. J Biol Chem. 2005;280:20126–20131. doi: 10.1074/jbc.M500292200. [DOI] [PubMed] [Google Scholar]
- 311.Kirchner KA. Insulin increases loop segment chloride reabsorption in the euglycemic rat. Am J Physiol. 1988;255:F1206–F1213. doi: 10.1152/ajprenal.1988.255.6.F1206. [DOI] [PubMed] [Google Scholar]
- 312.Kiritoshi S, Nishikawa T, Sonoda K, Kukidome D, Senokuchi T, Matsuo T, Matsumura T, Tokunaga H, Brownlee M, Araki E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes. 2003;52:2570–2577. doi: 10.2337/diabetes.52.10.2570. [DOI] [PubMed] [Google Scholar]
- 313.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du YS, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–31749. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 314.Klahr S, Morrissey J, Hruska K, Wang S, Chen Q. New approaches to delay the progression of chronic renal failure. Kidney Int Suppl. 2002:23–26. doi: 10.1046/j.1523-1755.61.s80.16.x. [DOI] [PubMed] [Google Scholar]
- 315.Kobayashi E, Sasamura H, Mifune M, Shimizu-Hirota R, Kuroda M, Hayashi M, Saruta T. Hepatocyte growth factor regulates proteoglycan synthesis in interstitial fibroblasts. Kidney Int. 2003;64:1179–1188. doi: 10.1046/j.1523-1755.2003.00202.x. [DOI] [PubMed] [Google Scholar]
- 316.Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes. 2008;57:714–723. doi: 10.2337/db07-1241. [DOI] [PubMed] [Google Scholar]
- 317.Komers R, Allen TJ, Cooper ME. Role of endothelium-derived nitric oxide in the pathogenesis of the renal hemodynamic changes of experimental diabetes. Diabetes. 1994;43:1190–1197. doi: 10.2337/diab.43.10.1190. [DOI] [PubMed] [Google Scholar]
- 318.Komers R, Anderson S. Paradoxes of nitric oxide in the diabetic kidney. Am J Physiol Renal Physiol. 2003;284:F1121–F1137. doi: 10.1152/ajprenal.00265.2002. [DOI] [PubMed] [Google Scholar]
- 319.Komers R, Cooper ME. Acute renal hemodynamic effects of ACE inhibition in diabetic hyperfiltration: Role of kinins. Am J Physiol. 1995;268:F588–F594. doi: 10.1152/ajprenal.1995.268.4.F588. [DOI] [PubMed] [Google Scholar]
- 320.Komers R, Lindsley JN, Oyama TT, Allison KM, Anderson S. Role of neuronal nitric oxide synthase (NOS1) in the pathogenesis of renal hemodynamic changes in diabetes. Am J Physiol Renal Physiol. 2000;279:F573–F583. doi: 10.1152/ajprenal.2000.279.3.F573. [DOI] [PubMed] [Google Scholar]
- 321.Komers R, Lindsley JN, Oyama TT, Schutzer WE, Reed JF, Mader SL, Anderson S. Immunohistochemical and functional correlations of renal cyclooxygenase-2 in experimental diabetes. J Clin Invest. 2001;107:889–898. doi: 10.1172/JCI10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Komers R, Oyama TT, Beard DR, Anderson S. Effects of systemic inhibition of Rho kinase on blood pressure and renal hemodynamics in diabetic rats. Br J Pharmacol. 2010 doi: 10.1111/j.1476-5381.2010.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Komers R, Oyama TT, Beard DR, Tikellis C, Xu B, Lotspeich DF, Anderson S. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int. 2010 doi: 10.1038/ki.2010.428. [DOI] [PubMed] [Google Scholar]
- 324.Komers R, Oyama TT, Chapman JG, Allison KM, Anderson S. Effects of systemic inhibition of neuronal nitric oxide synthase in diabetic rats. Hypertension. 2000;35:655–661. doi: 10.1161/01.hyp.35.2.655. [DOI] [PubMed] [Google Scholar]
- 325.Komers R, Schutzer W, Xue H, Oyama TT, Lindsley JN, Anderson S. Effects of p38 mitogen-activated protein kinase inhibition on blood pressure, renal hemodynamics, and renal vascular reactivity in normal and diabetic rats. Transl Res. 2007;150:343–349. doi: 10.1016/j.trsl.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 326.Komers R, Schutzer WE, Reed JF, Lindsley JN, Oyama TT, Buck DC, Mader SL, Anderson S. Altered endothelial nitric oxide synthase targeting and conformation and caveolin-1 expression in the diabetic kidney. Diabetes. 2006;55:1651–1659. doi: 10.2337/db05-1595. [DOI] [PubMed] [Google Scholar]
- 327.Komers R, Zdychova J, Cahova M, Kazdova L, Lindsley JN, Anderson S. Renal cyclooxygenase-2 in obese Zucker (fatty) rats. Kidney Int. 2005;67:2151–2158. doi: 10.1111/j.1523-1755.2005.00320.x. [DOI] [PubMed] [Google Scholar]
- 328.Kontessis PS, Jones SL, Barrow SE, Stratton PD, Alessandrini P, De CS, Ritter JM, Viberti GC. Effect of selective inhibition of thromboxane synthesis on renal function in diabetic nephropathy. J Lab Clin Med. 1993;121:415–423. [PubMed] [Google Scholar]
- 329.Koop K, Eikmans M, Baelde HJ, Kawachi H, de HE, Paul LC, Bruijn JA. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol. 2003;14:2063–2071. doi: 10.1097/01.asn.0000078803.53165.c9. [DOI] [PubMed] [Google Scholar]
- 330.Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, Sugimoto T, Yasuda H, Kashiwagi A, Ways DK, King GL, Kikkawa R. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 2000;14:439–447. doi: 10.1096/fasebj.14.3.439. [DOI] [PubMed] [Google Scholar]
- 331.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–126. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 333.Koya D, Lee IK, Ishii H, Kanoh H, King GL. Prevention of glomerular dysfunction in diabetic rats by treatment with d-alpha-tocopherol. J Am Soc Nephrol. 1997;8:426–435. doi: 10.1681/ASN.V83426. [DOI] [PubMed] [Google Scholar]
- 334.Krepinsky J. Mechanical stretch-induced signal transduction in cultured mesangial cells. Methods Mol Biol. 2009;466:205–221. doi: 10.1007/978-1-59745-352-3_15. [DOI] [PubMed] [Google Scholar]
- 335.Kriz W, Lehir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 336.Krolewski AS. Genetics of diabetic nephropathy: Evidence for major and minor gene effects. Kidney Int. 1999;55:1582–1596. doi: 10.1046/j.1523-1755.1999.00371.x. [DOI] [PubMed] [Google Scholar]
- 337.Ku CH, White KE, Dei CA, Hayward A, Webster Z, Bilous R, Marshall S, Viberti G, Gnudi L. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes. 2008;57:2824–2833. doi: 10.2337/db08-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Kuan CJ, Al-Douahji M, Shankland SJ. The cyclin kinase inhibitor p21WAF1, CIP1 is increased in experimental diabetic nephropathy: Potential role in glomerular hypertrophy. J Am Soc Nephrol. 1998;9:986–993. doi: 10.1681/ASN.V96986. [DOI] [PubMed] [Google Scholar]
- 339.Kumar AM, Gupta RK, Spitzer A. Intracellular sodium in proximal tubules of diabetic rats. Role of glucose. Kidney Int. 1988;33:792–797. doi: 10.1038/ki.1988.69. [DOI] [PubMed] [Google Scholar]
- 340.Landau D, Israel E, Rivkis I, Kachko L, Schrijvers BF, Flyvbjerg A, Phillip M, Segev Y. The effect of growth hormone on the development of diabetic kidney disease in rats. Nephrol Dial Transplant. 2003;18:694–702. doi: 10.1093/ndt/gfg142. [DOI] [PubMed] [Google Scholar]
- 341.Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 342.Lang F, Gorlach A, Vallon V. Targeting SGK1 in diabetes. Expert Opin Ther Targets. 2009;13:1303–1311. doi: 10.1517/14728220903260807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lang F, Klingel K, Wagner CA, Stegen C, Warntges S, Friedrich B, Lanzendorfer M, Melzig J, Moschen I, Steuer S, Waldegger S, Sauter M, Paulmichl M, Gerke V, Risler T, Gamba G, Capasso G, Kandolf R, Hebert SC, Massry SG, Broer S. Deranged transcriptional regulation of cell-volume-sensitive kinase hSGK in diabetic nephropathy. Proc Natl Acad Sci U S A. 2000;97:8157–8162. doi: 10.1073/pnas.97.14.8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Langham RG, Kelly DJ, Cox AJ, Thomson NM, Holthofer H, Zaoui P, Pinel N, Cordonnier DJ, Gilbert RE. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: Effects of angiotensin converting enzyme inhibition. Diabetologia. 2002;45:1572–1576. doi: 10.1007/s00125-002-0946-y. [DOI] [PubMed] [Google Scholar]
- 345.Langham RG, Kelly DJ, Maguire J, Dowling JP, Gilbert RE, Thomson NM. Over-expression of platelet-derived growth factor in human diabetic nephropathy. Nephrol Dial Transplant. 2003;18:1392–1396. doi: 10.1093/ndt/gfg177. [DOI] [PubMed] [Google Scholar]
- 346.Lansang MC, Price DA, Laffel LM, Osei SY, Fisher ND, Erani D, Hollenberg NK. Renal vascular responses to captopril and to candesartan in patients with type 1 diabetes mellitus. Kidney Int. 2001;59:1432–1438. doi: 10.1046/j.1523-1755.2001.0590041432.x. [DOI] [PubMed] [Google Scholar]
- 347.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 348.Lau C, Sudbury I, Thomson M, Howard PL, Magil AB, Cupples WA. Salt-resistant blood pressure and salt-sensitive renal autoregulation in chronic streptozotocin diabetes. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1761–R1770. doi: 10.1152/ajpregu.90731.2008. [DOI] [PubMed] [Google Scholar]
- 349.Lawson ML, Sochett EB, Chait PG, Balfe JW, Daneman D. Effect of puberty on markers of glomerular hypertrophy and hypertension in IDDM. Diabetes. 1996;45:51–55. doi: 10.2337/diab.45.1.51. [DOI] [PubMed] [Google Scholar]
- 350.Lee CM, Robinson LJ, Michel T. Oligomerization of endothelial nitric oxide synthase. Evidence for a dominant negative effect of truncation mutants. J Biol Chem. 1995;270:27403–27406. doi: 10.1074/jbc.270.46.27403. [DOI] [PubMed] [Google Scholar]
- 351.Lee MJ, Feliers D, Mariappan MM, Sataranatarajan K, Mahimainathan L, Musi N, Foretz M, Viollet B, Weinberg JM, Choudhury GG, Kasinath BS. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am J Physiol Renal Physiol. 2007;292:F617–F627. doi: 10.1152/ajprenal.00278.2006. [DOI] [PubMed] [Google Scholar]
- 352.Lee YJ, Han HJ. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3{beta}, Snail1, and {beta}-catenin in renal proximal tubule cells. Am J Physiol Renal Physiol. 2009 doi: 10.1152/ajprenal.00475.2009. [DOI] [PubMed] [Google Scholar]
- 353.Leighton PA, Mitchell KJ, Goodrich LV, Lu X, Pinson K, Scherz P, Skarnes WC, Tessier-Lavigne M. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature. 2001;410:174–179. doi: 10.1038/35065539. [DOI] [PubMed] [Google Scholar]
- 354.Lemley KV. Diabetes and chronic kidney disease: Lessons from the Pima Indians. Pediatr Nephrol. 2008;23:1933–1940. doi: 10.1007/s00467-008-0763-8. [DOI] [PubMed] [Google Scholar]
- 355.Lemley KV, Abdullah I, Myers BD, Meyer TW, Blouch K, Smith WE, Bennett PH, Nelson RG. Evolution of incipient nephropathy in type 2 diabetes mellitus. Kidney Int. 2000;58:1228–1237. doi: 10.1046/j.1523-1755.2000.00223.x. [DOI] [PubMed] [Google Scholar]
- 356.Leroy P, Mostov KE. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell. 2007;18:1943–1952. doi: 10.1091/mbc.E06-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Lervang HH, Jensen S, Brochner-Mortensen J, Ditzel J. Early glomerular hyperfiltration and the development of late nephropathy in type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1988;31:723–729. doi: 10.1007/BF00274773. [DOI] [PubMed] [Google Scholar]
- 358.Levine JH, Buse MG, Leaming AB, Raskin P. Effect of streptozotocin-induced diabetes on renal ornithine decarboxylase activity. Diabetes. 1980;29:532–535. doi: 10.2337/diab.29.7.532. [DOI] [PubMed] [Google Scholar]
- 359.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 360.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 361.Lewis MP, Fine LG, Norman JT. Pexicrine effects of basement membrane components on paracrine signaling by renal tubular cells. Kidney Int. 1996;49:48–58. doi: 10.1038/ki.1996.7. [DOI] [PubMed] [Google Scholar]
- 362.Leyssac PP, Karlsen FM, Skott O. Role of proximal tubular reabsorption for the intrarenal control of GFR. Kidney Int Suppl. 1991;32:S132–S135. [PubMed] [Google Scholar]
- 363.Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009;175:1380–1388. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, Johnson RJ, Lan HY. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004;18:176–178. doi: 10.1096/fj.02-1117fje. [DOI] [PubMed] [Google Scholar]
- 365.Li JJ, Kwak SJ, Jung DS, Kim JJ, Yoo TH, Ryu DR, Han SH, Choi HY, Lee JE, Moon SJ, Kim DK, Han DS, Kang SW. Podocyte biology in diabetic nephropathy. Kidney Int Suppl. 2007:S36–S42. doi: 10.1038/sj.ki.5002384. [DOI] [PubMed] [Google Scholar]
- 366.Li JM, Shah AM. ROS generation by nonphagocytic NADPH oxidase: Potential relevance in diabetic nephropathy. J Am Soc Nephrol. 2003;14:S221–S226. doi: 10.1097/01.asn.0000077406.67663.e7. [DOI] [PubMed] [Google Scholar]
- 367.Li Y, Tan X, Dai C, Stolz DB, Wang D, Liu Y. Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:1907–1918. doi: 10.1681/ASN.2008090930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest. 2003;112:503–516. doi: 10.1172/JCI17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20:2493–2502. doi: 10.1681/ASN.2008111186. [DOI] [PubMed] [Google Scholar]
- 370.Lin CL, Wang FS, Hsu YC, Chen CN, Tseng MJ, Saleem MA, Chang PJ, Wang JY. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes. 2010;59:1915–1925. doi: 10.2337/db09-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Lindenmeyer MT, Kretzler M, Boucherot A, Berra S, Yasuda Y, Henger A, Eichinger F, Gaiser S, Schmid H, Rastaldi MP, Schrier RW, Schlondorff D, Cohen CD. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol. 2007;18:1765–1776. doi: 10.1681/ASN.2006121304. [DOI] [PubMed] [Google Scholar]
- 373.Lindenmeyer MT, Rastaldi MP, Ikehata M, Neusser MA, Kretzler M, Cohen CD, Schlondorff D. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol. 2008;19:2225–2236. doi: 10.1681/ASN.2007121313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 375.Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Lorenz JN, Schnermann J, Brosius FC, Briggs JP, Furspan PB. Intra-cellular ATP can regulate afferent arteriolar tone via ATP-sensitive K+ channels in the rabbit. J Clin Invest. 1992;90:733–740. doi: 10.1172/JCI115945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Luik PT, Hoogenberg K, Van Der Kleij FG, Beusekamp BJ, Kerstens MN, De Jong PE, Dullaart RP, Navis GJ. Short-term moderate sodium restriction induces relative hyperfiltration in normotensive normoalbuminuric Type I diabetes mellitus. Diabetologia. 2002;45:535–541. doi: 10.1007/s00125-001-0763-8. [DOI] [PubMed] [Google Scholar]
- 378.Ma RCW, Tam CHT, Wang Y, Luk AO, Hu C, Yang X, Lam V, Chan AWH, Ho JSK, Chow CC, Tong PCY, Jia W, Ng MCY, So WY, Chan JCN. Genetic variants of the protein kinase C-{beta} 1 gene and development of end-stage renal disease in patients with type 2 diabetes. JAMA. 2010;304:881–889. doi: 10.1001/jama.2010.1191. [DOI] [PubMed] [Google Scholar]
- 379.Madala Halagappa VK, Tiwari S, Riazi S, Hu X, Ecelbarger CM. Chronic candesartan alters expression and activity of NKCC2, NCC, and ENaC in the obese Zucker rat. Am J Physiol Renal Physiol. 2008;294:F1222–F1231. doi: 10.1152/ajprenal.00604.2007. [DOI] [PubMed] [Google Scholar]
- 380.Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia. 2009;52:691–697. doi: 10.1007/s00125-009-1268-0. [DOI] [PubMed] [Google Scholar]
- 381.Magen D, Sprecher E, Zelikovic I, Skorecki K. A novel missense mutation in SLC5A2 encoding SGLT2 underlies autosomal-recessive renal glucosuria and aminoaciduria. Kidney Int. 2005;67:34–41. doi: 10.1111/j.1523-1755.2005.00053.x. [DOI] [PubMed] [Google Scholar]
- 382.Magri CJ, Fava S. The role of tubular injury in diabetic nephropathy. Eur J Intern Med. 2009;20:551–555. doi: 10.1016/j.ejim.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 383.Makino H, Miyamoto Y, Sawai K, Mori K, Mukoyama M, Nakao K, Yoshimasa Y, Suga S. Altered gene expression related to glomerulogenesis and podocyte structure in early diabetic nephropathy of db/db mice and its restoration by pioglitazone. Diabetes. 2006;55:2747–2756. doi: 10.2337/db05-1683. [DOI] [PubMed] [Google Scholar]
- 384.Manotham K, Tanaka T, Matsumoto M, Ohse T, Miyata T, Inagi R, Kurokawa K, Fujita T, Nangaku M. Evidence of tubular hypoxia in the early phase in the remnant kidney model. J Am Soc Nephrol. 2004;15:1277–1288. doi: 10.1097/01.asn.0000125614.35046.10. [DOI] [PubMed] [Google Scholar]
- 385.Marcus RG, England R, Nguyen K, Charron MJ, Briggs JP, Brosius FC., III Altered renal expression of the insulin-responsive glucose transporter GLUT4 in experimental diabetes mellitus. Am J Physiol. 1994;267:F816–F824. doi: 10.1152/ajprenal.1994.267.5.F816. [DOI] [PubMed] [Google Scholar]
- 386.Marcussen N. Atubular glomeruli and the structural basis for chronic renal failure. Lab Invest. 1992;66:265–284. [PubMed] [Google Scholar]
- 387.Maric C, Sullivan S. Estrogens and the diabetic kidney. Gend Med. 2008;5(Suppl A):S103–S113. doi: 10.1016/j.genm.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Marshall CB, Shankland SJ. Cell cycle and glomerular disease: A minireview. Nephron Exp Nephrol. 2006;102:e39–e48. doi: 10.1159/000088400. [DOI] [PubMed] [Google Scholar]
- 389.Marshall JD, Mu JL, Cheah YC, Nesbitt MN, Frankel WN, Paigen B. The AXB and BXA set of recombinant inbred mouse strains. Mamm Genome. 1992;3:669–680. doi: 10.1007/BF00444361. [DOI] [PubMed] [Google Scholar]
- 390.Martineau LC, McVeigh LI, Jasmin BJ, Kennedy CR. p38 MAP kinase mediates mechanically induced COX-2 and PG EP4 receptor expression in podocytes: Implications for the actin cytoskeleton. Am J Physiol Renal Physiol. 2004;286:F693–F701. doi: 10.1152/ajprenal.00331.2003. [DOI] [PubMed] [Google Scholar]
- 391.Mattar AL, Fujihara CK, Ribeiro MO, de NG, Zatz R. Renal effects of acute and chronic nitric oxide inhibition in experimental diabetes. Nephron. 1996;74:136–143. doi: 10.1159/000189293. [DOI] [PubMed] [Google Scholar]
- 392.Mauer M, Drummond K. The early natural history of nephropathy in type 1 diabetes: I. Study design and baseline characteristics of the study participants. Diabetes. 2002;51:1572–1579. doi: 10.2337/diabetes.51.5.1572. [DOI] [PubMed] [Google Scholar]
- 393.Mauer SM, Brown DM, Steffes MW, Azar S. Studies of renal autoregulation in pancreatectomized and streptozotocin diabetic rats. Kidney Int. 1990;37:909–917. doi: 10.1038/ki.1990.65. [DOI] [PubMed] [Google Scholar]
- 394.Mauer SM, Steffes MW, Azar S, Brown DM. Effects of sorbinil on glomerular structure and function in long-term-diabetic rats. Diabetes. 1989;38:839–846. doi: 10.2337/diab.38.7.839. [DOI] [PubMed] [Google Scholar]
- 395.Mauer SM, Steffes MW, Azar S, Sandberg SK, Brown DM. The effects of Goldblatt hypertension on development of the glomerular lesions of diabetes mellitus in the rat. Diabetes. 1978;27:738–744. doi: 10.2337/diab.27.7.738. [DOI] [PubMed] [Google Scholar]
- 396.Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74:1143–1155. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Mbanya JC, Thomas TH, Taylor R, Alberti KG, Wilkinson R. Increased proximal tubular sodium reabsorption in hypertensive patients with type 2 diabetes. Diabet Med. 1989;6:614–620. doi: 10.1111/j.1464-5491.1989.tb01238.x. [DOI] [PubMed] [Google Scholar]
- 398.McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J Biol Chem. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- 399.Meehan WP, Buchanan TA, Hsueh W. Chronic insulin administration elevates blood pressure in rats. Hypertension. 1994;23:1012–1017. doi: 10.1161/01.hyp.23.6.1012. [DOI] [PubMed] [Google Scholar]
- 400.Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, Schiffer M, Mengel M, Lindschau C, Leitges M, Haller H. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol. 2007;18:1190–1198. doi: 10.1681/ASN.2005070694. [DOI] [PubMed] [Google Scholar]
- 401.Meier M, Park JK, Overheu D, Kirsch T, Lindschau C, Gueler F, Leitges M, Menne J, Haller H. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes. 2007;56:346–354. doi: 10.2337/db06-0891. [DOI] [PubMed] [Google Scholar]
- 402.Meininger CJ, Marinos RS, Hatakeyama K, Martinez-Zaguilan R, Rojas JD, Kelly KA, Wu G. Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem J. 2000;349:353–356. doi: 10.1042/0264-6021:3490353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Menne J, Meier M, Park JK, Boehne M, Kirsch T, Lindschau C, Ociepka R, Leitges M, Rinta-Valkama J, Holthofer H, Haller H. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int. 2006;70:1456–1462. doi: 10.1038/sj.ki.5001830. [DOI] [PubMed] [Google Scholar]
- 404.Menne J, Park JK, Boehne M, Elger M, Lindschau C, Kirsch T, Meier M, Gueler F, Fiebeler A, Bahlmann FH, Leitges M, Haller H. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes. 2004;53:2101–2109. doi: 10.2337/diabetes.53.8.2101. [DOI] [PubMed] [Google Scholar]
- 405.Michaelis OE, Patrick DH, Hansen CT, Canary JJ, Werner RM, Carswell N. Insulin-independent diabetes mellitus (type II). Spontaneous hypertensive/NIH-corpulent rat. Am J Pathol. 1986;123:398–400. [PMC free article] [PubMed] [Google Scholar]
- 406.Miller JA. Renal responses to sodium restriction in patients with early diabetes mellitus. J Am Soc Nephrol. 1997;8:749–755. doi: 10.1681/ASN.V85749. [DOI] [PubMed] [Google Scholar]
- 407.Miller J, Bogdonoff M. Antidiuresis associated with administration of insulin. J Appl Physiol. 1954;6:509–512. doi: 10.1152/jappl.1954.6.8.509. [DOI] [PubMed] [Google Scholar]
- 408.Miracle CM, Rieg T, Mansoury H, Vallon V, Thomson SC. Ornithine decarboxylase inhibitor eliminates hyperresponsiveness of the early diabetic proximal tubule to dietary salt. Am J Physiol Renal Physiol. 2008;295:F995–F1002. doi: 10.1152/ajprenal.00491.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Miyata T, de Strihou CY. Diabetic nephropathy: A disorder of oxygen metabolism? Nat Rev Nephrol. 2010;6:83–95. doi: 10.1038/nrneph.2009.211. [DOI] [PubMed] [Google Scholar]
- 410.Miyata T, van Ypersele de SC, Ueda Y, Ichimori K, Inagi R, Onogi H, Ishikawa N, Nangaku M, Kurokawa K. Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: Biochemical mechanisms. J Am Soc Nephrol. 2002;13:2478–2487. doi: 10.1097/01.asn.0000032418.67267.f2. [DOI] [PubMed] [Google Scholar]
- 411.Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin Nephrol. 2004;24:354–365. doi: 10.1016/j.semnephrol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 412.Mogensen CE. Early glomerular hyperfiltration in insulin-dependent diabetics and late nephropathy. Scand J Clin Lab Invest. 1986;46:201–206. doi: 10.3109/00365518609083660. [DOI] [PubMed] [Google Scholar]
- 413.Mogensen CE, Andersen MJ. Increased kidney size and glomerular filtration rate in early juvenile diabetes. Diabetes. 1973;22:706–712. doi: 10.2337/diab.22.9.706. [DOI] [PubMed] [Google Scholar]
- 414.Mogensen CE, Christensen CK. Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med. 1984;311:89–93. doi: 10.1056/NEJM198407123110204. [DOI] [PubMed] [Google Scholar]
- 415.Mogensen CE, Christensen CK, Vittinghus E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes. 1983;32(Suppl 2):64–78. doi: 10.2337/diab.32.2.s64. [DOI] [PubMed] [Google Scholar]
- 416.Mohan S, Reddick RL, Musi N, Horn DA, Yan B, Prihoda TJ, Natarajan M, Abboud-Werner SL. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest. 2008;88:515–528. doi: 10.1038/labinvest.2008.23. [DOI] [PubMed] [Google Scholar]
- 417.Monkawa T, Hiromura K, Wolf G, Shankland SJ. The hypertrophic effect of transforming growth factor-beta is reduced in the absence of cyclin-dependent kinase-inhibitors p21 and p27. J Am Soc Nephrol. 2002;13:1172–1178. doi: 10.1097/01.asn.0000013162.29833.45. [DOI] [PubMed] [Google Scholar]
- 418.Morrisey K, Steadman R, Williams JD, Phillips AO. Renal proximal tubular cell fibronectin accumulation in response to glucose is polyol pathway dependent. Kidney Int. 1999;55:160–167. doi: 10.1046/j.1523-1755.1999.00248.x. [DOI] [PubMed] [Google Scholar]
- 419.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13:3005–3015. doi: 10.1097/01.asn.0000039661.06947.fd. [DOI] [PubMed] [Google Scholar]
- 420.Myers BD, Nelson RG, Williams GW, Bennett PH, Hardy SA, Berg RL, Loon N, Knowler WC, Mitch WE. Glomerular function in Pima Indians with noninsulin-dependent diabetes mellitus of recent onset. J Clin Invest. 1991;88:524–530. doi: 10.1172/JCI115335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95:34–42. doi: 10.1210/jc.2009-0473. [DOI] [PubMed] [Google Scholar]
- 422.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson MA, Johnson RJ, Croker B. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 423.Nakagawa T, Sato W, Sautin YY, Glushakova O, Croker B, Atkinson MA, Tisher CC, Johnson RJ. Uncoupling of vascular endothelial growth factor with nitric oxide as a mechanism for diabetic vasculopathy. J Am Soc Nephrol. 2006;17:736–745. doi: 10.1681/ASN.2005070759. [DOI] [PubMed] [Google Scholar]
- 424.Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Ebihara I, Koide H. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15:1379–1383. doi: 10.1093/ndt/15.9.1379. [DOI] [PubMed] [Google Scholar]
- 425.Nangaku M. Mechanisms of tubulointerstitial injury in the kidney: Final common pathways to end-stage renal failure. Intern Med. 2004;43:9–17. doi: 10.2169/internalmedicine.43.9. [DOI] [PubMed] [Google Scholar]
- 426.Nangaku M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 427.Nangaku M, Miyata T, Sada T, Mizuno M, Inagi R, Ueda Y, Ishikawa N, Yuzawa H, Koike H, van Ypersele de SC, Kurokawa K. Antihypertensive agents inhibit in vivo the formation of advanced glycation end products and improve renal damage in a type 2 diabetic nephropathy rat model. J Am Soc Nephrol. 2003;14:1212–1222. doi: 10.1097/01.asn.0000062961.76776.c1. [DOI] [PubMed] [Google Scholar]
- 428.Nasu T, Maeshima Y, Kinomura M, Hirokoshi-Kawahara K, Tanabe K, Sugiyama H, Sonoda H, Sato Y, Makino H. Vasohibin-1, a negative feedback regulator of angiogenesis, ameliorates renal alterations in a mouse model of diabetic nephropathy. Diabetes. 2009;58:2365–2375. doi: 10.2337/db08-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Nath KA, Croatt AJ, Hostetter TH. Oxygen consumption and oxidant stress in surviving nephrons. Am J Physiol. 1990;258:F1354–F1362. doi: 10.1152/ajprenal.1990.258.5.F1354. [DOI] [PubMed] [Google Scholar]
- 430.Negrete H, Studer RK, Craven PA, DeRubertis FR. Role for transforming growth factor beta in thromboxane-induced increases in mesangial cell fibronectin synthesis. Diabetes. 1995;44:335–339. doi: 10.2337/diab.44.3.335. [DOI] [PubMed] [Google Scholar]
- 431.Nejsum LN, Kwon TH, Marples D, Flyvbjerg A, Knepper MA, Frokiaer J, Nielsen S. Compensatory increase in AQP2, p-AQP2, and AQP3 expression in rats with diabetes mellitus. Am J Physiol Renal Physiol. 2001;280:F715–F726. doi: 10.1152/ajprenal.2001.280.4.F715. [DOI] [PubMed] [Google Scholar]
- 432.Nelson RG, Bennett PH, Beck GJ, Tan M, Knowler WC, Mitch WE, Hirschman GH, Myers BD. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med. 1996;335:1636–1642. doi: 10.1056/NEJM199611283352203. [DOI] [PubMed] [Google Scholar]
- 433.Ng YY, Huang TP, Yang WC, Chen ZP, Yang AH, Mu W, Nikolic-Paterson DJ, Atkins RC, Lan HY. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998;54:864–876. doi: 10.1046/j.1523-1755.1998.00076.x. [DOI] [PubMed] [Google Scholar]
- 434.Nguyen G, Delarue F, Berrou J, Rondeau E, Sraer JD. Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen. Kidney Int. 1996;50:1897–1903. doi: 10.1038/ki.1996.511. [DOI] [PubMed] [Google Scholar]
- 435.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Nguyen TQ, Tarnow L, Andersen S, Hovind P, Parving HH, Gold-schmeding R, van Nieuwenhoven FA. Urinary connective tissue growth factor excretion correlates with clinical markers of renal disease in a large population of type 1 diabetic patients with diabetic nephropathy. Diabetes Care. 2006;29:83–88. doi: 10.2337/diacare.29.1.83. [DOI] [PubMed] [Google Scholar]
- 437.Nguyen TQ, Tarnow L, Jorsal A, Oliver N, Roestenberg P, Ito Y, Parving HH, Rossing P, van Nieuwenhoven FA, Goldschmeding R. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care. 2008;31:1177–1182. doi: 10.2337/dc07-2469. [DOI] [PubMed] [Google Scholar]
- 438.Niehof M, Borlak J. HNF4 alpha and the Ca-channel TRPC1 are novel disease candidate genes in diabetic nephropathy. Diabetes. 2008;57:1069–1077. doi: 10.2337/db07-1065. [DOI] [PubMed] [Google Scholar]
- 439.Nieuwdorp M, Mooij HL, Kroon J, Atasever B, Spaan JA, Ince C, Holleman F, Diamant M, Heine RJ, Hoekstra JB, Kastelein JJ, Stroes ES, Vink H. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55:1127–1132. doi: 10.2337/diabetes.55.04.06.db05-1619. [DOI] [PubMed] [Google Scholar]
- 440.Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, Meijers JC, Holleman F, Hoekstra JB, Vink H, Kastelein JJ, Stroes ES. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55:480–486. doi: 10.2337/diabetes.55.02.06.db05-1103. [DOI] [PubMed] [Google Scholar]
- 441.Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 442.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 443.Nishikimi T, Akimoto K, Wang X, Mori Y, Tadokoro K, Ishikawa Y, Shimokawa H, Ono H, Matsuoka H. Fasudil, a Rhokinase inhibitor, attenuates glomerulosclerosis in Dahl salt-sensitive rats. J Hypertens. 2004;22:1787–1796. doi: 10.1097/00004872-200409000-00024. [DOI] [PubMed] [Google Scholar]
- 444.Nishikimi T, Koshikawa S, Ishikawa Y, Akimoto K, Inaba C, Ishimura K, Ono H, Matsuoka H. Inhibition of Rhokinase attenuates nephrosclerosis and improves survival in salt-loaded spontaneously hypertensive stroke-prone rats. J Hypertens. 2007;25:1053–1063. doi: 10.1097/HJH.0b013e3280825440. [DOI] [PubMed] [Google Scholar]
- 445.Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl. 2007:S49–S53. doi: 10.1038/sj.ki.5002386. [DOI] [PubMed] [Google Scholar]
- 446.Norman JT, Orphanides C, Garcia P, Fine LG. Hypoxia-induced changes in extracellular matrix metabolism in renal cells. Exp Nephrol. 1999;7:463–469. doi: 10.1159/000020625. [DOI] [PubMed] [Google Scholar]
- 447.O’Bryan GT, Hostetter TH. The renal hemodynamic basis of diabetic nephropathy. Semin Nephrol. 1997;17:93–100. [PubMed] [Google Scholar]
- 448.O’Donnell MP, Kasiske BL, Daniels FX, Keane WF. Effects of nephron loss on glomerular hemodynamics and morphology in diabetic rats. Diabetes. 1986;35:1011–1015. doi: 10.2337/diab.35.9.1011. [DOI] [PubMed] [Google Scholar]
- 449.O’Hagan M, Howey J, Greene SA. Increased proximal tubular reabsorption of sodium in childhood diabetes mellitus. Diabet Med. 1991;8:44–48. doi: 10.1111/j.1464-5491.1991.tb01515.x. [DOI] [PubMed] [Google Scholar]
- 450.O’Hare JA, Ferriss JB, Brady D, Twomey B, O’Sullivan DJ. Exchangeable sodium and renin in hypertensive diabetic patients with and without nephropathy. Hypertension. 1985;7:II43–II48. doi: 10.1161/01.hyp.7.6_pt_2.ii43. [DOI] [PubMed] [Google Scholar]
- 451.Ohishi K, Carmines PK. Superoxide dismutase restores the influence of nitric oxide on renal arterioles in diabetes mellitus. J Am Soc Nephrol. 1995;5:1559–1566. doi: 10.1681/ASN.V581559. [DOI] [PubMed] [Google Scholar]
- 452.Ohishi K, Okwueze MI, Vari RC, Carmines PK. Juxtamedullary microvascular dysfunction during the hyperfiltration stage of diabetes mellitus. Am J Physiol. 1994;267:F99–105. doi: 10.1152/ajprenal.1994.267.1.F99. [DOI] [PubMed] [Google Scholar]
- 453.Ohno M, Cooke JP, Dzau VJ, Gibbons GH. Fluid shear stress induces endothelial transforming growth factor beta-1 transcription and production. Modulation by potassium channel blockade. J Clin Invest. 1995;95:1363–1369. doi: 10.1172/JCI117787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Ohtomo S, Nangaku M, Izuhara Y, Takizawa S, Strihou CY, Miyata T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol Dial Transplant. 2008;23:1166–1172. doi: 10.1093/ndt/gfm715. [DOI] [PubMed] [Google Scholar]
- 455.Okamura DM, Himmelfarb J. Tipping the redox balance of oxidative stress in fibrogenic pathways in chronic kidney disease. Pediatr Nephrol. 2009;24:2309–2319. doi: 10.1007/s00467-009-1199-5. [DOI] [PubMed] [Google Scholar]
- 456.Oku A, Ueta K, Arakawa K, Ishihara T, Nawano M, Kuronuma Y, Matsumoto M, Saito A, Tsujihara K, Anai M, Asano T, Kanai Y, Endou H. T-1095, an inhibitor of renal Na+-glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes. 1999;48:1794–1800. doi: 10.2337/diabetes.48.9.1794. [DOI] [PubMed] [Google Scholar]
- 457.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of ACEI and ARB. Kidney Int. 2002;61:186–194. doi: 10.1046/j.1523-1755.2002.00123.x. [DOI] [PubMed] [Google Scholar]
- 459.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 460.O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 461.Orphanides C, Fine LG, Norman JT. Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int. 1997;52:637–647. doi: 10.1038/ki.1997.377. [DOI] [PubMed] [Google Scholar]
- 462.Ortola FV, Ballermann BJ, Anderson S, Mendez RE, Brenner BM. Elevated plasma atrial natriuretic peptide levels in diabetic rats. Potential mediator of hyperfiltration. J Clin Invest. 1987;80:670–674. doi: 10.1172/JCI113120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Osorio H, Bautista R, Rios A, Franco M, Santamaria J, Escalante B. Effect of treatment with losartan on salt sensitivity and SGLT2 expression in hypertensive diabetic rats. Diabetes Res Clin Pract. 2009;86:e46–e49. doi: 10.1016/j.diabres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 464.Osterby R. Glomerular structural changes in type 1 (insulin-dependent) diabetes mellitus: Causes, consequences, and prevention. Diabetologia. 1992;35:803–812. doi: 10.1007/BF00399925. [DOI] [PubMed] [Google Scholar]
- 465.Osterby R, Asplund J, Bangstad HJ, Nyberg G, Rudberg S, Viberti GC, Walker JD. Neovascularization at the vascular pole region in diabetic glomerulopathy. Nephrol Dial Transplant. 1999;14:348–352. doi: 10.1093/ndt/14.2.348. [DOI] [PubMed] [Google Scholar]
- 466.Osterby R, Parving HH, Hommel E, Jorgensen HE, Lokkegaard H. Glomerular structure and function in diabetic nephropathy. Early to advanced stages. Diabetes. 1990;39:1057–1063. doi: 10.2337/diab.39.9.1057. [DOI] [PubMed] [Google Scholar]
- 467.Ott C, Iwanciw D, Graness A, Giehl K, Goppelt-Struebe M. Modulation of the expression of connective tissue growth factor by alterations of the cytoskeleton. J Biol Chem. 2003;278:44305–44311. doi: 10.1074/jbc.M309140200. [DOI] [PubMed] [Google Scholar]
- 468.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46:1153–1160. doi: 10.1007/s00125-003-1155-z. [DOI] [PubMed] [Google Scholar]
- 470.Palm F, Connors SG, Mendonca M, Welch WJ, Wilcox CS. Angiotensin II type 2 receptors and nitric oxide sustain oxygenation in the clipped kidney of early Goldblatt hypertensive rats. Hypertension. 2008;51:345–351. doi: 10.1161/HYPERTENSIONAHA.107.097832. [DOI] [PubMed] [Google Scholar]
- 471.Palm F, Fasching A, Hansell P, Kallskog O. Nitric oxide originating from NOS1 controls oxygen utilization and electrolyte transport efficiency in the diabetic kidney. Am J Physiol Renal Physiol. 2010;298:F416–F420. doi: 10.1152/ajprenal.00229.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Palm F, Friederich M, Carlsson PO, Hansell P, Teerlink T, Liss P. Reduced nitric oxide in diabetic kidneys due to increased hepatic arginine metabolism: Implications for renomedullary oxygen availability. Am J Physiol Renal Physiol. 2008;294:F30–F37. doi: 10.1152/ajprenal.00166.2007. [DOI] [PubMed] [Google Scholar]
- 473.Palm F, Teerlink T, Hansell P. Nitric oxide and kidney oxygenation. Curr Opin Nephrol Hypertens. 2009;18:68–73. doi: 10.1097/MNH.0b013e32831c4cdf. [DOI] [PubMed] [Google Scholar]
- 474.Palmisano JJ, Lebovitz HE. Renal function in black Americans with type II diabetes. J Diabet Complications. 1989;3:40–44. doi: 10.1016/0891-6632(89)90009-3. [DOI] [PubMed] [Google Scholar]
- 475.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care. 2008;31(Suppl 2):S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 476.Park CW, Kim HW, Ko SH, Chung HW, Lim SW, Yang CW, Chang YS, Sugawara A, Guan Y, Breyer MD. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55:885–893. doi: 10.2337/diabetes.55.04.06.db05-1329. [DOI] [PubMed] [Google Scholar]
- 477.Park CW, Zhang Y, Zhang X, Wu J, Chen L, Cha DR, Su D, Hwang MT, Fan X, Davis L, Striker G, Zheng F, Breyer M, Guan Y. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69:1511–1517. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- 478.Parving HH, Christiansen JS, Noer I, Tronier B, Mogensen CE. The effect of glucagon infusion on kidney function in short-term insulin-dependent juvenile diabetics. Diabetologia. 1980;19:350–354. doi: 10.1007/BF00280519. [DOI] [PubMed] [Google Scholar]
- 479.Parving HH, Lehnert H, Brochner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 480.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 481.Pedersen SB, Flyvbjerg A, Gronbaek H, Richelsen B. Increased ornithine decarboxylase activity in kidneys undergoing hypertrophy in experimental diabetes. Mol Cell Endocrinol. 1992;86:67–72. doi: 10.1016/0303-7207(92)90176-7. [DOI] [PubMed] [Google Scholar]
- 482.Pedersen SB, Flyvbjerg A, Richelsen B. Inhibition of renal ornithine decarboxylase activity prevents kidney hypertrophy in experimental diabetes. Am J Physiol. 1993;264:C453–C456. doi: 10.1152/ajpcell.1993.264.2.C453. [DOI] [PubMed] [Google Scholar]
- 483.Pelikanova T, Smrckova I, Krizova J, Stribrna J, Lanska V. Effects of insulin and lipid emulsion on renal haemodynamics and renal sodium handling in IDDM patients. Diabetologia. 1996;39:1074–1082. doi: 10.1007/BF00400657. [DOI] [PubMed] [Google Scholar]
- 484.Peng F, Wu D, Gao B, Ingram AJ, Zhang B, Chorneyko K, McKenzie R, Krepinsky JC. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes. 2008;57:1683–1692. doi: 10.2337/db07-1149. [DOI] [PubMed] [Google Scholar]
- 485.Peng F, Zhang B, Ingram AJ, Gao B, Zhang Y, Krepinsky JC. Mechanical stretch-induced RhoA activation is mediated by the RhoGEF Vav2 in mesangial cells. Cell Signal. 2010;22:34–40. doi: 10.1016/j.cellsig.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 486.Perico N, Benigni A, Gabanelli M, Piccinelli A, Rog M, De RC, Remuzzi G. Atrial natriuretic peptide and prostacyclin synergistically mediate hyperfiltration and hyperperfusion of diabetic rats. Diabetes. 1992;41:533–538. doi: 10.2337/diab.41.4.533. [DOI] [PubMed] [Google Scholar]
- 487.Petermann AT, Pippin J, Durvasula R, Pichler R, Hiromura K, Monkawa T, Couser WG, Shankland SJ. Mechanical stretch induces podocyte hypertrophy in vitro. Kidney Int. 2005;67:157–166. doi: 10.1111/j.1523-1755.2005.00066.x. [DOI] [PubMed] [Google Scholar]
- 488.Petermann AT, Pippin J, Krofft R, Blonski M, Griffin S, Durvasula R, Shankland SJ. Viable podocytes detach in experimental diabetic nephropathy: Potential mechanism underlying glomerulosclerosis. Nephron Exp Nephrol. 2004;98:e114–e123. doi: 10.1159/000081555. [DOI] [PubMed] [Google Scholar]
- 489.Pfaff IL, Vallon V. Protein kinase C beta isoenzymes in diabetic kidneys and their relation to nephroprotective actions of the ACE inhibitor lisinopril. Kidney Blood Press Res. 2002;25:329–340. doi: 10.1159/000066789. [DOI] [PubMed] [Google Scholar]
- 490.Pfaff IL, Wagner HJ, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta1 and betaII in rat kidney. J Am Soc Nephrol. 1999;10:1861–1873. doi: 10.1681/ASN.V1091861. [DOI] [PubMed] [Google Scholar]
- 491.Phillips AO, Baboolal K, Riley S, Grone H, Janssen U, Steadman R, Williams J, Floege J. Association of prolonged hyperglycemia with glomerular hypertrophy and renal basement membrane thickening in the Goto Kakizaki model of non-insulin-dependent diabetes mellitus. Am J Kidney Dis. 2001;37:400–410. doi: 10.1053/ajkd.2001.21322. [DOI] [PubMed] [Google Scholar]
- 492.Phillips AO, Morrisey K, Steadman R, Williams JD. Decreased degradation of collagen and fibronectin following exposure of proximal cells to glucose. Exp Nephrol. 1999;7:449–462. doi: 10.1159/000020624. [DOI] [PubMed] [Google Scholar]
- 493.Phillips AO, Steadman R. Diabetic nephropathy: The central role of renal proximal tubular cells in tubulointerstitial injury. Histol Histopathol. 2002;17:247–252. doi: 10.14670/HH-17.247. [DOI] [PubMed] [Google Scholar]
- 494.Phillips AO, Steadman R, Morrisey K, Martin J, Eynstone L, Williams JD. Exposure of human renal proximal tubular cells to glucose leads to accumulation of type IV collagen and fibronectin by decreased degradation. Kidney Int. 1997;52:973–984. doi: 10.1038/ki.1997.419. [DOI] [PubMed] [Google Scholar]
- 495.Phillips AO, Steadman R, Topley N, Williams JD. Elevated D-glucose concentrations modulate TGF-beta 1 synthesis by human cultured renal proximal tubular cells. The permissive role of platelet-derived growth factor. Am J Pathol. 1995;147:362–374. [PMC free article] [PubMed] [Google Scholar]
- 496.Phillips AO, Topley N, Steadman R, Morrisey K, Williams JD. Induction of TGF-beta 1 synthesis in D-glucose primed human proximal tubular cells by IL-1 beta and TNF alpha. Kidney Int. 1996;50:1546–1554. doi: 10.1038/ki.1996.470. [DOI] [PubMed] [Google Scholar]
- 497.Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, Hess JF. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13:18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 498.Picard N, Baum O, Vogetseder A, Kaissling B, Le HM. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130:141–155. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Pieper GM. Review of alterations in endothelial nitric oxide production in diabetes: Protective role of arginine on endothelial dysfunction. Hypertension. 1998;31:1047–1060. doi: 10.1161/01.hyp.31.5.1047. [DOI] [PubMed] [Google Scholar]
- 500.Pieper GM. Enhanced, unaltered and impaired nitric oxide-mediated endothelium-dependent relaxation in experimental diabetes mellitus: Importance of disease duration. Diabetologia. 1999;42:204–213. doi: 10.1007/s001250051140. [DOI] [PubMed] [Google Scholar]
- 501.Pollock CA, Lawrence JR, Field MJ. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am J Physiol. 1991;260:F946–F952. doi: 10.1152/ajprenal.1991.260.6.F946. [DOI] [PubMed] [Google Scholar]
- 502.Posch K, Simecek S, Wascher TC, Jurgens G, Baumgartner-Parzer S, Kostner GM, Graier WF. Glycated low-density lipoprotein attenuates shear stress-induced nitric oxide synthesis by inhibition of shear stress-activated L-arginine uptake in endothelial cells. Diabetes. 1999;48:1331–1337. doi: 10.2337/diabetes.48.6.1331. [DOI] [PubMed] [Google Scholar]
- 503.Prabhakar SS. Tetrahydrobiopterin reverses the inhibition of nitric oxide by high glucose in cultured murine mesangial cells. Am J Physiol Renal Physiol. 2001;281:F179–F188. doi: 10.1152/ajprenal.2001.281.1.F179. [DOI] [PubMed] [Google Scholar]
- 504.Price DA, De’Oliveira JM, Fisher ND, Williams GH, Hollenberg NK. The state and responsiveness of the renin-angiotensin-aldosterone system in patients with type II diabetes mellitus. Am J Hypertens. 1999;12:348–355. [PubMed] [Google Scholar]
- 505.Price DA, Porter LE, Gordon M, Fisher ND, De’Oliveira JM, Laffel LM, Passan DR, Williams GH, Hollenberg NK. The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol. 1999;10:2382–2391. doi: 10.1681/ASN.V10112382. [DOI] [PubMed] [Google Scholar]
- 506.Price GJ, Berka JL, Werther GA, Bach LA. Cell-specific regulation of mRNAs for IGF-I and IGF-binding proteins-4 and -5 in streptozotocin-diabetic rat kidney. J Mol Endocrinol. 1997;18:5–14. doi: 10.1677/jme.0.0180005. [DOI] [PubMed] [Google Scholar]
- 507.Qi W, Chen X, Zhang Y, Holian J, Mreich E, Gilbert RE, Kelly DJ, Pollock CA. High glucose induces macrophage inflammatory protein-3 alpha in renal proximal tubule cells via a transforming growth factor-beta 1 dependent mechanism. Nephrol Dial Transplant. 2007;22:3147–3153. doi: 10.1093/ndt/gfm365. [DOI] [PubMed] [Google Scholar]
- 508.Raats CJ, Van Den Born J, Berden JH. Glomerular heparan sulfate alterations: Mechanisms and relevance for proteinuria. Kidney Int. 2000;57:385–400. doi: 10.1046/j.1523-1755.2000.00858.x. [DOI] [PubMed] [Google Scholar]
- 509.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–3434. doi: 10.2337/diabetes.54.12.3427. [DOI] [PubMed] [Google Scholar]
- 510.Rasch R. Tubular lesions in streptozotocin-diabetic rats. Diabetologia. 1984;27:32–37. doi: 10.1007/BF00253498. [DOI] [PubMed] [Google Scholar]
- 511.Rasch R, Dorup J. Quantitative morphology of the rat kidney during diabetes mellitus and insulin treatment. Diabetologia. 1997;40:802–809. doi: 10.1007/s001250050752. [DOI] [PubMed] [Google Scholar]
- 512.Rasch R, Norgaard JO. Renal enlargement: Comparative autoradiographic studies of 3H-thymidine uptake in diabetic and uninephrectomized rats. Diabetologia. 1983;25:280–287. doi: 10.1007/BF00279944. [DOI] [PubMed] [Google Scholar]
- 513.Rastaldi MP, Ferrario F, Giardino L, Dell’Antonio G, Grillo C, Grillo P, Strutz F, Muller GA, Colasanti G, D’Amico G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62:137–146. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- 514.Rebsomen L, Khammar A, Raccah D, Tsimaratos M. C-Peptide effects on renal physiology and diabetes. Exp Diabetes Res. 2008;2008:281–536. doi: 10.1155/2008/281536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Reddy MA, Li SL, Sahar S, Kim YS, Xu ZG, Lanting L, Natarajan R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J Biol Chem. 2006;281:13685–13693. doi: 10.1074/jbc.M511425200. [DOI] [PubMed] [Google Scholar]
- 516.Redon J, Cifkova R, Laurent S, Nilsson P, Narkiewicz K, Erdine S, Mancia G. Mechanisms of hypertension in the cardiometabolic syndrome. J Hypertens. 2009;27:441–451. doi: 10.1097/HJH.0b013e32831e13e5. [DOI] [PubMed] [Google Scholar]
- 517.Regele HM, Fillipovic E, Langer B, Poczewki H, Kraxberger I, Bittner RE, Kerjaschki D. Glomerular expression of dystroglycans is reduced in minimal change nephrosis but not in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2000;11:403–412. doi: 10.1681/ASN.V113403. [DOI] [PubMed] [Google Scholar]
- 518.Ren JL, Pan JS, Lu YP, Sun P, Han J. Inflammatory signaling and cellular senescence. Cell Signal. 2009;21:378–383. doi: 10.1016/j.cellsig.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Ries M, Basseau F, Tyndal B, Jones R, Deminiere C, Catargi B, Combe C, Moonen CW, Grenier N. Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level-dependent. J Magn Reson Imaging. 2003;17:104–113. doi: 10.1002/jmri.10224. [DOI] [PubMed] [Google Scholar]
- 520.Rincon-Choles H, Vasylyeva TL, Pergola PE, Bhandari B, Bhandari K, Zhang JH, Wang W, Gorin Y, Barnes JL, Abboud HE. ZO-1 expression and phosphorylation in diabetic nephropathy. Diabetes. 2006;55:894–900. doi: 10.2337/diabetes.55.04.06.db05-0355. [DOI] [PubMed] [Google Scholar]
- 521.Riser BL, Cortes P. Connective tissue growth factor and its regulation: A new element in diabetic glomerulosclerosis. Ren Fail. 2001;23:459–470. doi: 10.1081/jdi-100104729. [DOI] [PubMed] [Google Scholar]
- 522.Riser BL, Cortes P, Denichilo M, Deshmukh PV, Chahal PS, Mohammed AK, Yee J, Kahkonen D. Urinary CCN2 (CTGF) as a possible predictor of diabetic nephropathy: Preliminary report. Kidney Int. 2003;64:451–458. doi: 10.1046/j.1523-1755.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- 523.Riser BL, Cortes P, Yee J. Modelling the effects of vascular stress in mesangial cells. Curr Opin Nephrol Hypertens. 2000;9:43–47. doi: 10.1097/00041552-200001000-00008. [DOI] [PubMed] [Google Scholar]
- 524.Riser BL, Cortes P, Zhao X, Bernstein J, Dumler F, Narins RG. Intraglomerular pressure and mesangial stretching stimulate extra-cellular matrix formation in the rat. J Clin Invest. 1992;90:1932–1943. doi: 10.1172/JCI116071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Riser BL, Denichilo M, Cortes P, Baker C, Grondin JM, Yee J, Narins RG. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol. 2000;11:25–38. doi: 10.1681/ASN.V11125. [DOI] [PubMed] [Google Scholar]
- 526.Ritz E. Metabolic syndrome and kidney disease. Blood Purif. 2008;26:59–62. doi: 10.1159/000110566. [DOI] [PubMed] [Google Scholar]
- 527.Rizkalla B, Forbes JM, Cao Z, Boner G, Cooper ME. Temporal renal expression of angiogenic growth factors and their receptors in experimental diabetes: Role of the renin-angiotensin system. J Hypertens. 2005;23:153–164. doi: 10.1097/00004872-200501000-00026. [DOI] [PubMed] [Google Scholar]
- 528.Robben JH, Fenton RA, Vargas SL, Schweer H, Peti-Peterdi J, Deen PM, Milligan G. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009;76:1258–1267. doi: 10.1038/ki.2009.360. [DOI] [PubMed] [Google Scholar]
- 529.Roestenberg P, van Nieuwenhoven FA, Joles JA, Trischberger C, Martens PP, Oliver N, Aten J, Hoppener JW, Goldschmeding R. Temporal expression profile and distribution pattern indicate a role of connective tissue growth factor (CTGF/CCN-2) in diabetic nephropathy in mice. Am J Physiol Renal Physiol. 2006;290:F1344–F1354. doi: 10.1152/ajprenal.00174.2005. [DOI] [PubMed] [Google Scholar]
- 530.Romero M, Ortega A, Izquierdo A, Lopez-Luna P, Bosch RJ. Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-beta(1) and p27(Kip1): Implications for diabetic nephropathy. Nephrol Dial Transplant. 2010;25:2447–2457. doi: 10.1093/ndt/gfq104. [DOI] [PubMed] [Google Scholar]
- 531.Rosenberger C, Khamaisi M, Abassi Z, Shilo V, Weksler-Zangen S, Goldfarb M, Shina A, Zibertrest F, Eckardt KU, Rosen S, Heyman SN. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008;73:34–42. doi: 10.1038/sj.ki.5002567. [DOI] [PubMed] [Google Scholar]
- 532.Ruan X, Zheng F, Guan Y. PPARs and the kidney in metabolic syndrome. Am J Physiol Renal Physiol. 2008;294:F1032–F1047. doi: 10.1152/ajprenal.00152.2007. [DOI] [PubMed] [Google Scholar]
- 533.Rudberg S, Persson B, Dahlquist G. Increased glomerular filtration rate as a predictor of diabetic nephropathy—an 8-year prospective study. Kidney Int. 1992;41:822–828. doi: 10.1038/ki.1992.126. [DOI] [PubMed] [Google Scholar]
- 534.Russo LM, Sandoval RM, Campos SB, Molitoris BA, Comper WD, Brown D. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J Am Soc Nephrol. 2009;20:489–494. doi: 10.1681/ASN.2008050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Ruster C, Bondeva T, Franke S, Forster M, Wolf G. Advanced glycation end-products induce cell cycle arrest and hypertrophy in podocytes. Nephrol Dial Transplant. 2008;23:2179–2191. doi: 10.1093/ndt/gfn085. [DOI] [PubMed] [Google Scholar]
- 536.Ruster C, Bondeva T, Franke S, Tanaka N, Yamamoto H, Wolf G. Angiotensin II upregulates RAGE expression on podocytes: Role of AT2 receptors. Am J Nephrol. 2009;29:538–550. doi: 10.1159/000191467. [DOI] [PubMed] [Google Scholar]
- 537.Ruster C, Wolf G. The role of chemokines and chemokine receptors in diabetic nephropathy. Front Biosci. 2008;13:944–955. doi: 10.2741/2734. [DOI] [PubMed] [Google Scholar]
- 538.Saad S, Stevens VA, Wassef L, Poronnik P, Kelly DJ, Gilbert RE, Pollock CA. High glucose transactivates the EGF receptor and up-regulates serum glucocorticoid kinase in the proximal tubule. Kidney Int. 2005;68:985–997. doi: 10.1111/j.1523-1755.2005.00492.x. [DOI] [PubMed] [Google Scholar]
- 539.Sabbatini M, Sansone G, Uccello F, Giliberti A, Conte G, Andreucci VE. Early glycosylation products induce glomerular hyperfiltration in normal rats. Kidney Int. 1992;42:875–881. doi: 10.1038/ki.1992.363. [DOI] [PubMed] [Google Scholar]
- 540.Sackmann H, Tran-Van T, Tack I, Hanaire-Broutin H, Tauber JP, Ader JL. Renal functional reserve in IDDM patients. Diabetologia. 1998;41:86–93. doi: 10.1007/s001250050871. [DOI] [PubMed] [Google Scholar]
- 541.Sagami I, Daff S, Shimizu T. Intra-subunit and inter-subunit electron transfer in neuronal nitric-oxide synthase: Effect of calmodulin on het-erodimer catalysis. J Biol Chem. 2001;276:30036–30042. doi: 10.1074/jbc.M104123200. [DOI] [PubMed] [Google Scholar]
- 542.Sakharova OV, Taal MW, Brenner BM. Pathogenesis of diabetic nephropathy: Focus on transforming growth factor-beta and connective tissue growth factor. Curr Opin Nephrol Hypertens. 2001;10:727–738. doi: 10.1097/00041552-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 543.Santer R, Calado J. Familial renal glucosuria and SGLT2: From a mendelian trait to a therapeutic target. Clin J Am Soc Nephrol. 2010;5:133–141. doi: 10.2215/CJN.04010609. [DOI] [PubMed] [Google Scholar]
- 544.Sarafidis PA, Bakris GL. The antinatriuretic effect of insulin: An unappreciated mechanism for hypertension associated with insulin resistance? Am J Nephrol. 2007;27:44–54. doi: 10.1159/000098955. [DOI] [PubMed] [Google Scholar]
- 545.Sassy-Prigent C, Heudes D, Mandet C, Belair MF, Michel O, Perdereau B, Bariety J, Bruneval P. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49:466–475. doi: 10.2337/diabetes.49.3.466. [DOI] [PubMed] [Google Scholar]
- 546.Satchell SC, Tooke JE. What is the mechanism of microalbuminuria in diabetes: A role for the glomerular endothelium? Diabetologia. 2008;51:714–725. doi: 10.1007/s00125-008-0961-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Satoh M, Fujimoto S, Haruna Y, Arakawa S, Horike H, Komai N, Sasaki T, Tsujioka K, Makino H, Kashihara N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2005;288:F1144–F1152. doi: 10.1152/ajprenal.00221.2004. [DOI] [PubMed] [Google Scholar]
- 548.Satriano J, Mansoury H, Deng A, Sharma K, Vallon V, Blantz RC, Thomson SC. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am J Physiol Cell Physiol. 2010;299:C374–C380. doi: 10.1152/ajpcell.00096.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Satriano J, Vallon V. Primary kidney growth and its consequences at the onset of diabetes mellitus. Amino Acids. 2006;31:1–9. doi: 10.1007/s00726-006-0326-x. [DOI] [PubMed] [Google Scholar]
- 550.Scandling JD, Myers BD. Glomerular size-selectivity and microalbuminuria in early diabetic glomerular disease. Kidney Int. 1992;41:840–846. doi: 10.1038/ki.1992.129. [DOI] [PubMed] [Google Scholar]
- 551.Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin Sci (Lond) 2005;109:143–159. doi: 10.1042/CS20050025. [DOI] [PubMed] [Google Scholar]
- 552.Schambelan M, Blake S, Sraer J, Bens M, Nivez MP, Wahbe F. Increased prostaglandin production by glomeruli isolated from rats with streptozotocin-induced diabetes mellitus. J Clin Invest. 1985;75:404–412. doi: 10.1172/JCI111714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Schelling JR, Abboud HE, Nicholas SB, Pahl MV, Sedor JR, Adler SG, Arar NH, Bowden DW, Elston RC, Freedman BI, Goddard KA, Guo X, Hanson RL, Ipp E, Iyengar SK, Jun G, Kao WH, Kasinath BS, Kimmel PL, Klag MJ, Knowler WC, Nelson RG, Parekh RS, Quade SR, Rich SS, Saad MF, Scavini M, Smith MW, Taylor K, Winkler CA, Zager PG, Shah VO. Genome-wide scan for estimated glomerular filtration rate in multi-ethnic diabetic populations: The Family Investigation of Nephropathy and Diabetes (FIND) Diabetes. 2008;57:235–243. doi: 10.2337/db07-0313. [DOI] [PubMed] [Google Scholar]
- 554.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten DP, Mundel P, Bottinger EP. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S13–S18. doi: 10.1046/j.1523-1755.2000.07703.x. [DOI] [PubMed] [Google Scholar]
- 556.Schmidt AM, Hori O, Cao R, Yan SD, Brett J, Wautier JL, Ogawa S, Kuwabara K, Matsumoto M, Stern D. RAGE: A novel cellular receptor for advanced glycation end products. Diabetes. 1996;45(Suppl 3):S77–S80. doi: 10.2337/diab.45.3.s77. [DOI] [PubMed] [Google Scholar]
- 557.Schmidt AM, Yan SD, Wautier JL, Stern D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999;84:489–497. doi: 10.1161/01.res.84.5.489. [DOI] [PubMed] [Google Scholar]
- 558.Schnackenberg CG, Wilcox CS. The SOD mimetic tempol restores vasodilation in afferent arterioles of experimental diabetes. Kidney Int. 2001;59:1859–1864. doi: 10.1046/j.1523-1755.2001.0590051859.x. [DOI] [PubMed] [Google Scholar]
- 559.Schneider M, Van GK, Fraisl P, Kiss J, Aragones J, Mazzone M, Mairbaurl H, De BK, Jeoung NH, Mollenhauer M, Georgiadou M, Bishop T, Roncal C, Sutherland A, Jordan B, Gallez B, Weitz J, Harris RA, Maxwell P, Baes M, Ratcliffe P, Carmeliet P. Loss or silencing of the PHD1 prolyl hydroxylase protects livers of mice against ischemia/reperfusion injury. Gastroenterology. 2010;138:1143–1154. doi: 10.1053/j.gastro.2009.09.057. [DOI] [PubMed] [Google Scholar]
- 560.Schnermann J, Briggs J. Concentration-dependent sodium chloride transport as the signal in feedback control of glomerular filtration rate. Kidney Int Suppl. 1982;12:S82–S89. [PubMed] [Google Scholar]
- 561.Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun. 2005;338:617–626. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 562.Scholey JW, Meyer TW. Control of glomerular hypertension by insulin administration in diabetic rats. J Clin Invest. 1989;83:1384–1389. doi: 10.1172/JCI114026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Scholl-Burgi S, Santer R, Ehrich JH. Long-term outcome of renal glu-cosuria type 0: The original patient and his natural history. Nephrol Dial Transplant. 2004;19:2394–2396. doi: 10.1093/ndt/gfh366. [DOI] [PubMed] [Google Scholar]
- 564.Scholz H, Kurtz A. Role of protein kinase C in renal vasoconstriction caused by angiotensin II. Am J Physiol. 1990;259:C421–C426. doi: 10.1152/ajpcell.1990.259.3.C421. [DOI] [PubMed] [Google Scholar]
- 565.Schoonmaker GC, Fallet RW, Carmines PK. Superoxide anion curbs nitric oxide modulation of afferent arteriolar ANG II responsiveness in diabetes mellitus. Am J Physiol Renal Physiol. 2000;278:F302–F309. doi: 10.1152/ajprenal.2000.278.2.F302. [DOI] [PubMed] [Google Scholar]
- 566.Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem. 2004;279:30369–30374. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- 567.Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320:1161–1165. doi: 10.1056/NEJM198905043201801. [DOI] [PubMed] [Google Scholar]
- 568.Segev Y, Landau D, Rasch R, Flyvbjerg A, Phillip M. Growth hormone receptor antagonism prevents early renal changes in nonobese diabetic mice. J Am Soc Nephrol. 1999;10:2374–2381. doi: 10.1681/ASN.V10112374. [DOI] [PubMed] [Google Scholar]
- 569.Senthil D, Choudhury GG, McLaurin C, Kasinath BS. Vascular endothelial growth factor induces protein synthesis in renal epithelial cells: A potential role in diabetic nephropathy. Kidney Int. 2003;64:468–479. doi: 10.1046/j.1523-1755.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 570.Seyer-Hansen K. Renal hypertrophy in experimental diabetes: Some functional aspects. J Diabet Complications. 1987;1:7–10. doi: 10.1016/s0891-6632(87)80018-1. [DOI] [PubMed] [Google Scholar]
- 571.Seyer-Hansen K, Hansen J, Gundersen HJ. Renal hypertrophy in experimental diabetes. A morphometric study. Diabetologia. 1980;18:501–505. doi: 10.1007/BF00261707. [DOI] [PubMed] [Google Scholar]
- 572.Shah SV, Baliga R, Rajapurkar M, Fonseca VA. Oxidants in chronic kidney disease. J Am Soc Nephrol. 2007;18:16–28. doi: 10.1681/ASN.2006050500. [DOI] [PubMed] [Google Scholar]
- 573.Shankland SJ, Wolf G. Cell cycle regulatory proteins in renal disease: Role in hypertrophy, proliferation, and apoptosis. Am J Physiol Renal Physiol. 2000;278:F515–F529. doi: 10.1152/ajprenal.2000.278.4.F515. [DOI] [PubMed] [Google Scholar]
- 574.Shantz LM. Transcriptional and translational control of ornithine decarboxylase during Ras transformation. Biochem J. 2004;377:257–264. doi: 10.1042/BJ20030778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 576.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–F1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- 577.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44:1139–1146. doi: 10.2337/diab.44.10.1139. [DOI] [PubMed] [Google Scholar]
- 579.Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101:1142–1147. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Silbiger S, Crowley S, Shan Z, Brownlee M, Satriano J, Schlondorff D. Nonenzymatic glycation of mesangial matrix and prolonged exposure of mesangial matrix to elevated glucose reduces collagen synthesis and proteoglycan charge. Kidney Int. 1993;43:853–864. doi: 10.1038/ki.1993.120. [DOI] [PubMed] [Google Scholar]
- 581.Singh DK, Winocour P, Farrington K. Mechanisms of disease: The hypoxic tubular hypothesis of diabetic nephropathy. Nat Clin Pract Nephrol. 2008;4:216–226. doi: 10.1038/ncpneph0757. [DOI] [PubMed] [Google Scholar]
- 582.Singh P, Deng A, Weir MR, Blantz RC. The balance of angiotensin II and nitric oxide in kidney diseases. Curr Opin Nephrol Hypertens. 2008;17:51–56. doi: 10.1097/MNH.0b013e3282f29a8b. [DOI] [PubMed] [Google Scholar]
- 583.Sitte N, Merker K, von ZT, Grune T. Protein oxidation and degradation during proliferative senescence of human MRC-5 fibroblasts. Free Radic Biol Med. 2000;28:701–708. doi: 10.1016/s0891-5849(99)00279-8. [DOI] [PubMed] [Google Scholar]
- 584.Skott P, Hother-Nielsen O, Bruun NE, Giese J, Nielsen MD, Beck-Nielsen H, Parving HH. Effects of insulin on kidney function and sodium excretion in healthy subjects. Diabetologia. 1989;32:694–699. doi: 10.1007/BF00274259. [DOI] [PubMed] [Google Scholar]
- 585.Skott P, Vaag A, Bruun NE, Hother-Nielsen O, Gall MA, Beck-Nielsen H, Parving HH. Effect of insulin on renal sodium handling in hyperinsulinaemic type 2 (non-insulin-dependent) diabetic patients with peripheral insulin resistance. Diabetologia. 1991;34:275–281. doi: 10.1007/BF00405088. [DOI] [PubMed] [Google Scholar]
- 586.Skrha J, Perusicova J, Pont’uch P, Oksa A. Glycosaminoglycan sulodexide decreases albuminuria in diabetic patients. Diabetes Res Clin Pract. 1997;38:25–31. doi: 10.1016/s0168-8227(97)00076-4. [DOI] [PubMed] [Google Scholar]
- 587.Sochor M, McLean P. Changes in ornithine decarboxylase in kidney in experimental diabetes. Correlation with severity of diabetes and effects of unilateral nephrectomy. Enzyme. 1980;25:289–296. doi: 10.1159/000459269. [DOI] [PubMed] [Google Scholar]
- 588.Sommer M, Wolf G. Rosiglitazone increases PPARgamma in renal tubular epithelial cells and protects against damage by hydrogen peroxide. Am J Nephrol. 2007;27:425–434. doi: 10.1159/000105125. [DOI] [PubMed] [Google Scholar]
- 589.Song J, Hu X, Riazi S, Tiwari S, Wade JB, Ecelbarger CA. Regulation of blood pressure, the epithelial sodium channel (ENaC), and other key renal sodium transporters by chronic insulin infusion in rats. Am J Physiol Renal Physiol. 2006;290:F1055–F1064. doi: 10.1152/ajprenal.00108.2005. [DOI] [PubMed] [Google Scholar]
- 590.Song J, Knepper MA, Verbalis JG, Ecelbarger CA. Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am J Physiol Renal Physiol. 2003;285:F1125–F1137. doi: 10.1152/ajprenal.00143.2003. [DOI] [PubMed] [Google Scholar]
- 591.Soulis T, Thallas V, Youssef S, Gilbert RE, McWilliam BG, Murray-McIntosh RP, Cooper ME. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia. 1997;40:619–628. doi: 10.1007/s001250050725. [DOI] [PubMed] [Google Scholar]
- 592.Soulis-Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Retardation by aminoguanidine of development of albuminuria, mesangial expansion, and tissue fluorescence in streptozocin-induced diabetic rat. Diabetes. 1991;40:1328–1334. doi: 10.2337/diab.40.10.1328. [DOI] [PubMed] [Google Scholar]
- 593.Sourris KC, Morley AL, Koitka A, Samuel P, Coughlan MT, Penfold SA, Thomas MC, Bierhaus A, Nawroth PP, Yamamoto H, Allen TJ, Walther T, Hussain T, Cooper ME, Forbes JM. Receptor for AGEs (RAGE) blockade may exert its renoprotective effects in patients with diabetic nephropathy via induction of the angiotensin II type 2 (AT2) receptor. Diabetologia. 2010;53:2442–2451. doi: 10.1007/s00125-010-1837-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Stackhouse S, Miller PL, Park SK, Meyer TW. Reversal of glomerular hyperfiltration and renal hypertrophy by blood glucose normalization in diabetic rats. Diabetes. 1990;39:989–995. doi: 10.2337/diab.39.8.989. [DOI] [PubMed] [Google Scholar]
- 595.Stanton BA, Kaissling B. Regulation of renal ion transport and cell growth by sodium. Am J Physiol. 1989;257:F1–F10. doi: 10.1152/ajprenal.1989.257.1.F1. [DOI] [PubMed] [Google Scholar]
- 596.Steinke JM, Sinaiko AR, Kramer MS, Suissa S, Chavers BM, Mauer M. The early natural history of nephropathy in Type 1 Diabetes: III. Predictors of 5-year urinary albumin excretion rate patterns in initially normoalbuminuric patients. Diabetes. 2005;54:2164–2171. doi: 10.2337/diabetes.54.7.2164. [DOI] [PubMed] [Google Scholar]
- 597.Stenvinkel P, Bolinder J, Alvestrand A. Effects of insulin on renal haemodynamics and the proximal and distal tubular sodium handling in healthy subjects. Diabetologia. 1992;35:1042–1048. doi: 10.1007/BF02221679. [DOI] [PubMed] [Google Scholar]
- 598.Stenvinkel P, Ottosson-Seeberger A, Alvestrand A, Bolinder J. Effect of insulin on renal sodium handling and renal haemodynamics in insulin-dependent (type 1) diabetes mellitus patients. Acta Diabetol. 1995;32:230–234. doi: 10.1007/BF00576255. [DOI] [PubMed] [Google Scholar]
- 599.Strutz F, Muller GA. Renal fibrosis and the origin of the renal fibroblast. Nephrol Dial Transplant. 2006;21:3368–3370. doi: 10.1093/ndt/gfl199. [DOI] [PubMed] [Google Scholar]
- 600.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Muller GA, Neilson EG. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–1728. doi: 10.1046/j.1523-1755.2002.00333.x. [DOI] [PubMed] [Google Scholar]
- 602.Sugimoto H, Shikata K, Matsuda M, Kushiro M, Hayashi Y, Hiragushi K, Wada J, Makino H. Increased expression of endothelial cell nitric oxide synthase (ecNOS) in afferent and glomerular endothelial cells is involved in glomerular hyperfiltration of diabetic nephropathy. Diabetologia. 1998;41:1426–1434. doi: 10.1007/s001250051088. [DOI] [PubMed] [Google Scholar]
- 603.Sung SH, Ziyadeh FN, Wang A, Pyagay PE, Kanwar YS, Chen S. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol. 2006;17:3093–3104. doi: 10.1681/ASN.2006010064. [DOI] [PubMed] [Google Scholar]
- 604.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 605.Tabatabai NM, Sharma M, Blumenthal SS, Petering DH. Enhanced expressions of sodium-glucose cotransporters in the kidneys of diabetic Zucker rats. Diabetes Res Clin Pract. 2009;83:e27–e30. doi: 10.1016/j.diabres.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin FI. Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes. 1994;43:1046–1051. doi: 10.2337/diab.43.8.1046. [DOI] [PubMed] [Google Scholar]
- 607.Takahashi H, Ichihara A, Kaneshiro Y, Inomata K, Sakoda M, Takemitsu T, Nishiyama A, Itoh H. Regression of nephropathy developed in diabetes by (Pro)renin receptor blockade. J Am Soc Nephrol. 2007;18:2054–2061. doi: 10.1681/ASN.2006080820. [DOI] [PubMed] [Google Scholar]
- 608.Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111:3229–3235. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116:774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- 610.Tamsma JT, Van Der Woude FJ, Lemkes HH. Effect of sulphated glycosaminoglycans on albuminuria in patients with overt diabetic (type 1) nephropathy. Nephrol Dial Transplant. 1996;11:182–185. [PubMed] [Google Scholar]
- 611.Tang S, Leung JC, Abe K, Chan KW, Chan LY, Chan TM, Lai KN. Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo. J Clin Invest. 2003;111:515–527. doi: 10.1172/JCI16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D’Agati VD. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 613.Thomas MC, Burns WC, Cooper ME. Tubular changes in early diabetic nephropathy. Adv Chronic Kidney Dis. 2005;12:177–186. doi: 10.1053/j.ackd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 614.Thomas MC, Forbes JM, Cooper ME. Advanced glycation end products and diabetic nephropathy. Am J Ther. 2005;12:562–572. doi: 10.1097/01.mjt.0000178769.52610.69. [DOI] [PubMed] [Google Scholar]
- 615.Thomson S, Bao D, Deng A, Vallon V. Adenosine formed by 5′-nucleotidase mediates tubuloglomerular feedback. J Clin Invest. 2000;106:289–298. doi: 10.1172/JCI8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Thomson SC, Blantz RC. Glomerulotubular balance, tubuloglomerular feedback, and salt homeostasis. J Am Soc Nephrol. 2008;19:2272–2275. doi: 10.1681/ASN.2007121326. [DOI] [PubMed] [Google Scholar]
- 617.Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest. 2001;107:217–224. doi: 10.1172/JCI10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Thomson SC, Deng A, Komine N, Hammes JS, Blantz RC, Gabbai FB. Early diabetes as a model for testing the regulation of juxtaglomerular NOS I. Am J Physiol Renal Physiol. 2004;287:F732–F738. doi: 10.1152/ajprenal.00340.2003. [DOI] [PubMed] [Google Scholar]
- 619.Thomson SC, Deng A, Wead L, Richter K, Blantz RC, Vallon V. An unexpected role for angiotensin II in the link between dietary salt and proximal reabsorption. J Clin Invest. 2006;116:1110–1116. doi: 10.1172/JCI26092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, Cooper ME, Thomas MC. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 621.Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, Cooper ME. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41:392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- 622.Tiwari S, Halagappa VK, Riazi S, Hu X, Ecelbarger CA. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J Am Soc Nephrol. 2007;18:2661–2671. doi: 10.1681/ASN.2006121410. [DOI] [PubMed] [Google Scholar]
- 623.Tiwari S, Nordquist L, Halagappa VK, Ecelbarger CA. Trafficking of ENaC subunits in response to acute insulin in mouse kidney. Am J Physiol Renal Physiol. 2007;293:F178–F185. doi: 10.1152/ajprenal.00447.2006. [DOI] [PubMed] [Google Scholar]
- 624.Tiwari S, Riazi S, Ecelbarger CA. Insulin’s impact on renal sodium transport and blood pressure in health, obesity, and diabetes. Am J Physiol Renal Physiol. 2007;293:F974–F984. doi: 10.1152/ajprenal.00149.2007. [DOI] [PubMed] [Google Scholar]
- 625.Tiwari S, Sharma N, Gill PS, Igarashi P, Kahn CR, Wade JB, Ecelbarger CM. Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc Natl Acad Sci U S A. 2008;105:6469–6474. doi: 10.1073/pnas.0711283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Tolins JP, Shultz PJ, Raij L, Brown DM, Mauer SM. Abnormal renal hemodynamic response to reduced renal perfusion pressure in diabetic rats: Role of NO. Am J Physiol. 1993;265:F886–F895. doi: 10.1152/ajprenal.1993.265.6.F886. [DOI] [PubMed] [Google Scholar]
- 627.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, Meer E, Peti-Peterdi J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. 2008;118:2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Tomiyama H, Kushiro T, Abeta H, Kurumatani H, Taguchi H, Kuga N, Saito F, Kobayashi F, Otsuka Y, Kanmatsuse K. Blood pressure response to hyperinsulinemia in salt-sensitive and salt-resistant rats. Hypertension. 1992;20:596–600. doi: 10.1161/01.hyp.20.5.596. [DOI] [PubMed] [Google Scholar]
- 629.Townley DJ, Avery BJ, Rosen B, Skarnes WC. Rapid sequence analysis of gene trap integrations to generate a resource of insertional mutations in mice. Genome Res. 1997;7:293–298. doi: 10.1101/gr.7.3.293. [DOI] [PubMed] [Google Scholar]
- 630.Trevisan R, Bruttomesso D, Vedovato M, Brocco S, Pianta A, Mazzon C, Girardi C, Jori E, Semplicini A, Tiengo A, Del PS. Enhanced responsiveness of blood pressure to sodium intake and to angiotensin II is associated with insulin resistance in IDDM patients with microalbu-minuria. Diabetes. 1998;47:1347–1353. doi: 10.2337/diab.47.8.1347. [DOI] [PubMed] [Google Scholar]
- 631.Troncoso Brindeiro CM, Fallet RW, Lane PH, Carmines PK. Potassium channel contributions to afferent arteriolar tone in normal and diabetic rat kidney. Am J Physiol Renal Physiol. 2008;295:F171–F178. doi: 10.1152/ajprenal.00563.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Trujillo A, Eggena P, Barrett J, Tuck M. Renin regulation in type II diabetes mellitus: Influence of dietary sodium. Hypertension. 1989;13:200–205. doi: 10.1161/01.hyp.13.3.200. [DOI] [PubMed] [Google Scholar]
- 633.Tsiani E, Lekas P, Fantus IG, Dlugosz J, Whiteside C. High glucose-enhanced activation of mesangial cell p38 MAPK by ET-1, ANG II, and platelet-derived growth factor. Am J Physiol Endocrinol Metab. 2002;282:E161–E169. doi: 10.1152/ajpendo.2002.282.1.E161. [DOI] [PubMed] [Google Scholar]
- 634.Tuck ML, Sambhi MP, Levin L. Hyporeninemic hypoaldosteronism in diabetes mellitus. Studies of the autonomic nervous system’s control of renin release. Diabetes. 1979;28:237–241. doi: 10.2337/diab.28.3.237. [DOI] [PubMed] [Google Scholar]
- 635.Tucker BJ, Anderson CM, Thies RS, Collins RC, Blantz RC. Glomerular hemodynamic alterations during acute hyperinsulinemia in normal and diabetic rats. Kidney Int. 1992;42:1160–1168. doi: 10.1038/ki.1992.400. [DOI] [PubMed] [Google Scholar]
- 636.Tucker BJ, Mendonca MM, Blantz RC. Contrasting effects of acute insulin infusion on renal function in awake nondiabetic and diabetic rats. J Am Soc Nephrol. 1993;3:1686–1693. doi: 10.1681/ASN.V3101686. [DOI] [PubMed] [Google Scholar]
- 637.Tucker BJ, Rasch R, Blantz RC. Glomerular filtration and tubular reabsorption of albumin in preproteinuric and proteinuric diabetic rats. J Clin Invest. 1993;92:686–694. doi: 10.1172/JCI116638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care. 2005;28:2686–2690. doi: 10.2337/diacare.28.11.2686. [DOI] [PubMed] [Google Scholar]
- 639.Tuttle KR, Bruton JL. Effect of insulin therapy on renal hemody-namic response to amino acids and renal hypertrophy in non-insulin-dependent diabetes. Kidney Int. 1992;42:167–173. doi: 10.1038/ki.1992.274. [DOI] [PubMed] [Google Scholar]
- 640.Tuttle KR, Bruton JL, Perusek MC, Lancaster JL, Kopp DT, DeFronzo RA. Effect of strict glycemic control on renal hemodynamic response to amino acids and renal enlargement in insulin-dependent diabetes mellitus. N Engl J Med. 1991;324:1626–1632. doi: 10.1056/NEJM199106063242304. [DOI] [PubMed] [Google Scholar]
- 641.Tuttle KR, McGill JB, Haney DJ, Lin TE, Anderson PW. Kidney outcomes in long-term studies of ruboxistaurin for diabetic eye disease. Clin J Am Soc Nephrol. 2007;2:631–636. doi: 10.2215/CJN.00840207. [DOI] [PubMed] [Google Scholar]
- 642.Ueta K, Ishihara T, Matsumoto Y, Oku A, Nawano M, Fujita T, Saito A, Arakawa K. Long-term treatment with the Na+-glucose cotransporter inhibitor T-1095 causes sustained improvement in hyperglycemia and prevents diabetic neuropathy in Goto-Kakizaki Rats. Life Sci. 2005;76:2655–2668. doi: 10.1016/j.lfs.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 643.Uriu K, Kaizu K, Hashimoto O, Komine N, Etoh S. Acute and chronic effects of thromboxane A2 inhibition on the renal hemody-namics in streptozotocin-induced diabetic rats. Kidney Int. 1994;45:794–802. doi: 10.1038/ki.1994.105. [DOI] [PubMed] [Google Scholar]
- 644.US Renal Data System. USRDS 2008 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2008. [Google Scholar]
- 645.Vallon V, Albinus M, Blach D. Effect of KATP channel blocker U37883A on renal function in experimental diabetes mellitus in rats. J Pharmacol Exp Ther. 1998;286:1215–1221. [PubMed] [Google Scholar]
- 646.Vallon V, Blantz R, Thomson S. The salt paradox and its possible implications in managing hypertensive diabetic patients. Curr Hypertens Rep. 2005;7:141–147. doi: 10.1007/s11906-005-0089-x. [DOI] [PubMed] [Google Scholar]
- 647.Vallon V, Blantz RC, Thomson S. Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am J Physiol. 1995;269:F876–F883. doi: 10.1152/ajprenal.1995.269.6.F876. [DOI] [PubMed] [Google Scholar]
- 648.Vallon V, Blantz RC, Thomson S. Glomerular hyperfiltration and the salt paradox in early type 1 diabetes mellitus: A tubulo-centric view. J Am Soc Nephrol. 2003;14:530–537. doi: 10.1097/01.asn.0000051700.07403.27. [DOI] [PubMed] [Google Scholar]
- 649.Vallon V, Grahammer F, Richter K, Bleich M, Lang F, Barhanin J, Volkl H, Warth R. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J Am Soc Nephrol. 2001;12:2003–2011. doi: 10.1681/ASN.V12102003. [DOI] [PubMed] [Google Scholar]
- 650.Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K, Lang F. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci U S A. 2005;102:17864–17869. doi: 10.1073/pnas.0505860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Vallon V, Huang DY, Deng A, Richter K, Blantz RC, Thomson S. Salt-sensitivity of proximal reabsorption alters macula densa salt and explains the paradoxical effect of dietary salt on glomerular filtration rate in diabetes mellitus. J Am Soc Nephrol. 2002;13:1865–1871. doi: 10.1097/01.asn.0000016441.41118.57. [DOI] [PubMed] [Google Scholar]
- 652.Vallon V, Kirschenmann D, Wead LM, Lortie MJ, Satriano J, Blantz RC, Thomson SC. Effect of chronic salt loading on kidney function in early and established diabetes mellitus in rats. J Lab Clin Med. 1997;130:76–82. doi: 10.1016/s0022-2143(97)90061-5. [DOI] [PubMed] [Google Scholar]
- 653.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev. 2006;86:901–940. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 654.Vallon V, Osswald H. Dipyridamole prevents diabetes-induced alterations of kidney function in rats. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:217–222. doi: 10.1007/BF00169840. [DOI] [PubMed] [Google Scholar]
- 655.Vallon V, Osswald H, Blantz RC, Thomson S. Potential role of luminal potassium in tubuloglomerular feedback. J Am Soc Nephrol. 1997;8:1831–1837. doi: 10.1681/ASN.V8121831. [DOI] [PubMed] [Google Scholar]
- 656.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22:104–112. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–2576. doi: 10.1681/ASN.V10122569. [DOI] [PubMed] [Google Scholar]
- 658.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol. 2009;297:F704–F712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Vallon V, Schroth J, Satriano J, Blantz RC, Thomson SC, Rieg T. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiol. 2009;111:30–38. doi: 10.1159/000208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Vallon V, Schwark JR, Richter K, Hropot M. Role of Na(+)/H(+) exchanger NHE3 in nephron function: Micropuncture studies with S3226, an inhibitor of NHE3. Am J Physiol Renal Physiol. 2000;278:F375–F379. doi: 10.1152/ajprenal.2000.278.3.F375. [DOI] [PubMed] [Google Scholar]
- 661.Vallon V, Sharma K. Sodium-glucose transport: Role in diabetes mellitus and potential clinical implications. Curr Opin Nephrol Hypertens. 2010;19:425–431. doi: 10.1097/MNH.0b013e32833bec06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Vallon V, Thomson S. Inhibition of local nitric oxide synthase increases homeostatic efficiency of tubuloglomerular feedback. Am J Physiol. 1995;269:F892–F899. doi: 10.1152/ajprenal.1995.269.6.F892. [DOI] [PubMed] [Google Scholar]
- 663.Vallon V, Traynor T, Barajas L, Huang YG, Briggs JP, Schnermann J. Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J Am Soc Nephrol. 2001;12:1599–1606. doi: 10.1681/ASN.V1281599. [DOI] [PubMed] [Google Scholar]
- 664.Vallon V, Wead LM, Blantz RC. Renal hemodynamics and plasma and kidney angiotensin II in established diabetes mellitus in rats: Effect of sodium and salt restriction. J Am Soc Nephrol. 1995;5:1761–1767. doi: 10.1681/ASN.V5101761. [DOI] [PubMed] [Google Scholar]
- 665.Vallon V, Wyatt AW, Klingel K, Huang DY, Hussain A, Berchtold S, Friedrich B, Grahammer F, Belaiba RS, Gorlach A, Wulff P, Daut J, Dalton ND, Ross J, Jr, Flogel U, Schrader J, Osswald H, Kandolf R, Kuhl D, Lang F. SGK1-dependent cardiac CTGF formation and fibrosis following DOCA treatment. J Mol Med. 2006;84:396–404. doi: 10.1007/s00109-005-0027-z. [DOI] [PubMed] [Google Scholar]
- 666.Van Den Born J, Pisa B, Bakker MA, Celie JW, Straatman C, Thomas S, Viberti GC, Kjellen L, Berden JH. No change in glomerular heparan sulfate structure in early human and experimental diabetic nephropathy. J Biol Chem. 2006;281:29606–29613. doi: 10.1074/jbc.M601552200. [DOI] [PubMed] [Google Scholar]
- 667.Van Den Heuvel LP, Assink K, Willemsen M, Monnens L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2) Hum Genet. 2002;111:544–547. doi: 10.1007/s00439-002-0820-5. [DOI] [PubMed] [Google Scholar]
- 668.Vargas SL, Toma I, Kang JJ, Meer EJ, Peti-Peterdi J. Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J Am Soc Nephrol. 2009;20:1002–1011. doi: 10.1681/ASN.2008070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Vasylyeva TL, Ferry RJ., Jr Novel roles of the IGF-IGFBP axis in etiopathophysiology of diabetic nephropathy. Diabetes Res Clin Pract. 2007;76:177–186. doi: 10.1016/j.diabres.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Veelken R, Hilgers KF, Hartner A, Haas A, Bohmer KP, Sterzel RB. Nitric oxide synthase isoforms and glomerular hyperfiltration in early diabetic nephropathy. J Am Soc Nephrol. 2000;11:71–79. doi: 10.1681/ASN.V11171. [DOI] [PubMed] [Google Scholar]
- 672.Velasquez MT, Kimmel PL, Michaelis OE. Animal models of spontaneous diabetic kidney disease. FASEB J. 1990;4:2850–2859. doi: 10.1096/fasebj.4.11.2199283. [DOI] [PubMed] [Google Scholar]
- 673.Verbeke P, Perichon M, Friguet B, Bakala H. Inhibition of nitric oxide synthase activity by early and advanced glycation end products in cultured rabbit proximal tubular epithelial cells. Biochim Biophys Acta. 2000;1502:481–494. doi: 10.1016/s0925-4439(00)00071-5. [DOI] [PubMed] [Google Scholar]
- 674.Vervoort G, Veldman B, Berden JH, Smits P, Wetzels JF. Glomerular hyperfiltration in type 1 diabetes mellitus results from primary changes in proximal tubular sodium handling without changes in volume expansion. Eur J Clin Invest. 2005;35:330–336. doi: 10.1111/j.1365-2362.2005.01497.x. [DOI] [PubMed] [Google Scholar]
- 675.Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, Villaggio B, Gianiorio F, Tosetti F, Weiss U, Traverso P, Mji M, Deferrari G, Garibotto G. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2008;295:F1563–F1573. doi: 10.1152/ajprenal.90302.2008. [DOI] [PubMed] [Google Scholar]
- 676.Vestri S, Okamoto MM, De Freitas HS, Aparecida Dos SR, Nunes MT, Morimatsu M, Heimann JC, Machado UF. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182:105–112. doi: 10.1007/s00232-001-0036-y. [DOI] [PubMed] [Google Scholar]
- 677.Viberti GC, Benigni A, Bognetti E, Remuzzi G, Wiseman MJ. Glomerular hyperfiltration and urinary prostaglandins in type 1 diabetes mellitus. Diabet Med. 1989;6:219–223. doi: 10.1111/j.1464-5491.1989.tb01150.x. [DOI] [PubMed] [Google Scholar]
- 678.Vidotti DB, Arnoni CP, Maquigussa E, Boim MA. Effect of long-term type 1 diabetes on renal sodium and water transporters in rats. Am J Nephrol. 2008;28:107–114. doi: 10.1159/000109967. [DOI] [PubMed] [Google Scholar]
- 679.Vitalone MJ, O’Connell PJ, Jimenez-Vera E, Yuksel A, Wavamunno M, Fung CL, Chapman JR, Nankivell BJ. Epithelial-to-mesenchymal transition in early transplant tubulointerstitial damage. J Am Soc Nephrol. 2008;19:1571–1583. doi: 10.1681/ASN.2007050580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Vora JP, Dolben J, Williams JD, Peters JR, Owens DR. Impact of initial treatment on renal function in newly-diagnosed type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1993;36:734–740. doi: 10.1007/BF00401144. [DOI] [PubMed] [Google Scholar]
- 681.Vora JP, Oyama TT, Thompson MM, Anderson S. Interactions of the kallikrein-kinin and renin-angiotensin systems in experimental diabetes. Diabetes. 1997;46:107–112. doi: 10.2337/diab.46.1.107. [DOI] [PubMed] [Google Scholar]
- 682.Vukovich TC, Schernthaner G. Decreased protein C levels in patients with insulin-dependent type I diabetes mellitus. Diabetes. 1986;35:617–619. doi: 10.2337/diab.35.5.617. [DOI] [PubMed] [Google Scholar]
- 683.Wada T, Pippin JW, Terada Y, Shankland SJ. The cyclin-dependent kinase inhibitor p21 is required for TGF-beta1-induced podocyte apoptosis. Kidney Int. 2005;68:1618–1629. doi: 10.1111/j.1523-1755.2005.00574.x. [DOI] [PubMed] [Google Scholar]
- 684.Wahab NA, Yevdokimova N, Weston BS, Roberts T, Li XJ, Brinkman H, Mason RM. Role of connective tissue growth factor in the pathogenesis of diabetic nephropathy. Biochem J. 2001;359:77–87. doi: 10.1042/0264-6021:3590077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685.Wahren J, Ekberg K, Johansson J, Henriksson M, Pramanik A, Johansson BL, Rigler R, Jornvall H. Role of C-peptide in human physiology. Am J Physiol Endocrinol Metab. 2000;278:E759–E768. doi: 10.1152/ajpendo.2000.278.5.E759. [DOI] [PubMed] [Google Scholar]
- 686.Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, Laping NJ, Chen S. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol. 2007;293:F1657–F1665. doi: 10.1152/ajprenal.00274.2007. [DOI] [PubMed] [Google Scholar]
- 687.Wang B, Herman-Edelstein M, Koh P, Burns W, Jandeleit-Dahm K, Watson A, Saleem M, Goodall GJ, Twigg SM, Cooper ME, Kantharidis P. E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-beta. Diabetes. 2010;59:1794–1802. doi: 10.2337/db09-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Wang S, Denichilo M, Brubaker C, Hirschberg R. Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int. 2001;60:96–105. doi: 10.1046/j.1523-1755.2001.00776.x. [DOI] [PubMed] [Google Scholar]
- 689.Wang SN, Hirschberg R. Tubular epithelial cell activation and interstitial fibrosis. The role of glomerular ultrafiltration of growth factors in the nephrotic syndrome and diabetic nephropathy. Nephrol Dial Transplant. 1999;14:2072–2074. doi: 10.1093/ndt/14.9.2072. [DOI] [PubMed] [Google Scholar]
- 690.Wang SN, Hirschberg R. Growth factor ultrafiltration in experimental diabetic nephropathy contributes to interstitial fibrosis. Am J Physiol Renal Physiol. 2000;278:F554–F560. doi: 10.1152/ajprenal.2000.278.4.F554. [DOI] [PubMed] [Google Scholar]
- 691.Wang SN, Lapage J, Hirschberg R. Glomerular ultrafiltration of IGF-I may contribute to increased renal sodium retention in diabetic nephropathy. J Lab Clin Med. 1999;134:154–160. doi: 10.1016/s0022-2143(99)90120-8. [DOI] [PubMed] [Google Scholar]
- 692.Wang XX, Jiang T, Levi M. Nuclear hormone receptors in diabetic nephropathy. Nat Rev Nephrol. 2010;6:342–351. doi: 10.1038/nrneph.2010.56. [DOI] [PubMed] [Google Scholar]
- 693.Wang XX, Jiang T, Shen Y, Adorini L, Pruzanski M, Gonzalez FJ, Scherzer P, Lewis L, Miyazaki-Anzai S, Levi M. The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am J Physiol Renal Physiol. 2009;297:F1587–F1596. doi: 10.1152/ajprenal.00404.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Wang Y, Rangan GK, Tay YC, Wang Y, Harris DC. Induction of monocyte chemoattractant protein-1 by albumin is mediated by nuclear factor kappaB in proximal tubule cells. J Am Soc Nephrol. 1999;10:1204–1213. doi: 10.1681/ASN.V1061204. [DOI] [PubMed] [Google Scholar]
- 695.Weigert C, Brodbeck K, Klopfer K, Haring HU, Schleicher ED. Angiotensin II induces human TGF-beta 1 promoter activation: Similarity to hyperglycaemia. Diabetologia. 2002;45:890–898. doi: 10.1007/s00125-002-0843-4. [DOI] [PubMed] [Google Scholar]
- 696.Weigert C, Brodbeck K, Lehmann R, Haring HU, Schleicher ED. Overexpression of glutamine:fructose-6-phosphate-amidotransferase induces transforming growth factor-beta1 synthesis in NIH-3T3 fibroblasts. FEBS Lett. 2001;488:95–99. doi: 10.1016/s0014-5793(00)02395-4. [DOI] [PubMed] [Google Scholar]
- 697.Weinstein AM. Osmotic diuresis in a mathematical model of the rat proximal tubule. Am J Physiol. 1986;250:F874–F884. doi: 10.1152/ajprenal.1986.250.5.F874. [DOI] [PubMed] [Google Scholar]
- 698.Wen L, Huang JK, Blackshear PJ. Rat ornithine decarboxylase gene. Nucleotide sequence, potential regulatory elements, and comparison to the mouse gene. J Biol Chem. 1989;264:9016–9021. [PubMed] [Google Scholar]
- 699.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, Stein G, Bierhaus A, Liliensiek B, Arnold B, Nawroth PP, Stern DM, D’Agati VD, Schmidt AM. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Wijnhoven TJ, Lensen JF, Wismans RG, Lamrani M, Monnens LA, Wevers RA, Rops AL, van der Valg, Berden JH, Van Den Heuvel LP, van Kuppevelt TH. In vivo degradation of heparan sulfates in the glomerular basement membrane does not result in proteinuria. J Am Soc Nephrol. 2007;18:823–832. doi: 10.1681/ASN.2006070692. [DOI] [PubMed] [Google Scholar]
- 701.Wijnhoven TJ, Lensen JF, Wismans RG, Lefeber DJ, Rops AL, van der Valg J, Berden JH, Van Den Heuvel LP, van Kuppevelt TH. Removal of heparan sulfate from the glomerular basement membrane blocks protein passage. J Am Soc Nephrol. 2007;18:3119–3127. doi: 10.1681/ASN.2007020198. [DOI] [PubMed] [Google Scholar]
- 702.Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, Schmidt HH. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci U S A. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Wilkes BM. Reduced glomerular angiotensin II receptor density in diabetes mellitus in the rat: Time course and mechanism. Endocrinology. 1987;120:1291–1298. doi: 10.1210/endo-120-4-1291. [DOI] [PubMed] [Google Scholar]
- 704.Wilkes BM, Kaplan R, Mento PF, Aynedjian HS, Macica CM, Schlondorff D, Bank N. Reduced glomerular thromboxane receptor sites and vasoconstrictor responses in diabetic rats. Kidney Int. 1992;41:992–999. doi: 10.1038/ki.1992.151. [DOI] [PubMed] [Google Scholar]
- 705.Wiseman MJ, Saunders AJ, Keen H, Viberti G. Effect of blood glucose control on increased glomerular filtration rate and kidney size in insulin-dependent diabetes. N Engl J Med. 1985;312:617–621. doi: 10.1056/NEJM198503073121004. [DOI] [PubMed] [Google Scholar]
- 706.Wolf G. Cell cycle regulation in diabetic nephropathy. Kidney Int Suppl. 2000;77:S59–S66. doi: 10.1046/j.1523-1755.2000.07710.x. [DOI] [PubMed] [Google Scholar]
- 707.Wolf G, Mueller E, Stahl RA, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta. J Clin Invest. 1993;92:1366–1372. doi: 10.1172/JCI116710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Wolf G, Schanze A, Stahl RA, Shankland SJ, Amann K. p27(Kip1) Knockout mice are protected from diabetic nephropathy: Evidence for p27(Kip1) haplotype insufficiency. Kidney Int. 2005;68:1583–1589. doi: 10.1111/j.1523-1755.2005.00570.x. [DOI] [PubMed] [Google Scholar]
- 709.Wolf G, Schroeder R, Thaiss F, Ziyadeh FN, Helmchen U, Stahl RA. Glomerular expression of p27Kip1 in diabetic db/db mouse: Role of hyperglycemia. Kidney Int. 1998;53:869–879. doi: 10.1111/j.1523-1755.1998.00829.x. [DOI] [PubMed] [Google Scholar]
- 710.Wolf G, Schroeder R, Zahner G, Stahl RA, Shankland SJ. High glucose-induced hypertrophy of mesangial cells requires p27(Kip1), an inhibitor of cyclin-dependent kinases. Am J Pathol. 2001;158:1091–1100. doi: 10.1016/S0002-9440(10)64056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Wolf G, Schroeder R, Ziyadeh FN, Thaiss F, Zahner G, Stahl RA. High glucose stimulates expression of p27Kip1 in cultured mouse mesangial cells: Relationship to hypertrophy. Am J Physiol. 1997;273:F348–F356. doi: 10.1152/ajprenal.1997.273.3.F348. [DOI] [PubMed] [Google Scholar]
- 712.Wolf G, Ziyadeh FN. Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 1999;56:393–405. doi: 10.1046/j.1523-1755.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- 713.Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br J Nutr. 2003;89:3–9. doi: 10.1079/BJN2002763. [DOI] [PubMed] [Google Scholar]
- 714.Woods LL, Mizelle HL, Hall JE. Control of renal hemodynamics in hyperglycemia: Possible role of tubuloglomerular feedback. Am J Physiol. 1987;252:F65–F73. doi: 10.1152/ajprenal.1987.252.1.F65. [DOI] [PubMed] [Google Scholar]
- 715.Woolf AS, Gnudi L, Long DA. Roles of angiopoietins in kidney development and disease. J Am Soc Nephrol. 2009;20:239–244. doi: 10.1681/ASN.2008020243. [DOI] [PubMed] [Google Scholar]
- 716.Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280:F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]
- 717.Wright EM, Martin MG, Turk E. Familial glucose-galactose malabsorption and hereditary renal glycosuria. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 2001. pp. 4891–4980. [Google Scholar]
- 718.Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch. 2004;447:510–518. doi: 10.1007/s00424-003-1063-6. [DOI] [PubMed] [Google Scholar]
- 719.Wu D, Peng F, Zhang B, Ingram AJ, Kelly DJ, Gilbert RE, Gao B, Krepinsky JC. PKC-beta1 mediates glucose-induced Akt activation and TGF-beta1 upregulation in mesangial cells. J Am Soc Nephrol. 2009;20:554–566. doi: 10.1681/ASN.2008040445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 721.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: Cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Xu ZG, Ryu DR, Yoo TH, Jung DS, Kim JJ, Kim HJ, Choi HY, Kim JS, Adler SG, Natarajan R, Han DS, Kang SW. P-Cadherin is decreased in diabetic glomeruli and in glucose-stimulated podocytes in vivo and in vitro studies. Nephrol Dial Transplant. 2005;20:524–531. doi: 10.1093/ndt/gfh642. [DOI] [PubMed] [Google Scholar]
- 723.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A. 1993;90:1814–1818. doi: 10.1073/pnas.90.5.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Yamamoto Y, Maeshima Y, Kitayama H, Kitamura S, Takazawa Y, Sugiyama H, Yamasaki Y, Makino H. Tumstatin peptide, an inhibitor of angiogenesis, prevents glomerular hypertrophy in the early stage of diabetic nephropathy. Diabetes. 2004;53:1831–1840. doi: 10.2337/diabetes.53.7.1831. [DOI] [PubMed] [Google Scholar]
- 725.Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, Nejak K, Liu Y. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest. 2002;110:1525–1538. doi: 10.1172/JCI16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Yang ZZ, Zhang AY, Yi FX, Li PL, Zou AP. Redox regulation of HIF-1alpha levels and HO-1 expression in renal medullary interstitial cells. Am J Physiol Renal Physiol. 2003;284:F1207–F1215. doi: 10.1152/ajprenal.00017.2002. [DOI] [PubMed] [Google Scholar]
- 727.Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, Suske G, Rabbani N, Thornalley PJ, Sarthy VP, Hammes HP, Brownlee M. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem. 2007;282:31038–31045. doi: 10.1074/jbc.M704703200. [DOI] [PubMed] [Google Scholar]
- 728.Yip JW, Jones SL, Wiseman MJ, Hill C, Viberti G. Glomerular hyperfiltration in the prediction of nephropathy in IDDM: A 10-year follow-up study. Diabetes. 1996;45:1729–1733. doi: 10.2337/diab.45.12.1729. [DOI] [PubMed] [Google Scholar]
- 729.Yoon G, Kim HJ, Yoon YS, Cho H, Lim IK, Lee JH. Iron chelation-induced senescence-like growth arrest in hepatocyte cell lines: Association of transforming growth factor beta1 (TGF-beta1)-mediated p27Kip1 expression. Biochem J. 2002;366:613–621. doi: 10.1042/BJ20011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 730.Yoshioka T, Ichikawa I, Fogo A. Reactive oxygen metabolites cause massive, reversible proteinuria and glomerular sieving defect without apparent ultrastructural abnormality. J Am Soc Nephrol. 1991;2:902–912. doi: 10.1681/ASN.V24902. [DOI] [PubMed] [Google Scholar]
- 731.You G, Lee WS, Barros EJ, Kanai Y, Huo TL, Khawaja S, Wells RG, Nigam SK, Hediger MA. Molecular characteristics of Na(+)-coupled glucose transporters in adult and embryonic rat kidney. J Biol Chem. 1995;270:29365–29371. doi: 10.1074/jbc.270.49.29365. [DOI] [PubMed] [Google Scholar]
- 732.Young BA, Johnson RJ, Alpers CE, Eng E, Gordon K, Floege J, Couser WG, Seidel K. Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int. 1995;47:935–944. doi: 10.1038/ki.1995.139. [DOI] [PubMed] [Google Scholar]
- 733.Young BA, Maynard C, Boyko EJ. Racial differences in diabetic nephropathy, cardiovascular disease, and mortality in a national population of veterans. Diabetes Care. 2003;26:2392–2399. doi: 10.2337/diacare.26.8.2392. [DOI] [PubMed] [Google Scholar]
- 734.Zatz R, Dunn BR, Meyer TW, Anderson S, Rennke HG, Brenner BM. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest. 1986;77:1925–1930. doi: 10.1172/JCI112521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735.Zatz R, Meyer TW, Rennke HG, Brenner BM. Predominance of hemodynamic rather than metabolic factors in the pathogenesis of diabetic glomerulopathy. Proc Natl Acad Sci U S A. 1985;82:5963–5967. doi: 10.1073/pnas.82.17.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 738.Zerbini G, Bonfanti R, Meschi F, Bognetti E, Paesano PL, Gianolli L, Querques M, Maestroni A, Calori G, Del MA, Fazio F, Luzi L, Chiumello G. Persistent renal hypertrophy and faster decline of glomerular filtration rate precede the development of microalbuminuria in type 1 diabetes. Diabetes. 2006;55:2620–2625. doi: 10.2337/db06-0592. [DOI] [PubMed] [Google Scholar]
- 739.Zhang PL, Mackenzie HS, Troy JL, Brenner BM. Effects of an atrial natriuretic peptide receptor antagonist on glomerular hyperfiltration in diabetic rats. J Am Soc Nephrol. 1994;4:1564–1570. doi: 10.1681/ASN.V481564. [DOI] [PubMed] [Google Scholar]
- 740.Zhang SX, Wang JJ, Lu K, Mott R, Longeras R, Ma JX. Therapeutic potential of angiostatin in diabetic nephropathy. J Am Soc Nephrol. 2006;17:475–486. doi: 10.1681/ASN.2005020217. [DOI] [PubMed] [Google Scholar]
- 741.Zhang Y, Deb DK, Kong J, Ning G, Wang Y, Li G, Chen Y, Zhang Z, Strugnell S, Sabbagh Y, Arbeeny C, Li YC. Long-term therapeutic effect of vitamin D analog doxercalciferol on diabetic nephropathy: Strong synergism with AT1 receptor antagonist. Am J Physiol Renal Physiol. 2009;297:F791–F801. doi: 10.1152/ajprenal.00247.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Zhang Y, Peng F, Gao B, Ingram AJ, Krepinsky JC. Mechanical strain-induced RhoA activation requires NADPH oxidase-mediated ROS generation in caveolae. Antioxid Redox Signal. 2010;13:959–973. doi: 10.1089/ars.2009.2908. [DOI] [PubMed] [Google Scholar]
- 743.Zhang Z, Sun L, Wang Y, Ning G, Minto AW, Kong J, Quigg RJ, Li YC. Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney Int. 2008;73:163–171. doi: 10.1038/sj.ki.5002572. [DOI] [PubMed] [Google Scholar]
- 744.Zhang Z, Zhang Y, Ning G, Deb DK, Kong J, Li YC. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc Natl Acad Sci U S A. 2008;105:15896–15901. doi: 10.1073/pnas.0803751105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Zheng S, Noonan WT, Metreveli NS, Coventry S, Kralik PM, Carlson EC, Epstein PN. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes. 2004;53:3248–3257. doi: 10.2337/diabetes.53.12.3248. [DOI] [PubMed] [Google Scholar]
- 747.Ziyadeh FN, Han DC. Involvement of transforming growth factor-beta and its receptors in the pathogenesis of diabetic nephrology. Kidney Int Suppl. 1997;60:S7–11. [PubMed] [Google Scholar]
- 748.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitrans-forming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Ziyadeh FN, Simmons DA, Snipes ER, Goldfarb S. Effect of myoinositol on cell proliferation and collagen transcription and secretion in proximal tubule cells cultured in elevated glucose. J Am Soc Nephrol. 1991;1:1220–1229. doi: 10.1681/ASN.V1111220. [DOI] [PubMed] [Google Scholar]
- 750.Ziyadeh FN, Snipes ER, Watanabe M, Alvarez RJ, Goldfarb S, Haverty TP. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am J Physiol. 1990;259:F704–F714. doi: 10.1152/ajprenal.1990.259.4.F704. [DOI] [PubMed] [Google Scholar]
- 751.Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. doi: 10.2174/157339908783502370. [DOI] [PubMed] [Google Scholar]
- 752.Zou AP, Li N, Cowley AW., Jr Production and actions of superoxide in the renal medulla. Hypertension. 2001;37:547–553. doi: 10.1161/01.hyp.37.2.547. [DOI] [PubMed] [Google Scholar]