

Abstract
Triple positive breast cancers overexpress both the human epidermal growth factor receptor 2 (HER2) oncogene and the hormonal receptors (HR) to estrogen and progesterone. These cancers represent a unique therapeutic challenge because of a bidirectional cross-talk between the estrogen receptor alpha (ERα) and HER2 pathways leading to tumor progression and resistance to targeted therapy. Attempts to combine standard of care HER2-targeted drugs with antihormonal agents for the treatment of HR+/HER2+ breast cancer yielded encouraging results in preclinical experiments but did improve overall survival in clinical trial. In this review, we dissect multiple mechanisms of therapeutic resistance typical of HR+/HER2+ breast cancer, summarize prior clinical trials of targeted agents, and describe novel rational drug combinations that include antihormonal agents, HER2-targeted drugs, and CDK4/6 inhibitors for treatment of the HR+/HER2+ breast cancer subtype.
1. Triple Positive Breast Cancer Displays Increased Resistance to Targeted Therapy
Breast cancer is the most frequently diagnosed cancer, excluding skin malignancies, and a second leading cause of cancer death in women in the United States [1]. Approximately 20% of breast cancers overexpress the human epidermal growth factor receptor 2 (HER2), a transmembrane tyrosine kinase receptor mediating cell growth, differentiation, and survival [2]. HER2-positive (HER2+) breast tumors are more aggressive and historically have been associated with poorer outcomes compared to HER2-negative (HER2-) tumors, although the introduction of HER2-targeted therapies has allowed for significant improvements in survival of patients with HER2+ breast cancer [3–5].
Approximately half of HER2+ breast tumors overexpress hormone receptors (HR) [6, 7]. These cancers represent a therapeutic challenge because of a bidirectional cross-talk between HR and HER2 pathways leading to tumor progression and resistance to targeted therapies [8, 9]. HR+/HER2+ tumors are sometimes called “triple positive,” if HER2 and both estrogen receptor (ER) and progesterone receptor (PR) are expressed. Less frequently, HR+/HER2+ tumors express only one of the hormonal receptors (either ER or PR). In the University of Colorado Denver clinical database, among 114 HR+/HER2+ cases, 71% were triple positive, 21% were ER+/PR-/HER2+, and 8% were ER-/PR+/HER2+. Differences in clinical behavior between HER2 amplified tumors that are ER+/PR+, ER+/PR-, or ER-/PR+ are not well studied. It is assumed that even if only one of the hormonal receptors is expressed, tumor proliferation is driven by HR signaling along with HER2 pathway, which may lead to a resistant phenotype. Another complicating factor is that HR+/HER2+ breast tumors are heterogeneous at a molecular level and HR status does not completely recapitulate this heterogeneity: 40-50% of these tumors belong to HER2-enriched PAM50 molecular subtype, while the rest are classified as luminal A or B subtype [10, 11]. Intrinsic molecular subtypes of HR+/HER2+ breast cancer may affect therapeutic sensitivity [11, 12], as discussed below.
Multiple studies have shown that HR expression confers resistance to HER2-targeted therapies [8, 13, 14]. In HR+/HER2+ breast cancer cell lines, ER signaling and ER transcriptional activity are upregulated following treatment with HER2-targeted agents trastuzumab and lapatinib and ER functions as the key survival pathway reducing sensitivity to HER2-blockade [13, 15, 16]. The same phenomenon is seen in patients on neoadjuvant treatment. Multiple prospective clinical trials demonstrated that pCR rates in patients with HR+/HER2+ breast tumors are 1.5 – 2.5 times lower compared to patients with HR-/HER2+ tumors, regardless of HER2-targeted and chemotherapeutic agents administered (Table 1). Preclinical and clinical studies in HER2+ metastatic breast cancer confirmed that expression of HR is associated with reduced responsiveness to trastuzumab [13, 14], and combinations of antihormonal and HER2-targeted agents led to progression free survival (PFS) benefits in some trials [17–19].
Table 1.
Pathologic complete response after neoadjuvant chemotherapy in patients with HER2-positive breast cancer stratified by hormonal receptor status.
| Clinical trial | Chemotherapy and HER2-targeted agents | Outcome |
pCR rate HR+/HER2+ |
pCR rate HR-/HER2+ |
Reference |
|---|---|---|---|---|---|
| NeoSphere | Docetaxel Carboplatin Trastuzumab |
ypT0a | 20.0% | 36.8% | [32] |
|
| |||||
| NeoSphere | Docetaxel Carboplatin Trastuzumab Pertuzumab |
ypT0 | 26.0% | 63.2% | [32] |
|
| |||||
| NeoALTO | Paclitaxel Lapatinib Trastuzumab |
ypT0 | 41.6% | 61.3% | [33] |
|
| |||||
| NOAH | Doxorubicin Paclitaxel Cyclophosphamide Methotrexate Fluorouracil Trastuzumab |
ypT0 ypN0b |
30.0% | 51.2% | [12] |
|
| |||||
| ACOSOG Z1041 | 5-fluorouracil Epirubicin Cyclophosphamide Paclitaxel Trastuzumab |
ypT0 | 47.6% | 70.4% | [34] |
a–ypT0: pathologic complete response in the breast (absence of invasive neoplastic cells); b–ypN0: pathologic complete response in the axillary lymph nodes (absence of invasive neoplastic cells).
Similarly, HER2 overexpression is a major determinant of resistance to endocrine therapy [7, 20–22]. HR+ breast cancer cell lines that are sensitive to tamoxifen acquire tamoxifen resistance after transfection with the HER2 oncogene [20, 22]. Analysis of tumor samples from postmenopausal patients with stages II and III HR+ breast cancer treated on two-independent neoadjuvant endocrine therapy trials showed that HR+/HER2+ tumors had significantly higher histologic grade and Ki-67 and significantly less suppression of Ki-67 after treatment with tamoxifen or an aromatase inhibitor (AI) compared with HR+/HER2- tumors. These tumors display continued estrogen-independent proliferation despite ongoing endocrine therapy [23]. Results of two-adjuvant therapy clinical trials (Breast International Group 1-98 study and the Arimidex or Tamoxifen Alone or in combination study) demonstrated that HER2+ status is associated with a significantly higher relapse rate, regardless of whether the adjuvant antihormonal therapy administered was tamoxifen or an AI [24, 25]. Similarly, studies in metastatic breast cancer demonstrated decreased responses to antihormonal therapy in patients with HR+/HER2+ tumors [23, 25–28].
NCCN guidelines (version 4.2017) suggest several options for an initial treatment of HR+/HER2+ metastatic disease. Chemotherapy with a taxane plus trastuzumab and pertuzumab remains a preferred frontline regimen based on the CLEOPATRA clinical trial [5]. NCCN included the antibody-drug conjugate TDM-1 as one of the frontline options after considering the results of the MARIANNE trail [29]. Other options include single agent endocrine therapy (for patients with bone or soft tissue metastases, or asymptomatic minimal visceral disease), or dual combinations of antihormonal and HER2-targeted agents [30]. Although the preferred combination chemotherapy approach is highly effective, it is associated with multiple side effects. Single agent antihormonal therapy generally has a poor efficacy in patients with HR+/HER2+ breast cancer, resulting in PFS of 3-4 months [19, 31]. Dual combinations of HER2-targeted and antihormonal agents demonstrated efficacy in phase II clinical trials [17, 18]; however, they did not improve overall survival in randomized phase III clinical trials [19, 31]. Therefore, there is an unmet clinical need to develop a more effective chemotherapy-free approaches based on novel targeted drug combinations for patients with HR+/HER2+ breast cancer.
Below we summarize current approaches to targeted therapy in HR+/HER2+ breast cancer, highlight drug resistance mechanisms, and focus on CDK4/6 inhibitors as promising agents that may counteract therapeutic resistance in patients with HR+/HER2+ breast cancer.
2. Dual Blockade: Combining Antihormonal and HER2-Targeted Agents
Preclinical modeling in breast tumor cell lines and murine xenografts demonstrated synergy of HER2-targeted agents combined with endocrine therapy in suppressing growth of HR+/HER2+ breast tumors [13, 59]. However, translation of these exciting preclinical results into human clinical trials has not been straightforward.
Neoadjuvant randomized phase III clinical trial NSABP B-52 explored the concept of dual targeting of HER2 and HR pathways combined with chemotherapy, with the goal of improving pCR rates in patients with HR+/HER2+ early breast cancer. In this trial, 308 women were randomized to receive neoadjuvant chemotherapy with docetaxel, carboplatin, trastuzumab, and pertuzumab (n = 154), or the same chemotherapy plus endocrine therapy with estrogen deprivation (n = 157). The pCR rates were numerically better in the estrogen deprivation arm comparing to control (46% versus 41%); however, the difference did not reach statistical significance (p = 0.39). A subgroup analysis looking at patients by menopausal status showed no significant difference for premenopausal (46% versus 44%) or postmenopausal women (45% versus 38%) [75].
Phase II neoadjuvant trial PAMELA enrolled 151 patients with stage I-IIIA HER2+ breast cancer [10]. The trial was specifically designed to test the hypothesis that PAM50 tumor molecular subtypes will determine response to targeted therapy. All patients received lapatinib and trastuzumab for 18 weeks. Additionally, patients with HR+/HER2+ disease received daily letrozole or tamoxifen. The overall pCR rate in the breast was 30.2% (40.2% in HER2-enriched tumors irrespective of HR status versus 10.0% in non-HER2-enriched tumors). HR status lost its association with pCR once intrinsic molecular subtypes were taken into account in the multivariable model. Therefore, this trial suggested that the HER2-enriched subtype is a predictor of anti-HER2 sensitivity, regardless of HR status [10, 11]. One striking peculiarity of the trial results was the low pCR rate in patients with luminal tumors despite dual HR and HER2 blockade.
In metastatic settings, the eLEcTRA trial compared efficacy of letrozole combined with trastuzumab (N = 26) versus letrozole alone (N = 31) as a frontline treatment [17]. Median time to progression was 3.3 months in the letrozole group compared to 14.1 months in the trastuzumab and letrozole group. Clinical benefit rate was 39% compared to 65% in the single agent letrozole versus dual combination. The trial showed that the combination of letrozole and trastuzumab may be a safe and effective treatment option. However, although this was a randomized trial, the sample size was quite small.
Results of two larger randomized phase III clinical trials combining antihormonal therapy with HER2-targeted agents for metastatic breast cancer have been reported [19, 31]. The TAnDEM trial evaluated the benefit of adding trastuzumab to anastrozole as a frontline therapy in 207 patients with HR+/HER2+ metastatic breast cancer. Median PFS was 4.8 months for the combination group versus 2.4 months for the anastrozole monotherapy group, with a hazard ratio of 0.63 (p = 0016; 95% CI, 0.47 to 0.84). In patients with centrally confirmed HR+ tumors, median PFS was 5.6 versus 3.8 month in the trastuzumab plus anastrozole and anastrozole alone arms, respectively (p = 0.006). The overall response rate (ORR) was significantly higher with the combination treatment compared with anastrozole alone (20.3% v 6.8%; p = 018). The clinical benefit rate (CBR) was also higher in patients in the combination arm compared with the anastrozole arm (42.7% v 27.9%; p = 0.026). No statistically significant improvement in overall survival (OS) was demonstrated (28.5 v 23.9 months for dual combination versus monoagent letrozole; p = 0.325) [31].
Similarly, in the EGF30008 study, anti-HER2 tyrosine kinase inhibitor lapatinib was combined with letrozole and compared to letrozole plus placebo in 219 patients with HR+ metastatic breast cancer. In the HER2+ subgroup, the addition of lapatinib reduced risk of disease progression, with a hazard ratio of 0.71 (P = 0.019; 95% CI, 0.53 to 0.96) and median PFS of 8.2 versus 3.0 months. The ORR was also higher in the combination therapy group (28% v 15%; P = 0.021). CBR was significantly greater for lapatinib plus letrozole (48% v 29%; odds ratio 0.4; 95%CI, 0.2 to 0.8; P = 0.03). These benefits did not translate into an improvement in median OS (33.3 v 32.3 months) [19].
The effect of combined HR and HER2 blockade was further evaluated in the PERTAIN randomized phase II clinical trial. In this trial, 258 postmenopausal patients with metastatic HR+/HER2+ breast cancer who did not receive prior systemic chemotherapy for metastatic disease were randomized to receive a combination of trastuzumab and an AI (anastrozole or letrozole), or trastuzumab plus pertuzumab and an AI. Fifty-seven percent of patients initially received 18-24 weeks of induction chemotherapy with docetaxel or paclitaxel in combination with HER2-targeted agents. The addition of pertuzumab led to a statistically significant increase of median PFS from 15.8 months to 18.9 months (trastuzumab + AI versus trastuzumab + pertuzumab + AI, HR 0.65, 95%CI 0.48–0.89; p = 0.007) [18]. These results are drastically different from the TAnDEM trial, where patients on trastuzumab and an AI had a median PFS of 4.8 months [31]. One potential explanation could be that the TAnDEM trial enrolled “all comers” for a frontline targeted therapy, while in the PERTAIN trial more than half of the patients, potentially those with more aggressive disease, received induction chemotherapy prior to going on targeted therapy maintenance. Patients who did not receive induction chemotherapy had much better outcomes with the HER2 and HR blockade compared to the TAnDEM trial. However, it is not clear if these patients actually had less aggressive disease, because the decision of whether or not to administer induction chemotherapy was at the discretion of the treating physician; therefore, selection bias might have been introduced. Pertuzumab, trastuzumab and an AI combination was well tolerated, making this an attractive treatment option for a selected patient population. The trial clearly demonstrated that pertuzumab and trastuzumab maintenance is better than trastuzumab alone. The PERTAIN study did not address the question of whether the addition of endocrine therapy to dual HER2-blockade further improves efficacy, because both randomization groups received an AI.
Although the TAnDEM and EGF30008 studies showed statistically significant improvement of PFS in patients with HR+/HER2+ tumors with the addition of HER2-targeted agents to endocrine therapy, these phase III trials were not practice changing because PFS benefits were small and no benefits in OS were demonstrated. What is not known is whether these results were due to patient selection, limitations of specific HER2-targeted agents, inherent resistance mechanisms that were not counteracted by antihormonal and HER2-targeted therapy, or a combination of all these factors. The PERTAIN trial showed feasibility of a frontline multiagent targeted therapy approach in selected patients, although these results may not be fully applicable to the overall population of patients with HR+/HER2+ disease. New rationally designed combinations of targeted agents for patients with HR+/HER2+ breast cancer are warranted.
3. CDK4/6 Inhibitors Synergize with Antihormonal and HER2-Targeted Agents
Inhibition of the cyclin D1-CDK4/6 complex emerged as a promising therapeutic strategy in breast cancer. In a pivotal study of the CDK4/6 inhibitor palbociclib, Finn and colleagues [37] compared baseline gene expression profiles from breast cancer cell lines highly sensitive or resistant to palbociclib. HR+ cell lines, including those with HER2 amplification, were the most sensitive, and there was a significant overlap between the gene expression profiles associated with palbociclib sensitivity and that which distinguished a luminal breast cancer subtype [37]. In preclinical studies, palbociclib was active against both luminal A and luminal B tumors and synergized with both tamoxifen and anti-HER2 agents (trastuzumab, lapatinib, and TDM-1) providing a potent addition to antihormonal and HER2-targeted therapies [37, 76, 77]. Additionally, another CDK4/6 inhibitor, abemaciclib, showed significant activity in HER2+ preclinical models, supporting the hypothesis that CDK4/6 inhibitors may resensitize resistant tumors to the HER2 blockade [78].
Palbociclib was FDA approved in patients with HR+ metastatic breast cancer based on the results of the PALOMA-2 randomized phase II clinical trial, which showed marked improvement in median PFS in women who received palbociclib and letrozole versus letrozole alone (26.1 versus 7.5 months) [38]. Additionally, palbociclib demonstrated remarkable activity in the second-line metastatic setting in combination with fulvestrant in the PALOMA-3 clinical trial, resulting in more than a doubling of median PFS (9.2 months palbociclib with fulvestrant versus 3.8 months placebo with fulvestrant; HR 0.42; P <0.001) [39]. Comparable efficacy in patients with HR+/HER2-metastatic breast cancer was demonstrated for ribociclib and letrozole combination in the MONALEESA-2 trial [79] and for abemaciclib and antihormonal therapy combination in the MONARCH-2 and MONARCH-3 clinical trials [80, 81]. Notably, abemaciclib has a remarkable single agent activity [82] and documented efficacy in the central nervous system metastatic disease [83].
Taken together, this data suggest that it is reasonable to combine HER2-targeted agents with synergistic combinations of CDK4/6 inhibitors and antihormonal agents for the treatment of patients with HR+/HER2+ breast cancer. Multiple clinical trials have shown synergy of CDK4/6 inhibitors with endocrine therapy, and large amounts of preclinical data support synergy of CDK4/6 inhibitors with HER2-targeted therapies. Triple targeting of HR, HER2, and CDK4/6 pathways is a promising approach that has a strong preclinical rationale. This approach is now being tested in clinical trials.
4. Triple Blockade of HR, HER2, and Cell Cycle Checkpoints: Signaling Rationale and Ongoing Clinical Trials
Activation of cyclin D1 and CDK4/6 plays a significant role in the tumorigenesis of HR+/HER2+ breast cancer [37, 76, 84]. Mitogenic signaling from HER2 and HR receptors converges at cell cycle checkpoints and results in the synergistic increase of cyclin D1 expression (Figure 1). Specifically, HER2/MAPK kinase signaling activates E2F transcription factors, leading to the transcription of CCND1 gene encoding cyclin D1, while active estrogen receptor alpha (ERα) in complex with FOXA1 transcription factor intensifies CCND1 transcription through an estradiol-responsive enhancer [85]. CCND1 gene located on chromosome 11q13 is amplified in ~15% of breast cancers [86, 87]. However, cyclin D1 is overexpressed at the protein levels in ~50% of breast cancers in the presence or absence of gene amplification [88]. The difference in the frequency of CCND1 gene amplification and protein overexpression can be explained at least in part by the CCND1 promoter activation by aberrant mitogenic signaling in tumors with HER2 amplification or overexpression [89]. Consistent with this data, the frequency of cyclin D1 overexpression is two times higher in luminal B versus luminal A tumors (58% versus 29%) [90], because many of luminal B tumors are HER2 amplified. Amplification or overexpression of cyclin D1 is strongly associated with short survival in breast cancer patients [91].
Figure 1.
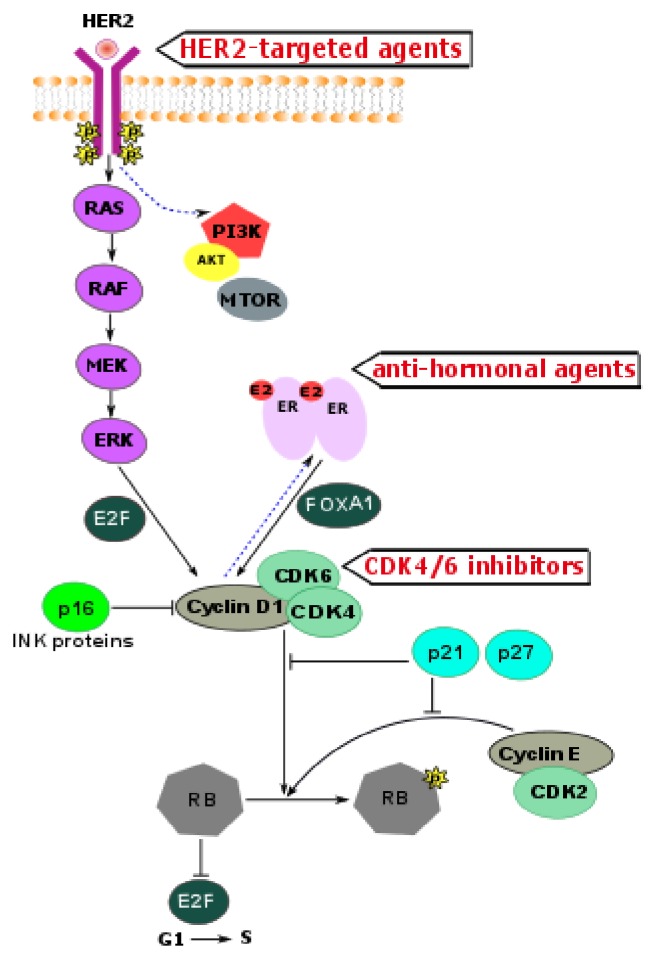
HR and HER2 signaling converge at cell cycle checkpoints.
Cyclin D1–CDK4/6 complex phosphorylates the retinoblastoma protein (RB). Controlled phosphorylation and deactivation of RB is essential for progression of the cell cycle from G1 to S phase [88]. Activity of the cyclin D1–CDK4/6 is counteracted by tumor suppressor protein p16 and other INK-family proteins. However, p16 is frequently inactivated in breast tumors [85]. Notably, cyclin D1 associates with ERα and the steroid receptor coactivator increasing transcriptional activity of ERα [92–94].
In addition to being catalytically active, the cyclin D1–CDK4/6 complex sequesters the cell cycle inhibitors p21 and p27, thereby promoting the activation of another key component of G1 to S transition: the cyclin E–CDK2 complex. This complex can further phosphorylate RB, leading to full saturation of all phosphorylation sites. Hyperphosphorylated RB loses its inhibitory effect on the E2F transcription program, allowing G1 to S transition [95, 96]. Because cyclin E itself is an E2F target gene, cyclin E may reinforce its own expression. Once cyclin E–CDK2 becomes active, RB phosphorylation is rendered partially independent of the mitogenic control that regulates cyclin D1 expression [97]. Additionally, CDK2 phosphorylates ERα, providing a positive feedback loop and further increasing ERα transcriptional activity [98].
Considering the synergistic effects of ERα signaling and HER2 overexpression on cell cycle checkpoints, as well as multiple feedback loops between these pathways, there is a strong signaling rationale to combine CDK4/6 inhibitors with HER2 inhibitors and antihormonal agents for treatment of HR+/HER2+ breast cancer. Multiple clinical trials are ongoing in Europe and the United States to test triple combinations of HR, HER2, and CDK4/6 inhibitors (Table 2).
Table 2.
Ongoing clinical trials combining CDK4/6 inhibitors with HER2-targeted and antihormonal agents in patients with HR+/HER2+ breast cancer.
| Clinical trial | Therapeutic agents | Phase |
Line of therapy Disease stage |
Location |
|---|---|---|---|---|
| NCT03054363 a | (i) Palbociclib (ii) Tucatinib (iii) Letrozole |
Ib/II | 1st line and beyond Stage IV |
United States ABRCC consortium |
|
| ||||
|
NCT02947685 (PATINA) |
(i) Palbociclib (ii) Trastuzumab (iii) Pertuzumab (iv) Any AIb or fulvestrant |
III | 1st line maintenance after 4-8 cycles of induction chemotherapy Stage IV |
United States Alliance foundation |
|
| ||||
|
NCT02907918 (PALTAN) |
(i) Palbociclib (ii) Trastuzumab (iii) Letrozole |
II | Neo-adjuvant therapy Stage II, III |
United States |
|
| ||||
|
NCT02675231 (MonarcHER2) |
(i) Abemaciclib (ii) Trastuzumab (iii) Fulvestrant |
II | 3rd line and beyond Stage IV |
Worldwide |
|
| ||||
|
NCT02448420 (PATRICIA) |
(i) Palbociclib (ii) Trastuzumab (iii) Letrozole |
II | 3rd to 5th line of therapy Stage IV |
Spain SOLTI group |
|
| ||||
|
NCT02530424 (NA-PHER2) |
(i) Palbociclib (ii) Trastuzumab (iii) Pertuzumab (iv) Fulvestrant |
II | Neo-adjuvant therapy Stage I-III |
Italy |
|
| ||||
| NCT03304080 | (i) Palbocilib (ii) Trastuzumab (iii) Pertuzumab (iv) Anastrozole |
I/II | 1st line Stage IV |
United States |
a: clinical trial number in the US National Institutes of Health clinical trial database; b–AI: aromatase inhibitor.
The goal of these clinical trials is to develop an effective and safe targeted therapy-based regimen which will overcome multiple drug resistance mechanisms typical of HR+/HER2+ breast cancer, resulting in the improved response to neoadjuvant treatment in early disease, as well as prolonged PFS and improved quality of life in metastatic settings. Potentially overlapping side effects of the drugs in combination could be of concern, especially diarrhea, which is a side effect of CDK4/6 inhibitors and many HER2-targeted agents. However, overall safety and toxicity profiles of CDK4/6 inhibitors, HER2-targeted drugs, and antihormonal agents are favorable, and the majority of side effects are nonoverlapping. Targeted drug combinations are attractive from patient perspective, because they are expected to be tolerated significantly better compared to chemotherapy-based combinations.
5. Potential Mechanisms of Resistance to HR and HER2 and Cell Cycle Checkpoint Inhibition
In the design of rational drug combinations, it is critical to evaluate pathways of resistance to each targeted agent and identify potential cross-resistance mechanisms. Mathematical modeling of tumor clonal evolution demonstrated that even a single genetic alteration conferring resistance to two-targeted agents may decrease efficacy of treatment with these agents given in combination. Similarly, once metastases have been established with significant tumor burden, the probability of mutation(s) that will confer multidrug resistance increases and treatment is doomed to fail [99]. Will the combinations of HER2-targeted agents with antihormonal drugs and CDK4/6 inhibitors avoid this problem? To answer this question, we performed a literature search looking for the mechanisms of resistance to antihormonal agents, HER2 inhibitors, and CDK4/6 inhibitors in published preclinical and clinical studies. Our goal was to identify candidate single mechanism, which could confer resistance to all three drugs in combination. This exploratory analysis was not intended to provide guidance in clinical management, but rather to provide a framework for further studies. The results of our findings are summarized in Table 3.
Table 3.
Potential pathways of resistance to antihormonal agents, HER2 inhibitors and CDK4/6 inhibitors.
| Mechanisms | Resistance to therapy | Counteracted by |
|---|---|---|
| Cell cycle checkpoints pathway | ||
|
| ||
| Cyclin D1 amplification or overexpression | Endocrine therapy (P, C)a [35, 36] | CDK4/6 inhibitors [37–40] |
|
| ||
| CDK4 amplification | CDK4/6 inhibitors (P) [41, 42] | To be studied |
|
| ||
| CDK6 amplification | CDK4/6 inhibitors (P)b [43] | To be studied |
|
| ||
| Cyclin E1 amplification or overexpression | Endocrine therapy (P, C) [44] HER2 inhibitors (P, C) [45] CDK4/6 inhibitors (P) [46] |
CDK2 inhibitors [45] |
|
| ||
| Cyclin E2 amplification or overexpression | Endocrine therapy (P) [47] | CDK2 inhibitors [47] |
|
| ||
| RB loss | CDK4/6 inhibitors (P) [37, 48] | To be studied |
|
| ||
| p21 loss | Endocrine therapy (P, C) [49, 50] CDK4/6 inhibitors (P) [51] |
To be studied |
|
| ||
| p27 loss | Endocrine therapy (P, C) [50, 52] CDK4/6 inhibitors (P) [51] |
To be studied |
|
| ||
|
| ||
| ESR1 activating mutations | Endocrine therapy (P, C) [53–55] | CDK4/6 inhibitors [56] mTOR inhibitors [57] Fulvestrant [56] Novel ER antagonists [58] |
|
| ||
| MAPK kinase pathway | ||
|
| ||
| HER2 amplification | Endocrine therapy (P, C) [7, 20–28] | HER2 inhibitors [13, 59] |
|
| ||
| HER2 truncation (p95HER2) or mutations in the extracellular domain | HER2-targeted antibodies (P, C) [60, 61] | HER2 small molecule inhibitors [61] |
|
| ||
| HER3 amplification | HER2 inhibitors [62] | HER3 inhibitors [63–65] |
|
| ||
| C-MYC overexpression | Endocrine therapy (P) [66] | CDK1 inhibitors [67, 68] |
|
| ||
| PI3K/AKT/mTOR pathway | ||
|
| ||
| PI3K pathway activation | Endocrine therapy (P, C) [69] HER2 inhibitors (P) [62, 70, 71] |
PI3K inhibitors [72] mTOR inhibitors [62, 71, 73] CDK4/6 inhibitors [74] |
As evident in Table 3, there are multiple potential mechanisms of resistance to single agents and significantly fewer pathways of resistance to dual and triple targeted drug combinations. Cyclin E1 amplification or overexpression potentially could confer resistance to all three drugs in our combination of interest (anti-endocrine agents, HER2-targeted drugs, and CDK4/6 inhibitors). Amplification of CCNE1 gene encoding cyclin E1 is uncommon in HR+/HER2+ cancer subtype [100], although overexpression of cyclin E1 is a more common event [101]. Cyclin E1 amplification and overexpression have been shown to mediate resistance to antiendocrine and HER2-targeted agents in patients [44, 45]. Resistance to CDK4/6 inhibitors due to cyclin E1 overexpression has been demonstrated in preclinical models [46]; however, it was not yet confirmed in the clinic. Whether cyclin E1 amplification or overexpression could modulate patient response to CKD4/6 inhibitors remains to be determined. Assessment of cyclin E1 levels in clinical studies of HR, HER2, and CDK4/6 inhibitors is certainly of great interest.
CDK2 inhibitors might provide the ability to target tumors driven by cyclin E1 amplification [45, 102]. Several nonselective CDK inhibitors capable of targeting CDK2 were tested in clinical trials; however, clinical development was stopped because of toxicity [103]. Selective CDK2 inhibitors are not yet clinically available, although there is a great interest in development of this compounds.
Based on nonoverlapping resistance mechanisms, other rational drug combinations for treatment of HR+/HER2+ breast cancer may be suggested (for example, a combination of antihormonal drugs with HER2-targeted agents and PI3K or mTOR inhibitors). Some of these combinations are now being tested in clinical trials, which is outside of the scope of this paper.
In conclusion, triple combination targeting therapy (HER2 and CDK4/6 inhibitors combined with antihormonal therapy) remains promising in HR+/HER2+ breast cancer, given largely nonoverlapping resistance mechanisms to the agents in this combination. An overlapping mechanism of resistance to all three drugs probably exists (cyclin E overexpression). Assessment of this potential resistance mechanisms should be performed in the ongoing clinical trials.
6. Concluding Remarks
Breast tumors with HR expression and HER2 amplification represent a therapeutic challenge because they utilize multiple oncogenic drivers and pathways of therapeutic resistance. Based on the vast amount of preclinical and clinical data, the concept of triple targeting of HR, HER2, and CDK4/6 pathways simultaneously is a logical approach. Therapy with a triple combination of agents blocking HR, HER2, and CDK4/6 is supported by a strong signaling rationale and is feasible from a toxicity standpoint. With a majority of the resistance mechanisms being nonoverlapping, this promising combination has a reasonable potential to be effective and to overcome targeted therapy resistance. Multiple clinical trials are underway to test triple combinations of antihormonal therapy with HER2 and CDK4/6-targeted agents in locally advanced and metastatic HR+/HER2+ breast cancer, offering new hope to patients with this challenging disease.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- 1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta: American Cancer Society; 2015.
- 2.Owens M. A., Horten B. C., Da Silva M. M. HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clinical Breast Cancer. 2004;5(1):63–69. doi: 10.3816/CBC.2004.n.011. [DOI] [PubMed] [Google Scholar]
- 3.Slamon D. J., Leyland-Jones B., Shak S., et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England Journal of Medicine. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 4.Verma S., Miles D., Gianni L., et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. The New England Journal of Medicine. 2012;367(19):1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baselga J., Cortés J., Kim S.-B., et al. Pertuzumab plustrastuzumab plus docetaxel for metastatic breast cancer. The New England Journal of Medicine. 2012;366(2):109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slamon D. J., Clark G. M., Wong S. G., Levin W. J., Ullrich A., McGuire W. L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3026048. [DOI] [PubMed] [Google Scholar]
- 7.Prat A., Baselga J. The role of hormonal therapy in the management of hormonal-receptor-positive breast cancer with co-expression of HER2. Nature Clinical Practice Oncology. 2008;5(9):531–542. doi: 10.1038/ncponc1179. [DOI] [PubMed] [Google Scholar]
- 8.Montemurro F., Di Cosimo S., Arpino G. Human epidermal growth factor receptor 2 (her2)-positive and hormone receptor-positive breast cancer: New insights into molecular interactions and clinical implications. Annals of Oncology. 2013;24(11):2715–2724. doi: 10.1093/annonc/mdt287.mdt287 [DOI] [PubMed] [Google Scholar]
- 9.Cortes J., Baselga J. How to treat hormone receptor-positive, human epidermal growth factor receptor 2-amplified breast cancer. Journal of Clinical Oncology. 2009;27(33):5492–5494. doi: 10.1200/JCO.2009.23.8089. [DOI] [PubMed] [Google Scholar]
- 10.Llombart-Cussac A., Cortés J., Paré L., et al. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): an open-label, single-group, multicentre, phase 2 trial. The Lancet Oncology. 2017;18(4):545–554. doi: 10.1016/S1470-2045(17)30021-9. [DOI] [PubMed] [Google Scholar]
- 11.Prat A., Pascual T., Adamo B. Intrinsic molecular subtypes of HER2+ breast cancer. Oncotarget . 2017;8(43):73362–73363. doi: 10.18632/oncotarget.20629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prat A., Bianchini G., Thomas M., et al. Research-Based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2- Positive breast cancer in the NOAH Study. Clinical Cancer Research. 2014;20(2):511–521. doi: 10.1158/1078-0432.CCR-13-0239. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y.-C., Morrison G., Gillihan R., et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2- positive breast cancers -- role of estrogen receptor and HER2 reactivation. Breast Cancer Research. 2011;13(6, article no. R121) doi: 10.1186/bcr3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montemurro F., Rossi V., Cossu Rocca M., et al. Hormone-receptor expression and activity of trastuzumab with chemotherapy in HER2-positive advanced breast cancer patients. Cancer. 2012;118(1):17–26. doi: 10.1002/cncr.26162. [DOI] [PubMed] [Google Scholar]
- 15.Xia W., Bacus S., Hegde P., et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proceedings of the National Acadamy of Sciences of the United States of America. 2006;103(20):7795–7800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins D. C., Cocchiglia S., Tibbitts P., et al. Growth factor receptor/steroid receptor cross talk in trastuzumab-treated breast cancer. Oncogene. 2015;34(4):525–530. doi: 10.1038/onc.2013.586. [DOI] [PubMed] [Google Scholar]
- 17.Huober J., Fasching P. A., Barsoum M., et al. Higher efficacy of letrozole in combination with trastuzumab compared to letrozole monotherapy as first-line treatment in patients with HER2-positive, hormone-receptor-positive metastatic breast cancer - Results of the eLEcTRA trial. The Breast. 2012;21(1):27–33. doi: 10.1016/j.breast.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Arpino G., Ferrero J.-M., de la Haba-Rodriguez J., et al. Primary analysis of PERTAIN: A randomized, two-arm, open-label, multicenter phase II trial assessing the efficacy and safety ofpertuzumab given in combination withtrastuzumab plus an aromatase inhibitor in first-line patients with HER2-positive and hormone receptor-positive metastatic or locally advanced breast cancer. Cancer Research. 2016;2017(77):S3–S4. [Google Scholar]
- 19.Johnston S., Pippen J., Jr., Pivot X., et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor—positive metastatic breast cancer. Journal of Clinical Oncology. 2009;27(33):5538–5546. doi: 10.1200/jco.2009.23.3734. [DOI] [PubMed] [Google Scholar]
- 20.Benz C. C., Scott G. K., Sarup J. C., et al. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Research and Treatment. 1992;24(2):85–95. doi: 10.1007/BF01961241. [DOI] [PubMed] [Google Scholar]
- 21.Pietras R. J., Arboleda J., Reese D. M., et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10(12):2435–2446. [PubMed] [Google Scholar]
- 22.Shou J., Massarweh S., Osborne C. K., et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. Journal of the National Cancer Institute. 2004;96(12):926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 23.Ellis M. J., Tao Y., Young O., et al. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. Journal of Clinical Oncology. 2006;24(19):3019–3025. doi: 10.1200/JCO.2005.04.3034. [DOI] [PubMed] [Google Scholar]
- 24.Viale G., Regan M., Dell'Orto P., et al. Central Review of ER, PgR and HER2 in BIG 1-98 Evaluating Letrozole vs. Letrozole Followed by Tamoxifen vs. Tamoxifen Followed by Letrozole as Adjuvant Endocrine Therapy for Postmenopausal Women with Hormone Receptor-Positive Breast Cancer. Cancer Research. 2014;69(Suppl. 24, article 76) doi: 10.1158/0008-5472.SABCS-09-76. [DOI] [Google Scholar]
- 25.Dowsett M., Ebbs S. R., Dixon J. M., et al. Biomarker changes during neoadjuvant anastrozole, tamoxifen, or the combination: Influence of hormonal status and HER-2 in breast cancer - A study from the IMPACT trialists. Journal of Clinical Oncology. 2005;23(11):2477–2492. doi: 10.1200/JCO.2005.07.559. [DOI] [PubMed] [Google Scholar]
- 26.Lipton A., Ali S. M., Leitzel K., et al. Serum HER-2/neu and response to the aromatase inhibitor letrozole versus tamoxifen. Journal of Clinical Oncology. 2003;21(10):1967–1972. doi: 10.1200/JCO.2003.09.098. [DOI] [PubMed] [Google Scholar]
- 27.Lipton A., Ali S. M., Leitzel K., et al. Elevated serum HER-2/neu level predicts decreased response to hormone therapy in metastatic breast cancer. Journal of Clinical Oncology. 2002;20(6):1467–1472. doi: 10.1200/JCO.20.6.1467. [DOI] [PubMed] [Google Scholar]
- 28.Houston S. J., Plunkett T. A., Barnes D. M., Smith P., Rubens R. D., Miles D. W. Overexpression of c-erbB2 is an independent marker of resistance to endocrine therapy in advanced breast cancer. British Journal of Cancer. 1999;79(7-8):1220–1226. doi: 10.1038/sj.bjc.6690196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez E. A., Barrios C., Eiermann W., et al. Trastuzumab Emtansine With or Without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2–Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. Journal of Clinical Oncology. 2017;35(2):141–148. doi: 10.1200/JCO.2016.67.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Breast Cancer (Version 4.2017). https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed March 1, 2018.
- 31.Kaufman B., Mackey J. R., Clemens M. R., et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: Results from the randomized phase III TAnDEM study. Journal of Clinical Oncology. 2009;27(33):5529–5537. doi: 10.1200/JCO.2008.20.6847. [DOI] [PubMed] [Google Scholar]
- 32.Gianni L., Pienkowski T., Im Y.-H., et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. The Lancet Oncology. 2012;13(1):25–32. doi: 10.1016/S1470-2045(11)70336-9. [DOI] [PubMed] [Google Scholar]
- 33.Baselga J., Bradbury I., Eidtmann H. Erratum: Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): A randomised, open-label, multicentre, phase 3 trial (The Lancet (2012) 379 (633-640)) The Lancet. 2012;379(9816):p. 616. doi: 10.1016/S0140-6736(12)60268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buzdar A. U., Suman V. J., Meric-Bernstam F., et al. Fluorouracil, epirubicin, and cyclophosphamide (FEC-75) followed by paclitaxel plus trastuzumab versus paclitaxel plus trastuzumab followed by FEC-75 plus trastuzumab as neoadjuvant treatment for patients with HER2-positive breast cancer (Z1041): A randomised, controlled, phase 3 trial. The Lancet Oncology. 2013;14(13):1317–1325. doi: 10.1016/S1470-2045(13)70502-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudas M., Lehnert M., Huynh A., et al. Cyclin D1 expression in breast cancer patients receiving adjuvant tamoxifen-based therapy. Clinical Cancer Research. 2008;14(6):1767–1774. doi: 10.1158/1078-0432.CCR-07-4122. [DOI] [PubMed] [Google Scholar]
- 36.Kenny F. S., Hui R., Musgrove E. A., et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clinical Cancer Research. 1999;5(8):2069–2076. [PubMed] [Google Scholar]
- 37.Finn R. S., Dering J., Conklin D., et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Research. 2009;11(5, article no. R77) doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finn R. S., Crown J. P., Lang I., et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. The Lancet Oncology. 2015;16(1):25–35. doi: 10.1016/s1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 39.Turner N. C., et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. The New England Journal of Medicine. 2015;373(3):209–219. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 40.Finn R. Abstract MS1-2: Targeting CDK 4/6 in breast cancer. Cancer Research. 2016;76(4 Supplement):MS1-2–MS1-2. doi: 10.1158/1538-7445.SABCS15-MS1-2. [DOI] [Google Scholar]
- 41.Cen L., Carlson B. L., Schroeder M. A., et al. P16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro-Oncology. 2012;14(7):870–881. doi: 10.1093/neuonc/nos114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olanich M. E., Sun W., Hewitt S. M., Abdullaev Z., Pack S. D., Barr F. G. CDK4 amplification reduces sensitivity to CDK4/6 inhibition in fusion-positive rhabdomyosarcoma. Clinical Cancer Research. 2015;21(21):4947–4959. doi: 10.1158/1078-0432.CCR-14-2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang C., Li Z., Bhatt T., et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36(16):2255–2264. doi: 10.1038/onc.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akli S., Bui T., Wingate H., et al. Low-molecular-weight cyclin E can bypass letrozole-induced G1 arrest in human breast cancer cells and tumors. Clinical Cancer Research. 2010;16(4):1179–1190. doi: 10.1158/1078-0432.CCR-09-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scaltriti M., Eichhorn P. J., Cortés J., et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proceedings of the National Acadamy of Sciences of the United States of America. 2011;108(9):3761–3766. doi: 10.1073/pnas.1014835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herrera-Abreu M. T., Palafox M., Asghar U., et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Research. 2016;76(8):2301–2313. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caldon C. E., Sergio C. M., Kang J., et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Molecular Cancer Therapeutics. 2012;11(7):1488–1499. doi: 10.1158/1535-7163.MCT-11-0963. [DOI] [PubMed] [Google Scholar]
- 48.Konecny G. E., Winterhoff B., Kolarova T., et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clinical Cancer Research. 2011;17(6):1591–1602. doi: 10.1158/1078-0432.ccr-10-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abukhdeir A. M., Vitolo M. I., Argani P., et al. Tamoxifen-stimulated growth of breast cancer due to p21 loss. Proceedings of the National Acadamy of Sciences of the United States of America. 2008;105(1):288–293. doi: 10.1073/pnas.0710887105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cariou S., Donovan J. C. H., Flanagan W. M., Milic A., Bhattacharya N., Slingerland J. M. Down-regulation of p21WAF1/CIP1 or p27KiP1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proceedings of the National Acadamy of Sciences of the United States of America. 2000;97(16):9042–9046. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dean J. L., Thangavel C., McClendon A. K., Reed C. A., Knudsen E. S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene. 2010;29(28):4018–4032. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 52.Stendahl M., Nilsson S., Wigerup C., et al. P27Kip1 is a predictive factor for tamoxifen treatment response but not a prognostic marker in premenopausal breast cancer patients. International Journal of Cancer. 2010;127(12):2851–2858. doi: 10.1002/ijc.25297. [DOI] [PubMed] [Google Scholar]
- 53.Merenbakh-Lamin K., Ben-Baruch N., Yeheskel A., et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Research. 2013;73(23):6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 54.Robinson D. R., Wu Y.-M., Vats P., et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature genetics. 2013;45(12):1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeselsohn R., Yelensky R., Buchwalter G. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical Cancer Research. 2014;20(7):1757–1767. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fribbens C., O'Leary B., Kilburn L., et al. Plasma ESR1 Mutations and the treatment of estrogen receptor-Positive advanced breast cancer. Journal of Clinical Oncology. 2016;34(25):2961–2968. doi: 10.1200/JCO.2016.67.3061. [DOI] [PubMed] [Google Scholar]
- 57.Chandarlapaty S., Chen D., He W., et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncology. 2016;2(10):1310–1315. doi: 10.1001/jamaoncol.2016.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toy W., Weir H., Razavi P., et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discovery. 2017;7(3):277–287. doi: 10.1158/2159-8290.CD-15-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arpino G., Gutierrez C., Weiss H., et al. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. Journal of the National Cancer Institute. 2007;99(9):694–705. doi: 10.1093/jnci/djk151. [DOI] [PubMed] [Google Scholar]
- 60.Scaltriti M., Rojo F., Ocaña A., et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. Journal of the National Cancer Institute. 2007;99(8):628–638. doi: 10.1093/jnci/djk134. [DOI] [PubMed] [Google Scholar]
- 61.Rexer B. N., Arteaga C. L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Critical Reviews in Oncogenesis. 2012;17(1):1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amin D. N., Ruiz-Saenz A., Gulizia N., Moasser M. M. Chemical probing of HER2-amplified cancer cells identifies TORC2 as a particularly effective secondary target for combination with lapatinib. Oncotarget . 2015;6(38):41123–41133. doi: 10.18632/oncotarget.5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang J., Wang S., Lyu H., et al. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Molecular Cancer. 2013;12(1, article no. 134) doi: 10.1186/1476-4598-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schoeberl B., Faber A. C., Li D., et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Research. 2010;70(6):2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schoeberl B., Kudla A., Masson K., et al. Systems biology driving drug development: from design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121) NPJ Systems Biology and Applications. 2017;3(1, article 16034) doi: 10.1038/npjsba.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venditti M., Iwasiow B., Orr F. W., Shiu R. P. C. C-myc gene expression alone is sufficient to confer resistance to antiestrogen in human breast cancer cells. International Journal of Cancer. 2002;99(1):35–42. doi: 10.1002/ijc.10269. [DOI] [PubMed] [Google Scholar]
- 67.Goga A., Yang D., Tward A. D., Morgan D. O., Bishop J. M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nature Medicine. 2007;13(7):820–827. doi: 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- 68.Kang J., Sergio C. M., Sutherland R. L., Musgrove E. A. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer. 2014;14(1, article no. 32) doi: 10.1186/1471-2407-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fox E. M., Arteaga C. L., Miller T. W. Abrogating endocrine resistance by targeting ERα and PI3K in breast cancer. Frontiers in Oncology. 2012;2 doi: 10.3389/fonc.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campbell M. R., Moasser M. M. HER targeting in HER2-Negative breast cancers: Looking for the HER3 Positive. Clinical Cancer Research. 2015;21(13):2886–2888. doi: 10.1158/1078-0432.CCR-14-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amin D. N., Sergina N., Ahuja D., et al. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Science Translational Medicine. 2010;2(16, article 16ra7) doi: 10.1126/scitranslmed.3000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller T. W., Balko J. M., Arteaga C. L. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. Journal of Clinical Oncology. 2011;29(33):4452–4461. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baselga J., Campone M., Piccart M., et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England Journal of Medicine. 2012;366(6):520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cristofanilli M., Turner N. C., Bondarenko I., et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. The Lancet Oncology. 2016;17(4):425–439. doi: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- 75.Rimawi M., Cecchini R., Rastogi P., et al. Abstract S3-06: A phase III trial evaluating pCR in patients with HR+, HER2-positive breast cancer treated with neoadjuvant docetaxel, carboplatin, trastuzumab, and pertuzumab (TCHP) +/- estrogen deprivation: NRG Oncology/NSABP B-52. Cancer Research. 2017;77(Suppl. 4):S3–S6. doi: 10.1158/1538-7445.SABCS16-S3-06. [DOI] [Google Scholar]
- 76.Witkiewicz A. K., Cox D., Knudsen E. S. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes & Cancer. 2014;5(7-8):261–272. doi: 10.18632/genesandcancer.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nikolai B. C., Lanz R. B., York B., et al. HER2 signaling drives DNA anabolism and proliferation through SRC-3 phosphorylation and E2F1-regulated genes. Cancer Research. 2016;76(6):1463–1475. doi: 10.1158/0008-5472.CAN-15-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tolaney S. M., et al. A phase II randomized study to compareabemaciclib plustrastuzumab with or withoutfulvestrant to standard of care chemotherapy plustrastuzumab in hormone receptor positive, HER2-positive advanced breast cancer (monarcHER) Journal of Clinical Oncology. 2017;35(Suppl. 15):S1109–S1109. [Google Scholar]
- 79.Hortobagyi G. N., Stemmer S. M., Burris H. A., et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. The New England Journal of Medicine. 2016;375(18):1738–1748. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 80.Sledge G. W., Toi M., Neven P., et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. Journal of Clinical Oncology. 2017;35(25):2875–2884. doi: 10.1200/JCO.2017.73.7585. [DOI] [PubMed] [Google Scholar]
- 81.Goetz M. P., Toi M., Campone M., et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. Journal of Clinical Oncology. 2017;35(32):3638–3646. doi: 10.1200/JCO.2017.75.6155. [DOI] [PubMed] [Google Scholar]
- 82.Dickler M. N., Tolaney S. M., Rugo H. S., et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, n patients with refractory HR+/HER2- metastatic breast cancer. Clinical Cancer Research. 2017;23(17):5218–5224. doi: 10.1158/1078-0432.CCR-17-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tolaney S. M., Lin N. U., Thornton D., et al. Abemaciclib for the treatment of brain metastases (BM) secondary to hormone receptor positive (HR+), HER2 negative breast cancer. Journal of Clinical Oncology. 2017;35(Suppl. 15):1019–1019. [Google Scholar]
- 84.Musgrove E. A., Sutherland R. L. Biological determinants of endocrine resistance in breast cancer. Nature Reviews Cancer. 2009;9(9):631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 85.Geradts J., Wilson P. A. High frequency of aberrant p16(INK4A) expression in human breast cancer. The American Journal of Pathology. 1996;149(1):15–20. [PMC free article] [PubMed] [Google Scholar]
- 86.Gillett C., Fantl V., Smith R., et al. Amplification and Overexpression of Cyclin D1 in Breast Cancer Detected by Immunohistochemical Staining. Cancer Research. 1994;54(7):1812–1817. [PubMed] [Google Scholar]
- 87.Ormandy C. J., Musgrove E. A., Hui R., Daly R. J., Sutherland R. L. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Research and Treatment. 2003;78(3):323–335. doi: 10.1023/A:1023033708204. [DOI] [PubMed] [Google Scholar]
- 88.Arnold A., Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. Journal of Clinical Oncology. 2005;23(18):4215–4224. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]
- 89.Citri A., Skaria K. B., Yarden Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Experimental Cell Research. 2003;284(1):54–65. doi: 10.1016/S0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 90.Cadoo K. A., Gucalp A., Traina T. A. Palbociclib: An evidence-based review of its potential in the treatment of breast cancer. Breast Cancer : Targets and Therapy. 2014;6:123–133. doi: 10.2147/BCTT.S46725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chin K., DeVries S., Fridlyand J., et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10(6):529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 92.Zwijsen R. M. L., Wientjens E., Klompmaker R., Van der Sman J., Bernards R., Michalides R. J. A. M. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88(3):405–415. doi: 10.1016/S0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
- 93.Zwijsen R. M. L., Buckle R. S., Hijmans E. M., Loomans C. J. M., Bernards R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes & Development. 1998;12(22):3488–3498. doi: 10.1101/gad.12.22.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Casimiro M. C., Wang C., Li Z., et al. Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Molecular Endocrinology. 2013;27(9):1415–1428. doi: 10.1210/me.2013-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang L., Wang J., Blaser B. W., et al. Pharmacologic inhibition of CDK4/6: Mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood. 2007;110(6):2075–2083. doi: 10.1182/blood-2007-02-071266. [DOI] [PubMed] [Google Scholar]
- 96.Shapiro G. I. Cyclin-dependent kinase pathways as targets for cancer treatment. Journal of Clinical Oncology. 2006;24(11):1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 97.Hwang H. C., Clurman B. E. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 98.Zhou W., Srinivasan S., Nawaz Z., Slingerland J. M. ERα, SKP2 and E2F-1 form a feed forward loop driving late ERα targets and G1 cell cycle progression. Oncogene. 2014;33(18):2341–2353. doi: 10.1038/onc.2013.197. [DOI] [PubMed] [Google Scholar]
- 99.Bozic I., Reiter J. G., Allen B., et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2, article e00747 doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson T. R., Yu J., Lu X., et al. The molecular landscape of high-risk early breast cancer: comprehensive biomarker analysis of a phase III adjuvant population. NPJ Breast Cancer. 2016;2(1, article 16022) doi: 10.1038/npjbcancer.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sørlie T., Tibshirani R., Parker J. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Acadamy of Sciences of the United States of America. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Asghar U., Witkiewicz A. K., Turner N. C., Knudsen E. S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature Reviews Drug Discovery. 2015;14(2):130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lapenna S., Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nature Reviews Drug Discovery. 2009;8(7):547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]