

SUMMARY
Lysine 2-hydroxyisobutyrylation (Khib) is an evolutionarily conserved and widespread histone mark like lysine acetylation (Kac). Here we report that p300 functions as a lysine 2-hyroxyisobutyryltransferase to regulate glycolysis in response to nutritional cues. We discovered that p300 differentially regulates the Khib and Kac on distinct lysine sites, with only 6 out of the 149 p300-targeted Khib sites overlapping with the 693 p300-targeted Kac sites. We demonstrated that diverse cellular proteins, particularly glycolytic enzymes, are targeted by p300 for Khib but not for Kac. Specifically, deletion of p300 significantly reduces Khib levels on several p300-dependent, Khib-specific sites on key glycolytic enzymes including ENO1, decreasing their catalytic activities. Consequently, p300 deficient cells have impaired glycolysis and are hypersensitive to glucose depletion-induced cell death. Our study reveals a p300-catalyzed, Khib-specific molecular mechanism that regulates cellular glucose metabolism, and further indicate that p300 has an intrinsic ability to select short-chain acyl-CoA-dependent protein substrates.
Keywords: p300, lysine 2-hydroxyisobutyrylation, glycolysis, cell survival
eTOC Blurb
Huang et al. show that p300-dependent, lysine 2-hydroxyisobutyrylation-specific modification of glycolytic enzymes promotes glycolysis and increases cell survival in response to glucose depletion.
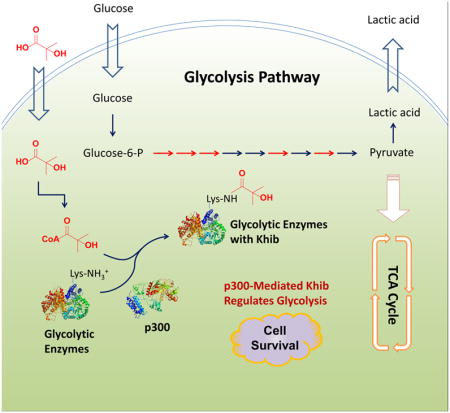
INTRODUCTION
Glucose is the major carbon source for energy production and biosynthesis in most cells of the body, and tight regulation of systemic glucose homeostasis in response to hormonal, nutritional, and environmental cues is critical for survival (Carey et al., 2013; Girard, 1995; Han et al., 2016). Protein post-translational modifications (PTMs), such as phosphorylation and lysine acetylation (Kac), are known to swiftly modulate the activities of key glycolytic enzymes in response to acute nutritional signals. Emerging evidence suggests that host glucose metabolism can also be regulated by gut bacteria through microbe specific production of metabolites (such as short-chain fatty acids) and lipopolysaccharides (Morrison and Preston, 2016; Schroeder and Backhed, 2016; Tremaroli and Backhed, 2012). However, the molecular mechanisms underlying PTMs and the host-environment interactions that affect cellular glucose metabolism remain unclear.
2-Hydroxyisobutyrate (hib), also known as α-hydroxyisobutyrate, is a short-chain fatty acid that has been detected at micromolar concentrations in a variety of bio-fluids in humans, including blood, urine, saliva, and feces (Bouatra et al., 2013; Dame et al., 2015; Guneral and Bachmann, 1994; Hoffmann et al., 1993; Husek et al., 2016; Psychogios et al., 2011). In particular, it is detected at high levels in the urine of obese patients and is associated with the presence of specific gut microbiota (Calvani et al., 2010; Li et al., 2008). Intriguingly, in both humans and microorganisms, 2-hydroxyisobutyrate is a key intermediate metabolite during degradation of methyl tert-butyl ether (MTBE) (Dekant et al., 2001; Francois et al., 2002; Lopes Ferreira et al., 2006; Steffan et al., 1997), a widely used gasoline additive and the second most common contaminant of urban aquifers in the US, yet our knowledge on the impacts of this environmental metabolite on human health is still limited. Our recent study has shown that 2-hydroxyisobutyrate is a precursor for the synthesis of 2-hydroxyisobutyryl-CoA that mediates a widespread histone mark, lysine 2-hydroxyisobutyrylation (Khib) (Dai et al., 2014). This new chemical modification poses a unique chemical structure and distinguished genomic distributions that are different from the well-studied histone Kac and lysine methylation (Kme) marks, and is associated with active gene transcription (Dai et al., 2014). These observations raise the possibility that 2-hydroxyisobutyrate may directly mediate environmental influences on the epigenome and other biological processes through protein Khib. However, how this modification is regulated remains unclear, and the biological consequences of this modification are almost completely unknown.
In this report, we identified p300 as a “writer” for Khib in mammalian cells. Using a Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-based quantitative proteomics approach, we identified 4239 unique Khib sites in wild type (WT) and p300 knock out (KO) HCT116 cells, including 149 sites significantly decreased in response to deletion of p300. Remarkably, only 6 out of the 149 sites overlapped with the Kac sites mediated by p300, suggesting a unique regulatory mechanism for p300 to differentially target either Khib or Kac. Moreover, the p300-mediated Khib directly targets the glycolytic enzymes in human cells and is essential for maintenance of homeostatic glucose metabolism and cell survival in response to glucose restriction. Our findings illuminate the landscape of the Khib in mammalian cells and reveal an important role of p300 in regulation of glycolysis through Khib.
RESULTS
p300 Catalyzes Khib on Histones
Khib is a widespread histone mark like histone Kac (Dai et al., 2014), raising the possibility that certain histone lysine acetyltransferases (KATs) may catalyze the generation of this modification on histones. p300 is a transcriptional co-activator that regulates gene transcription through acylation of histones (Dancy and Cole, 2015). Given that p300 is able to catalyze multiple types of lysine acylations such as acetylation and crotonylation (Sabari et al., 2015), we hypothesized that it has a catalytic activity toward Khib.
To test this hypothesis, we sought to determine whether alterations of p300 expression levels could change histone Khib levels in cells. Consistent with the well-known KAT activity of p300, the Kac levels on histones were decreased upon deletion of p300 (Figure 1A), while increased upon transient overexpression of wild-type p300 but not its catalytically deficient mutant (Figure 1B), providing a positive control. Notably, the Khib levels on core histones, as detected by a previously well-characterized specific Pan-Khib antibody (Dai et al., 2014) and various site-specific antibodies against different histone Khib marks, followed a similar pattern in response to changes of cellular p300 levels (Figures 1A and 1B). Further immunofluorescence assay indicated that p300 overexpression was associated with increased nuclear levels of H4K8hib in U2OS cells (Figure 1C), suggesting that p300 can positively regulate the Khib levels in cells. To confirm these observations, we treated different human cell lines with two p300 inhibitors, A485, a newly reported potent p300 inhibitor (Lasko et al., 2017), and C646, one of the most well-known p300 inhibitors (Bowers et al., 2010), respectively. As shown in Figure 1D, inhibition of p300 by A485 dose-dependently reduced both Kac and Khib levels in HCT116 cells (Figure 1D, top panels). Interestingly, the Khib levels on global histones in HEK293T cells decreased in a dose-dependent manner, while the Kac levels did not show obvious changes upon A485 treatment (Figure 1D, bottom panels). High doses of C646 decreased both Khib and Kac levels in HeLa cells, yet only slightly decreased Kac but not Khib in HEK293T cells (Figure S1A). These observations strongly suggest that p300 is capable of catalyzing both Khib and Kac reactions on histones in cells, and different p300 inhibitors affect one activity more than the other but in a cell-type specific manner.
Figure 1. p300 Catalyzes Histone Khib.

(A) p300 deficiency impairs both Khib and Kac of histones in cells. Histone Khib and Kac levels were analyzed in WT and p300 KO HCT116 cells by immuno-blotting with indicated antibodies.
(B) Overexpression of WT but not a mutant p300 increases both Khib and Kac levels on histones. HEK293T cells were transfected with either empty vector (V), or constructs expressing Flag-p300 (WT) or Flag-p300D1399Y (Mut) for 48 hours. The indicated histone modification marks were analyzed by immuno-blotting.
(C) Overexpression of WT p300 enhances both Khib and Kac of histones in U2OS cells. U2OS cells transfected with WT p300 were immuno-stained with anti-H4K8hib and anti-H3K9ac antibodies, respectively. Please note that cells overexpressing FLAG-p300 have higher levels of H4K8hib (left) and H3K9ac (right) than untransfected cells. Bars, 50 μm.
(D) Inhibition of p300 dose-dependently reduces Khib and Kac levels of histones in cells. HCT116 and HEK293T cells were treated with a p300 inhibitor A485 for 24 hours, and the Khib and Kac levels of histones were analyzed by immuno-blotting with indicated antibodies.
(E) p300 2-hydroxyisobutyrates histone octamers in actively transcribed chromatin in vitro. p300-dependent in vitro transcription system was set up as described in STAR Methods, and the Khib and Kac levels of histones were analyzed by immuno-blotting with indicated antibodies.
(F) p300-mediated histone 2-hydroxyisobutyration activates p53-dependent transcription in vitro. p300-dependent in vitro transcription system was set up using WT histones and K-R mutant histones. RNA products were visualized by autoradiography.
See also Figure S1.
To validate that p300 indeed directly catalyzes Khib modification on histones thereby regulating gene transcription, we took advantage of a cell-free p53-dependent transcription system wherein p300-catalyzed acylation on recombinant chromatin can stimulate in vitro transcription (Figure S1B) (Tang et al., 2013). Acetyl-CoA (Ac-CoA) and 2-hydroxyisobutyryl-CoA (Hib-CoA) were added separately in this system, with Ac-CoA as a positive control. As shown in Figure 1E, p300 increased the Kac levels on H3K27 (K3K27ac) and the Khib levels on H3K18 and H4K8 (H3K18hib and H4K8hib) only when p53 and respective coenzymes were added together with recombinant chromatin, indicating that p300 is able to directly acetylate or 2-hydroxyisobutyrylate histones on actively transcribed chromatin in vitro. Furthermore, in line with the observation that Khib of histones is associated with active gene transcription (Dai et al., 2014), this p300-mediated Khib of histones directly activated p53-dependent transcription in vitro in a WT histone dependent manner, as replacement of the wild type histone H3 or H4 with corresponding K-to-R mutations inhibited p300-driven transcription (Figure 1F). Therefore, p300-mediated Khib of histones is important for p300-driven transcriptional activation. Taken together, these findings demonstrate that p300 is not only a histone acetyltransferase but also a histone 2-hydroxyisobutyryltransferase in vitro and in a number of different human cell lines.
p300 2-Hydroxyisobutyrylates and Acetylates Distinct Sets of Substrate Proteins
Given that p300 showed a catalytic activity towards both Khib and Kac on histones, we next sought to investigate whether p300 could also mediate Khib on non-histone proteins, as it has been shown that p300 can shuttle between the nucleus and cytosol and has a broad substrate specificity (Dancy and Cole, 2015). Indeed, deletion of p300 in HCT116 cells substantially reduced both Khib and Kac levels on a number of non-histone proteins (Figure 2A). Adding 2-hydroxyisobutyrate (NaHib) into the culture medium dose-dependently increased Khib on various proteins (histones and non-histone proteins) in WT HCT116 cells (Figure 2B, WT), yet the protein Khib levels in p300 KO cells only increased slightly with no obvious dose-dependence upon treatment with NaHib (Figure 2B, KO), confirming the importance of p300 in mediating Khib modification on various proteins. The Khib transferase activities of other HATs may contribute to the slight increase of Khib in p300 KO cells after the NaHib treatment.
Figure 2. Deletion of p300 alters Khib levels on various protein substrates.

A) Deletion of p300 reduces Kac and Khib levels on various non-histone proteins. Total cell lysate from WT and p300 KO HCT116 cells were analyzed for Khib and Kac levels by immuno-blotting with indicated pan anti-Khib or anti-Kac antibody.
(B) 2-hydroxyisobutyrate dose-dependently increases total Khib levels on various cellular proteins in part through p300. WT and p300 KO HCT116 cells were treated with 2-hydroxyisobutyrate (Na-Hib) at indicated concentrations for 24 hours. Please note that Na-Hib treatment dose-dependently increased the Khib levels in WT cells, but this trend was blunted in p300 KO cells.
(C) p300 deficiency leads to systemic reduction of Khib and Kac on a number of protein substrates. The scatter plots show the ratio of Khib and Kac peptides in p300 KO vs WT cells in relation to average peptide intensities.
(D) p300 catalyzes Khib and Kac on distinct lysine residues in a variety of protein substrates. The Venn diagrams show overlap between p300-targeted Khib and Kac sites.
(E) The consensus sequence logos show enrichment of amino acid residues among the p300-targeted Khib and Kac sites.
To better understand the landscape of p300-mediated Khib in comparison with that of Kac in human cells, we quantified Khib and Kac proteomes in WT and p300 KO HCT116 cells using a global SILAC-based assay in conjunction with immunoaffinity enrichment with confirmed Pan-anti-Khib or anti-Kac antibody and MS analysis (Figure S2A). We identified a total of 4239 unique Khib sites on 1459 proteins and 3682 unique Kac sites on 1887 proteins, respectively, with varied numbers of modification sites on each substrate (Table S1, Figure S2B and S2C). Among the 4239 identified Khib sites, 973 sites were detected in the pellet fractions and 284 of these sites exclusively existed in the pellet fractions, suggesting that the pellet of NETN-based cell lysis is a valuable source for PTMs identification. Intriguingly, although some protein substrates bear both Kac and Khib sites, about 82% of the Khib sites did not overlap with Kac sites, suggesting that Kac and Khib pathways have unique regulatory mechanisms (Figure S2D). Furthermore, in contrast to the reported lysine malonylated and succinylated proteins which are mainly located in mitochondria (Park et al., 2013; Rardin et al., 2013), most of the identified acetylated and 2-hydroxyisobutylated proteins were exclusively nuclear, or partially nuclear or cytoplasmic (Figure S2E). This observation is consistent with the notion that p300 is a predominantly nuclear protein that can be shuttled to cytosol (Dancy and Cole, 2015).
To identify p300-targted Khib and Kac sites, we first quantified the relative abundance of Khib and Kac sites between WT and p300 KO HCT116 cells, then normalized the detected abundances to the expression of their corresponding proteins. The analysis identified 149 Khib sites and 693 Kac sites whose normalized abundance was decreased by more than 50% in p300 KO cells compared to WT cells (mean ratio of KO/WT < 0.5 or only identified in WT) (Figure 2C, Table S2). The proteins containing these lysine sites were therefore considered as potential p300-targeted substrates. Please note that we also detected 11 Khib sites whose normalized abundance was increased by more than 2 folds in addition to 149 decreased Khib sites in p300 KO cells, possibly due to an indirect regulation of diverse cellular processes, including the activities of HDACs, by p300 (Dancy and Cole, 2015).
Strikingly, only 6 out of the 149 p300-targeted Khib sites overlapped with the 693 p300-targeted Kac sites (Figure 2D), suggesting that p300 has different substrate selectivity toward Khib and Kac. This low overlap between p300-targeted Khib and Kac sites further implies that p300 may have different sequence preferences toward these two PTMs. In support of this notion, flanking sequence analysis (Colaert et al., 2009) of all the p300-targeted Khib or Kac sites revealed that the flanking sequence motif of Khib substrates had over-representation of hydrophobic amino acids alanine and isoleucine at −1, −2, −3, −4, and +2 position, while positively charged amino acids lysine and arginine were enriched at −5, −6, +5, and +6 positions, and arginine at −1 position was largely depleted (Figure 2E, Khib). In contrast, the negatively charged glutamic acid and aspartic acid residues were enriched at −1 position in the Kac flanking motif. Moreover, lysine was preferred from +2 to +6 positions, while arginine was largely depleted from −1 to −4 positions (Figure 2E, Kac). These results indicate that p300-mediated Khib and Kac target distinct sequences in proteins.
p300-mediated Khib Specifically Targets Metabolic Pathways, Particularly Glycolytic Enzymes
To explore the functional impact of p300-mediated Kac and Khib, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Kanehisa and Goto, 2000) on potential p300-targeted substrates identified in Figures 2C, 2D, and Table S2. As shown in Figure 3A, although both p300-mediated modifications impacted the spliceosome and ribosome, p300-mediated Kac primarily targeted proteins involved in RNA biology, whereas p300-mediated Khib was selectively enriched in multiple metabolism-related pathways, such as carbon metabolism and biosynthesis of amino acids, as well as glycolysis/gluconeogenesis. Glycolysis converts glucose into pyruvate by a cascade of 10 key enzyme-catalyzed reactions (Lunt and Vander Heiden, 2011). Remarkably, 5 out of the 10 glycolytic enzymes were 2-hydroxyisobutylated by p300, including glucose-6-phosphate isomerase (GPI), ATP-dependent 6-phosphofructokinase muscle type (PFKM), fructose-bisphosphate aldolase A (ALDOA), phosphoglycerate kinase 1 (PGK1), and alpha/gamma-enolase (ENO1/2) (Figures 3B and 3C).
Figure 3. p300-mediated Khib specifically targets glycolytic enzymes.

(A) p300-catalyzed Khib has a unique impact on cellular metabolism. The bar graphs show KEGG pathways enriched in p300-targeted Khib and Kac proteomes.
(B) Deletion of p300 reduces Khib levels on 5 key glycolytic enzymes. The bar graphs show the KO/WT ratio of p300-targeted Khib sites on the key enzymes in the glycolysis pathway.
(C) The full MS and MS/MS spectra of ENO1 K281hib peptide for its identification and quantification. The b and y ions indicate peptide backbone fragment ions containing the N and C terminals, respectively.
(D) p300-mediated Khib specifically target glycolytic enzymes. Interaction Network of p300-regulated Khib Proteome Based on STRING Database (v10). The network is visualized in Cytoscape, and the size of the node is positively corresponded to the number of Khib sites per protein.
See also Table S2.
Consistent with the enriched KEGG pathways, the protein interaction networks of p300-regulated Khib proteins based on STRING database (Szklarczyk et al., 2015) showed several functional clusters, including glycolysis/gluconeogenesis (Figure 3D).
p300 Deficiency Impairs Glycolysis through Khib of Key Glycolytic Enzymes
The specific enrichment of p300-targeted Khib proteins in the metabolic pathways suggests that p300 may modulate cellular metabolic homeostasis through Khib. To test this possibility, we first investigated whether p300 deficiency could lead to defects in any metabolic pathways through a large scale unbiased metabolomics analysis. Systematic analysis of 364 detectable metabolites from WT and p300 KO HCT116 cells revealed that p300 deficiency resulted in alteration in a number of central metabolic pathways, including glycolysis, pentose phosphate pathway (PPP), TCA cycle, and glutamine metabolism when cultured in complete DMEM-based medium (Figures 4A, S3A, and Table S3). Further analyses using the MetaboAnalyst pointed out that glycolysis or gluconeogenesis was the most significantly down-regulated metabolic pathway in p300 KO HCT116 cells (Figures 4B, S3B, and S3C). The relative intracellular abundance of many key metabolites related to glycolysis, such as pyruvate, ATP, and NADH, were all significantly reduced in p300 KO HCT116 cells compared to WT cells (Figure 4C). Consistently, p300 KO HCT116 cells had reduced glucose consumption and lactate production compared to WT HCT116 cells (Figure 4D). Seahorse analyses further confirmed that p300 KO cells displayed reduced glycolysis, glycolytic capacity, and glycolytic reserve compared to WT cells (Figures 4E, 4F, and S3D). These defects in glycolysis were even more evident after depletion of glucose from the culture medium (Figure 4G). Specifically, 6 hours after glucose depletion, all detectable intermediate metabolites in the glycolysis pathway were significantly reduced in p300 KO HCT116 cells compared to WT cells in the same medium, particularly those on the nodes catalyzed by the 5 hypo-2-hydroxyisobutyrylated enzymes, GPI, PFK, ALDO, PGK1, and ENO1 (Figure 4G, highlighted in blue boxes). As expected from the reduction of glycolysis, the intracellular glucose was significantly accumulated in p300 KO cells in this glucose depletion condition (Figure 4G, Glucose). Therefore, p300 deficiency in HCT116 cells impairs glycolysis.
Figure 4. p300 Deficiency Results in Defective Glycolysis.

(A) p300 KO HCT116 cells have altered glycolysis, TCA cycle, and glutamine metabolism. WT and p300 KO HCT116 cells cultured in complete medium were used for metabolomics analysis. The relative abundance of metabolites involved in glycolysis, TCA cycle, PPP, and glutamine pathway was displayed by the heat map (n=3 independent experiments).
(B) Deletion of p300 significantly impairs glycolysis or gluconeogenesis. The metabolites significantly reduced in p300 KO cells (fold change: KO/WT < 0.5, and p<0.05) were analyzed by pathway enrichment analysis and pathway topology analysis in the Pathway Analysis module of MetaboAnalyst 3.0, and the pathway impact scores of the top ten pathways were shown.
(C) p300 KO HCT116 cells have reduced levels of many metabolites related to glycolysis when cultured in complete medium. The relative amounts of indicated metabolites were quantified by metabolomics (n=3, *p<0.05, values are expressed as mean ±SEM).
(D) p300 KO HCT116 cells have reduced glucose consumption and lactate production when cultured in complete medium. The decrease of glucose and increase of lactate in culture medium were measured for indicated times (n=4, *p<0.05, values are expressed as mean ±SEM).
(E) p300 KO HCT116 cells have reduced glycolytic activities. The glycolysis activity was analyzed by the Seahorse analyzer as described in STAR Methods (n=4 technical repeats, values are expressed as mean ±SEM).
(F) p300 KO HCT116 cells have reduced glycolysis, glycolytic capacity, and glycolytic reserve (n=4 independent experiments from 2 pairs of WT and p300 KO HCT116 cells, *p<0.05, values are expressed as mean ±SEM).
(G) p300 KO HCT116 cells display reduced glycolysis after 6 hours of glucose depletion. WT and p300 KO HCT116 cells were cultured in medium without glucose for 6 hours, and the relative amounts of metabolites were quantified by metabolomics analysis (n=3). Metabolites not detected in the experiment were designated inside the parentheses. Five hypo-2-hydroxyisobutyrylated proteins in p300 KO cells were highlighted with blue boxes.
We next investigated whether the defective glycolysis observed in p300 deficient cells is due to hypo-2-hydroxyisobutyrylation of glycolytic enzymes. Compared with WT HCT116 cells, p300 KO HCT116 cells had comparable expression levels of the hypo-2-hydroxyisobutyrylated enzymes (Figures S4A and S4B). However, the activities of two of these enzymes (that have the established activity assays available), PFK and ENO, were significantly reduced in the total cell lysates from p300 KO HCT116 cells when measured in vitro (Figure 5A), supporting the idea that the activities of these enzymes are regulated at the posttranslational level, possibly by p300-mediated Khib. Consistent with this possibility, recombinant p300 protein directly 2-hydroxyisobutyrylated recombinant PFKM (Figure 5B, left panels) and immuno-purified HA-ENO1 from p300 KO cells (Figure 5B, right panels) in the presence of 2-hydroxyisobutyrylate-CoA in a cell-free system, whereas the Khib levels of endogenous PFKM were reduced in p300 KO cells (Figure S4C). In the case of ENO1, the glycolytic enzyme that catalyzes the conversion of 2-phsophoglycerate (2-P Glycerate) to phosphoenolpyruvate, both hypo-2-hydroxyisobutyrylated lysine residues (K228 and K281) were on its surface (Figure S4D), making them accessible to p300-mediated dynamic modifications. In support of the possibility that p300 activates glycolytic enzymes through Khib, p300-mediated in vitro Khib modification of ENO1 directly activated its enzymatic activity when measured in vitro (Figure 5C). Additionally, the Khib levels of ENO1 are sensitive to nutrient availability. The interaction between ENO1 and p300 as well as the Khib levels on ENO1 were increased in WT HCT116 cells shortly after glucose depletion then gradually went down (Figure 5D). Prolonged glucose deprivation, on the other hand, resulted in reduced Khib and degradation of ENO1 protein (Figure 5D).
Figure 5. p300 Regulates the Activities of Key Glycolytic Enzymes through Khib.

(A) Both PFK and ENO1 enzymes display reduced activities in p300 KO HCT116 cells. WT and p300 KO HCT116 cells were cultured in complete medium. The activities of PFK and ENO1 from total cell lysates were measured using colorimetric assay kits (n=3, *p<0.05, values are expressed as mean ±SEM).
(B) p300 2-hydroxyisobutyrylates PKFM and ENO1 in vitro. Immuno-purified HA-ENO1 proteins from p300 KO HCT116 cells and recombinant PFKM proteins were in vitro 2-hydroxyisobutyrylated by recombinant p300 as described in the STAR Methods.
(C) p300-mediated Khib activates ENO1 in vitro. Immuno-purified HA-ENO1 proteins from p300 KO HCT116 cells were in vitro 2-hydroxyisobutyrylated by recombinant p300, and their activities were then analyzed in vitro as described in the STAR Methods (n=3, *p<0.05, values are expressed as mean ±SEM). Please note that in addition to Khib on K228 and K281, ENO1 also had a number of Kac sites that were dynamically regulated by p300, including K228, K335, and K343 (Table S2). It appeared that p300-mediated Kac on these sites also activated ENO1 in vitro.
(D) Khib levels of ENO1 are dynamically regulated upon glucose depletion in cells. The Khib levels of HA-ENO1 and the interaction between ENO1 and p300 were analyzed at different time points after glucose depletion.
(E) An ENO1 mutant that cannot be 2-hydroxyisobutyrylated has reduced enzymatically activity when expressed in WT HCT116 cells where endogenous ENO1 was knocked down by shRNAs. The HA-ENO1 proteins were immuno-purified by anti-HA and the enzymatic activity of purified proteins were analyzed in vitro as described in the STAR Methods (n=3, values are expressed as mean ±SEM).
(F) HCT116 cells expressing K281R ENO1 have reduced glycolysis. The glycolysis activity was analyzed by the Seahorse analyzer (n=5 technical repeats, values are expressed as mean ±SEM).
(G) Overexpression of K281R ENO1 decreases glycolysis and glycolytic capacity in cells (n=3 independent experiments, *p<0.05, values are expressed as mean ±SEM).
See also Figure S4.
Finally, to directly assess the importance of Khib in modifying the activities of glycolytic enzymes thereby influencing glycolysis, we overexpressed WT ENO1 and an ENO1 mutant (K281R) that cannot be 2-hydroxyisobutyrylated at a major Khib site (K281) into WT HCT116 cells. In contrast to the K228 site of ENO1 that can be modified by p300 through both Kac and Khib (Table S2), the K281 site in ENO1 was not found to be acetylated in either WT or p300 KO HCT116 cells in our SILAC analysis, therefore was dynamically regulated by p300 through Khib but not Kac. Consistently, the K281R mutant HA-ENO1 protein had reduced Khib but not Kac levels when expressed in WT cells (Figure S4E), and immuno-purified HA-ENO1 K281R protein had about 4-fold reduction in its Vmax and roughly 3-fold reduction in its Km when 2-P Glycerate was used as the substrate in vitro compared with WT HA-ENO1 protein (Figure 5E). Furthermore, the HA-ENO1 K281R mutant protein had a comparable stability as the WT HA-ENO1 protein when overexpressed in the WT HCT116 cells (Figure S4F), and overexpression of this mutant protein was sufficient to reduce glycolysis (Figures 5F and 5G). Therefore, 2-hydroxyisobutyrylation of ENO1, the most downstream hypo-2-hydroxyisobutyrylated glycolytic enzyme in p300 KO HCT116 cells (Figure 4G), is an important element in p300-mediated regulation of glycolysis. Taken together, our observations indicate that p300-mediated Khib of glycolytic enzymes is critical for modulation of glycolysis in response to changes of nutrient availability.
p300 Mediates Nutritional Regulation of Cell Survival through Glycolysis
As the major carbon source for energy production and biosynthesis, glucose is essential for cell proliferation, growth, and survival. Reduced glycolysis in p300 KO HCT116 cells (Figures 4 and 5) raised the possibility that p300-deficient cells may be sensitive to glucose depletion-induced defects in cell proliferation and survival. In line with this possibility, p300 KO HCT116 cells displayed a reduced proliferation rate when cultured in complete medium containing 25 mM glucose (Figure 6A, Complete). Switching into a culture medium containing no glucose but not no glutamine led to dramatic loss of p300 KO cells within 24 hours compared to WT cells (Figures 6A and 6B). Further analysis indicated that p300 KO HCT116 cells were hypersensitive to glucose depletion-induced cell death compared with WT HCT116 cells (Figures 6C and 6D). In contrast, both WT and p300 KO HCT116 cells had a comparable sensitivity to glutamine depletion-induced cell death (Figures 6C and 6D), indicating that p300 specifically regulates the sensitivity to glucose depletion.
Figure 6. p300 Deficiency leads to hyper-sensitivity to glucose depletion.

(A) p300 KO HCT116 cells have reduced proliferation in different culture media. Cells were plated in the complete medium and switched to the indicated medium at day 1 (n=3, *p<0.05, values are expressed as mean ±SEM).
(B) p300 KO HCT116 cells are sensitive to glucose but not glutamine depletion. Cells were plated in the complete medium and switched to the indicated medium 24 hours later, and were then analyzed after 24 hour-incubation at the indicated media. Bar, 200 μm.
(C–D) p300 KO HCT116 cells display reduced cell survival and enhanced cell death upon glucose depletion. Cells were cultured as in (A) in indicated medium, and cell death was analyzed by FACS as described in STAR Methods (n=3, *p<0.05, values are expressed as mean ±SEM).
(E–F) HCT116 cells expressing K281R ENO1 have reduced cell survival upon glucose depletion. WT and p300 KO HCT116 cells, and WT HCT116 cells expressing WT ENO1 or K281R ENO1 proteins were incubated in complete medium or glucose-free medium for 24 hours, the cell survival was measured by the WST-1 reagent (n=3, *p<0.05, values are expressed as mean ±SEM). Bar in (E), 100 μm.
(G–H) Adding back pyruvate in culture medium rescues p300 deficiency-induced sensitivity to glucose depletion. WT and p300 KO HCT116 cells were cultured in complete medium, no glucose medium, or no glucose medium plus 1 mM pyruvate for 24 hours. Bar in (G), 100 μm.
To test whether the hypersensitivity of p300 KO cells to glucose depletion is due to diminished 2-hydroxyisobutyrylation of glycolytic enzymes, we analyzed the survival of HCT116 cells with overexpression of WT or K281R mutant ENO1 in response to glucose depletion. As shown in Figures 6E and 6F, WT HCT116 cells overexpressing the K281R mutant ENO1 experienced increased cell death after 24-hour depletion of glucose. This observation, together with K281R ENO1-induced defective glycolysis (Figures 5F and 5G), strongly suggests that hypo-2-hydroxyisobutyrylation of ENO1-induced defective glycolysis is sufficient to induce cell death in response to glucose withdrawal. Furthermore, supplementation of 1 mM pyruvate, the end product of cellular glycolytic pathway, into the culture medium rescued glucose depletion-induced cell death in p300 KO HCT116 cells (Figures 6G and 6H), indicating that p300 deficiency induced hyper-sensitivity to glucose depletion is due to defective glycolysis.
The HCT116 cells used in our study carry a heterozygous deletion in the exon 31 of the p300 gene, resulting in a truncated protein (Bryan et al., 2002; Iyer et al., 2004). To rule out the possible interference of this truncated p300 protein in our observations, we investigated whether p300 regulates cellular glucose metabolism through Khib in human cells only carrying the wild type p300 gene. As shown in Figure 7A, CRISPR/Cas9-mediated deletion of p300 in HEK293T cells, a human cell line with the wild type p300 gene, reduced the ENO1 activity (Figure 7B), decreased glycolysis (Figures 7C and 7D), and increased the sensitivity of HEK293T cells to glucose depletion induced cell death (Figures 7E, 7F, and S5A, Vector transfected WT and KO HEK293T cells), indicating that p300 deficiency in cells bearing the wild type p300 gene also leads to deficiency in glucose metabolism. Also in line with observations in HCT116 cells, overexpression of the K281R mutant ENO1 in WT HEK293T cells significantly reduced cell survival upon glucose depletion (Figures 7E, 7F, and S5A, WT HEK293T/K281R ENO1). Moreover, overexpression of neither WT nor K281R mutant ENO1 alter the hypersensitivity of p300 KO HEK293T cells to glucose depletion (Figures 7E, 7F, and S5A, KO HEK293T WT or K281R ENO1), indicating WT ENO1 behaves like the K281R mutant in the absence of p300, and further supporting the notion that p300-mediated 2-hydroxyisobutyrylation of ENO1 K281 is important in regulating glucose metabolism.
Figure 7. p300-mediated 2-hydroxyisobutyrylation regulates glycolysis in HEK293T cells.

(A) Deletion of p300 in HEK293T cells by the CRISPR/Cas9 gene editing technology. The protein levels of p300 in WT and p300 KO HEK293T cells were analyzed by immuno-blotting.
(B) p300 KO HEK293T cells have reduced activity of ENO1 (n=3, values are expressed as mean ±SEM).
(C) p300 KO HEK293T cells have reduced glycolysis. Representative ECAR profile from WT and p300 KO HEK293T cells is shown (n=5 technical repeats, values are expressed as mean ±SEM).
(D) p300 KO HEK293T cells have reduced glycolysis, glycolytic capacity, and glycolytic reserve (n=4 independent experiments, *p<0.05, values are expressed as mean ±SEM).
(E–F) p300 KO HEK293T cells are hypersensitive to glucose depletion. WT and p300 KO HEK293T cells transfected with constructs expressing indicated proteins were plated in the complete medium and switched to the indicated medium 24 hours later, and were then analyzed after 24 hour-incubation at the indicated media (n=3, values are expressed as mean ±SEM). Bar in (E), 100 μm.
(G) The ENO1 K281R has reduced enzymatically activity when expressed in HEK293T cells where endogenous ENO1 was knocked down by shRNAs. The HA-ENO1 proteins were immuno-purified by anti-HA beads from HEK293T cells and the enzymatic activity of purified proteins were analyzed in vitro as described (n=3, values are expressed as mean ±SEM).
(H) HEK293T cells expressing K281R ENO1 have reduced glycolysis (n=5 technical repeats, values are expressed as mean ±SEM).
See also Figure S5.
To further assess the contribution of 2-hydroxyisobutyrylation of ENO1 to this metabolic regulation in HEK293T cells, we generated HEK293T cells expressing endogenous levels of WT or K281R ENO1 by first overexpressing WT or K281R ENO1 into HEK293T cells, then knocking down the expression of endogenous ENO1 using shRNAs against the 3′ UTR of the endogenous ENO1 gene (Figures S5B and S5C). Consistent with our previous observations in Figure 5E, HA-ENO1 K281R protein immuno-purified from shENO1 HEK293T cells had reduced Vmax and Km compared to WT HA-ENO1 proteins (Figures 7G and S5C). More importantly, shENO1 HEK293T cells expressing this mutant protein displayed a reduced rate of glycolysis compared to shENO1 HEK293T cells expressing WT HA-ENO1 protein (Figure 7H), indicating that hypo-2-hydroxyisobutyrylation of ENO1 on K281 is sufficient to impair glycolysis in HEK293T cells. ShRNAs-mediated knockdown of p300 in SW620, another human cell line carrying the wild type p300 gene (Bryan et al., 2002), also resulted in hypersensitivity to glucose depletion induced cell death (Figures S5D–S5G). Collectively, our findings demonstrate that p300 mediates nutritional regulation of cell survival through 2-hydroxyisobutyrylation of glycolytic enzymes in various human cells.
DISCUSSION
In the past decade, a panel of short-chain and long-chain lysine acylations (or lipid lysine acylations) has been identified. While structurally similar, these lipids are generated by completely different metabolic pathways. Emerging evidence suggest that lipid lysine acylations are associated with not only cellular functions, such as gene transcription and metabolism, but also physiology and diseases (Goudarzi et al., 2016; Lin et al., 2012; Sabari et al., 2015; Sabari et al., 2017). Functional studies of these modification pathways require knowledge of their regulatory enzymes (writers and erasers), substrates, and binding proteins. A significant progress has been made in the identification of deacylation enzymes in the past several years, and the classically annotated HDACs were shown to have activities for not only acetylation but also other acetylation-independent acylations (Anderson et al., 2017; Bheda et al., 2016; Hirschey and Zhao, 2015; Jiang et al., 2013; Li et al., 2016; Sabari et al., 2017; Tasselli et al., 2017; Wei et al., 2017). However, characterization of acyltransferase lags far behind that of deacylation enzymes. While it was suggested that spontaneous chemical reaction is likely the major mechanism for lysine acylation reactions in mitochondria where there is no known acyltransferase and its pH is high (~8.0) (Casey et al., 2010), evidence indicates that enzyme-catalyzed reaction is mainly responsible for lysine acylations in cytosol and nuclei (Dancy and Cole, 2015; Liu et al., 2017; Sabari et al., 2015; Tang et al., 2013; Xu et al., 2017). In this study, we identified p300 as a “writer” for one of the most less-characterized lysine acylations, Khib. We discovered that p300 is capable of selectively catalyzing Khib and Kac on distinct protein substrates, and further uncovered a vital role of Khib of glycolytic enzymes in p300-mediated regulation of cellular glucose metabolism and cell survival in response to changes of nutrient availability. Our findings indicate that p300 has an intrinsic ability to select short-chain acyl-CoA-dependent substrate proteins, and offer new avenues to study the diverse functions of Khib in glycolysis related pathophysiology.
A long-standing question in the field of lysine acylations is the differences and specificity between Kac and other lysine acylations. Earlier studies on acyltransferases were mainly focused on histones in the context of chromatin localization and transcription (Sabari et al., 2017). Some classically annotated acetyltransferases, such as p300, CBP and MOF, can use a broad range of acyl-CoAs and thus catalyze reactions for both acetylation and other short-chain acylations (Chen et al., 2007; Dancy and Cole, 2015; Liu et al., 2017; Sabari et al., 2015). This observation raises an intriguing hypothesis that these enzymes have little selectivity and the difference between acetylation and other acylations are mainly controlled by the concentration (and/or availability) of short-chain-CoAs. Surprisingly, our quantitative proteomics data demonstrate that p300-targeted Khib proteome is very different from p300-targeted Kac proteome in terms of substrate proteins and substrate peptide sequences (Figures 2D and 2E). Thus, there is an internal regulatory element for p300 to select substrates for either lysine acetylation or other acylations.
What is the mechanism for p300 to control its substrate specificity among diverse acylations? It is highly likely that multiple possibilities exist for this large protein. A recent structural study on p300 in complex with acyl-CoA variants has shown that the aliphatic portions of p300-bound short-chain acyl-CoAs could be positioned in the binding tunnel of the lysine substrate, sterically clashing with the lysine substrate binding (Kaczmarska et al., 2017). Therefore, it is predicted that the lysine substrates need to remodel the acyl-CoAs into a conformation compatible with acyl-chain transfer. Different acyl-CoAs, as a result, require remodeling from distinct lysine substrates in order to accommodate in a hydrophobic pocket in the active site of p300. The size of the pocket and its aliphatic nature thus offer a possible alternative strategy for p300 to select acyl-CoA and its corresponding substrates. In addition, p300 could exist in different protein sub-complexes that may have unique substrate preference. Finally, p300 can be modified by acetylation, phosphorylation, and methylation (Dancy and Cole, 2015). It is conceivable that these modification profiles may modulate the structure of p300 that in turn changes its specificity.
It is still not clear how Khib regulates the activity of glycolytic enzymes. Since ENO1 has two Khib sites on the surface (Figure S4D), it is likely that replacing the positive charged lysine side chains with the bulky hydrophilic 2-hydroxyisobutyryl-groups will directly interfere with the binding of co-factors or introduce conformational changes that lead to altered substrate binding and turnover. PGK1, on the other hand, has a p300-targeted Khib site (K216hib) located within 5 residues of a key residue K220. Acetylation on K220 of PGK1 was reported to inhibit PGK1 activity by disrupting the binding with its substrate ADP (Wang et al., 2015). K116hib of PGK1, therefore, may also affect the ADP binding. Further biochemical and structural studies will help to dissect the exact mechanisms of Khib-mediated regulation of individual metabolic enzymes.
In summary, through quantitative proteomics, global metabolomics, and functional metabolic analysis, we discovered p300-mediated Khib as a molecular mechanism to regulate cellular glucose metabolism. We also find that p300 has different targets for Khib and Kac. Our study paves the way for future investigations on the diverse functions of Khib in mediating environmental influence on metabolism and metabolic diseases.
STAR Methods
Key Resources Table
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiaoling Li (lix3@niehs.nih.gov).
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-acetyllysine antibody (clone Kac-01)(anti-Pan Kac) | PTM Biolabs | Cat# PTM-101 |
| Rabbit polyclonal anti-acetyllysine antibody (anti-Pan Kac) | PTM Biolabs | Cat# PTM-105 |
| Rabbit polyclonal anti-2-hydroxyisobutyryllysine antibody (anti-Pan Khib) | PTM Biolabs | Cat# PTM-801 |
| Rabbit polyclonal anti-acetyl-Histone H3(Lys9) antibody | PTM Biolabs | Cat# PTM-112 |
| Mouse monoclonal anti-acetyl-Histone H3 (Lys18) antibody (clone 9E1) | PTM Biolabs | Cat# PTM-158 |
| Mouse monoclonal anti-acetyl-Histone H3 (Lys27) antibody (clone 12G5) | PTM Biolabs | Cat# PTM-160 |
| Rabbit polyclonal anti-acetyl-Histone H4(Lys8) antibody | PTM Biolabs | Cat# PTM-120 |
| Mouse monoclonal anti-2-hydroxyisobutyryl-Histone H3 (Lys18) antibody | PTM Biolabs | Cat# PTM-882 |
| Rabbit polyclonal anti-2-hydroxyisobutyryl-Histone H4(Lys8) antibody | PTM Biolabs | Cat# PTM-805 |
| Rabbit polyclonal anti-Histone H3 antibody | Abcam | Cat# ab1791 |
| Rabbit polyclonal anti-p300 antibody | Santa Cruz Biotechnology | Cat# sc-584 |
| Rabbit polyclonal anti-p300 antibody | Bethyl Laboratories | Cat# A300-358A |
| Mouse monoclonal anti-Flag antibody (clone M2) | Sigma-Aldrich | Cat# F1804 |
| Rabbit monoclonal anti-ENO1 antibody (clone EPR10864(B)) | Abcam | Cat# ab155955 |
| Rabbit polyclonal anti-PFK1 antibody | Santa Cruz Biotechnology | Cat# sc-67028 |
| Rabbit polyclonal anti-PFKM antibody | Bethyl Laboratories | Cat# A304-255A |
| Mouse monoclonal anti-HA antibody (clon F7) | Santa Cruz Biotechnology | Cat# sc-7392 |
| Rabbit polyclonal anti-HA antibody | Santa Cruz Biotechnology | Cat# sc-805 |
| Mouse monoclonal anti-Actin antibody (clon C4) | Millipore | Cat# MAB1501 |
| Bacterial and Virus Strains | ||
| One Shot BL21 (DE3) Competent Cells | Invitrogen | Cat# C600003 |
| Recombinant lentivirus pseudotyped with VSVG (for all Sigma Mission shRNA cloned in pLKO.1) | NIEHS Viral Core Facility | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-hydroxyisobutyryl-CoA | Synthesized (Huang et al., 2018) | N/A |
| acetyl-CoA | Sigma-Aldrich | Cat# A2056 |
| C646 | Sigma-Aldrich | Cat# SML0002 |
| A485 | Gift from Dr. Philip A. Cole at Johns Hopkins School of Medicine | |
| Recombinant human p300 protein | Active Motif | Cat# 31124 |
| Recombinant Human Phosphofructokinase 1/PFKM/PFK-X Protein (C-6His) | Novoprotein Scientific | Cat# C140 |
| Pyruvate Kinase/Lactic Dehydrogenase enzymes from rabbit muscle | Sigma-Aldrich | Cat# P0294 |
| D-(+)-2-Phosphoglyceric Acid Sodium | Santa Cruz Biotechnology | Cat# sc-218005 |
| Critical Commercial Assays | ||
| PFK activity assay kit | Sigma-Aldrich | Cat# MAK093 |
| ENO1 activity assay kit | Abcam | Cat# ab117994 |
| FITC Annexin V Apoptosis Detection Kit I | BD Biosciences | Cat# 556547 |
| Cell Proliferation Reagent WST-1 | Roche Applied Science | Cat# 05015944001 |
| QuikChange™ Multi Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200514-5 |
| Deposited Data | ||
| Mass spectrometry proteomics data | This paper | ProteomeXchange Consortium id: PXD008525 |
| The source file for unprocessed and uncompressed immuno-blots used in the figures | This paper | Mendeley Dataset: doi:10.17632/cszpvms38z.1 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL-247 |
| HEK293T | ATCC | CRL-3216 |
| HeLa | ATCC | CCL-2 |
| U2OS | ATCC | HTN-96 |
| SW620 | ATCC | CCL-227 |
| Oligonucleotides | ||
| HsENO1-F: ACCCAAAGAGGATCGCCAAG (qPCR) | This paper | N/A |
| HsENO1-R: CCCGAACGATGAGACACCAT (qPCR) | This paper | N/A |
| HsPFKM-F: GGTGCCCGTGTCTTCTTTGT (qPCR) | This paper | N/A |
| HsPFKM-R: AAGCATCATCGAAACGCTCTC (qPCR) | This paper | N/A |
| HsSGLT1-F: TACCTGAGGAAGCGGTTTGGA (qPCR) | This paper | N/A |
| HsSGLT1-R: CGAGAAGATGTCTGCCGAGA (qPCR) | This paper | N/A |
| HsGLUT1-F: TCTGGCATCAACGCTGTCTTC (qPCR) | This paper | N/A |
| HsGLUT1-R: CGATACCGGAGCCAATGGT (qPCR) | This paper | N/A |
| HsGLUT2-F: GCTGCTCAACTAATCACCATGC (qPCR) | This paper | N/A |
| HsGLUT2-R: TGGTCCCAATTTTGAAAACCCC (qPCR) | This paper | N/A |
| HsGPI-F: CAAGGACCGCTTCAACCACTT (qPCR) | This paper | N/A |
| HsGPI-R: TCCAGGATGGGTGTGTTTGACC (qPCR) | This paper | N/A |
| HsPFKL-F: GCTGGGCGGCACTATCATT (qPCR) | This paper | N/A |
| HsPFKL-R: TCAGGTGCGAGTAGGTCCG (qPCR) | This paper | N/A |
| HsALDOA-F: CAGGGACAAATGGCGAGACTA (qPCR) | This paper | N/A |
| HsALDOA-R: GGGGTGTGTTCCCCAATCTT (qPCR) | This paper | N/A |
| HsPGK1-F: GAACAAGGTTAAAGCCGAGCC (qPCR) | This paper | N/A |
| HsPGK1-R: GTGGCAGATTGACTCCTACCA (qPCR) | This paper | N/A |
| HsLDHA-F: TTGACCTACGTGGCTTGGAAG (qPCR) | This paper | N/A |
| HsLDHA-R: GGTAACGGAATCGGGCTGAAT (qPCR) | This paper | N/A |
| HsLDHB-F: CCTCAGATCGTCAAGTACAGTCC (qPCR) | This paper | N/A |
| HsLDHB-R: ATCACGCGGTGTTTGGGTAAT (qPCR) | This paper | N/A |
| HsGAPDH-F: CCACTCCTCCACCTTTGAC (qPCR) | This paper | N/A |
| HsGAPDH-R: ACCCTGTTGCTGTAGCCA (qPCR) | This paper | N/A |
| 18S rRNA-F: CGTCTGCCCTATCAACTTTCG (qPCR) | This paper | N/A |
| 18S rRNA-R: TGCCTTCCTTGGATGTGGTAG (qPCR) | This paper | N/A |
| HsENO1 K281R mutagenesis (forward): ctggctgacctgtacaggtccttcatcaaggac | This paper | N/A |
| HsENO1 K281R mutagenesis (reverse): gtccttgatgaaggacctgtacaggtcagccag | This paper | N/A |
| Recombinant DNA | ||
| pCRISPR-CG01-p300 (human) (Target sequence: CATCGCTGGCGGACGCCGAG) | GeneCopoeia | Cat# HCP000995-CG01-1-B |
| p300shRNA-3 (human) (Target sequence: CCCGGTGAACTCTCCTATAAT) | Sigma-Aldrich | Cat# TRCN0000039886 |
| p300shRNA-5 (human) (Target sequence: TCAATAATGCCTGGTTATATA) | Sigma-Aldrich | Cat# TRCN0000231135 |
| shENO1-3′ UTR 1 (human) (Target sequence:CGTACCGCTTCCTTAGAACTT) | Sigma-Aldrich | Cat# TRCN0000029324 |
| shENO1-3′ UTR 2 (human) (Target sequence:CGTACCGCTTCCTTAGAACTT) | Sigma-Aldrich | Cat# TRCN0000293182 |
| pCMV3-HA-ENO1 | Sino Biological | Cat# HG11554-NY |
| pCMV3-HA-ENO1 K281R | This paper | N/A |
| pCMV-Flag-p300 | Gift from Dr. Wei Gu at Columbia University | N/A |
| pCMV-Flag-p300-DY (D1399Y) | Gift from Dr. Wei Gu at Columbia University | N/A |
| Software and Algorithms | ||
| Prism v7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Microsoft Office Excel | Microsoft | |
| Maxquant v1.3.0.5 | Dr. Matthias Mann group at Max-Planck-Institut für Biochemie | http://www.biochem.mpg.de/5111795/maxquant |
| clusterProfiler | A package in R | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| STRING v10 | Online database | https://string-db.org/cgi/input.pl |
| Cytoscape v3.2.1 | National Institute of General Medical Sciences | http://www.cytoscape.org/ |
| MetaboAnalyst 3.0 | Dr. Jianguo Xia’s lab at McGill University | http://www.metaboanalyst.ca/ |
Experimental Model and Subject Details
Cell Lines
WT and p300 KO HCT116 cells have been described previously (Iyer et al., 2004). Another pair of WT and p300 KO HCT116 cells, and WT and p300 KO HEK293T cells were generated using the standard Crispr/Cas9 technology with a CRISPR All-in-one sgRNA expression clone in mammalian pCRISPR-CG01 vector (GeneCopoeia, Catalog #: HCP000995-CG01-1-B, target site: CATCGCTGGCGGACGCCGAG), and deletion of p300 were confirmed by immuno-blotting analysis.
Control and p300 shRNA SW620 cells were generated using lentiviruses containing control shRNA vector or shRNA constructs expressing shRNA specifically against human p300 (Sigma, p300shRNA-3: TRCN0000039886, target sequence: CCCGGTGAACTCTCCTATAAT; p300shRNA-5: TRCN0000231135, target sequence: TCAATAATGCCTGGTTATATA), and deletion of p300 were confirmed by immuno-blotting analysis.
To knock down the expression of endogenous ENO1 while allowing reexpression of exogenous WT and K281R HA-ENO1 in HCT116 cells and HEK293T cells, two shRNA constructs targeting the 3′UTR of the human ENO1 gene (target sequence: CGTACCGCTTCCTTAGAACTT) were purchased from Sigma (TRCN0000029324 and TRCN0000293182). Lentiviral particles were then prepared to stably infect HCT116 and HEK293T cells pre-overexpressing WT or K281R HA-ENO1. The deletion of endogenous ENO1 and expression of exogenous WT and K281R HA-ENO1 were confirmed by real-time PCR using primers targeting 3′ UTR or the coding region of ENO1.
HCT116 (ATCC CCL-247), HEK293T (ATCC CRL-3216), HeLa (ATCC CCL-2), U2OS (ATCC HTN-96), and SW620 (ATCC CCL-227) cells used in this study were all maintained at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM) without pyruvate containing 10% fetal bovine serum (FBS). No mycoplasma contamination was detected using a MycoAlert™ Mycoplasma Detection Kit (Lonza, LT07-118).
Method Details
Immunofluorescence
U2OS cells (with or without WT p300 overexpression) grown on coverslips were washed with phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4) and then fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. After rinse with PBS twice, cells were permeabilized with 0.5% Triton X-100 at room temperature for 10 min and blocked with 3% BSA at room temperature for 60 min. Then the cells were incubated with primary antibodies indicated in 1% BSA at 4°C overnight with gentle shaking. After washing with PBS for 10 min twice at room temperature, the cells were incubated with appropriate secondary antibodies (with Rhodamine Red-X or FITC conjugated, Invitrogen) in 1% BSA for 60 min at room temperature followed by washing with PBS for 10 min twice. Immuno-labeled cells were analyzed with an EVOS FL Cell Imaging System (Life Technologies).
In vitro p300-mediated chromatin-based transcription
Chromatin assembly and chromatin-based transcription was performed as described (An and Roeder, 2004) with minor modifications. The various steps were performed as follows (Figure S1B): (1) activator binding: p53 (10 ng) was incubated with recombinant chromatin template (40 ng) in the presence of ACF1, ISWI, and NAP1 in reaction buffer for 20 min at 27°C; (2) chromatin-based histone acetylation: p300 (20 ng) with either acetyl-CoA (20 μM) or 2-hydroxyisobutyryl-CoA (20 μM) were added to the reactions and incubated at 30°C for 30 min; (3) transcription: 5.0 μl (10 mg/ml) of HeLa nuclear extract and nucleotide mixture with 10 μCi of [α-32P] UTP (3000 Ci/mmol; PerkinElmer) were sequentially added to the reaction for 30 min at 30°C; (4) finally, the radiolabeled RNA was purified and resolved on 5% polyacrylamide (19:1) with 8.0 M urea gel and analyzed by autoradiography.
SILAC sample preparation
WT and p300 KO HCT116 cells (Iyer et al., 2004) were cultured in lysine-free DMEM supplemented with 10% dialyzed FBS, and with “heavy” lysine (13C615N2-Lys) or “light” lysine (12C614N2-Lys) (100 mg/L, Cambridge Isotope Laboratories, Tewksbury, MA). Cells were grown for more than seven generations before being harvested, to achieve more than 98% labeling efficiency (based on MS analysis). The “heavy” and “light” cells were harvested, washed with cold phosphate-buffered saline (PBS), and sonicated for three minutes on ice using a sonic dismembrator (Model 500, Fisher Scientific, Hampton, NH) in NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 0.5% Nonidet P-40, pH 8.0). After centrifugation (18,000g) at 4 °C for 10 min, the supernatant was collected and the pellet was further lysed by urea lysis buffer (8 M urea, 2 mM EDTA, 3 μM Trichostatin A, 50 mM Nicotinamide, and 5 mM DTT). Equal amounts of proteins from the NETN-soluble or re-solubilized pellet fractions of WT and p300 KO HCT116 cells were mixed and precipitated using trichloroacetic acid. The resulting protein precipitate (5 mg of NETN-soluble proteins and 1 mg of proteins from re-solubilized pellet) were digested with trypsin (Promega Corp., Madison, WI), initially at 1:50 trypsin-to-protein ratio (weight to weight) overnight followed by the second digestion at 1:100 trypsin-to-protein ratio (weight to weight) for 4 hours. The tryptic digest was reduced with 10 mM DTT for 1 h at 37 °C and alkylated with 20 mM iodoacetamide for 45 min at room temperature in darkness. The excess iodoacetamide was blocked by 20 mM cysteine.
Peptide fractionation and immunoaffinity enrichment
The resulting proteolytic peptides from NETN-soluble fraction were separated on preparative HPLC into 6 fractions using Agilent 300 Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length) followed by drying by SpeedVac (ThermoFisher Scientific, Waltham, MA). Pan anti-Kac or pan anti-Khib antibody (PTM Biolabs, Chicago, IL, catalogue number PTM-105 or PTM-501) was first immobilized to pre-washed protein A agarose beads (GE Healthcare Biosciences, Pittsburgh, PA) at a density of 4 mg of antibody per mL drained beads. Then the peptide powder was dissolved in NETN buffer and incubated with 20 μl of antibody-immobilized protein A beads at 4 °C overnight with gentle shaking. After incubation, the beads were washed four times with NETN buffer and twice with ddH2O. The bound Kac or Khib peptides were eluted from the beads with 0.1% trifluoroacetic acid, and the eluted peptides were desalted by C18 ZipTip (EMD Millipore, Billerica, MA).
HPLC/MS/MS analysis
The enriched peptides obtained above were dissolved in 0.1% formic acid in water and loaded onto a reversed-phase microcapillary column (10 cm length with 75 μm inner diameter) packed in-house with Reprosil 100 C18 resin (3 μm particle size, Dr. Maisch GmbH, Beim Brückle, Germany). The loaded samples were separated using a gradient of 5% to 80% HPLC buffer B (0.1% formic acid in 90% acetonitrile, v/v) in buffer A (0.1% formic acid in water, v/v) at a flow rate of 200 nL/min over 60 min by an EASY-nLC 1000 UPLC (ThermoFisher Scientific, Waltham, MA). The samples were analyzed by a Q Exactive™ hybrid quadrupole-orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA). A data-dependent procedure that alternated between one full mass scan followed by the top 15 most intense precursor ions was applied with a 25 s dynamic exclusion. Intact peptides were detected with a resolution of 70,000, and the tandem mass spectra were acquired with a mass resolution of 17,500 at 27% normalized collision energy.
Database search and data filter criteria
The acquired MS/MS data was searched by MaxQuant with integrated Andromeda search engine (v.1.3.0.5) (Cox and Mann, 2008; Cox et al., 2009). All the data were searched against UniProt Human protein database (88,277 entries, http://http://www.uniprot.org). Trypsin was specified as cleavage enzyme allowing a maximum of 2 missing cleavages. Cysteine carbamidomethylation was specified as fixed modification. Methionine oxidation, protein N-terminal acetylation, lysine acetylation, and lysine 2-hydroxyisobutyrylation were specified as variable modifications. FDR thresholds for protein, peptide and modification site were specified at 1%. The following peptides were considered as false positives and removed from our list: peptides identified from reverse or contaminant protein sequences, peptides with score below 40, site localization probability below 0.75, Kac or Khib sites on peptide C-terminus unless the peptide C-terminal was also the corresponding protein C-terminal.
To ensure that the Kac and Khib level change in p300 KO cells is not caused by the protein level change, we quantified the protein expression levels. Briefly, the proteolytic peptides obtained in previous step were separated on preparative HPLC into 20 fractions, or separated into 12 fractions with SDS-PAGE. These samples were analyzed using the same procedures for the Kac and Khib peptides quantification. Then all the ratios of quantified Kac and Khib peptides were normalized by the ratios of their corresponding protein expression levels.
Bioinformatics Analysis
Pathway enrichment analysis was performed using a hypergeometric test in clusterProfiler package in R (Yu et al., 2012) and the FDR threshold was specified at 0.05. The protein-protein interaction networks of p300-regulated Khib proteome were determined using STRING database (v10, http://www.string-db.org/) and visualized in Cytoscape (v.3.2.1) (Shannon et al., 2003; Szklarczyk et al., 2015).
Metabolomics Analysis
To quantitatively analyze metabolic profiles in WT and p300 KO HCT116 cells (Iyer et al., 2004), cells were seeded at a density of 200,000 cells per well in 6 well plates. After overnight incubation in complete growth medium, the old medium was removed and cells were washed with 1 ml PBS before the addition of 2 ml of high glucose DMEM medium or glucose free DMEM medium for 6 hours. After 6 hours incubation, metabolites were extracted and analyzed as described previously (Liberti et al., 2017; Liu et al., 2014).
Seahorse Analysis
To analyze glycolysis in real-time, Seahorse analyses were performed on a Seahorse XF Analyzer according to manufacturer’s instructions (Agilent Technologies, Santa Clara, CA). Briefly, cells were plated in a 24-well assay plate the day before the analysis in the complete medium to ensure 80–90% confluence on next day, then washed and incubated in a freshly prepared XF assay medium containing 2 mM glutamate. Final glycolysis (ECAR) was normalized to total cell protein contents.
In vitro Kac and Khib Assay
Recombinant human p300 proteins were purchased from Active Motif (Carlsbad, CA, catalogue number 31124). For each reaction, 200 ng of p300 protein, 2.5 μg of immuno-purified HA-tagged protein from p300 KO cells, and 10 μM of CoA were added in the reaction buffer (50 mM Tris-CI, pH 8.0, 10% glycerol, 100 nM TSA, 5 mM Nicotinamide, 0.1 mM EDTA, 1 mM DTT and 1× proteinase inhibitor cocktail). The reaction mixtures were incubated at 30 °C for 1 hour, followed by ENO1 activity assay or addition of SDS sample buffer. The levels of Kac and Khib were detected by SDS-PAGE and immunoblotting analysis. Acetyl-CoA (Ac-CoA) was purchased from Sigma (St. Louis, MO, A2056), and 2-hydroxyisobutyryl-CoA (hib-CoA) was synthesized using a previously described method (Huang et al., 2018).
Activity Assays for PFK and ENO1
The activities of PFK and ENO1 in total cell lysate were measured using colorimetric assay kits from Sigma (St. Louis, MO, MAK093) and Abcam (Cambridge, MA, ab117994), respectively. The kinetics of immuno-purified WT and K281R mutant ENO1 (from WT HCT116 or HEK293T cells with endogenous ENO1 knocked down by shRNAs) were measured in vitro using a pyruvate kinase/L-lactate dehydrogenase coupled reaction. Briefly, an equal amount of purified WT and K281R ENO1 were incubated in the reaction buffer containing 100 mM triethanolamine, pH 7.4, 0.2 mM NADH, 30 mM MgSO4, 120 mM KCl, 1.75 mM ADP, 10 units pyruvate kinase, 15 units L-lactate dehydrogenase, and 0–5 mM 2-P glycerate. The rate of NADH oxidation was monitored by absorbance at 340 nm for 10 minutes at the room temperature. The initial reaction rates at each concentration of 2-P glycerate were calculated from the first 3–5 min reaction in triplicates. The Vmax and Km were then calculated using the double-reciprocal plot of the Michaelis-Menten equation.
Cell Apoptosis and Viability Assays
To analyze cell death in WT and p300 KO cells, cells cultured in different medium for 48 hours. The cells were then washed with PBS and stained with FITC-Annexin V and PI according to the manufacturer’s instructions (BD Bioscience Annexin V Kit). Apoptotic cells and dead cells were determined by flow cytometry analysis (Figure 6C and 6D). Cell viability in Figure 6F was determined with Cell Proliferation Reagent WST-1 (Roche Applied Science) according to the manufacturer’s protocol.
Quantification and Statistical Analysis
Values are expressed as mean ± standard error of mean (SEM) from at least three independent experiments or biological replicate, unless otherwise indicated in the figure legend. Significant differences between the means were analyzed by the two-tailed, unpaired, Student’s t-test, and differences were considered significant at *p< 0.05. Data were analyzed using Prism Software 7.0 (GraphPad) or Microsoft Office Excel (Version 16.10).
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD008525.
The source file for unprocessed and uncompressed immuno-blots used in the figures is available in the following dataset: http://dx.doi.org/10.17632/cszpvms38z.1.
Supplementary Material
Table S1. List of the identified Khib and Kac sites, Related to Figure 2.
Table S3. Metabolomic analysis of the influence of p300 on metabolism in HCT116 cells, Related to Figure 4.
HIGHLIGHTS.
p300 is a “writer” for lysine 2-hydroxyisobutyrylation.
p300 has distinct profiles of lysine 2-hydroxyisobutyrylation and acetylation.
p300 regulates glycolysis through 2-hydroxyisobutyrylation of glycolytic enzymes.
p300 mediates nutritional regulation of cell survival through glycolysis.
Acknowledgments
We thank Drs. Paul Wade and Jason Williams, and members of the Li and Zhao laboratories for critical reading of the manuscript; The p300 WT and KO HCT116 cell lines were originally constructed by Professor Carlos Caldas at University of Cambridge. This research was supported by the Intramural Research Program of National Institute of Environmental Health Sciences of the NIH to X. Li (Z01 ES102205), and by NIH grants to Y. Z. (DK107868 and GM115961).
Footnotes
AUTHOR CONTRIBUTIONS
H. H. analyzed the quantitative proteomics, performed the Khib transferase activity assay of p300 in cultured cells, and wrote the manuscript. M. J. and S. T. analyzed p300-mediated Khib in regulation of glycolysis and cell survival. X. Liu and J. W. L. analyzed metabolites by metabolomics and critically reviewed the manuscript. Z. T., M. S., and R. G. R. were involved in cell-free transcription assay. S. Q. performed immunoaffinity enrichment for SILAC analysis. Y. Z. and X. Li designed and coordinated the study, analyzed data, and wrote the manuscript.
DECLARATION OF INTERESTS
Y. Z. is on the science advisory board of PTM Biolabs.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An W, Roeder RG. Reconstitution and transcriptional analysis of chromatin in vitro. Methods Enzymol. 2004;377:460–474. doi: 10.1016/S0076-6879(03)77030-X. [DOI] [PubMed] [Google Scholar]
- Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, Wagner GR, Thompson JW, Madsen AS, Green MF, et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017;25:838–855. e815. doi: 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bheda P, Jing H, Wolberger C, Lin H. The Substrate Specificity of Sirtuins. Annu Rev Biochem. 2016;85:405–429. doi: 10.1146/annurev-biochem-060815-014537. [DOI] [PubMed] [Google Scholar]
- Bouatra S, Aziat F, Mandal R, Guo AC, Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P, et al. The human urine metabolome. PLoS One. 2013;8:e73076. doi: 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan EJ, Jokubaitis VJ, Chamberlain NL, Baxter SW, Dawson E, Choong DY, Campbell IG. Mutation analysis of EP300 in colon, breast and ovarian carcinomas. Int J Cancer. 2002;102:137–141. doi: 10.1002/ijc.10682. [DOI] [PubMed] [Google Scholar]
- Calvani R, Miccheli A, Capuani G, Tomassini Miccheli A, Puccetti C, Delfini M, Iaconelli A, Nanni G, Mingrone G. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int J Obes (Lond) 2010;34:1095–1098. doi: 10.1038/ijo.2010.44. [DOI] [PubMed] [Google Scholar]
- Carey M, Kehlenbrink S, Hawkins M. Evidence for central regulation of glucose metabolism. J Biol Chem. 2013;288:34981–34988. doi: 10.1074/jbc.R113.506782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, Debernardi A, Buchou T, Rousseaux S, Jin F, et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- Dame ZT, Aziat F, Mandal R, Krishnamurthy R, Bouatra S, Borzouie S, Guo AC, Sajed T, Deng L, Lin H, et al. The human saliva metabolome. Metabolomics. 2015 doi: 10.1007/s11306-015-0840-5. [DOI] [Google Scholar]
- Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115:2419–2452. doi: 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekant W, Bernauer U, Rosner E, Amberg A. Biotransformation of MTBE, ETBE, and TAME after inhalation or ingestion in rats and humans. Res Rep Health Eff Inst. 2001:29–71. discussion 95–109. [PubMed] [Google Scholar]
- Francois A, Mathis H, Godefroy D, Piveteau P, Fayolle F, Monot F. Biodegradation of methyl tert-butyl ether and other fuel oxygenates by a new strain, Mycobacterium austroafricanum IFP 2012. Appl Environ Microbiol. 2002;68:2754–2762. doi: 10.1128/AEM.68.6.2754-2762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard J. Hormonal Control of Glucose Metabolism. Diabetes. 1995;35:1–22. [Google Scholar]
- Goudarzi A, Zhang D, Huang H, Barral S, Kwon OK, Qi S, Tang Z, Buchou T, Vitte AL, He T, et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol Cell. 2016;62:169–180. doi: 10.1016/j.molcel.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guneral F, Bachmann C. Age-related reference values for urinary organic acids in a healthy Turkish pediatric population. Clin Chem. 1994;40:862–866. [PubMed] [Google Scholar]
- Han HS, Kang G, Kim JS, Choi BH, Koo SH. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med. 2016;48:e218. doi: 10.1038/emm.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Zhao Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol Cell Proteomics. 2015;14:2308–2315. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann GF, Meier-Augenstein W, Stockler S, Surtees R, Rating D, Nyhan WL. Physiology and pathophysiology of organic acids in cerebrospinal fluid. J Inherit Metab Dis. 1993;16:648–669. doi: 10.1007/BF00711898. [DOI] [PubMed] [Google Scholar]
- Huang H, Luo Z, Qi S, Huang J, Xu P, Wang X, Gao L, Li F, Wang J, Zhao W, et al. Landscape of the regulatory elements for lysine 2-hydroxyisobutyrylation pathway. Cell Res. 2018;28:111–125. doi: 10.1038/cr.2017.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husek P, Svagera Z, Hanzlikova D, Rimnacova L, Zahradnickova H, Opekarova I, Simek P. Profiling of urinary amino-carboxylic metabolites by in-situ heptafluorobutyl chloroformate mediated sample preparation and gas chromatography-mass spectrometry. J Chromatogr A. 2016;1443:211–232. doi: 10.1016/j.chroma.2016.03.019. [DOI] [PubMed] [Google Scholar]
- Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, Brindle K, Aparicio S, Caldas C. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci U S A. 2004;101:7386–7391. doi: 10.1073/pnas.0401002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarska Z, Ortega E, Goudarzi A, Huang H, Kim S, Marquez JA, Zhao Y, Khochbin S, Panne D. Structure of p300 in complex with acyl-CoA variants. Nat Chem Biol. 2017;13:21–29. doi: 10.1038/nchembio.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–132. doi: 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L, et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun. 2016;7:12235. doi: 10.1038/ncomms12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A. 2008;105:2117–2122. doi: 10.1073/pnas.0712038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017;26:1–12. doi: 10.1016/j.cmet.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. 2012;7:947–960. doi: 10.1021/cb3001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Cluntun AA, Mentch SJ, Locasale JW. A strategy for sensitive, large scale quantitative metabolomics. J Vis Exp. 2014 doi: 10.3791/51358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wei W, Liu Y, Yang X, Wu J, Zhang Y, Zhang Q, Shi T, Du JX, Zhao Y, et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017;3:17016. doi: 10.1038/celldisc.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes Ferreira N, Labbe D, Monot F, Fayolle-Guichard F, Greer CW. Genes involved in the methyl tert-butyl ether (MTBE) metabolic pathway of Mycobacterium austroafricanum IFP 2012. Microbiology. 2006;152:1361–1374. doi: 10.1099/mic.0.28585-0. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189–200. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, et al. The human serum metabolome. PLoS One. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58:203–215. doi: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BO, Backhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22:1079–1089. doi: 10.1038/nm.4185. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan RJ, McClay K, Vainberg S, Condee CW, Zhang D. Biodegradation of the gasoline oxygenates methyl tert-butyl ether, ethyl tert-butyl ether, and tert-amyl methyl ether by propane-oxidizing bacteria. Appl Environ Microbiol. 1997;63:4216–4222. doi: 10.1128/aem.63.11.4216-4222.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Chen WY, Shimada M, Nguyen UT, Kim J, Sun XJ, Sengoku T, McGinty RK, Fernandez JP, Muir TW, et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154:297–310. doi: 10.1016/j.cell.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasselli L, Zheng W, Chua KF. SIRT6: Novel Mechanisms and Links to Aging and Disease. Trends Endocrinol Metab. 2017;28:168–185. doi: 10.1016/j.tem.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Wang S, Jiang B, Zhang T, Liu L, Wang Y, Wang Y, Chen X, Lin H, Zhou L, Xia Y, et al. Insulin and mTOR Pathway Regulate HDAC3-Mediated Deacetylation and Activation of PGK1. PLoS Biol. 2015;13:e1002243. doi: 10.1371/journal.pbio.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Liu X, Chen J, Gao S, Lu L, Zhang H, Ding G, Wang Z, Chen Z, Shi T, et al. Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017;27:898–915. doi: 10.1038/cr.2017.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wan J, Zhan J, Li X, He H, Shi Z, Zhang H. Global profiling of crotonylation on non-histone proteins. Cell Res. 2017;27:946–949. doi: 10.1038/cr.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the identified Khib and Kac sites, Related to Figure 2.
Table S3. Metabolomic analysis of the influence of p300 on metabolism in HCT116 cells, Related to Figure 4.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD008525.
The source file for unprocessed and uncompressed immuno-blots used in the figures is available in the following dataset: http://dx.doi.org/10.17632/cszpvms38z.1.