

Abstract
Long-term, low-grade inflammation does not seem to be a risk factor for venous thromboembolism. The impact of acute inflammation, regardless of cause, on risk of venous thromboembolism is scarcely studied. We aimed to investigate the impact of acute inflammation, assessed by C-reactive protein, on short-term risk of venous thromboembolism. We conducted a case-crossover study of patients with venous thromboembolism (n=707) recruited from a general population. Information on triggers and C-reactive protein levels were retrieved from hospital records during the 90 days before the event (hazard period) and in four preceding 90-day control periods. Conditional logistic regression was used to obtain β coefficients for change in natural log (ln) transformed C-reactive protein from control to hazard periods and to determine corresponding odds ratios for venous thromboembolism. Median C-reactive protein was 107 mg/L in the hazard period, and ranged from 7 mg/L to 16 mg/L in the control periods. The level of C-reactive protein was 58% (95% CI 39-77%) higher in the hazard period than in the control periods. A one-unit increase in ln-C-reactive protein was associated with increased risk of venous thromboembolism (OR 1.79, 95% CI 1.48-2.16). The risk estimates were only slightly attenuated after adjustment for immobilization and infection. In stratified analyses, ln-C-reactive protein was associated with increased risk of venous thromboembolism in cases with (OR 1.55, 95% CI 1.01-2.38) and without infection (OR 1.77, 95% CI 1.22-2.57). In conclusion, we found that acute inflammation, assessed by C-reactive protein, was a trigger for venous thromboembolism.
Introduction
Venous thromboembolism (VTE), consisting of deep vein thrombosis (DVT) and pulmonary embolism (PE), is a multicausal disease associated with substantial morbidity and mortality.1 Contrary to arterial thrombotic disease, there has been no decline in the incidence of VTE during the last decades.2,3 Thus, there is an unmet need for improved risk stratification and prevention of VTE.
Chronic inflammation is recognized as part of the pathophysiological process in arterial thrombosis,4 but its role in venous thrombosis has been less clear.5,6 Inflammatory biomarkers such as high-sensitivity C-reactive protein (hs-CRP) can predict long-term risk of arterial cardiovascular disease, but have not been associated with risk of VTE in prospective studies with long-term follow up.7–9 However, in studies with shorter follow up time, inflammatory markers such as hs-CRP and neutrophil to lymphocyte ratio were associated with increased risk of VTE.10,11
Several conditions associated with increased risk of VTE, including cancer, acute infections, autoimmune diseases and obesity, share the feature of inflammation.12–15 Even though chronic diseases such as autoimmune diseases carry an increased VTE risk, the risk of VTE is more pronounced during disease flare-ups, where inflammation is predominant.14 Acute infection triggers an acute inflammatory response, and several studies have found an increased risk of VTE associated with infections.16,17 We have previously investigated the role of infection during hospitalization in a case-crossover design, and found that acute infection was a frequent and strong trigger for VTE, also after adjustment for immobilization and other transient risk factors.18 Taken together, this points towards an association between inflammation and VTE which is dependent on the degree of the inflammatory response within a shorter time perspective than as observed in arterial thrombotic disease.
The objective of this study was to investigate the role of acute inflammation, assessed by CRP, as a trigger for VTE using a case-crossover design. In this study design, each case serves as his or her own control, and the design is therefore well suited for studying transient risk factors.19 We hypothesized that increased CRP, independent of cause, was a trigger of VTE.
Methods
For an extensive description of the methods, please see the Online Supplementary Methods. We conducted a case-crossover study including all incident VTE cases (n=707) diagnosed among the participants of the fourth Tromsø Study during 1994-2012. The study was approved by the regional ethics committee, and all participants provided informed written consent. A hazard period of 90 days preceding the incident VTE was compared to four preceding 90-day control periods. To avoid carry-over effects, we included a 90-day washout period between the hazard and control periods (Figure 1). For every VTE case, trained medical personnel searched the hospital medical records for relevant risk factors, diagnostic procedures, surgical and medical treatment, laboratory test results and diagnoses during hospital admissions, day care and outpatient clinic visits in any of the hazard or control periods. We did not have access to medical records from general practice. A transient risk factor, or trigger, was defined by its presence during the defined 90-day period. If an exposure occurred over several days, it was considered to have occurred if any of the days of exposure fell within the specified 90-day time period.
Figure 1.

Case-crossover study design. Relevant risk factors and levels of C-reactive protein were recorded for each case of venous thromboembolism, in the 90-day hazard period prior to the event and in four preceding 90-day control periods, separated by a 90-day washout period.
CRP was analyzed in serum with a particle-enhanced immunoturbidimetric assay. CRP measurements from the last two days before the date of VTE were not included in the analyses to avoid reverse causation, as CRP in these cases could be caused by an inflammatory response to the VTE itself. If a participant had several CRP-measurements during a control or hazard period, the maximum CRP value for each period was used.
Statistical analyses were carried out using STATA version 14.0 (Stata corporation, College station, Texas, USA). Natural log (ln) transformation was used for CRP to achieve normal distributions. Only cases who had their CRP measured in both the hazard and a control period were included in the main analyses. Since CRP was measured upon request, this would yield the most conservative risk estimate. We used conditional logistic regression to obtain β coefficients with 95% confidence intervals (CI) for change in ln-CRP from control to hazard periods, and to calculate odds ratios (ORs) with 95% CI per one-unit change in ln-CRP. The analyses were adjusted for immobilization and infection in two different models. Further, we performed analyses comparing CRP in the hazard period with each individual control period, to investigate whether time to event influenced the association between acute inflammation and VTE.
In the main analyses, we included only hazard and control periods in which CRP had been measured. The risk estimates from this conservative approach could be underestimations, as subjects with no hospital contact during a hazard or control period, or with a hospital contact without a CRP measurement, most likely had a low CRP at that time. To address this concern, we performed sensitivity analyses where missing CRP values were set at the lower reported cut-off level of 5 mg/L. We also performed sensitivity analyses where we included only those CRP-measurements performed more than seven days before the date of VTE, to address potential bias due to reverse causation.
Results
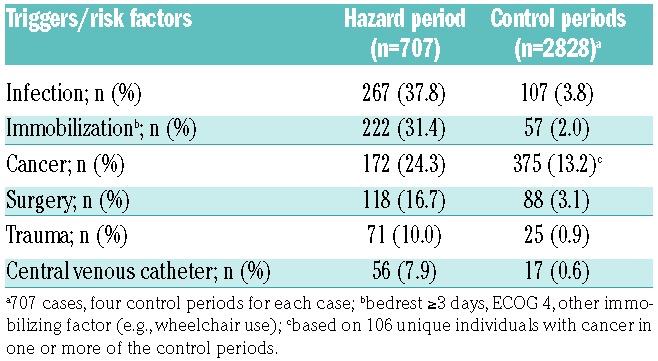
In total, 707 incident VTEs were identified, of which there were 408 DVTs and 299 PEs (with or without concurrent DVT). The median age at time of VTE-diagnosis was 71 years, and 53.6% were women. Moreover, 416 (58.8%) VTEs were not related to recent hospitalization, 135 (19.1%) occurred during hospitalization, and 156 (22.1%) were diagnosed within 30 days after hospitalization (Table 1). Infection was the most common risk factor in the hazard period, recorded in 267 (37.8%) of the periods, followed by immobilization (31.4%), and cancer (24.3%). The distribution of VTE risk factors and triggers in the hazard- and control periods are shown in Table 2. Prophylactic treatment with low-molecular weight heparin was prescribed in 138 (19.5%) of the 707 hazard periods, and in 78 (2.8%) of the 2828 control periods.
Table 1.
Characteristics of study participants at the time of venous thromboembolism (VTE) diagnosis.

Table 2.
Triggers and risk factors for venous thromboembolism.
In total, after exclusion of hospital contacts the last two days before VTE-diagnosis, 1283 hospital contacts were registered during the hazard period and control periods (Table 3). The number of hospital contacts was higher in the periods closest to the VTE, increasing from 165, 172, 187 and 199, respectively, in the control periods, to 560 contacts in the hazard period. CRP was measured in 298 cases during the hazard period, and in 75, 72, 86 and 96 cases during the four control periods. Median CRP was highest in the hazard period (CRP 107 mg/L), and ranged from 7 mg/L to 16 mg/L in the control periods (Table 3).
Table 3.
Hospital contacts and measurements of C-reactive protein (CRP) in hazard (H) and control periods (C1-C4).

Based on β coefficients for ln-CRP obtained from logistic regression analyses, the mean CRP level was 58% (95% CI 39-77%) higher in hazard than in control periods. After adjustment for immobilization, the CRP level was 51% (95% CI 31-70%) higher, and after adjustment for infection the CRP level was 40% (95% CI 19-61%) higher in the hazard than in the control periods (Table 4). This corresponded to a 1.8-fold increased estimated VTE risk per unit increase in ln-CRP (OR 1.79, 95% CI 1.48-2.16), which were only slightly attenuated after adjustment for immobilization (OR 1.66, 95% CI 1.37-2.02) and for infection (OR 1.50, 95% CI 1.21-1.85).
Table 4.
Association of C-reactive proteina with risk of venous thromboembolism

In analyses stratified for infection, the mean CRP level was 57% (95% CI 20-94%) higher in the hazard versus control periods in those without infection and 44% (95% CI 1-87%) higher in those with infection. Estimated increase in VTE risk according to a one-unit increase in ln-CRP was 1.8-fold (OR 1.77, 95% CI 1.22-2.57) in those without infection and 1.6-fold in those with infection (OR 1.55, 95% CI 1.01-2.38). Adjustment for immobilization revealed similar results (Table 4).
Sensitivity analyses restricted to CRP measurements that were conducted more than 7 days before the date of VTE-diagnosis yielded essentially similar results (Online Supplementary Table S1). In sensitivity analyses where missing CRP values were set to 5 mg/L, the estimated VTE risk per unit increase in ln-CRP was 2.4-fold increased (OR 2.36, 95% CI 2.14-2.61) (Online Supplementary Table S2).
When comparing CRP levels in the hazard period to each control period (C1-4) separately, there was no trend for change in CRP level according to time between control and hazard period (the CRP level was increased by 65%, 77%, 42% and 60%, respectively) (Table 5). The estimated risk of VTE by ln-CRP was 1.9-fold increased when comparing the hazard period with C1, 2.2-fold increased when compared with C2, 1.5-fold increased when compared with C3, and 1.8-fold increased when compared with C4.
Table 5.
Association of C-reactive proteina with risk of venous thromboembolism.

Discussion
In this case-crossover study including 707 incident VTEs, we found that acute inflammation, assessed by increase in CRP, was a trigger for VTE. The association remained after adjustment for immobilization and for infection. In stratified analyses, inflammation assessed by CRP was associated with increased risk of VTE also in cases without infection. The strength of the estimated risk of VTE by CRP remained similar when separately compared to the different control periods.
Clinically, DVT often presents with the cardinal signs of inflammation; i.e., redness, swelling, heat, pain and disturbance of function. In a case-control study investigating inflammatory markers, patients with DVT had significantly higher levels of inflammatory markers, including CRP, than controls.20 Based on these results the authors suggested that inflammation was a consequence rather than a cause of VTE. However, the fact that inflammation is a consequence of VTE does not exclude the possibility that inflammation can be a cause of the disease. Previous prospective studies on the association between inflammation and VTE have shown conflicting results. In a cohort of healthy men followed for more than 8 years, baseline hs-CRP was associated with increased risk of arterial but not venous thrombosis.9 Similarly, two long-term population-based cohorts reported no association between inflammatory markers and VTE.7,8 In contrast, studies with shorter follow-up time have shown an association between CRP and incident VTE. In a population-based case-cohort study with 515 VTE-cases and 1505 controls, an association between baseline CRP and VTE was only present in cases suffering a VTE within the first year after baseline.10 We found a similar time-dependent pattern between another inflammation marker, the neutrophil to lymphocyte ratio (NLR), and VTE risk in the Tromsø study cohort.11 There was no association between NLR and risk of VTE after a median follow-up time of 17.7 years, but when follow-up time was restricted to the first 3 years, those with the highest baseline NLR had a 2.4-fold increased risk of VTE. Taken together, these studies suggest that acute and augmented inflammation rather than longstanding, low-grade inflammation is associated with VTE risk.
Accordingly, in this case-crossover study, we found that acute inflammation assessed by CRP was associated with increased risk of VTE. Acute infection, a strong trigger of inflammation, is a risk factor for VTE, and higher CRP-levels are expected in patients with acute infections.16,17,21 After adjustment for infection, and in analyses stratified for infection, increased serum levels of CRP were still associated with increased VTE risk, also in cases without infection. Thus, our findings suggest that inflammatory responses caused by non-infectious conditions, such as cancer, surgery, acute medical conditions and trauma, can partly explain the VTE risk related to these conditions. Immobilization may accompany these conditions, and thereby act as a confounder for the observed association. However, in our study the risk estimates remained essentially similar after adjustment for immobilization.
The risk of VTE has been shown to be highest the first two weeks following an infection, and to gradually decline thereafter.16,17 In our study, there was no trend of a change in risk estimates according to time between the hazard and control periods. This further supports that acute inflammation of short duration is more important for the VTE risk. Some chronic inflammatory conditions, such as autoimmune disorders and rheumatic diseases, also carry increased risk of VTE. However, the risk of VTE in patients with inflammatory bowel disease, for example, is especially high during disease flare-ups, where acute inflammation dominates.22 Furthermore, in a population-based cohort study on VTE risk in patients with psoriasis and rheumatoid arthritis (RA), patients with severe psoriasis and RA-patients in need of a disease modifying anti-rheumatic drug (DMARD) had higher estimated risk of VTE than those not prescribed DMARDs.23
Inflammation and coagulation are closely linked.24 Inflammation can be triggered by infection, tissue injury or tissue stress and malfunction.25 Of these triggers, inflammation induced by infection has been best characterized. Extensive crosstalk exists between the coagulation and the complement cascades, and complement activation enhances coagulation through increased tissue factor (TF) expression and by inhibition of fibrinolysis.26 Anticoagulant activity by the protein C- pathway is down-regulated by inflammatory cytokines.27 TF expression increases in response to inflammatory cytokines and through recruitment from microvesicles and monocytes induced by P-selectin.28 Activated neutrophils secrete neutrophil extracellular traps (NETs), composed of proteins and decondensed chromatin.29 In addition to an important role in neutralizing and killing microbes, NETs also contribute to coagulation and platelet aggregation.30 NET formation occurs not only in response to infection, but also in sterile inflammatory processes and in metastatic cancer.30
CRP is an acute-phase protein rapidly synthesized mainly in the liver under control by inflammatory cytokines, and CRP levels cease rapidly when the stimuli for production is diminished.31 CRP has no diurnal variation, is unaffected by eating, and drugs reducing CRP typically also affect the underlying acute-phase stimulus.31 CRP is commonly used as a marker of inflammation in clinical practice. Since our study was based on clinical data, other markers of inflammation and coagulation were not available as these were only occasionally measured. CRP is therefore well suited to serve as a marker of inflammation, a process linked to coagulation through several pathways as described above. In addition to its role in innate immunity and complement activation,31 CRP has been found to have prothrombotic effects in some studies.32–34 Due to methodological issues, especially the possibility of contamination of CRP preparations with bacterial lipopolysaccharides, controversy regarding a direct role of CRP in thrombosis still exists.35
Our study has both strengths and limitations. The case-crossover design is suitable for studying transient risk factors, as potential fixed confounders are mainly controlled for through the design. Further, the VTE-cases were derived from a large, population-based cohort with high attendance rate, and all VTE events were symptomatic and validated. All hospital care in the region is provided by a single hospital, facilitating the completeness of the VTE registry. However, some VTE cases might have been clinically diagnosed and treated without hospital contact, and some cases of PE presenting as sudden death might have been misclassified. As each subject serves as his or her own control, such potential cases would most likely not affect our results. Our study was limited to information from hospital records, as we did not have access to data from general practice. In most cases, a high CRP level measured in general practice will increase the likelihood of the patient to be referred to hospital. We cannot find any reason why hospital referral praxis should differ in hazard versus control periods. Our data source was the hospital medical records of VTE-cases, and CRP measurements were made according to each clinician’s preference. As each case serves as his or her own control in the case-crossover design, only those who had a CRP measurement in both the hazard period and in one or more control periods were included in the analyses. We can assume that CRP levels were lower in periods where the clinician did not find a reason to have CRP measured than in periods where CRP was measured. As more CRP measurements were done in hazard than in control periods, this might have diluted the risk estimates. Sensitivity analyses where missing CRP values were set to 5 mg/L resulted in higher risk estimates than in the main analyses. We did not adjust for therapeutic agents that could affect CRP-levels, such as statins, steroids and other immunosuppressive drugs, as we wanted to investigate the impact of inflammation on VTE regardless of the cause of the inflammation. CRP levels measured the last two days before the date of VTE were omitted to avoid reverse causation. However, we cannot be certain that elevated CRP was not caused by the incident VTE more than two days before the date of VTE diagnosis. To address this, we performed sensitivity analyses excluding CRP measurements the last seven days before the date of VTE-diagnosis, and the results were essentially similar.
In conclusion, acute inflammation assessed by CRP was a trigger for VTE in this case-crossover study, also in cases with inflammatory triggers other than infection.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/7/1245
References
- 1.Rosendaal FR. Venous thrombosis: a multi-causal disease. Lancet. 1999; 353(9159): 1167–1173. [DOI] [PubMed] [Google Scholar]
- 2.Mannsverk J, Wilsgaard T, Mathiesen EB, et al. Trends in modifiable risk factors are associated with declining incidence of hospitalized and nonhospitalized acute coronary heart disease in a population. Circulation. 2016;133(1):74–81. [DOI] [PubMed] [Google Scholar]
- 3.Arshad N, Isaksen T, Hansen JB, Braekkan SK. Time trends in incidence rates of venous thromboembolism in a large cohort recruited from the general population. Eur J Epidemiol. 2017;32(4):299–305. [DOI] [PubMed] [Google Scholar]
- 4.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zacho J, Tybjaerg-Hansen A, Nordestgaard BG. C-reactive protein and risk of venous thromboembolism in the general population. Arterioscler Thromb Vasc Biol. 2010; 30(8):1672–1678. [DOI] [PubMed] [Google Scholar]
- 6.Riva N, Donadini MP, Ageno W. Epidemiology and pathophysiology of venous thromboembolism: similarities with atherothrombosis and the role of inflammation. Thromb Haemost. 2015; 113(6):1176–1183. [DOI] [PubMed] [Google Scholar]
- 7.Hald EM, Braekkan SK, Mathiesen EB, et al. High-sensitivity C-reactive protein is not a risk factor for venous thromboembolism: the Tromso study. Haematologica. 2011;96(8):1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai AW, Cushman M, Rosamond WD, et al. Coagulation factors, inflammation markers, and venous thromboembolism: the longitudinal investigation of thromboembolism etiology (LITE). Am J Med. 2002;113(8):636–642. [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336(14):973–979. [DOI] [PubMed] [Google Scholar]
- 10.Quist-Paulsen P, Naess IA, Cannegieter SC, et al. Arterial cardiovascular risk factors and venous thrombosis: results from a population-based, prospective study (the HUNT 2). Haematologica. 2010;95(1):119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimnes G, Horvei LD, Tichelaar V, Braekkan SK, Hansen JB. Neutrophil to lymphocyte ratio and future risk of venous thromboembolism and mortality-The Tromso Study. Haematologica. 2016; 101(10):e401–e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heit JA. Epidemiology of venous thromboembolism. Nat Rev Cardiol. 2015; 12(8):464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. [DOI] [PubMed] [Google Scholar]
- 14.Tichelaar YI, Kluin-Nelemans HJ, Meijer K. Infections and inflammatory diseases as risk factors for venous thrombosis. A systematic review. Thromb Haemost. 2012; 107(5):827–837. [DOI] [PubMed] [Google Scholar]
- 15.Samad F, Ruf W. Inflammation, obesity, and thrombosis. Blood. 2013;122(20):3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt M, Horvath-Puho E, Thomsen RW, Smeeth L, Sorensen HT. Acute infections and venous thromboembolism. J Intern Med. 2012;271(6):608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367(9516):1075–1079. [DOI] [PubMed] [Google Scholar]
- 18.Grimnes G, Isaksen T, Tichelaar YIGV, Braekkan SK, Hansen J-B. Acute infection as a trigger for incident venous thromboembolism: Results from a population-based case-crossover study. Res Pract Thromb Haemost. 2018;2(1):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maclure M. The case-crossover design: a method for studying transient effects on the risk of acute events. Am J Epidemiol. 1991;133(2):144–153. [DOI] [PubMed] [Google Scholar]
- 20.Roumen-Klappe EM, den Heijer M, van Uum SH, van der Ven-Jongekrijg J, van der Graaf F, Wollersheim H. Inflammatory response in the acute phase of deep vein thrombosis. J Vasc Surg. 2002;35(4):701–706. [DOI] [PubMed] [Google Scholar]
- 21.Rogers MA, Levine DA, Blumberg N, Flanders SA, Chopra V, Langa KM. Triggers of hospitalization for venous thromboembolism. Circulation. 2012;125(17):2092–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grainge MJ, West J, Card TR. Venous thromboembolism during active disease and remission in inflammatory bowel disease: a cohort study. Lancet. 2010; 375(9715):657–663. [DOI] [PubMed] [Google Scholar]
- 23.Ogdie A, Kay McGill N, Shin DB, et al. Risk of venous thromboembolism in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a general population-based cohort study. Eur Heart J. 2017. April 20 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemost. 2011; 9 Suppl 1:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008; 454(7203):428–435. [DOI] [PubMed] [Google Scholar]
- 26.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esmon CT. The impact of the inflammatory response on coagulation. Thromb Res. 2004;114(5-6):321–327. [DOI] [PubMed] [Google Scholar]
- 28.Esmon CT. Molecular circuits in thrombosis and inflammation. Thromb Haemost. 2013;109(3):416–420. [DOI] [PubMed] [Google Scholar]
- 29.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–147. [DOI] [PubMed] [Google Scholar]
- 30.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003; 111(12):1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh U, Devaraj S, Jialal I. C-reactive protein decreases tissue plasminogen activator activity in human aortic endothelial cells: evidence that C-reactive protein is a procoagulant. Arterioscler Thromb Vasc Biol. 2005;25(10):2216–2221. [DOI] [PubMed] [Google Scholar]
- 33.Devaraj S, Yun JM, Adamson G, Galvez J, Jialal I. C-reactive protein impairs the endothelial glycocalyx resulting in endothelial dysfunction. Cardiovasc Res. 2009;84(3):479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102(18):2165–2168. [DOI] [PubMed] [Google Scholar]
- 35.Ridker PM. From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118(1): 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.