

Abstract
Filter aided sample preparation (FASP) is becoming a central method for proteomic sample cleanup and peptide generation prior to LC-MS analysis. We previously adapted this method to a 96-well filter plate, and applied to prepare protein digests from cell lysate and body fluid samples in a high throughput quantitative manner. While the 96FASP approach is scalable and can handle multiple samples simultaneously, two key advantages compared to single FASP, it is also time-consuming. The centrifugation-based liquid transfer on the filter plate takes 3~5 times longer than single filter. To address this limitation, we now present a quick 96FASP (named q96FASP) approach that, relying on the use of filter membranes with a large MWCO size (~30 kDa), significantly reduces centrifugal times. We show that q96FASP allows the generation of protein digests derived from whole cell lysates and body fluids in a quality similar to that of the single FASP method. Processing a sample in multiple wells in parallel, we observed excellent experimental repeatability by label-free quantitation approach. We conclude that the q96FASP approach promises to be a promising cost- and time-effective method for shotgun proteomics and will be particularly useful in large scale biomarker discovery studies.
Keywords: FASP, 96-well Filter plate, Quantitative proteomics, Klebsiella pneumoniae, Human urine, Applications
Introduction
Efficient and effective sample preparation is central to LC-MS based proteomics [1]. While system suitability assessment and experimental design need to be considered for a successful proteomic experiment [2-4], sample cleanup and protein digestion are two primary components of the analysis [1]. Traditional gel-based approaches to prepare samples are laborious and time-consuming [1, 5]. Gel-free approaches such as precipitation with organic solvents prior to in-solution digestion do not recover complete proteomes [6, 7]. In-solution digestion aided by proteolytic enzyme-compatible mild detergents bypasses the precipitation step, but is limited to a few reliable chemicals that fail to solubilize all aggregating and transmembrane proteins [1, 8]. By contrast, filter-aided sample preparation (FASP) offers a versatile approach that allows gel-free sample processing and membrane-based protein digestion [9]. FASP is based on the use of membrane filters that allow using detergents (e.g., sodium dodecyl sulfate, SDS) for cell lysis and solubilization of protein mixtures with subsequent ultrafiltration-based steps to remove the detergents and other substances interfering with optimal enzymatic digestion (e.g., salts and small molecules). Following protein digestion, FASP offers the additional advantage of further peptide cleanup due to the fact that the latter pass the membrane while other macromolecules are retained on the filter membrane in the final centrifugation step. FASP has thus been widely applied to various types of samples including body fluids and lysates of animal or plant tissues, fungi and bacteria [10-15]. Further developments of FASP methods have also been reported, such as MED-FASP and eFASP to improve peptide recovery and efficiency [16, 17], iFSP to accommodate chemical labeling reactions [18], microwave-assisted FASP to speed up on-filter digestion [19], abFASP to analyze affinity-purified protein complexes [20], endoProteoFASP to study the peptidome [21], and other adaptations to investigate specific subsets of modified proteins/peptides [22, 23]. Among them, 96FASP is a pioneering method that first extended the application from individual filters to multi-well filter plates [24]. The 96FASP method allows processing multiple samples in parallel while not sacrificing experimental repeatability. Highly parallel sample processing is of particular interest in clinical proteomics or biomarker discovery where large numbers of samples are usually screened to identify candidate markers or potential therapeutic targets [2, 25, 26]. The development of this method offered reliable semi-automated digestion of samples in batches with reduced sample handling, less time to experiment completion when a large batch is processed, and lower cost of shotgun proteomic sample preparation [24].
A noted disadvantage of the reported 96FASP method is the lengthy filtration time [5, 27, 28], Each spin step in the protocol takes 45 to 90 min while single filter-based FASP can be completed in one fourth of the time. With over ten centrifugation steps in the entire workflow, experiments may not be completed in a one-workday time frame, thus reducing its broad appeal. Single FASP usually applies 30-kDa molecular weight cutoff (MWCO) filters and centrifuges at over 10,000 x g during sample preparation, whereas the 96-well filter plate features 10-kDa cutoff and can maximally centrifuge at less than 3,000 × g. These fundamental differences contribute to less effective ultrafiltration [24]. In addition, the high viscosity of the primary depletion solution, 8 M urea, extends the centrifugation time and makes the 96FASP approach even less appealing. Therefore, a new version of 96FASP approach that is entirely comparable with single FASP is desirable. We recently noticed that two types of 96-well filter plates with larger pore size (~30 kDa) are commercially available, one is the Pall AcroPrep Advance 96-well Omega filter plate, and the other one is the Millipore MultiScreen-PCR96 filter plate. The latter plate has been applied for PCR product purification [29, 30]. To our knowledge, there are no publications reporting the evaluation of these products for proteomic sample preparation. In this study, we use different type of sample sources, including Klebsiella pneumoniae whole cell lysate and human urine samples, to systemically investigate the possible application. Our rationale to choose these samples is to gain insights into several ongoing projects that are related to urine proteomics and urinary pathogens [31, 32]. We compare their performance with single FASP, and attempt to demonstrate their broad application for quantitative proteome analyses.
Materials and methods
Bacterial cell isolation, culture, and lysis and urine sample preparation
A K. pneumoniae strain that represented a clinical isolated (KPC27) was used to grow bacterial cells in suspension culture in Luria Broth media at 37°C. At two time points, 4 hour and 16 hour, the cells were collected as exponential and stationary phase, respectively. Two separated culture experiments were performed as biological replicates. The cells were first centrifuged at 5,000 x g for 10 minutes at 4°C. The pellets were washed twice with a 10-fold volume of ice-cold PBS, then lysed in several steps: (1) resuspension of the pellet in a solution of 1% SDS, 5 mM EDTA, and 50 mM DTT (1:5 volume ratio); (2) vortexing a few times followed by heating for 3 min at 95°C; (3) sonication of the lysate in a Misonex sonicator water bath at amplitude 6 in 10 cycles (30 sec on/30 sec off) in ice water; (4) a repeated heating step. After cooling down to room temperature, the samples were centrifuged at 16,000 g for 30 min. An aliquot of the K. pneumoniae lysate supernatant was taken to determine the protein concentration using the BCA assay as described before [24]. For protein digestion on the filter plates, all the 96FASP procedures [24] were followed except that the time for each centrifugation step was shortened to ~ 20 min. For digestion using single-device FASP, YM30 filters (Microcon, Millipore) were used. Around 50 μg total protein was used for each FASP experiment. After digestion, the peptides were desalted following spinnable StageTip protocol [33], and stored in −80°C until further usage. An illustrative workflow of q96FASP approach is depicted in Supporting Information Figure S-1.
Urine sample was processed following protocols published previously [32]. In brief, around 20 ml urine were first concentrated using Amicon Ultra-15 centrifugal filter device (10 kDa MWCO, Fisher Scientific). The protein concentration was measured using the Bradford Protein Assay Kit [24]. Aliquots (~50 μg protein) were boiled with 1% SDS (w/v) and 50 mM DTT at 95°C for 10 minutes, then loaded onto 96-well filter plate. The following processing steps were similar to the procedure described above. As a note, the urine specimens were deidentified and considered as medical waste from an on-going project [32]. Therefore, the study was exempted from human subject protocols and bioethical review [32].
LC-MS/MS, protein identification and quantitation
LC-MS/MS analysis was performed using an Ultimate 3000-nano LC system coupled to a Q-Exactive mass spectrometer (Thermo Scientific). The experimental and data acquisition methods were previously described in detail [24]. Briefly, peptides were separated over a 150 min gradient from 2% to 80% (120 min to 35%, 10 min to 80%) in buffer B (0.1% formic acid in acetonitrile) at a flow rate of 200 nl/min in an in-house packed column (75 μm x 15 cm, 3.0 μm ReproSil-Pur C18-AQ media). The MS survey scans were acquired at a resolution of 70,000 over a mass range of m/z 250-1,800. In each cycle, the ten most intense ions were subjected to high-energy collisional dissociation (HCD) applying a normalized collision energy of 27%. The MS/MS scans were performed at a resolution of 17,500.
The MS raw data were processed using the Proteome Discoverer platform (version 1.4, Thermo Scientific) and Sequest HT algorithm. A database that contained protein sequences from the bacterial strain UHKPC27 (5,642 sequences) and from a non-redundant human proteome (20,195 sequences; reviewed sequences only; version 2015_06) acquired from UniProt knowledgebase was used to computationally identify peptides and proteins. Search parameters included (1) two missed tryptic cleavages, (2) oxidation (M), protein N-terminal acetylation and deamidation (N, Q) as variable modifications, and (3) carbamidomethylation (C) as a fixed modification. The minimum peptide length is seven amino acids. MS and MS/MS ion tolerances were set at 10 ppm and 0.02 Da, respectively. The FDR was estimated using the integrated Percolator tool. Only protein hits identified with a 1% FDR threshold were accepted. For protein quantification, the MaxQuant and Andromeda software suite (version 1.4.2.0) was used. We accepted most of the default settings provided in this software [32]. Both the label-free quantitation (LFQ) and the intensity-based absolute quantitation (iBAQ) tools were enabled. The MaxLFQ algorithm generates relative quantification using the integrated MS1 peak areas from high resolution MS data [34]. The iBAQ tool sums the intensities of all the peptide ions for a given protein divided by the number of predicted peptides [35]. It is capable of quantifying proteins within the same proteome when the in-depth coverage is adequately achieved [36]. In this study, only proteins that were quantified by at least two unique peptides were used for analysis. The clustering and correlation analyses were performed in Perseus environment (version 1.5.0.15) using embedded functions. Before analysis, the LFQ intensities generated by MaxQuant were log (base 2) transformed, and then imputed with missing values by default settings in Perseus. All LC-MS/MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD006137. Detailed protein/peptide identifications were implemented in Supplemental Tables.
To calculate the isoelectric point (pI), molecular weight (MW) and Grand average of hydropathy (GRAVY) score of proteins or peptides, we used the online tools http://www.gravy-calculator.de and http://web.expasy.org/compute_pi/.
Results and Discussion
q96FASP plates centrifuge equally quick to single FASP filter
In most published reports using the FASP method, two types of filter membranes have largely been used: one is cellulose acetate-based such as the Vivacon 500 filter (Sartorius) [37-41] and the other is regenerated cellulose-based such as the Microcon filter (Millipore) [42-45]. Although the chemistry of the filter materials differs slightly, both membranes are compatible with high molarity urea solutions and allow fast liquid transfer. The types of membrane filters reported here, the MultiScreen-PCR96 filter (referred as PCR filter in the manuscript) and the Pall AcroPrep 96-well filter (referred as the Pall filter in the manuscript), are manufactured with an undisclosed material and modified polyethersulfone media, respectively. Both membranes have a pore size around 30 kDa MWCO. First, we set to assess the time required to filter a 8M urea solution through the plates centrifuged at the manufacturer recommended speed. In our experiments, both plates allowed filtration of 200 μl urea solution in 20 min using a bench-top plate-adapted centrifuge with swinging-bucket rotors at the centrifugal force of 2,500 g. An equivalent experiment with 200 μl ABC buffer took ~15 min, which was much faster than the one using a 10 kDa filter plate [24]. Thus, the sample processing time was entirely comparable with that of single FASP. We then assessed whether removal of the detergent from an 4% SDS solution from the two membranes could be achieved. We indeed did not observe any SDS signals during LC-MS analysis [24].
q96FASP plates perform as well as single FASP filter
We investigated whether the two 96well filter plates allowed the identification of proteins in numbers observed for single FASP filter devices. Our pre-experiment data showed that both the PCR and Pall plates generated hundreds of protein identifications in a single LC-MS run. We proceeded to more quantitative investigations. We used a K. pneumoniae strain UHKPC27 cell lysate as the sample source (referred to as Kp lysate from here on). Aliquots of the Kp lysate were processed in five wells of each of the q96FASP plates, and in five single FASP filters (YM30). The LC-MS/MS experiments identified 1083 ± 43, 1065 ± 41 and 1107 ± 79 proteins from PCR, Pall and YM30 filters, respectively. The peptide identification numbers were 5,851 ± 424, 5,922 ± 244 and 6,050 ± 356, respectively (Figure 1). These data suggest that the quality of the sample processing, including protein and peptide recovery for LC-MS/MS, was equally good in the two types of 96-well filter plates compared to the single FASP filter device. For quantitative proteomics, both label-based and label-free approaches have been employed previously. Although labeling-based methods have been the gold standard for quantification, they inherently require extra preparation steps, and can be significantly uneconomical for the studies involving large sample cohort [34]. This is often true for clinical proteomics or biomarker discovery studies [46]. Label-free approach by its nature is economical and in principle is universal to any type of sample. In this study, we utilized such method, MaxLFQ, which integrates delayed normalization and maximal peptide ratio extraction strategies to determine proteome-wide quantitation [34]. The LFQ data derived from MaxQuant that suggest protein quantities of Kp lysates were evaluated and visualized in the heat map (Figure 2). The result indicated a high similarity of protein abundances across all five experiments for each of the three types of filters. The required number of unique peptides per protein was set at minimally two peptides present in at least two of the five replicates of each method. We also required the proteins to be quantified in three or more experiments of total 15. In total, 857 proteins were quantified and displayed in the heat map. The overall similarity of the proteomic quantification in Kp lysates was confirmed by pairwise Pearson correlation analysis (Supporting Information Figure S-2). The correlation r values are generally greater than 0.9 for wells from intra-filter comparisons and greater than 0.8 for inter-filter comparisons.
Figure 1.
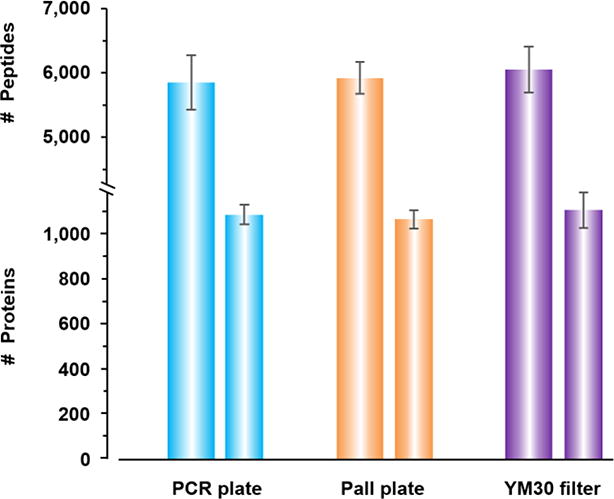
Comparison of protein and peptide identification by q96FASP plates (PCR and Pall) and single FASP filter (YM30). Five replicates were used for the plot.
Figure 2.
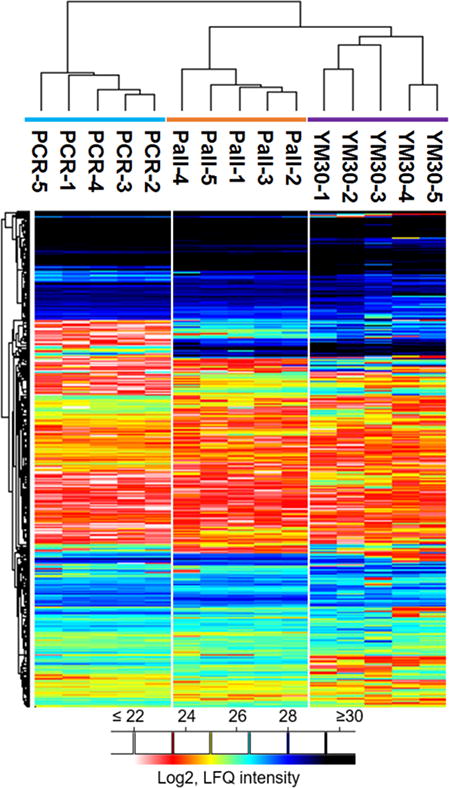
Quantitative overview of the performance of q96FASP plates and single FASP filter. Log2-transformed LFQ intensity values obtained from MaxQuant were loaded into Perseus software, and unsupervised hierarchical clustering using euclidean distance was applied. The proteins were filtered to eliminate those that were detected in only one run or quantified by single peptide. In total, 857 proteins were displayed in the plot.
As the q96FASP plates and single filter FASP device contain membranes with different chemistries, the same number of identification may not reflect the same group of identifications. We investigated if the products retained varying subsets of the entire proteome. We combined the search results of all the replicates, and compared the overlap of protein and peptide identifications. The Venn diagrams (Figure 3) showed that around 82% of the proteins and 57% of the peptides were common to each membrane. On average, 5% of protein identification and 21% of peptide identification are unique to one of the filters. We then determined the Gene Ontology (GO) cellular compartment annotations of the identified proteins. The ten most abundant GO terms with high significance (p < 10−5) were listed side-by-side (Figure 3). Only minor differences were noted for the majority of the GO terms. The slightly increased numbers of cell wall and plasma membrane proteins in the PCR 96qFASP plates were not statistically significant.
Figure 3.
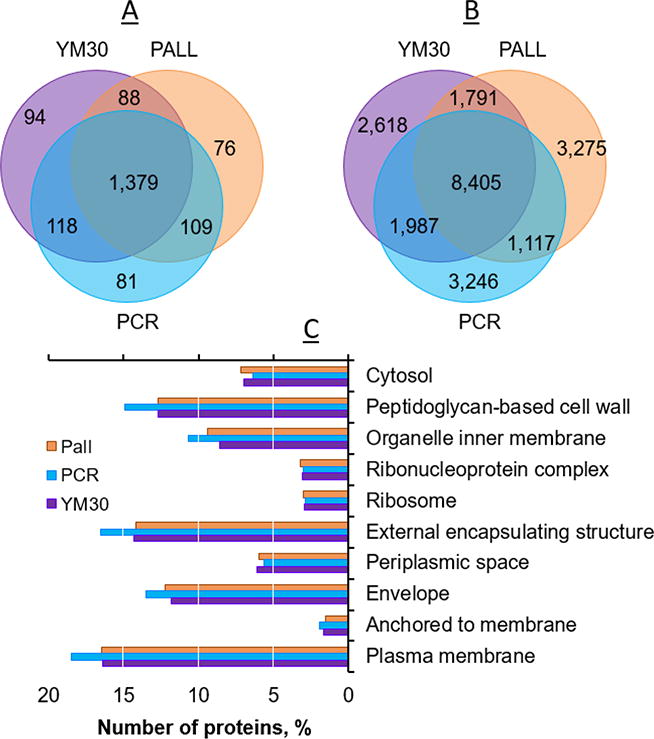
Venn diagrams showing the overlap analyses of protein (A) and peptide (B) identifications by the three filters. Histogram (C) shows the top10 most populated GO cellular compartment terms for the proteins identified by each method.
To further understand filter-specific differences, we investigated the physicochemical properties of the identifications on the protein and peptide level, respectively. As shown in Figure 4, we compared the pI, molecular weight (or mass), and GRAVY score. The latter is used to measure overall protein or peptide hydrophobicity [47]. The three filter devices did not show proteomic differences as it pertains to these biophysical chemical traits. The PCR plate showed only a slight preference for proteins with a low pI.
Figure 4.
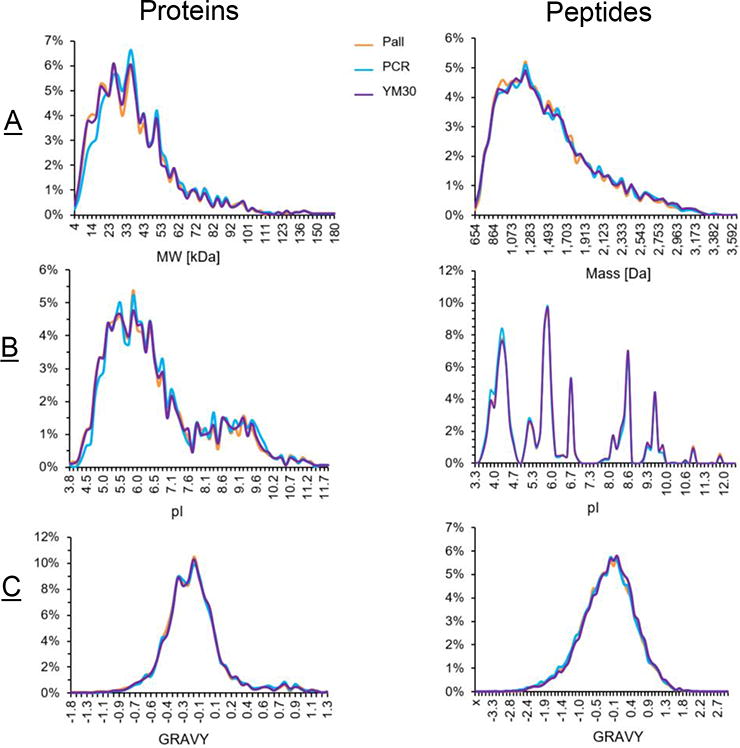
Comparisons of physicochemical properties of the proteins (left) and peptide (right) identified by the three filters. The protein molecular weight (MW) or peptide mass (A), pI (B) and GRAVY scores (C) were compared.
Missed cleavages have an ultimate effect on label-free protein quantitation, which uses combined information of all the identified peptides belonging to a protein. Therefore, we investigated if the q96FASP plates would affect the on-membrane digestion. The data showed that the identified peptides containing one missed tryptic cleavage were 15.9%, 20.1% and 16.9% for PCR plate, Pall plate and FASP filter, respectively. Our data are consistent with a previous report [48]. The peptides that contain two missed cleavages were less than 2% as it pertains to all three filter devices. The minimal variations of the digestion efficiency between q96FASP plates and single FASP filter suggest that the q96FASP approach has no visible side effects on on-filter tryptic digestion. In summary, the data presented above suggested that the new q96FASP approach dramatically reduces the spin times required for sample concentration and liquid transfer. It is fairly reproducible and shows no altered representation on the protein or peptide level compared to the single FASP method and no reduction in depth of proteome coverage, as shown in Figure 2. Slight variations among the methods according to side-by-side correlation analysis (r < 0.9; Supplemental Figure S2) may be related to differences in the membrane chemistry, protein and peptide binding differences, and/or potential losses based on protein and filter pore size. Different manufacturing of the filter membranes may also contribute to differences in protein retention and peptide passage through filters. We observed higher variations on peptide level than on protein level (Figure 5 and Supporting Information Figure S-3). Our data suggest that, if high protein coverage is desirable, the use of all three devices may improve proteome coverage due to some complementarity in the peptide identification data used. Quantitative proteome may benefit more from such combination. For instance, when the data from both q96FASP plates and FASP filter are combined, the number of proteins that could be reliably quantify increased to 1,086, which was almost a 30% increase comparing to either approach alone.
Figure 5.
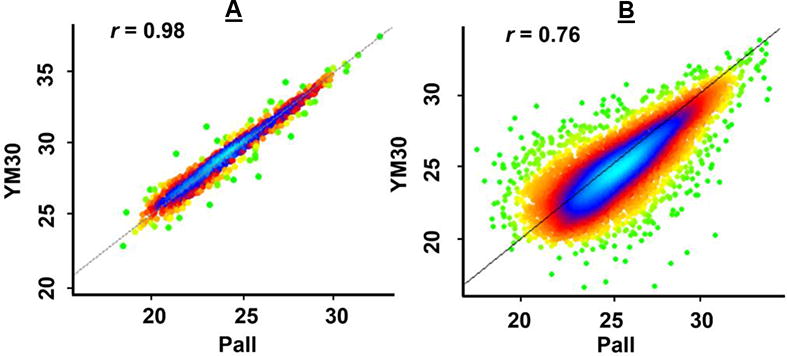
Pearson correlation analyses of q96FASP plate and single FASP filter. The density scatter plots between Pall and YM30 filter were shown as representative (Figure S-3 for details). Both proteins (A) and peptides (B) identified by the two filters were correlated. Combined results of five replicates of each filter were used for the analysis. The correlation r values were shown in the plot.
Analysis of the proteome of carbapenem-resistant K. pneumoniae, an infectious agent resistant to nearly all antibiotic drugs
We analyzed the clinical isolate UHKPC27 of K. pneumoniae. The strain associated with a urinary tract infection was recently sequenced [49]. The isolate was grown in LB media to both exponential and stationary phases. Two biological replicates were generated for each phase. We analyzed the samples with two technical replicates using single-run LC-MS/MS. Proteomic studies of different cell growth conditions of uropathogenic E. coli and S. aureus have been reported [50, 51], which indicated that broad quantitative changes on the proteome level may occur during the shift from growing to non-growing conditions. Stationary phase S. aureus cells were found to be extremely difficult to kill by antibiotics [52]. Global quantitative investigation may identify proteins that attentively cope with environmental or conditional changes, therefore help discover therapeutic makers. In our study, about 1,490 and 1,488 proteins were identified from exponential and stationary phases, respectively, and 1,275 proteins were commonly identified in both conditions. In total, 1,703 unique protein groups were identified, equivalent to one third of the predicted proteome (5,642 sequences, UniProt Taxon identifier 1284813) [49].
To quantify the proteins and compare their abundance levels across the two growth conditions, we used the MaxLFQ-based label-free approach as described above. To ensure maximal accuracy, we required the proteins to be identified in at least two runs in one of the groups (Exp or Stat), and overall in at least three runs. In total, 818 proteins were retained, and their intensities (after Log2 transformation) were displayed in the heat map. Both biological and technical replicates generally showed high similarities, indicating good to excellent experimental reproducibility. Two samples t-test led to 99 proteins with significant changes (fold change ≥ 2, p value ≤ 0.01), including 63 proteins up-regulated in the stationary phase and 36 proteins up-regulated in the exponential phase (Figure 6). Gene Ontology (GO) analysis indicated that the biological process GO terms over-represented in stationary phase were oxidation reduction, amine and acetyl-CoA catabolic processes, aerobic respiration and response to osmotic stress were. Those over-represented in the exponential phase were glycerol and G3P metabolic processes, translation, glucoside transport and carbohydrate catabolic process. Extensive reprogramming of gene expression was reported to take place upon transition from exponential (or growing) to stationary (or non-growing) phase [50, 51, 53]. Our findings are consistent with previous reports. In addition, using human urine as a different sample type, we further demonstrated the q96FASP application for body fluid samples. We processed the urine to assess data quality and differences among the two types of q96FASP plates. About 300~400 proteins were identified in Pall and PCR plate. The heat map in Supporting Information Figure S-4 shows the profiles of the urinary proteome obtained using q96FASP plates. The urinary proteome spans over five orders of magnitude, a dynamic range similar to that reached by the 96FASP method [24].
Figure 6.
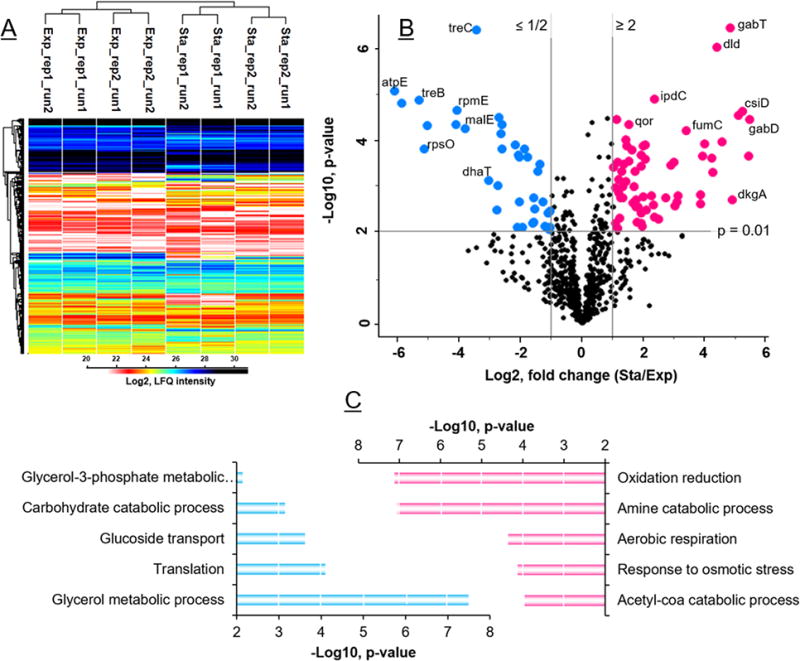
Quantitative proteomics of K. pneumoniae cells growing in exponential and stationary phases. The heat map (A) shows the unsupervised hierarchical clustering analysis of 818 proteins identified from two biological replicates (rep1 & rep2) and two technical replicates (run1 & run2) of each condition. Volcano plot (B) depicts the same set of proteins with fold changes (x-axis) and significance (y-axis). Histograms (C) show the top5 most significant GO biological process terms of each growth condition.
Conclusions
In this study, we demonstrate a technically improved process for highly parallel proteomic sample preparation which we call q96FASP. The membrane filter plates we used featured two different filter chemistries and a pore size with around 30-kDa cutoff. The method solved the issue observed with a previously developed 96FASP approach: slow liquid transfer times at the allowable centrifugal force. The new method is now completely comparable with the conventional single FASP method. Direct comparisons of the two methods show that q96FASP provides as many protein identifications as the single FASP device using a bacterial lysate. The method shows no bias towards proteins or peptides with distinct biophysical or chemical properties. Although the majority of identifications is shared, there is still a relatively large number of them, particularly on the peptide level, are unique to each method, which suggests that combined approach may lead to deeper proteome coverage than single method. A comparative analysis of two K. pneumoniae growth states using label-free quantitation revealed that biologically meaningful conclusions could be drawn. Urinary sample analysis using q96FASP indicates its potential in clinical proteomics and biomarker discovery studies. While the manuscript was in preparation, we noticed a similar study released very recently [54], in which the authors investigated the possibility of Pall 30K filter plate for high throughput proteomics sample preparation. All the results and conclusions were consistent with what have presented in this study. Meanwhile, in addition to the comprehensive quantitation assessed here, we further demonstrated its applications for the analysis of the dynamic proteome of carbapenem-resistant K. pneumoniae strain as well as urine proteome. Such demonstrations were solely present in our study. We also provided more options of the q96FASP filter plate by using both Pall filter plate and Millipore MultiScreen PCR plate. The PCR plate has been utilized for genomic studies [29, 30], but has never been tested before for proteomic sample preparation. The plate tends to have appealing utilities in the sample preparation for multi-omics studies (e.g., genomics, transcriptomics, proteomics) [55, 56]. Overall, we believe the data present here will make the q96FASP approach much appealing for high throughput quantitative proteome analysis.
Supplementary Material
Significance.
High throughput sample processing is of particular interests for quantitative proteomics. The previously developed 96FASP is high throughput and appealing, however it is time-consuming in the context of centrifugation-based liquid transfer (~1.5 hour per spin). This study presents a truly high throughput sample preparation method based on large cut-off 96-well filter plate, which shortens the spin time to ~20 min. To our knowledge, this is the first multi-well method that is entirely comparable with conventional FASP. This study thoroughly examined two types of filter plates and performed side-by-side comparisons with single FASP. Two types of samples, whole cell lysate of a UTI (urinary tract infection)-associated Klebsiella pneumoniae cell and human urine, were tested which demonstrated its capability for quantitative proteomics. The q96FSAP approach makes the filter plate-based approach more appealing for protein biomarker discovery projects, and could be broadly applied to large scale proteomics analysis.
Acknowledgments
This work was supported in part by grants NIH-1R01GM103598 (from National Institute of General Medical Sciences) and 1DP3DK094343-01 (from National Institute of Diabetes and Digestive and Kidney). We thank the Ruggles Family Foundation for a contribution to fund the mass spectrometry system, and Meredith Wright (JCVI) for providing the UHKPC27 strain. We also thank Joseph Kramer and Susan Gigler (EMD Millipore Chemicals) for technical support, and Dr. Feng Yan (GSK Vaccines, Rockville, MD) for helpful discussion. The PCR and Pall plates utilized in this study are generous gifts from Millipore and Pall Corporation, respectively.
Footnotes
Conflict of Interest Disclosure
The authors declare no competing financial interest.
References
- 1.Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem Rev. 2013;113(4):2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Percy AJ, Byrns S, Pennington SR, Holmes DT, Anderson NL, Agreste TM, Duffy MA. Clinical translation of MS-based, quantitative plasma proteomics: status, challenges, requirements, and potential. Expert Rev Proteomics. 2016;13(7):673–684. doi: 10.1080/14789450.2016.1205950. [DOI] [PubMed] [Google Scholar]
- 3.Coorssen J, Yergey A. Proteomics Is Analytical Chemistry: Fitness-for-Purpose in the Application of Top-Down and Bottom-Up Analyses. Proteomes. 2015;3(4):440–453. doi: 10.3390/proteomes3040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoofnagle AN. Quantitative Clinical Proteomics by Liquid Chromatography–Tandem Mass Spectrometry: Assessing the Platform. Clin Chem. 2010;56(2):161–164. doi: 10.1373/clinchem.2009.134049. [DOI] [PubMed] [Google Scholar]
- 5.Berger ST, Ahmed S, Muntel J, Cuevas Polo N, Bachur R, Kentsis A, Steen J, Steen H. MStern Blotting–High Throughput Polyvinylidene Fluoride (PVDF) Membrane-Based Proteomic Sample Preparation for 96-Well Plates. Mol Cell Proteomics. 2015;14(10):2814–2823. doi: 10.1074/mcp.O115.049650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Botelho D, Wall MJ, Vieira DB, Fitzsimmons S, Liu F, Doucette A. Top-Down and Bottom-Up Proteomics of SDS-Containing Solutions Following Mass-Based Separation. J Proteome Res. 2010;9(6):2863–2870. doi: 10.1021/pr900949p. [DOI] [PubMed] [Google Scholar]
- 7.Tran JC, Doucette AA. Gel-Eluted Liquid Fraction Entrapment Electrophoresis: An Electrophoretic Method for Broad Molecular Weight Range Proteome Separation. Anal Chem. 2008;80(5):1568–1573. doi: 10.1021/ac702197w. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J-Y, Dann GP, Shi T, Wang L, Gao X, Su D, Nicora CD, Shukla AK, Moore RJ, Liu T, Camp Ii DG, Smith RD, Qian W-J. Simple Sodium Dodecyl Sulfate-Assisted Sample Preparation Method for LC-MS-Based Proteomics Applications. Anal Chem. 2012;84(6):2862–2867. doi: 10.1021/ac203394r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 10.Han D, Moon S, Kim Y, Kim J, Jin J, Kim Y. In-depth proteomic analysis of mouse microglia using a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics. 2013;13(20):2984–2988. doi: 10.1002/pmic.201300091. [DOI] [PubMed] [Google Scholar]
- 11.Vertommen A, Møller ALB, Cordewener JHG, Swennen R, Panis B, Finnie C, America AHP, Carpentier SC. A workflow for peptide-based proteomics in a poorly sequenced plant: A case study on the plasma membrane proteome of banana. J Proteomics. 2011;74(8):1218–1229. doi: 10.1016/j.jprot.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 12.George IS, Fennell AY, Haynes PA. Protein identification and quantification from riverbank grape, Vitis riparia: Comparing SDS-PAGE and FASP-GPF techniques for shotgun proteomic analysis. Proteomics. 2015;15(17):3061–3065. doi: 10.1002/pmic.201500085. [DOI] [PubMed] [Google Scholar]
- 13.Nagaraj N, Alexander Kulak N, Cox J, Neuhauser N, Mayr K, Hoerning O, Vorm O, Mann M. System-wide Perturbation Analysis with Nearly Complete Coverage of the Yeast Proteome by Single-shot Ultra HPLC Runs on a Bench Top Orbitrap. Mol Cell Proteomics. 2012;11(3):M111.013722. doi: 10.1074/mcp.M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nel AJ, Garnett S, Blackburn JM, Soares NC. Comparative reevaluation of FASP and enhanced FASP methods by LC-MS/MS. J Proteome Res. 2015;14(3):1637–1642. doi: 10.1021/pr501266c. [DOI] [PubMed] [Google Scholar]
- 15.Hernandez-Valladares M, Aasebø E, Mjaavatten O, Vaudel M, Bruserud Ø, Berven F, Selheim F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol Proced Online. 2016;18(1):13. doi: 10.1186/s12575-016-0043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiśniewski JR, Mann M. Consecutive Proteolytic Digestion in an Enzyme Reactor Increases Depth of Proteomic and Phosphoproteomic Analysis. Anal Chem. 2012;84(6):2631–2637. doi: 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
- 17.Erde J, Loo RRO, Loo JA. Enhanced FASP (eFASP) to Increase Proteome Coverage and Sample Recovery for Quantitative Proteomic Experiments. J Proteome Res. 2014;13(4):1885–1895. doi: 10.1021/pr4010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDowell GS, Gaun A, Steen H. iFASP: Combining Isobaric Mass Tagging with Filter-Aided Sample Preparation. J Proteome Res. 2013;12(8):3809–3812. doi: 10.1021/pr400032m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Xie L, Gunawardena HP, Khatun J, Maier C, Spitzer W, Leerkes M, Giddings MC, Chen X. GOFAST: an integrated approach for efficient and comprehensive membrane proteome analysis. Anal Chem. 2012;84(21):9008–9014. doi: 10.1021/ac300134e. [DOI] [PubMed] [Google Scholar]
- 20.Huber ML, Sacco R, Parapatics K, Skucha A, Khamina K, Müller AC, Rudashevskaya EL, Bennett KL. abFASP-MS: Affinity-Based Filter-Aided Sample Preparation Mass Spectrometry for Quantitative Analysis of Chemically Labeled Protein Complexes. J Proteome Res. 2014;13(2):1147–1155. doi: 10.1021/pr4009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trindade F, Amado F, Gomes PS, Vitorino R. endoProteoFASP: a novel FASP approach to profile salivary peptidome and disclose salivary proteases. Talanta. 2015;132:486–93. doi: 10.1016/j.talanta.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Wisniewski JR, Prus G. Homogenous Phase Enrichment of Cysteine-Containing Peptides for Improved Proteome Coverage. Anal Chem. 2015;87(13):6861–6867. doi: 10.1021/acs.analchem.5b01215. [DOI] [PubMed] [Google Scholar]
- 23.Zielinska DF, Gnad F, Wiśniewski JR, Mann M. Precision Mapping of an In Vivo N-Glycoproteome Reveals Rigid Topological and Sequence Constraints. Cell. 2010;141(5):897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Yu Y, Suh MJ, Sikorski P, Kwon K, Nelson KE, Pieper R. Urine Sample Preparation in 96-Well Filter Plates for Quantitative Clinical Proteomics. Anal Chem. 2014;86(11):5470–5477. doi: 10.1021/ac5008317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frantzi M, Bhat A, Latosinska A. Clinical proteomic biomarkers: relevant issues on study design & technical considerations in biomarker development. Clin Transl Med. 2014;3(1):7. doi: 10.1186/2001-1326-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Din S, Lennon AM, Arnott IDR, Hupp T, Satsangi J. Technology Insight: the application of proteomics in gastrointestinal disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4(7):372–385. doi: 10.1038/ncpgasthep0872. [DOI] [PubMed] [Google Scholar]
- 27.Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol. 2014;10(10):757. doi: 10.15252/msb.20145625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cominetti O, Núñez Galindo A, Corthésy J, Oller Moreno S, Irincheeva I, Valsesia A, Astrup A, Saris WHM, Hager J, Kussmann M, Dayon L. Proteomic Biomarker Discovery in 1000 Human Plasma Samples with Mass Spectrometry. J Proteome Res. 2016;15(2):389–399. doi: 10.1021/acs.jproteome.5b00901. [DOI] [PubMed] [Google Scholar]
- 29.Hertzberg M, Aspeborg H, Schrader J, Andersson A, Erlandsson R, Blomqvist K, Bhalerao R, Uhlén M, Teeri TT, Lundeberg J, Sundberg B, Nilsson P, Sandberg G. A transcriptional roadmap to wood formation. Proc Natl Acad Sci U S A. 2001;98(25):14732–14737. doi: 10.1073/pnas.261293398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramadan N, Flockhart I, Booker M, Perrimon N, Mathey-Prevot B. Design and implementation of high-throughput RNAi screens in cultured Drosophila cells. Nat Protocols. 2007;2(9):2245–2264. doi: 10.1038/nprot.2007.250. [DOI] [PubMed] [Google Scholar]
- 31.Yu Y, Kwon K, Tsitrin T, Bekele S, Sikorski P, Nelson KE, Pieper R. Characterization of Early-Phase Neutrophil Extracellular Traps in Urinary Tract Infections. PLoS Pathog. 2017;13(1):e1006151. doi: 10.1371/journal.ppat.1006151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Y, Sikorski P, Smith M, Bowman-Gholston C, Cacciabeve N, Nelson KE, Pieper R. Comprehensive Metaproteomic Analyses of Urine in the Presence and Absence of Neutrophil-Associated Inflammation in the Urinary Tract. Theranostics. 2017;7(2):238–252. doi: 10.7150/thno.16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Smith M, Pieper R. A spinnable and automatable StageTip for high throughput peptide desalting and proteomics. Protocol Exchange. 2014 doi: 10.1038/protex.2014.033. [DOI] [Google Scholar]
- 34.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol Cell Proteomics. 2014;13(9):2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 36.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins. Mol Cell Proteomics. 2012;11(3):M111.014050. doi: 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiśniewski JR, Zielinska DF, Mann M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal Biochem. 2011;410(2):307–309. doi: 10.1016/j.ab.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Pieper R. Urinary Pellet Sample Preparation for Shotgun Proteomic Analysis of Microbial Infection and Host–Pathogen Interactions. In: Posch A, editor. Proteomic Profiling. Springer; New York: 2015. pp. 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kramer T, Greco TM, Enquist LW, Cristea IM. Proteomic Characterization of Pseudorabies Virus Extracellular Virions. J Virol. 2011;85(13):6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, Matic I. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat Chem Biol. 2016;12(12):998–1000. doi: 10.1038/nchembio.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruschi M, Santucci L, Ravera S, Candiano G, Bartolucci M, Calzia D, Lavarello C, Inglese E, Ramenghi LA, Petretto A, Ghiggeri GM, Panfoli I. Human urinary exosome proteome unveils its aerobic respiratory ability. J Proteomics. 2016;136:25–34. doi: 10.1016/j.jprot.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Wiśniewski JR. Filter-Aided Sample Preparation: The Versatile and Efficient Method for Proteomic Analysis. In: Shukla AK, editor. Methods in Enzymology. Academic Press; 2017. pp. 15–27. [DOI] [PubMed] [Google Scholar]
- 43.Ren J, Zhang M-J, Li T-M, Zhang J-E, Lin R, Chen S, Luo M, Dong M-Q. Quantitative Proteomics of Sleep-Deprived Mouse Brains Reveals Global Changes in Mitochondrial Proteins. PLoS One. 2016;11(9):e0163500. doi: 10.1371/journal.pone.0163500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glatter T, Ahrné E, Schmidt A. Comparison of Different Sample Preparation Protocols Reveals Lysis Buffer-Specific Extraction Biases in Gram-Negative Bacteria and Human Cells. J Proteome Res. 2015;14(11):4472–4485. doi: 10.1021/acs.jproteome.5b00654. [DOI] [PubMed] [Google Scholar]
- 45.Han D, Jin J, Woo J, Min H, Kim Y. Proteomic analysis of mouse astrocytes and their secretome by a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics. 2014;14(13-14):1604–1609. doi: 10.1002/pmic.201300495. [DOI] [PubMed] [Google Scholar]
- 46.Decramer S, de Peredo AG, Breuil B, Mischak H, Monsarrat B, Bascands JL, Schanstra JP. Urine in Clinical Proteomics. Mol Cell Proteomics. 2008;7(10):1850–1862. doi: 10.1074/mcp.R800001-MCP200. [DOI] [PubMed] [Google Scholar]
- 47.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 48.Wiśniewski JR. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal Chem. 2016;88(10):5438–5443. doi: 10.1021/acs.analchem.6b00859. [DOI] [PubMed] [Google Scholar]
- 49.Wright MS, Suzuki Y, Jones MB, Marshall SH, Rudin SD, van Duin D, Kaye K, Jacobs MR, Bonomo RA, Adams MD. Genomic and Transcriptomic Analyses of Colistin-Resistant Clinical Isolates of Klebsiella pneumoniae Reveal Multiple Pathways of Resistance, Antimicrob. Agents Chemother. 2015;59(1):536–543. doi: 10.1128/AAC.04037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Becher D, Hempel K, Sievers S, Zühlke D, Pané-Farré J, Otto A, Fuchs S, Albrecht D, Bernhardt J, Engelmann S, Völker U, van Dijl JM, Hecker M. A Proteomic View of an Important Human Pathogen – Towards the Quantification of the Entire Staphylococcus aureus Proteome. PLoS One. 2009;4(12):e8176. doi: 10.1371/journal.pone.0008176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King T, Kocharunchitt C, Gobius K, Bowman JP, Ross T. Physiological response of Escherichia coli O157:H7 Sakai to dynamic changes in temperature and water activity as experienced during carcass chilling. Mol Cell Proteomics. 2016;15(11):3331–3347. doi: 10.1074/mcp.M116.063065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503(7476):365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otto A, Bernhardt J, Meyer H, Schaffer M, Herbst FA, Siebourg J, Mäder U, Lalk M, Hecker M, Becher D. Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nature Communications. 2010;1:137. doi: 10.1038/ncomms1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Potriquet J, Laohaviroj M, Bethony J, Mulvenna J. A modified FASP protocol for high-throughput preparation of protein samples for mass spectrometry. bioRxiv. 2016 doi: 10.1371/journal.pone.0175967. doi: https://doi.org/10.1101/084533. [DOI] [PMC free article] [PubMed]
- 55.Yoon SH, Han MJ, Jeong H, Lee CH, Xia XX, Lee DH, Shim JH, Lee SY, Oh TK, Kim JF. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012;13(5):R37. doi: 10.1186/gb-2012-13-5-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu NQ, ter Huurne M, Nguyen LN, Peng T, Wang S-Y, Studd JB, Joshi O, Ongen H, Bramsen JB, Yan J, Andersen CL, Taipale J, Dermitzakis ET, Houlston RS, Hubner NC, Stunnenberg HG. The non-coding variant rs1800734 enhances DCLK3 expression through long-range interaction and promotes colorectal cancer progression. Nat Commun. 2017;8:14418. doi: 10.1038/ncomms14418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.