

Abstract
Pluripotent stem cells may serve as an alternative source of beta-like cells for replacement therapy of type 1 diabetes; however, the beta-like cells generated in many differentiation protocols are immature. The maturation of endogenous beta cells involves an increase in insulin expression starting in late gestation and a gradual acquisition of the abilities to sense glucose and secrete insulin by week 2 after birth in mice; however, what molecules regulate these maturation processes are incompletely known. In this study, we aim to identify small molecules that affect immature beta cells. A cell-based assay, using pancreatic beta-like cells derived from murine embryonic stem (ES) cells harboring a transgene containing an insulin 1-promoter driven enhanced green fluorescent protein reporter, was used to screen a compound library (NIH Clinical Collection-003). Cortisone, a glucocorticoid, was among five positive hit compounds. Quantitative reverse transcription–polymerase chain reaction analysis revealed that glucocorticoids enhance the gene expression of not only insulin 1 but also glucose transporter-2 (Glut2; Slc2a2) and glucokinase (Gck), two molecules important for glucose sensing. Mifepristone, a pharmacological inhibitor of glucocorticoid receptor (GR) signaling, reduced the effects of glucocorticoids on Glut2 and Gck expression. The effects of glucocorticoids on ES-derived cells were further validated in immature primary islets. Isolated islets from 1-week-old mice had an increased Glut2 and Gck expression in response to a 4-day treatment of exogenous hydrocortisone in vitro. Gene deletion of GR in beta cells using rat insulin 2 promoter-driven Cre crossed with GRflox/flox mice resulted in a reduced gene expression of Glut2, but not Gck, and an abrogation of insulin secretion when islets were incubated in 0.5 mM d-glucose and stimulated by 17 mM d-glucose in vitro. These results demonstrate that glucocorticoids positively regulate glucose sensors in immature murine beta-like cells.
Keywords: : small molecule screening, immature beta cells, murine embryonic stem cells, glucose sensors, postnatal young islets
Introduction
Pancreatic beta cells are required for maintaining glucose homeostasis by secreting insulin when they encounter high concentrations of glucose in blood after feeding. Glucose-stimulated insulin secretion is not observed immediately at birth. Rather, beta cells undergo gradual maturation processes, including an increase of beta cell mass and insulin expression in beta cells starting at late gestation [1,2] and the development of glucose-responsive insulin secretion comparable to that of their adult counterparts by 2 weeks after birth in mice [3]. Immature islets are characterized by a lower responsive threshold to glucose (∼2.8 mM) [3], compared with adult islets (5–7 mM) [4].
The maturation of beta cells can be controlled by transcription factors in a cell-autonomous manner; for example, by the transcription factor v-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA) [5]. MafA is capable of binding directly to the insulin promoter and enhancing insulin gene expression [6]. Knockout of MafA in the pancreas of mice does not affect the architecture of islets [7], which contain beta cells at the core and glucagon-secreting alpha cells at the periphery. The beta-to-alpha cell ratio is not affected by deletion of MafA either [7]. However, MafA-knockout mice progressively become diabetic due to lowered expression of glucose transporter-2 (Glut2) [7]. Overexpression of MafA delivered by an adeno-viral vector in cultured islets from 2-day-old rats enhances glucokinase (Gck) expression and glucose-stimulated insulin secretion [8]. Glut2 [9] and Gck [10] are two key glucose sensors in beta cells. These studies demonstrate that MafA is critically important for beta cell maturation. However, little is known about the noncell autonomous molecules that regulate beta cell maturation.
Recent studies have demonstrated that insulin-expressing beta-like cells can be efficiently generated from human pluripotent stem cells in vitro [11,12]. However, those beta-like cells do not match their adult counterparts in glucose-stimulated insulin secretion [11]. In this study, we began to explore whether immature beta cells can be affected by small molecules. We reasoned that the practicality of a small molecule, compared to a transcription factor, would provide an advantage in delivery to cells to affect the maturation process in vitro.
We started our investigation by performing a cell-based screen of a library containing 440 compounds that are presently on the market or have undergone Phase 1 clinical trial. A murine embryonic stem (ES) cell line (designated MIP-EGFP) [13], which contains a transgene with an enhanced green fluorescent protein (EGFP) reporter driven by the murine insulin 1 promoter, was used as the cellular platform. An increase in fluorescent signal indicates an increase in activity of insulin 1 promoter. Surprisingly, we found that cortisone, an agonist of the glucocorticoid receptor (GR; encoded by Nr3c1), caused an increase in the EGFP signal in the primary screen.
GR is a transcription factor that belongs to a large family of nuclear hormone receptors and plays multiple roles in development, immune response, and metabolism [14]. In adults, administration of GR agonists inhibits insulin secretion from beta cells and causes insulin resistance in peripheral tissues [15]. During mouse development, inactivation of GR in pancreatic progenitor cells leads to increased beta cell mass, suggesting that GR inhibits the differentiation of early pancreatic progenitors into beta cells [16]. However, deletion of GR in the committed beta cells in the mouse embryos does not change beta cell mass or beta-to-alpha cell ratio [16,17], demonstrating a minimal effect of GR in the differentiated beta cells. However, whether GR affects maturation of beta cells has not been determined.
Interestingly, during secondary analyses of the hit compounds we observed that cortisone enhanced expression not only of Insulin 1 but also of Glut2 and Gck. We therefore hypothesized that glucocorticoid signaling positively affects glucose sensors in immature beta cells. In this study, we show that glucocorticoids increase the gene expression of Glut2 and Gck in murine ES cell-derived cells, as well as in immature 1-week-old islets from mice. Genetic knockout of GR in insulin-expressing beta cells of 1-week-old islets reduced Glut2 gene expression and abrogated glucose-responsive insulin secretion in vitro. These results suggest a previously unknown role of glucocorticoid signaling in glucose sensing in immature murine pancreatic beta cells. In this study, murine cells were used as a model system because of our extensive experiences in differentiation of murine ES cells into pancreatic-like cells in vitro [18–21], as well as the availability of plentiful reagents and transgenic models in mice. Since many signaling pathways that control embryonic and postnatal development are commonly shared among mice and humans, we anticipate that the glucose-sensing molecules of beta-like cells derived from human pluripotent stem cells are likely to be affected by glucocorticoids as well.
Materials and Methods
Murine ES cell lines
The MIP-EGFP ES cell line (in C57Bl/6 background) was derived as reported [13] and used in the chemical screening experiments. The Ngn3-EGFP ES cell line (in 129 background) contains an EGFP reporter gene driven by the endogenous Neurogenin (Ngn) 3 promoter [22]. MIP-EGFP, Ngn3-EGFP, and the nontransgenic R1 (in 129 background) [23] ES cell lines were used for secondary analyses. These ES cell lines were routinely checked for mycoplasma contamination.
Cell-based screening of compounds
Murine ES cells were differentiated into embryoid bodies (EBs) in suspension culture for 6 days followed by plating into two-dimensional attachment culture (in six-well plates) to generate beta-like cells as reported [18–20] (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd). For screening experiments, day-6 EBs were plated into flat-bottom, black-walled, 96-well plates (Corning Costar, Tewksbury, MA; cat. no. 3603) at a density of 10 EBs per well and incubated at 37°C for an additional 12 days (to yield day-18 cells). Based on our prior results [18], day 16 was the preferred stage of cells for starting the screening experiment; however, day-18 cells were used because of scheduling issues. A total of 440 compounds from the NIH Clinical Collection (NCC-003) (Supplementary Table S1) were prepared as 10 mM stock solutions in dimethyl sulfoxide (DMSO). Using a Biomek FX liquid handler (Beckman Coulter, Brea, CA), compounds were diluted 1:100 in water, and subsequently 2 μL of each of the diluted solutions was added to day-18 cells for an overall 1:10,000 dilution (the final concentration of each compound was 1 μM). The plates were returned to the incubator for 9 days, with the media supplemented with the test compounds refreshed every 3 days. DMSO was used as the vehicle control (n = 8 per experiment). A total of two screening experiments, each done in technical duplicate, were conducted. After 9 days, plates were scanned on an ImageXpressUltra (Molecular Devices, Sunnyvale, CA) equipped with a 488 nm argon laser to detect the EGFP signal. The green fluorescence levels were recorded and subsequently analyzed using a custom threshold from the MetaXpress software (Molecular Devices). The top 1% hits were selected for subsequent analyses.
Quantitative reverse transcription–polymerase chain reaction
These methods were the same as previously reported [18]. The internal control used in all experiments was β-Actin. TaqMan probes used in this study are listed in Supplementary Table S2.
Mice
Both sexes of postnatal CD-1 [24] and C57Bl/6J mice were used in the experiments associated with Figure 6. A total of 12–16 young mice were obtained for each experiment. Beta cell-specific GR knockout mice (both sexes) were generated by mating homozygous GRflox/flox (Jax mice stock no. 021021) [25] with rat insulin 2 promoter (RIP)-driven Cre+/− mice (Jax mice stock no. 003573) [26]. Breeding was conducted by the Animal Resource Center of the City of Hope. Genotyping was performed by tail sampling 2–3 days before dissection. Eight-day-old GR β-cell-KO mice (GRflox/flox; RIP-Cre+/−) and control littermates (GRflox/flox; RIP-Cre−/−) (both sexes; 4–8 mice per group per experiment) were used for the studies associated with Figure 7. All mice were maintained under specific pathogen-free conditions, and animal experiments were conducted using protocols approved by the Institutional Animal Care and Use Committee at City of Hope.
FIG. 6.

Postnatal young islets express GR and GLUT2 and respond to HC in enhancing Glut2 and Gck expression. (A) Postnatal day (P) 3, P8, and adult pancreases were fixed in formalin, paraffin-embedded, sectioned, and stained with insulin and GR antibodies. Scale bar = 50 μm. (B) Islets were isolated from P5, P8, and adult mice and gene expression analyzed by qRT-PCR. Data represent mean ± SD from three independent experiments. Significance was determined by one-way ANOVA. **** indicates P < 0.0001 compared to either P5 or P8 islets. (C) P7 islets were isolated and cultured in the presence of HC for 1 or 4 days. At the end of the culture, gene expression was analyzed by qRT-PCR using β-Actin as internal control. Data represent fold change compared to the DMSO control (designated as “0’). Data represent mean ± SD of four independent experiments. Significance was determined by one-way ANOVA. *, **, and *** indicate P < 0.05, P < 0.01, and P < 0.001, respectively, compared to the DMSO control.
FIG. 7.

Islets from mice with beta cell-specific knockout of GR have reduced gene expression of Glut2 and do not exhibit glucose-stimulated insulin secretion in vitro. P8 islets were isolated from mice that had beta cell-specific knockout of GR (GR β-cell-KO) or from control (Con) littermates. (A) Gene expression was analyzed by qRT-PCR using β-Actin as an internal control and expressed as fold change compared to the control mice. Data represent mean ± SD from six independent experiments. Significance was determined by t-test. *** indicates P < 0.001 compared to control. n.s. indicates not significant. (B) P8 islets were sequentially stimulated with low (0.5 mM) followed by high (17.0 mM) concentrations of d-glucose. C-PEPTIDE concentrations in the media were determined by enzyme linked immunosorbent assay. Stimulation index is defined as the C-PEPTIDE concentration in high glucose divided by C-PEPTIDE concentration in low glucose. Data represent mean ± standard error of the mean from three independent experiments. Significance was determined by t-test. * Indicates P < 0.05.
Isolation of one-week-old islets
Mice were routinely euthanized at ∼10 a.m. Pancreases were dissected, minced for 2–3 min with spring scissors, and incubated in Dulbecco's phosphate buffered saline (Corning Cellgro) containing 1.5 mg/mL Collagenase B (Roche Applied Science, Penzberg, Germany) at 37°C for 6–8 min with gentle pipetting after the first 3 min. The lightly digested tissue was washed twice with Hank's balanced salt solution (HBSS; Corning Cellgro) containing 0.5% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) and resuspended in Histopaque 1100 (Sigma-Aldrich). An equal volume of HBSS/0.5% BSA was carefully layered on top. Density centrifugation was performed using a Sorvall ST 40r bench top centrifuge (Thermo Fisher Scientific, Waltham, MA) at 1,400 rpm for 25 min at 4°C, with the brakes turned off. Islets, in the resulting middle phase, were carefully collected by a Pasteur pipette. The islet-enriched fraction was washed twice with HBSS/0.5% BSA and allowed to recover overnight in 60 mm dish in RPMI 1640 supplemented with 10% fetal calf serum (FCS; Tissue Culture Biologicals, Long Beach, CA) and 11 mM d-glucose before subsequent culture with hydrocortisone (HC) or glucose challenge in vitro.
Isolation of adult islets
Islets from adult mice (2–4 month old) were isolated as described [27].
Briefly, pancreases were distended by injecting a solution containing 2.5 mg/mL Collagenase V (Sigma-Aldrich) through the common bile duct, dissected and digested at 37°C for ∼9 min. Following digestion, islets were separated from the nonislet tissue using Histopaque 1077 (Sigma-Aldrich). Adult islets were further purified by hand picking under the visualization of a light microscope.
Culture of islets
Enriched islets were randomly aliquoted (three wells per group) in 24-well plate and incubated in a medium containing Dulbecco's modified Eagle's medium (glucose free; Corning Cellgro), 15% Knockout Serum Replacement (Invitrogen, Carlsbad, CA), 5.0 mM d-glucose (Corning Cellgro), and 100 μg/mL aECM-lam [28] at 37°C for 1 or 4 days, in the presence of HC or DMSO control.
In vitro glucose-stimulated C-peptide secretion
Islets were randomly aliquoted (three wells per group) in 96-well plate and incubated at 37°C for 2 h in Krebs-Ringer buffered solution (KRBS; 129 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5 mM NaHCO3, 10 mM HEPES, and 0.1% BSA) containing 10% FCS and 0.5 mM d-glucose. Subsequently, islets were washed twice with KRBS containing 2% FCS and 0.5 mM d-glucose and sequentially incubated with 0.5 and 17.0 mM d-glucose (37°C, 0.15 mL/well, 1.5 h), starting with islets from GR β-cell-KO followed by control mice. The concentration of the released C-peptide in the buffer was measured using the murine C-peptide ELISA Kit, U-Type (Shibayaji Co., Gunma, Japan), which has a detection limit of 30 pg/mL. Stimulation was expressed as the fold change of C-peptide concentration in buffer from the second (high glucose) treatment compared with the first (low glucose) treatment of the same well.
Cortisone, analogs, and other compounds
All small molecules used in this report were purchased from Sigma-Aldrich and dissolved in DMSO to 10 mM stock concentration before use.
Immunohistochemical staining
The methods were the same as previously reported [29]. Formalin-fixed, paraffin-embedded cells from murine ES cell-derived cells or postnatal pancreas were processed for sections and staining. To ensure the specificity of the primary antibodies, positive control and negative control tissues were tested first. Subsequently, isotype control antibodies or omission of the primary antibodies were used in parallel to the primary antibodies in each staining experiment. Antibodies used are listed in Supplementary Table S3.
Western blot analysis
Cells were lysed on ice in RIPA buffer (Thermo Fisher Scientific) containing 50 mM TRIS, pH 7.4, 150 mM NaCl, 0.1 mM ethylenediaminetetraacetic acid, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) and supplemented with phosphatase inhibitor cocktail III (Calbiochem/Merck, Bad Soden, Germany) and complete protease inhibitor (Roche, Mannheim, Germany). Extracts were sedimented at 10,000 g for 15 min at 4°C to remove insoluble material. Protein concentrations were determined using the Pierce® BCA protein assay (Thermo Fisher Scientific). Samples (10 μg) of proteins were boiled in 2× sample buffer (Bio-Rad, Hercules, CA), separated by SDS-polyacrylamide gel electrophoresis on a 10% Laemmli gel, transferred to a polyvinylidene difluoride membrane (Bio-Rad), incubated with primary and secondary antibodies, and developed using enhanced luminol chemiluminescence (ECL Kit; Bio-Rad). Images were detected using the luminescent image analyzer SERIES 2000A Processor (TI-BA Enterprises, Rochester, NY). Antibodies used are listed in Supplementary Table S4.
Statistical analysis
Significance was determined by GraphPad Prism 7 software using one-way analysis of variance followed by Dunnett's multiple comparison test when three or more groups were compared or t-tests (unpaired, two-tailed) when two groups were compared. p < 0.05 was considered significant.
Results
Cell-based assay for screening of small molecules that enhance expression of the insulin1-EGFP reporter
Our laboratory previously established an in vitro differentiation system to drive murine ES cells into beta-like cells [18–20]. In those studies, overexpression of MafA between days 16 and 25 using an inducible murine ES cell line resulted in cells with enhanced expression of Insulin 1, Glut2, and Gck and increased glucose-stimulated insulin secretion [18]. These results indicated that cells in this time window were capable of undergoing maturation and were suitable targets for the chemical screen used in this study.
MIP-EGFP ES cells were differentiated into EBs, seeded in 96-well plates, and grown into day-18 cells (Supplementary Fig. S1). Small molecules from the NIH Clinical Collection-003 were added at a final concentration of 1 μM (n = 2 wells per molecule), and cells were cultured for an additional 9 days. Negative control wells (n = 8) received the vehicle DMSO (0.01%). EGFP fluorescence intensity was recorded at the end of the culture. A hit rate of 1% was preselected for our screening [30]. Results showed that the top 1% compounds from our screening had an average EGFP intensity that was at least 4.57-fold higher than the DMSO controls (Fig. 1A).
FIG. 1.

Screening of the NIH Clinical Collection (NCC-003) leads to the identification of five hit compounds that increase Insulin 1 expression in mES cell-derived pancreatic-like cells. (A) In the primary screening, the top five compounds showed more than fourfold increases in insulin1-EGFP reporter signals, compared to DMSO controls. (B) Secondary analyses, using MIP-EGFP mES-derived cells and commercially-available compounds, confirmed the increased expression of Insulin 1. Other genes, including Insulin 2, Glut2, and Gck, were examined as well. Day-18 cells (in six-well plates) derived from murine MIP-EGFP ES cells were cultured in the presence of designated compounds for 9 days and gene expression analyzed by qRT-PCR using β-Actin as the internal control. Data represent fold change compared to the DMSO vehicle control (designated as “0”). Note that cortisone (but not the other compounds) enhanced the expression of both Glut2 and Gck. Data represent mean ± SD of three independent experiments. Significance was determined by one-way ANOVA analysis. (C) Summary of the effects of cortisone on Glut2 and Gck expression. Data represent mean ± SD of four independent experiments. Significance was determined by t-test. *, **, ***, and **** indicate P < 0.05, P < 0.01, P < 0.001, and P < 0.0001, respectively, compared to the DMSO control. mES, murine embryonic stem; EGFP, enhanced green fluorescent protein; DMSO, dimethyl sulfoxide; qRT-PCR, quantitative reverse transcription–polymerase chain reaction; SD, standard deviation; ANOVA, analysis of variance; GR, glucocorticoid receptor; Glut2, glucose transporter-2; Gck, glucokinase.
Secondary analyses using commercially-available compounds confirmed that all five hits enhanced the expression of Insulin 1 in MIP-EGFP ES cells (Fig. 1B), consistent with the Insulin1-EGFP reporter results (Fig. 1A). The highest (80-fold) increase of Insulin 1, compared to DMSO control, was induced by Idarubicin (Fig. 1B). Interestingly, in the primary screen the green fluorescence signal induced by Idarubicin was very high (∼2,600-fold above the control) (Fig. 1A). Idarubicin belongs to the family of anthracycline drugs that are known to emit green fluorescence [31]. Indeed, under a fluorescence microscope we observed green fluorescence emission for Idarubicin solution in the absence of cells (not shown), suggesting that the autofluorescence of Idarubicin contributes to the strong fluorescence signal.
Among the hits, only cortisone enhanced both Glut2 and Gck expression compared to controls (Fig. 1B). The effects of cortisone on Glut2 and Gck were confirmed in four independent experiments (Fig. 1C). To determine if the effects of cortisone were cell line specific, we tested murine Ngn3-EGFP ES cells [19], which have a genetic background different from that of MIP-EGFP cells. Cortisone also enhanced Glut2 and Gck expression in Ngn3-EGFP ES cells (Fig. 2A). Together, these results identified cortisone as a promising subject for further study.
FIG. 2.
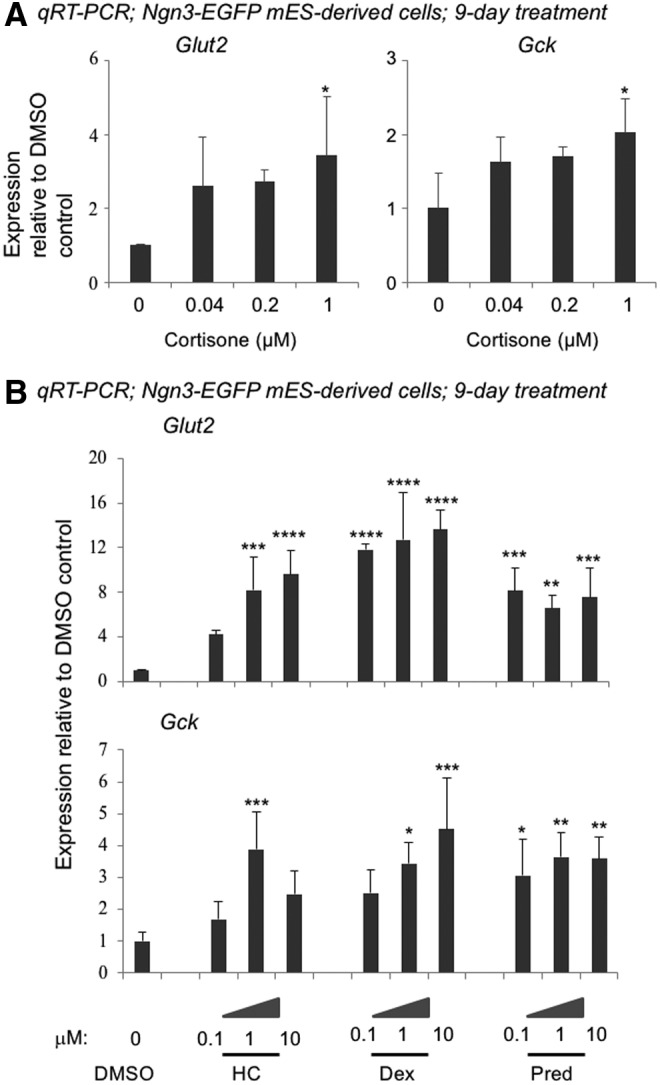
Effects of cortisone and other glucocorticoids on Glut2 and Gck gene expression in Ngn3-EGFP mES-derived cells. Day-18 cells derived from the Ngn3-EGFP mES cells were incubated with cortisone (A) or three other glucocorticoids (B) for a total of 9 days. The resulting cells were analyzed by qRT-PCR using β-Actin as the internal control. Data represent fold change compared to the DMSO vehicle control (designated as “0”). Data represent mean ± SD of three independent experiments. Significance was determined by one-way ANOVA. *, **, ***, and **** indicate P < 0.05, P < 0.01, P < 0.001, and P < 0.0001, respectively, compared to the DMSO control. Ngn, neurogenin.
Other glucocorticoids also enhance Glut2 and Gck expression in murine ES cell-derived cells
In addition to cortisone, we tested the effects of three other glucocorticoids-hydrocortisone (HC), dexamethasone (Dex), and prednisolone (Pred) (Fig. 3A). Expression of both Glut2 and Gck was increased by all three compounds in MIP-EGFP ES cell-derived cells (Fig. 3A), demonstrating that these effects are not unique to cortisone. Similarly in Ngn3-EGFP ES cells, HC, Dex, and Pred increased expression of Glut2 and Gck (Fig. 2B), showing again that the effects are not cell line specific. Because HC (also known as cortisol) is the natural glucocorticoid hormone secreted by the adrenal gland, this compound, rather than cortisone, was selected for subsequent studies.
FIG. 3.

Glucocorticoids enhance expression of Glut2 and Gck in mES cell-derived beta-like cells through GR signaling. Day-18 cells derived from mES cells were cultured in the presence of designated glucocorticoids for 9 (A, B) or 6 (C, D) days. (A) qRT-PCR analyses showed that glucocorticoids enhanced the expression of Glut2 and Gck in MIP-EGFP mES-derived cells. β-Actin was used as the internal control. Data represent fold change compared to the DMSO vehicle control (designated as “0”). Data represent mean ± SD of three independent experiments. Significance was determined by one-way ANOVA. *, **, ***, and **** indicate P < 0.05, P < 0.01, P < 0.001, and P < 0.0001, respectively, compared to the DMSO control. (B) Mifepristone, a GR antagonist, inhibited the effects of HC on the expression of Glut2 and Gck. Data represent mean ± SD of three independent experiments. Significance was determined by one-way ANOVA. * and ** indicate P < 0.05 and P < 0.01, respectively. (C) Western blot analysis showed that protein expression of Glut2 was enhanced by HC. β-ACTIN was the loading control. (D) Double immunofluorescence staining showed that C-PEPTIDE expressing cells coexpressed GLUT2 after HC treatment of R1 mES-derived cells. Scale bar = 50 μm. HC, hydrocortisone; Dex, dexamethasone; Pred, prednisolone.
Pharmacological inhibition of GR signaling reduces HC-induced Glut2 and Gck expression in murine ES-derived cells
To test whether HC enhances expression of Glut2 and Gck through GR signaling in day-18 cells, mifepristone [32], a GR antagonist, was added at increasing concentrations to cells cultured in 1 μM HC for 9 days (Fig. 3B). A dose-dependent inhibition of Glut2 and Gck by mifepristone was observed (Fig. 3B), suggesting that the effects of HC were dependent on GR signaling.
Protein expression of GLUT2 is enhanced by HC in murine ES-derived cells
Time course analyses demonstrated that expression of Glut2 and Gck was elevated in MIP-EGFP ES-derived cells after 6 days of HC treatment (Fig. 4A); therefore, this time point was used for subsequent analyses. Glut2 expression was confirmed by western blot analysis and found to be enhanced by HC after 6 days of treatment in MIP-EGFP ES-derived cells (Fig. 3C).
FIG. 4.

Time course analyses on the effects of HC on Glut2 and Gck expression. Day-18 cells from (A) MIP-EGFP or (B) R1 mES cells received 1 or 10 μM HC or DMSO control for 3, 6, or 9 days. The resulting cells were analyzed by qRT-PCR using β-Actin as the internal control. Six days of HC treatment, compared to the DMSO control, were sufficient to induce higher expression of Glut2 and Gck in both ES cell lines. Therefore, in some subsequent experiments we used an endpoint of 6 days post-HC incubation. Data represent mean ± SD of three independent experiments. Significance was determined by one-way ANOVA. * indicates P < 0.05 compared to the DMSO control.
Hepatocytes are known to express Glut2 [33]. To ensure that Glut2 is expressed by beta-like cells derived from murine ES cells, we performed double immunostaining of C-PEPTIDE (a marker for de novo synthesized insulin) and Glut2 (Fig. 3D). R1 murine ES cells were used in these experiments because this cell line does not carry an EGFP transgene. Time course analysis in R1 ES cells confirmed that expression of Glut2 and Gck was enhanced in the presence of HC after 6 days (Fig. 4B). In addition, GR protein was detected in the C-peptide positive cells in day-18 cultures (Fig. 5), confirming that murine ES-derived cells are equipped to respond to glucocorticoids. GLUT2 became detectable in C-PEPTIDE expressing cells treated with 1 μM HC for 6 days, but not in those treated with the DMSO control (Fig. 3D). These results confirm that HC increases the expression of GLUT2 in beta-like cells derived from murine ES cells in vitro.
FIG. 5.

mES-derived pancreatic-like cells express GR. Day-18 cells derived from R1 mES cells were fixed, paraffin-embedded, sectioned, and stained with rabbit anti-C-PEPTIDE and mouse anti-GR antibodies. Scale bar = 50 μm.
Beta cells of young mice express lower levels of GR and Glut2 and show different patterns of protein localization compared with adult beta cells
To further validate the findings from the murine ES cell system, we studied primary islets of young mice. Double immunostaining of formalin-fixed, paraffin-embedded pancreatic tissues confirmed the presence of GR proteins in beta cells of postnatal day (P)3, P8, and adult mice (Fig. 6A). Interestingly, GR was localized mostly in the cytoplasm at P3, both cytoplasm and nucleus at P8, and mostly nucleus in the adult beta cells. GR messages were detected in isolated islets from P5, P8, and adult mice, with higher levels of GR in adult compared with P5 and P8 islets (Fig. 6B). In this study, P5 islets instead of P3 islets were isolated because P3 islets were difficult to separate cleanly from the acinar tissues (not shown). The significance of the differential expression and localization of GR in young versus adult islets is presently unknown, but may suggest a stage-specific function of GR in beta cells.
It has been previously shown that Glut2 messages are increased in adult islets compared with those in the postnatal young mice [34,35]; here we confirmed this finding showing higher gene expression of Glut2 in adult compared to P5 and P8 islets (Fig. 6B). Double immunostaining of GLUT2 and insulin revealed that, in the P3 and P8 beta cells, GLUT2 was found mostly in the cytoplasm, with a subpopulation displaying higher intensity of GLUT2 on the plasma membrane (Fig. 6A). In contrast, GLUT2 localized mostly on cell surface of the adult beta cells (Fig. 6A). These results are consistent to prior reports on GLUT2 [34–37] and suggest that a lower Glut2 gene expression and a protein localization at the cytoplasm are associated with beta cell immaturity.
HC enhances gene expression of Glut2 and Gck in postnatal young islets in vitro
The results above (Fig. 6A) demonstrate that primary young beta cells are equipped with GR to respond to HC. To further test this, islets were isolated from 1-week-old mice using density centrifugation, allowed to recover overnight and cultured in the presence of HC or vehicle for 1 or 4 days (Fig. 6C). Expression of Glut2 and Gck was significantly increased by day 4 in islets treated with HC compared to vehicle (Fig. 6C), demonstrating that primary immature beta cells respond to glucocorticoids by increasing Glut2 and Gck gene expression.
Genetic knockout of GR in beta cells of one-week-old mice results in a reduced Glut2 but not Gck gene expression
To further test whether the expression of Glut2 and Gck requires GR in endogenous beta cells of 1-week-old mice, we deleted the GR gene in insulin-expressing cells by crossing GRflox/flox mice [38] with rat insulin 2 promoter (RIP)-Cre+/− [26] mice (Supplementary Fig. S2). One-week-old GRflox/flox;RIP-Cre+/− (GR β-cell-KO) mice appeared normal, consistent to prior reports [16,17]. Islets and acinar cells (Supplementary Fig. S3) were isolated from pancreases of 1-week-old GR β-cell-KO mice and GRflox/flox;RIP-Cre−/− littermates (controls), allowed to recover overnight and analyzed by quantitative reverse transcription–polymerase chain reaction. Expression of GR was reduced but not absent from the islet cell preparation in GR β-cell-KO mice, compared to controls (Fig. 7A). In GRflox/flox mice [25], exon 3 of GR, which contains the DNA-binding domain, is flanked by the loxP sequences and deleted in the GR β-cell-KO mice. The TaqMan probes that we used recognize GR at the junction of exons 2 and 3. Therefore, the GR mRNAs detected in the islet preparation of GR β-cell-KO mice were most likely due to the presence of other endocrine cells and the potential contamination of some acinar cells. Expression of Glut2, but not Gck, was decreased in the GR knockout islets compared to the controls (Fig. 7A), suggesting that GR is required for Glut2 but not Gck expression in primary young islets. The significance of the lack of Gck reduction in the absence of GR is not clear but may be due to a compensatory effect by other nuclear receptors [39].
GR is required for insulin secretion by young islets incubated in 0.5 mM and stimulated with 17 mM d-glucose in vitro
Finally, to test whether the islets of GR β-cell-KO mice were functional, isolated islets were subjected to sequential incubations with low (0.5 mM) and high (17.0 mM) concentrations of d-glucose in vitro. The concentration of 0.5 mM d-glucose was used because, as mentioned, young beta cells have a lower glucose threshold (∼2.8 mM) for stimulation compared with adult beta cells [3]. The amount of C-PEPTIDE released to the buffer was determined by enzyme linked immunosorbent assay. The stimulation index (ie, the ratio of concentrations of C-PEPTIDE in the medium measured at high and low concentrations of d-glucose) was determined. Control young islets exhibited a positive response to glucose as shown by a stimulation index of ∼2 (Fig. 7B). In contrast, islets from GR β-cell-KO mice did not secrete C-PEPTIDE in response to high concentrations of glucose, as indicated by a stimulation index near 1. These results demonstrate that GR is required for the postnatal young beta cells to secrete insulin in response to high concentrations of d-glucose in vitro.
Discussion
A rise in glucocorticoid concentrations in plasma during late gestation in multiple species [40] and between postnatal days 5 and 15 in mice [41] has led to the hypothesis that glucocorticoid signaling is important for the maturation of organs critical for survival after birth. Indeed, glucocorticoid signaling affects the maturation of several organs and cell types [40]. For example, glucocorticoids enhance the maturation of perinatal lung epithelium by promoting the expression of surfactant [42], induce myofibril assembly and systolic function of fetal heart [43], and increase amylase expression and secretion in pancreatic acinar cells in postnatal young rats [44].
In this study, we have discovered that beta cell maturation is also positively affected by glucocorticoid signaling. Specifically, exogenous HC is sufficient to increase gene expression of Glut2 and Gck, two molecules critical for glucose sensing, both in ES-derived beta-like cells and primary young islets. In addition, we found that genetic deletion of GR in the 1-week-old beta cells reduced Glut2 but not Gck expression, demonstrating that GR is critically important for Glut2 expression in young beta cells.
The increased expression of Glut2 in young islets induced by exogenous HC was unlikely to be an in vitro artifact because increases in expression of the Glut2 gene from the neonate to adult islets are known to occur in vivo [34,35] and confirmed in this study (Fig. 6B). In addition, we found 1 μM (36 μg/100 mL) exogenous HC to be sufficient to increase Glut2 and Gck expression by murine ES-derived beta-like cells and 1-week-old islets; this dose is above average but within the physiological range among newborn babies [45]. Taken together, our results demonstrate a positive role of glucocorticoid signaling in enhancing glucose sensing in immature beta cells. The enhancement of glucose sensing in immature beta cells, however, may not be sufficient to induce fully mature insulin secretion, because secretion requires additional steps, such as opening of ion channels, membrane depolarization, and exocytosis [46]. Further studies are needed to test the effects of glucocorticoids and GR on the later steps of insulin secretion in young beta cells. In addition, whether glucocorticoid signaling is sufficient to raise the glucose threshold in young beta cells to the adult level awaits further investigation.
Our results are surprising in lieu of the extensive literature demonstrating the inhibitory effects of glucocorticoids on adult beta cells [15]. The reason as to why glucocorticoid signaling might elicit different outcomes in young beta cells versus their adult counterparts is currently unknown, but we note that mature (but not immature) dendritic cells are susceptible to apoptotic signals induced by glucocorticoids due to the expression of different translational isoforms of GR [47].
Glucocorticoid signaling is known to regulate metabolism in adults [15]. The positive effects of glucocorticoids on immature beta (this study), acinar [44], and liver [40] cells—cell types important for metabolism, implicate the roles of glucocorticoid signaling in general metabolism of suckling mice. We speculate that feeding schedules in suckling mice may affect glucocorticoid signaling [48,49], which in turn may affect the peripheral clocks in the pancreas and liver [50] that regulate the expression of genes controlling metabolism.
We anticipate that GR agonists may enhance the efficacy of in vitro-generated beta-like cells derived from human pluripotent stem cells by enabling these cells to better sense glucose. Human pluripotent stem cell-derived pancreatic progenitor cells [51] are currently being tested (ClinicalTrial.gov; ID: NCT02239354) for cell replacement therapy in type 1 diabetes patients, in whom beta cells are destroyed by autoimmune attacks. As mentioned, beta-like cells can now be efficiently generated from human pluripotent stem cells in vitro [11,12], but they are relatively immature [11]. Interestingly, in their in vitro differentiation protocols for human pluripotent stem cells some laboratories have already used glucocorticoids to increase insulin-expressing cells [52] and glucose-stimulated insulin release [53]; however, the mechanisms underlying those results have remained unclear. The current study provides mechanistic insight into the potential effects of glucocorticoid signaling in immature human beta-like cells.
Supplementary Material
Acknowledgments
The authors thank Donna Isbell and Kelley Carpenter from the Animal Research Center of City of Hope for assistance in breeding the GR β-cell KO mice. The authors also thank Dr. Manami Hara for providing MIP-EGFP ES cells. This work was supported, in part, by the National Institutes of Health (R01DK081587 and R01DK099734 to H.T.K. and P30CA33572 to City of Hope). Support from the Oxnard Foundation, Ella Fitzgerald Foundation and the Wanek Family Project of Type 1 Diabetes to H.T.K. is also gratefully acknowledged. N.G. was supported by a predoctoral fellowship as part of an institutional grant to City of Hope from the California Institute for Regenerative Medicine (CIRM). Work at California Institute of Technology was supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program to M.T.K. and by National Science Foundation grant DMR 1506483 to D.A.T. The sponsor did not participate in the study design, collection, analysis, and interpretation of data.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, Sieweke M. and Stein R. (2007). MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A 104:3853–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kakita K, Giddings SJ, Rotwein PS. and Permutt MA. (1983). Insulin gene expression in the developing rat pancreas. Diabetes 32:691–696 [DOI] [PubMed] [Google Scholar]
- 3.Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A. and Melton DA. (2012). Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 30:261–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ensinck JW, Laschansky EC, Vogel RE. and D'Alessio DA. (1991). Effect of somatostatin-28 on dynamics of insulin secretion in perfused rat pancreas. Diabetes 40:1163–1169 [DOI] [PubMed] [Google Scholar]
- 5.Hang Y. and Stein R. (2011). MafA and MafB activity in pancreatic beta cells. Trends Endocrinol Metab 22:364–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olbrot M, Rud J, Moss LG. and Sharma A. (2002). Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A 99:6737–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, et al. (2005). MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol 25:4969–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A. and Bonner-Weir S. (2011). Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 54:583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guillam MT, Hummler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, Schmidt A, Deriaz N. and Thorens B. (1997). Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 17:327–330 [DOI] [PubMed] [Google Scholar]
- 10.Matschinsky FM. (1990). Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes 39:647–652 [DOI] [PubMed] [Google Scholar]
- 11.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O'Dwyer S, Quiskamp N, Mojibian M, et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32:1121–1133 [DOI] [PubMed] [Google Scholar]
- 12.Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D. and Melton DA. (2014). Generation of functional human pancreatic beta cells in vitro. Cell 159:428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milewski WM, Temple KA, Wesselschmidt RL. and Hara M. (2009). Generation of embryonic stem cells from mouse insulin I promoter-green fluorescent protein transgenic mice and characterization in a teratoma model. In Vitro Cell Dev Biol Anim 45:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicolaides NC, Charmandari E, Chrousos GP. and Kino T. (2014). Recent advances in the molecular mechanisms determining tissue sensitivity to glucocorticoids: novel mutations, circadian rhythm and ligand-induced repression of the human glucocorticoid receptor. BMC Endocr Disord 14:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose AJ. and Herzig S. (2013). Metabolic control through glucocorticoid hormones: an update. Mol Cell Endocrinol 380:65–78 [DOI] [PubMed] [Google Scholar]
- 16.Gesina E, Tronche F, Herrera P, Duchene B, Tales W, Czernichow P. and Breant B. (2004). Dissecting the role of glucocorticoids on pancreas development. Diabetes 53:2322–2329 [DOI] [PubMed] [Google Scholar]
- 17.Valtat B, Dupuis C, Zenaty D, Singh-Estivalet A, Tronche F, Breant B. and Blondeau B. (2011). Genetic evidence of the programming of beta cell mass and function by glucocorticoids in mice. Diabetologia 54:350–359 [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Chai J, Singh L, Kuo CY, Jin L, Feng T, Marzano S, Galeni S, Zhang N, et al. (2011). Characterization of an in vitro differentiation assay for pancreatic-like cell development from murine embryonic stem cells: detailed gene expression analysis. Assay Drug Dev Technol 9:403–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ku HT, Chai J, Kim YJ, White P, Purohit-Ghelani S, Kaestner KH. and Bromberg JS. (2007). Insulin-expressing colonies developed from murine embryonic stem cell-derived progenitors. Diabetes 56:921–929 [DOI] [PubMed] [Google Scholar]
- 20.Ku HT, Zhang N, Kubo A, O'Connor R, Mao M, Keller G. and Bromberg JS. (2004). Committing embryonic stem cells to early endocrine pancreas in vitro. Stem Cells 22:1205–1217 [DOI] [PubMed] [Google Scholar]
- 21.Winkler M, Trieu N, Feng T, Jin L, Walker S, Singh L. and Ku HT. (2011). A quantitative assay for insulin-expressing colony-forming progenitors. J Vis Exp (57):e3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee CS, Perreault N, Brestelli JE. and Kaestner KH. (2002). Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev 16:1488–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson E, Bradley A, Kuehn M. and Evans M. (1986). Germ-line transmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature 323:445–448 [DOI] [PubMed] [Google Scholar]
- 24.Jin L, Feng T, Shih HP, Zerda R, Luo A, Hsu J, Mahdavi A, Sander M, Tirrell DA, Riggs AD. and Ku HT. (2013). Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc Natl Acad Sci U S A 110:3907–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mittelstadt PR, Monteiro JP. and Ashwell JD. (2012). Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest 122:2384–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD. and Magnuson MA. (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274:305–315 [DOI] [PubMed] [Google Scholar]
- 27.Omori K, Kobayashi E, Komatsu H, Rawson J, Agrawal G, Parimi M, Oancea AR, Valiente L, Ferreri K, et al. (2016). Involvement of a proapoptotic gene (BBC3) in islet injury mediated by cold preservation and rewarming. Am J Physiol Endocrinol Metab 310:E1016–E1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghazalli N, Mahdavi A, Feng T, Jin L, Kozlowski MT, Hsu J, Riggs AD, Tirrell DA. and Ku HT. (2015). Postnatal pancreas of mice contains tripotent progenitors capable of giving rise to duct, acinar, and endocrine cells in vitro. Stem Cells Dev 24:1995–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin L, Gao D, Feng T, Tremblay J, Quijano J, Wedeken L, Luo A, Hsu J, Mahdavi A, et al. (2016). Cells with surface expression of CD133 high CD71 low are enriched for tripotent colony-forming progenitor cells in adult murine pancreas. Stem Cell Res 16:40–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ilouga PE. and Hesterkamp T. (2012). On the prediction of statistical parameters in high-throughput screening using resampling techniques. J Biomol Screen 17:705–712 [DOI] [PubMed] [Google Scholar]
- 31.Karukstis KK, Thompson EH, Whiles JA. and Rosenfeld RJ. (1998). Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys Chem 73:249–263 [DOI] [PubMed] [Google Scholar]
- 32.Jung-Testas I. and Baulieu EE. (1983). Inhibition of glucocorticosteroid action in cultured L-929 mouse fibroblasts by RU 486, a new anti-glucocorticosteroid of high affinity for the glucocorticosteroid receptor. Exp Cell Res 147:177–182 [DOI] [PubMed] [Google Scholar]
- 33.Thorens B. (2015). GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58:221–232 [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Flores M, Zueco J, Arenas J. and Blázquez E. (2002). Expression of glucose transporter-2, glucokinase and mitochondrial glycerolphosphate dehydrogenase in pancreatic islets during rat ontogenesis. Eur J Biochem 269:119–127 [DOI] [PubMed] [Google Scholar]
- 35.Navarro-Tableros V, Fiordelisio T, Hernandez-Cruz A. and Hiriart M. (2007). Physiological development of insulin secretion, calcium channels, and GLUT2 expression of pancreatic rat beta-cells. Am J Physiol Endocrinol Metab 292:292:E1018–E1029 [DOI] [PubMed] [Google Scholar]
- 36.Thorens B, Gerard N. and Deriaz N. (1993). GLUT2 surface expression and intracellular transport via the constitutive pathway in pancreatic beta cells and insulinoma: evidence for a block in trans-Golgi network exit by brefeldin A. J Cell Biol 123:1687–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M. and Marth JD. (2005). Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123:1307–1321 [DOI] [PubMed] [Google Scholar]
- 38.Brewer JA, Khor B, Vogt SK, Muglia LM, Fujiwara H, Haegele KE, Sleckman BP. and Muglia LJ. (2003). T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med 9:1318–1322 [DOI] [PubMed] [Google Scholar]
- 39.Huang P, Chandra V. and Rastinejad F. (2010). Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol 72:247–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fowden AL, Li J. and Forhead AJ. (1998). Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc 57:113–122 [DOI] [PubMed] [Google Scholar]
- 41.Taves MD, Plumb AW, Sandkam BA, Ma C, Van Der Gugten JG, Holmes DT, Close DA, Abraham N. and Soma KK. (2015). Steroid profiling reveals widespread local regulation of glucocorticoid levels during mouse development. Endocrinology 156:511–522 [DOI] [PubMed] [Google Scholar]
- 42.Odom MJ, Snyder JM, Boggaram V. and Mendelson CR. (1988). Glucocorticoid regulation of the major surfactant associated protein (SP-A) and its messenger ribonucleic acid and of morphological development of human fetal lung in vitro. Endocrinology 123:1712–1720 [DOI] [PubMed] [Google Scholar]
- 43.Rog-Zielinska EA, Craig MA, Manning JR, Richardson RV, Gowans GJ, Dunbar DR, Gharbi K, Kenyon CJ, Holmes MC, et al. (2015). Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: a role for PGC-1alpha. Cell Death Differ 22:1106–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Werlin SL. and Stefaniak J. (1982). Maturation of secretory function in rat pancreas. Pediatr Res 16:123–125 [DOI] [PubMed] [Google Scholar]
- 45.Stevens JF. (1970). Plasma cortisol levels in the neonatal period. Arch Dis Child 45:592–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rorsman P, Braun M. and Zhang Q. (2012). Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 51:300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao Y, Bender IK, Konstantinidis SC, Shin CMJ, Cidlowski RP, Cidlowski JA, Schleimer RP. and Lu NZ. (2013). Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood 121:1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schutz G. and Schibler U. (2000). Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289:2344–2347 [DOI] [PubMed] [Google Scholar]
- 49.Le Minh N, Damiola F, Tronche F, Schutz G. and Schibler U. (2001). Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J 20:7128–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F. and Schibler U. (2000). Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev 14:2950–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz TC. (2015). Concise review: manufacturing of pancreatic endoderm cells for clinical trials in Type 1 diabetes. Stem Cells Transl Med 4:927–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunisada Y, Tsubooka-Yamazoe N, Shoji M. and Hosoya M. (2012). Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem Cell Res 8:274–284 [DOI] [PubMed] [Google Scholar]
- 53.Takeuchi H, Nakatsuji N. and Suemori H. (2014). Endodermal differentiation of human pluripotent stem cells to insulin-producing cells in 3D culture. Sci Rep 4:4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.