

Abstract
Somatic mutations have been studied extensively in the context of cancer. Recent studies have demonstrated that high-throughput sequencing data can be used to detect somatic mutations in non-tumor cells. Analysis of such mutations allows us to better understand the mutational processes in normal cells, explore cell lineage in development, and examine potential associations with age-related disease. Here, we describe approaches for characterizing somatic mutations in normal and non-tumor disease tissues. We discuss several experimental designs and common pitfalls in somatic mutation detection, as well as more recent developments, such as phasing and linked-read technology. With the dramatically increasing numbers of samples undergoing genome sequencing, bioinformatic analysis will enable the characterization of somatic mutations and their impact in non-cancer tissues.
Keywords: mosaicism, cell lineage, single-nucleotide variants, phasing, linked-reads
Somatic mosaicism and challenges in detecting mosaic variants
Genomes from individuals of the same species differ from one another due to a constant influx of genetic mutation and recombination. Single nucleotide variants (SNVs), copy number variants (CNVs), transposable element (TE) insertions, and other structural variants (SVs) (see Glossary) are common types of genetic variation. Population-level heterogeneity generally arises due to germline mutations that occur before the formation of the zygote and are inherited by all cells in the offspring. However, heterogeneity within an individual may also exist due to somatic mutations that occur post-zygotically and exist only in a sub-population of cells. The genetic heterogeneity resulting from somatic mutations is known as somatic mosaicism. Recent papers have attempted to characterize somatic mosaicism [1]; yet the extent to which it exists, whether specific regions of the genome and nucleotide contexts are more susceptible to it, and how it impacts normal cellular function remain open questions.
In bulk sequencing data, somatic mutations have variant allele fractions (VAFs) that deviate from those typical of germline mutations (~0.5/1 for heterozygous/homozygous). The VAF of a somatic mutation depends both on the prevalence of the mutation, which is largely driven by how early the mutation occurs in development, and the heterogeneity of the tissue selected for sequencing. For example, if a mutation occurs during the first cell division and every cell produces the same number of descendants, the VAF would be ~0.25 in an unbiased sample (Figure 1, Key Figure). On the other extreme, if a mutation is uniquely acquired in a post-mitotic cell, the VAF would be infinitesimal (if bulk sequencing with 1 million cells, the VAF would be ~0.5 × 10−6). In general, somatic mutations occurring earlier during development attain higher VAFs than those occurring later. However, asymmetry in the developmental cell lineage tree [2], heterogeneity in selective pressure across tissues [3], and technical factors (such low read depth, sequencing error, and misalignment) can violate this principle.
Figure 1. Detecting somatic mosaicism in the genome through various sequencing strategies.
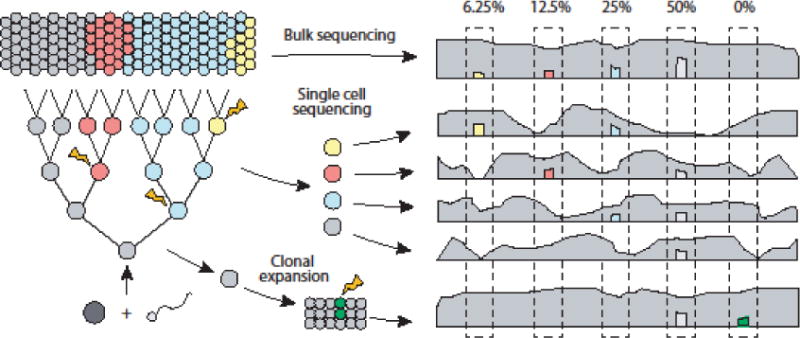
Somatic mutations arise during development and propagate to a sub-population of cells (blue: 50% of cells; red: 25%; yellow: 12.5%). With bulk sequencing, these somatic mutations are expected to be approximately half of the sub-population frequency. Lower frequency somatic mutations require higher sequencing depth to maintain detection sensitivity. With single cell sequencing, somatic mutations can be detected as heterozygous variants that occur in a subset of cells. The ability to detect variants is dependent on uniformity of coverage and allelic balance in genome amplification, as well as picking cells that contain variants. Clonal expansion followed by bulk sequencing does not suffer from the problems associated with single cell sequencing, but artifactual mutations that occur early during expansion (green) can be difficult to distinguish from mutations in the original cell.
A great deal of work has been done to develop algorithms for detecting somatic mutations in cancer. However, the VAFs of functionally-relevant cancer mutations tend to be higher than those in normal cells due to the selective advantage conferred by those mutations in proliferating cells. Thus, many popular algorithms for cancer are not focused on detecting very low VAF events (e.g., < 5% [4]), and comprehensive detection of somatic mutations at arbitrarily small VAFs in normal cells requires alternative methods. Additionally, somatic mutations in cancer are typically identified by the tumor-normal design, in which tumor tissue is compared to noncancerous (“normal”) tissue from the same individual to determine the mutations unique to the tumor. For non-cancer samples, mosaic variants arising early in embryogenesis are often shared among many tissues. This makes it difficult to identify a clear normal cellular subpopulation that can serve as a matched control. With careful selection of tissue specimens, however, it is possible to derive an accurate list of mosaic mutations that allows lineage analysis of cells in an individual. For example, Lodato et al. [5] analyzed heart and brain tissues, which develop from the mesoderm and ectoderm respectively, to find mosaic mutations informative of brain cell lineage; Behjati et al. [6] compared endoderm-derived gastrointestinal tissues to mouse tail, which consists of both mesodermal and ectodermal tissues, to find early embryonic mutations. The locations of selected specimens within a larger tissue can also be relevant: Martincorena et al. [7] utilized ultra-deep sequencing of multiple nearby fine biopsies to infer spatial patterns and rates of mosaicism in human skin.
In this review, we will provide an overview of somatic mutation analysis in normal cells. We will first cover the various platforms and experimental designs including bulk sequencing and single cell sequencing. Then we will describe strategies for detecting variants such as haplotype phasing, as well as common pitfalls encountered in these analyses.
Strategies for profiling mosaic variants
Whole-genome sequencing (WGS), whole-exome sequencing (WES), and targeted panels offer trade-offs between the types of detectable variants and the range of detectable VAFs. WGS produces the most uniform read depth across the genome and enables detection of most types of somatic mutations, including structural variants. However, detection is limited to relatively high VAF mutations, as the high sequencing depth required to detect low VAF mutations remains prohibitively expensive [8]. If attention can be restricted to specific loci, a customized panel can be constructed (e.g., amplicon-seq or targeted hybridization methods) and sequenced at very high depth (e.g., >100,000×). WES offers a compromise between WGS and small panels by targeting the ~1-2% of the genome that codes for proteins and need not be custom-designed.
Characterizing variants at the single cell level
Unlike bulk sequencing strategies that pool DNA from thousands or millions of cells, single cell sequencing attempts to sequence the DNA of only one cell. The advantage is that rare mosaic mutations can be more easily detected: if present in a diploid region of the chosen cell, the mutation will be present on one of two alleles, regardless of its frequency in the surrounding tissue (Figure 1, Key Figure). This shifts the technical difficulties associated with low frequency away from variant detection and onto the cell selection process. To estimate the overall frequency of each mutation in the tissue, multiple single cells must be sequenced, which can be expensive, laborious, and confounded by sampling bias. Hybrid experimental designs integrating bulk (either WGS or targeted) and single cell approaches can address many of these issues. For example, somatic mutations discovered in bulk can be confirmed by single cell data, and frequencies for somatic mutations discovered in single cells can be estimated from bulk sequencing.
A common strategy to produce sufficient input DNA for next-generation sequencing from a single cell is clonal expansion, in which a cell is expanded in culture until enough cells exist to perform standard bulk sequencing [6, 9–13]. However, additional mutations—especially SNVs— are continuously acquired during expansion and must be differentiated from mutations that existed in the founding cell. This is often addressed by discarding low AF candidate mutations, because in vitro mutations acquired after the first mitosis should be present at <25% VAF if cell division in culture is approximately symmetric. However, this symmetry assumption could be violated by variability in cell cycle lengths and the potential for selectively advantageous mutations in vitro, so careful analysis is warranted. It has also been shown that in vitro mutations can be characterized by mutational signatures that correlate with increasing culture time [6, 10]. An additional concern is that only a subset of the isolated single cells may successfully expand into colonies, possibly reflecting differences in cell fitness, tolerance to handling and cell culture or stochastic effects. Thus, studies relying exclusively on clonal expansion might not provide an accurate picture of tissue heterogeneity due to biased loss of specific cell types. For post-mitotic cell types (e.g., neurons), clonal expansion is not directly applicable. Encouragingly, a recent study demonstrated that adult neurons in mice could be clonally expanded and sequenced after inducing totipotency via single cell nuclear transfer (SCNT) [14]. However, SCNT is labor intensive, notoriously inefficient and may be even further affected by selection biases.
Another widely-used approach to produce enough DNA from a single cell is to apply whole genome amplification (WGA) [15–17] followed directly by sequencing. This approach has been used both in cancer [18–20] and in development [5, 21–24]. Several methods for WGA are available and represent different tradeoffs between genomic coverage, amplification uniformity and artifact load, and are reviewed elsewhere [15]. Because cell culture is unnecessary, WGA-based methodologies enjoy significant cost savings in both labor and reagents and can be directly applied to post-mitotic cells (such as neurons) and cells that are difficult to culture. The technical simplicity of WGA has also made it an attractive technology for scaling to handle hundreds or thousands of cells simultaneously [25, 26]. However, the disadvantage of WGA is the introduction of considerable amplification bias and allelic imbalance, which can produce artifacts that can be difficult to distinguish from true mutations. Research to improve variant calling in spite of these amplification artifacts is ongoing. It was recently demonstrated [27] that good specificity can be achieved for SNV detection for candidate somatic mutations that can be linked to nearby germline heterozygous variants (about 20% of the candidates, if using standard Illumina sequencing; more discussions below).
Mosaic mutation calling
Approaches for filtering germline variants
In cancer applications, mutation callers are often designed to simultaneously evaluate data from tumor and matched normal tissue from the same individual to discard mutations with any support in the normal tissue [4, 28–31]. Germline variants can also be filtered out by querying public variation databases or by using a “panel of normals” (PON) consisting of unrelated individuals (Figure 2). A recent study estimated that common variants in the public dbSNP database account for ~95% of germline SNVs in a typical human genome [32–34]; however, there is also evidence that aggressive exclusion of all polymorphic sites in dbSNP could lead to considerable false negative rates [35]. If the PON samples are processed and analyzed in the same way as the tumor, the PON approach can better control for systematic artifacts, such as those due to misalignment. For removing germline variants, it has been estimated that a PON consisting of at least 400 individuals would be necessary to reach the accuracy of having a matched normal sample [36]. Matched normal tissue sequencing, PON approaches and population databases are often combined to achieve high specificity.
Figure 2. Different strategies for detecting and filtering somatic mutations.
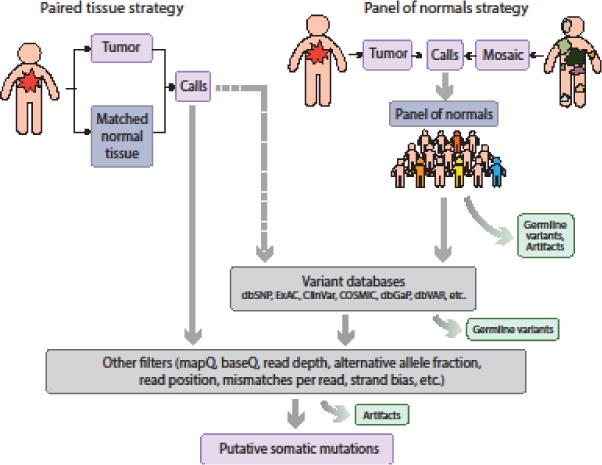
Somatic variant callers for a tumor tissue often require a matched normal tissue from the same individual. However, this strategy is not possible when matched normal tissue is unavailable. For somatic mosaicism in a non-tumor tissue, a matched ‘normal’ may not exist, as mutations of interest may be shared across tissues. Whenever matched normal tissue is unavailable, germline variants as well as some artifacts can be removed by querying public variation databases or by constructing a ‘panel of normals’ from sequencing data of unrelated individuals. Additional filters can be applied to further remove artifacts.
Applying these same strategies to detecting somatic mutations in non-tumor samples is problematic, as there is no clear “normal” tissue to use as a reference. When another tissue from the same individual is used as a reference, a true somatic mutation can be present in the reference sample if the mutation occurred in a common ancestor to both selected tissues. A large panel of other individuals may be used, with all samples processed in the same way as the sample of interest. But because the somatic mutation rate in non-cancer samples is much lower than in cancer [37–39], studies of somatic mosaicism are substantially less tolerant of false positives. More sophisticated algorithms and a series of stringent filters are necessary for detecting somatic mutations with higher accuracy. One example is MosaicHunter [40], which aims to detect mosaic SNVs without a matched normal using a Bayesian approach.
Some have applied germline variant callers, such as the Genome Analysis Toolkit (GATK) HaplotypeCaller, to detect mosaic SNVs [41]. One approach is to search for ‘heterozygous’ mutations and then to distinguish somatic mutations from germline mutations using a VAF threshold or other ad hoc heuristics. To increase sensitivity for low VAF variants, one could set the ‘ploidy’ in GATK HaplotypeCaller to be high, which lowers the expected VAF for a heterozygous variant [42]. However, a straightforward application of a germline caller is unlikely to yield sufficient sensitivity.
A parent-offspring trio analysis greatly increases the accuracy of variant detection, as mosaic mutations arising post-zygotically in a child are unlikely to be shared by the parents. Recently, four groups [41–44] studying autism spectrum disorders successfully detected mosaic SNVs by WES of parent-offspring trios using various approaches. However, even after removing variants present in either parent, mosaic SNV validation rates remained modest (~10-40%; more on validation later). Each study found it necessary to apply additional filters to reduce false positives, and in some cases it was necessary to exclude families with excess candidate mutations altogether. It was also apparent that the detection sensitivity and accuracy of many tools were diminished for mosaic SNVs with VAF < 0.10.
Detection of mosaic structural variations
Somatic CNV detection in cancer is complicated by clonal heterogeneity as well as experimental and technical noise, thus requiring sophisticated computational approaches [45]. Detecting mosaic CNVs in non-tumor samples is challenging because the amplitude of the copy number change may be small and matched normal samples are frequently unavailable. Some success in identifying mosaic CNVs has been achieved in single cell sequencing data [21, 22] or WES data [46], but they are limited to large, megabase-scale CNVs. Combined haplotyping (described later) of CNVs with SNVs or pedigree-based analyses appear to be the most promising strategies for detecting mosaic CNVs [47, 48].
Structural variation consists of many types of variants with varying length scales, including deletions, insertions, translocations, and TE insertions (SVs encompass events that result in CNVs and copy-neutral events). A survey of existing SV callers can be found elsewhere [49]. Less progress has been made in detecting mosaic SVs, as most somatic SV methods in cancer require matched normal data [50–54]. A recent method called MrMosaic can detect mosaic SVs without any matched normal by using deviations in coverage and allele fraction at polymorphic SNV sites [46]. However, MrMosaic can only detect insertions, deletions and loss-of-heterozygosity events, and does not identify specific breakpoints.
TEs are DNA sequences that can be copied and re-inserted into the genome. Although most TE activity in somatic tissue is repressed, some TEs are active during early embryogenesis and in germline cells. TEs have been shown to play important roles in many cancers [55–57], and there is some evidence that TEs may contribute to neuronal diversity, although the rate of such insertions has been shown to be much lower than initially proposed [58]. Detection of mosaic TEs from bulk data is difficult unless the insertion occurred early in development and has a high VAF. Alternative approaches include L1-insertion profiling [59] or WGS [23] for single cells.
Alternative technologies
Although short-read sequencing has matured considerably, it still suffers from alignment issues (especially in repetitive regions) and has limited power to detect complex structural variants. In particular, detecting very low VAF variants requires relying on as few supporting reads as possible, and even the smallest error rate in sequencing introduces potential artifacts. Read misalignments, which can create artifacts with many supporting reads, are often very difficult to differentiate from true somatic mutations. One recent technology with potential to alleviate issues related to short-read alignment is linked-read sequencing, in which fragments derived from the same long DNA molecule share a unique barcode [60]. These short fragments are then sequenced using standard short-read platforms, and the barcodes are used to stitch the reads into long sequences representing the original DNA molecule. Linked-read sequencing incurs additional cost for library construction but provides new opportunities for haplotype construction, detecting complex structural variants and extending mutation detection into repetitive regions of the genome. Single-molecule sequencing chemistries from PacBio and Oxford Nanopore also offer similar advantages, but their relatively high per-base error rate and cost do not make them competitive for large-scale profiling at this point.
Methods have also been designed to reduce the rate of sequencing artifacts to an order of magnitude below the somatic mutation rate by sequencing both the forward and reverse DNA strands [61, 62]. The mutations identified represent a random sampling of mosaics from the cell population and can provide estimates for somatic mutation rates and spectra. In theory, these methods can detect mutations present on only a single DNA molecule with reasonable specificity. In practice, a small fraction of the genome can be assayed and higher VAF mutations are more likely to be sampled.
Increasing accuracy by haplotype phasing
A haplotype is the sequence of alleles on one chromosome that are inherited from a single parent and haplotype phasing—sometimes simply referred to as haplotyping or phasing—is the process of identifying alleles that are co-located on the same chromosome. Haplotype phasing is informative in several applications, including correlating genetic variation with disease, detecting genotyping error, inferring evolutionary history, and examining the effect of cis-regulatory elements on gene expression [63]. Phasing is beneficial for somatic mutation detection because true mosaic events create a new haplotype with a consistent allele sequence, whereas artifacts often associate with haplotypes non-specifically.
Read-based phasing for mosaic SNVs
Traditional germline phasing methods infer haplotypes by taking advantage of segregation patterns in related individuals [63] or models of genetic recombination and mutation in a large population [63, 64]. However, these methods require genotype data from several individuals and depend on genetic inheritance, so they are of little use when phasing de novo somatic mutations. Sequencing data enable a different approach to phasing by exploiting the direct, physical evidence of linkage provided by reads (or read pairs) that span multiple variants. This ‘read-based’ phasing approach does not rely on inheritance and can be easily applied to data from a single individual; however, it is only effective when consecutive variant loci are close enough to be covered by a single sequencing read (or read pair). Since the read (or library fragment) length determines the maximum linkable inter-variant distance, the effectiveness of this approach depends considerably on the choice of sequencing platform [65, 66].
Spontaneously-arising mosaic mutations are extremely unlikely to affect more than one haplotype, so true mosaics that can be linked to a nearby germline heterozygous variant should be associated with only one of the two germline alleles. In bulk sequencing data of diploid organisms, a pair of SNVs consisting of a mosaic mutation and a germline heterozygote should therefore produce three haplotypes (Figure 3a,b), whereas certain types of artifacts (e.g., misalignment, sequencing errors at homopolymers, sample contamination) would associate with both alleles, generating additional haplotypes. Most candidate mosaic mutations with two or four apparent haplotypes can be safely rejected. This approach has been successfully applied in various studies [2, 42, 43] and specialized mutation-and-linkage callers have been developed [67]. Although only a small set (~10-30%) of candidate mosaic events are sufficiently close to be linked to germline SNPs, the retained mutations are typically of higher quality. However, it is important to note that a significant fraction of variants with three haplotypes may still be false positives [42], most likely due to misalignment (Figure 3c). This is most prominent in repetitive regions, but must also be guarded against in non-repetitive regions.
Figure 3. Overview of read-based mosaic phasing scenarios.
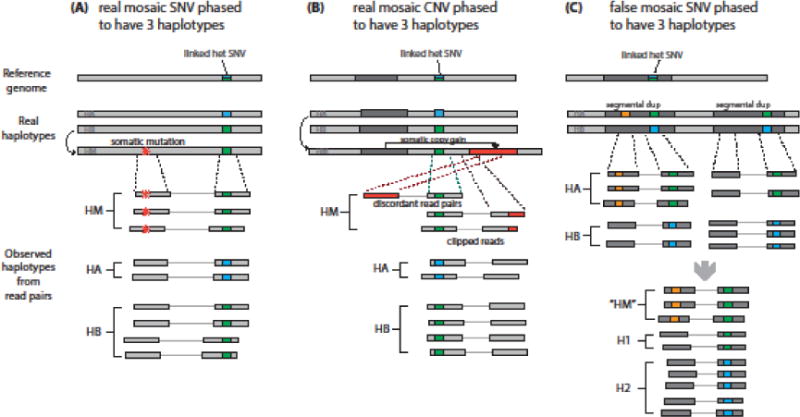
Read-based phasing can help identify true somatic mosaic mutations by examining the relationship between germline heterozygous variants and putative somatic mutations. However, some patterns of false-positives can confound this method. (A) If a real mosaic SNV (red star) arises near a heterozygous SNP, it will always be found in conjunction with one of the two SNP alleles (green) and will never appear on reads with the other allele (blue). This generates three haplotypes in bulk sequencing (HM for mosaic haplotype, in addition to HA and HB). (B) Similarly, a true mosaic CNV will phase with one allele of a nearby heterozygous SNP, resulting in three haplotypes. (C) Segmental duplications can cause a germline variant (orange) occurring on one duplicated segment to phase to a nearby heterozygous SNP occurring on both segments as if it were somatic, resulting in a false-positive identification.
The linkage principle can be extended to two or more nearby germline heterozygotes to reduce the probability that an artifact associates only with a single germline allele by chance. However, the number of potential haplotypes increases exponentially with the number of heterozygotes considered, which quickly leads to computational issues. An algorithm called LocHap [68] models the number of haplotypes at several SNVs in small genomic regions and defines regions with three or more haplotypes to contain mosaic events. However, because consideration of all possible haplotypes is computationally expensive, it disregards regions with more than three SNVs.
Phasing for single cells, structural variants, and the use of linked-reads
For single cell data, read-based phasing is particularly attractive because standard variant callers have difficulty distinguishing true mutations from the relatively large number of artifactual mutations that arise in genome amplification. Although only ~20% of the total candidate mutations in single cell WGS data can be phased, that subset can be used to infer the genome-wide mutation rate and to characterize the sequence features of the mutational processes. Recently, a method called LiRA was developed based on this idea [27] and applied to neuron WGS data to demonstrate that aging and neurodegeneration are associated with increased rate of mutation in the brain and to infer the source of those mutations [24].
The prevalence of SVs in healthy individuals is still under active investigation [69]. Current SV phasing methods are limited to germline events and often rely on data from multiple sequencing strategies [70]. Mosaic SVs can, in principle, be phased to a germline SNV in a manner analogous to mosaic SNVs by regarding the inferred SV breakpoint as a point event. As in SNV detection, artifactual mosaic SVs are likely to link to both of the germline alleles (Figure 3b).
Longer sequencing reads increase the power of read-based haplotyping by increasing the fraction of the genome that can be physically linked to germline heterozygous sites [65, 71] and by improving alignment to repetitive regions. Currently, the applicability of long read platforms to mosaic mutation detection is limited due to high cost and low per-base pair accuracy. However, an effective compromise may be provided by recent linked-read sequencing platforms that retain much of the long-range linkage information while achieving error rates similar to standard short-read sequencing. Several programs specializing in the phasing of linked reads are now available [60, 72, 73] and additional developments are likely to play an important role in future investigations of mosaic mutations.
Pitfalls in detection of mosaic variants
The search for mosaic mutations can be confounded by many factors, and claims of mosaic mutation discovery should be made cautiously. Several artifact sources that may lead to false positive mosaic calls are discussed below and summarized in Figure 4.
Figure 4. False positive mosaic calls can arise from multiple sources.
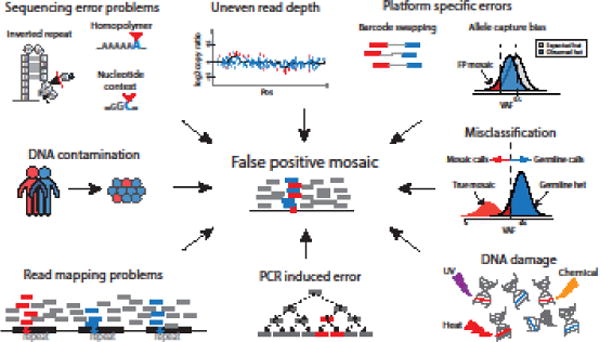
Clockwise from top left: inverted repeats, homopolymers, and some specific nucleotide contexts are common locations of sequencing error. Uneven read coverage can cause false positive calls of mosaic CNVs. Platform-specific errors from targeted sequencing methods may result in underestimated VAF for germline variants; barcode swapping can lead to the spread of false positive signals in multiplexed samples. Germline mutations with low VAF due to read sampling bias can be misclassified as somatic. DNA damage can induce artifactual single-base substitutions during sample handling and library preparation. PCR errors are also common and will propagate in subsequent PCR steps. Misalignment, especially within repetitive regions of the genome, contributes to a large proportion of false positive calls. Cross-individual contamination may lead to false positives.
DNA contamination
DNA contamination—whether by other samples or artificial constructs—can occur at several steps during sample handling and sequencing. DNA contamination by other human subjects is perhaps the most dangerous: it was recently estimated that 1.5% contamination by another human source is a common occurrence and produces roughly 0.2 erroneous somatic mutation calls per megabase in tumor-normal experiments [74], a considerable burden given that somatic mutation rate estimates in various cancers roughly range from 0.1 to 100 SNVs per megabase [1]. In principle, if genotypes for the contaminating individual are known, then putative somatic mutations coinciding with known genotypes in the contaminant should be treated with suspicion; if the contaminant is unknown, common variants from population databases can serve as an approximate substitute. Several algorithms can quantify contamination from sequencing data when the source is unknown [74–76] or known [77–80]. Some somatic mutation callers can be adjusted to compensate for contamination [4, 29, 75, 81], but it is also reasonable to remove candidate mutations at known polymorphic sites (with the associated loss of sensitivity in mind) or to exclude highly contaminated samples altogether.
DNA damage
Low levels of DNA damage frequently occur during routine sample handling and storage. Many sources of DNA damage have been identified: for example, ultraviolet radiation can create pyrimidine dimers [82], high temperature increases the rate of spontaneous cytosine deamination resulting in C>T transitions [83], reactive oxygen species can induce 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxoG) which can mis-pair with A [84], and ionizing radiation can cause double-stranded DNA breaks [85]. These types of infrequent damage often go unnoticed in germline variant analyses, but they become much more prominent when low VAF somatic mutations are of interest. It was recently found that the majority of low VAF G>T/C>A somatic mutations in an exome dataset was likely caused by oxidative damage during library construction [43, 61, 86] and that similar damage is widespread in WGS samples [87]. Single cell sequencing experiments are especially vulnerable to DNA damage prior to amplification because a single base lesion affects a quarter of the original DNA strands. Indeed, pronounced effects have been observed when single cells are lysed by heat treatment [88]. Added care in sample handling and during routine benchwork may help to prevent damage to some extent, but investigators should remain wary, as the full spectrum of damage-inducing processes is unknown.
Read mapping problems
Improperly aligned reads are responsible for a large fraction of false positive variant calls, especially for the low VAF cases. Misalignment or non-unique alignment often occurs near an indel or in repetitive regions of the genome, such as centromeres or telomeres. Although repetitive regions are estimated to account for nearly half of the human genome [89], they pose such a great a challenge for mutation detection that they are often excluded from analysis [2, 43, 90]. Reads can also be misplaced due to limitations of the reference genome, which lacks any representation of genetic variation. Emerging long- and linked-read technologies will be needed to mitigate alignment issues. Ultimately, de novo assembly that does not rely on a reference genome will be needed, however it is not yet feasible for routine analysis [72, 73, 91–93].
Sequencing artifacts
While tolerable for germline variant calling, the per-base error rates intrinsic to sequencing platforms (~0.3% are miscalls according to one estimate [94]) are high relative to the rate of somatic mutation. If miscalls were produced independently, they would essentially be supported by only a single sequencing read, and thus removed. However, artifacts are frequently reproduced by factors that increase local error density, e.g., homopolymer runs and high GC content) [94–98], early amplification errors [99–101], uneven capture efficiency [102], and incorrect sample assignment in multiplexed sequencing runs [103]. Technical replicates can provide a modicum of internal control [104], but true low VAF mutations may also be less reproducible due to sampling bias.
Validation methods for mosaic mutations
Because false positive mosaic mutations can arise from so many sources, confirmation using an orthogonal technology is essential. Available methodologies offer trade-offs in cost, effort, and scalability [105, 106]. A popular method is droplet digital PCR (ddPCR), which can achieve sensitivity as low as 0.001% VAF by performing millions of fluorescently-labeled PCR reactions in nanoliter-sized droplets and measuring the fraction of fluorescent droplets [107]. A disadvantage of ddPCR, however, is that it is less scalable since PCR probes must designed for every candidate mutation and options for target multiplexing are currently limited [108]. Another approach is multiplexed confirmation of many candidates using deep sequencing, either through unique molecular barcodes that aid in artifact removal [109] or by sheer sequencing depth [110–113]. Mutations with VAF as low as 0.1% have been confirmed using these techniques [109, 110]; similarly, mutations with relatively high VAFs can be distinguished from heterozygous germline ones when high sequencing depth allows for a more precise estimate of their VAFs. Ideally, multiple tissues from the same individual should be examined to confirm the somatic nature of a mutation. Single cell sequencing may also provide confirmation for candidates identified in bulk sequencing; however, given sampling noise, a large number of cells may be necessary to capture the cells carrying the mutation of interest for low VAF mutations.
Concluding remarks and future perspectives
Somatic mutations are now being implicated in a growing number of diseases. As our understanding of mutagenic processes in normal cells increases, we will be able to better delineate the extent of somatic mosaicism in healthy individuals and their potential contribution to a wide range of diseases (see Outstanding Questions).
Outstanding Questions.
What is the role of somatic mosaicism in human evolution and human diseases?
What are the best bioinformatic approaches for identifying somatic mutations, especially when matched controls are not available? Could we use haplotype phasing to improve variant identification?
What are the common artifacts that confound detection of mosaic variants and how do we mitigate their effect? Which methods should be used to validate mosaic mutations?
Although methods for detection and validation of somatic mutation have long been studied in cancer research, characterization of mutation in non-tumor cells presents new challenges due to (i) the orders-of-magnitude lower mutation rates and (ii) extremely low frequency of the majority of variants, in the absence of selection. Many of the artifacts we described—sample contamination, damage to DNA in vitro, read misalignment, sequencing instrument errors and platform biases—tend to occur at low allele frequencies and vastly outnumber mosaic mutations. Whereas germline sequencing is typically done at ~30× (thus an average of ~15 reads supporting a heterozygous variant), the same level evidence for a low frequency somatic variant would require an amount of sequencing that is currently impractical unless confined to a small region.
Thus, bioinformatics algorithms that incorporate refined filtering criteria will be key for improved sensitivity and specificity in mutation detection. Recent advances in machine-learning algorithms, for instance, offer the possibility that various features related to the supporting reads and their configurations could be combined more efficiently for higher prediction accuracy. Experimental and computational methods are still being developed for single-cell approaches, but they will be essential for detailed analysis of how mutations arise de novo.
Trends.
Somatic mosaicism resulting from postzygotic mutations has been shown to contribute to many diseases including brain-related disorders, in addition to cancer. Emerging data also suggest that mosaicism is common in healthy individuals.
Mutations occurring late in development have very low allele fractions, and their detection requires specialized algorithms and filters that can remove artifacts that arise in sample handling, DNA sequencing, and analysis.
Emerging technologies, such as single-cell sequencing and linked-read sequencing, allow for improved phasing of variants, thus increasing detection accuracy.
GLOSSARY
- Allele dropout/allelic imbalance
A difference in the sequencing of two alleles caused by differential amplification (allelic imbalance) or the amplification failure (allele dropout) of one allele. A frequent source of error in single cell DNA-sequencing
- Amplification bias
The differential amplification of a region of DNA relative to another, resulting in unequal coverage across the genome
- Bulk sequencing
The sequencing of DNA extracted from a large number of cells from the same individual
- Copy number variant (CNV)
A section of the genome that has a different number of repeats or copies as compared to the reference genome
- Coverage
The number of reads overlapping a region in DNA sequencing, also known as depth. Sequence coverage can also refer to the average number of reads covering loci in the entire genome
- Haplotype
A segment of DNA that is inherited as a block from a single parent
- Indel
The insertion or deletion of a small sequence of DNA (1 - 50 bp) in the genome, affecting fewer bases than a structural variant
- Phasing
The process of statistical estimation of an individual’s haplotypes using the variants in their genome, also called haplotype estimation
- Single nucleotide variant (SNV)
A single nucleotide in an individual’s genome that differs from the reference nucleotide
- Somatic mutation
A change to an individual’s genome that arises during its lifetime as opposed to being inherited, also called a postzygotic mutation
- Somatic mosaicism
The existence in an individual of at least two genetically distinct populations of cells, arising from somatic mutation(s)
- Structural variant (SV)
A rearrangement of the genome affecting a region greater than 50bp. Structural variation consists of many types of variants with varying length scales, including deletions, insertions, translocations, and TE insertions. SVs encompass events that result in CNVs and copy-neutral events
- Transposable Element (TE)
A sequence of DNA that, either by an RNA intermediate (retrotransposons) or a DNA intermediate (DNA transposons), can relocate within a genome. Some active TEs include L1s and Alu elements
- Variant allele fraction (VAF)
The fraction of sequencing reads in a sample corresponding to the non-reference allele. For bulk sequencing data, this is an estimate of the frequency of DNA molecules carrying the variant
- Whole genome amplification
The amplification of a single genome, or a similarly limited amount of DNA, to generate sufficient DNA for sequencing. Necessary for single cell DNA sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349(6255):1483–9. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 2.Ju YS, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543(7647):714–718. doi: 10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martincorena I, et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell. 2017;171(5):1029–1041 e21. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lodato MA, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350(6256):94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behjati S, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513(7518):422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martincorena I, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sims D, et al. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–32. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 9.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blokzijl F, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538(7624):260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abyzov A, et al. One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin. Genome Res. 2017;27(4):512–523. doi: 10.1101/gr.215517.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bae T, et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science. 2018;359(6375):550–555. doi: 10.1126/science.aan8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hazen JL, et al. The Complete Genome Sequences, Unique Mutational Spectra, and Developmental Potency of Adult Neurons Revealed by Cloning. Neuron. 2016;89(6):1223–1236. doi: 10.1016/j.neuron.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizutani E, et al. Generation of cloned mice from adult neurons by direct nuclear transfer. Biol Reprod. 2015;92(3):81. doi: 10.1095/biolreprod.114.123455. [DOI] [PubMed] [Google Scholar]
- 15.Gawad C, et al. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175–88. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 16.Hou Y, et al. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. Gigascience. 2015;4:37. doi: 10.1186/s13742-015-0068-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang L, et al. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu Rev Genomics Hum Genet. 2015;16:79–102. doi: 10.1146/annurev-genom-090413-025352. [DOI] [PubMed] [Google Scholar]
- 18.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–60. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang CZ, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522(7555):179–84. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voet T, et al. Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res. 2013;41(12):6119–38. doi: 10.1093/nar/gkt345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McConnell MJ, et al. Mosaic copy number variation in human neurons. Science. 2013;342(6158):632–7. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evrony GD, et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron. 2015;85(1):49–59. doi: 10.1016/j.neuron.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lodato MA, et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2017 doi: 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vitak SA, et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat Methods. 2017;14(3):302–308. doi: 10.1038/nmeth.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan F, et al. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat Biotechnol. 2017;35(7):640–646. doi: 10.1038/nbt.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bohrson CL, et al. Linked-read analysis identifies mutations in single-cell DNA sequencing data. bioRxiv. 2017 doi: 10.1038/s41588-019-0366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–5. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 30.Ewing AD, et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat Methods. 2015;12(7):623–30. doi: 10.1038/nmeth.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alioto TS, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6:10001. doi: 10.1038/ncomms10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung H, et al. Systematic investigation of cancer-associated somatic point mutations in SNP databases. Nat Biotechnol. 2013;31(9):787–9. doi: 10.1038/nbt.2681. [DOI] [PubMed] [Google Scholar]
- 36.Hiltemann S, et al. Discriminating somatic and germline mutations in tumor DNA samples without matching normals. Genome Res. 2015;25(9):1382–90. doi: 10.1101/gr.183053.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26(8):345–52. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahbari R, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48(2):126–133. doi: 10.1038/ng.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson IR, et al. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013;14(10):703–18. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang AY, et al. MosaicHunter: accurate detection of postzygotic single-nucleotide mosaicism through next-generation sequencing of unpaired, trio, and paired samples. Nucleic Acids Res. 2017;45(10):e76. doi: 10.1093/nar/gkx024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim ET, et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci. 2017;20(9):1217–1224. doi: 10.1038/nn.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freed D, Pevsner J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS Genet. 2016;12(9):e1006245. doi: 10.1371/journal.pgen.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dou Y, et al. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum Mutat. 2017;38(8):1002–1013. doi: 10.1002/humu.23255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krupp DR, et al. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am J Hum Genet. 2017;101(3):369–390. doi: 10.1016/j.ajhg.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xi R, et al. A survey of copy-number variation detection tools based on high-throughput sequencing data. Curr Protoc Hum Genet. 2012 doi: 10.1002/0471142905.hg0719s75. Chapter 7, Unit7 19. [DOI] [PubMed] [Google Scholar]
- 46.King DA, et al. Detection of structural mosaicism from targeted and whole-genome sequencing data. Genome research. 2017;27(10):1704–1714. doi: 10.1101/gr.212373.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su SY, et al. Inferring combined CNV/SNP haplotypes from genotype data. Bioinformatics. 2010;26(11):1437–45. doi: 10.1093/bioinformatics/btq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palta P, et al. Haplotype phasing and inheritance of copy number variants in nuclear families. PLoS One. 2015;10(4):e0122713. doi: 10.1371/journal.pone.0122713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guan P, Sung WK. Structural variation detection using next-generation sequencing data: A comparative technical review. Methods. 2016;102:36–49. doi: 10.1016/j.ymeth.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 50.Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153(4):919–29. doi: 10.1016/j.cell.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye K, et al. Systematic discovery of complex insertions and deletions in human cancers. Nature medicine. 2016;22(1):97–104. doi: 10.1038/nm.4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8(8):652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai Z, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44(11):e108. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee E, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tubio JM, et al. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345(6196):1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helman E, et al. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014;24(7):1053–63. doi: 10.1101/gr.163659.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evrony GD, et al. Resolving rates of mutation in the brain using single-neuron genomics. Elife. 2016;5 doi: 10.7554/eLife.12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evrony GD, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151(3):483–96. doi: 10.1016/j.cell.2012.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng GX, et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol. 2016;34(3):303–11. doi: 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109(36):14508–13. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoang ML, et al. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc Natl Acad Sci U S A. 2016;113(35):9846–51. doi: 10.1073/pnas.1607794113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 2011;12(10):703–14. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delaneau O, et al. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9(2):179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 65.Edge P, et al. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 2017;27(5):801–812. doi: 10.1101/gr.213462.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delaneau O, et al. Haplotype estimation using sequencing reads. Am J Hum Genet. 2013;93(4):687–96. doi: 10.1016/j.ajhg.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramu A, et al. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods. 2013;10(10):985–7. doi: 10.1038/nmeth.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sengupta S, et al. Ultra-fast local-haplotype variant calling using paired-end DNA-sequencing data reveals somatic mosaicism in tumor and normal blood samples. Nucleic Acids Res. 2016;44(3):e25. doi: 10.1093/nar/gkv953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sudmant PH, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chaisson MJ, et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. bioRxiv. 2017;193144 doi: 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuleshov V, et al. Whole-genome haplotyping using long reads and statistical methods. Nat Biotechnol. 2014;32(3):261–266. doi: 10.1038/nbt.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weisenfeld NI, et al. Direct determination of diploid genome sequences. Genome Res. 2017;27(5):757–767. doi: 10.1101/gr.214874.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seo JS, et al. De novo assembly and phasing of a Korean human genome. Nature. 2016;538(7624):243–247. doi: 10.1038/nature20098. [DOI] [PubMed] [Google Scholar]
- 74.Cibulskis K, et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27(18):2601–2. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Flickinger M, et al. Correcting for Sample Contamination in Genotype Calling of DNA Sequence Data. Am J Hum Genet. 2015;97(2):284–90. doi: 10.1016/j.ajhg.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jun G, et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet. 2012;91(5):839–48. doi: 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim S, et al. Virmid: accurate detection of somatic mutations with sample impurity inference. Genome Biol. 2013;14(8):R90. doi: 10.1186/gb-2013-14-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su X, et al. PurityEst: estimating purity of human tumor samples using next-generation sequencing data. Bioinformatics. 2012;28(17):2265–6. doi: 10.1093/bioinformatics/bts365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bergmann EA, et al. Conpair: concordance and contamination estimator for matched tumor-normal pairs. Bioinformatics. 2016;32(20):3196–3198. doi: 10.1093/bioinformatics/btw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee S, et al. NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res. 2017;45(11):e103. doi: 10.1093/nar/gkx193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sinha RP, Hader DP. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1(4):225–36. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 83.Fryxell KJ, Zuckerkandl E. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol Biol Evol. 2000;17(9):1371–83. doi: 10.1093/oxfordjournals.molbev.a026420. [DOI] [PubMed] [Google Scholar]
- 84.Evans MD, et al. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567(1):1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 85.Helleday T, et al. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15(9):585–98. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costello M, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41(6):e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen L, et al. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 2017;355(6326):752–756. doi: 10.1126/science.aai8690. [DOI] [PubMed] [Google Scholar]
- 88.Dong X, et al. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods. 2017;14(5):491–493. doi: 10.1038/nmeth.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13(1):36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang AY, et al. Postzygotic single-nucleotide mosaicisms in whole-genome sequences of clinically unremarkable individuals. Cell Res. 2014;24(11):1311–27. doi: 10.1038/cr.2014.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012;22(3):549–56. doi: 10.1101/gr.126953.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Iqbal Z, et al. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet. 2012;44(2):226–32. doi: 10.1038/ng.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Snyder MW, et al. Haplotype-resolved genome sequencing: experimental methods and applications. Nat Rev Genet. 2015;16(6):344–58. doi: 10.1038/nrg3903. [DOI] [PubMed] [Google Scholar]
- 94.Ross MG, et al. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14(5):R51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dohm JC, et al. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36(16):e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meacham F, et al. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12:451. doi: 10.1186/1471-2105-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Allhoff M, et al. Discovering motifs that induce sequencing errors. BMC Bioinformatics. 2013;14(Suppl 5):S1. doi: 10.1186/1471-2105-14-S5-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakamura K, et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39(13):e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Keohavong P, Thilly WG. Fidelity of DNA polymerases in DNA amplification. Proc Natl Acad Sci U S A. 1989;86(23):9253–7. doi: 10.1073/pnas.86.23.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Walsh PS, et al. Preferential PCR amplification of alleles: mechanisms and solutions. PCR Methods Appl. 1992;1(4):241–50. doi: 10.1101/gr.1.4.241. [DOI] [PubMed] [Google Scholar]
- 101.Brodin J, et al. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS One. 2013;8(7):e70388. doi: 10.1371/journal.pone.0070388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lelieveld SH, et al. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum Mutat. 2015;36(8):815–22. doi: 10.1002/humu.22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sinha R, et al. Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. bioRxiv. 2017;125724 [Google Scholar]
- 104.Robasky K, et al. The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet. 2014;15(1):56–62. doi: 10.1038/nrg3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McConnell MJ, et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science. 2017;356(6336) doi: 10.1126/science.aal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Campbell IM, et al. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015;31(7):382–92. doi: 10.1016/j.tig.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hindson CM, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10(10):1003–5. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McDermott GP, et al. Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Anal Chem. 2013;85(23):11619–27. doi: 10.1021/ac403061n. [DOI] [PubMed] [Google Scholar]
- 109.Hiatt JB, et al. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23(5):843–54. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu X, et al. Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of “de novo” SCN1A Mutations in Children with Dravet Syndrome. Hum Mutat. 2015;36(9):861–72. doi: 10.1002/humu.22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Froyen G, et al. Validation and application of a custom-designed targeted next-generation sequencing panel for the diagnostic mutational profiling of solid tumors. PloS one. 2016;11(4):e0154038. doi: 10.1371/journal.pone.0154038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nikiforova MN, et al. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. The Journal of Clinical Endocrinology & Metabolism. 2013;98(11):E1852–E1860. doi: 10.1210/jc.2013-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Izawa K, et al. Detection of base substitution-type somatic mosaicism of the NLRP3 gene with> 99.9% statistical confidence by massively parallel sequencing. DNA research. 2012;19(2):143–152. doi: 10.1093/dnares/dsr047. [DOI] [PMC free article] [PubMed] [Google Scholar]