

Abstract
Here, we address the function of protein phosphatase 6 (PP6) loss on K‐ras‐initiated tumorigenesis in keratinocytes. To do so, we developed tamoxifen‐inducible double mutant (K‐ras G12D‐expressing and Ppp6c‐deficient) mice in which K‐ras G12D expression is driven by the cytokeratin 14 (K14) promoter. Doubly‐mutant mice showed early onset tumor formation in lips, nipples, external genitalia, anus and palms, and had to be killed by 3 weeks after induction by tamoxifen, while comparably‐treated K‐ras G12D‐expressing mice did not. H&E‐staining of lip tumors before euthanasia revealed that all were papillomas, some containing focal squamous cell carcinomas. Immunohistochemical analysis of lips of doubly‐mutant vs K‐ras G12D mice revealed that cell proliferation and cell size increased approximately 2‐fold relative to K‐ras G12D‐expressing mutants, and epidermal thickness of lip tissue greatly increased relative to that seen in K‐ras G12D‐only mice. Moreover, AKT phosphorylation increased in K‐ras G12D‐expressing/Ppp6c‐deficient cells, as did phosphorylation of the downstream effectors 4EBP1, S6 and GSK3, suggesting that protein synthesis and survival signals are enhanced in lip tissues of doubly‐mutant mice. Finally, increased numbers of K14‐positive cells were present in the suprabasal layer of doubly‐mutant mice, indicating abnormal keratinocyte differentiation, and γH2AX‐positive cells accumulated, indicating perturbed DNA repair. Taken together, Ppp6c deficiency enhances K‐ras G12D‐dependent tumor promotion.
Keywords: AKT pathway, K‐ras, mouse keratinocyte, protein phosphatase 6, tumor initiation and promotion
1. INTRODUCTION
Protein phosphorylation by protein kinases plays essential roles in signaling pathways that regulate normal cell function, and kinase dysregulation leads to various pathologies, including cancer. Cancer‐related signaling by kinases has been well analyzed, but information relevant to the role of serine/threonine phosphoprotein phosphatases (PPP) in tumor formation remains limited.1, 2 Protein phosphatase 6 (PP6) is a PPP consisting of a catalytic (Ppp6c) and regulatory subunits.3 In mammalian cell lines, diverse phenotypes seen following siRNA‐based Ppp6c depletion suggest that PP6 regulates mitosis by dephosphorylating aurora kinase A,4 activates DNA‐PK to sensitize cells to ionizing radiation,5, 6 and is required for homology‐directed double strand repair.7 PP6 also regulates NFκB signaling by suppressing IkBε degradation in response to TNFα8 and inactivates TAK1.9
Recent DNA sequencing screens relevant to human cancer highlight the importance of PP6 in tumorigenesis. Somatic mutation of Ppp6c is reported in approximately 10% of malignant melanomas,10, 11 and approximately 20% of skin basal cell carcinomas.12 In melanoma samples, Ppp6c mutations are often accompanied by loss of heterozygosity (LOH) and occur in genomic hot‐spots associated with protein function or stability,13 suggesting that Ppp6c loss is associated with tumor formation. Another important finding is that melanoma tissues harboring Ppp6c mutations constitute a different subset than those containing mutations in pTEN, a major melanoma suppressor, although both subtypes contain B‐raf or N‐ras mutations.10
We previously assessed Ppp6c function in vivo using a skin carcinogenesis mouse model engineered by Hayashi et al14. Given that Ppp6c null mice exhibit early embryonic lethality,15 that group engineered mice homozygous for a loxP‐targeted allele on chromosome 4, which, when crossed with K14Cre tam mice, undergo Cre‐dependent Ppp6c deletion in cytokeratin 14‐positive keratinocytes.14 In normal skin, a single treatment with 7,12‐dimethylbenz(a)anthracene (DMBA) does not produce papillomas in the absence of tumor promoters16; however, we subsequently reported that a single DMBA application was sufficient to generate papillomas in Ppp6c‐deficient skin, suggesting that Ppp6c loss predisposes cells to DMBA‐induced papilloma formation.14 Using the same mice, Kato et al17 reporte that Ppp6c loss in mouse keratinocytes increases susceptibility to UVB‐induced carcinogenesis. These latter 2 studies were the first to report that Ppp6c loss‐of‐function acts as a tumor promoter in mice.
K‐ras activating mutations occur in one‐third of all human cancers.18 Although oncogenic K‐ras functions in tumor initiation and progression, the use of mouse models to define how K‐ras targets and activates downstream effectors has been limited. Here, we examined the effect of Ppp6c loss on K‐ras‐induced proliferation in the context of keratinocyte tumorigenesis. We report that Ppp6c deficiency enhances K‐ras G12D‐dependent tumor promotion.
2. MATERIALS AND METHODS
2.1. Generation of mice with inducible Kras G12D expression and Ppp6c deletion
Mouse strains, K14‐CreER tam (the Jackson Laboratory) and Ppp6c flox/flox mice have been described.14 LSL‐K‐ras G12D (a strain of C57BL/6) mice were obtained from the Jackson Laboratory. K14‐CreER tam mice were crossed with LSL‐K‐ras G12D mice to generate K14‐CreER tam /LSL‐K‐ras G12D mice, which were then bred with Ppp6c flox/flox mice to generate K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/+ mice. These mice were further crossed with Ppp6c flox/+ to generate mice of the following 3 genotypes: K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox, K14‐CreER tam /LSL‐K‐ras G12D and K14‐CreER tam mice. Littermate controls were used in experiments. Ppp6c +/− mice were generated as described.15 All animal experiments were performed with the approval of the Miyagi Cancer Center Research Institute Animal Care and Use Committee.
2.2. 4‐Hydroxytamoxifen treatment
4‐Hydroxytamoxifen (4HT) was purchased from Toronto Research Chemicals (North York, Canada). Eight‐week‐old female mice under general isoflurane anesthesia were administered 4HT by ip injection of 0.5 mg 4HT/mouse/d for 5 consecutive days. Control mice were ip‐injected with solvent (corn oil) on the same schedule.
2.3. PCR genotype analysis
To confirm excision of the loxP‐flanked transcription stop cassette from the LSL‐K‐ras G12D allele in DNA from mice treated with 4HT, we used the following primers: (i) 5′‐GTCTTTCCCCAGCACAGTGC‐3′, (ii) 5′‐CTCTTGCCTACGCCAGCT‐3′ and (iii) 5′‐AGCTAGCCACCATGGCTTGAGTAAGTCTGCA‐3′ (Figure 1A). Cre‐mediated deletion of the Ppp6c floxed allele was confirmed as described,14 using primers (iv) 5′‐TATCACGAGGCCCTTTCG‐3′ and (v) 5′‐TAGTGAACCTCTTCGAGG‐3′ (Figure 1A).
Figure 1.
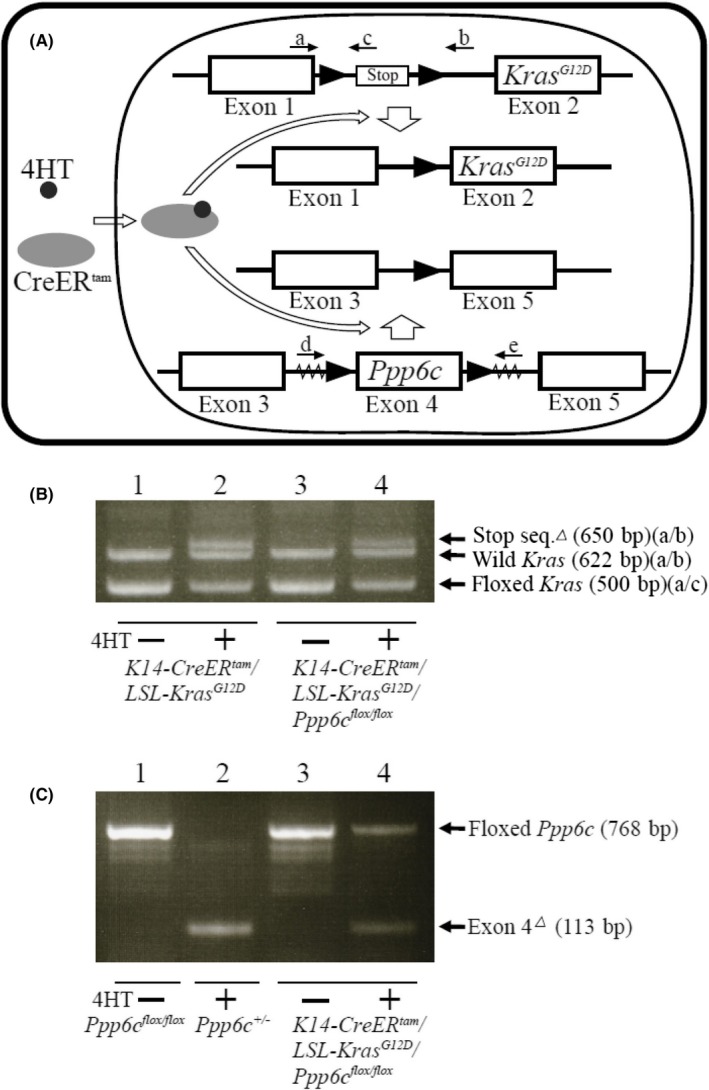
CreER
tam‐mediated KrasG12D expression and Ppp6c disruption. A, Schematic representation of a K‐ras floxed allele and deletion of the transcription stop sequence by activated CreER
tam. Positions of primers a, b and c are indicated. Schematic representation of the Ppp6c‐floxed allele and deletion of exon 4 by activated CreER
tam. Positions of primers d and e are indicated.  indicates loxP sequence
indicates loxP sequence  (indicates exon sequence. Serrated region indicates vector sequence. B, PCR analysis of genomic DNA to confirm removal of the transcription stop sequence using primers a, b and c. Lanes 1 and 2: genomic DNA obtained from lips of 4HT‐untreated (lane1) and 4HT‐treated (lane 2) K14‐CreER
tam
/LSL‐K‐ras
G12D mice. Lanes 3 and 4: genomic DNA obtained from lips of 4HT‐untreated (lane 3) and 4HT‐treated (lane 4) K14‐CreER
tam
/LSL‐K‐ras
G12D
/Ppp6c
flox/flox mice. Fragments of 650, 622 and 500 bp correspond to the stop sequence‐deleted allele (amplified with primers a and b), wild‐type allele (amplified with primers a and b) and floxed alleles (amplified with primers a and c), respectively. C, PCR analysis of genomic DNA to detect the exon 4‐deleted Ppp6c allele using primers d and e. Lane 1: genomic DNA from lips of Ppp6c
flox/flox mice. Lane 2: genomic DNA from lips of Ppp6c
+/− mice. Lanes 3 and 4: genomic DNA from lips of 4HT‐untreated (lane 3) and 4HT‐treated (lane 4) K14‐CreER
tam
/LSL‐K‐ras
G12D
/Ppp6c
flox/flox mice. Fragments of 768 and 113 bp correspond to floxed and exon 4‐deleted alleles, respectively
(indicates exon sequence. Serrated region indicates vector sequence. B, PCR analysis of genomic DNA to confirm removal of the transcription stop sequence using primers a, b and c. Lanes 1 and 2: genomic DNA obtained from lips of 4HT‐untreated (lane1) and 4HT‐treated (lane 2) K14‐CreER
tam
/LSL‐K‐ras
G12D mice. Lanes 3 and 4: genomic DNA obtained from lips of 4HT‐untreated (lane 3) and 4HT‐treated (lane 4) K14‐CreER
tam
/LSL‐K‐ras
G12D
/Ppp6c
flox/flox mice. Fragments of 650, 622 and 500 bp correspond to the stop sequence‐deleted allele (amplified with primers a and b), wild‐type allele (amplified with primers a and b) and floxed alleles (amplified with primers a and c), respectively. C, PCR analysis of genomic DNA to detect the exon 4‐deleted Ppp6c allele using primers d and e. Lane 1: genomic DNA from lips of Ppp6c
flox/flox mice. Lane 2: genomic DNA from lips of Ppp6c
+/− mice. Lanes 3 and 4: genomic DNA from lips of 4HT‐untreated (lane 3) and 4HT‐treated (lane 4) K14‐CreER
tam
/LSL‐K‐ras
G12D
/Ppp6c
flox/flox mice. Fragments of 768 and 113 bp correspond to floxed and exon 4‐deleted alleles, respectively
2.4. Histology and immunohistochemistry
After perfusion with PBS to remove blood, lip tissue from tumor and control mice was dissected, fixed overnight in buffered 3.7% formalin at room temperature, transferred to sequential 70%, 80%, 90% and 100% methanol, and embedded in paraffin. Sections were stained with H&E and examined microscopically by 2 pathologists. Images were acquired using an Olympus BX53 microscope (Tokyo, Japan). Unstained serial sections were used for immunohistochemistry with the following antibodies: goat antibody against mouse cytokeratin 14 (sc‐17104, Santa Cruz Biotechnology, Texas, USA); rabbit antibody against mouse Ki67 (790‐4286, Roche, Basel, Switzerland); rabbit antibody against mouse γH2AX (#9718, Cell Signaling Technology, Danvers, USA); rabbit antibody against mouse p‐Akt (ab38449, Abcam, Cambridge, UK); rabbit antibody against mouse p‐Erk1/2 (#4370, Cell Signaling Technology); rabbit antibody against mouse p‐Erk1/2 (#9101, Cell Signaling Technology); rabbit antibody against mouse p‐4EBP1 (#2855, Cell Signaling Technology); rabbit antibody against mouse p‐S6 protein (#4858, Cell Signaling Technology); and rabbit antibody against mouse p‐GSK3 (#9331, Cell Signaling Technology). Immunohistochemistry was performed using the Ventana Discovery XT system (Roche Tissue Diagnostics K. K. Basel, Switzerland).
2.5. Statistical analysis
Kaplan‐Meier survival curves and corresponding statistical analysis, as well as log‐rank tests, were done using Prism version 7 (GraphPad Software, San Diego, California, USA). Other statistical significance was determined using Student's t test. P < .05 served as the cut‐off for significance.
3. RESULTS
3.1. Development of keratinocyte‐specific K‐ras G12D‐expressing and Ppp6c‐deficient mice
To assess the effects of PP6 loss on activated K‐ras‐induced tumorigenesis in keratinocytes, we established K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice (exhibiting epidermal‐specific tamoxifen‐inducible K‐ras G12D expression plus Ppp6c deficiency) and K14‐CreER tam /LSL‐K‐ras G12D mice (which showed only epidermal‐specific tamoxifen‐inducible K‐ras G12D expression) (Figure 1A). We injected 8‐week‐old double mutant K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox or single‐mutant K14‐CreER tam /LSL‐K‐ras G12D female mice with 4HT daily for 5 days and then killed them 12 days after the first injection. We then used PCR to examine genomic DNA from lip skin of both groups to confirm Cre‐induced recombination of the floxed K‐ras allele (Figure 1B) and floxed Ppp6c alleles (Figure 1C).
For the floxed K‐ras allele, PCR analysis of lip skin genomic DNA of untreated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice and K14‐CreER tam /LSL‐K‐ras G12D mice using primers a and c revealed a 500‐bp band derived from the floxed allele (Figure 1A,B). Comparable DNA from both lines of 4HT‐treated mice showed an additional 650‐bp fragment when amplified with primers a and b, indicating excision of the stop signal and K‐ras G12D expression (Figure 1A,B). A 622‐bp PCR fragment representing the wild‐type K‐ras allele was also seen in every sample amplified by primers a and b.
For the floxed Ppp6c allele, PCR analysis of lip skin genomic DNA from untreated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice using primers d and e showed a 768‐bp band (Figure 1A,C) derived from the floxed allele. However, genomic DNA from lip skin of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice showed an additional 113‐bp fragment, indicative of exon 4 deletion (Figure 1A,C).
3.2. Development of tumors in K14‐CreER tam /LSL‐K‐ras G12 /Ppp6c flox/flox mice
We next examined the effect of Ppp6c deletion on Kras G12D expression in a mouse model. To do so, we used 8‐week‐old K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox, K14‐CreER tam /LSL‐K‐ras G12D and K14CreER tam female mice injected for 5 days with 4HT. 4HT‐treated K14‐CreER tam mice survived for more than a year, indicating that 4HT itself does not decrease survival (data not shown). Given that preliminary analysis showed that 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice die approximately 20 days after the first 4HT injection, we killed mice when they had lost 20% of their body weight. All 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice were killed within 20 days (Figure 2A). By contrast, 80% of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D mice were alive at 100 days (Figure 2A), indicating that Ppp6c loss accelerates death of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D mice (log‐rank test, P < .001). 4HT‐treated K14‐CreER tam /Ppp6c flox/flox mice survived for longer than a year without overt defects, as described.14
Figure 2.
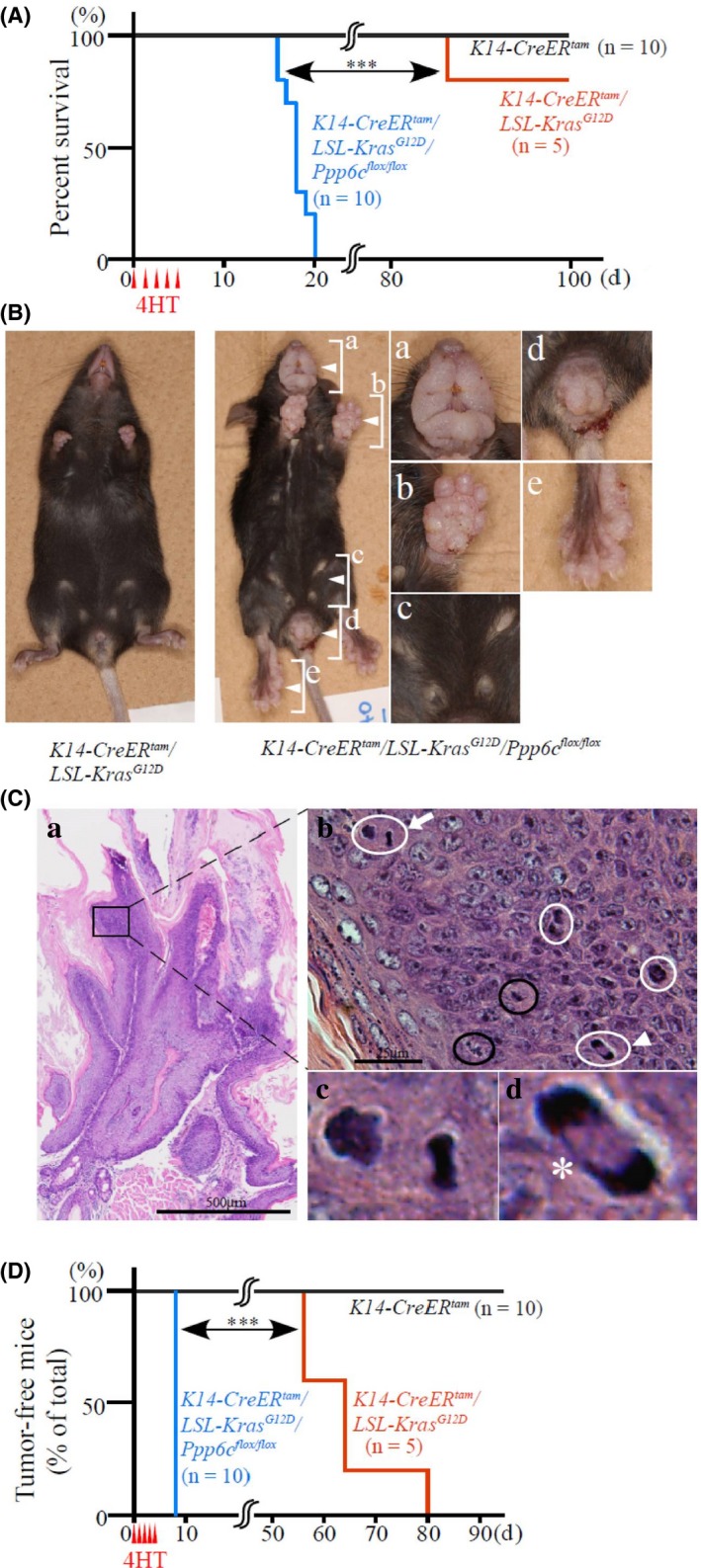
K‐rasG12D plus Ppp6c deficiency driven by cytokeratin 14 promoter‐induced tumor formation and early death. K14‐CreER tam mice (n = 10), K14‐CreER tam /LSL‐K‐ras G12D mice (n = 5) and K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice (n = 10) were administered 4HT IPip for 5 d and then examined macroscopically daily until day 20, and then every 4 d. A, Percentage of surviving mice. ***P < .001. B, Macroscopic appearance of representative K14‐CreER tam /LSL‐K‐ras G12D (left) and K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox (right) female mice at day 18 after the first 4HT injection. Panels at right show higher magnification views of lip (Ba), palm of forelimb (Bb), nipples (Bc), external genitalia and anus (Bd), and palm of hindlimb (Be). C, Histological appearance of lip tissue shown in Ba. Note that panel Ca shows hyperkeratotic neoplastic lesion with a wide stromal stalk, while the higher magnification view shown in Cb indicates a hypercellular area in that region (b). In the latter, neoplastic squamous cells exhibit severe nuclear atypia with 6 mitoses, including 2 normal (circled in black) and 4 abnormal cells (circled in white). Abnormal mitoses indicated by an arrow and arrowhead are shown in respective higher magnification images (c) and (d). A chromosomal bridge (*) is also indicated (Cd). D, Percentage of mice free of macroscopic lip tumors. H&E staining. ***P < .001
Figure 2B (lower) shows a representative 4HT‐injected K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mouse 18 days after the first injection; that animal exhibits tumors on lips, nipples, external genitalia, anus and palms. By contrast, by 20 days, a representative 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D mouse does not exhibit tumors (Figure 2B, upper). Ten independent 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice developed similar tumors. When we autopsied mice to determine cause of death, we found that the stomachs of most mice contained less food content than did stomachs of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D mice. No tumors were found in organs such as the oral cavity and esophagus, which contains cytokeratin 14‐expressing basal epithelium. Other intra‐abdominal and thoracic organs appeared free of tumors or metastasis based on macroscopic examination. These observations suggest that large lip tumors interfered with feeding and mice became asthenic. Microscopic analysis of lip tumor tissue from the mouse shown in Figure 2B (lower) indicated hyperkeratotic papillomatous proliferative lesions (Figure 2Ca). It is noteworthy that a region of squamous cell carcinoma in situ (CIS) within this lesion (Figure 2Cb) contains 6 mitoses, including 4 abnormal mitoses (Figure 2Cb‐d). All 7 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice killed between days 16 to 20 (Figure 2A) developed hyperkeratotic papillomaous proliferative lesions in lip tissue, and 100% of these lesions contained CIS regions.
The timing of onset of lip tumors, as identified by visual inspection, is shown in Figure 2D. Tumors starting appeared at 56 days in 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D mice but appeared within 8 days in 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice, suggesting that tumor promotion by K‐ras G12D is due to Ppp6c loss (log‐rank test, P < .001). In contrast, tumors did not emerge on lips of 4HT‐treated K14‐CreER tam /Ppp6c flox/flox mice for a year (data not shown), in agreement with a previous finding that dorsal skin of K14CreER tam /Ppp6c flox/flox mice treated with 4HT painting is normal.14 These findings confirm that the tumor‐promoting effect of Ppp6c deficiency appears in the presence of K‐ras G12D.
3.3. Ppp6c deficiency enhances K‐ras G12D ‐induced cell proliferation
To assess cell proliferation in Ppp6c‐deficient K‐ras G12D‐expressing mice, we injected mice with 4HT, killed them 12 days later and then compared lip tissues histochemically with those from K14‐CreER tam /LSL‐K‐ras G12D and K14‐CreER tam mice. Representative tissue samples (Figure 3A) show comparable H&E staining of lips from 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D (middle) and K14‐CreER tam (upper) mice, indicating minimal effects of cytokeratin 14 driven K‐ras G12D expression. However, 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice showed epidermal hyperplasia (acanthosis) and hyperkeratosis (Figure 3Ac,f). We then examined cell size among the 3 genotypes, and for comparison purposes, calculated the area of basal layer cells (Figure 3Ad‐f). The ratios of cell size in K14‐CreER tam /LSL‐K‐ras G12D and K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice to cell size seen in K14‐CreER tam mice were 1.0 and 2.3, showing that the double mutation increases keratinocyte size in the basal layer (Figure 3B).
Figure 3.
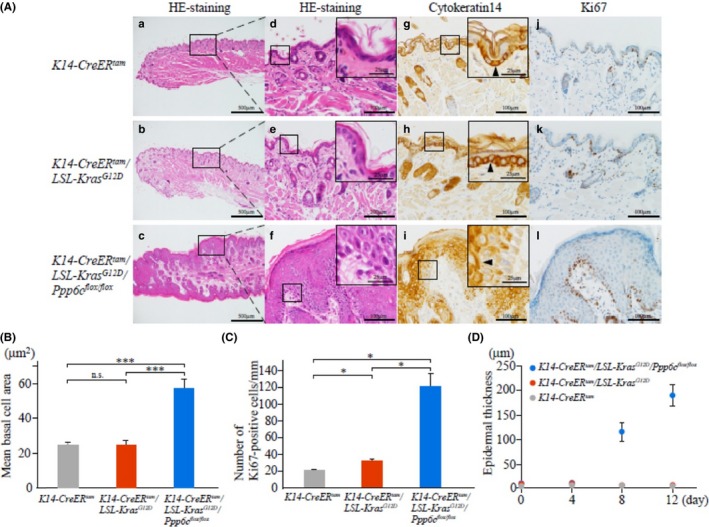
Ppp6c deficiency plus KrasG12D overexpression driven by cytokeratin 14 promoter increases keratinocyte cell proliferation and size as well as epidermal thickness. A, Appearance of lips of 4HT‐treated mice. H&E staining at low (a‐c) and high (d‐f) magnification, and immunohistochemistry using anti‐cytokeratin 14 (g‐i) and anti‐Ki67 (j‐l) antibodies. 4HT‐treated mice as described in Figure 2 were killed 12 d after the first 4HT injection. Samples from K14‐CreER tam (upper), K14‐CreER tam /LSL‐K‐ras G12D (middle) and K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox (lower) mice are shown. Scale bar: 500 μm (low magnification) or 100 μm (high magnification). Insets in (d‐i) represent higher magnification images of corresponding boxed areas depicting basal membrane. Arrowheads of anti‐cytokeratin 14 samples show cytokeratin 14‐positive cells. Scale bar: 25 μm. B, Basal cell size in mouse lip skin (left). The area of 1 cell in H&E‐stained samples shown in (A) was measured using Image J 1.57t (NIH, USA). Data are mean values of 100 cells ± SE. ***P < .001. n.s., non‐significant. C, The number of Ki67‐positive cells in mouse lip skin, counted using samples shown in (A). In each sample, Ki67‐positive cells of approximately 1‐mm width in epidermis were counted. Data are means derived from 4 areas in independent samples ± SE. *P < .05. D, Epidermal thickness in mouse lip tissue at indicated times after first 4HT injection. Data are means ± SE derived from 10 areas of 1 representative mouse
Because K‐ras G12D induction in these studies is driven by the cytokeratin 14 promoter, we assessed cytokeratin 14 localization (Figure 3Ag‐i). As reported, cytokeratin 14 was detected in basal cells of the epidermis in control K14‐CreER tam (Figure 3Ag) and K14‐CreER tam /LSL‐K‐ras G12D cells (Figure 3Ah)19; however, in double mutants, cytokeratin 14 was not detected in the basal layer but, rather, was distributed in the suprabasal layer, where the number of cytokeratin 14‐positive cells increased (Figure 3Ai), suggestive of perturbed keratinocyte differentiation. When we counted cells that were positive for the proliferation marker Ki67 (Figure 3Aj‐l) we found that the ratio of this number in 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D and K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice to that of control K14‐CreER tam mice was 1.6 and 6.1, respectively (Figure 3C), indicating that cell proliferation induced by K‐ras G12D is accelerated by Ppp6c deficiency.
To examine lip epidermis growth, we measured tissue thickness at 4, 8 and 12 days after 4HT injection. Lip thickness of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D was almost the same as that of control K14‐CreER tam mice within 12 days (Figure 3D); however, the epidermal thickness of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice increased dramatically 12 days after 4HT injection (Figure 3D). Specifically, the ratio of lip thickness of K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox to that of K14‐CreER tam /LSL‐K‐ras G12D mice was 2.3, 24 and 42, at days 4, 8 and 12, respectively (Figure 3D).
3.4. K‐ras G12D‐positive/Ppp6c‐deficient lip skin tissues show enhanced AKT phosphorylation and γH2AX levels
To define molecular mechanisms underlying early onset papilloma formation in lips of 4HT‐treated K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice, we evaluated signaling downstream of K‐ras. Because ERK and AKT are the major K‐ras effectors governing cell proliferation and growth,18 we examined their phosphorylation levels. As shown in Figure 4, 12 days after 4HT treatment we observed enhanced AKT phosphorylation at residue T308 in lip tissues from K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox compared to K14‐CreER tam /LSL‐K‐ras G12D and control K14‐CreER tam mice. However, ERK phosphorylation at T202/Y204 did not differ significantly among 4HT‐treated mice of all 3 genotypes, suggesting that Ppp6c deficiency has a more predominant effect on AKT phosphorylation. Because AKT activates the cell cycle,20 enhanced cell cycle progression seen in doubly‐mutant lip skin tissues (Figure 3C) could be attributable to activated AKT.
Figure 4.
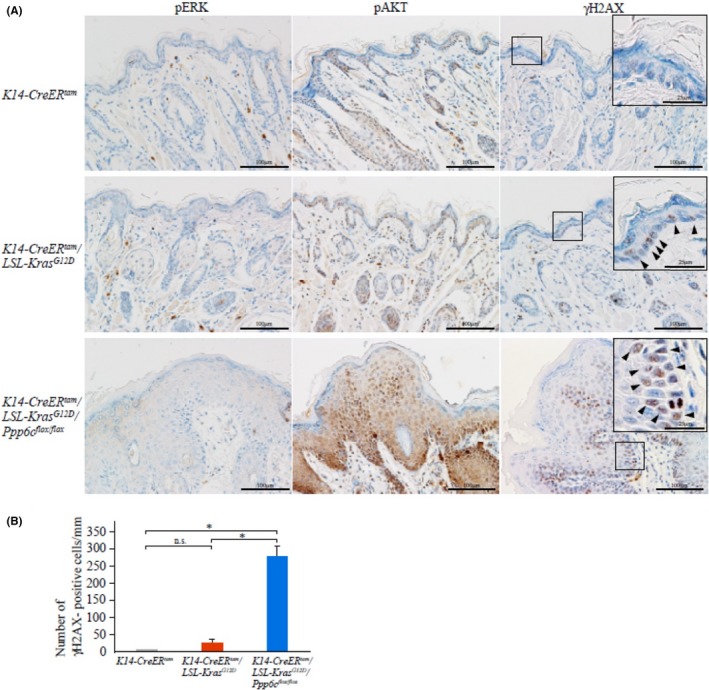
Accumulation of phospho‐AKT‐positive and γH2AX‐positive cells in doubly mutant (KrasG12D plus Ppp6c deficiency) keratinocytes. A, Immunohistochemistry of lip samples shown in Figure 3A using phospho‐ERK (T202/Y204), phosphoƒ‐AKT (T308) and γH2AX (S139) antibodies. Scale bar: 100 μm. Arrowheads in higher magnification insets of anti‐γH2AX samples show γH2AX‐positive cells. Scale bar: 25 μm. B, The number of γH2AX‐positive cells in lip epidermis. γH2AX‐positive cells were counted using samples shown in (A). Staining was evaluated in γH2AX‐positive epidermal cells of 1‐mm width. Data are means ± SE derived from 4 areas in independent samples. *P < .05. n.s., non‐significant
We next examined the effect of Ppp6c deficiency on DNA repair, as it is reported that activated K‐ras induces DNA breaks by inducing reactive oxygen species,21 and PP6 plays a key role in homology‐directed repair.22 To monitor double‐strand breaks (DSB) in epidermis, we evaluated the expression of γH2AX, a DSB marker. As shown in Figure 4, by 12 days after 4HT treatment, lip tissues of K14‐CreER tam /LSL‐K‐ras G12D /Ppp6c flox/flox mice showed numerous γH2AX‐positive cells in a diffuse pattern in the epidermis. Specifically, the ratio of the number of γH2AX‐positive cells in the K‐ras G12D positive and doubly‐mutant (K‐ras G12D positive/Ppp6c‐deficient) lip to wild type was 6.9 and 76, respectively (Figure 4B), strongly suggesting that DNA repair is greatly disturbed in doubly‐mutant keratinocytes and that Ppp6c loss may enhance DNA breakage primed by mutant K‐ras G12D.
3.5. K‐ras G12D‐positive/Ppp6c‐deficient lip skin tissues show enhanced activation of AKT‐dependent pathways
Activated AKT reportedly functions in both cell cycle progression and growth control.20 As a downstream target of AKT associated with cell growth, we examined phosphorylation of 4EBP1 at T37/46 (Figure 5), which is a priming phosphorylation site resulted in activation of cap‐dependent translation.20 We observed phosphorylated 4EBP1 in almost all epidermal tissues in doubly‐mutant (K‐ras G12D positive/Ppp6c‐deficient) mice. Moreover, when we analyzed phosphorylation of S6 protein at S235/236 by immunohistochemistry, that pattern was similar to that of phosphorylated 4EBP1. These findings suggest that protein synthesis is activated in lip epidermis of doubly‐mutant mice. Finally, we assessed phosphorylation of GSK3, an AKT target and key effector of AKT signaling.20 As shown in Figure 5, phosphorylation of GSK3 at S21/9 was upregulated in almost all epidermal tissues in doubly‐mutant mice.
Figure 5.
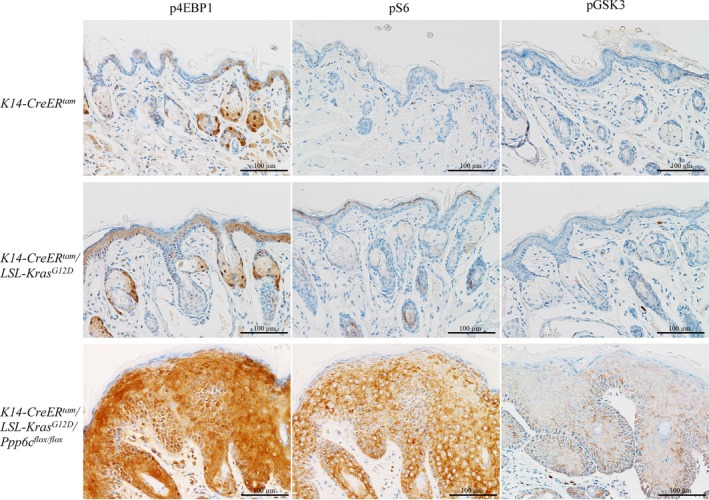
Accumulation of phospho‐4EBP1, phospho‐S6 and phospho‐GSK3 in doubly mutant (KrasG12D expression with Ppp6c deficiency) keratinocytes. Immunohistochemistry of lip samples shown in Figure 3A, using phospho‐4EBP1 (T37/46), phospho‐S6 (S235/236) and phospho‐GSK3 (S21/9) antibodies. Scale bar: 100 μm
4. DISCUSSION
The purpose of this study was to examine the effect of Ppp6c loss‐of‐function on mutant K‐ras‐induced tumorigenesis. To do so, we developed mice with an epidermal‐specific inducible double mutation (combining K‐ras G12D expression with Ppp6c loss) for comparison with K‐rasG12D‐expressing mice. Within 12 days of induction, lip tissues of double mutants displayed a hyperplastic epidermis (Figure 3Af), and on the day of euthanasia, which ranged from 16 to 20 days after induction, animals developed papillomas, some containing focal areas of SCC (Figure 2Cb). SCC represents the promotion stage in progression of skin carcinogenesis (ie from normal skin, to hyperplastic epidermis, to papilloma, and then to SCC).16 Single mutant mice harboring only the K‐ras G12D transgene did not exhibit these phenotypes, providing evidence that Ppp6c deficiency accelerates mutant K‐ras‐initiated tumorigenesis in skin tissues. Immunohistochemical analyses of lip tissues further revealed that Ppp6c deficiency significantly enhanced AKT phosphorylation initiated by K‐rasG12D (Figure 4A), whereas an effect of Ppp6c deficiency on ERK phosphorylation was absent or minimal, suggesting that AKT itself is a PP6 substrate. It is known that AKT phosphorylated at T308 is a PP2A substrate, but whether PP6 can dephosphorylate this site is not yet known. Some phosphopeptide motifs reportedly become hyper‐phosphorylated in mitotic, Ppp6c‐depleted HeLa cells,23 but the sequence motif around AKT phospho‐T308 does not fit these. Thus, more information relevant to motifs recognized by PP6 is required. Interestingly, we observed greater accumulation of phospho‐4EBP1 and phospho‐S6, both AKT downstream targets, than that of phospho‐AKT, suggesting that these proteins are also PP6 substrates. The identify of novel PP6 substrates regulating AKT‐pathway remains to be determined.
Epidermal lip tissues from double mutants were approximately 40‐fold thicker than comparable tissues of K‐rasG12D mice by 12 days after induction by tamoxifen treatment (Figure 3D). Cell proliferation and cell size in these tissues were both upregulated, likely in response to activated AKT. Phosphorylation of 4EBP1 and S6, both downstream of AKT, in these tissues was also enhanced, indicating upregulated protein synthesis. Phosphorylation of GSK3, a direct AKT substrate that functions in cell survival signaling, was also upregulated in these tissues. Taken together, selective activation of AKT by Ppp6c deficiency in the presence of K‐rasG12D may promote epidermal hyperplasia by enhancing both cell proliferation and survival.
K‐ras mutations reportedly increase generation of mitochondrial reactive oxygen species in pancreatic acinar cells,24 and overexpression of mutant N‐ras increases the frequency of DNA damage, leading to increased frequency of error‐prone repair of DSB by nonhomologous end‐joining.25 Our findings suggest that similar mechanisms may operate in mouse skin. Figure 4B shows that mutated K‐ras‐induced epidermal γH2AX accumulation, an effect significantly enhanced by Ppp6c deficiency, suggesting perturbed DNA stability in doubly mutant tissues. Given reports that PP6 functions in homology directed repair via a DNA‐PK‐dependent mechanism5, 6 these data strongly suggest that K‐ras‐induced DNA breaks are not fully repaired in conditions of PP6 deficiency, supporting the idea that Ppp6c deficiency accelerates K‐rasG12D‐initiated genomic instability.
K‐ras is frequently mutated in pancreatic ductal adenocarcinoma (PDA) (approximately 90%), colon and rectal carcinoma (CRC) (approximately 50%) and lung adenocarcinoma (LAC) (approximately 25%).18 Numerous anti‐Ras reagents have been devised as treatment for PDA, CRC or LAC, but successful therapies remain elusive.18 It has been shown that in some pathologies, Ppp6c is downregulated epigenetically. For example, NF‐κB‐induced miR‐31 downregulates Ppp6c and promotes epidermis hyperproliferation in psoriasis.26 Ppp6c downregulation by miR‐373 may explain why miR‐373 expression can promote proliferation of hepatocellular carcinoma cells.27 Thus, loss of Ppp6c function by upregulation of miRNA that bind to the Ppp6c 3′UTR may accelerate formation of K‐ras primed PDA, CRC and LAC.
In conclusion, we have defined synergy between Ppp6c deficiency and expression of mutant K‐ras in skin tumorigenesis. Highly activated AKT seen in double mutant cells likely plays an important role in epidermal hyperplasia. Future studies should address effects of Ppp6c loss on K‐ras‐initiated tumors in cells other than keratinocytes or in the presence of other activated oncogenes.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
ACKNOWLEDGMENTS
We thank Professor Takuji Tanaka for pathological examination. We thank Yoshimi Sakamoto and Kuniko Komuro for technical assistance. We thank Dr Elise Lamar for English editing.
Kurosawa K, Inoue Y, Kakugawa Y, et al. Loss of protein phosphatase 6 in mouse keratinocytes enhances K‐ras G12D‐driven tumor promotion. Cancer Sci. 2018;109:2178–2187. https://doi.org/10.1111/cas.13638
Funding information
JSPS KAKENHI Grant Numbers 16K19745 to YI, 17K16876 to MN, 15K20237 to YM and 26430130 to HS; Research Grant from the Princess Takamatsu Cancer Research Fund to HS; Cooperative Research Project Program of Joint Usage/Research Center at the Institute of Development, Aging and Cancer, Tohoku University to HS; Nara Women's University Intramural Grant for Project Research to TW.
REFERENCES
- 1. Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468‐484. [DOI] [PubMed] [Google Scholar]
- 2. Brautigan DL. Protein Ser/Thr phosphatases–the ugly ducklings of cell signalling. FEBS J. 2013;280:324‐345. [DOI] [PubMed] [Google Scholar]
- 3. Ziembik MA, Bender TP, Larner JM, Brautigan DL. Functions of protein phosphatase‐6 in NF‐kappaB signaling and in lymphocytes. Biochem Soc Trans. 2017;45:693‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T‐loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol. 2010;191:1315‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mi J, Dziegielewski J, Bolesta E, Brautigan DL, Larner JM. Activation of DNA‐PK by ionizing radiation is mediated by protein phosphatase 6. PLoS ONE. 2009;4:e4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, Larner JM. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase‐6 to DNA‐dependent protein kinase. J Biol Chem. 2012;287:9230‐9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhong J, Liao J, Liu X, et al. Protein phosphatase PP6 is required for homology‐directed repair of DNA double‐strand breaks. Cell Cycle. 2011;10:1411‐1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stefansson B, Brautigan DL. Protein phosphatase 6 subunit with conserved Sit4‐associated protein domain targets IkappaBepsilon. J Biol Chem. 2006;281:22624‐22634. [DOI] [PubMed] [Google Scholar]
- 9. Kajino T, Ren H, Iemura S, et al. Protein phosphatase 6 down‐regulates TAK1 kinase activation in the IL‐1 signaling pathway. J Biol Chem. 2006;281:39891‐39896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonilla X, Parmentier L, King B, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016;48:398‐406. [DOI] [PubMed] [Google Scholar]
- 13. Hammond D, Zeng K, Espert A, et al. Melanoma‐associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora‐A. J Cell Sci. 2013;126:3429‐3440. [DOI] [PubMed] [Google Scholar]
- 14. Hayashi K, Momoi Y, Tanuma N, et al. Abrogation of protein phosphatase 6 promotes skin carcinogenesis induced by DMBA. Oncogene. 2015;34:4647‐4655. [DOI] [PubMed] [Google Scholar]
- 15. Ogoh H, Tanuma N, Matsui Y, et al. The protein phosphatase 6 catalytic subunit (Ppp6c) is indispensable for proper post‐implantation embryogenesis. Mech Dev. 2016;139:1‐9. [DOI] [PubMed] [Google Scholar]
- 16. Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi‐stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4:1350‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kato H, Kurosawa K, Inoue Y, et al. Loss of protein phosphatase 6 in mouse keratinocytes increases susceptibility to ultraviolet‐B‐induced carcinogenesis. Cancer Lett. 2015;365:223‐228. [DOI] [PubMed] [Google Scholar]
- 18. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amen N, Mathow D, Rabionet M, et al. Differentiation of epidermal keratinocytes is dependent on glucosylceramide:ceramide processing. Hum Mol Genet. 2013;22:4164‐4179. [DOI] [PubMed] [Google Scholar]
- 20. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park MT, Kim MJ, Suh Y, et al. Novel signaling axis for ROS generation during K‐Ras‐induced cellular transformation. Cell Death Differ. 2014;21:1185‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S. γH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013;753:24‐40. [DOI] [PubMed] [Google Scholar]
- 23. Rusin SF, Schlosser KA, Adamo ME, Kettenbach AN. Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Sci Signal. 2015;8:rs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liou GY, Doppler H, DelGiorno KE, et al. Mutant KRas‐induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14:2325‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rassool FV, Gaymes TJ, Omidvar N, et al. Reactive oxygen species, DNA damage, and error‐prone repair: a model for genomic instability with progression in myeloid leukemia? Cancer Res. 2007;67:8762‐8771. [DOI] [PubMed] [Google Scholar]
- 26. Yan S, Xu Z, Lou F, et al. NF‐κB‐induced microRNA‐31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nat Commun. 2015;6:7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu N, Liu X, Xu X, et al. MicroRNA‐373, a new regulator of protein phosphatase 6, functions as an oncogene in hepatocellular carcinoma. FEBS J. 2011;278:2044‐2054. [DOI] [PubMed] [Google Scholar]