

Abstract
IDH‐mutant gliomas are classified into astrocytic or oligodendroglial tumors by 1p/19q status in the WHO 2016 classification, with the latter presenting with characteristic morphology and better prognosis in general. However, the morphological and genetic features within each category are varied, and there might be distinguishable subtypes. We analyzed 170 WHO grade II‐IV gliomas resected in our institution. 1p/19q status was analyzed by microsatellite analysis, and genetic mutations were analyzed by next‐generation sequencing and Sanger sequencing. For validation, the Brain Lower Grade Glioma dataset of The Cancer Genome Atlas was analyzed. Of the 42 grade III IDH‐mutated gliomas, 12 were 1p‐intact/19q‐intact (anaplastic astrocytomas [AA]), 7 were 1p‐intact/19q‐loss (AA), and 23 showed 1p/19q‐codeletion (anaplastic oligodendrogliomas). Of the 88 IDH‐wild type glioblastomas (GBMs), 14 showed 1p‐intact/19q‐loss status. All of the seven 1p‐intact/19q‐loss AAs harbored TP53 mutation, but no TERT promotor mutation. All 19q‐loss AAs had regions presenting oligodendroglioma‐like morphology, and were associated with significantly longer overall survival compared to 19q‐intact AAs (P = .001). This tendency was observed in The Cancer Genome Atlas Lower Grade Glioma dataset. In contrast, there was no difference in overall survival between the 19q‐loss GBM and 19q‐intact GBM (P = .4). In a case of 19q‐loss AA, both oligodendroglial morphology and 19q‐loss disappeared after recurrence, possibly indicating correlation between 19q‐loss and oligodendroglial morphology. We showed that there was a subgroup, although small, of IDH‐mutated astrocytomas harboring 19q‐loss that present oligodendroglial morphology, and also were associated with significantly better prognosis compared to other 19q‐intact astrocytomas.
Keywords: 19q, astrocytoma, neuropil‐like island, oligodendroglioma, WHO 2016
1. INTRODUCTION
Gliomas consist of various groups of tumors presenting with a wide range of morphologies, molecular genetic alterations, and clinical behaviors. Classification and diagnosis of gliomas are important steps for proper management of patients with this disease, including prediction of prognosis and choice of treatment. For almost a century, this has been done based almost solely on microscopic histology with clinical information. However, accumulation of information on molecular alterations of gliomas has been changing this process significantly over the past 20‐30 years, and the recently updated WHO classification of central nervous system tumors officially incorporated molecular information in the definition of diffuse gliomas.1 One of the most notable is the definition of oligodendroglial tumors: now the oligodendroglioma is defined as diffusely infiltrative glioma with IDH1 or IDH2 mutation and codeletion of chromosomal arms 1p and 19q. Diffuse astrocytoma is defined as diffuse glioma with IDH1 or IDH2 mutation without 1p/19q codeletion, that most likely harbors TP53 and ATRX mutations. Difference in the clinical behavior has been confirmed in multiple studies showing that the 1p/19q codeleted gliomas show significantly better prognosis in both WHO grade II and III tumors.2, 3, 4 In general, morphological features and genetic alterations correlate well, with the majority of 1p/19q codeleted tumors presenting classic oligodendroglioma morphology with isomorphic round nuclei with artefactually swollen clear cytoplasm and delicate branching capillary network, whereas non‐codeleted IDH‐mutant tumors tend to show features of well‐differentiated (grade II) or moderately anaplastic (grade III) fibrillary astrocytes. However, morphological variations are significant, and there are some cases with oligodendroglial morphology without 1p/19q codeletion, as well as the reverse. In the current WHO classification, molecular information trumps morphology, designating IDH‐mutant, oligodendroglioma‐like tumors without 1p/19q codeletion as diffuse astrocytoma.
In a previous study comparing the histological features and molecular genetics, we noticed that there was relatively small number of cases of non‐codeleted diffuse gliomas presenting oligodendroglial morphological features, and those tend to have 19q‐loss only.5 In this study, we investigated those tumors further, including morphology, other genetic alterations, and clinical prognosis.
2. MATERIALS AND METHODS
2.1. Patients
Tumor samples were obtained at surgery carried out at the Dokkyo Medical University Hospital (Mibu, Japan) during the period of 2001‐2015. Histological diagnosis was made according to the latest WHO 2016 classification.6 We analyzed 170 diffuse gliomas histologically compatible with WHO grades II‐IV, which included 19 IDH‐mutated anaplastic astrocytomas (AA), 23 IDH‐mutated, 1p/19q codeleted anaplastic oligodendrogliomas (AO), and 88 IDH‐wild type glioblastomas (GBM). There was no case harboring only 19q‐loss without 1p‐loss among the WHO grade II gliomas analyzed (n = 33). There were seven WHO grade III gliomas with IDH‐wild type, and five WHO grade IV GBMs with IDH‐mutation. Given the still existing controversy on whether those tumors can be considered to be established entities, they were not included in the study. Overall survival (OS) was calculated as the period from the date of surgery to the date of death or latest follow‐up. Median age was 46.5 years for grade III tumors and 62 years for grade IV tumors. There were 26 male patients (62%) with grade III tumors, and 50 male patients (57%) with grade IV tumors. The median follow‐up period was 66.6 months for grade III tumors and 12.2 months for grade IV tumors. Most of the AAs (15/19) were treated with extended local radiation therapy concomitant with temozolomide, and most of the AOs (16/23) were initially treated with upfront PAV (procarbazine, ACNU, and vincristine) therapy. For the recurrence of AO, extended local radiation therapy concomitant with temozolomide was carried out. There was no therapeutic difference in AA between the 19q‐intact tumor and 19q‐loss tumor except for four cases. Clinical profiles of all analyzed tumors are presented in Table 1.
Table 1.
Profiles of all analyzed patients with WHO grade II‐IV gliomas (n = 130)
| AA, 19q‐intact | AA, 19q‐loss | AO | GBM, 19q‐intact | GBM, 19q‐loss | |
|---|---|---|---|---|---|
| n | 12 | 7 | 23 | 74 | 14 |
| Age, years | |||||
| Median (SD) | 43 (14) | 40 (11) | 49 (15) | 62 (15) | 63 (12) |
| Sex, male | |||||
| n (%) | 9 (75) | 6 (86) | 11 (48) | 50 (68) | 10 (71) |
| Tumor side | |||||
| Right | 7 | 4 | 13 | 36 | 8 |
| Left | 0 | 3 | 9 | 29 | 6 |
| Bilateral | 3 | 0 | 1 | 6 | 0 |
| Midline | 2 | 0 | 0 | 3 | 0 |
| Tumor location | |||||
| Frontal | 6 | 7 | 14 | 34 | 6 |
| Temporal | 1 | 0 | 2 | 15 | 3 |
| Parietal | 1 | 0 | 3 | 12 | 5 |
| Occipital | 1 | 0 | 0 | 2 | 0 |
| Thalamus | 1 | 0 | 0 | 5 | 0 |
| Brain stem | 1 | 0 | 0 | 3 | 0 |
| Othera | 1 | 0 | 4 | 3 | 0 |
| Extent of resection | |||||
| GTR | 7 | 5 | 15 | 41 | 12 |
| PR | 1 | 2 | 5 | 10 | 1 |
| Biopsy | 4 | 0 | 3 | 23 | 1 |
| Adjuvant therapy (first line) | |||||
| RT + TMZ | 9 | 6 | 5 | 49 | 12 |
| RT + PAV | 1 | 0 | 1 | 17 | 2 |
| PAV | 0 | 1 | 15 | 0 | 0 |
| RT only | 1 | 0 | 1 | 5 | 0 |
| Otherb | 1 | 0 | 1 | 3 | 0 |
| Genetic status | |||||
| IDH | Mutant | Mutant | Mutant | Wild type | Wild type |
| 1p | Intact | Intact | Loss | Intact | Intact |
| 19q | Intact | Loss | Loss | Loss | Intact |
AA, anaplastic astrocytoma; AO, anaplastic oligodendroglioma; GBM, glioblastoma; GTR, gross total removal; PR, partial removal.
Includes pineal lesion, insula, and involvement of the several lobes.
Includes temozolomide (TMZ) alone, radiation therapy (RT) with procarbazine, ACNU, and vincristine (PAV), or best supportive care.
2.2. DNA extraction
The tumor samples were frozen in liquid nitrogen immediately after resection and stored at −80°C until use. DNA from the tumor tissue was extracted using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer's protocol. Constitutional DNA used as a control was extracted from peripheral blood. Written informed consent was obtained from all patients involved. All experiments using human samples were approved by the ethics committee at the Dokkyo Medical University (Nos. 1431 and 24053).
2.3. Microsatellite analysis
Microsatellite analysis was undertaken using the Genetic Analyzer 310 (Applied Biosystems, Waltham, MA, USA) following the manufacturer's protocol. The following microsatellite markers spanning the whole chromosome arms of 1p and 19q, as well as markers located at the commonly deleted 10q region, were used to detect loss of heterozygosity: D1S1166(1p11), D1S495(1p13), D1S207(1p21), D1S435(1p21), D1S2835(1p31.3), D1S2797(1p32), D1S2892(1p33), D1S2657(1p34), D1S2647(1p36.1), D1S402(1p36.1), and D1S244(1p36.22) for 1p; D19S919(19q12), D19S220(19q13.13), D19S420(19q13.2), D19S219(19q13.3), D19S112(19q13.3), D19S412(19q13.33), and D19S596(19q13.33) for 19q; and D10S1680(10q23), D10S185(10q23‐24), and D10S587(10q26) for 10q.
2.4. Mutation analysis
Mutations in IDH1/2, TERT promoter, and TP53 were detected by direct Sanger sequencing. For IDH mutation, codon 132 of IDH1 and codon 172 of IDH2, the well‐described hotspots, were sequenced. For TERT promoter, C228T and C250T mutations were surveyed by sequencing the PCR products. For TP53, exons 4‐8 were sequenced after PCR amplification. Primer sequences are listed in Table S1. Mutational analysis of tumor‐related genes was undertaken by next‐generation sequencing using the Ion Torrent System in which 20 ng DNA was used for multiplex PCR of a panel covering 93 brain tumor‐related genes including CIC and FUBP1 (NCC Brain Tumor Panel; Thermo Fisher Scientific). Subsequent processing was carried out according to the manufacturer's protocol. Sequencing was done on the Ion PI Chip V3 using the Ion Proton System (Thermo Fisher Scientific). Data analysis was carried out using the Torrent Suite version 5.0 software (Thermo Fisher Scientific). The mutational genes meeting the following conditions were listed: covered at least 250 times, mutant allele frequencies were ≥30%, and registered as missense mutation in the COSMIC database (https://cancer.sanger.ac.uk/cosmic).
2.5. The Cancer Genome Atlas data
The clinical, mutation, and copy number variation data of 516 cases from the Brain Lower Grade Glioma project were obtained from The Cancer Genome Atlas (TCGA) datasets using the cBioPortal (http://www.cbioportal.org). The 1p/19q status was determined by downloading the copy number data, and cases with <−0.3 log2 value of mean copy number of either 1p or 19q were scored as 1p‐loss or 19q‐loss, respectively. Only cases with loss of the long arm of chromosome 19 without loss of the short arm of chromosome 1 were considered as 19q‐loss astrocytomas.
2.6. Statistical analysis
The log–rank test was used to compare OS. Fisher's exact test was used to compare two groups. The SPSS version 23 (IBM, New York, NY, USA) software was used for the statistical analyses. Differences were considered significant if P < .05.
3. RESULTS
3.1. 19q‐loss AA has better prognosis than 19q‐intact AA
On multiple marker microsatellite analysis, 19q‐loss without 1p‐loss was detected in 7/19 AAs and in 14/88 GBMs. All 23 AOs had 1p/19q codeletion by definition. Overall survival for 1p‐intact/19q‐loss AA was significantly longer than 1p‐intact/19q‐intact AA (104.9 months vs 44.2 months; P = .001, log–rank test) (Figure 1). Overall survival of 1p‐intact/19q‐loss AA was similar to that of 1p/19q codeleted AO (P = .93, log–rank test) (Figure 1A). There was no difference in OS between 1p‐intact/19q‐loss GBMs and other GBMs (18 months vs 29 months; P = .40, log–rank test; (Figure 1B). These results indicated that IDH‐mutated, 1p‐intact/19q‐loss AAs had significantly better prognosis compared to other IDH‐mutated AAs, but similar association of 19q‐loss with better prognosis was not observed within the IDH‐wild type GBMs. Although not included in the survival analysis, there was one case of GBM with IDH‐mutation and 1p‐intact/19q‐loss. This patient died 11.9 months after surgery, without showing any sign of better prognosis than other GBMs.
Figure 1.
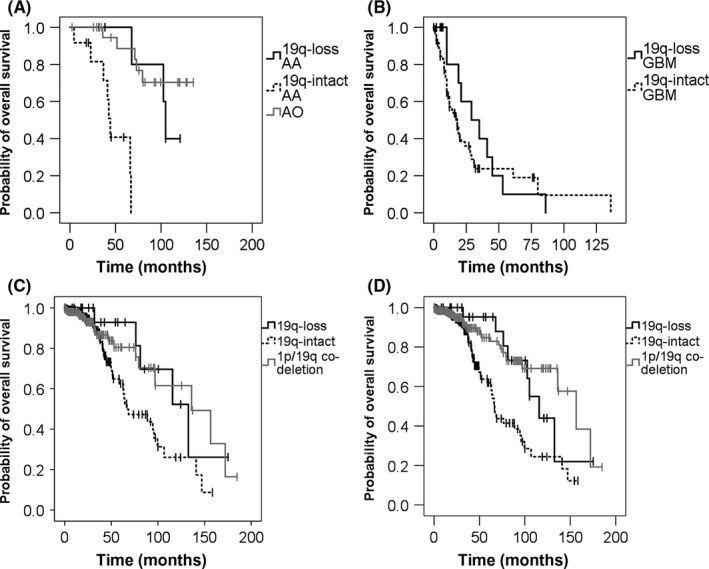
Kaplan–Meier estimates of overall survival (OS) by 1p/19q status in 170 WHO grade II‐IV gliomas. Vertical bars indicate censored cases. A, Survival for WHO grade III IDH‐mutant glioma. OS for 19q‐loss anaplastic astrocytomas (AA; n = 7), 19q‐intact AA (n = 12), and 1p/19q codeleted anaplastic oligodendrogliomas (AO; n = 23) were plotted. OS of 19q‐loss AA was significantly longer than 19q‐intact AA (P = .001, log–rank test), and was not significantly worse than that of anaplastic oligodendrogliomas (P = .93, log–rank test). B, Survival for WHO grade IV IDH‐wild type glioblastomas (GBM). OS for 19q‐loss GBM (n = 14) and 19q‐intact GBM (n = 74) were compared, but there was no significant difference (P = .4, log–rank test). C, Survival for lower grade glioma of The Cancer Genome Atlas cases by 19q status. WHO grade II and III astrocytomas harboring both IDH mutation and TP53 mutation with 19‐loss (n = 23) and 19q‐intact group (n = 204). WHO grade II and III oligodendrogliomas harboring both IDH mutation and TP53 wild‐type with 1p/19q codeletion (n = 159). D, Survival of lower grade gliomas by 19q status in the combined cases of The Cancer Genome Atlas and our series. When combined, OS for the 19q‐loss group (n = 30) was significantly longer than the 19q‐intact group (n = 226) (P = .015, log–rank test). 1p/19q codeletion group (n = 200) is also shown
3.2. Histological characteristics of 19q‐loss AA
We analyzed the histological characteristics of the subgroup of 19p‐loss AA. All seven tumors had distinct regions consisting of tumor cells with uniformly round nuclei and perinuclear halo, the histological features for oligodendroglioma (Figure 2A). Furthermore, 4/7 (57%) cases showed neuropil‐like islands in the oligodendroglioma‐like regions that were positively stained by neuronal markers such as synaptophysin (Syp), class III beta‐tubulin (TUJ1), and neuronal nuclear antigen (NeuN). Oligodendrocyte transcription factor (Olig2) was positive, and glial fibrillary acidic protein (GFAP) was negative in such regions (Figure 2B). By contrast, 3/12 (25%) of 19q‐intact AA showed oligodendroglioma‐like morphology, indicating that 19q‐loss AA tended to show oligodendroglial morphology more frequently than 19q‐intact AA (P = .003, Fisher's exact test). Neuropil‐like islands were not observed in any of the AO with 1p/19q codeletion.
Figure 2.
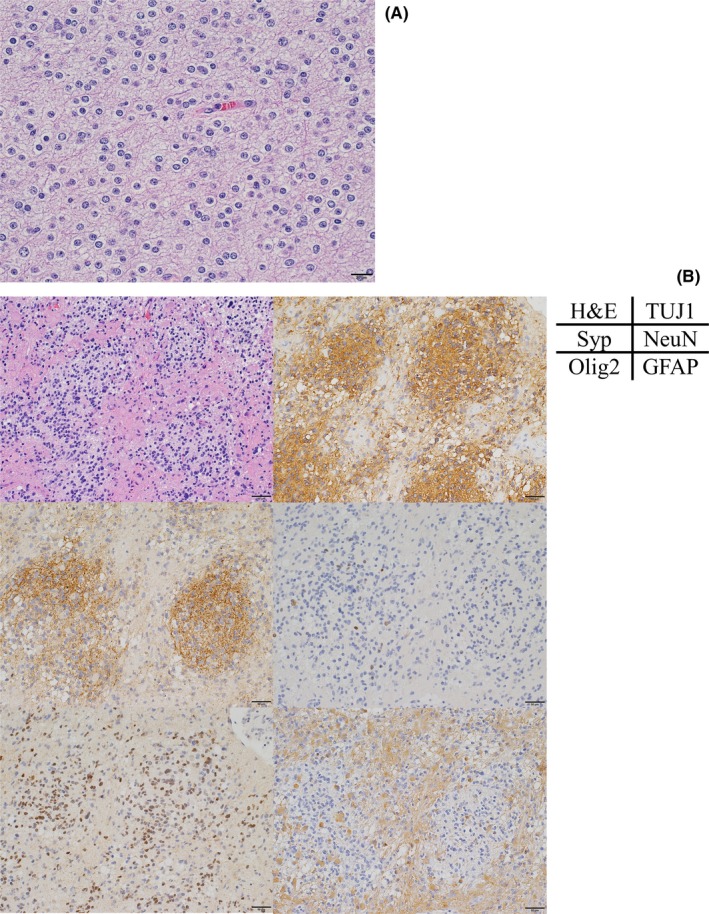
Typical findings of oligodendroglioma‐like cells and neuropil‐like islands in 19q‐loss anaplastic astrocytomas. A, H&E staining of case 4 (scale bar, 20 μm). Uniformly round‐shaped nuclear and perinuclear halos were observed. B, Representative region of a neuropil‐like island (case 7). H&E staining and immunostaining for class III beta‐tubulin (TUJ1), synaptophysin (Syp), neuronal nuclear antigen (NeuN), oligodendrocyte transcription factor (Olig2), and glial fibrillary acidic protein (GFAP) (scale bar, 50 μm) are shown. Cells in the neuropil‐like island were positive for TUJ1, Syp, NeuN, and Olig2, but negative for GFAP
3.3. Genetics of 19q‐loss AA
The deletion map of all microsatellite markers in the 19q‐loss AAs indicated that most of informative markers on 19q were deleted in all cases except for Case 4, in which the partial loss at the telomeric end of 19q was observed (Figure 3). Mutational analysis showed that all IDH‐mutated, 1p‐intact/19q‐loss AAs harbored TP53 mutations, and ATRX mutation was detected in 2/7 (29%). However, TERT promotor mutation, CIC mutation or FUBP1 mutation was not detected in any of the seven tumors (Table 2). These results confirmed that those tumors were molecular genetically compatible with the diagnosis of astrocytomas. No other genes commonly mutated in gliomas were detected, except that mutation of SMARCB1, MSH6, and LRP1B were detected in case 1, and mutation of PIK3CA was detected in case 7.
Figure 3.
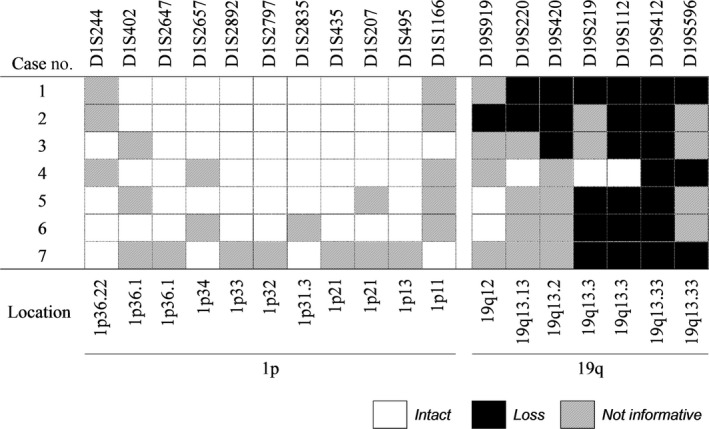
Deletion map of microsatellite markers in all 1p‐intact/19q‐loss anaplastic astrocytomas assessed in this study
Table 2.
Clinical, histological, and genetic profile of all 19q‐loss gliomas
| Case no. | Age, years | Sex | Classification | WHO grade | Side | Location | IDHmut | 1p | 19q | 10q | p53mut | TERTmut | Oligodendroglial component | Neuropil‐like island | Adjuvant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 29 | M | AA, IDHmut | III | L | Fr | R132H | Intact | Loss | Intact | F113del | Wt | + | − | RT + TMZ |
| 2 | 33 | M | AA, IDHmut | III | R | Fr | R132H | Intact | Loss | Intact | G266R | Wt | + | − | RT + TMZ |
| 3 | 40 | M | AA, IDHmut | III | L | Fr | R132H | Intact | Loss | Intact | H179N | Wt | + | + | RT + TMZ |
| 4 | 40 | M | AA, IDHmut | III | R | Fr | R132H | Intact | Partial loss | Intact | R273C | Wt | + | + | RT + TMZ |
| 5 | 50 | M | AA, IDHmut | III | R | Fr | R132H | Intact | Loss | Intact | D49H | Wt | + | + | RT + TMZ |
| 6 | 56 | F | AA, IDHmut | III | R | Fr | R132H | Intact | Loss | Loss | C141Y | Wt | + | − | RT + TMZ |
| 7 | 57 | M | AA, IDHmut | III | L | Fr | R132H | Intact | Loss | Intact | P80 fs | Wt | + | + | PAV |
| 8 | 40 | M | GBM, IDHwt | IV | L | Fr | Wt | Intact | Loss | Intact | Nd | Nd | + | − | RT + TMZ |
| 9 | 51 | M | GBM, IDHwt | IV | L | Fr | Wt | Intact | Loss | Loss | Nd | Nd | + | − | RT + TMZ |
| 10 | 52 | M | GBM, IDHwt | IV | R | Fr | Wt | Intact | Loss | Loss | Nd | Nd | − | − | RT + TMZ |
| 11 | 55 | F | GBM, IDHwt | IV | R | P | Wt | Intact | Loss | Loss | Nd | Nd | − | − | RT + TMZ |
| 12 | 58 | F | GBM, IDHwt | IV | L | P | Wt | Intact | Loss | Loss | Nd | Nd | + | − | RT + TMZ |
| 13 | 59 | F | GBM, IDHwt | IV | R | T | Wt | Intact | Loss | Loss | Nd | Nd | − | − | RT + TMZ |
| 14 | 60 | M | GBM, IDHwt | IV | R | T | Wt | Intact | Loss | Intact | Nd | Nd | + | − | RT + TMZ |
| 15 | 66 | M | GBM, IDHwt | IV | R | Fr | Wt | Intact | Loss | Loss | Nd | Nd | + | − | RT + PAV |
| 16 | 66 | M | GBM, IDHwt | IV | R | Fr | Wt | Intact | Loss | Loss | Nd | Nd | + | − | RT + PAV |
| 17 | 71 | M | GBM, IDHwt | IV | L | P | Wt | Intact | Loss | Intact | Nd | Nd | − | − | RT + TMZ |
| 18 | 76 | M | GBM, IDHwt | IV | L | P | Wt | Intact | Loss | Partial loss | Nd | Nd | − | − | RT + TMZ |
| 19 | 77 | M | GBM, IDHwt | IV | R | Fr | Wt | Intact | Loss | Intact | Nd | Nd | + | − | RT + TMZ |
| 20 | 78 | M | GBM, IDHwt | IV | R | T | Wt | Intact | Loss | Loss | Nd | Nd | + | − | RT + TMZ |
| 21 | 83 | F | GBM, IDHwt | IV | L | P | Wt | Intact | Loss | Loss | Nd | Nd | − | − | RT + TMZ |
AA, anaplastic astrocytoma; F, female; Fr, frontal lobe; GBM, glioblastoma; L, left; M, male; mut, mutant; Nd, no data; P, parietal lobe; PAV, procarbazine, ACNU, vincristine; R, right; RT, radiation therapy; T, temporal lobe; TMZ, temozolomide; Wt, wild type;
3.4. Morphological change accompanied with loss of 19q‐loss at recurrence
In one case (case 3), we could obtain samples at recurrence. This patient was a 40‐year‐old man with a left frontal glioma. Primary tumor was IDH‐mutated, 1p‐intact/19q‐loss AA with oligodendroglioma‐like regions and neuropil‐like islands. There was a TP53 mutation, but no TERT promotor mutation. After gross total resection, the patient underwent radiation therapy (60 Gy) with temozolomide, and the tumor recurred after 2 years. The recurrent tumor was resected again, and molecular examination showed that the IDH1 mutation and TP53 mutation were maintained, but the 19q‐loss was not observed (Figure 4A). Notably, the oligodendroglioma‐like morphology observed in the primary tumor was not evident in the recurrent tumor (Figure 4B). This case suggested that there might be an association between the oligodendroglial morphology and 19q‐loss.
Figure 4.
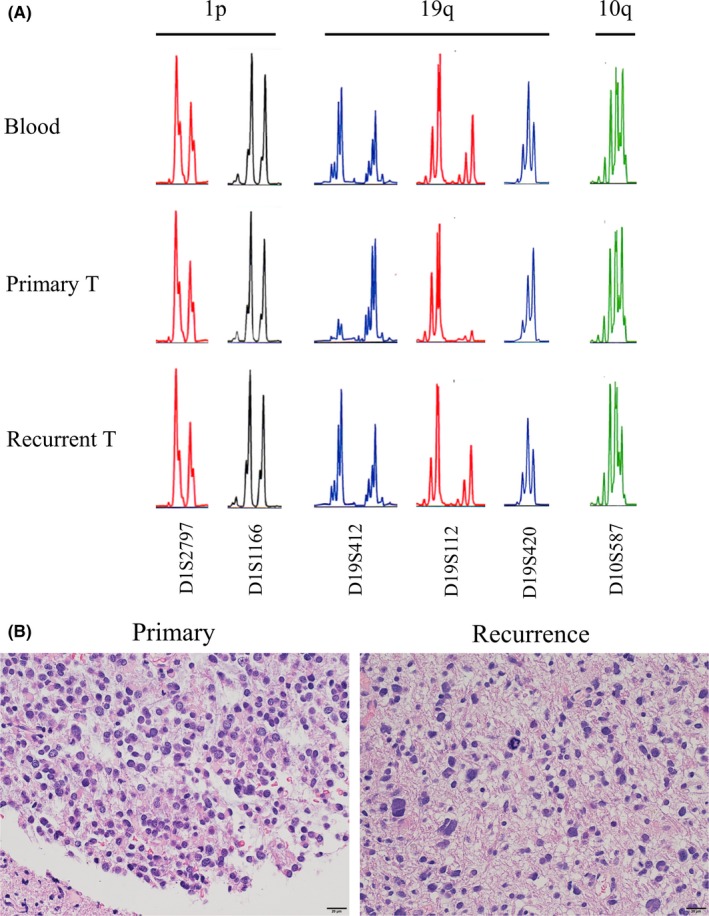
Alteration of the 19q status and histology between the primary and recurrent tumors (T) in case 3, a 40‐y‐old man with a left frontal glioma. A, Microsatellite analysis showing 1p/19q/10q loss of heterozygosity status of primary tumor and recurrent tumor compared with the constitutional DNA. Results of representative primers are shown. Primary tumor showed 1p‐intact, 19q‐loss, and 10q‐intact. Recurrent tumor showed 1p/19q‐intact, losing the19q‐loss status, and also showed new 10q‐loss. B, H&E staining (scale bar, 20 μm). Change in histological features between the primary and recurrent tumors is shown. Oligodendroglioma‐like cells observed in the primary tumor disappeared after recurrence
3.5. Validation analysis on TCGA dataset
Of the 516 cases included in the Brain Lower Glioma project in TCGA datasets, 228 cases with both IDH mutation and TP53 mutation were further analyzed. Of those, 100 were scored as WHO grade II, 99 as grade III, and grades were not available in the remaining 29 cases. Considering the inherent ambiguity of the grading as well as the fact that there was no significant difference in the prognosis between grade II and III tumors in this TCGA dataset (P = .16, log–rank test), we analyzed both grade II and III astrocytomas in this validation study, and compared to our series including grade II tumors, although none of those had 19q‐loss‐only status. In our grade II cohort, 18/33 had 1p19q codeletion, and the remaining 10 were included in the analysis as 19q‐intact tumors.
Of the 228 cases from TCGA dataset, one case had 1p/19q codeletion and was removed from the survival analysis. Of the remaining 227 cases, 23 (10.1%) had 19q‐loss without 1p‐loss, and 204 had intact 19q. Overall survival of the 19q‐loss‐only patients showed tendency to be better, but fell slightly short of reaching statistical significance (P = .052, Figure 1C). However, when the analysis was carried out on combined patients including our series (23 + 7 19q‐loss‐only vs 204 + 22 19q‐intact patients), the 19q‐loss‐only astrocytomas showed significantly longer OS than 19q‐intact astrocytomas (115.5 months vs 66.1 months; P = .015, log–rank test) (Figure 1D).
As for the genetic profile of the analyzed TCGA cohort, TERT mutation was not observed in the 19q‐loss group and in only one case in the 19q‐intact group. ATRX mutation was observed in 18 cases (78%) of 19q‐loss group and 143 cases (70%) of 19q‐intact group. CIC mutation was observed in one case in the 19q‐loss group and in two cases in the 19q‐intact group.
The survival data of 1p/19q codeleted tumors were also analyzed to compare with the survival of 19q‐loss and 19q‐intact tumors. Of the 186 cases showing both IDH mutation and TP53 wild‐type in TCGA datasets, 159 cases with 1p/19q codeletion were extracted and its survival curve was added as the 1p/19q codeletion group in Figure 1(C). In the combined analysis of our series and TCGA datasets, our 18 cases of WHO grade II oligodendroglioma and 23 cases of WHO grade III AO with 1p/19q codeletion were added to the 1p/19q codeletion group in Figure 1(D).
4. DISCUSSION
Our data presented here suggest the possibility that the IDH‐mutated, 1p‐intact/19q‐loss diffuse gliomas might constitute a subgroup of AA that appear like, and behave more like, oligodendrogliomas, although molecular genetics clearly classifies those into the entity of AA in the current WHO classification, with TP53 mutations and without TERT mutations. This association of 19q‐loss with oligodendroglial morphology and better prognosis did not extend to the IDH‐wild type glioblastomas.
There are a few previous reports discussing possible associations between 19q‐loss and prognosis. Brat et al7 reported that 19q‐loss was observed more frequently in the long survival group than in the short survival group in AA. This result was compatible with our results, although IDH mutation was not evaluated in their study. In terms of grade IV gliomas, several studies have looked into the genetic alterations in glioblastoma with oligodendroglioma component, but neither 1p‐loss nor 19q‐loss was associated with appearance of oligodendroglioma component or better prognosis.8, 9
Underlying mechanisms for this possible association between 19q‐loss and prognosis or morphology in IDH‐mutated gliomas are not clear. In fact, molecular mechanisms of the biological effect caused by 1p/19q codeletion in oligodendrogliomas is yet unknown. Cytogenetic studies revealed that unbalanced translocation between chromosome 1 and 19 resulting in the loss of der(1;19)(p10;q10) was the mechanism of the codeletion.10 However, despite extensive past efforts looking for specific genes primarily targeted by 1p‐loss and 19q‐loss in oligodendrogliomas, those genes have not been pinpointed. Analysis of commonly deleted regions of 19q in astrocytoma had narrowed the region to 19q13.3, but none of the candidate genes within this region showed constant mutation.11 Whole exome sequencing identified FUBP1 on 1p31.1 and CIC on 19q13.2 as the most frequently mutated genes on 1p and 19q in oligodendrogliomas, but the mutation rates remain 50%‐60% at most,12 and meticulous studies on mutational landscape have shown that mutations of those two genes can be heterogeneous within a tumor, indicating mutations of those genes are later events following the translocation.13, 14 In our 19q‐loss AAs, 19q13.3 was deleted in all cases including the case with partial loss. However, CIC mutation and other common mutational genes on 19q was not observed in any, indicating these morphological and clinical features of 1p‐intact/19q‐loss tumors were not associated with CIC mutation or any other specific gene on 19q.
The neuropil‐like islands observed in the oligodendroglioma‐like regions of our 19q‐loss AAs might represent neuronal differentiation that is frequently observed in oligodendroglioma.15, 16, 17, 18, 19, 20, 21, 22 Immunopositivity for neuronal markers is also in line with this notion. Kamoun et al23 reported that comprehensive expression analysis revealed overexpression of the genes implicated in neurogenesis in a subset of 1p/19q codeleted oligodendroglioma. Our findings might suggest that 19q‐loss could be more important for neuronal differentiation in gliomas.
Loss of oligodendroglial morphology accompanied with loss of 19q‐loss observed in the recurrent tumor in case 3 is of potential interest. A possible interpretation would be that the dominant clone with 19q‐loss at the primary region showing oligodendroglioma‐like morphology was more sensitive to chemo/radiotherapy, resulting in selection of the 19q‐intact clone that was more resistant to the treatment, and had given rise to the recurrence. Although still being a single case, it appears to corroborate the notion that tumors with 19q‐loss may confer better prognosis.
Although the 19‐loss‐only astrocytomas appear to constitute only <10% of lower grade astrocytomas, and hence pose limitations to our study, the validation study on the independent TCGA dataset was in line with our notion.
In conclusion, we showed that there appears to be a subgroup of IDH‐mutated astrocytomas harboring 19q‐loss and TP53 mutation, which can present morphology similar to oligodendroglial tumors, and also show significantly better prognosis compared to other AAs with IDH mutation. The 19q‐loss might be associated with those features, although the molecular mechanisms are not known.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We are grateful to the patients for donating their tissue for research and acknowledge the excellent technical assistance of Kayoko Iwata at the laboratory of Dokkyo Medical University. We also thank Dr. Yoshiko Nakano at the National Cancer Center Japan for her expert assistance in the analysis of next‐generation sequencing. This work was supported by The Japan Agency for Medical Research and Development (Project for Development of Innovative Research on Cancer Therapeutics to A.M. and Project for Cancer Research and Therapeutic Evolution to A.M.), and Dokkyo Medical University (No. 2017‐07 to R.O.).
Otani R, Uzuka T, Higuchi F, et al. IDH‐mutated astrocytomas with 19q‐loss constitute a subgroup that confers better prognosis. Cancer Sci. 2018;109:2327–2335. https://doi.org/10.1111/cas.13635
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803‐820. [DOI] [PubMed] [Google Scholar]
- 2. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473‐1479. [DOI] [PubMed] [Google Scholar]
- 3. Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long‐term results of RTOG 9402. J Clin Oncol. 2013;31:337‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long‐term follow‐up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31:344‐350. [DOI] [PubMed] [Google Scholar]
- 5. Ueki K, Nishikawa R, Nakazato Y, et al. Correlation of histology and molecular genetic analysis of 1p, 19q, 10q, TP53, EGFR, CDK4, and CDKN2A in 91 astrocytic and oligodendroglial tumors. Clin Cancer Res. 2002;8:196‐201. [PubMed] [Google Scholar]
- 6. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumours of the central nervous system, 4th edn, Revised, Vol 1 edn. Lyon: World Health Organization; 2016. [Google Scholar]
- 7. Brat DJ, Seiferheld WF, Perry A, et al. Analysis of 1p, 19q, 9p, and 10q as prognostic markers for high‐grade astrocytomas using fluorescence in situ hybridization on tissue microarrays from Radiation Therapy Oncology Group trials. Neuro Oncol. 2004;6:96‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Li S, Chen L, et al. Glioblastoma with an oligodendroglioma component: distinct clinical behavior, genetic alterations, and outcome. Neuro Oncol. 2012;14:518‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laxton RC, Popov S, Doey L, et al. Primary glioblastoma with oligodendroglial differentiation has better clinical outcome but no difference in common biological markers compared with other types of glioblastoma. Neuro Oncol. 2013;15:1635‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852‐9861. [DOI] [PubMed] [Google Scholar]
- 11. Hartmann C, Johnk L, Kitange G, et al. Transcript map of the 3.7‐Mb D19S112‐D19S246 candidate tumor suppressor region on the long arm of chromosome 19. Cancer Res. 2002;62:4100‐4108. [PubMed] [Google Scholar]
- 12. Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453‐1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458‐468. [DOI] [PubMed] [Google Scholar]
- 14. Aihara K, Mukasa A, Nagae G, et al. Genetic and epigenetic stability of oligodendrogliomas at recurrence. Acta Neuropathol Commun. 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirose T, Nobusawa S, Nakazato Y, Sasaki A. A case of oligodendroglioma with prominent neuronal differentiation. Hum Pathol. 2013;44:2353‐2359. [DOI] [PubMed] [Google Scholar]
- 16. Mukasa A, Ueki K, Ge X, et al. Selective expression of a subset of neuronal genes in oligodendroglioma with chromosome 1p loss. Brain Pathol. 2004;14:34‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM. Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol. 2002;61:947‐955. [DOI] [PubMed] [Google Scholar]
- 18. Takeuchi H, Kubota T, Kitai R, Matsuda K, Hashimoto N, Sato K. Chromosome 1p and 19q deletions in malignant glioneuronal tumors with oligodendroglioma‐like component. J Neurooncol. 2009;91:33‐38. [DOI] [PubMed] [Google Scholar]
- 19. Vajtai I, Arnold M, Vassella E. Oligodendroglioma with neurocytic differentiation and characteristic loss of heterozygosity on chromosomes 1p and 19q. Acta Neuropathol. 2005;110:520‐522. [DOI] [PubMed] [Google Scholar]
- 20. Vyberg M, Ulhoi BP, Teglbjaerg PS. Neuronal features of oligodendrogliomas–an ultrastructural and immunohistochemical study. Histopathology. 2007;50:887‐896. [DOI] [PubMed] [Google Scholar]
- 21. Wharton SB, Chan KK, Hamilton FA, Anderson JR. Expression of neuronal markers in oligodendrogliomas: an immunohistochemical study. Neuropathol Appl Neurobiol. 1998;24:302‐308. [DOI] [PubMed] [Google Scholar]
- 22. Bielle F, Ducray F, Mokhtari K, et al. Tumor cells with neuronal intermediate progenitor features define a subgroup of 1p/19q co‐deleted anaplastic gliomas. Brain Pathol. 2017;27:567‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kamoun A, Idbaih A, Dehais C, et al. Integrated multi‐omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co‐deleted gliomas. Nat Commun. 2016;7:11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials