

Abstract
Immunological checkpoint blockade therapies benefit a limited population of cancer patients. We have previously shown that vaccine immunotherapy with Toll‐like receptor (TLR)3‐adjuvant and tumor antigen overcomes anti‐programmed death ligand‐1 (PD‐L1) resistance in mouse tumor models. In the present study, 4 different ovalbumin (OVA)‐expressing tumor cell lines were implanted into syngeneic mice and subjected to anti‐tumor immunotherapy using ARNAX and whole OVA protein. ARNAX is a TLR3‐specific agonist that does not activate the mitochondrial antiviral‐signaling protein (MAVS) pathway, and thus does not induce systemic inflammation. Dendritic cell priming and proliferative CTL were induced by ARNAX + OVA, but complete remission was achieved only in a PD‐L1‐low cell line of EG7. Addition of anti‐PD‐L1 antibody to the ARNAX + OVA therapy brought complete remission to another PD‐L1‐high subline of EG7. Tumor shrinkage but not remission was observed in MO5 in that regimen. We analyzed tumor cells and tumor‐infiltrating immune cells to identify factors associated with successful ARNAX vaccine therapy. Tumors that responded to ARNAX therapy expressed high levels of MHC class I and low levels of PD‐L1. The tumor‐infiltrating immune cells in ARNAX‐susceptible tumors contained fewer immunosuppressive myeloid cells with low PD‐L1 expression. Combination with anti‐PD‐L1 antibody functioned not only within tumor sites but also within lymphoid tissues, augmenting the therapeutic efficacy of the ARNAX vaccine. Notably, ARNAX therapy induced memory CD8+ T cells and rejection of reimplanted tumors. Thus, ARNAX vaccine + anti‐PD‐L1 therapy enabled permanent remission against some tumors that stably present antigens.
Keywords: cancer immunotherapy, memory CD8+ T cell, PD‐L1 blockade, Toll‐like receptor 3 adjuvant, tumor microenvironment
Abbreviations
- CBA
cytometric bead array
- DC
dendritic cell
- DLN
draining lymph nodes
- IFNAR
interferon alpha receptor
- IFN
interferon
- MDSC
myeloid‐derived suppressor cell
- MFI
mean fluorescence intensity
- PD‐1
programmed death‐1
- PD‐L1
programmed death ligand‐1
- TAA
tumor‐associated antigen
- TAM
tumor‐associated macrophage
- TCM
central memory T cells
- TEFF
effector T cells
- TEM
effector memory T cells
- TICAM‐1
Toll‐interleukin‐1 receptor domain‐containing adaptor molecule‐1
- TLR
Toll‐like receptor
1. INTRODUCTION
Cancer is a leading cause of death in humans, and peptide vaccine immunotherapies have been proposed as a new therapeutic modality that harnesses the immune system to attack malignant cells. Since then, rapid exploration into this field has been conducted using new technologies that allow the detection of tumorigenic mutations, TAA and tumor‐specific CTL. In fact, immunological checkpoint blockade therapies have led to complete remission in some progressive cancers when appropriate antigens are expressed.1, 2, 3, 4, 5, 6
Responses to PD‐1/PD‐L1 blockade therapies are associated with pre‐existing tumor‐reactive CD8+ T cells and non‐synonymous mutational burden in tumors.1, 4, 5, 7 As the generation of tumor‐specific CTL and the number of cancer neoantigens differ between tumor types, and as T‐cell infiltration is regulated by the tumor microenvironment,8, 9, 10 only approximately 20%‐30% of solid tumors in patients have responded to PD‐1/PD‐L1‐targeted therapies.11, 12 PD‐L1 levels in both tumor and host cells affect the efficacy of PD‐1/PD‐L1 blockade therapy.13, 14, 15, 16, 17 Furthermore, even if tumor‐specific CTL are generated, they often become exhausted and acquire epigenetic stability that limits the durability of reinvigoration by PD‐1 blockade during continuous antigen stimulation.18
In PD‐1 blockade‐unresponsive tumors, fewer tumor‐specific CTL appear to be induced by intrinsic factors. We have previously shown that forced tumor‐specific CTL induction by vaccine immunotherapy TAA and the TLR3‐specific adjuvant, ARNAX, efficiently eradicates tumors in PD‐1/PD‐L1 blockade‐unresponsive mouse tumor models.19, 20 Combined ARNAX and TAA treatment induces tumor‐specific CTL, facilitates T‐cell infiltration into tumors, modulates the tumor microenvironment to establish Th1‐type anti‐tumor immunity, and leads to tumor regression without inflammation. However, the therapeutic efficacy of the ARNAX vaccine depended on the tumor type.20 In the present study, we analyzed the tumor microenvironment in ARNAX‐sensitive and ‐resistant mouse tumor models to uncover factors that influence the efficacy of TLR3 adjuvant therapy. Our data show that tumor‐specific memory CTL are generated in ARNAX‐sensitive tumors, and that they inhibit the tumor growth of reimplanted tumors.
2. MATERIALS AND METHODS
2.1. Mice
WT C57BL/6J mice were purchased from CLEA Japan (Tokyo, Japan). Ticam1 −/− mice were bred in our laboratory.21 Ifnar −/− mice were kindly provided by Dr T. Taniguchi (Tokyo University, Tokyo, Japan). Tlr3 −/− and Myd88 −/− were kindly provided by Dr S. Akira (Osaka University, Osaka, Japan). All mice were back‐crossed >8 times to C57BL/6 background and maintained under specific pathogen‐free conditions in the animal faculty of the Hokkaido University Graduate School of Medicine. All animal research protocols for this work were reviewed and approved by the Animal Safety Center (#17‐0096) of Hokkaido University, Japan.
2.2. Cells
EG7 (ATCC® CRL‐2113™) was purchased from ATCC (Manassas, VA, USA) and cultured in RPMI 1640 supplemented with 10% heat‐inactivated FBS (catalog number: SH30910.03; Thermo Scientific, Waltham, MA, USA), 10 mmol/L HEPES (15630‐080; Gibco, Gaithersburg, MD, USA), 1 mmol/L sodium pyruvate (11360‐070; Gibco), 55 μmol/L 2‐mercaptoethanol (21985‐023; Gibco), 100 IU penicillin/100 μg/mL streptomycin (15070‐063; Gibco) and 0.5 mg/mL G418 (04 727 894 001; Roche, Basel, Switzerland). PD‐L1hi EG7 (sgPd‐l1‐transfected EG7) cells were prepared as previously described.22 MO523 was kindly provided by Dr H. Udono (Okayama University, Japan) and was cultured in RPMI 1640 supplemented with 10% heat‐inactivated FBS, 100 IU penicillin/100 μg/mL streptomycin and 0.1 mg/mL G418. LLC‐OVA24 was kindly provided by Dr T. Nishimura and Dr H. Kitamura (Hokkaido University, Japan) and was cultured in Iscove's Modified Dulbecco's Medium (12440053; Gibco) supplemented with 10% FBS, 55 μmol/L 2‐mercaptoethanol, 100 IU penicillin/100 μg/mL streptomycin and 0.1 mg/mL G418.
2.3. Reagents and antibodies
ARNAX having 120 and 140 bp dsRNA (named ARNAX‐120 and ARNAX‐140, respectively) were synthesized as described19 by GeneDesign, Inc. (Osaka, Japan). TLR3 agonistic activity of ARNAX‐120 was comparable to that of ARNAX‐140 (Figure S1). Poly(I:C) (27‐4732‐01) was purchased from GE Healthcare Life Sciences; recombinant mouse IFN‐γ (575302) was from BioLegend (San Diego, CA, USA); EndoGrade® Ovalbumin (OVA) (321001) was from Hyglos; OVA (H2Kb‐SL8) tetramer (TS‐5001‐P) and OVA257‐264 peptide (SIINFEKL: SL8) (TS‐5001‐P) were from MBL. Anti‐PD‐L1 antibody (Ab) (clone: 10F.9G2, catalog number: BE0101) and rat IgG2b isotype control Ab (LTF‐2, BE0090) were purchased from Bio X Cell. Abs used for flow cytometry analysis are listed in Table S1.
2.4. Tumor challenge and ARNAX therapy
The backs of mice were shaved and s.c. injected with 2 × 106 WT EG7 (PD‐L1lo EG7), PD‐L1hi EG7, MO5 and LLC‐OVA cells, respectively. Tumor volume was calculated by using the formula: tumor volume [mm3] = 0.52 × (long diameter [mm]) × (short diameter [mm])2. PBS, 10 μg ARNAX‐120 or ‐140 and 100 μg OVA were s.c. injected around the tumor when the tumor volume reached 500‐600 mm3. For combination therapy with ARNAX + OVA and anti‐PD‐L1 Ab, 200 μg isotype control Ab or anti‐PD‐L1 Ab was ip injected into mice on the same day of PBS or ARNAX + OVA injection. After the first Ab injection, subsequent Ab treatment was carried out 3‐5 times every 2 or 3 days. Mice were killed when tumor volume reached 2500 mm3. For the EG7 reimplantation model, EG7 cells were reimplanted into mice in which complete EG7 tumor regression was induced by ARNAX + OVA treatment. EG7 cells were reimplanted near the first implantation site.
2.5. Gene expression analysis of tumor cell lines
PD‐L1lo EG7, PD‐L1hi EG7, MO5 and LLC‐OVA were seeded in a 24‐well plate. PBS, 10 μg/mL ARNAX‐140 or 100 U/mL IFN‐γ was added to each well. For gene expression analysis, cells were lysed with TRIzol reagent (15596018; Invitrogen, Carlsbad, CA, USA) 4 hours after incubation and total RNA was prepared following the manufacturer's instructions. RT‐PCR was carried out using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Real‐time PCR was carried out using a StepOne Real‐Time PCR System (4368813; Applied Biosystems). Sequences of primers used in this study are shown in Table S2. Levels of target mRNAs were normalized to Gapdh and fold‐induction of transcripts was calculated using the ΔΔCT method. Surface expression of PD‐L1 and MHC class I on tumor cells was analyzed by flow cytometry 24 hours after ARNAX or IFN‐γ stimulation.
2.6. Analysis of tumor microenvironment
After killing mice at the indicated time points, tumor tissues were harvested. Tumors were finely minced and treated with 0.05 mg/mL collagenase I (C0130‐100MG; Sigma‐Aldrich, St Louis, MO, USA), 0.05 mg/mL collagenase IV (C5138‐1G; Sigma‐Aldrich), 0.025 mg/mL hyaluronidase (H6254‐500MG; Sigma‐Aldrich) and 0.01 mg/mL DNase I (10 104 159 001; Roche) in HBSS (H9269‐500ML; Sigma‐Aldrich) at room temperature for 15 minutes. Cells in the tumor microenvironment were analyzed by FACS AriaII (BD Biosciences, Franklin Lakes, NJ, USA).
2.7. Statistical analysis
P‐values were calculated by the following statistical analyses. In cases of tumor volume and FACS analysis, 1‐way analysis of variance (ANOVA) with Bonferroni's test or Kruskal‐Wallis test with Dunn's multiple comparison was carried out for multiple comparisons. Student's t test was carried out for comparison between 2 groups. Error bars represent the SD or SEM between samples. In the case of survival rate analysis, log‐rank test with Bonferroni's test was carried out for multiple comparisons.
3. RESULTS
3.1. Efficacy of ARNAX vaccine therapy depends on tumor cell type
The tumor microenvironment is quite different in various tumor types. Therefore, we assessed the efficacy of ARNAX vaccine therapy in 4 different tumor‐bearing mouse models, WT EG7 (PD‐L1lo EG7; OVA‐expressing EL4 lymphoma), PD‐L1hi EG7,22 MO5 (OVA‐expressing B16 melanoma),23 and LLC‐OVA (OVA‐expressing Lewis lung carcinoma).24 Tumor‐bearing mice were treated with PBS or ARNAX + whole OVA protein. ARNAX vaccine therapy induced potent tumor regression in PD‐L1lo EG7 and partial tumor growth retardation in MO5, but did not show a significant effect in LLC‐OVA‐bearing mice (Figure 1A‐C). We previously showed that there was marked infiltration of CD8+ T cells into tumors following ARNAX vaccine therapy in PD‐L1lo or hi EG7‐ and MO5‐bearing mice.20 In contrast, only a few infiltrating CD8+ T cells were detected in LLC‐OVA‐bearing mice, even in the ARNAX vaccine therapy group (Figure 1D, left panel). Additionally, the proportion of OVA‐specific CD8+ T cells among all intratumor CD8+ T cells was only slightly increased by ARNAX in LLC‐OVA compared with PD‐L1lo or hi EG7 and MO5 tumors (Figure 1D, right panel).20 These results suggest that the efficacy of the ARNAX vaccine depends on tumor type.
Figure 1.
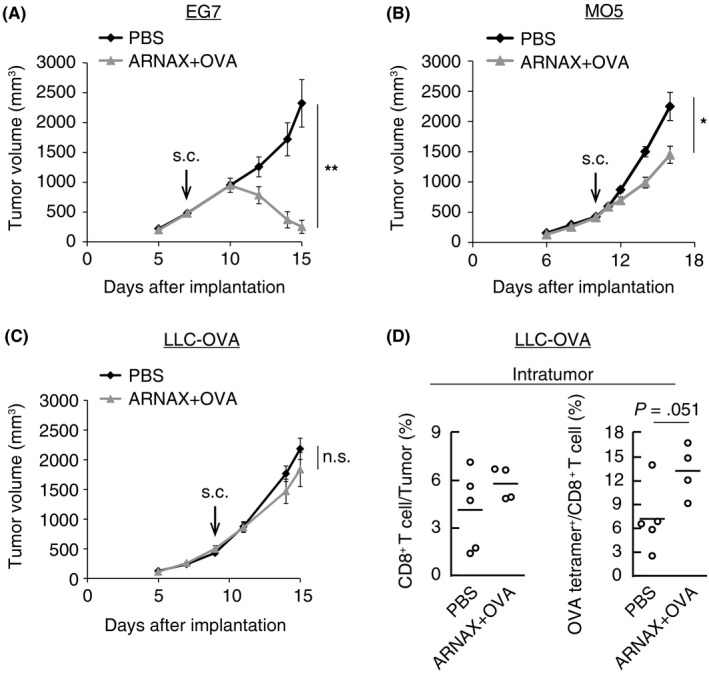
Efficacy of ARNAX + tumor‐associated antigen (TAA) therapy depends on tumor type. A, WT EG7 (PD‐L1lo EG7)‐bearing mice were s.c. given PBS or 10 μg ARNAX‐120 and 100 μg ovalbumin (OVA) 7 d after tumor implantation. PD‐L1, programmed death ligand‐1. B, MO5‐bearing mice were s.c. given PBS or 10 μg ARNAX‐140 + OVA 10 d after tumor implantation. C, LLC‐OVA‐bearing mice were s.c. given PBS or 10 μg ARNAX‐140 + OVA 9 d after tumor implantation. A‐C, Tumor sizes were evaluated in each group. Error bars indicate means ± SEM; n = 4‐6 per group. Student's t test was carried out to analyze statistical significance; *P < .05, **P < .01, n.s., not significant. D, LLC‐OVA‐bearing mice treated as in (C) were killed 15 d after tumor implantation. Proportions of CD8+ T cells in tumors and OVA‐specific cells among intratumor CD8+ T cells were analyzed by flow cytometry. Student's t test was carried out to analyze statistical significance
To elucidate the factors that influence responsiveness to ARNAX therapy, we assessed PD‐L1 and MHC class I levels in tumor cells, as these proteins affect CTL‐dependent anti‐tumor response.25 PD‐L1lo EG7, MO5 and LLC‐OVA cells expressed PD‐L1 at low or intermediate levels, and PD‐L1hi EG7 expressed PD‐L1 highly (Figure 2A). PD‐L1lo or hi EG7 and LLC‐OVA cells expressed high levels of MHC class I molecules on their cell surfaces, whereas MO5 scarcely expressed it (Figure 2A). ARNAX stimulation did not affect the expression levels of these molecules on any cell type examined (Figure 2A). As IFN‐γ is an inducer of PD‐L1 and MHC class I25 and is secreted into the tumor microenvironment when ARNAX therapy succeeds,20 we assessed tumor cell response to IFN‐γ. PD‐L1 expression was upregulated by IFN‐γ in MO5 and LLC‐OVA cells but not in PD‐L1lo or hi EG7 cells. Additionally, MHC class I expression was potently upregulated in MO5 cells, but only marginally increased in PD‐L1lo or hi EG7 and LLC‐OVA cells, suggesting a high susceptibility of MO5 cells to INF‐γ signaling (Figure 2B).
Figure 2.
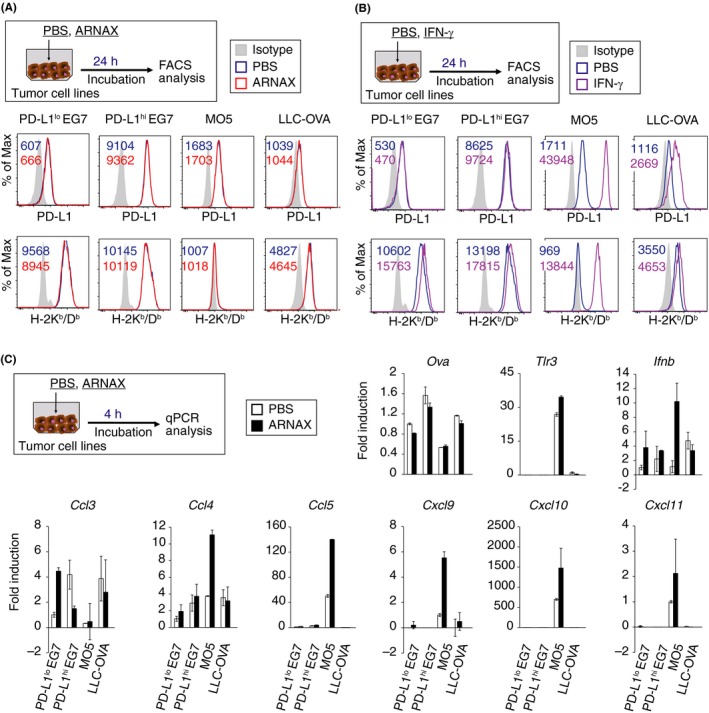
MO5 cells directly respond to ARNAX and interferon (IFN)‐γ stimulation. (A, B) PD‐L1lo EG7, PD‐L1hi EG7, MO5 and LLC‐ovalbumin (OVA) cells were cultured in the presence or absence of 10 μg/mL ARNAX‐140 (A) and 100 U/mL IFN‐γ (B). After 24‐h incubation, PD‐L1 and H‐2Kb/Db expression were analyzed by flow cytometry. Numbers in the histogram indicate mean fluorescence intensity. PD‐L1, programmed death ligand‐1. (C) PD‐L1lo EG7, PD‐L1hi EG7, MO5 and LLC‐OVA cells were cultured in the presence or absence of ARNAX‐140. After 4‐h incubation, mRNA expression was analyzed by qPCR. Error bars show mean ± SD
In MO5‐bearing mice, ARNAX + OVA immunization successfully induced OVA‐specific CD8+ T‐cell proliferation and IFN‐γ secretion in tumor tissues,20 which may affect the phenotype of MO5 cells. Although ARNAX has an ability to induce DC‐mediated activation of natural killer (NK) cells,19 it remains unexamined as to whether NK cells serve as effector cells against MHC class I‐low MO5 tumor at the early time point of ARNAX therapy. All tumor types expressed the Ova gene, but MO5 cells expressed relatively low Ova levels (Figure 2C). Only MO5 cells expressed Tlr3 and Ifnb, which were directly induced by ARNAX stimulation. Basal levels of T‐cell chemoattractant genes, including Ccl5 and Cxcl9, 10, and 11 were higher in MO5 cells than in EG7 or LLC‐OVA cells. Moreover, Ccl4 and 5, and Cxcl9‐11 were upregulated following ARNAX stimulation in MO5 cells, indicating that MO5 cells tended to be more phenotypically affected by ARNAX therapy. These data suggest that MHC class I gene levels and intrinsic tumor cell responses affect the responsiveness to ARNAX therapy.
3.2. Immunosuppressive myeloid cells highly infiltrate into LLC‐OVA tumor
Composition of tumor‐infiltrating immune cells influences the immune status of the tumor microenvironment. TAM and MDSC establish a potent immunosuppressive microenvironment.26, 27, 28 We evaluated the percentage of tumor‐infiltrating immune cells, especially myeloid cells in PD‐L1lo EG7‐, MO5‐ and LLC‐OVA‐bearing mice. Among these tumor models, LLC‐OVA showed the highest immune cell infiltration in tumors (Figure S2A). The proportion of the CD11b+Ly6G+ population, which includes granulocyte‐like MDSC, was highest 6 days after ARNAX vaccine therapy in the LLC‐OVA model (Figure S2B). Although the proportion of the CD11b+Ly6C+ population, which includes monocytic MDSC, among intratumor immune cells was almost the same among the 3 tumor models, their proportion in tumors was higher in the LLC‐OVA than in the PD‐L1lo EG7, or MO5 models (Figure S2A,C). TAM also highly infiltrated into LLC‐OVA tumors (Figure S2D). These results showed that LLC‐OVA tumors contained abundant immunosuppressive myeloid cells, which appeared to inhibit ARNAX‐induced CTL infiltration.
3.3. Immune cells in lymphoid and tumor tissues highly express PD‐L1
Programmed death ligand‐1 expression on tumor cells or host cells can disturb anti‐tumor immunity.13, 14, 15, 16 Thus, we assessed PD‐L1 expression on immune and tumor cells in each tumor model (Figure 3). In the case of PD‐L1lo or hi EG7 tumors (CD45.2+), we used CD45.1 congenic mice as the host to distinguish tumor and immune cells with CD45.1 and CD45.2 markers. Various immune cells in lymphoid tissues expressed PD‐L1, and their levels of expression were almost identical between each tumor model (Figure 3, Figure S3). PD‐L1 expression on immune cells in lymphoid tissues, but not in tumors, was slightly upregulated 24 hours after giving ARNAX + OVA and, in particular, the increase was most prominent on macrophages in DLN. PD‐L1 levels on immune cells in tumor tissues were much higher than those on tumor and mesenchymal cells in all tumor models. Notably, intratumor myeloid DC and TAM showed high PD‐L1 expression in MO5 and LLC‐OVA models, which were higher than those in the PD‐L1lo or hi EG7 model. Hence, PD‐L1 expression on immune cells both in lymphoid tissues and in tumors critically contributes to immune suppression.
Figure 3.
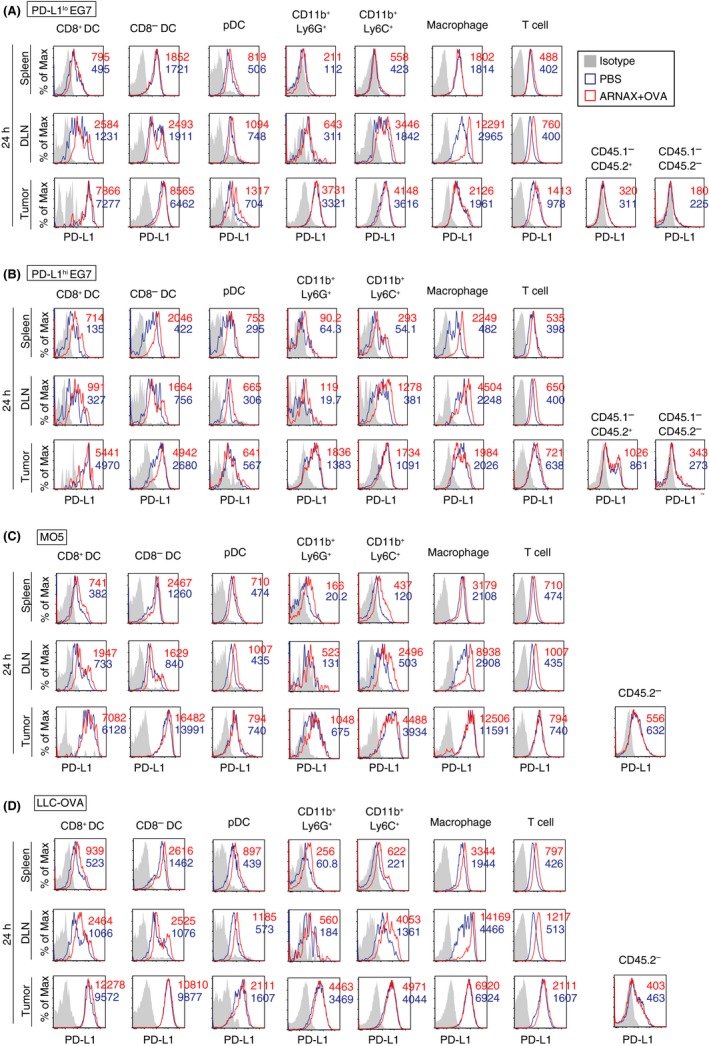
Programmed death ligand‐1 (PD‐L1) expression on intratumor myeloid cells is different in each tumor type. A‐D, PD‐L1lo EG7 and PD‐L1hi EG7‐bearing CD45.1 congenic mice (C57BL/6) and MO5‐ and LLC‐ovalbumin (OVA)‐bearing WT mice (CD45.2) were treated with PBS or 10 μg ARNAX‐140 + OVA on day 7 (PD‐L1lo or hi EG7‐bearing mice) or day 10 (MO5‐ and LLC‐OVA‐bearing mice). After 24 h, spleens, draining lymph nodes (DLN) and tumor tissues were harvested, and cell suspensions were mixed in each group. PD‐L1 expression levels on immune cells in spleen, DLN and tumor were evaluated. In the case of PD‐L1lo or hi EG7 tumors, tumor cells (CD45.2) and host immune cells (CD45.1) were distinguished using CD45.1 and CD45.2 markers. PD‐L1 expression on intratumor non‐immune cells (the CD45.1−CD45.2+ population in EG7 tumors = tumor cells; the CD45.1−CD45.2− population in EG7 tumors = mesenchymal cells; and the CD45.2− population in MO5 and LLC‐OVA tumors = tumor cells and mesenchymal cells) were also analyzed. Results in PD‐L1lo EG7‐ (A), PD‐L1hi EG7‐ (B), MO5‐ (C) and LLC‐OVA‐bearing mice (D) are shown. Numbers in the histogram indicate mean fluorescence intensity
3.4. Combined ARNAX vaccine and PD‐L1 blockade improves therapeutic outcomes
Programmed death ligand‐1 expression in lymphoid tissues and within tumors may attenuate the anti‐tumor responses of ARNAX therapy in both the priming and effector phases. To assess the contribution of PD‐L1 blockade in the priming phase, OVA‐specific CTL expansion in the spleen was evaluated in tumor‐free mice immunized with ARNAX + OVA with or without PD‐L1 blockade. ARNAX + OVA‐induced OVA‐specific CD8+ T‐cell expansion and IFN‐γ production were enhanced by the addition of anti‐PD‐L1 Ab (Figure 4). These data indicate that PD‐L1 contributes to suppression of CTL induction/proliferation at the priming phase; thus, we conclude that ARNAX‐induced CTL expansion is augmented by PD‐L1 blockade.
Figure 4.
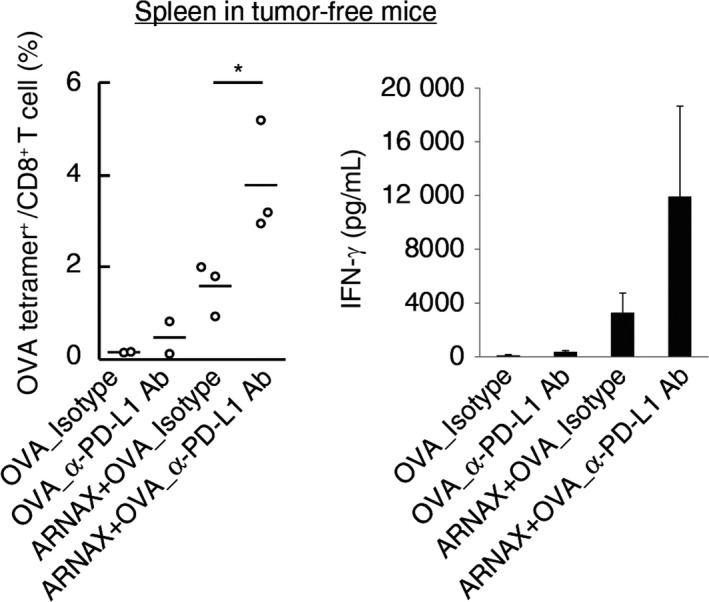
Programmed death ligand‐1 (PD‐L1) blockade augments ARNAX‐induced antigen‐specific CTL expansion in the priming phase. A, B, Tumor‐free mice were s.c. given ovalbumin (OVA) or 60 μg ARNAX‐140 + OVA on day 0. Isotype control or anti‐PD‐L1 Ab was given ip on days 0, 2, 4 and 6. On day 7, spleens were harvested. A, OVA‐specific CD8+ T‐cell proliferation in the spleen was evaluated with the tetramer assay. Student's t test was carried out to analyze statistical significance between ARNAX + OVA_Isotype group and ARNAX + OVA_anti‐PD‐L1 Ab group; *P < .05. B, Splenocytes were cultured in the presence of 100 nmol/L SL8 peptide for 3 d. Interferon (IFN)‐γ concentrations in the culture media were measured using the Cytometric Bead Array. Error bars indicate means ± SD; n = 2‐3 per group. Student's t test was carried out to analyze statistical significance between the ARNAX + OVA_Isotype and ARNAX + OVA_anti‐PD‐L1 Ab groups
To further develop our ARNAX vaccine strategy, combination therapy with anti‐PD‐L1 Ab was carried out. Tumor growth rates and survival times were evaluated in PD‐L1lo EG7‐, PD‐L1hi EG7‐, and MO5‐bearing mouse models. In the PD‐L1lo EG7 model, ARNAX + OVA induced potent tumor regression even without anti‐PD‐L1 Ab, and complete tumor regression was observed in 40% of mice. Addition of the anti‐PD‐L1 Ab failed to produce additional improvements (Figure 5A). There was no significant difference in survival rate between ARNAX + OVA and ARNAX + OVA + anti‐PD‐L1 Ab, although survival time appears to be prolonged with combination therapy. In the PD‐L1hi EG7 model, anti‐PD‐L1 Ab augmented tumor regression from the ARNAX vaccine, and 60% of mice survived >60 days. In the end, complete tumor regression was observed in 40% of mice treated with ARNAX + OVA + anti‐PD‐L1 Ab vs 20% of mice treated with anti‐PD‐L1 Ab alone (Figure 5B). We have previously shown that the frequencies of OVA‐specific CD8+ T cells in spleen following ARNAX + OVA therapy were comparable between PD‐L1lo EG7‐bearing mice and PD‐L1hi EG7‐bearing mice in the presence or absence of anti‐PD‐L1 Ab.20 Thus, tumor cell‐expressing PD‐L1 does not affect the priming phase in DLN but acts in the effector phase. In the MO5 model, tumor growth suppression and survival times were improved by ARNAX + OVA + anti‐PD‐L1 Ab compared with ARNAX vaccine or anti‐PD‐L1 Ab as monotherapies (Figure 5C). These results indicate the effectiveness of combination therapy with ARNAX vaccine and PD‐L1 blockade.
Figure 5.
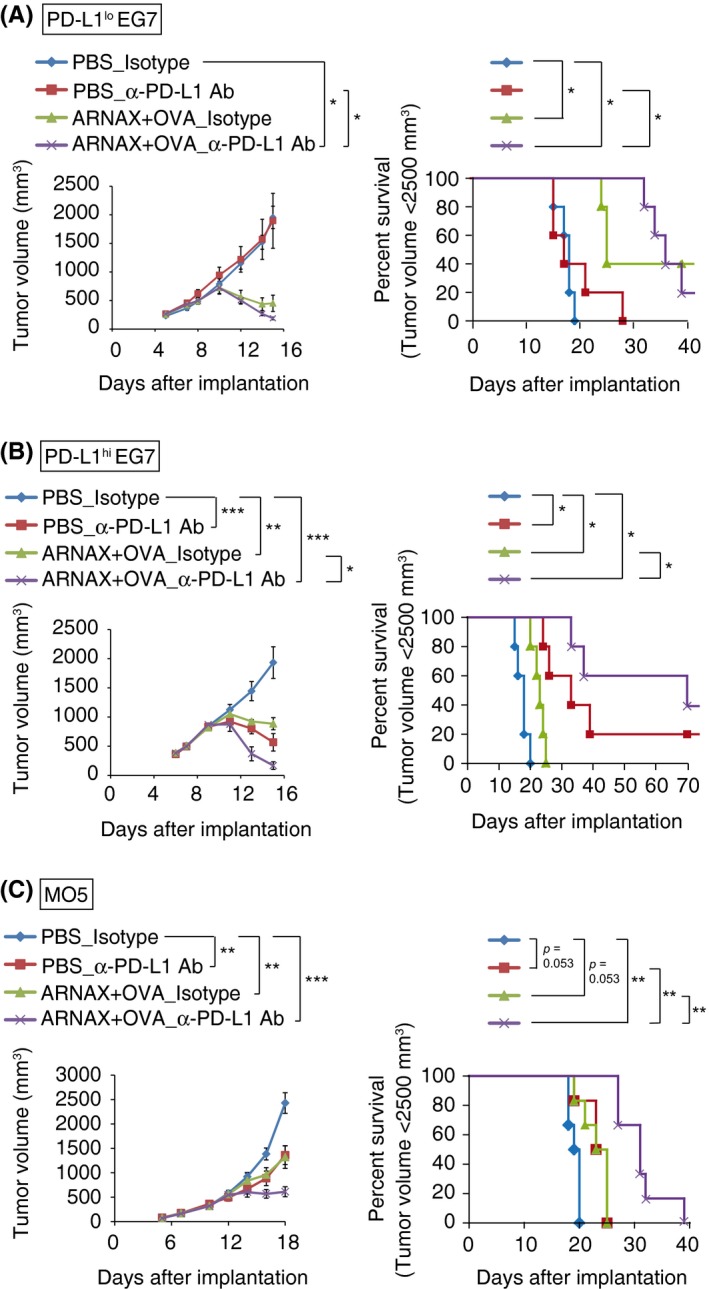
ARNAX + tumor‐associated antigen (TAA) enhances the therapeutic efficacy of programmed death ligand‐1 (PD‐L1) blockade and prolongs survival times. A, B, PD‐L1lo EG7‐ or PD‐L1hi EG7‐bearing mice were s.c. given PBS or 10 μg ARNAX‐120 + ovalbumin (OVA) 7 d after tumor implantation. Isotype control Ab or anti‐PD‐L1 Ab was ip given on days 7, 10, 13 and 16. PD‐L1, programmed death ligand‐1. C, MO5‐bearing mice were s.c. given PBS or 50 μg ARNAX‐140 + OVA 10 d after tumor implantation. Isotype control Ab or anti‐PD‐L1 Ab was ip given on days 10, 12, 14, 16, 18 and 20. (Left of A‐C) Tumor sizes were evaluated in each group. Error bars indicate means ± SEM; n = 5‐6 per group. Kruskal‐Wallis test with Dunn's multiple comparison test (A) and 1‐way analysis of variance (ANOVA) with Bonferroni's test (B,C) were carried out to analyze statistical significance; *P < .05, **P < .01 ***P < .001. (Right of A‐C) Mice were killed when tumor volumes reached 2500 mm3, and survival data were analyzed. Log‐rank test with Bonferroni's test were carried out to analyze statistical significance; *P < .05, **P < .01
3.5. ARNAX vaccine therapy induces memory CD8+ T‐cell formation
We next assessed the formation of functional memory CD8+ T cells by ARNAX. Each CD8+ T‐cell subset was defined from the following molecular expression patterns: TEFF, CD44+CD62LloCD127−CD8+; naive T cells, CD44−CD62LhiCD8+; TCM, CD44+CD62LhiCD8+; and TEM, CD44+CD62LloCD127+CD8+.29, 30, 31, 32 Then, PD‐L1lo EG7‐bearing mice were treated with ARNAX + OVA, and 11 days after complete tumor regression, PD‐L1lo EG7 cells were reimplanted into the mice (Figure 6A). Although non‐immunized mice could not reject tumor growth, the immunized mice completely rejected the second set of tumor cells (Figure 6B). After the reimplantation of EG7, proportions of OVA‐specific CD8+ T cells and TEM were significantly higher in spleens of the reimplantation group than in the first‐time implantation group. However, the proportions of TEFF, and naive and TCM were similar between the 2 groups (Figure 6C).
Figure 6.
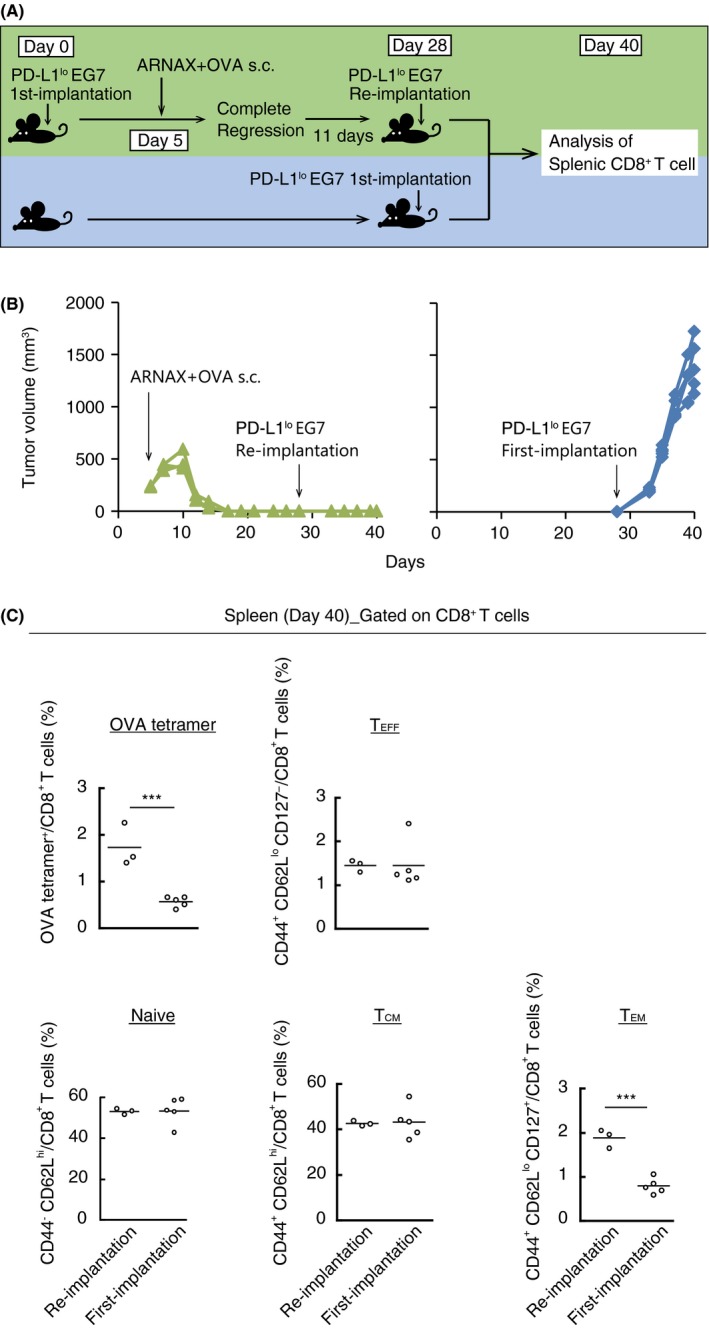
ARNAX + tumor‐associated antigen (TAA) induces memory CD8+ T cells that cause rejection of reimplanted tumor cells. A, Scheme of each treatment for PD‐L1lo EG7‐bearing mice is shown. PD‐L1lo EG7 were reimplanted into mice in which complete tumor regression was induced by 10 μg ARNAX‐140 + ovalbumin (OVA) treatment. B, Tumor sizes of individual mice were evaluated in the EG7 reimplantation group (green line) and EG7 first‐time implantation group (blue line). C, Mice were killed on day 40. Proportions of OVA‐specific cells, TEFF (CD44+CD62LloCD127−), naive T cells (CD44−CD62Lhi), TCM (CD44+CD62Lhi) and TEM (CD44+CD62LloCD127+) among splenic CD8+ T cells were analyzed by flow cytometry; n = 3‐5 per group. Student's t test was carried out to analyze statistical significance; ***P < .001
We further analyzed the formation of memory CD8+ T cells following combined ARNAX + OVA and anti‐PD‐L1 Ab therapy in PD‐L1lo and PD‐L1hi EG7‐bearing mouse models. Six weeks after complete tumor regression from each treatment, PD‐L1lo or PD‐L1hi EG7 cells were reimplanted into the mice (Figure S4A). Although the first‐time implantation group could not reject tumor growth, reimplantation groups rejected tumor cells regardless of treatment regimen in both the PD‐L1lo and PD‐L1hi EG7 models (Figure S4B,C). After the reimplantation of PD‐L1lo or PD‐L1hi EG7 cells, OVA‐specific CD8+ T cells and TEM were elevated in the spleens (Figure S4D,E). Thus, it is likely that combination therapy induces effector CD8+ T cells, which differentiate into memory CD8+ T cells once tumors are controlled and tumor antigens are cleared.
4. DISCUSSION
The goal of the present study was to unveil factors that affect vaccine therapy for cancer unresponsive to PD‐1/PD‐L1 blockade therapy. We have previously shown that ARNAX therapy relieves resistance to PD‐L1 blockade and evokes anti‐tumor immunity without systemic inflammatory cytokinemia in tumor‐bearing mouse models.20 Herein, we showed that ARNAX therapy induced complete remission in some PD‐L1lo EG7‐bearing mice. When combined with anti‐PD‐L1 Ab, ARNAX therapy also induced complete remission in some PD‐L1hi EG7‐bearing mice. Thus, vaccine therapy enables TAA‐positive tumors to regress depending on PD‐L1 expression status. ARNAX vaccine therapy accomplishes partial remission and prolonged survival in the MO5 model, although it does not attain remission in mice with LLC‐OVA.
PD‐L1 is expressed on many human cancer cells and its expression is induced by cytokines, including IFN‐γ and various oncogenic signaling pathways that show increased activity as a result of genetic mutations.33 Moreover, a common aberrant structural variation in the 3′‐untranslated region of the PD‐L1 mRNA induces PD‐L1 overexpression.22 Level of PD‐L1 expression on MO5 and LLC‐OVA cells within tumors was higher than that of PD‐L1lo EG7 cells (right panels of Figure 3), and IFN‐γ potently upregulated PD‐L1 expression in MO5 and LLC‐OVA cells (Figure 2). PD‐L1 levels on tumor‐infiltrating immune cells were consistently high in the MO5 and LLC‐OVA models. Thus, high PD‐L1 levels on both tumor and immune cells decrease TAA‐dependent tumor shrinkage, although other factors still remain undetectable in the effectiveness of ARNAX therapy in MO5 and LLC‐OVA models.
MO5 expressed functional TLR3, and the expression of CTL chemoattractants was directly upregulated by ARNAX (Figure 2). A higher number of CD8+ T cells infiltrated into MO5 tumor tissues in ARNAX + OVA vaccine therapy.20 Direct chemokine production from tumor cells during ARNAX therapy facilitated CTL tumor infiltration in MO5‐bearing mice, which enhanced the efficacy of PD‐1 blockade (Figure 5).
Pan‐genomic analyses across clinical human cancers has shown that the somatic mutational burden is highest in lung carcinoma and melanoma, whereas the frequency is low in lymphoma.8, 9, 10 Mutations associated with antigenicity, such as TAA and MHC class I, particularly impact immune evasion. Kaluza et al. showed that an adoptive transfer of OT‐I cells into B16‐OVA‐bearing mice induced loss of the Ova transgene in tumor cells and tumor regrowth after transient shrinkage.34 Loss of β2‐microglobulin, which is a component of MHC class I molecules, was also observed in metastatic melanoma cells derived from patients who had received immunotherapy.35 Frequency of genetic mutations controlled by ARNAX‐induced CTL activation may have been one of the factors affecting therapeutic success.
A high rate of immunosuppressive myeloid cell infiltration into the tumor microenvironment also disturbs antitumor immune responses. Varn et al. showed that intratumor CD8+ T‐cell‐high/macrophage‐low patients survived longer relative to CD8+ T‐cell‐high/macrophage‐high patients in several different cancer types.10 A large number of TAM and MDSC in the tumor microenvironment could be a predisposition for resistance to ARNAX therapy in the LLC‐OVA model.
It is generally accepted that in patients with non‐inflammatory tumors (termed “cold tumors”), endogenous CTL‐priming stimuli are lacking and only a small population of tumor‐specific CTL exist within the tumor. Immunization of ARNAX and whole OVA protein can induce both OVA‐specific CD4+ and CD8+ T cells, the outcome of which is quite different from the vaccine using SL8 killer peptide. Previous RNA‐seq analysis in EG7 tumor clearly showed upregulation of genes related to the activation of antigen‐presenting cells (APC), CD4+ T cells and CD8+ T cells following ARNAX + OVA therapy.20 Thus, multiple CD4+ and CD8+ T‐cell epitopes are required for successful ARNAX vaccine therapy to induce effective CTL expansion in the priming phase and allow CTL infiltration into tumors without inflammation. However, immune cells also express PD‐L1 in both lymphoid and tumor tissues (Figure 3). PD‐L1 blockade not only reinvigorated exhausted CTL into the effector phase, but also promoted ARNAX‐induced CTL expansion in the priming phase (Figure 4). Thus, combination therapy with ARNAX and PD‐1 blockade is a rational strategy to overcome the unresponsiveness to PD‐1/PD‐L1 blockade therapy.
In ARNAX‐sensitive PD‐L1lo and PD‐L1hi EG7 models, tumor‐specific memory CD8+ T cells were established in mice once tumors were eradicated, and these mice also rejected reimplanted tumors (Figure 6, Figure S4). Development of tumor‐specific effector CD8+ T cells to memory CD8+ T cells is important for preventing tumor relapse. Insufficient tumor eradication with PD‐1 blockade may induce re‐exhaustion of tumor‐specific T cells that have no ability to be reinvigorated during persistent antigen stimulation,36, 37 resulting in tumor recurrence. Thus, strong induction of CTL may be essential for the clearance of tumor antigens following functional memory T‐cell formation. Indeed, a recent paper demonstrated that vaccine immunotherapy with a mixture of long peptides derived from tumor neoantigens and poly(I:C) induced durable antitumor immunity and complete remission in melanoma patients.38 However, our experimental system does not exactly reflect a clinical tumor recurrence. Human tumors individually differ in their genomic background, rendering immune response to tumor variable. Thus, the preventive activity of ARNAX therapy on tumor recurrence should be individually investigated in patients.
In summary, we established models of different tumor types to identify characteristics that affect the efficacy of ARNAX vaccine therapy. Our data showed that tumor cell phenotypes, such as high MHC class I and TAA expression, low PD‐L1 expression and a low mutation rate could be advantageous for the success of ARNAX therapy. ARNAX vaccine therapy is effective for tumors with poor TAM and MDSC infiltration, and those with low PD‐L1 expression on tumor and/or immune cells. PD‐L1 blockade functions in both the priming and effector phases and enhanced the efficacy of ARNAX therapy. Strikingly, ARNAX established memory CD8+ T cells, which facilitates the prediction of applicable tumor types for ARNAX vaccine therapy.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We are grateful to Drs H. Takaki, and K. Funami in our laboratory for their valuable discussions, Dr H. Udono (Okayama University, Okayama, Japan) for providing MO5 cells, Dr T. Taniguchi (Tokyo University, Tokyo, Japan) for Ifnar −/− mice, Dr S. Akira (Osaka University, Osaka, Japan) for Tlr3 −/− and Myd88 −/− mice, and Drs M. Kasahara (Hokkaido University), M. Fukushima and H. Nishimura (Translational Research Informatics Center, Kobe) for their support of our study. This work was supported, in part, by a Grant‐in‐Aid for Research Activity Start‐Up under Grant number 17H06484 and Grants‐in‐Aid from the Drug Discovery Support Promotion Project from Japan Agency for Medical Research and Development (AMED) under Grant number 16nk0101327h0002. We had non‐profit support from Nobelpharma Co., Ltd (Tokyo, Japan) and AID Co., Ltd (Tokyo, Japan) through the university‐company contract, which we acknowledge gratefully.
Takeda Y, Yoshida S, Takashima K, et al. Vaccine immunotherapy with ARNAX induces tumor‐specific memory T cells and durable anti‐tumor immunity in mouse models. Cancer Sci. 2018;109:2119–2129. https://doi.org/10.1111/cas.13649
Funding information
Japan Agency for Medical Research and Development (Grant/Award Number: 16nk0101327h0002), Japan Society for the Promotion of Science (Grant/Award Number: 17H06484).
Contributor Information
Tsukasa Seya, Email: seya-tu@pop.med.hokudai.ac.jp.
Misako Matsumoto, Email: matumoto@pop.med.hokudai.ac.jp.
REFERENCES
- 1. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 2014;371:2189‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science. 2015;350:207‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang AC, Postow MA, Orlowski RJ, et al. T‐cell invigoration to tumour burden ratio associated with anti‐PD‐1 response. Nature. 2017;545:60‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69‐74. [DOI] [PubMed] [Google Scholar]
- 8. Brown SD, Warren RL, Gibb EA, et al. Neo‐antigens predicted by tumor genome meta‐analysis correlate with increased patient survival. Genome Res. 2014;24:743‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varn FS, Wang Y, Mullins DW, Fiering S, Cheng C. Systematic pan‐cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res. 2017;77:1271‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alsaab HO, Sau S, Alzhrani R, et al. PD‐1 and PD‐L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noguchi T, Ward JP, Gubin MM, et al. Temporally distinct PD‐L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. 2017;5:106‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lau J, Cheung J, Navarro A, et al. Tumour and host cell PD‐L1 is required to mediate suppression of anti‐tumour immunity in mice. Nat Commun. 2017;8:14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Juneja VR, McGuire KA, Manguso RT, et al. PD‐L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD‐L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology. 2017;6:e1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Curiel TJ, Wei S, Dong H, et al. Blockade of B7‐H1 improves myeloid dendritic cell‐mediated antitumor immunity. Nat Med. 2003;9:562‐567. [DOI] [PubMed] [Google Scholar]
- 18. Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD‐1 blockade. Science. 2016;354:1160‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsumoto M, Tatematsu M, Nishikawa F, et al. Defined TLR3‐specific adjuvant that induces NK and CTL activation without significant cytokine production in vivo. Nat Commun. 2015;6:6280. [DOI] [PubMed] [Google Scholar]
- 20. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3‐specific adjuvant relieves innate resistance to PD‐L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. 2017;19:1874‐1887. [DOI] [PubMed] [Google Scholar]
- 21. Akazawa T, Ebihara T, Okuno M, et al. Antitumor NK activation induced by the Toll‐like receptor 3‐TICAM‐1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci USA. 2007;104:252‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD‐L1 expression through 3′‐UTR disruption in multiple cancers. Nature. 2016;534:402‐406. [DOI] [PubMed] [Google Scholar]
- 23. Ryu MS, Woo MY, Kwon D, et al. Accumulation of cytolytic CD8+ T cells in B16‐melanoma and proliferation of mature T cells in TIS21‐knockout mice after T cell receptor stimulation. Exp Cell Res. 2014;327:209‐221. [DOI] [PubMed] [Google Scholar]
- 24. Yokouchi H, Chamoto K, Wakita D, et al. Tetramer‐blocking assay for defining antigen‐specific cytotoxic T lymphocytes using peptide‐MHC tetramer. Cancer Sci. 2006;97:148‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Šmahel M. PD‐1/PD‐L1 blockade therapy for tumors with downregulated MHC class I expression. Int J Mol Sci. 2017;18:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gabrilovich DI, Ostrand‐Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Achyut BR, Arbab AS. Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. Onco Targets Ther. 2016;9:1047‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hargadon KM. Tumor‐altered dendritic cell function: implications for anti‐tumor immunity. Front Immunol. 2013;4:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T‐cell memory. Blood. 2010;115:1678‐1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016;28:28‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Purushe J, Sun H, He S, Zhang Y. Transcriptional regulation of T cell heterogeneity and tumor immunity. J Immunol Res Ther. 2016;1:49‐62. [Google Scholar]
- 32. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD‐L1 checkpoint. Immunity. 2018;48:434‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. 2012;131:844‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta2‐microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88:100‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schietinger A, Philip M, Krisnawan VE, et al. Tumor‐specific T cell dysfunction is a dynamic antigen‐driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45:389‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials