

Abstract
Pancreatic cancer is known for its dismal prognosis despite efforts to improve therapeutic outcome. Recently, cancer nanomedicine, application of nanotechnology to cancer diagnosis and treatment, has gained interest for treatment of pancreatic cancer. The enhanced permeability and retention (EPR) effect that promotes selective accumulation of nanometer‐sized molecules within tumors is the theoretical rationale of treatment. However, it is clear that EPR may be insufficient in pancreatic cancer as a result of stromal barriers within the tumor microenvironment (TME). These limit intratumoral accumulation of macromolecules. The TME and stromal barriers inside it consist of various stromal cell types which interact both with each other and with tumor cells. We are only beginning to understand the complexities of the stromal barriers within the TME and its functional consequences for nanomedicine. Understanding the complex crosstalk between barrier stromal cells is challenging because of the difficulty of modeling pancreatic cancer TME. Here we provide an overview of stromal barriers within the TME. We also describe the preclinical models, both in vivo and in vitro, developed to study them. We furthermore discuss the critical gaps in our understanding, and how we might formulate a better strategy for using nanomedicine against pancreatic cancer.
Keywords: drug delivery, nanomedicine, pancreatic cancer, stromal barrier, tumor microenvironment
Abbreviations
- ABC
accelerated blood clearance
- CAST
cancer stroma targeting therapy
- EPR
enhanced permeability and retention
- GEMM
genetically engineered murine model
- PDGF
platelet‐derived growth factor
- PDX
patient‐derived xenograft
- PSC
pancreatic stellate cell
- TGF‐β
transforming growth factor‐β
- TME
tumor microenvironment
1. INTRODUCTION
Pancreatic cancer has a dismal prognosis despite intensive research over the last several decades.1 A recent development in treatment is approval of albumin‐bound nab‐paclitaxel in combination with gemcitabine. This prolongs median survival from 6.7 months (for gemcitabine alone) to 8.5 months.2 The basic assumption behind nab‐paclitaxel is the EPR effect, first proposed in 1986.3 The EPR hypothesis suggests that tumor neovasculature is immature with underdeveloped lymphatic drainage. This leads to increased leakage of macromolecules from blood vessels (enhanced permeability) and accumulation of leaked macromolecules (enhanced retention).
The EPR effect is the theoretical basis of cancer nanomedicine4 and its application to diagnosis and/or treatment. However, nanomedicine has yet to reach its full potential. It is becoming increasingly clear that a major hurdle is the existence of a heterogeneous TME5, 6 in which there exists complex tumor‐stromal crosstalk.7, 8 We therefore aim to provide an overview of the TME and its importance to nanomedicine efficacy in pancreatic cancer. Importantly, we will refer to the biological and physical obstacles that the TME poses to nanomedicine penetration of the tumor as “stromal barriers”. We also look at preclinical models of pancreatic cancer and stress the importance of developing clinically relevant models sufficiently recapitulating the characteristics of the TME to promote and accelerate studies on stromal barriers.
2. STROMAL BARRIERS TO DRUG DELIVERY WITHIN THE TME IN PANCREATIC CANCER
The TME consists of various stromal cell types,9 and these prevent penetration of nanotherapeutic agents into tumors, thus limiting efficacy.10 Stromal barriers consist both of the cells and their secreted products. For this reason, it is informative to analyze the stromal tissue architecture of the cancer in question. Pancreatic cancer is characterized by fibrosis, and a nanotherapeutic agent, given i.v., must first extravasate and pass through a thick, fibrous tissue to locate a tumor target (Figure 1).
Figure 1.
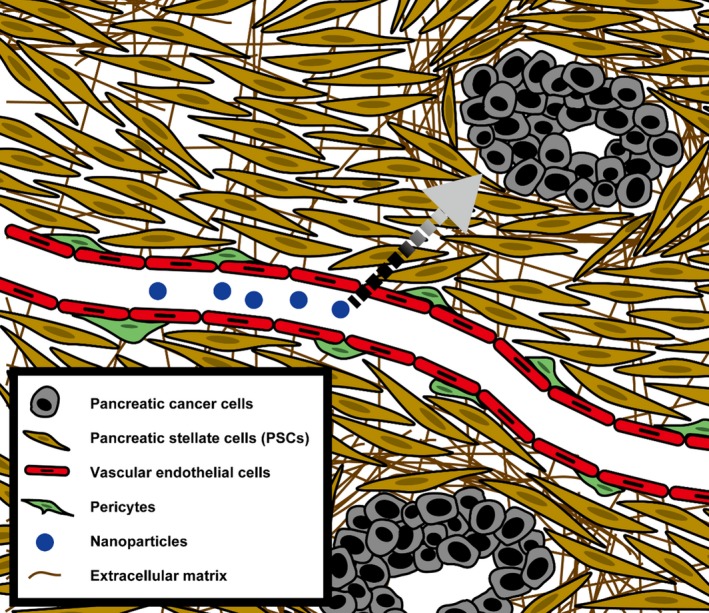
Pancreatic cancer microenvironment. The tumor microenvironment (TME) consists of numerous cell types and extracellular matrix, which collectively affect drug delivery. Pancreatic cancer is notably characterized by fibrosis separating cancer cells from blood vessels. The dotted arrow shows the path that an i.v. given nanoparticle must travel to reach cancer cells and achieve its effects
We have previously shown, in a murine BxPC‐3 xenograft model of pancreatic cancer, that pharmacological inhibition of TGF‐β signaling reduced pericyte coverage and increased intratumoral accumulation of nanotherapeutic agent. This improved efficacy11, 12: extending the known role of pericytes in physiological vessel stabilization13 to hindrance of nanoparticle extravasation. Furthermore, histopathological analyses of various human cancers showed that variable pericyte coverage correlated significantly with chemotherapeutic response. For example, we observed prominent pericyte coverage in pancreatic cancer in clear contrast to colon cancer. The latter cancer lacks pericyte coverage and is generally more responsive to chemotherapy.14, 15 We also found that while the CT26 colon carcinoma model with little pericyte coverage shows optimal intratumoral nanoparticle accumulation when treated with the angiogenesis inhibitor Sorafenib, the BxPC‐3 model responds only to TGF‐β inhibition.16 This suggests that vessel architecture, notably in relation to pericyte coverage, is an important determinant of nanotherapeutic efficacy. Thus, understanding and exploiting abnormalities in vessel architecture may lead to better accumulation of nanotherapeutic agents in pancreatic cancers (Figure 2).
Figure 2.
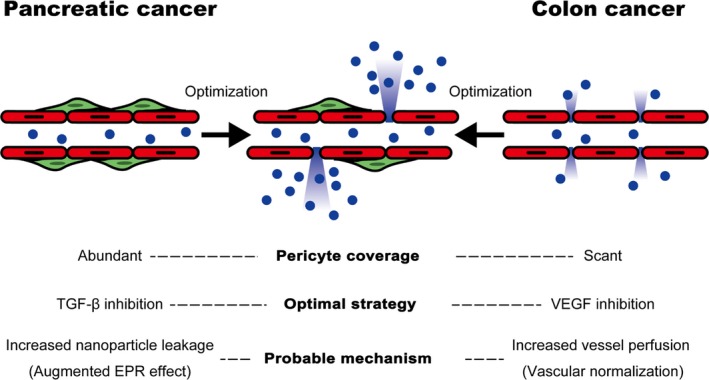
Pericyte coverage of intratumoral vessels affects vascular function and nanoparticle extravasation. Intratumoral vessels with varying levels of pericyte coverage show different profiles regarding nanoparticle extravasation: pancreatic cancer vessels have abundant pericyte coverage. Transforming growth factor beta (TGF‐β) inhibition reduces pericyte coverage and results in increased nanoparticle leakage possibly as a result of augmented enhanced permeability and retention (EPR) effect. Colon cancer, characterized by vessels with little pericyte coverage, is shown for comparison. Unlike pancreatic cancer, the optimal strategy to increase nanoparticle extravasation is vascular endothelial growth factor (VEGF) inhibition. This increases perfusion by normalization of the vasculature
Furthermore, Smith et al have reported varying responses to anti‐angiogenic agents in tumors relative to vascular and stromal architecture. They provide a conceptual framework to explain these variations,17 and suggest that tumors can be divided into those with a tumor‐vessel phenotype (blood vessels are distributed among cancer cells), and those with a stromal‐vessel phenotype (blood vessels are embedded within stroma surrounding cancer cells). Only the first type is responsive to anti‐angiogenic drugs. Pancreatic cancer, with its characteristic, desmoplastic morphology, is of the stromal‐vessel phenotype,18, 19 and thus requires an alternative targeting strategy (such as TGF‐β inhibition) to facilitate nanomedicine penetration of the tumor.
Furthermore, recent reports show that fibrotic stroma—consisting of PSC and secreted ECM components such as collagen and hyaluronan20, 21—also constitute a barrier to drug delivery.18, 22, 23, 24 Fibrosis is thus considered a target in improving drug delivery in pancreatic cancer.25 However, the way in which fibrotic elements block drug delivery is unclear: it may involve physical obstruction as a result of increased ECM deposition,24, 26, 27 decreased stromal vessel density,18, 28 and vessel compression and collapse 22, 23 (Figure 3).
Figure 3.
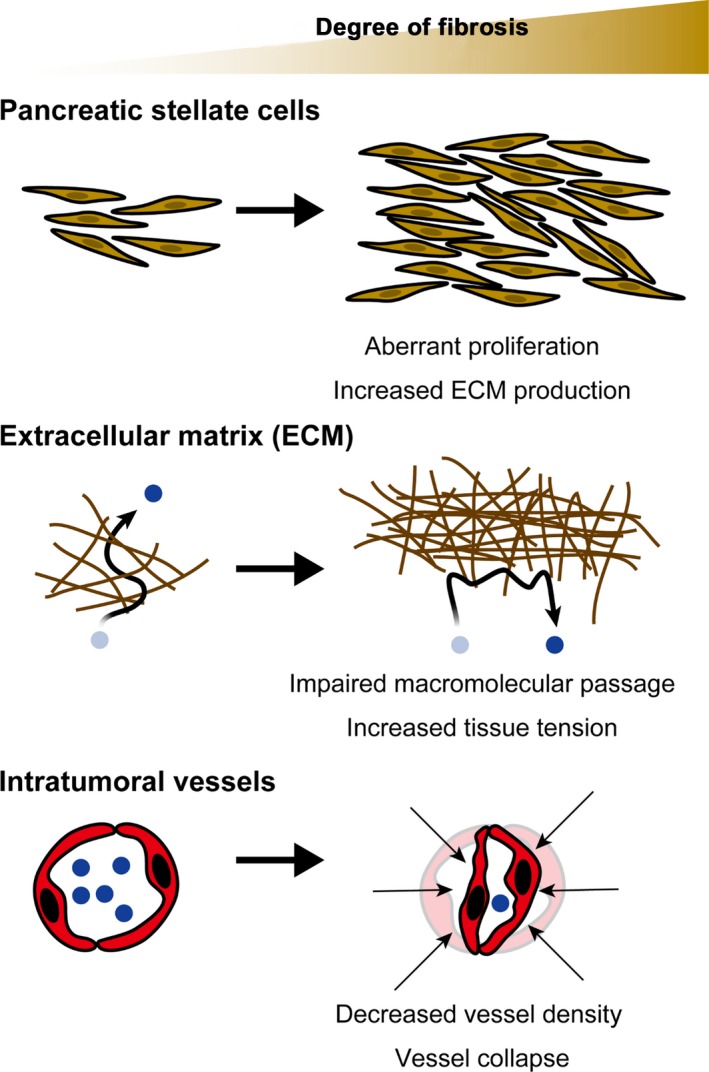
Effect of fibrosis on delivery of nanotherapeutics. Pancreatic cancer has prominent fibrosis. Pancreatic stellate cells are the main constituent of fibrosis in pancreatic cancer, and readily produce extracellular matrix (ECM) components such as collagen. The abundantly produced ECM is a physical barrier to macromolecules. It also increases tissue tension resulting in vascular compression and collapse, thus reducing intratumoral perfusion and nanotherapeutic delivery
3. PRECLINICAL IN VIVO MODELS OF PANCREATIC CANCER
The presence of stromal barriers within the TME of pancreatic cancer that limit therapeutic efficacy underscores the need for preclinical models that recapitulate the essential components of the TME. Here, and in the following section, we describe current models and the steps being taken towards development of new models. This topic has recently also been reviewed elsewhere.29, 30
The current way of demonstrating efficacy of a particular formulation of nanomedicine is in vivo, usually with mice. Murine pancreatic cancer models can be generally divided into cell‐line‐based xenograft models, GEMM, and PDX models (Table 1). Cell‐line‐based xenograft models are generated by inoculation of pancreatic cancer cell lines, and have been widely used, mostly for their relative ease of use. Although xenografts, generated by inoculation of the BxPC‐3 cell line, show prominent fibrosis—especially when given together with fibroblast growth factor‐2 as we have reported24—most cell‐line‐based xenografts fail to show appreciable levels of fibrosis. In contrast, GEMM, that rely on recapitulating mutations observed in human pancreatic cancer in genes such as Kras, in combination with Tp53, Cdkn2a, Smad4, and Tgfbr2, more closely mirror the histopathology of human pancreatic cancer.31, 32, 33, 34, 35 However, they usually take more time to fully develop and thus are more time‐consuming than cell‐line‐based xenografts. Another method, now gaining momentum, is the PDX model. This engrafts tumor specimens from patients. PDX models of pancreatic cancer reportedly retain and/or recapitulate many features of human disease,36, 37, 38, 39 and have enabled large‐scale screening in mice.40 However, engraftment efficiency is not uniform and has been associated with adverse clinicopathological features in the patient of origin, which may confound results.41
Table 1.
Frequently used animal models of pancreatic cancer
| Animal model | Advantages | Drawbacks |
|---|---|---|
| Cell‐line‐based xenografts |
Easy to perform, especially when s.c. inoculated Short experiment duration Comparison with in vitro results is intuitive |
Often lacks stroma Immunological involvement is difficult to assess because of use of immunodeficient mice Cell lines may not faithfully represent cancer cell population found in human primary tumors |
| Genetically engineered mouse models (GEMM) |
Recapitulates human histopathology well Well characterized and reproducible |
Expensive Time‐consuming |
| Patient‐derived xenograft (PDX) models |
Recapitulates human histopathology well Patient‐specific mechanisms may be addressed |
Engraftment efficiency is not high and may select for aggressive tumors Labor intensive Expensive Reproducibility may become a concern as a result of patient specificity |
4. PRECLINICAL IN VITRO MODELS OF PANCREATIC CANCER
An exciting new development is introduction of in vitro models with 3D culture techniques such as spheroid culture,42, 43 organotypic/organoid culture,44, 45, 46, 47 and layer‐by‐layer ECM nanofilm coating‐based culture19, 48 (Figure 4). The advantage, compared to conventional culture (ie, cells cultured 2D on plastic), is that it facilitates modeling of complex intercellular and/or cell‐ECM interactions.
Figure 4.
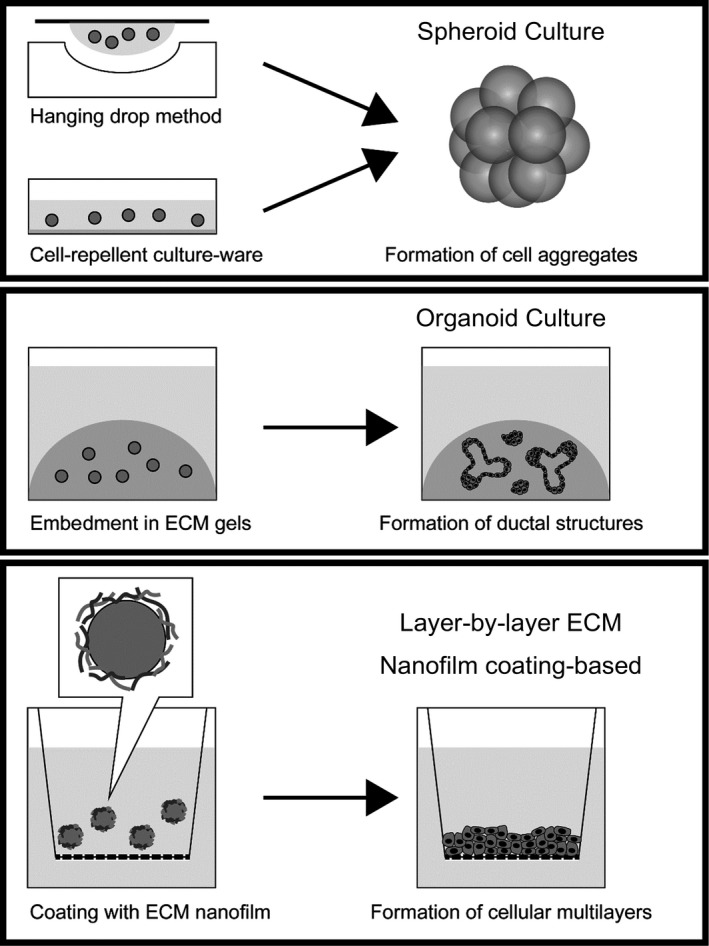
Various 3D culture methods. Spheroid culture is achieved through the hanging drop method or through use of cell‐repellent culture‐ware. Organoid culture is achieved by embedment of cells in extracellular matrix (ECM) gels. Layer‐by‐layer ECM nanofilm coating‐based culture is achieved by creating an ECM nanofilm on the surface of cells prior to cell seeding. Multiple cell types may be mixed in these 3D culture methods to generate in vitro models of pancreatic cancer tumor microenvironment. For simplicity, only a single cell type is depicted in this figure
Spheroid culture is usually made with cell‐repellent culture‐ware43 and/or the hanging drop method.42 In this way, 3D cultures of both pancreatic cancer cells and PSC have been generated, and used to analyze tumor‐stromal interaction and nanoparticle penetration. Organoid cultures use ECM gels in which cell lines or cells/tissues obtained from surgery or biopsy are embedded. These have been used both for analyses in vitro, and transplanted into mice to model the histopathology of human disease.44 We have used the layer‐by‐layer ECM nanofilm coating method, that sequentially builds a nanofilm of ECM components on the cell surface,49 to construct models of the desmoplastic reaction in pancreatic cancer and assess nanoparticle penetration of fibrotic tissue.19, 48 The technique also allows generation of open‐ended, vascular networks within 3D tissue, and may be used to analyze nanoparticle extravasation from the vasculature.50, 51
For the present, in vitro 3D tissue studies cannot fully replace in vivo studies. However, they will complement in vivo studies when molecular scale analysis is required and high spatiotemporal resolution may be cumbersome or challenging. They may also help interpret the complex crosstalk within the TME.
5. GAPS IN OUR UNDERSTANDING OF STROMAL BARRIERS: FUTURE DIRECTIONS
Here, we discuss major gaps in our understanding of stromal barriers within TME, and examine how to fill these. Although the EPR effect has been a useful guide to nanotherapeutics, our understanding of nanoparticle extravasation is limited. For example, we know that extravasation and subsequent intratumoral accumulation depend on nanoparticle size,12 but we do not know why. Furthermore, although the EPR effect suggests static pores within tumor neovessels, Stirland et al52 report that nanoparticles of the same size, injected at different times, do not colocalize. We also know that dynamic bursts, or nano‐eruptions, in tumor blood vessels lead to accumulation of nanoparticles within the tumor,53 but we do not know how nano‐eruptions occur. However, their existence does suggest that the static image of leaky tumor vessels conjured by the EPR hypothesis needs to be modified. Notably, with greater understanding of genotypic and phenotypic alterations in tumor endothelial cells,54, 55 the relationship between such alterations, and occurrence of nano‐eruptions, is a highly interesting question. The 3D models detailed above50, 51 may help us better understand this complexity.
Another important question concerns the role of fibrotic stroma in pancreatic cancer and how this may be therapeutically overcome. A number of reports show that simple ablation of fibrotic cells within pancreatic cancer results in disease progression.28, 56, 57 Thus, a more sophisticated strategy of reprogramming fibrotic cells into a tumor‐suppressive phenotype may be required.58, 59 This is consistent with genomic and transcriptomic analyses reporting molecularly distinct subtypes of pancreatic cancer,60, 61, 62 with distinct stromal expression signatures that serve as prognostic indices independent of tumor cells.63 Furthermore, we have found that pancreatic cancer patients with high positivity for PDGF receptor‐β in stroma have a worse prognosis.64 Although studies assessing the importance of these distinct subtypes to nanomedicine have not yet been carried out, it may be a good approach, given increased expression of collagen proteins in those “activated” stromal subtypes with worse prognosis63 and our knowledge of the role of PDGF signaling in fibrosis.65 However, Laklai et al66 have also emphasized the importance of tissue tension in pancreatic cancer prognosis. Experimental conditions with varying levels of fibrosis, which presumably demonstrate different tissue tension as well, must thus be compared with caution. Indeed, mechanical forces, such as interstitial fluid pressure and tissue tension, are not easily manipulated experimentally as independent variables and consequently remain largely under‐studied.
The long‐term safety and efficacy of nanomedicine is also an urgent issue. For example, nanotherapeutic formulations often use an outer coating of PEG to increase biocompatibility, but PEGylated nanoparticles are more rapidly cleared after repeated injections: a process known as ABC.67, 68 The ABC phenomenon is immunological—caused by generation of IgM antibodies—and may be clinically problematic in multiple dose treatment.69 Furthermore, research on cobalt‐chromium nanoparticles indicates possible DNA damage propagated across cellular barriers.70, 71 Therefore, immunogenicity and mutagenicity of nanoparticles, the mechanisms by which they develop, and their biological/clinical consequences for the patient, are all topics that require further study.
Finally, in addition to passive targeting through the EPR effect, the development of active targeting by specific markers within TME for intratumoral accumulation of nanotherapeutics also requires study.72, 73 A number of candidate targets for pancreatic cancer is under consideration and has been reviewed elsewhere.74 Furthermore, uptake and intracellular behavior of nanoparticles by individual cells must also be tailored to prevent premature degradation and to facilitate optimal therapeutic effect (a concept known as “subcellular targeting”).75, 76 However, as with passive targeting by EPR, stromal barriers within TME will likely hinder passage of actively targeted nanotherapeutics. This is especially the case following antibody conjugation for active targeting, which further increases the size of nanotherapeutic formulation. Matsumura recently proposed the CAST strategy to circumvent this difficulty. Using antibodies, it first targets extracellular components, such as collagen IV, insoluble fibrin, and tissue factor, within stroma to make a scaffold from which conjugated low‐molecular cytotoxic agents can be released and diffuse freely.27
6. CONCLUSIONS
The existence of stromal barriers within TME which limit therapeutic efficacy of nanomedicine is now clearly established. However, how these barriers develop and how they hinder therapy is far from clear. Both increased knowledge of the complex crosstalk within TME, and preclinical models that accurately show the complexity of TME, in vivo and in vitro, are needed to advance research and treatment of pancreatic cancer.
CONFLICT OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
The authors would like to thank and acknowledge their debt to past and current lab members and collaborators. The authors also thank Michael W. Miller for editorial work. This work was financially supported by Okayama University, Health Labour Sciences Research Grant, the Japan Society for the Promotion of Science (JSPS) through the “Funding Program for World‐Leading Innovative R&D on Science and Technology (FIRST Program)” initiated by the Council for Science and Technology Policy (CSTP) and Grant‐in‐Aid for Scientific Research (KAKENHI; 17016011, 19790282, 23790433, and 26293119), Japan Foundation for Applied Enzymology, Kato Memorial Bioscience Foundation, the Pancreas Research Foundation, and the Yasuda Medical Foundation.
Tanaka HY, Kano MR. Stromal barriers to nanomedicine penetration in the pancreatic tumor microenvironment. Cancer Sci. 2018;109:2085–2092. https://doi.org/10.1111/cas.13630
Funding information
Okayama University, Health Labour Sciences Research Grant, Kato Memorial Bioscience Foundation, Pancreas Research Foundation, Japan Foundation for Applied Enzymology, Yasuda Memorial Medical Foundation, Japan Society for the Promotion of Science (FIRST Program, ‘KAKENHI 17016011, 19790282, 23790433, and 26293119’)
REFERENCES
- 1. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039‐1049. [DOI] [PubMed] [Google Scholar]
- 2. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387‐6392. [PubMed] [Google Scholar]
- 4. Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25‐37. [DOI] [PubMed] [Google Scholar]
- 5. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2016;17:20‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 8. Tape CJ, Ling S, Dimitriadi M, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell. 2016;165:910‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591‐5596. [DOI] [PubMed] [Google Scholar]
- 10. Kano MR. Nanotechnology and tumor microcirculation. Adv Drug Deliv Rev. 2014;74:2‐11. [DOI] [PubMed] [Google Scholar]
- 11. Kano MR, Bae Y, Iwata C, et al. Improvement of cancer‐targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF‐beta signaling. Proc Natl Acad Sci USA. 2007;104:3460‐3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub‐100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815‐823. [DOI] [PubMed] [Google Scholar]
- 13. Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193‐215. [DOI] [PubMed] [Google Scholar]
- 14. Zhang L, Nishihara H, Kano MR. Pericyte‐coverage of human tumor vasculature and nanoparticle permeability. Biol Pharm Bull. 2012;35:761‐766. [DOI] [PubMed] [Google Scholar]
- 15. Kamei R, Tanaka HY, Kawano T, et al. Regulation of endothelial Fas expression as a mechanism of promotion of vascular integrity by mural cells in tumors. Cancer Sci. 2017;108:1080‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kano MR, Komuta Y, Iwata C, et al. Comparison of the effects of the kinase inhibitors imatinib, sorafenib, and transforming growth factor‐β receptor inhibitor on extravasation of nanoparticles from neovasculature. Cancer Sci. 2009;100:173‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith NR, Baker D, Farren M, et al. Tumor stromal architecture can define the intrinsic tumor response to VEGF‐targeted therapy. Clin Cancer Res. 2013;19:6943‐6956. [DOI] [PubMed] [Google Scholar]
- 18. Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hosoya H, Kadowaki K, Matsusaki M, et al. Engineering fibrotic tissue in pancreatic cancer: a novel three‐dimensional model to investigate nanoparticle delivery. Biochem Biophys Res Commun. 2012;419:32‐37. [DOI] [PubMed] [Google Scholar]
- 20. Erkan M, Adler G, Apte MV, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61:172‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144:1210‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakai S, Iwata C, Tanaka HY, et al. Increased fibrosis and impaired intratumoral accumulation of macromolecules in a murine model of pancreatic cancer co‐administered with FGF‐2. J Control Release. 2016;230:109‐115. [DOI] [PubMed] [Google Scholar]
- 25. Erkan M, Hausmann S, Michalski CW, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9:454‐467. [DOI] [PubMed] [Google Scholar]
- 26. Diop‐Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA. 2011;108:2909‐2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsumura Y. Cancer stromal targeting (CAST) therapy. Adv Drug Deliv Rev. 2012;64:710‐719. [DOI] [PubMed] [Google Scholar]
- 28. Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hwang C‐I, Boj SF, Clevers H, Tuveson DA. Preclinical models of pancreatic ductal adenocarcinoma. J Pathol. 2016;238:197‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feigin ME, Tuveson DA. Challenges and opportunities in modeling pancreatic cancer. Cold Spring Harb Symp Quant Biol. 2016;81:231‐235. [DOI] [PubMed] [Google Scholar]
- 31. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469‐483. [DOI] [PubMed] [Google Scholar]
- 32. Izeradjene K, Combs C, Best M, et al. KrasG12D and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11:229‐243. [DOI] [PubMed] [Google Scholar]
- 33. Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16Ink4a and the p19Arf‐p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947‐5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437‐450. [DOI] [PubMed] [Google Scholar]
- 35. Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas‐specific blockade of transforming growth factor‐β signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147‐3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roife D, Dai B, Kang Y, et al. Ex vivo testing of patient‐derived xenografts mirrors the clinical outcome of patients with pancreatic ductal adenocarcinoma. Clin Cancer Res. 2016;22:6021‐6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Go KL, Delitto D, Judge SM, et al. Orthotopic patient‐derived pancreatic cancer xenografts engraft into the pancreatic parenchyma, metastasize, and induce muscle wasting to recapitulate the human disease. Pancreas. 2017;46:813‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delitto D, Pham K, Vlada AC, et al. Patient‐derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. Am J Pathol. 2015;185:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martinez‐Garcia R, Juan D, Rausell A, et al. Transcriptional dissection of pancreatic tumors engrafted in mice. Genome Med. 2014;6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao H, Korn JM, Ferretti S, et al. High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318‐1325. [DOI] [PubMed] [Google Scholar]
- 41. Pergolini I, Morales‐Oyarvide V, Mino‐Kenudson M, et al. Tumor engraftment in patient‐derived xenografts of pancreatic ductal adenocarcinoma is associated with adverse clinicopathological features and poor survival. PLoS ONE. 2017;12:e0182855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ware MJ, Keshishian V, Law JJ, et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials. 2016;108:129‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Priwitaningrum DL, Blondé J‐BG, Sridhar A, et al. Tumor stroma‐containing 3D spheroid arrays: a tool to study nanoparticle penetration. J Control Release. 2016;244:257‐268. [DOI] [PubMed] [Google Scholar]
- 44. Boj SF, Hwang C‐I, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Di Maggio F, Arumugam P, Delvecchio FR, et al. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology. 2016;16:995‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carapuça EF, Gemenetzidis E, Feig C, et al. Anti‐stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J Pathol. 2016;239:286‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Öhlund D, Handly‐Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsusaki M, Komeda M, Mura S, et al. Desmoplastic reaction in 3D‐pancreatic cancer tissues suppresses molecular permeability. Adv Healthc Mater. 2017;6:1700057. [DOI] [PubMed] [Google Scholar]
- 49. Matsusaki M, Case CP, Akashi M. Three‐dimensional cell culture technique and pathophysiology. Adv Drug Deliv Rev. 2014;74:95‐103. [DOI] [PubMed] [Google Scholar]
- 50. Hikimoto D, Nishiguchi A, Matsusaki M, Akashi M. High‐throughput blood‐ and lymph‐capillaries with open‐ended pores which allow the transport of drugs and cells. Adv Healthc Mater. 2016;5:1969‐1978. [DOI] [PubMed] [Google Scholar]
- 51. Chetprayoon P, Matsusaki M, Yokoyama U, Tejima T, Ishikawa Y, Akashi M. Use of three‐dimensional arterial models to predict the in vivo behavior of nanoparticles for drug delivery. Angew Chem Int Ed Engl. 2016;55:4461‐4466. [DOI] [PubMed] [Google Scholar]
- 52. Stirland DL, Matsumoto Y, Toh K, Kataoka K, Bae YH. Analyzing spatiotemporal distribution of uniquely fluorescent nanoparticles in xenograft tumors. J Control Release. 2016;227:38‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsumoto Y, Nichols JW, Toh K, et al. Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery. Nat Nanotechnol. 2016;11:533‐538. [DOI] [PubMed] [Google Scholar]
- 54. Hida K, Ohga N, Akiyama K, Maishi N, Hida Y. Heterogeneity of tumor endothelial cells. Cancer Sci. 2013;104:1391‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2:a006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Özdemir BC, Pentcheva‐Hoang T, Carstens JL, et al. Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee JJ, Perera RM, Wang H, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci USA. 2014;111:E3091‐E3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor‐mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chronopoulos A, Robinson B, Sarper M, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47‐52. [DOI] [PubMed] [Google Scholar]
- 62. Waddell N, Pajic M, Patch A‐M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor‐ and stroma‐specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuzawa S, Kano MR, Einama T, Nishihara H. PDGFRβ expression in tumor stroma of pancreatic adenocarcinoma as a reliable prognostic marker. Med Oncol. 2012;29:2824‐2830. [DOI] [PubMed] [Google Scholar]
- 65. Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255‐273. [DOI] [PubMed] [Google Scholar]
- 66. Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dams ET, Laverman P, Oyen WJ, et al. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. 2000;292:1071‐1079. [PubMed] [Google Scholar]
- 68. Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J Control Release. 2003;88:35‐42. [DOI] [PubMed] [Google Scholar]
- 69. Ishida T, Ichihara M, Wang X, Kiwada H. Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J Control Release. 2006;115:243‐250. [DOI] [PubMed] [Google Scholar]
- 70. Bhabra G, Sood A, Fisher B, et al. Nanoparticles can cause DNA damage across a cellular barrier. Nat Nanotechnol. 2009;4:876‐883. [DOI] [PubMed] [Google Scholar]
- 71. Sood A, Salih S, Roh D, et al. Signalling of DNA damage and cytokines across cell barriers exposed to nanoparticles depends on barrier thickness. Nat Nanotechnol. 2011;6:824‐833. [DOI] [PubMed] [Google Scholar]
- 72. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti‐cancer drug delivery. J Control Release. 2010;148:135‐146. [DOI] [PubMed] [Google Scholar]
- 74. Yu X, Zhang Y, Chen C, Yao Q, Li M. Targeted drug delivery in pancreatic cancer. Biochim Biophys Acta. 2010;1805:97‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rajendran L, Knölker H‐J, Simons K. Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov. 2010;9:29‐42. [DOI] [PubMed] [Google Scholar]
- 76. Murakami M, Cabral H, Matsumoto Y, et al. Improving drug potency and efficacy by nanocarrier‐mediated subcellular targeting. Sci Transl Med. 2011;3:64ra2. [DOI] [PubMed] [Google Scholar]