

Abstract
Ninety percent of hepatocarcinoma (HCC) develops in a chronically damaged liver. Interactions between non‐tumor stromal components, especially macrophages, and cancer cells are still incompletely understood. Our aim was to determine whether a chronically injured liver represents a favorable environment for the seeding and growth of HCC cells, and to evaluate the potential roles of macrophages infiltrated within the tumor. HCC cells were injected into the liver in healthy mice (healthy liver group [HL]) and in mice chronically treated with carbon tetrachloride (CCl4) for 7 weeks (CCl4 7w group). Livers were examined for the presence of tumor 2 weeks post‐injection. Tumor and non‐tumor tissues were analyzed for macrophage infiltration, origin (monocytes‐derived vs resident macrophages) and polarization state, and MMP production. Fifty‐three percent of mice developed neoplastic lesion in the HL group whereas a tumor lesion was found in all livers in the CCl4 7w group. Macrophages infiltrated more deeply the tumors of the CCl4 7w group. Evaluation of factors involved in the recruitment of macrophages and of markers of their polarization state was in favor of prominent infiltration of M2 pro‐tumor monocyte‐derived macrophages inside the tumors developing in a chronically injured liver. MMP‐2 and ‐9 production, attributed to M2 pro‐tumor macrophages, was significantly higher in the tumors of the CCl4 7w group. In our model, chronic liver damage promotes cancer development. Our results suggest that an injured background favors the infiltration of M2 pro‐tumor monocyte‐derived macrophages. These secrete MMP‐2 and MMP‐9 that promote tumor progression.
Keywords: hepatocarcinoma, inflammation, liver fibrosis, macrophage, matrix metalloproteinase
Abbreviations
- Arg‐1
Arginase‐1
- CCL17
chemokine (C‐C motif) ligand 17
- CCL2
chemokine (C‐C motif) ligand 2
- CCl4
carbon tetrachloride
- CCL5
chemokine (C‐C motif) ligand 5
- HCC
hepatocellular carcinoma
- IL‐6
interleukin 6
- LW/BW
liver to body weight ratio
- M‐CSF
macrophage colony stimulating factor
- MR
Mannose receptor
- MT1‐MMP
membrane type 1 metalloproteinase
- NASH
non‐alcoholic steatohepatitis
- SR
Scavenger receptor
- TAM
tumor‐associated macrophage
- TGF‐β1
transforming growth factor beta 1
- TV
tumor volume
1. INTRODUCTION
Hepatocellular carcinoma (HCC) represents the main form of primary liver cancer.1 Worldwide incidence is increasing mainly as a result of increased prevalence of advanced hepatitis C infection and NASH‐related cirrhosis.2 Despite major efforts to better understand the mechanisms involved in the development and progression of HCC, the prognosis is still poor.
As in many other cancers, complex interactions exist between hepatic tumor cells and the non‐tumor stroma. The latter comprises non‐malignant cells such as immune cells, endothelial cells and fibroblasts, components of the ECM, and soluble factors.3 This dynamic system the distribution and composition of which vary in space and time is defined as the peritumoral microenvironment. This notion is of particular interest in the field of HCC as more than 90% of liver cancers develop in chronically damaged tissue characterized by hepatocellular injury, inflammation and fibrosis and defining per se a pathological environment, supportive for the initiation and progression of HCC.3, 4 However, the nature of the interactions between stromal components and the cancer cells that benefit cancer growth are still incompletely understood.
Tumor‐associated macrophages are important components of the tumor microenvironment.5, 6 They may be tissue‐resident or bone‐marrow‐derived cells recruited from the bloodstream. Once located inside the tumor, macrophages undergo final differentiation depending on the environmental stimuli.7 Two polarization states of macrophages are described, the classically activated (M1) and the alternatively activated (M2) phenotypes. M2‐polarized macrophages are globally recognized as pro‐tumor macrophages.8, 9, 10 Although largely used, this dichotomous classification based on the expression of specific markers is likely too simplistic as it does not reflect cellular functions. As a consequence, both expression of markers and cellular functions have to be considered when describing tumor‐associated macrophage (TAM).
One of the pro‐tumor functions attributed to M2‐polarized TAM is the production of MMP. MMP are able to degrade virtually all compounds of the ECM but also to activate other MMP and growth factors by proteolysis.11 Besides their involvement in tissue remodeling, MMP are implicated in various physiological and pathological processes such as organ development and involution, inflammatory response, wound healing or cancer development and progression.12, 13
Among other MMP, MMP‐2, MMP‐9 and MT1‐MMP are frequently overexpressed in human tumors.14 MMP‐2 and MMP‐9 are gelatinases that cleave several types of collagen whereas MT1‐MMP contributes to the activation of pro‐MMP‐2. They are produced by a variety of cell types, including inflammatory cells, endothelial cells and fibroblasts.12, 13 Several studies have described the expression and/or activity of MMP‐2, ‐9 and MT1‐MMP in human HCC and established a link with aggressive behavior of liver cancer.15, 16, 17, 18, 19, 20, 21
The aim of the present study was to determine whether a chronically injured liver represents a favorable environment for liver tumor cell seeding and growth and to evaluate the potential roles of macrophage infiltration in the tumors.
2. MATERIALS AND METHODS
2.1. Mice and cell line
Seven‐week‐old male C57Bl/6 mice were purchased from Elevage Janvier (Le Genest‐Saint‐Isle, France). They were exposed to a 12‐hour light/12‐hour dark cycle, maintained at a constant temperature of 20‐22°C and received food and water ad libitum. Animal care was provided in accordance with the guidelines for humane care for laboratory animals established by the Université Catholique de Louvain in accordance with European regulations and in conformity with ARRIVE guidelines. The study protocol was approved by the university ethics committee (2012UCLMD026).
The murine Hepa 1‐6 cell line derives from the C57Bl/6 mouse hepatoma lesion and was obtained from the European Collection of Authenticated Cell Culture (ECACC, Salisbury, UK). Cells were maintained in DMEM high glucose, supplemented with 10% heat inactivated FBS, 1% penicillin and streptomycin, and 1% glutamine in 5% CO2 at 37°C. Confluent Hepa 1‐6 cells were collected by trypsinization (trypsin‐EDTA; Life Technologies, Inc.). After counting, the cells were suspended in PBS and kept on ice before intrahepatic injection.
2.2. In situ tumor model
Tumors were generated as previously described.22 Briefly, 1 × 106 Hepa 1‐6 cells suspended in 100 μL PBS were slowly injected directly into the left lobe of the liver, both in healthy mice (healthy liver group [HL]) and in mice treated with carbon tetrachloride (CCl4) for 7 weeks (injection of HCC cells 48 hours after the last CCl4 injection [CCl4 7w group]). As the number of mice developing a tumor in the HL group was expected to be low,23 15 mice were included in this group whereas 10 mice were included in the CCl4 7w group. In the latter, 1 mouse died during the induction of chronic liver injury and 1 mouse died within hours following injection of HCC cells.
All mice were killed 14 days after HCC cell injection. At the time of death, the liver was rapidly removed and examined for the presence of tumor (whitish nodule ≥ 1 mm). Tumor burden was evaluated by TV calculated as follows: TV (mm3) = (length × width2)/2 and by LW/BW.24 A piece of each tumor as well as of the non‐tumor parenchyma was immediately immersed in 4% formalin for histological analysis and the rest was snap‐frozen in liquid nitrogen and kept at −80°C until analysis.
2.3. Carbon tetrachloride‐induced liver fibrosis
Before tumor cell intrahepatic injection, a fibrotic regimen (ip, injection 800 μL/kg body weight of CCl4 diluted in corn oil, 3 times a week for 7 weeks) was applied to mice of the CCl4 7w group. To validate the CCl4‐induced chronic liver injury model, 6 mice were injected with CCl4 for 7 weeks and killed 48 hours after the last CCl4 injection, timing also chosen for the HCC cell injection in the CCl4 7w group as previously mentioned (Figure 1A). Histology and mRNA concentration in liver from mice injected with CCl4 and killed 48 hours after the last injection were analyzed and compared with data of healthy mice killed at the same time (n = 6).
Figure 1.
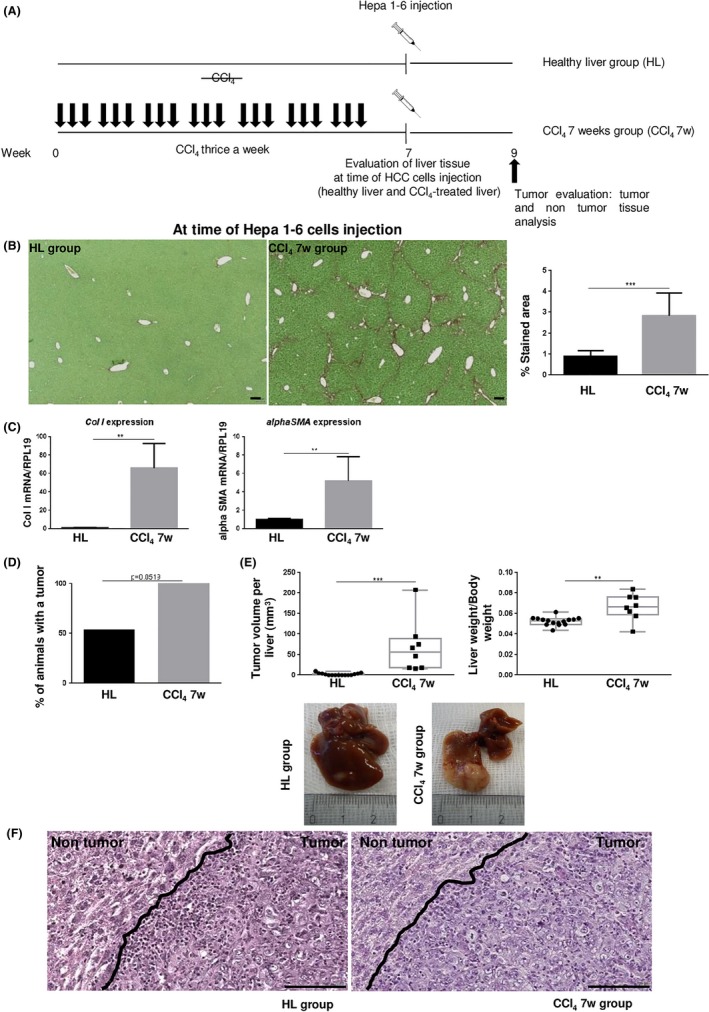
A, Comparison of hepatocellular carcinoma (HCC) cell growth when Hepa 1‐6 cells were injected into a healthy liver (HL group) and in mice pretreated with carbon tetrachloride (CCl4) for 7 wk (CCl4 7w group). B, Sirius red‐stained liver sections in CCl4‐treated (at time of HCC cell injection) and non‐treated mice. Scale bar, 100 μm. Collagen fibers were evaluated as percentage of stained area in the section (mean ± SD). C, Hepatic gene expression of Collagen I (Col I) and alphaSma (mean ± SD) at time of HCC cell injection. D,E, Percentage of animals with a tumor was calculated in the HL and CCl4 7w groups and tumor burden was evaluated by total tumor volume per liver (mm3) and the liver to body weight ratio. F, Representative H&E‐stained tumor and non‐tumor tissue sections. Scale bar, 100 μm. **P < .01; ***P < .001
2.4. Histology
Formalin‐fixed and paraffin‐embedded tissue was cut in 5‐μm‐thick sections. Each macroscopic tumor lesion was microscopically analyzed by H&E staining to confirm the tumor diagnosis.
Liver fibrosis quantification on non‐tumor parenchyma (percentage of stained area in the section) was carried out on Sirius red‐stained tissue using Tissue IA software (Leica Biosystems, Dublin, Ireland) after digitalization with a SCN400 slide scanner (Leica Biosystems, Wetzlar, Germany).
Macrophages were identified by F4/80 immunostaining using a primary rat anti‐mouse F4/80 monoclonal Ab (1:200, MCA497G, Clone A3‐1; AbD Serotec, Oxford, UK), a rabbit anti‐rat immunoglobulin (1:100, AI‐4001; Vector Laboratories, Burlingame, CA, USA), and then a goat anti‐rabbit streptavidin HRP‐conjugated Ab (En Vision K4003; Dako, Glostrup, Denmark). Peroxidase activity was seen with diaminobenzidine (DAB) and slides counterstained with hematoxylin.
2.5. RNA extraction, reverse transcription, RT‐qPCR
Total RNA was extracted from frozen liver samples using TRIzol Isolation Reagent (Life Technologies, Belgium). cDNA was synthesized from 1 μg RNA using a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Lennik, Belgium). Real‐time PCR analysis was carried out in duplicate with the StepOnePlus real‐time PCR System (Applied Biosystems) using SYBR green. Primer pairs for transcripts of interest were designed using primer express design software (Applied Biosystems) and are listed in Table 1. RPL19 mRNA was chosen as an invariant standard. Results are expressed as fold expression relative to expression in the control group using the ∆∆Ct method.
Table 1.
Sequences of primer pairs for transcripts of interest
| Gene | Forward | Reverse |
|---|---|---|
| Collagen I | TTCACCTACAGCACGCTTGT | TCTTGGTGGTTTTGTATTCGATGA |
| Alpha Sma | TCCTGACGCTGAAGTATCCGATA | GGTGCCAGATCTTTTCCATGTC |
| F4/80 | GATGAATTCCCGTGTTGTTGGT | ACATCAGTGTTCCAGGAGACACA |
| Ccl2 | CCACTCACCTGC TGCTACTCAT | CTGCTGGT GATCCTCTTGT |
| Mcsf | CAGCTGCTTCACCAAGGACT | TCATGGAAAGTTCGGACACA |
| Ccl5 | TCCAATCTTGCAGTCGTGTTTG | TCTGGGTTGGCACACACTTG |
| CD11b | GGGTCATTCGCTACGTAATTGG | TGTTCACCAGCTGGCTTAGATG |
| Fizz‐1 | ACTGCCTGTGCTTACTCGTTGA | TCCACCTCTTCATTCTTAGGACAGTT |
| Ym‐1 | GTCTGGCCCCTGGACATG | AGAGGGAAATGTCTCTGGTGACA |
| Scavenger receptor | CAGGTGTTATCTGCTCCGAGTTC | CCATGTACCATTGTAAAACACTTCAA |
| Mannose receptor | CTCGTGGATCTCCGTGACAC | GCAAATGGAGCCGTCTGTGC |
| Arg‐1 | CATGGGCAACCTGTGTCCTT | TCCTGGTACATCTGGGAACTTTC |
| Il6 | CTGCAA GAGACTTCCATCCAGTT | GAAG TAGGGAAGGC CGTGG |
| Ccl17 | AGTGCTGCCTGGATTACTTCAAAG | CTGGACAGTCAGAAACACGATGG |
| Il4 | GGAGATGGATGTGCCAAACG | CGAGCTCACTCTCTGTGGTGTT |
| Tgf beta1 | CCTGCAAGA CCATCGACATG | GAGCCTTAGTTTGGACAGGATCTG |
| Mmp9 | TGAATCATAGAGGAAGCCCATTACA | CGGAGTCCAGCGTTGCA |
| Mmp2 | GGACCCCGGTTTCCCTAA | CAGGTTATCAGGGATGGCATTC |
| Mt1‐mmp | TTTGGGCTTATCTGGGACAGA | AGTCAGGGTCACCCACAAAGA |
2.6. Transforming growth factor beta 1 determination
Free and total TGF‐β1 were quantitated in tumor parenchyma by ELISA as previously described.25, 26 Liver homogenates were prepared as described below. To assess the amount of total TGF‐β1, acid activation was carried out to release free TGF‐β1 from latent complex. Protein lysate (40 μL, equivalent to 200 μg protein) was acidified with 10 μL of 1N HCl at room temperature for 10 minutes, then neutralized with 10 μL of 1.2N NaOH/0.5 mol/L HEPES. Diluent reagent was added to both acid‐activated and native samples to a final dilution of 1:5. ELISA was carried out using TGF‐β1 Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA) following the manufacturer instructions.
2.7. Protein preparation and gelatin zymography
Frozen liver tissue samples were homogenized in ice‐cold lysis buffer (50 mmol/L HEPES, 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 10% glycerol, 0.1% Triton X‐100, 1 mmol/L DTT, 1 mmol/L NaF, 1 mmol/L PMSF, 0.1 mmol/L Na3VO4, 2 μg/mL aprotinin, 100 μg/mL leupeptin). The homogenates were centrifuged at 10 000 g for 5 minutes at 4°C and the supernatant was stored at −80°C. Protein content was measured using BSA as reference.
Latent and active forms of MMP‐2 and ‐9 were analyzed by gelatin‐substrate zymography carried out as described previously.27 Briefly, 15 μg protein samples were electrophoresed in an 8% polyacrylamide gel embedded with 500 μg/mL gelatin. After electrophoresis, gels were washed thrice in 2.5% Triton X‐100 for 20 minutes. Then, the gels were incubated overnight at 37°C in 50 mmol/L Tris‐HCl, pH 7.5, 5 mmol/L CaCl2, 0.02% NaN3, 1% Triton X‐100, 1 μmol/L ZnCl2, followed by Coomassie blue staining and destaining. MMP activities were quantified by gel densitometry. The activities were normalized according to those contained in a culture medium conditioned by human endometrial explants, used as standard in each gel.28
2.8. Statistical analysis
Data are presented as mean ± standard deviation (SD) in bar graphs, and median, 25th and 75th percentiles, minimum, and maximum in box and whisker graphs. Statistical analysis was done using Fischer's exact test for comparison of tumor development frequencies, non‐parametric Mann‐Whitney U‐test for simple comparison and 1‐way or 2‐way ANOVA for repeated measures followed by Bonferroni's post‐hoc correction. Statistical significance was assumed for P‐values <.05 (*P < .05; **P < .01; ***P < .001). GraphPad Prism software (San Diego, CA, USA) was used for graphs and statistics. IBM SPSS software (Armonk, NY, USA) was used for linear regression statistical analysis.
3. RESULTS
3.1. Chronic liver injury promotes HCC cell seeding and growth
We first asked the question as to whether chronic liver injury favors HCC cell seeding and growth. To induce a hepatic chronic injured state, we used the well‐known CCl4‐induced liver fibrosis model (Figure 1A). Mice were injected with CCl4 for 7 weeks. Animals killed 2 days after the last CCl4 injection showed significant liver fibrosis compared to animals not injected with CCl4 as evaluated by Sirius red quantification (Figure 1B). Moreover, Col I and alphaSma mRNA was significantly more expressed in CCl4‐treated mice compared to the healthy animals, confirming the activation of fibrogenesis (Figure 1C).
We compared tumor growth when Hepa 1‐6 cells were injected in a healthy liver (HL group) and when HCC cells were injected in mice pretreated with CCl4 for 7 weeks (CCl4 7w group). In both groups, liver tumor was evaluated 2 weeks after neoplastic cell injection (Figure 1A). In the HL group, 53% of mice (8/15) developed a neoplastic lesion whereas in the CCl4 7w group, a tumor lesion was found in all livers (8/8) (P = .0519) (Figure 1D). Total TV per liver and LW/BW calculation indicated that tumor burden was significantly higher in the CCl4 7w group compared to the HL group (Figure 1E). These results support that chronic liver injury favors cancer growth. Histological examination analysis of the tumor lesions showed similar aspects in both groups characterized by a non‐trabecular pattern of tumor cells, rather organization in clusters, and a peripheral inflammatory ring, surrounding each tumor lesion (Figure 1F).
3.2. Chronic liver injury promotes tumor infiltration by recruited macrophages
We used F4/80 immunohistochemistry and gene expression to assess tumor macrophage infiltration. In both groups, F4/80‐positive cells were similarly concentrated around the lesion in a tumor peripheral inflammatory ring. In contrast, although only a few F4/80‐positive cells were observed inside the cancer nodule in the HL group, clusters of tumor cells were surrounded by macrophages deeply infiltrating the neoplastic lesion in the CCl4 7w group (Figure 2A). Prominent tumor macrophage infiltration was confirmed by higher F4/80 mRNA expression in tumors of the CCl4 7w group (Figure 2B). These results show different macrophage behavior towards the tumor whether it develops on a naïve or a chronically wounded liver, which may then suggest distinct macrophage subpopulations inside the tumor.
Figure 2.
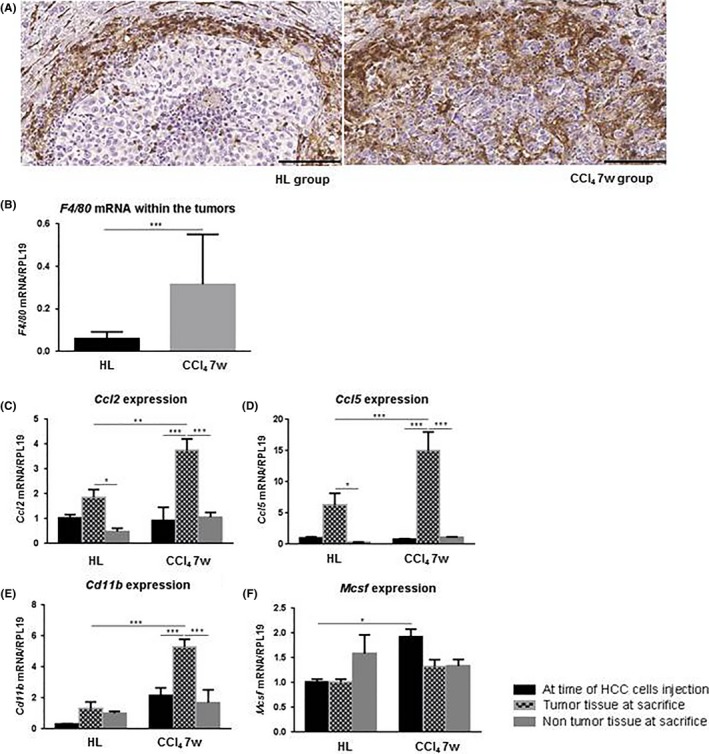
A, Macrophages were identified by F4/80 immunohistochemistry staining in the inflammatory peripheral ring and inside the tumor. Scale bar, 100 μm. B, Tumor gene expression of F4/80 (mean ± SD). C‐F, Gene expression of Ccl2, Ccl5, Cd11b, Mcsf (mean ± SD) at the time of hepatocellular carcinoma (HCC) cell injection (black bars), inside the tumor at time of death (grid pattern bars), and in the non‐tumor parenchyma at time of death (grey bars). *P < .05; **P < .01; ***P < .001. Carbon tetrachloride (CCl4) 7w, mice pretreated with CCl4 for 7 wk; HL, healthy liver group
Heterogeneity in the TAM resides partly in macrophage origin. Indeed, both tissue‐resident macrophages, the so‐called Kupffer cells in the liver, and blood‐recruited monocyte‐derived macrophages are involved in the tumor microenvironment.29 We investigated mRNA expression of soluble factors involved in the recruitment of monocyte‐derived macrophages CCL2, CCL5 and M‐CSF. mRNA expression of Cd11b, a marker of freshly infiltrating monocyte‐derived macrophages, was also analyzed. In both the HL and CCl4 7w groups, we observed significantly higher expression of Ccl2 and Ccl5 inside the tumor compared to the adjacent non‐tumor parenchyma (Figure 2C,D). Cd11b mRNA expression was also significantly higher in the tumor compared to the non‐tumor tissue but only in the CCl4 7w group (Figure 2E). The increased expression of Ccl2, Ccl5 and Cd11b was significantly more prominent inside the tumor of the CCl4 7w group compared to the tumors of the HL group (Figure 2C‐E). Moreover, in the CCl4 7w group, Ccl2, Ccl5 and Cd11b were more expressed inside the tumor than in the liver tissue in which the tumor cells were injected (Figure 2C‐E). These results are in favor of a tumor‐dependent recruitment of monocyte‐derived macrophages inside the neoplastic lesions developing in a chronically damaged hepatic tissue compared to tumors growing in a healthy liver. In contrast, although Mcsf expression was significantly higher in the chronically wounded parenchyma at the time of HCC cell injection in the CCl4 7w group, no difference was noticed between tumor tissues (Figure 2F). This may suggest that M‐CSF in the injured liver may initiate tumor monocyte recruitment.
3.3. Chronic liver injury promotes tumor infiltration by M2 polarized macrophages
Macrophages may be categorized based on their M1 or M2 polarization.30 TAM usually have an M2‐like phenotype, associated with pro‐tumor functions.7 We evaluated the M2 polarization state of the macrophages infiltrating the tumors by analyzing tumor gene expression of both M2 surface markers such as Fizz‐1, Ym‐1, SR, MR and Arg‐1, and M2‐produced cytokines and chemokines such as IL‐6 and CCL17.8 We also analyzed mRNA expression of IL‐4 and TGF‐β1), both soluble factors involved in the induction of macrophage M2 polarization.31, 32 Although no difference was observed inside the tumors regarding Sr, Mr and Arg‐1 mRNA expression, Fizz‐1 and Ym‐1 as well as Il6 and Ccl17 were significantly more expressed in tumors of the CCl4 7w group (Figure 3A). Similarly, Il4 and Tgfbeta1 mRNA were more expressed in tumors of the CCl4 7w group (Figure 3B). Moreover, active and total TGF‐β1, quantified in the tumor parenchyma by ELISA, were significantly higher in tumors growing in injured livers (Figure 3C). These results suggest that chronically damaged livers favor the polarization of recruited monocyte‐derived macrophages towards the M2 state.
Figure 3.
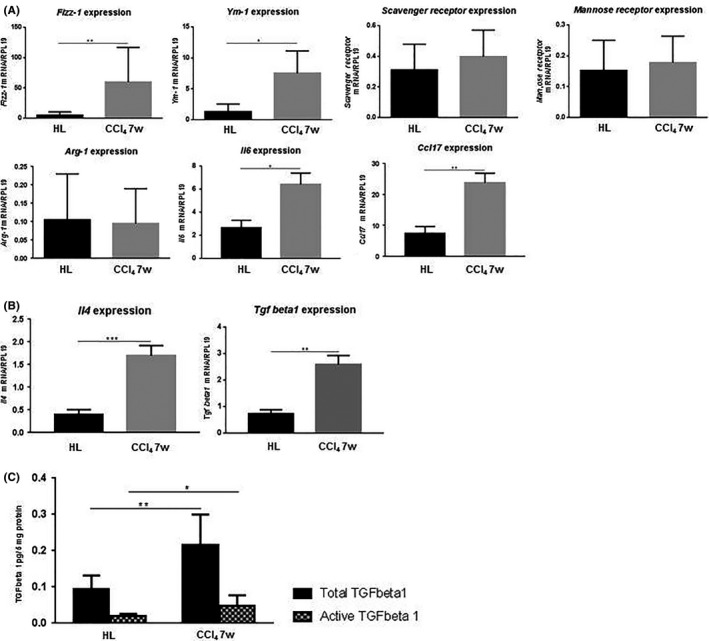
A, Tumor gene expression (mean ± SD) of Fizz‐1, Ym‐1, Scavenger receptor, Mannose receptor, Arginase‐1 (Arg‐1), interleukin‐6 (Il6), chemokine (C‐C motif) ligand 17 (Ccl17). B, Tumor gene expression (mean ± SD) of Il4 and transforming growth factor beta 1 (Tgf beta1). C, ELISA for total acid‐activated TGF beta1 (black bars) and active TGF beta1 (grid pattern bars) proteins inside the tumors (pg/5 mg protein, mean ± SD). *P < .05; **P < .01; ***P < .001. Carbon tetrachloride (CCl4) 7w, mice pretreated with CCl4 for 7 wk; HL, healthy liver group
3.4. Tumor‐associated macrophages favor tumor growth through increased production of MMP
We evaluated the production of MMP‐2, ‐9 and MT1‐MMP known to be supported by M2, pro‐tumor TAM.12 Gene expression of all 3 MMP was significantly upregulated in tumors of the CCl4 7w group (Figure 4A). Interestingly, a positive correlation was observed between TV and expression of Mmp2, ‐9 or Mt1‐mmp, suggesting the positive role of MMP‐2, ‐9 and MT1‐MMP in tumor expansion (Figure 4B).
Figure 4.
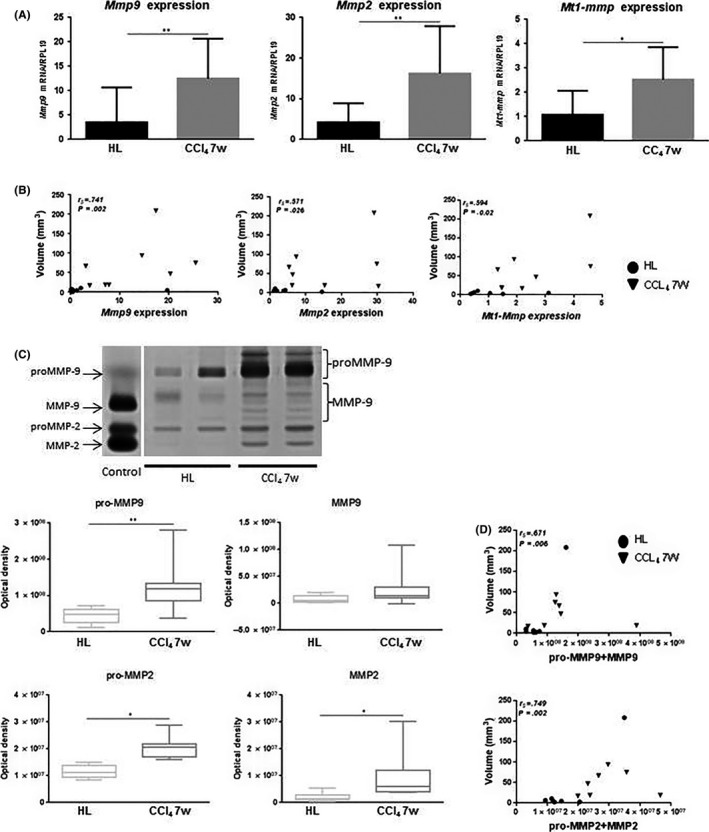
A, Tumor gene expression (mean ± SD) of Mmp9, ‐2 and membrane type 1 metalloproteinase (Mt1‐mmp). B, Correlation between tumor volume (mm3) and Mmp9, ‐2 and Mt1‐mmp mRNA expression (circles for HL data and inverted triangles for the carbon tetrachloride [CCl4] 7w group). Spearman rank correlation coefficient is indicated as r s. C, Zymogram showing latent and active forms of MMP‐2 and MMP‐9 in the tumors. MMP activities were quantified by gel densitometry. Three bands and 5 bands were considered for proMMP‐9 and MMP‐9, respectively. Only 1 zymolytic band was detected for proMMP‐2 and MMP‐2. D, Correlation between tumor volume (mm3) and the sum of the latent and active forms of MMP‐9 and MMP‐2 (circles for HL data and inverted triangles for the CCl4 7w group). Spearman rank correlation coefficient is indicated as r s. *P < .05; **P < .01. CCl4 7w, mice pretreated with CCl4 for 7 wk; HL, healthy liver group
MMP‐2 and ‐9 are initially secreted as pro‐enzymes and then activated into active enzymatic forms. Both the inactive and the active forms may be detected by zymography. We carried out zymography with protein extracts from tumor samples and quantified both the pro‐ and active forms of MMP‐2 and ‐9. Although only 1 zymolytic band was detected for proMMP‐2 and MMP‐2, several bands were detected for proMMP‐9 and MMP‐9, probably corresponding to sequentially cleaved pro‐ and active enzymes. We quantified 3 bands for the proMMP‐9 and 5 bands for the MMP‐9 and added up all 3 and all 5 bands to obtain a final quantification for proMMP‐9 and MMP‐9, respectively (Figure 4C). The proMMP‐9 zymogen as well as the proMMP‐2 and MMP‐2 proteins were significantly more expressed in tumors of the CCl4 7w group, confirming the mRNA expression results. These data were not confirmed for the active MMP‐9. A positive correlation was noticed between TV and the sum of the pro‐ and active forms for each MMP (Figure 4D).
3.5. Secretion and activity of MMP‐2 and ‐9 are dependent of tumor tissue and independent of the adjacent non‐tumor parenchyma
Matrix remodeling as a result of MMP activity is sustained in chronic liver wound healing. To define whether increased MMP activity in the tumors reflects the healing process in the host liver, we analyzed both MMP mRNA and protein expressions in liver tissue at time of HCC cell injection and in the non‐tumor adjacent parenchyma. In the HL group, no difference was observed in terms of Mmp2, Mmp9 and Mt1‐mmp gene expression between the tumor parenchyma and the liver tissue at the time of HCC cell injection or in the non‐tumor adjacent parenchyma. On the contrary, in the CCl4 7w group, all 3 Mmp gene expression was significantly more induced in the tumor compared with the parenchyma prior to HCC cell implantation or non‐tumor adjacent parenchyma at death (Figure 5A). Globally, zymography analysis carried out in the CCl4 7w group showed higher MMP protein expression in tumors compared to non‐tumor tissues but results were only statistically significant for pro‐ and active MMP‐2 between the tumor tissue and the non‐tumor adjacent parenchyma (Figure 5B). These results show that MMP production in the CCl4 7w group originates from the tumor and not from the surrounding tissue.
Figure 5.
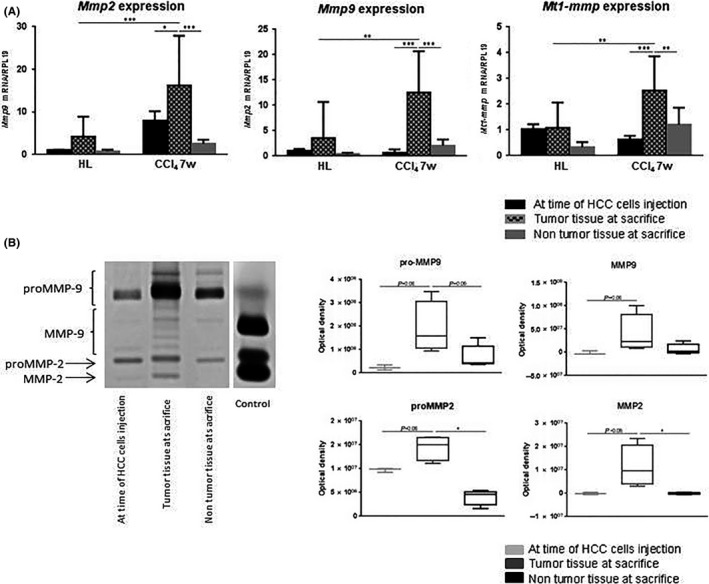
A, Gene expression of Mmp9, ‐2 and membrane type 1 metalloproteinase (Mt1‐mmp) (mean ± SD) at the time of hepatocellular carcinoma (HCC) cell injection (black bars), inside the tumor at time of death (grid pattern bars), and in the adjacent non‐tumor parenchyma at time of death (grey bars). B, Zymogram showing latent and active forms of MMP‐2 and MMP‐9 at the time of HCC cell injection (light grey box in the quantification), inside the tumor at time of death (dark grey box in the quantification), and in the non‐tumor parenchyma at time of death (black box in the quantification). *P < .05. Carbon tetrachloride (CCl4) 7w, mice pretreated with CCl4 for 7 wk; HL, healthy liver group
4. DISCUSSION
Chronic liver injury recapitulates a pathological ground permissive to cancer. The cancero‐pathogenic role of the pre‐existing damaged environment is supported by clinical observations. High liver stiffness as measured by transient elastography and reflecting advanced liver fibrosis correlates with an increased risk of HCC.33 The rate of late intrahepatic recurrence after resection is high, supporting an important role of the pre‐existing injured tissue in carcinogenesis.34 Moreover, gene expression profile of the non‐cancerous surrounding tissue predicts HCC prognosis as well as early and late recurrence.35, 36, 37, 38
In the present study, we showed that CCl4‐induced chronic liver injury promotes HCC cell seeding and growth. This result is consistent with previous studies using thioacetamide as a chronic liver injury inducer. Indeed, Kornek and colleagues described higher tumor volume and satellite lesions count when HCC cells were implanted in the livers of mice previously injected with thioacetamide and fed with alcohol.23 Similarly, the number of cancer nodules increased when neoplastic cells were injected in the spleen and migrated in the liver of mice pretreated with thioacetamide.39 In these studies, pro‐angiogenic23 and immunosuppressive impacts39 of chronic liver injury were incriminated to favor tumor spread. Compared to animal models where HCC grows in a healthy liver, such a type of neoplastic model permitting tumor development in a chronically injured liver resembling the human situation is expected to provide more clinically relevant data.
HCC is an inflammation‐related cancer.40 TAM represent the predominant type of leukocytes in liver cancer and influence each neoplastic stage.29 In a probably simplistic view, tumor macrophage density, usually evaluated by immunohistochemistry, is associated with poor prognosis in human HCC.29 The reality of TAM is undoubtedly more complex as mentioned by several studies suggesting that distinct populations may have different roles in tumorigenesis.29 In the liver, macrophages form a heterogeneous population. Some originate from tissue‐resident macrophages (Kupffer cells) whereas others are monocyte‐derived recruited cells. The relative roles of each population in hepatocarcinogenesis remains a burning and unresolved question difficult to answer.41 To date, one must remain careful in drawing definitive conclusions as different models were used to gain each piece of information. Although Kupffer cells may provide initial support to cancer cells,42, 43, 44 available data support that recruited cells have probably minor roles in early promotion of tumorigenesis. In the diethylnitrosamine model, enhanced monocyte recruitment in D6‐deficient mice does not impact tumor incidence or growth compared to wild‐type mice.45 Kang and colleagues showed that recruited monocytes rather contribute to the elimination of premalignant hepatocytes in an oncogene‐driven senescence model because the depletion of monocytes and neutrophils, but not the depletion of tissue‐resident macrophages, inhibits the clearance of premalignant cells.46
Recruited monocyte‐derived macrophages seem to gain their pro‐tumor functions, once a primary tumor is established, as a result of their plasticity. Indeed, compared to resident cells, recruited monocyte‐derived macrophages are thought to be more plastic, educable by the tumor environment, and prone to switch from an anti‐tumor to a pro‐tumor phenotype. These phenotypes are usually and roughly conceptualized in the M1/M2 polarization classification even though it is more and more admitted that the different functional patterns of macrophages exceeds the M1/M2 dichotomy.6 In a subcutaneous injection model, blockade of the CCL2/CCR2 axis reduces monocyte recruitment (and consequentially tumor macrophage infiltration), and inhibits in residual tumor macrophages the switch towards a pro‐tumor M2 phenotype, reducing tumor growth.47 Interestingly, Kupffer cells are a major source of CCL2 and, therefore, may participate in the recruitment and education of monocyte‐derived macrophages.44 In human HCC, the association between monocyte‐derived macrophages recruitment and a poor prognosis supports the leading role of recruited cells in HCC pathogenesis.47, 48
In our work, we characterized macrophage behavior when HCC cells were injected in a chronically injured liver. Compared to injection in healthy mice, we observed deeper infiltration of macrophages around clusters of tumor cells and gene/protein expression profiles inside the tumor favored not only recruited monocyte‐derived macrophages but also a M2 pro‐tumor phenotype. Although our results do not formally prove a cause‐effect relationship, they clearly support that the pro‐tumor role of chronic liver injury may be partially mediated by facilitated recruitment inside the tumors of monocyte‐derived macrophages that are more prone to an M2 phenotypic switch under the influence of the growing tumor cells and other elements of the microenvironment. Specific roles of Kupffer cells, already activated as a result of repeated CCl4 injections, were not investigated but one may expect that they play a role by producing CCL2 and helping recruitment of monocytes in this way. Although not evaluated in our study, other elements observed in fibrotic livers such as altered ECM and stiffness, and activated hepatic stellate cells may favor liver cancer development as reviewed elsewhere.49
MMP production is one of the numerous functions displayed by M2 pro‐tumor macrophages.6 By showing a higher production/activity of MMP‐2 and ‐9 in tumors developing in damaged livers, we showed the M2 pro‐tumor state of TAM in these lesions, favored by the underlying pathological fibrotic and inflammatory background. Different roles are attributed to MMP in neoplastic diseases such as promotion of growth signals and inhibition of apoptosis for cancer cells, induction of epithelial‐to‐mesenchymal transformation, enhanced metastasis development through degradation and remodeling of the ECM.12 In our work, tumor volume positively correlated with both MMP‐2 and ‐9 gene and protein expression, supporting the pro‐tumor roles of such enzymes and encouraging preclinical evaluation of specific MMP‐2 and/or MMP‐9 inhibitors in liver tumors. In 2015, the study of Elewa and colleagues argues in favor of an anti‐tumor effect of the targeting of MMP‐9 in thioacetamide‐induced HCC in rats. Unfortunately, conclusions cannot be drawn from this study as doxycycline, a non‐specific pan‐MMP inhibitor, was used and because the tumor impact was evaluated by animal survival and serum alpha‐fetoprotein level only.50
Besides the pro‐tumor roles of MMP and the potential therapeutic impact of their inhibition, our work also supports TAM targeting as a therapeutic strategy against cancer to improve the efficacy of current systemic therapy. Global depletion of macrophages by zoledronic acid or clodronate‐encapsulated liposomes has shown anti‐tumor impact in murine transplanted HCC models.51, 52 Specific inhibition of monocyte recruitment to the tumor and inhibition/reversion of tumor macrophage polarization may represent the next step to better therapeutic control of HCC. Of particular interest, cenicriviroc, an inhibitor of CCL2 monocyte recruitment to the liver, is currently being tested in clinical trials for the treatment of liver fibrosis in adult subjects with non‐alcoholic steatohepatitis.41 In the near future, it will be interesting to test such types of molecules in cancer liver.
CONFLICT OF INTEREST
Peter Stärkel received research grants from Abbvie Belgium, Gilead Belgium and BMS Belgium.
ACKNOWLEDGMENTS
This work was supported by grants from the D.G. Higher Education and Scientific Research of the French Community of Belgium (12/17‐047), the FRS‐FNRS (PDR T.1067.14) (Belgium) and unrestricted research grants from Gilead Belgium, Janssens Pharmaceutica Belgium, Abbvie Belgium and BMS Belgium. I.L. is a FRS‐FNRS senior research associate, P.H. is a FR‐FNRS research associate, and B.D. is a FRS‐FNRS PhD fellow.
Delire B, Henriet P, Lemoine P, Leclercq IA, Stärkel P. Chronic liver injury promotes hepatocarcinoma cell seeding and growth, associated with infiltration by macrophages. Cancer Sci. 2018;109:2141–2152. https://doi.org/10.1111/cas.13628
REFERENCES
- 1. de Martel C, Maucort‐Boulch D, Plummer M, Franceschi S. World‐wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology. 2015;62:1190‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol. 2013;87:227‐247. [DOI] [PubMed] [Google Scholar]
- 3. Hernandez‐Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem. 2006;387:349‐360. [DOI] [PubMed] [Google Scholar]
- 5. Yang L, Zhang Y. Tumor‐associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology. 2015;62:1304‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shirabe K, Mano Y, Muto J, et al. Role of tumor‐associated macrophages in the progression of hepatocellular carcinoma. Surg Today. 2012;42:1‐7. [DOI] [PubMed] [Google Scholar]
- 8. Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res. 2016;39:1588‐1596. [DOI] [PubMed] [Google Scholar]
- 9. Yeung OW, Lo CM, Ling CC, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. 2015;62:607‐616. [DOI] [PubMed] [Google Scholar]
- 10. Yang W, Lu Y, Xu Y, et al. Estrogen represses hepatocellular carcinoma (HCC) growth via inhibiting alternative activation of tumor‐associated macrophages (TAMs). J Biol Chem. 2012;287:40140‐40149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem. 1999;274:21491‐21494. [DOI] [PubMed] [Google Scholar]
- 12. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCawley LJ, Matrisian LM. Tumor progression: defining the soil round the tumor seed. Curr Biol. 2001;11:R25‐R27. [DOI] [PubMed] [Google Scholar]
- 14. Orlichenko LS, Radisky DC. Matrix metalloproteinases stimulate epithelial‐mesenchymal transition during tumor development. Clin Exp Metastasis. 2008;25:593‐600. [DOI] [PubMed] [Google Scholar]
- 15. Altadill A, Rodriguez M, Gonzalez LO, et al. Liver expression of matrix metalloproteases and their inhibitors in hepatocellular carcinoma. Dig Liver Dis. 2009;41:740‐748. [DOI] [PubMed] [Google Scholar]
- 16. Arii S, Mise M, Harada T, et al. Overexpression of matrix metalloproteinase 9 gene in hepatocellular carcinoma with invasive potential. Hepatology. 1996;24:316‐322. [DOI] [PubMed] [Google Scholar]
- 17. Theret N, Musso O, Turlin B, et al. Increased extracellular matrix remodeling is associated with tumor progression in human hepatocellular carcinomas. Hepatology. 2001;34:82‐88. [DOI] [PubMed] [Google Scholar]
- 18. Yamamoto H, Itoh F, Adachi Y, et al. Relation of enhanced secretion of active matrix metalloproteinases with tumor spread in human hepatocellular carcinoma. Gastroenterology. 1997;112:1290‐1296. [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto H, Itoh F, Adachi Y, et al. Messenger RNA expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human hepatocellular carcinoma. Jpn J Clin Oncol. 1999;29:58‐62. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Q, Chen X, Zhou J, et al. CD147, MMP‐2, MMP‐9 and MVD‐CD34 are significant predictors of recurrence after liver transplantation in hepatocellular carcinoma patients. Cancer Biol Ther. 2006;5:808‐814. [DOI] [PubMed] [Google Scholar]
- 21. Ashida K, Nakatsukasa H, Higashi T, et al. Cellular distribution of 92‐kd type IV collagenase/gelatinase B in human hepatocellular carcinoma. Am J Pathol. 1996;149:1803‐1811. [PMC free article] [PubMed] [Google Scholar]
- 22. Yao X, Hu JF, Daniels M, et al. A novel orthotopic tumor model to study growth factors and oncogenes in hepatocarcinogenesis. Clin Cancer Res. 2003;9:2719‐2726. [PubMed] [Google Scholar]
- 23. Kornek M, Raskopf E, Tolba R, et al. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro‐angiogenic and prometastatic factors in murine liver fibrosis. Liver Int. 2008;28:509‐518. [DOI] [PubMed] [Google Scholar]
- 24. Lee WC, Wang HC, Jeng LB, et al. Effective treatment of small murine hepatocellular carcinoma by dendritic cells. Hepatology. 2001;34:896‐905. [DOI] [PubMed] [Google Scholar]
- 25. Khan SA, Joyce J, Tsuda T. Quantification of active and total transforming growth factor‐beta levels in serum and solid organ tissues by bioassay. BMC Res Notes. 2012;5:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206‐213. [DOI] [PubMed] [Google Scholar]
- 27. Marbaix E, Donnez J, Courtoy PJ, Eeckhout Y. Progesterone regulates the activity of collagenase and related gelatinases A and B in human endometrial explants. Proc Natl Acad Sci USA. 1992;89:11789‐11793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brun JL, Galant C, Delvaux D, et al. Menstrual activity of matrix metalloproteinases is decreased in endometrium regenerating after thermal ablation. Hum Reprod. 2009;24:333‐340. [DOI] [PubMed] [Google Scholar]
- 29. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van OE, Van Ginderachter JA. Tissue‐resident versus monocyte‐derived macrophages in the tumor microenvironment. Biochim Biophys Acta. 2016;1865:23‐34. [DOI] [PubMed] [Google Scholar]
- 30. Edholm ES, Rhoo KH, Robert J. Evolutionary aspects of macrophages polarization. Results Probl Cell Differ. 2017;62:3‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889‐896. [DOI] [PubMed] [Google Scholar]
- 32. Yan W, Liu X, Ma H, et al. Tim‐3 fosters HCC development by enhancing TGF‐beta‐mediated alternative activation of macrophages. Gut. 2015;64:1593‐1604. [DOI] [PubMed] [Google Scholar]
- 33. Singh S, Fujii LL, Murad MH, et al. Liver stiffness is associated with risk of decompensation, liver cancer, and death in patients with chronic liver diseases: a systematic review and meta‐analysis. Clin Gastroenterol Hepatol. 2013;11:1573‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Imamura H, Matsuyama Y, Tanaka E, et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J Hepatol. 2003;38:200‐207. [DOI] [PubMed] [Google Scholar]
- 35. Budhu A, Forgues M, Ye QH, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99‐111. [DOI] [PubMed] [Google Scholar]
- 36. Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okamoto M, Utsunomiya T, Wakiyama S, et al. Specific gene‐expression profiles of noncancerous liver tissue predict the risk for multicentric occurrence of hepatocellular carcinoma in hepatitis C virus‐positive patients. Ann Surg Oncol. 2006;13:947‐954. [DOI] [PubMed] [Google Scholar]
- 38. Tsuchiya M, Parker JS, Kono H, Matsuda M, Fujii H, Rusyn I. Gene expression in nontumoral liver tissue and recurrence‐free survival in hepatitis C virus‐positive hepatocellular carcinoma. Mol Cancer. 2010;9:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang MC, Chang CP, Lei HY. Induction of liver fibrosis in a murine hepatoma model by thioacetamide is associated with enhanced tumor growth and suppressed antitumor immunity. Lab Invest. 2010;90:1782‐1793. [DOI] [PubMed] [Google Scholar]
- 40. Capece D, Fischietti M, Verzella D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor‐associated macrophages. Biomed Res Int. 2013;2013:187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300‐1312. [DOI] [PubMed] [Google Scholar]
- 42. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977‐990. [DOI] [PubMed] [Google Scholar]
- 43. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88‐dependent IL‐6 production. Science. 2007;317:121‐124. [DOI] [PubMed] [Google Scholar]
- 44. Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM‐1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72:3977‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schneider C, Teufel A, Yevsa T, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine‐induced liver cancer. Gut. 2012;61:1733‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature. 2011;479:547‐551. [DOI] [PubMed] [Google Scholar]
- 47. Li X, Yao W, Yuan Y, et al. Targeting of tumour‐infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157‐167. [DOI] [PubMed] [Google Scholar]
- 48. Kuang DM, Zhao Q, Peng C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD‐L1. J Exp Med. 2009;206:1327‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Affo S, Yu LX, Schwabe RF. The role of cancer‐associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol. 2017;12:153‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elewa MA, Al‐Gayyar MM, Schaalan MF, Abd El Galil KH, Ebrahim MA, El‐Shishtawy MM. Hepatoprotective and anti‐tumor effects of targeting MMP‐9 in hepatocellular carcinoma and its relation to vascular invasion markers. Clin Exp Metastasis. 2015;32:479‐493. [DOI] [PubMed] [Google Scholar]
- 51. Wang B, Li Q, Qin L, Zhao S, Wang J, Chen X. Transition of tumor‐associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 2011;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang W, Zhu XD, Sun HC, et al. Depletion of tumor‐associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16:3420‐3430. [DOI] [PubMed] [Google Scholar]