

Abstract
Long noncoding RNAs are involved in a variety of cellular functions. In particular, an increasing number of studies have revealed the functions of long noncoding RNA in various cancers; however, their precise roles and mechanisms of action remain to be elucidated. NORAD, a cytoplasmic long noncoding RNA, is upregulated by irradiation and functions as a potential oncogenic factor by binding and inhibiting Pumilio proteins (PUM1/PUM2). Here, we show that NORAD upregulates transforming growth factor‐β (TGF‐β) signaling and regulates TGF‐β‐induced epithelial‐to‐mesenchymal transition (EMT)‐like phenotype, which is a critical step in the progression of lung adenocarcinoma, A549 cells. However, PUM1 does not appear to be involved in this process. We thus focused on importin β1 as a binding partner of NORAD and found that knockdown of NORAD partially inhibits the physical interaction of importin β1 with Smad3, inhibiting the nuclear accumulation of Smad complexes in response to TGF‐β. Our findings may provide a new mechanism underlying the function of NORAD in cancer cells.
Keywords: epithelial‐to‐mesenchymal transition, long noncoding RNA, non‐small‐cell lung cancer, nuclear transport, transforming growth factor‐β
1. INTRODUCTION
According to its pathological classification, non‐small cell lung cancer (NSCLC) accounts for approximately 80% of all lung cancers. Despite the development of molecular target drugs in recent years, the long‐term survival rate remains poor. Thus, continued efforts are needed to understand the molecular mechanisms of lung cancer progression for the development of new targeted therapies.
Transforming growth factor‐β (TGF‐β) regulates various developmental and pathological processes. The receptor‐regulated class of Smad proteins (R‐Smads), such as Smad2 and Smad3, are directly phosphorylated by type I TGF‐β receptor kinase.1 Phosphorylated R‐Smads then form complexes with Smad4 and translocate to the nucleus. In the nucleus, Smad complexes act as a transcription factor to regulate the expression of target genes. Previous studies have shown that signal‐induced cytoplasmic‐to‐nuclear translocation of Smad3 is supported by multiple factors, including importin, exportin, and a direct interaction with nucleoporins. In particular, in the nuclear import step, previous studies revealed that TGF‐β‐induced Smad3 phosphorylation enhances its interaction with importin β1 and its nuclear accumulation in a Ran‐dependent manner.2, 3 In addition, another group recently showed that the nuclear import of Smad1 and Smad2/3 is mediated by importin 7 and 8.4
In cancer cells, TGF‐β paradoxically exerts both tumor‐suppressive and tumor‐promoting effects. In the early stage of cancer, TGF‐β promotes cell cycle arrest and apoptosis; however, in advanced stages, TGF‐β mediates tumor progression by promoting cell motility, invasive ability and epithelial‐to‐mesenchymal transition (EMT).5, 6 EMT is a dynamic process that mediates normal development. However, EMT also occurs in pathological contexts in tumor invasion and metastasis. During EMT, polarized epithelial cells alter their morphology and acquire the characteristics of mesenchymal cells. TGF‐β is a major inducer of EMT,7 and upon TGF‐β treatment, epithelial cells lose cell‐cell adhesion, change their morphology to an elongated spindle shape, and exhibit increased motility and invasiveness. EMT is also associated with resistance to apoptosis and chemotherapy. Therefore, a better understanding of TGF‐β signaling and EMT is important for the development of new therapeutic strategies.
An increasing number of studies have demonstrated the importance of long non‐coding RNAs (lncRNAs), which are RNAs longer than 200 bases that lack protein coding ability.8 LncRNAs were previously thought to be transcriptional junk, but an increasing volume of evidence describes their functions. Although the biological roles of the majority of lncRNAs remain unexplored, some have been suggested to act as guides, decoys or scaffolds. Mechanistically, cytoplasmic lncRNAs are known to modulate the activity or abundance of their interacting proteins or microRNAs (miRNAs). Several studies have shown that some lncRNAs play roles in tumor progression, invasion and metastasis.9, 10, 11 In particular, some lncRNAs are thought to be involved in TGF‐β signaling and TGF‐β‐induced EMT.12, 13, 14, 15, 16 However, the detailed molecular mechanisms of these lncRNAs are not fully understood.
Recently, the functions of a novel cytoplasmic lncRNA, NORAD (also known as LINC00657 or LOC647979), have been characterized. NORAD is expressed at 500‐1000 copies per cell, and it binds to and suppresses the activity of Pumilio proteins that bind specific mRNAs at their consensus sequence for downregulation. NORAD thus upregulates target transcripts of Pumilio proteins and plays a role in genomic stability.17, 18 In addition, NORAD is reported to be an oncogene, as its expression is associated with poor prognosis in breast and pancreatic cancer patients.19, 20 In pancreatic cancer cells, NORAD has been demonstrated to promote EMT and metastasis; however, the mechanisms underlying these events are not fully understood. In this study, we found that NORAD regulates TGF‐β signaling and TGF‐β‐induced EMT‐like phenotype independently of the involvement of Pumilio proteins in A549 lung adenocarcinoma cells. Our data reveal that NORAD regulates TGF‐β signaling by mediating the signal‐induced nuclear translocation of Smad complexes. Our findings may provide a potential molecular target for NSCLC.
2. MATERIALS AND METHODS
2.1. Cell culture
A549 cells were obtained from the JCRB cell bank. A549 cells were cultured in DMEM (#11965; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (#SH30910.03; Thermo Fisher Scientific), 100 U/mL penicillin G and 100 μg/mL streptomycin. Cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
2.2. Reagents and antibodies
Recombinant TGF‐β (TGF‐β3) was purchased from R&D Systems (Minneapolis, MN, USA). Phalloidin‐tetramethylrhodamine B isothiocyanate (#P1951) was obtained from Sigma‐Aldrich (St. Louis, MO, USA). Antibodies against Smad2 (#ab33875) and Smad3 (#ab40854) were purchased from Abcam (Cambridge, UK). Anti‐phospho‐Smad3 (Ser423/425; #9520) antibody was purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against Smad2/3 (#610843) was purchased from BD Biosciences (Franklin Lakes, NJ, USA). Anti‐HDAC1 (#2E10) and phospho‐Smad2 (Ser465/467; #04‐953) antibodies were obtained from Merck Millipore (Darmstadt, Germany). Antibody against c‐Myc (#017‐21871) was obtained from Wako Pure Chemical Industries (Osaka, Japan). Antibodies against FLAG (M2; #F3165) and α‐tubulin (#T6199) were obtained from Sigma‐Aldrich.
2.3. Plasmids
Plasmids encoding human PUM1, NORAD, Smad3 and importin β1 were constructed by PCR amplification. Fragments were subcloned into pcDNA3‐FLAG vector (PUM1 and importin β1), CSII‐CMV‐RfA (NORAD) or pcDNA3‐6xMyc vector (Smad3). All cDNAs were verified by sequencing.
2.4. Lentivirus production and infection
Lentiviral shRNA expression vectors were generated as described previously21 using the following oligonucleotides: #1, forward: GATCCCC GCGGTTGGTCTTCATTCTACCACGTGTGCTGTCCGTGGTAGAATGAAGACCAACCGC TTTTTGGAAAT, reverse: CTAGATTT CCAAAAA GCGGTTGGTCTTCATTCTACCACGGACAGCACACGTGGTAGAATGAAGACCAACCGC GGG; #2, forward: GATCCCC GCTAGTATCTCCATATCTAGAACGTGTGCTGTCCGTTCTAGATATGGAGATACTAGC TTTTTGGAAAT, reverse: CTAGATTTCCAAAAA GCTAGTATCTCCATATCTAGAACGGACAGCACACGTTCTAGATATGGAGATACTAGC GGG.
Lentiviral expression vectors were obtained from Dr Hiroyuki Miyoshi (RIKEN BioResource Center; current address Keio University, Tokyo, Japan). shRNAs were transferred into the lentivirus vector CS‐RfA‐EG through the pENTER‐4H1 vector. To produce lentivirus, HEK293FT cells were co‐transfected with the vector constructs CS‐RfA‐EG (for shRNAs), CSII‐CMV‐RfA (for NORAD expression), pCMV‐VSV‐G‐RSV‐Rev and pCAG‐HIVgp. Virus was collected, concentrated using the Lenti‐X Concentrator (Takara Bio, Shiga, Japan), and used to infect A549 cells.
2.5. Transfection of cDNA
Transient transfection into cells was performed using Lipofectamine 3000 Reagent (Thermo Fisher Scientific), as recommended by the manufacturer's protocol.
2.6. Immunoblotting
Cells were rinsed with ice‐cold PBS and lysed with lysis buffer (1% NP‐40, 150 mmol/L NaCl, 20 mmol/L Tris‐HCl [pH 7.5] and cOmplete EDTA‐free protease inhibitor [Roche Diagnostics, Basel, Switzerland]). After centrifugation at 20 400 g) and 4°C for 10 min, protein concentrations were estimated using the BCA Protein Assay Kit (Thermo Fisher Scientific). Total cell lysates were subjected to SDS polyacrylamide gel electrophoresis and transferred to Fluoro Trans W membranes (Pall, Port Washington, NY, USA). Immunoblotting was carried out using the indicated antibodies, and imaging was performed with an LAS‐4000 lumino‐image analyzer (FUJIFILM, Tokyo, Japan).
2.7. Immunoprecipitation
Cultured cells were lysed as described above. Immunoprecipitation was performed as previously described.22
2.8. RNA extraction and quantitative RT‐PCR
For mRNA detection, total RNA was extracted with the RNeasy Mini Kit (QIAGEN, Dusseldorf, Germany). First‐strand cDNAs were synthesized using PrimeScript II reverse transcriptase (Takara Bio). Quantitative RT‐PCR was performed with the StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific). All samples were run in duplicate, and results were averaged and normalized to the expression of GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase). Primer sequences are shown in Table S1.
2.9. Chamber migration assay
The migration assay was performed as described previously.23 After 8 hours of incubation, migrated cells were fixed and counted in images taken of randomly selected fields. The average number of cells was calculated from these images.
2.10. Dual‐luciferase assay
Cells were cultured in 12‐well plates and transfected with luciferase constructs (9xCAGA‐luc). For normalization, pGL4.75‐CMV‐Renilla was co‐transfected. Cells were lysed and used for luciferase assay using the Dual‐Luciferase Reporter Assay System (Promega, Fitchburg, WI, USA). Data were collected using Mithras LB‐940 (Berthold Technologies, Bad Wildbad, Germany).
2.11. ChIP
Cells were cultured in 10‐cm plates, and ChIP was performed using anti‐Smad2/3 antibody. ChIP‐qPCR was performed as previously described.22 All samples were run in duplicate, and results were averaged. Primer sequences are shown in Table S2.
2.12. RNA sequencing
Total RNA was extracted with the RNeasy Mini Kit (QIAGEN) from shRNA‐transfected A549 cells treated with TGF‐β for 24 hours. mRNA was purified using the Dynabeads mRNA DIRECT Purification Kit (Thermo Fisher Scientific). Libraries were prepared using the Ion Total RNA‐Seq Kit v2 according to the manufacturer's protocol and directionally sequenced with the Ion Proton (Thermo Fisher Scientific). Reads were aligned against the human genome (hg19) using TopHat2 (https://ccb.jhu.edu/software/tophat/). Differential expression was evaluated using the Cuffdiff function of Cufflinks (http://cufflinks.cbcb.umd.edu/). The raw sequencing data are available from Gene Expression Omnibus (GEO) (GSE109296).
2.13. Statistical analysis
Comparisons between the multiple experimental groups were made using one‐way ANOVA with the Holm‐Sidak test. Statistical analyses were conducted with Prism 6.
3. RESULTS
3.1. NORAD regulates transforming growth factor‐β signaling and expression of epithelial‐to‐mesenchymal transition‐related genes
To better understand the function of NORAD in lung cancer cells, we performed RNA‐seq in NORAD‐silenced A549, a human lung adenocarcinoma cell line, in the presence of TGF‐β to induce EMT. We then conducted Gene Set Enrichment Analysis (GSEA) of the RNA‐seq data. We found that a gene set upregulated by TGF‐β and a gene set related to EMT, both from the Molecular Signatures Database (MSigDB), were enriched among the genes expressed in control cells compared to those expressed in NORAD‐depleted cells (Figure 1A,B). Moreover, the depletion of NORAD resulted in more marked changes when TGF‐β‐induced genes in A549 cells were used as the gene set (Figure 1C). The effect of NORAD knockdown on the expression of each gene is provided in Table S3. We then reanalyzed published data (GSE79804) involving human osteosarcoma U2OS cells.18 The results of U2OS cells in which NORAD was overexpressed (Figure S1A,B) and depleted (Figure S1C,D) also indicated that NORAD positively regulates TGF‐β signaling and the EMT‐like phenotype. However, these data also suggested that the overexpression of PUM1 did not inhibit but rather enhanced TGF‐β signaling and EMT (Figure S1E,F). We confirmed by RT‐PCR that the expression of TGF‐β target genes, including SERPINE1 and SNAI1, was not significantly affected by overexpression of PUM1 in A549 cells, indicating that the function of NORAD in TGF‐β signaling and EMT is independent of Pumilio proteins (Figure S2).
Figure 1.
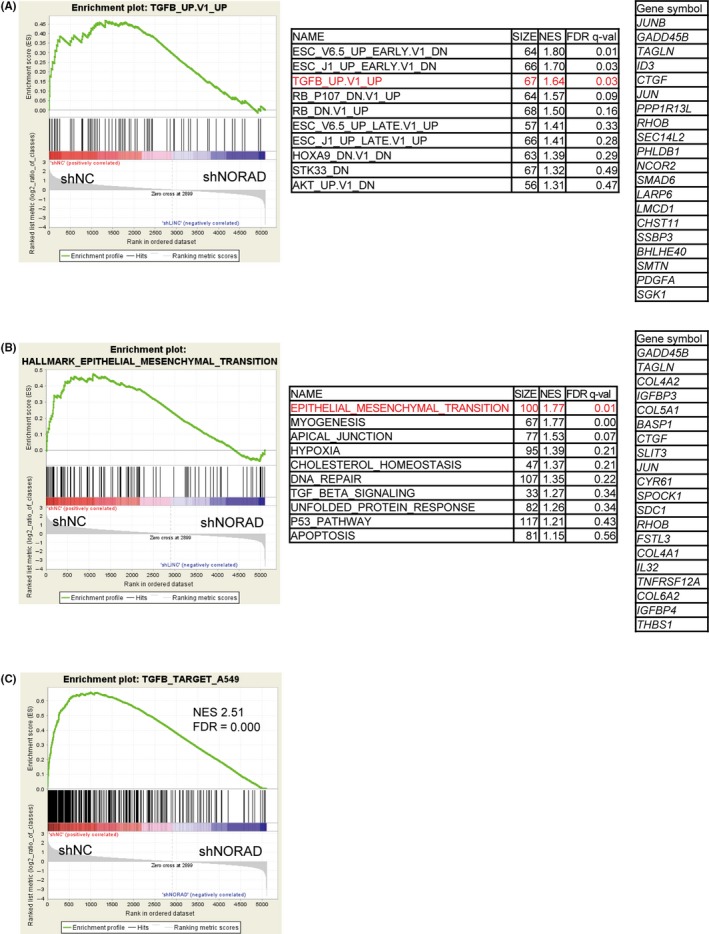
NORAD upregulates the expression of genes related to transforming growth factor‐β (TGF‐β) signaling and EMT in lung adenocarcinoma cells. A,B, Gene expression data obtained by RNA‐seq in A549 cells expressing NORAD‐targeted shRNA were used for gene set enrichment analysis. An expression cutoff of fragments per kilobase of exon per million mapped sequence reads (FPKM) value ≥10 was applied for evaluation. A list of the top most enriched oncogenic signature (A) and hallmark gene sets (B) (MSigDB) and gene lists upregulated in the negative control (shNC) relative to NORAD‐knockdown (shNORAD#1) cells are shown. The number of genes in each set, normalized enrichment score (NES), and false discovery rate (FDR) are shown for each gene set analyzed. (C) A gene set upregulated by TGF‐β in A549 cells was obtained from the RNA‐seq data, and the effect of NORAD knockdown on their expression levels was evaluated
3.2. NORAD upregulates the transforming growth factor‐β‐Smad pathway
Next, we verified that the knockdown of NORAD did not affect the mRNA expression of SMAD2 and SMAD3 by RT‐PCR (Figure 2A). However, it was revealed that several TGF‐β‐induced target genes and EMT marker genes (SERPINE1, SNAI1 and FN1) were significantly downregulated in NORAD‐depleted cells (Figure 2A). Of note, the expression of CDH1 was downregulated by NORAD shRNA in the absence of TGF‐β, and the inhibitory effect of TGF‐β on CDH1 expression was not affected by the presence of the shRNA, implying that NORAD affects some of the EMT phenotype in a TGF‐β‐independent manner. In contrast, stable overexpression of NORAD increased the expression of SMAD2, TGF‐β‐induced target genes and EMT marker genes (Figure S3A). For further confirmation, luciferase reporter assays using 9xCAGA‐luc, a Smad3‐specific reporter,24 were performed in NORAD‐depleted A549 cells. The results showed that NORAD knockdown significantly suppressed the TGF‐β‐induced 9xCAGA reporter activity (Figure 2B). Consistently, forced expression of NORAD increased the TGF‐β‐induced Smad3‐specific luciferase reporter activity (Figure S3B). We then performed ChIP‐qPCR analysis of Smad2/3. Depletion of NORAD reduced the enrichment of Smad2/3 in the SERPINE1 and ID1 promoter regions, although not statistically significant (Figure 2C). In contrast, NORAD shRNAs showed little effect on the TGF‐β‐induced C‐terminal phosphorylation of Smad2 and Smad3, indicating that NORAD regulates the activation of Smad signaling independently of their C‐terminal phosphorylation (Figure 2D).
Figure 2.
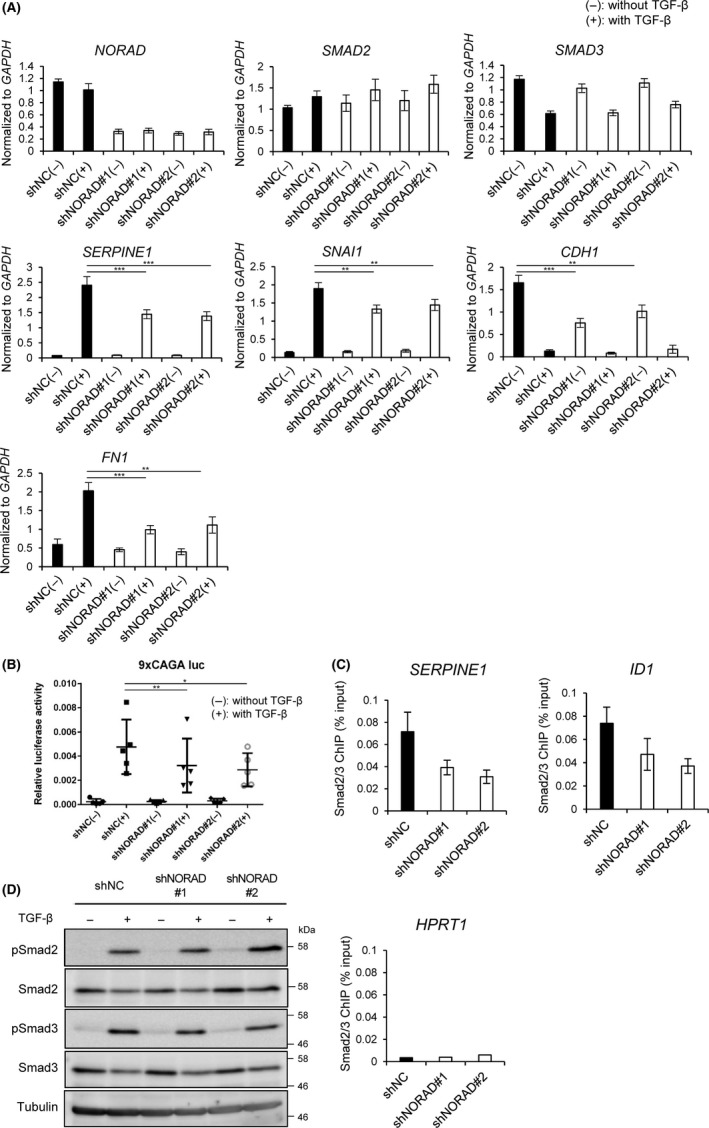
NORAD upregulates transforming growth factor‐β (TGF‐β)‐Smad signaling. A, A549 cells expressing the indicated shRNAs were treated with (+) or without (−) TGF‐β for 24 h. mRNA expression was quantified by quantitative RT‐PCR. Results are means ± SE of the means of 5 independent experiments. **P < .01; ***P < .001. B, A549 cells expressing the indicated shRNAs were transfected with luciferase reporter construct (9xCAGA‐luc) and treated with (+) or without (−) TGF‐β for 24 h. Cells were harvested and assayed for luciferase activities. Results are means ± SE of the means of 5 independent experiments. *P < .05; **P < .01. C, ChIP‐qPCR was performed in A549 cells expressing the indicated shRNAs and treated with TGF‐β for 1 h. Results are means ± SE of the means of 5 independent experiments (SERPINE1 and ID1). In the case of HPRT1, used as a negative control, standard deviations could not be calculated because some of the samples were below the detection limit. D, A549 cells were treated as in (A). Cells were then harvested for immunoblotting. The positions of the molecular weight markers are shown on the right
3.3. NORAD is important for nuclear transport of the transforming growth factor‐β‐induced Smad complexes
We next focused on TGF‐β‐induced nuclear‐cytoplasmic translocation of the Smad complexes. Nuclear/cytoplasmic fractionation showed that in response to TGF‐β, total and phosphorylated Smad2 was found in the nuclear fraction, but in NORAD‐depleted cells, cytoplasmic phosphorylated Smad2 and Smad3 were increased and nuclear total and phosphorylated Smad2 and Smad3 were reduced (Figure 3A). Consistent with these findings, the results of immunostaining for Smad2 showed that depletion of NORAD partially inhibited TGF‐β‐induced nuclear staining of Smad2 protein (Figure 3B). These results indicate that NORAD affects the TGF‐β‐induced nuclear translocation of the Smad complexes.
Figure 3.
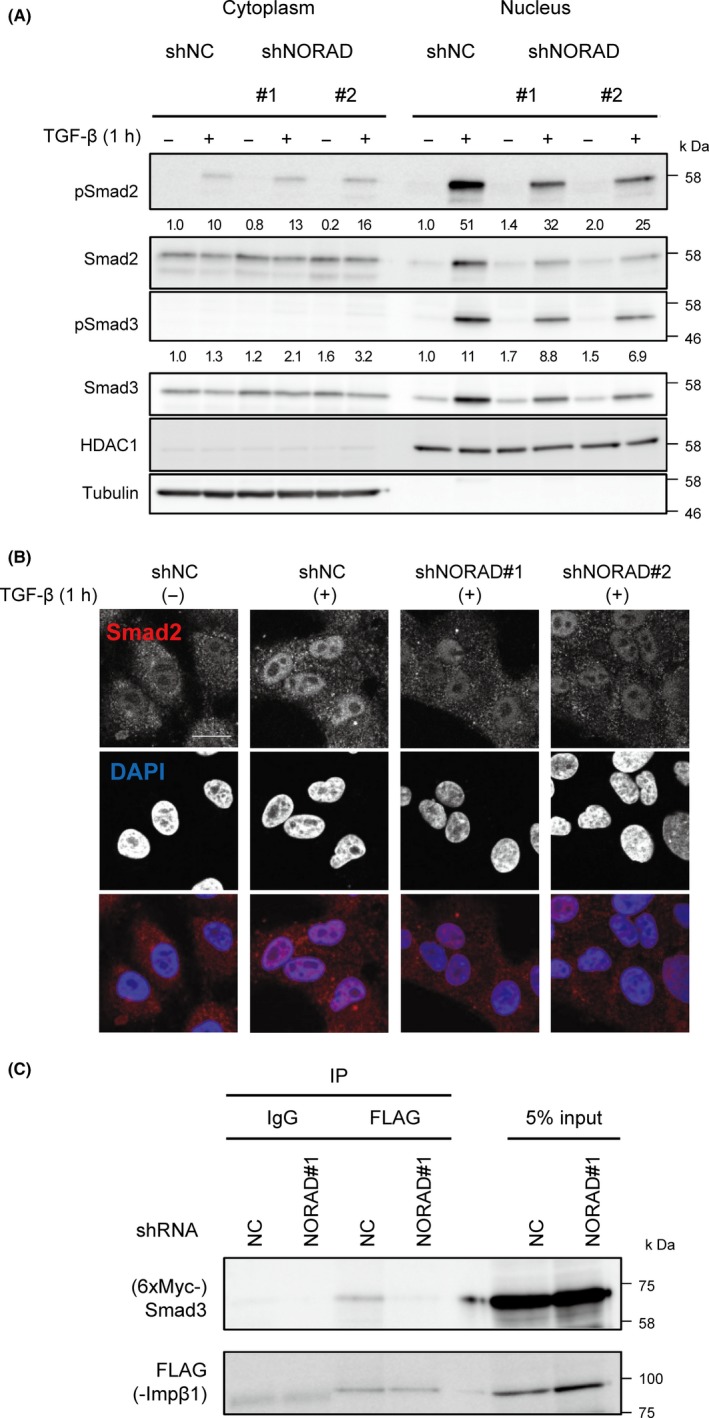
NORAD mediates transforming growth factor‐β (TGF‐β)‐induced nuclear transport of the Smad complexes. A, A549 cells expressing the indicated shRNAs were treated with (+) or without (−) to TGF‐β for 1 h. Cell lysates were fractionated into cytoplasmic and nuclear fractions, and immunoblotting was performed. The relative band intensities of the immunoblots of phosphorylated Smad2 and Smad3 (pSmad2 and pSmad3, respectively) after normalization with tubulin (cytoplasm) or HDAC1 (nucleus) are shown in the panel. The positions of the molecular weight markers are shown on the right. B, A549 cells were treated as in (A). Cells were then fixed and immunostained with the anti‐Smad2 antibody. Scale bar, 20 μm. C, A549 cells expressing the indicated shRNAs were transfected with expression plasmids for 6xMyc‐Smad3 and FLAG‐importin β1 and treated with TGF‐β for 1 h. Cell lysates were subjected to immunoprecipitation using an antibody against FLAG. Immunoprecipitates and 5% input extracts were immunoblotted with the antibodies against Smad3 or FLAG. The positions of the molecular weight markers are shown on the right
Nuclear transport of the Smad complexes is regulated by several pathways, including importin‐mediated nuclear import.2, 3, 4 To determine the role of NORAD in regulating these pathways, we attempted to identify its interacting proteins by examining a list of proteins found in NORAD RNA co‐precipitates which were identified by mass spectrometry.17, 18 Interestingly, this suggested that NORAD interacts with importin 7 and importin β1, both of which have been reported to be important for the nuclear import of phosphorylated Smads.2, 3, 4 In particular, the results of co‐immunoprecipitation assay showed that knockdown of NORAD partially inhibited interactions between Smad3 and importin β1 in TGF‐β‐treated A549 cells (Figure 3C). Together, these results suggest that the cytoplasmic lncRNA NORAD is involved in the formation of Smad3 and importin β1 complexes, which is one of the crucial steps in the signal‐induced nuclear translocation of the Smad complexes.
3.4. NORAD mediates transforming growth factor‐β‐induced epithelial‐to‐mesenchymal transition‐like phenotype in lung adenocarcinoma cells
Finally, we explored the effects of NORAD knockdown on tumor cell motility using a chamber migration assay. Knockdown of NORAD significantly suppressed cell migration compared to that in control cells (Figure 4A). Because the remodeling of actin filaments is necessary for completing EMT, we then performed phalloidin staining to visualize filamentous actin (F‐actin) formation. The results revealed that TGF‐β induced actin stress fiber formation in control cells. In contrast, NORAD‐depleted cells showed reduced stress fiber formation and a rounder morphology (Figure 4B), indicating that NORAD affects actin cytoskeletal reorganization and EMT‐like morphological changes induced by TGF‐β in these cells.
Figure 4.
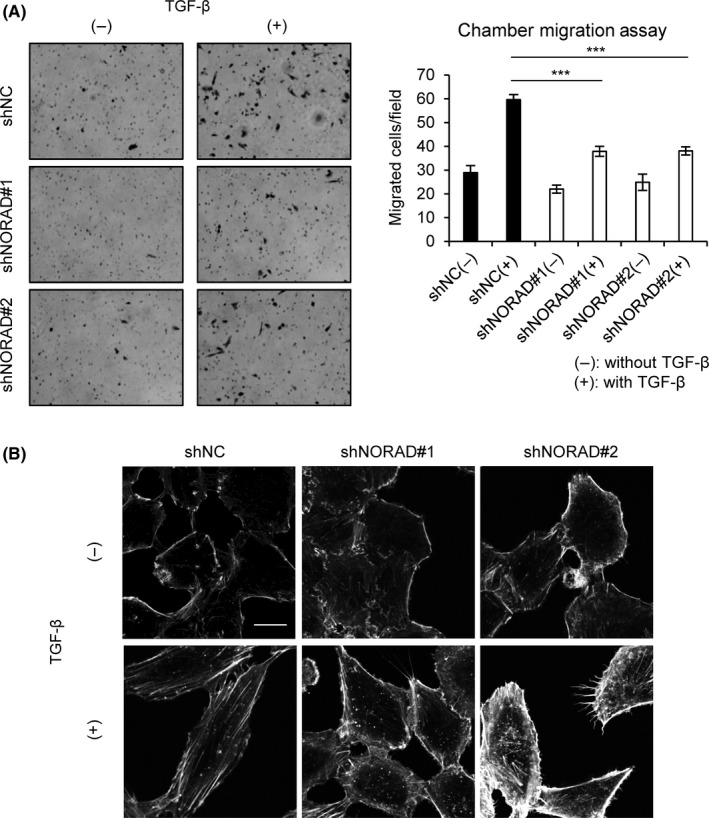
NORAD regulates cell migration and F‐actin formation. A, A549 cells expressing indicated shRNAs were treated with (+) or without (−) transforming growth factor‐β (TGF‐β) for 24 h. Cells were then seeded in upper chambers with collagen‐coated pores. Cells that migrated through the membrane were counted after 8 h of incubation. Representative fields are shown (left). Results are means ± SE of the means of 3 independent experiments (right). ***P < .001. B, A549 cells were treated as in (A). Cells were then fixed and stained with phalloidin–tetramethylrhodamine B isothiocyanate. Scale bar, 20 μm
4. DISCUSSION
Epithelial‐to‐mesenchymal transition has been shown to play important roles in the invasion and metastasis of epithelial cancers including NSCLC. In NSCLC, it has been reported that EMT markers are highly expressed in tumor cells at the peripheral leading edge when compared with centrally located cells.25 The EMT phenotype is also frequently associated with therapeutic resistance in lung adenocarcinoma cells.26, 27 Taken together, EMT leads to aggressive tumor behavior in NSCLC and may represent a potential target for cancer therapy.
Epithelial‐to‐mesenchymal transition causes epithelial cells to lose polarity and acquire properties of mesenchymal cells. Previous studies have demonstrated that TGF‐β induces EMT in lung adenocarcinoma cells, including A549 cells.28, 29 TGF‐β directly induces the expression of the EMT transcription factors Snail, Slug, ZEB1, ZEB2, Twist and E12/E47. In addition, previous reports have indicated that some lncRNAs, including lncRNA‐ATB, ANCR, lncRNA‐HIT and MEG3,12, 13, 14, 15, 16 play roles in TGF‐β‐induced EMT and TGF‐β signaling. Here, we identified the lncRNA NORAD as a new regulator of TGF‐β signaling and EMT‐like phonotype. Notably, GSEA analysis revealed that NORAD induced EMT‐related genes, and regulation of the expression of SERPINE1, SNAI1 and FN1 was confirmed by RT‐PCR. However, NORAD appears to regulate CDH1 expression in a TGF‐β‐independent manner, suggesting that NORAD regulates some, but not all, the EMT‐related genes, in the process of EMT.
NORAD, a highly conserved lncRNA, has been reported to act as a negative regulator of Pumilio proteins,17, 18 the RNA‐binding post‐transcriptional repressors of the specific mRNAs, and it has also been implicated in tumor progression. In breast cancer cells, knockout of NORAD suppresses tumor cell growth and proliferation.19 In pancreatic cancer patients, NORAD upregulation is correlated with poor overall survival.20 The same study also showed that the depletion of NORAD inhibits hypoxia‐induced EMT and metastasis in vitro and in vivo in pancreatic cancers, in which NORAD probably acts as a competitive endogenous RNA (ceRNA) to regulate RhoA through competition with hsa‐miR‐125a‐3p. In colorectal cancer patients, the expression of NORAD is positively related with EMT, metastasis and poor prognosis through serving as a ceRNA for miR‐202‐5p.30 Here, we showed a different potential mechanism by which NORAD regulates EMT. Our results demonstrated that NORAD positively regulates TGF‐β signaling and TGF‐β‐induced EMT‐like phenotype in lung adenocarcinoma cells in a Pumilio‐independent manner. Because our results indicated that NORAD also affects TGF‐β signaling in U2OS cells, it may widely function as a regulator of TGF‐β signaling.
In the present study, we focused on the role of NORAD in the signal‐induced nuclear translocation of the Smad complexes, which is an essential step for TGF‐β signaling. According to previously published mass spectrometry data,17, 18 NORAD has the ability to bind to importin β1. Although one study showed that importin β1 interacts with Smad3 but not with Smad2, another group reported that knockdown of importin β1 abrogates nuclear import of both Smad2 and Smad3.31 The results of the immunoprecipitation assay showed that NORAD appears to affect the interaction between Smad3 and importin β1 (Figure 3C). Several cytoplasmic RNAs have been reported to interact with cytoplasmic proteins and modulate their roles.32, 33 NORAD may, thus, regulate the nuclear transport of the Smad complexes by affecting importin β1‐Smad3 interactions, although its precise molecular mechanism remains to be elucidated. Because there is no evidence that NORAD interacts with Smad3 or other Smad proteins, the effect of NORAD on interactions between importin β1 and Smad3 may be transient or indirect. Moreover, it has been shown that importin β1 recognizes nuclear localization signals and is involved in pathways other than TGF‐β signaling, such as the STAT3 and p53 pathways.34, 35 It is, therefore, possible that NORAD has broad roles in maintaining nuclear translocation and signal transduction. Further studies on the roles of NORAD in TGF‐β signaling and EMT will facilitate our understanding of lncRNA biology in cancer.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
The authors are grateful to Keiko Yuki for technical assistance.
Kawasaki N, Miwa T, Hokari S, et al. Long noncoding RNA NORAD regulates transforming growth factor‐β signaling and epithelial‐to‐mesenchymal transition‐like phenotype. Cancer Sci. 2018;109:2211–2220. https://doi.org/10.1111/cas.13626
Funding information
KAKENHI grants‐in‐aid for Scientific Research (C) (15K06831, D.K.) and (S) (15H05774, K.M.) from the Japan Society for the Promotion of Science (JSPS); Research on Development of New Drugs (Japan Agency for Medical Research and Development [AMED]); research grant from the Princess Takamatsu Cancer Research Fund to D.K; Research Fellowship for Young Scientists (DC) and the Graduate Program for Leaders in Life Innovation from JSPS to N.K.
REFERENCES
- 1. Morikawa M, Derynck R, Miyazono K. TGF‐β and the TGF‐β family: context‐dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:pii: a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kurisaki A, Kose S, Yoneda Y, Heldin CH, Moustakas A. Transforming growth factor‐β induces nuclear import of Smad3 in an importin‐β1 and Ran‐dependent manner. Mol Biol Cell. 2001;12:1079‐1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xiao Z, Liu X, Lodish HF. Importin β mediates nuclear translocation of Smad 3. J Biol Chem. 2000;275:23425‐23428. [DOI] [PubMed] [Google Scholar]
- 4. Xu L, Yao X, Chen X, Lu P, Zhang B, Ip YT. Msk is required for nuclear import of TGF‐β/BMP‐activated Smads. J Cell Biol. 2007;178:981‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyazono K, Ehata S, Koinuma D. Tumor‐promoting functions of transforming growth factor‐β in progression of cancer. Ups J Med Sci. 2012;117:143‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katsuno Y, Lamouille S, Derynck R. TGF‐β signaling and epithelial‐mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76‐84. [DOI] [PubMed] [Google Scholar]
- 7. Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415‐424. [DOI] [PubMed] [Google Scholar]
- 8. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kondo Y, Shinjo K, Katsushima K. Long non‐coding RNAs as an epigenetic regulator in human cancers. Cancer Sci. 2017;108:1927‐1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354‐361. [DOI] [PubMed] [Google Scholar]
- 11. Arase M, Horiguchi K, Ehata S, et al. Transforming growth factor‐β‐induced lncRNA‐Smad7 inhibits apoptosis of mouse breast cancer JygMC(A) cells. Cancer Sci. 2014;105:974‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yuan JH, Yang F, Wang F, et al. A long noncoding RNA activated by TGF‐β promotes the invasion‐metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666‐681. [DOI] [PubMed] [Google Scholar]
- 13. Li Z, Dong M, Fan D, et al. LncRNA ANCR down‐regulation promotes TGF‐β‐induced EMT and metastasis in breast cancer. Oncotarget. 2017;8:67329‐67343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang J, Shao N, Ding X, et al. Crosstalk between transforming growth factor‐β signaling pathway and long non‐coding RNAs in cancer. Cancer Lett. 2016;370:296‐301. [DOI] [PubMed] [Google Scholar]
- 15. Richards EJ, Zhang G, Li ZP, et al. Long non‐coding RNAs (LncRNA) regulated by transforming growth factor (TGF) β: LncRNA‐HIT‐mediated TGFβ‐induced epithelial to mesenchymal transition in mammary epithelia. J Biol Chem. 2015;290:6857‐6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terashima M, Tange S, Ishimura A, Suzuki T. MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial‐mesenchymal transition in lung cancer cell lines. J Biol Chem. 2017;292:82‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S, Kopp F, Chang TC, et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016;164:69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tichon A, Gil N, Lubelsky Y, et al. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat Commun. 2016;7:12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu H, Li J, Koirala P, et al. Long non‐coding RNAs as prognostic markers in human breast cancer. Oncotarget. 2016;7:20584‐20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Wang X, Wen C, et al. Long noncoding RNA NORAD, a novel competing endogenous RNA, enhances the hypoxia‐induced epithelial‐mesenchymal transition to promote metastasis in pancreatic cancer. Mol Cancer. 2017;16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoshino Y, Nishida J, Katsuno Y, et al. Smad4 decreases the population of pancreatic cancer‐initiating cells through transcriptional repression of ALDH1A1. Am J Pathol. 2015;185:1457‐1470. [DOI] [PubMed] [Google Scholar]
- 22. Koinuma D, Tsutsumi S, Kamimura N, et al. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol Cell Biol. 2009;29:172‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ehata S, Hanyu A, Fujime M, et al. Ki26894, a novel transforming growth factor‐β type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 2007;98:127‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGFβ‐inducible elements in the promoter of human plasminogen activator inhibitor‐type 1 gene. EMBO J. 1998;17:3091‐3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahmood MQ, Ward C, Muller HK, Sohal SS, Walters EH. Epithelial mesenchymal transition (EMT) and non‐small cell lung cancer (NSCLC): a mutual association with airway disease. Med Oncol. 2017;34:45. [DOI] [PubMed] [Google Scholar]
- 26. Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial‐mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan X, Wu H, Han N, et al. Notch signaling and EMT in non‐small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saito RA, Watabe T, Horiguchi K, et al. Thyroid transcription factor‐1 inhibits transforming growth factor‐β‐mediated epithelial‐to‐mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 2009;69:2783‐2791. [DOI] [PubMed] [Google Scholar]
- 29. Isogaya K, Koinuma D, Tsutsumi S, et al. A Smad3 and TTF‐1/NKX2‐1 complex regulates Smad4‐independent gene expression. Cell Res. 2014;24:994‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J, Li XY, Hu P, Ding YS. LncRNA NORAD contributes to colorectal cancer progression by inhibition of miR‐202‐5p. Oncol Res. 2018. https://doi.org/10.3727/096504018X15190844870055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilkes MC, Repellin CE, Kang JH, Andrianifahanana M, Yin X, Leof EB. Sorting nexin 9 differentiates ligand‐activated Smad3 from Smad2 for nuclear import and transforming growth factor β signaling. Mol Biol Cell. 2015;26:3879‐3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF‐1α and lincRNA‐p21 modulates the Warburg effect. Mol Cell. 2014;53:88‐100. [DOI] [PubMed] [Google Scholar]
- 33. Liu B, Sun L, Liu Q, et al. A cytoplasmic NF‐κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27:370‐381. [DOI] [PubMed] [Google Scholar]
- 34. Khomenko T, Deng X, Ahluwalia A, et al. STAT3 and importins are novel mediators of early molecular and cellular responses in experimental duodenal ulceration. Dig Dis Sci. 2014;59:297‐306. [DOI] [PubMed] [Google Scholar]
- 35. Komlodi‐Pasztor E, Trostel S, Sackett D, Poruchynsky M, Fojo T. Impaired p53 binding to importin: a novel mechanism of cytoplasmic sequestration identified in oxaliplatin‐resistant cells. Oncogene. 2009;28:3111‐3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials