

Abstract
Angiogenesis and inflammation are crucial processes through which the tumor microenvironment (TME) influences tumor progression. In this study, we showed that peroxisome proliferator‐activated receptor γ (PPARγ) is not only expressed in CT26 and 4T1 tumor cell lines but also in cells of TME, including endothelial cells and tumor‐associated macrophages (TAM). In addition, we showed that rosiglitazone may induce tumor vessel normalization and reduce TAM infiltration. Additionally, 4T1 and CT26 tumor‐bearing mice treated with rosiglitazone in combination with radiotherapy showed a significant reduction in lesion size and lung metastasis. We reported that a single dose of 12 Gy irradiation strongly inhibits local tumor angiogenesis. Secretion of C‐C motif chemokine ligand 2 (CCL2) in response to local irradiation facilitates the recruitment of migrating CD11b+ myeloid monocytes and TAM to irradiated sites that initiate vasculogenesis and enable tumor recurrence after radiotherapy. We found that rosiglitazone partially decreases CCL2 secretion by tumor cells and reduces the infiltration of CD11b+ myeloid monocytes and TAM to irradiated tumors, thereby delaying tumor regrowth after radiotherapy. Therefore, combination of the PPARγ agonist rosiglitazone with radiotherapy enhances the effectiveness of radiotherapy to improve local tumor control, decrease distant metastasis risks and delay tumor recurrence.
Keywords: angiogenesis, CCL2, macrophage, PPARγ, radiotherapy
Abbreviations
- CCL2
C‐C motif chemokine ligand 2
- CFRT
conventional fractionated irradiation therapy
- HFRT
hypofractionated irradiation therapy
- HIF‐1α
hypoxia‐inducible factor 1α
- MDSC
myeloid‐derived suppressor cell
- PPARγ
peroxisome proliferator‐activated receptor γ
- RT
radiotherapy
- SABR
stereotactic ablative radiation therapy
- SBRT
stereotactic body radiation therapy
- TAM
tumor‐associated macrophage
- TAN
tumor‐associated neutrophil
- TME
tumor microenvironment
- VEGFR2
vascular endothelial growth factor receptor 2
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
As an indispensable treatment option, approximately 60% of newly diagnosed cancer patients radiotherapy (RT) for a neoadjuvant, definitive, adjuvant or palliative purpose as required.1 Recently, several preclinical investigations have highlighted changes in the tumor stroma or in the tumor microenvironment (TME) caused by irradiation that might induce proangiogenic and proinflammatory effects that lead to tumor recurrence.2, 3 Tumor angiogenesis and tumor‐promoting inflammation are regarded as 2 prominent hallmarks of cancer.4 Abnormal tumor vasculature impairs blood perfusion and the delivery of oxygen, and the resulting hypoxic TME helps tumor cells attain an increased invasive and metastatic capacity, and escape from host immune attack leading to radioresistance.5, 6 The TME also contains various inflammatory immune cells, chemokines and cytokines7; moreover, myeloid‐derived cells have been shown to play a pivotal part, both structurally and functionally, in tumor angiogenesis, progression and response to treatment.8, 9
Based on preclinical and clinical studies, stereotactic body radiation therapy (SBRT) or stereotactic ablative radiation therapy (SABR) has better antitumor efficacy than conventional fractionated radiation therapy. High single fraction doses (8‐16 Gy) induce endothelial cell dysfunction and apoptosis and cause serious damage to the tumor vasculature accompanied by indirect tumor cell death.10, 11, 12 Irradiation blocks local tumor angiogenesis, whereas vasculogenesis, as a backup pathway, recovers tumor blood supply to enable tumor regrowth or recurrence after RT. Proangiogenic myeloid‐derived cells such as tumor‐associated macrophage (TAM),13 Tie‐2‐expressing monocytes (TEM),14 CD11b+ myeloid cells15 and myeloid‐derived suppressor cell (MDSC) 16 participate in vasculogenesis.
In general, peroxisome proliferator‐activated receptor γ (PPARγ) is a ligand‐activated transcription factor that has anti‐inflammatory, antiangiogenic and antineoplastic activities based on preclinical data.17, 18 PPARγ deletion in mouse macrophages not only facilitates mammary tumor progression but also weakens the anti‐tumor effects of the PPARγ agonist rosiglitazone accompanied by increased infiltration of CD11b+ myeloid cells and TAM of proinflammatory and proangiogenic phenotypes.19 With the increasing knowledge of tumor angiogenesis and inflammation, antiangiogenic or anti‐inflammatory treatment modalities have emerged as novel research topics. PPARγ agonists have been intensively evaluated as a promising new therapeutic strategy in the treatment of cancer. However, their effect on the tumor vasculature and cancer‐related inflammation remains poorly explored. Angiogenesis and inflammation are crucial processes, through which the TME influences tumor radioresponse. The aim of the present study is to evaluate the synergistic effect of the PPARγ agonist rosiglitazone and RT.
2. MATERIALS AND METHODS
2.1. Cell culture and in vitro irradiation
The murine colon carcinoma cell line CT26, breast cancer cell line 4T1, RAW264.7 macrophages, HUVEC and THP‐1 cells were gifted from the State Key Laboratory of Biotherapy of West China Hospital. Cells were cultured in DMEM or RPMI 1640 (HyClone, Logan, UT, USA) containing 10% FBS (Roche, Basel, Switzerland) and 1% penicillin/streptomycin (HyClone) and incubated at 37°C in 5% CO2. Cells were irradiated (distance from X‐ray source to the target was 30 cm) at a dose rate of 4 Gy/min using an X ray‐generator (XRAD320; Precision X‐ray Inc., North Branford, CT, USA). Sham irradiation involved placing cell culture plates at a similar temperature for the length of irradiation.
2.2. Animals
Female BALB/c mice 6‐8 weeks of age were purchased from the Chinese Academy of Medical Science. The animals were maintained under specific pathogen‐free conditions and used according to Institutional Animal Care and Use Guidelines (no. SYXK2007‐008). All animal experiments were carried out under approved protocols of the institutional animal use and care committee.
2.3. Xenograft studies
CT26 and 4T1 cells were harvested and suspended in PBS. Each female BALB/c mouse was inoculated with 5 × 105 cells on the right proximal hind leg. When the tumor size reached approximately 50‐100 mm3 or 200‐300 mm3, the mice were randomly divided into 4‐6 groups for CT26 (n = 6 for each group) and 4T1 models (n = 6 for each group). To investigate the therapeutic effect of rosiglitazone combined with RT, the mice were given 100 mg/kg rosiglitazone (Selleck Chemicals, Houston, TX, USA) or vehicle (DMSO:PEG300:Tween80:ddH2O = 4:30:5:100) by oral gavage per day for 2 weeks. Before 6 or 12 Gy irradiation, each mouse was anesthetized and shielded by a lead box with only the tumor exposed. Tumor diameters were measured every 2 or 3 days using digital calipers, and the tumor volume was calculated by the formula: volume = length × width × width × 0.5.
2.4. Clonogenic survival assays
CT26 or 4T1 cells were plated in 6‐well plates at various densities and treated with 40 μmol/L rosiglitazone or vehicle 1 hour prior to irradiation at various doses.20 After 2 weeks of incubation, surviving colonies containing at least 50 cells were counted. Survival fraction was calculated, and then radiation dose‐survival curves, radiobiological parameters and sensitizing enhancement ratio were determined using the multi‐target single‐hit model as follows: S = 1 ‐ (1 ‐ e−D/D0)N, S = Survival fraction; e = natural logarithm; D0 = dose that decreases surviving fraction to 37%; D = dose; N = extrapolation number.
2.5. Endothelial cell tube formation assays
HUVEC were cultured for 24 hours with 5, 10, 20, 40 and 80 μmol/L rosiglitazone or vehicle. The cells were then harvested and plated at a density of 4 × 104 cells/well in 96‐well plates coated with 50 μL growth factor‐reduced Matrigel (BD Biosciences, San Jose, CA, USA). After 6 hours of incubation, tube formation was observed under an inverted microscope. Images were captured within 4 randomly selected fields. Total tube length and number of tubules were calculated with ImageJ software (NIH, Bethesda, MD, USA) for angiogenesis analysis.
2.6. Cell chemotaxis assays
Cell chemotaxis assays were carried out using 8.0 μm pore size transwells (Corning, Corning, NY, USA). CT26 cells were exposed to 12 Gy irradiation, and then the cells were cultured for 24 hours with 40 μmol/L rosiglitazone. Approximately 5 × 104 RAW264.7 cells were resuspended in serum‐free media and seeded in the upper chambers of 24‐well transwells. Serum‐free medium (no chemoattractant) in the lower chambers served as background control. To the remaining lower wells, 10 ng/mL recombinant murine CCL2 (PeproTech Inc., Rocky Hill, NJ, USA) was added. The relevant interventions were used as corresponding conditioned media. After 6 hours of incubation at 37°C, the migrated cells attached to the lower surface of the filters were stained with Wright's Stain Kit. Migrated cells were examined and counted in 5 random microscopic fields. Chemotaxis index was defined as the mean number of migrated RAW264.7 cells in response to conditioned medium as fold increase relative to serum‐free medium control.
2.7. Hypoxia studies
Approximately 200 μL pimonidazole (PIMO) hydrochloride (60 mg/kg; Hypoxyprobe, Inc., Burlington, MA, USA) was ip, injected 1 hour before killing the mice. Pimonidazole immunofluorescence staining was carried out according to the manufacturer's instructions.
2.8. In vivo tumor microvascular imaging and perfusion assays
For tumor microvascular imaging assays, each tumor‐bearing mouse was anesthetized, and 200 μL FITC‐dextran (100 mg/mL, 2000 kDa; Sigma‐Aldrich, St Louis, MO, USA) was i.v. injected for circulating for 2 minutes. Local tumor was scanned and imaged with a Nikon A1R MP+ multiphoton confocal microscope (Nikon Instruments Inc., New Orleans, LA, USA). For tumor microvascular perfusion assays, each tumor‐bearing mouse was given an i.v. injection of 200 μL FITC‐lectin (1 mg/mL; Sigma‐Aldrich) 15 minutes before death. Tumors were collected and directly frozen in liquid nitrogen until cryosectioning into 5‐μm sections.
2.9. Immunofluorescence, immunohistochemistry, immunoblotting and ELISA
Immunofluorescence, immunohistochemistry, immunoblotting and ELISA assays were carried out as described in Data S1.
2.10. Flow cytometry
Single cell suspensions (106 cells in 100 μL total volume) from CT26 tumor samples were incubated with 7‐AAD (BD Biosciences), anti‐mouse Fc‐block CD16/32 antibody (eBioscience, San Diego, CA, USA) and fluorescently labeled antibodies and incubated at 4°C for 30 minutes. The primary antibodies against cell surface markers F4/80 (BM8), CD45 (30‐F11), CD11b (M1/70), Ly‐6G (1A8) were obtained from BD Bioscience. Multicolor FACS analysis was carried out with a flow cytometer (BD FACS Array). Gating strategy for myeloid monocytes, TAM and tumor‐associated neutrophil (TAN) is presented in Figure S1. The data were analyzed using FlowJo v10.0 software (Treestar Inc., Ashland, OR, USA).
2.11. Lung metastasis quantification
Lungs from 4T1 tumor‐bearing mice were obtained 32 days after s.c. inoculation. H&E staining was used to quantify lung metastasis.
2.12. In vivo lung imaging
4T1 tumor‐bearing mice were anesthetized with isoflurane gas inhalation. Lung microcomputed tomography (CT) was done on a Quantum GX microCT Imaging System (standard 4 minutes, field of view = 36 mm), and used for 3‐D reconstruction with a volume‐rendering technique. Then, gross tumor volumes, lungs and bones were delineated on every section of the CT scans by the software Caliper Analyze.
2.13. Statistical analysis
Results are expressed as means ± SEM. Statistical tests were carried out using ANOVA and unpaired Student's t tests as required. Differences were considered statistically significant for P‐values <.05. All data were analyzed using SPSS v22.0 (IBM, Armonk, NY, USA).
3. RESULTS
3.1. Peroxisome proliferator‐activated receptor γ is not only expressed in tumor cells but also in cells of the TME, including tumor endothelial cells and TAM
To investigate the expression of the therapeutic target PPARγ, we first tested murine tumor cell lines CT26 and 4T1, murine macrophage RAW264.7 and HUVEC in vitro. Immunofluorescence staining and western blot indicated that all cell lines expressed the PPARγ protein (Figure 1A,B). Furthermore, PPARγ was expressed in tumor endothelial cells as well as in TAM from s.c. transplanted 4T1 and CT26 tumors (Figure 1C).
Figure 1.
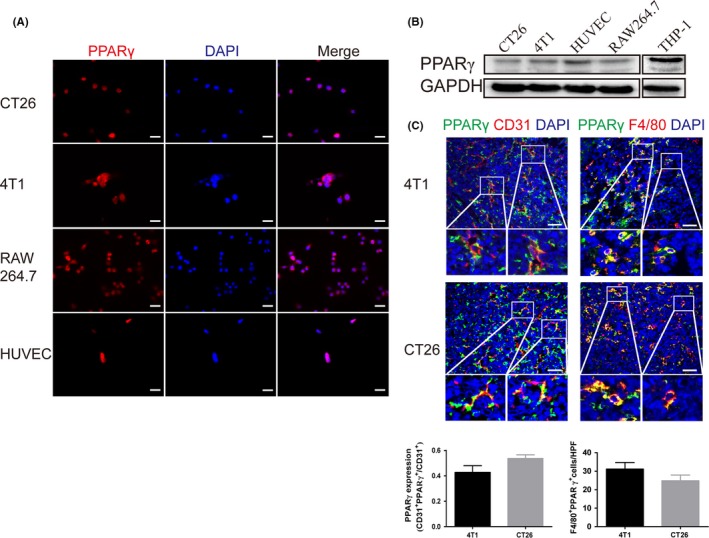
Peroxisome proliferator‐activated receptor γ (PPARγ) is expressed in tumor cells, endothelial cells and tumor‐associated macrophages (TAM). A, Immunofluorescence staining for PPARγ expression in cultured murine colon carcinoma cell line CT26, breast cancer cell line 4T1, macrophage line RAW264.7 and HUVEC. Scale bar, 20 μm. B, Western blot analysis of PPARγ expression in lysates from cultured 4T1 cells, CT26 cells, HUVEC and RAW264.7 macrophages. THP‐1 cells as positive controls. C, Double immunofluorescence staining for CD31 and PPARγ shows PPARγ expression in the endothelium of s.c. transplanted 4T1 and CT26 tumors. Double immunofluorescence staining for F4/80 and PPARγ indicates PPARγ expression in TAM of s.c. transplanted 4T1 and CT26 tumors. Scale bar, 50 μm
3.2. Rosiglitazone remodels the tumor vasculature, decreases intratumoral hypoxia and TAM infiltration and improves vessel perfusion
Abnormal tumor vasculatures lead to a hypoxic and immunosuppressive TME that facilitates tumor progression, invasion and radioresistance.6 Before treatment, we collected some tumor samples as pretreatment baseline. As the tumors progressed 20 days after tumor inoculation, great changes occur in the TME involving increased microvessel density and neovascularization, reduced pericyte coverage, poor microvessel perfusion, elevated hypoxia and TAM infiltration. However, rosiglitazone can reverse these changes (Figure 2). Overall, these data indicate that rosiglitazone treatment normalizes CT26 tumor vasculature and suppresses TAM recruitment.
Figure 2.
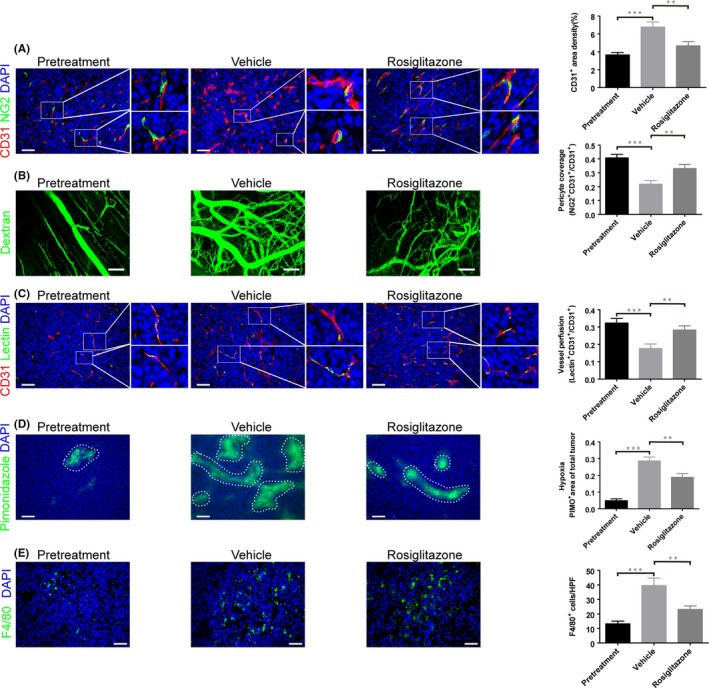
Rosiglitazone normalizes colon carcinoma vasculature and reduces tumor‐associated macrophage (TAM) infiltration. When CT26 tumor size approximately reached 50 mm3, the mice were given 100 mg/kg rosiglitazone or vehicle by oral gavage per day for 2 weeks. Then, the tumor samples were collected. A, Microvascular density and pericyte coverage were investigated by double immunofluorescence staining for CD31 and NG2. Scale bar, 50 μm. B, Multiphoton laser‐scanning microscopy images of CT26 tumor vasculature (green) stained with FITC‐dextran. Scale bar, 100 mm. C, Tumor vessel perfusion indicated by CD31‐positive endothelial cells stained red; FITC‐lectin perfused vessels are stained green. Scale bar, 50 μm. D, Representative micrographs show pimonidazole (PIMO)‐stained (green) CT26 tumor sections for hypoxia study. Scale bar, 100 μm. Hypoxic tumor regions are highlighted by dashed lines. E, Immunofluorescence staining for F4/80 showing TAM infiltration. Scale bar, 50 μm. Immunofluorescence images from (A to E) were captured within randomly selected fields (4‐6 fields per tumor, n = 6 mice). Data are presented mean ± SEM. **P < .01; ***P < .001
Furthermore, HUVEC were organized into network‐like structures in Matrigel in response to rosiglitazone, and this tube formation was inhibited by rosiglitazone in a concentration‐dependent method (Figure 3A‐C), indicating that rosiglitazone exerts antiangiogenic effects in vitro. The vascular endothelial growth factor (VEGF)/vascular endothelial growth factor receptor 2 (VEGFR2) signaling pathway is responsible for the survival, migration, vascular permeability and angiogenesis of endothelial cells. VEGF (50 ng/mL) significantly activated the VEGFR2/PI3K/Akt signaling pathway, whereas the combination of VEGF with 40 μmol/L rosiglitazone partially reversed this effect (Figure 3D). In addition, VEGF and MMP9 play an important role in angiogenesis. When CT26 and 4T1 cells were incubated with rosiglitazone for 48 hours, expression of VEGF and MMP9 proteins decreased (Figure 3E). In addition, hypoxia induced proangiogenic proteins including hypoxia‐inducible factor 1α (HIF‐1α), VEGF and MMP9, but rosiglitazone treatment downregulated the expression levels of these proteins compared with the vehicle group in vivo (Figure 3F).
Figure 3.
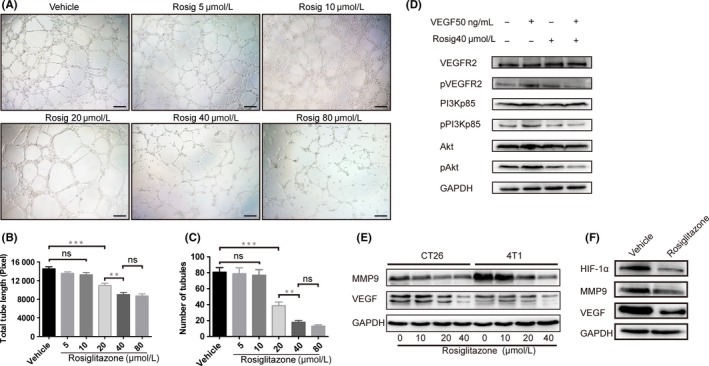
Peroxisome proliferator‐activated receptor γ (PPARγ) agonist rosiglitazone exerts antiangiogenic effects. A, Effect of rosiglitazone on tube formation by HUVEC. Scale bar, 100 pixels. Quantification of (B) total tube length and (C) number of tubules in bar graphs. Data are presented mean ± SEM (n = 3). **P < .01, *** P < .001; ns, non‐significant. D, Representative western blot shows the levels of total and phosphorylated vascular endothelial growth factor receptor 2 (VEGFR2), PI3K and Akt in HUVEC treated with 50 ng/mL vascular endothelial growth factor (VEGF) stimulation following 40 μmol/L rosiglitazone for 24 h. E, Representative western blot shows the expression of VEGF and MMP9 in CT26 and 4T1 cells treated with 0, 10, 20 and 40 μmol/L rosiglitazone for 48 h. F, Western blot analysis of hypoxia‐inducible factor 1α (HIF‐1α), MMP9 and VEGF protein expression in lysates from CT26 tumors treated with vehicle or 100 mg/kg rosiglitazone
3.3. Rosiglitazone combined with RT exerts a synergistically antitumor effect
To investigate the therapeutic potential of rosiglitazone combined with local RT, BALB/c mice were inoculated with CT26 and 4T1 cells. Compared with the vehicle treatment, rosiglitazone treatment mildly delayed the growth of s.c. transplanted CT26 and 4T1 tumor models. Furthermore, 6 Gy irradiation caused significant growth retardation of the irradiated tumors. However, tumor growth was markedly inhibited in response to RT combined with rosiglitazone (Figure 4A,B), indicating a synergistic interaction between the 2 treatments. The optimal schedule for combination treatment with rosiglitazone and RT was further investigated in CT26 tumor‐bearing mice. Irradiation (6 Gy) was given to the mice on either day 1 or day 5 of rosiglitazone treatment equivalent to 8 or 13 days after inoculation. No significant difference in tumor growth was observed between the groups on the 2 schedules; however, compared with radiation alone, rosiglitazone alone or the vehicle, the combination of rosiglitazone and irradiation markedly delayed tumor growth (Figure 4C).
Figure 4.
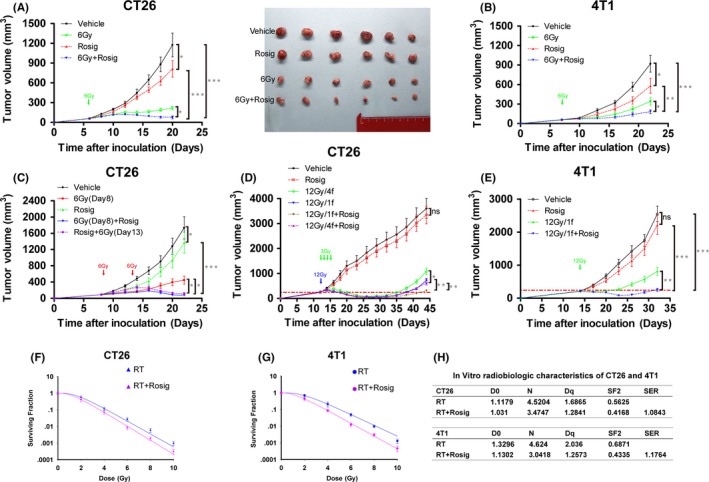
Rosiglitazone and radiotherapy exert a synergistic antitumor effect. A‐E, CT26 and 4T1 tumor growth curves. Data are presented mean ± SEM (n = 6 mice/group). Rosig, rosiglitazone. *P < .05, **P < .01, ***P < .001; ns, non‐significant. Radiation dose‐survival curves of (F) CT26 and (G) 4T1 tumor cells after radiotherapy (RT) or RT plus 40 μmol/L rosiglitazone (Rosig) treatment are obtained from the following multi‐target single‐hit model: S = 1 ‐ (1 ‐ e−D/D0)N. S = Survival fraction; e = natural logarithm; D0 = dose that decreases surviving fraction to 37%; D = dose; N = extrapolation number. Data are presented mean ± SEM (n = 3). H, Radiobiological parameters of CT26 and 4T1 cells
To test whether rosiglitazone could improve the antineoplastic effect of hypofractionated irradiation therapy (HFRT) or conventional fractionated irradiation therapy (CFRT), we used CT26 and 4T1 tumor models. We defined 12 Gy delivered in 1 fraction as HFRT, and 12 Gy delivered in 4 daily fractions of 3 Gy each as CFRT. As shown in Figure 4D, rosiglitazone had a slight effect on CT26 tumor growth; however, compared with vehicle treatment, HFRT or CFRT significantly slowed tumor progression. Treatment with a combination of HFRT or CFRT and rosiglitazone effectively suppressed tumor growth (Figure 4D). The efficacy was also validated in s.c. transplanted 4T1 tumors (Figure 4E). Next, radiation dose‐survival curves showed that rosiglitazone had a slight radiosensitizing effect on CT26 and 4T1 cells (Figure 4F‐H).
3.4. Rosiglitazone inhibits 4T1 tumor metastasis to the lungs
We investigated whether rosiglitazone could change the metastatic potential of 4T1 cells in vivo. Lung tissue samples were collected 32 days after s.c. tumor inoculation, and lung metastasis was finally confirmed by H&E staining. Interestingly, 100 mg/kg rosiglitazone markedly inhibited lung metastasis of 4T1 cells. Furthermore, no significant difference in lung metastasis was observed between the control and the 12 Gy (HFRT) group. Treatment with a combination of HFRT and rosiglitazone effectively suppressed lung metastasis, albeit without significant difference between the HFRT plus rosiglitazone group and the rosiglitazone alone group (Figure 5A,B). Chest CT scans also indicated similar results (Figure 5C). Because TAM have been proposed as important participants of the metastatic process, we also examined TAM infiltration in 4T1 primary tumors and lung metastases. Indeed, rosiglitazone evidently suppressed TAM infiltration into primary and metastatic sites (Figure 5D).
Figure 5.
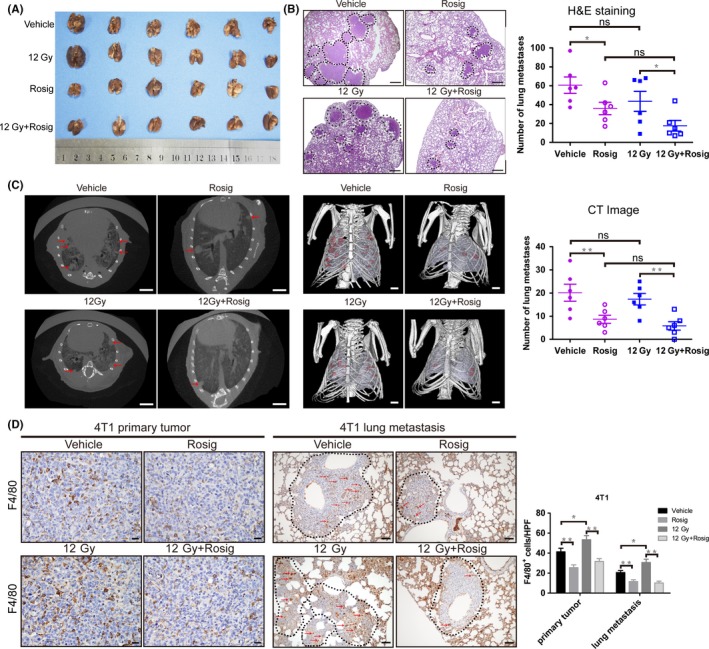
Rosiglitazone inhibits 4T1 tumor metastasis to the lung. A, Photographs of the lungs. B, Representative H&E‐stained lung sections and quantification of H&E‐stained lung metastasis (n = 6). Scale bar, 0.5 mm. Metastatic lung foci are highlighted by dashed lines. C, Representative microCT images of 4T1 mice (n = 6). Gross tumor volumes (red), lungs (pale purple) and bones (white) are delineated on every section of the CT scans. Scale bar, 2.5 mm. D, Immunohistochemical stain for F4/80 to indicate tumor‐associated macrophage infiltration in 4T1 primary tumor and lung metastasis (n = 6). Scale bars, 50 μm. Data are presented as mean ± SEM. Rosig, rosiglitazone. *P < .05, **P < .01; ns, non‐significant
3.5. Rosiglitazone reduces tumor‐derived CCL2 levels and restricts recruitment of myeloid cells or TAM to irradiated tumors, which delays tumor regrowth by disrupting vasculogenesis after HFRT
Hypofractionated irradiation therapy significantly delayed tumor progression. However, 2 weeks after HFRT, tumors in the HFRT group started to regrow. HFRT plus rosiglitazone was more effective in controlling tumor regrowth than was HFRT alone (Figure 6A). Next, we investigated the effect of HFRT on local angiogenesis. Two weeks after HFRT, the irradiated tumors had fewer endothelial cells and pericytes than the vehicle‐treated unirradiated tumors (Figure 6B). Thereby, 12 Gy HFRT significantly abrogated local angiogenesis, which contributes to tumor regression. However, when the tumors had completely relapsed 4 weeks after HFRT, endothelial cell number and pericyte coverage had nearly reverted to control values, indicating that vasculogenesis supported tumor recurrence. However, combined with HFRT alone, HFRT combined with rosiglitazone treatment effectively inhibited vasculogenesis and delayed tumor recurrence (Figure 6A,B).
Figure 6.
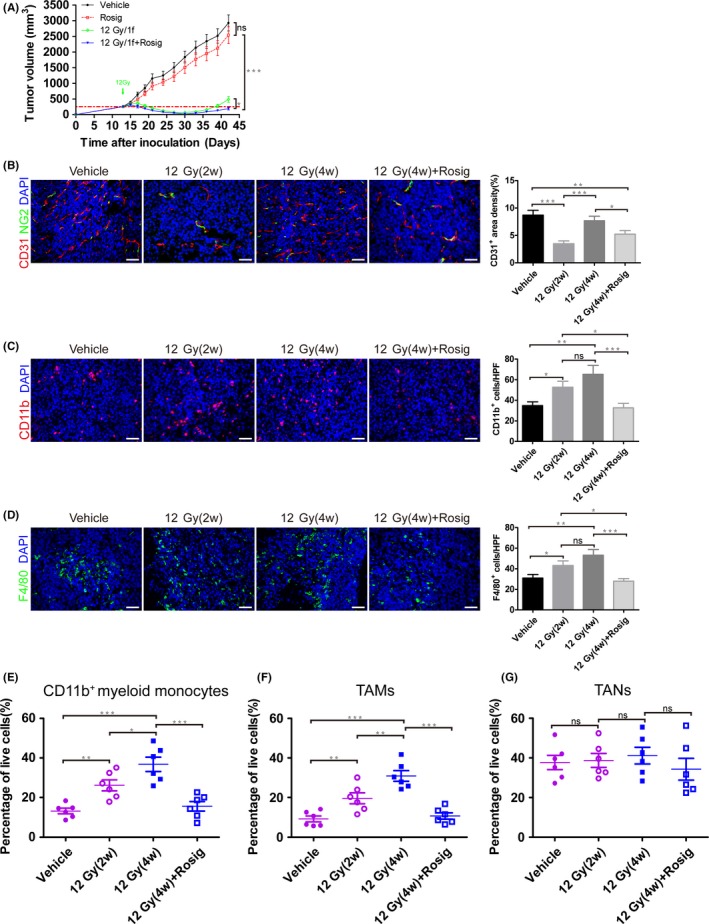
Rosiglitazone decreases the recruitment of myeloid monocytes and tumor‐associated macrophages (TAM) to irradiated tumor. A, CT26 tumor growth curve. B‐D, Immunofluorescence staining of CT26 tumor harvested from mice 2 wk after 12 Gy irradiation (12 Gy[2w]), recurrent tumors harvested from mice 4 wk after 12 Gy irradiation (12 Gy[4w]), and tumors harvested from mice 4 wk after 12 Gy irradiation plus daily 100 mg/kg rosiglitazone dosage for 14 consecutive days (12 Gy[4w] + Rosig). B, Microvascular density and pericyte coverage were investigated by double immunofluorescence staining for CD31 and NG2. C, Immunofluorescence staining for CD11b showing myeloid monocyte infiltration. D, Immunofluorescence staining for F4/80 showing TAM infiltration. B‐D, Scale bar, 50 μm. Immunofluorescence images from (B to D) were captured within randomly selected fields (4‐6 fields per tumor, n = 6 mice). E‐G, Flow cytometry analysis shows the effect of rosiglitazone on the infiltration of leukocyte subsets within CT26 tumors. Quantification of (E) myeloid monocytes, (F) TAM and (G) tumor‐associated neutrophils (TAN). Data are presented as mean ± SEM (n = 6). *P < .05, **P < .01, ***P < .001; ns, non‐significant
We also examined the infiltration of inflammatory cells involved in vasculogenesis. By immunofluorescence staining, tumors that relapsed after HFRT reflected a remodeled vasculature by increasing the recruitment of CD11b+ myeloid monocytes and TAM, which can be postponed by rosiglitazone (Figure 6C,D). In addition, using flow cytometry, we observed an increasing infiltration of leukocyte subsets within CT26 tumors after HFRT (Figure S1). Specifically, we found an increase in CD11b+ myeloid monocytes (Figure 6E) and TAM (Figure 6F), which can be inhibited by rosiglitazone. In contrast, we detected no change in TAN recruitment (Figure 6G).
CCL2 is an important chemoattractant involved in the recruitment of monocytes or TAM.21 We used ELISA to assess the potentiality of CT26 and 4T1 cells to produce CCL2 in response to irradiation. As expected, irradiation promoted CT26 and 4T1 cells to secrete CCL2 in a dose‐dependent method. However, compared with irradiation alone, irradiation combined with rosiglitazone treatment significantly reduced the level of CCL2 in both cell supernatant and serum (Figure 7A,B). Similarly, immunofluorescence staining for CCL2 showed that HFRT evidently upregulated CCL2 production, whereas rosiglitazone inhibited the expression of CCL2 in CT26 tumors after HFRT (Figure 7C). Next, Transwell chemotaxis assays were carried out to determine the importance of tumor‐derived CCL2 in recruiting TAM to the TME. Rosiglitazone inhibited the chemotactic migration of RAW264.7 macrophages (Figure 7D). Thus, rosiglitazone may reduce CCL2 secretion from tumors and reduces TAM infiltration to irradiated tumors.
Figure 7.
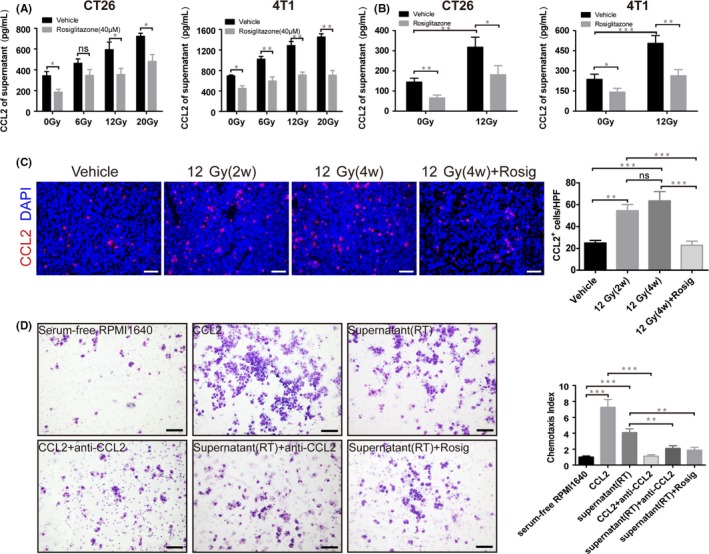
Rosiglitazone decreases the ability of CT26 and 4T1 cells to produce C‐C motif chemokine ligand 2 (CCL2) in response to radiotherapy and inhibits the chemotactic migration of RAW264.7 macrophages. CCL2 levels in culture (A) supernatant (n = 4) or (B) blood serum (n = 6) were determined by ELISA. C, Immunofluorescence staining for CCL2 from CT26 tumors. Scale bar, 50 μm. Immunofluorescence images were captured within randomly selected fields (4‐6 fields per tumor, n = 6 mice). D, Representative images and quantification of chemotactic migration of RAW264.7 macrophages (n = 3). Scale bar, 100 μm. Data are presented mean ± SEM. *P < .05, **P < .01, ***P < .001; ns, non‐significant
4. DISCUSSION
In the present study, we showed that the PPARγ agonist rosiglitazone exerts antiangiogenic and antineoplastic effects that inhibit the proliferative, invasive and metastatic potential of tumor cells (Figure 8A). RT displays prominent synergy with rosiglitazone that delays tumor recurrence by reducing the recruitment of myeloid monocytes and TAM to irradiated tumors and by disrupting vasculogenesis after RT (Figure 8B). Moreover, giving rosiglitazone does not affect the weight of CT26‐ or 4T1‐tumor bearing mice (Figure S2).
Figure 8.
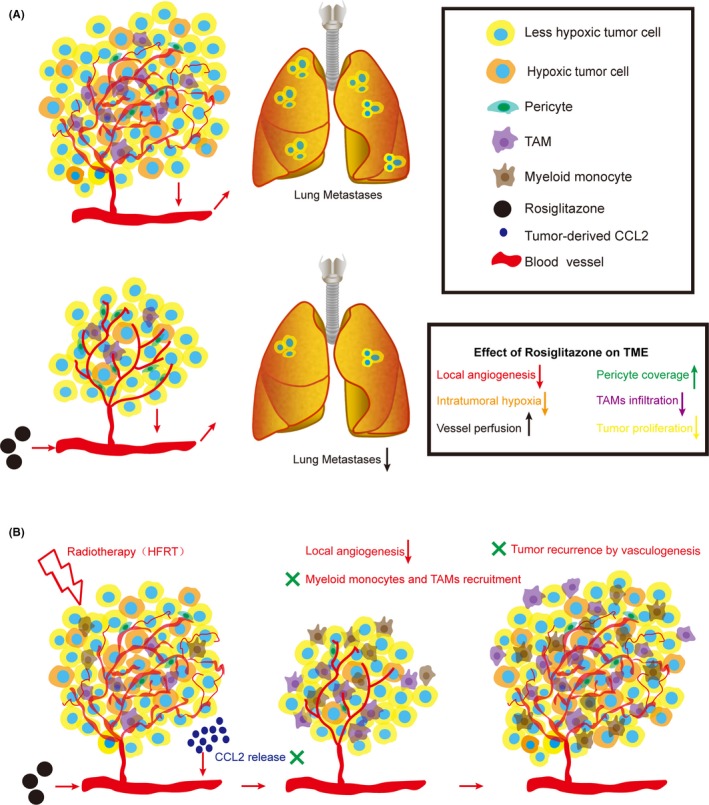
Model showing how rosiglitazone affects the tumor vasculature and inflammation and corresponding tumor progression, metastasis and recurrence. A, Rosiglitazone remodels the tumor vasculature, alleviates hypoxia, limits tumor‐associated macrophage (TAM) infiltrations and facilitates perfusion in tumor vessels, which delays tumor progression and decreases lung metastasis. CCL2, C‐C motif chemokine ligand 2; TME, tumor microenvironment. B, Hypofractionated irradiation therapy (HFRT) abrogates local angiogenesis to delay tumor growth. However, HFRT triggers the secretion of high levels of CCL2 from tumor cells to recruit CD11b+ myeloid monocytes and TAM to initiate vasculogenesis, and this process supports tumor recurrence after radiotherapy. Rosiglitazone, as an adjuvant therapy to HFRT, inhibits CCL2‐mediated tumor recurrence by disrupting the recruitment of CD11b+ myeloid monocytes and TAM and consequent vasculogenesis
Radioresponse is closely related to the TME. Recently, angiogenesis inhibitors, especially those targeting the VEGF signaling pathway, have made significant breakthroughs in cancer treatment from both clinical and preclinical perspectives.22 Consequently, angiogenesis inhibitors have been shown to improve the efficacy of RT,23, 24 thereby emerging as a novel therapeutic strategy. However, intrinsic or evasive resistance against antiangiogenic therapies is an intractable problem.25 Accordingly, the exploration of a novel antiangiogenic and therapeutic target is necessary and important.
More than 20 years have passed since the PPAR subgroups were first discovered.26 Rosiglitazone, one of the synthetic ligands of PPARγ with highly pharmacological specificity, selectivity and affinity, shows potentially antitumor and antiangiogenic effects.17 However, there are only a handful of studies investigating whether the antitumor effect of rosiglitazone is mediated only by its inhibitory effect on the tumor vasculature. Normalization of the tumor vasculature (an emerging concept in antiangiogenic therapy) leads to a more efficient delivery of blood and oxygen to cancer cells that can improve the antineoplastic efficacy of chemotherapy or RT.27 We discovered that rosiglitazone promotes vascular normalization, and these results may provide a fresh insight into the effect of rosiglitazone on improving radioresponse. We showed that rosiglitazone exerts these antiangiogenic effects by inhibiting the proangiogenic VEGF/VEGFR2 signaling pathway in vitro. VEGFA is the master regulator of angiogenesis, binding to VEGFR2 to stimulate the proliferation of endothelial cells through the RAS‐RAF‐MAPK‐ERK signaling pathway. VEGFA triggers endothelial cell migration, which is an integral component of angiogenesis.22 Hypoxia, a characteristic of solid tumors, is an important inducer of VEGF. In addition, rosiglitazone downregulates the expression of some key proangiogenic modulators such as HIF‐1α, VEGF and MMP9. Similar findings have been reported in another study in which another PPARγ ligand, troglitazone, was shown to downregulate the expression of MMP9 and MMP2 in leukemia K562 and HL‐60 cells in vitro.28 Rosiglitazone also decreased the expression levels of VEGF in human glioblastoma cells and murine Lewis lung carcinoma.29 The hypoxic TME elicits neovascularization in a HIF‐dependent method by mediating the recruitment of bone marrow‐derived cells to orthotopic glioblastomas.30 Further evidence indicates that MMP9‐expressing bone marrow‐derived myelomonocytes are sufficient to activate the angiogenic switch and are essential for the process of vasculogenesis; thus, these cells might become a novel therapeutic target for adjuvant therapy after HFRT.31 Therefore, it is reasonable to speculate that rosiglitazone delays vasculogenesis by reducing the production of these proangiogenic factors.
The synergistic interaction between rosiglitazone and RT is evident. When the tumor size reached approximately 50 mm3 in the early stage, the combination of rosiglitazone treatment and 6 Gy irradiation effectively controlled tumor growth. Furthermore, when 6 Gy irradiation was given either on day 1 or on day 5 of rosiglitazone treatment, no significant difference in tumor growth was observed between these 2 groups. When tumor size reached approximately 200‐300 mm3 in locally advanced stage, rosiglitazone alone showed limited antitumor efficacy. However, in comparison with HFRT or CFRT alone, combination of HFRT or CFRT with rosiglitazone effectively reduced tumor growth. Furthermore, the synergistic effect of HFRT plus rosiglitazone was superior to that of CFRT plus rosiglitazone.
Tumor‐promoting inflammation, a remarkable hallmark of cancer, has been regarded as a novel therapeutic target to improve the efficacy of RT.32 The potential anti‐inflammatory effects of PPARγ agonists lead to the speculation that some of the observed in vivo anti‐tumor effects are as a result of a reduction in tumor‐promoting inflammation. However, the mechanisms by which PPARγ ligands control tumor growth and exert anti‐inflammatory effects have not been rigorously investigated. Inflammatory immune cells, especially TAM, support tumor progression and angiogenesis, metastasis, immune suppression and regulate response to treatment.8, 9, 33 Tumor‐derived CCL2 is reported to be crucial for the recruitment of inflammatory monocytes and TAM to the TME.7 High numbers of TAM and inflammatory monocytes and high levels of CCL2 have been shown to be correlated with poor prognosis in some carcinomas, such as breast cancer, pancreatic ductal adenocarcinoma and hepatocellular carcinoma.34, 35, 36 Tumor‐derived CCL2 preferentially recruits inflammatory monocytes to promote lung metastases.37 CCL2 also stimulates the recruitment of metastasis‐associated macrophages (MAM) which is essential for extravasation and persistent growth of breast cancer cells.38, 39 Here, we showed that rosiglitazone inhibits CCL2 secretion by 4T1 cells and leads to a reduction in the number of infiltrating TAM at metastatic sites. It has been reported that the inhibition of macrophage PPARγ by rosiglitazone inhibits tumor cell proliferation in vitro. Furthermore, macrophage PPARγ is a crucial mediator of the antitumor effect of rosiglitazone in vivo. Deletion of macrophage PPARγ in mice not only facilitates mammary tumor progression but also weakens the antitumor effects of PPARγ agonists, and these effects are accompanied by an increase in the infiltration of CD11b+ myeloid cells and TAM with proinflammatory and proangiogenic phenotypes.19
Our findings indicate that tumor‐derived CCL2 mediates tumor recurrence after HFRT in s.c. transplanted CT26 tumor models. Similar results have been observed in pancreatic ductal adenocarcinoma.40 HFRT abrogates local angiogenesis that causes indirect tumor cell death. As expected, HFRT triggers the secretion of high levels of CCL2 from tumor cells, leading to the recruitment of CD11b+ myeloid monocytes and TAM. These cells are highly proangiogenic, which can initiate vasculogenesis and support tumor recurrence after RT.13, 31, 41 Therefore, the CCL2‐mediated inflammatory response can be considered a novel therapeutic target for improving the efficacy of RT.
In conclusion, we provide evidence that the PPARγ agonist rosiglitazone displays effective antineoplastic and anti‐metastatic potential. Moreover, rosiglitazone may promote normalization of the tumor vasculature and limit TAM infiltration, thereby providing a fresh perspective on delayed tumor progression. Importantly, the synergistic interaction between rosiglitazone and RT is evident, which indicates this combination as a powerful antitumor treatment. Rosiglitazone, as an adjuvant therapy to HFRT, inhibits CCL2‐mediated tumor recurrence by reducing the recruitment of CD11b+ myeloid monocytes and TAM and consequent vasculogenesis. Therefore, combination of the PPARγ agonist rosiglitazone with RT might represent a novel therapeutic strategy for improving efficacy of RT and for promoting local tumor control, decreasing distant metastasis risks and delaying tumor recurrence.
CONFLICT OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENT
This work was supported by National Natural Science Foundation of China (81672982, 81602670).
Huang G, Yin L, Lan J, et al. Synergy between peroxisome proliferator‐activated receptor γ agonist and radiotherapy in cancer. Cancer Sci. 2018;109:2243‐2255. https://doi.org/10.1111/cas.13650
Contributor Information
Jianxin Xue, Email: killercell@163.com.
You Lu, Email: radyoulu@hotmail.com.
REFERENCES
- 1. Jaffray DA. Image‐guided radiotherapy: from current concept to future perspectives. Nat Rev Clin Oncol. 2012;9:688‐699. [DOI] [PubMed] [Google Scholar]
- 2. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15:409‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin OA, Anderson RL, Narayan K, MacManus MP. Does the mobilization of circulating tumour cells during cancer therapy cause metastasis? Nat Rev Clin Oncol. 2017;14:32‐44. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 5. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393‐410. [DOI] [PubMed] [Google Scholar]
- 6. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature. 2008;454:436‐444. [DOI] [PubMed] [Google Scholar]
- 8. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618‐631. [DOI] [PubMed] [Google Scholar]
- 9. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447‐462. [DOI] [PubMed] [Google Scholar]
- 10. Garcia‐Barros M, Paris F, Cordon‐Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155‐1159. [DOI] [PubMed] [Google Scholar]
- 11. Park HJ, Griffin RJ, Hui S, Levitt SH, Song CW. Radiation‐induced vascular damage in tumors: implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS). Radiat Res. 2012;177:311‐327. [DOI] [PubMed] [Google Scholar]
- 12. Brown JM, Carlson DJ, Brenner DJ. The tumor radiobiology of SRS and SBRT: are more than the 5 Rs involved? Int J Radiat Oncol Biol Phys. 2014;88:254‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang W, Zhu XD, Sun HC, et al. Depletion of tumor‐associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16:3420‐3430. [DOI] [PubMed] [Google Scholar]
- 14. Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70:5679‐5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac‐1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. 2010;107:8363‐8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu T, Xie C, Ma H, et al. Gr‐1+ CD11b+ cells facilitate Lewis lung cancer recurrence by enhancing neovasculature after local irradiation. Sci Rep. 2014;4:4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator‐activated receptor gamma agonists. Lancet Oncol. 2004;5:419‐429. [DOI] [PubMed] [Google Scholar]
- 18. Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748‐759. [DOI] [PubMed] [Google Scholar]
- 19. Cheng WY, Huynh H, Chen PW, Pena‐Llopis S, Wan YH. Macrophage PPAR gamma inhibits Gpr132 to mediate the anti‐tumor effects of rosiglitazone. eLife. 2016;5:1‐20. pii: e18501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315‐2319. [DOI] [PubMed] [Google Scholar]
- 21. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferrara N, Adamis AP. Ten years of anti‐vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016;15:385‐403. [DOI] [PubMed] [Google Scholar]
- 23. Dings RP, Loren M, Heun H, et al. Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clin Cancer Res. 2007;13:3395‐3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin‐1, and matrix metalloproteinases. Cancer Cell. 2004;6:553‐563. [DOI] [PubMed] [Google Scholar]
- 25. Bergers G, Hanahan D. Modes of resistance to anti‐angiogenic therapy. Nat Rev Cancer. 2008;8:592‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645‐650. [DOI] [PubMed] [Google Scholar]
- 27. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58‐62. [DOI] [PubMed] [Google Scholar]
- 28. Liu J, Lu H, Huang R, et al. Peroxisome proliferator activated receptor‐gamma ligands induced cell growth inhibition and its influence on matrix metalloproteinase activity in human myeloid leukemia cells. Cancer Chemother Pharmacol. 2005;56:400‐408. [DOI] [PubMed] [Google Scholar]
- 29. Panigrahy D, Singer S, Shen LQ, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Du R, Lu KV, Petritsch C, et al. HIF1 alpha induces the recruitment of bone marrow‐derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahn GO, Brown JM. Matrix metalloproteinase‐9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow‐derived myelomonocytic cells. Cancer Cell. 2008;13:193‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239‐253. [DOI] [PubMed] [Google Scholar]
- 33. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404‐3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li X, Yao W, Yuan Y, et al. Targeting of tumour‐infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157‐167. [DOI] [PubMed] [Google Scholar]
- 37. Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature. 2011;475:222‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kitamura T, Qian BZ, Soong D, et al. CCL2‐induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis‐associated macrophages. J Exp Med. 2015;212:1043‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kalbasi A, Komar C, Tooker GM, et al. Tumor‐derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2017;23:137‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials