

Abstract
Invadopodia and podosomes are discrete, actin-based molecular protrusions that form in cancer cells and normal cells respectively in response to diverse signaling pathways and extracellular matrix cues. Although they participate in a host of different cellular processes, they share a common functional theme of controlling pericellular proteolytic activity, which sets them apart from other structures that function in migration and adhesion, including focal adhesions, lamellipodia, and filopodia. In this review, we highlight research that explores the function of these complex structures, including roles for podosomes in embryonic and postnatal development, in angiogenesis and remodeling of the vasculature, in maturation of the post-synaptic membrane, in antigen sampling and recognition, and in cell-cell fusion mechanisms, as well as the involvement of invadopodia at multiple steps of the metastatic cascade, and how all of this may apply in the treatment of human disease states. Finally, we explore recent research that implicates a novel role for exosomes and microvesicles in invadopodia-dependent and invadopodia-independent mechanisms of invasion, respectively.
Keywords: invadosome, invadopodia, podosomes, Tks5, MT1-MMP, metastasis, invasion, 3D growth, F-actin, degradation
Graphical Abstract
Invadosomes are recently discovered, highly complex molecular structures that couple actin polymerization with proteolytic activity to orchestrate extracellular matrix remodeling and invasion processes. In this review, we highlight novel research that delineates the functions of invadosomes, as well as how these molecular structures may be therapeutically targeted in various human diseases.
INTRODUCTION
Cellular invasion plays vital roles in homeostasis and is employed by many different cell types throughout the lifetime of the organism, including during embryonic development, immune surveillance and response, and wound healing [1, 2]. In cancer, tumor cells hijack the molecular components of normal cellular invasion to achieve degradation of the basement membrane and subsequent invasion into the extracellular matrix (ECM) and the underlying vasculature, which begins the metastatic cascade of aggressive tumors [3]. The ECM, which refers to both the basement membrane and the interstitial matrix, is a complex, yet dynamic structural component found in and between all tissues [4, 5]. Comprised of many different macromolecules, including fibrillar collagens, proteoglycans, glycoproteins and polysaccharides, the ECM is a molecular scaffold that provides underlying support to promote tissue boundaries, architecture and integrity [6]. During cellular invasion processes, cells must be able to degrade and remodel the ECM. One way in which cells accomplish this task is through podosomes and invadopodia (sometimes collectively referred to as “invadosomes”) [7], which are dynamic protrusions of the plasma membrane found in both normal (referred to as podosomes) and in cancer cells (referred to as invadopodia) [8–12].
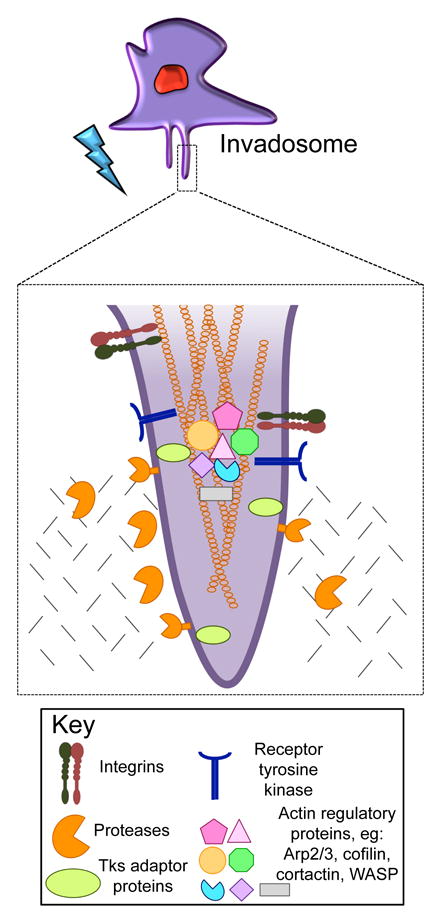
Podosomes and invadopodia are complex molecular structures comprised of a dense filamentous (F)-actin core containing actin-regulating proteins, including polymerization activators, filament crosslinkers, nucleators and binders, surrounded by proteins involved in regulation, adhesion and scaffolding, including integrins, kinases, GTPases, and adaptor proteins [7, 12, 13]. Key components of invadosomes include the scaffold protein Tks5, the actin regulators cortactin, Wiskott-Aldrich syndrome protein family members (WASP or N-WASP) and cofilin, and membrane type I matrix metalloprotease (MT1-MMP or MMP14).
Tks5, a Src substrate, is a scaffold protein that localizes to, and is required for invadosome formation [14–16]. It contains an amino-terminal Phox homology (PX) domain, which binds to phosphorylated phosphatidylinositol lipids [17], as well as five Src homology 3 (SH3) domains, small non-catalytic domains that facilitate protein-protein interactions through the recognition of proline-rich residues [18]. In vitro studies have demonstrated that Tks5 is necessary for the ability of both normal cells and tumor cells to degrade and invade [15, 16, 19], and it has been shown to interact with numerous actin-remodeling proteins, including N-WASP, Grb2 and Nck2 [20, 21], as well as proteases, including the ADAM-family proteases [17]. Unlike many other invadosome proteins, Tks5 is not found in other protrusions and adhesions (such as lamellipodia, filopodia and focal adhesions) [15, 17, 19]. Furthermore, expression of Tks5 in non-invasive cancer cells drives the formation of invadopodia [15]. There are at least 3 isoforms of Tks5: Tks5α (Tks5long), Tks5β, and Tks5short [22, 23]. Tks5β and Tks5short are initiated at distinct internal promoters, and lack the PX domain. Only Tks5α contributes to invadosome formation [15]; furthermore, cancer cell lines in culture predominantly express Tks5α [15]. In lung adenocarcinoma, the ratio of Tks5α to Tks5short expression increases with tumor progression, and is a predictor of worse outcome [23]. High Tks5α expression is also a predictor of poor survival in breast cancer, particularly for those with stage I and II tumors [24]. Other studies have also noted a correlation between Tks5 expression and decreased survival, although these studies did not differentiate the Tks5 isoforms [25, 26].
Mature podosomes and invadopodia are sites of pericellular proteolytic activity, resulting in ECM degradation. Most investigators consider that this focal proteolysis is diagnostic of the presence of invadosomes, although one recent paper has described MT1-MMP and Src-dependent proteolysis at focal adhesions (FAs) [27]. Three classes of proteases have been reported at invadosomes; zinc-regulated matrix metalloproteases (eg MMP2, MMP9, MT1-MMP and the ADAMs family of sheddases), cathepsin cysteine proteases (eg cathepsin B); and serine proteases (eg seprase and urokinase-type plasminogen activator, or uPA) [7, 28]. Of these, MT1-MMP, a transmembrane MMP [29], has often been described as a master regulator of invadosome function [30–36]. As well as ECM degradation and remodeling, pericellular proteases can function in the control of cell growth, apoptosis, and in cell-cell communications [37], through the release of growth factors that have a high affinity for matrix proteins (eg fibroblast growth factor, or FGF and transforming growth factor- β, or TGF-β) [37], direct cleavage and activation of growth factors (eg TGF-β and interleukin-1 β) [38], and cleavage of cell surface receptors (eg FGF receptor 1) [37, 38]. Whether the localization of proteases to invadosomes is required for these diverse functions is an important but unanswered question.
The invadosome is considered a distinct cellular structure from other actin-based structures such as filopodia, lamellipodia and FAs [7]. FAs are the sites of attachment to, and signaling by, the ECM [39, 40]. Lamellipodia are thin, sheet-like cellular protrusions that are found at the leading edge of a migratory cell and which contain a branched network of actin filaments [41, 42]. Filopodia, which are often found extending from the lamellipodial actin network, are thin protrusions that contain tightly packed, parallel bundles of F-actin, and have been implicated in probing the cell environment, in cell-cell adhesion, and in guidance towards chemoattractant gradients in neuronal growth cones [43]. All of these structures are involved in cell-ECM interaction, but, with the caveat mentioned above, proteolytic activity is usually confined to invadosomes. Indeed, the co-localization of F-actin, Tks5 and ECM degradation is often regarded as diagnostic for invadosomes (Figure 1). Colocalization of actin and other proteins such as talin or Arp2/3 is also often used, but we caution that these proteins are also found together in FAs [44] and lamellipodia [45, 46], respectively.
Figure 1. Normal cells and cancer cells form podosomes and invadopodia, respectively, and degrade a gelatin matrix.

(A) A simplified schematic of a cell with invadosomes on top of a fluorescently-labeled gelatin matrix. Using the proteolytic activity of its invadosomes, the cell is able to degrade the gelatin matrix. In order to visualize the degradation activity, the cells are fixed and stained with phalloidin, a fluorescently conjugated F-actin probe. The use of an appropriate fluorescent wavelength allows for the observation of the fluorescently labeled, degraded gelatin. Both of these events are illustrated in the zoomed-in image directly underneath the invadosome. The colocalization of F-actin punctae and areas of degraded matrix (see zoomed-in illustration) are indicative of invadosome activity. (B) THP-1 cells differentiated into macrophages by PMA and (C) BxPC-3 pancreatic adenocarcinoma cells were seeded onto FITC-labeled gelatin and allowed to degrade. In the left-most images, cells were stained with phalloidin. The middle images show areas of degraded FITC-gelatin. Podosomes are observed in (B) and invadopodia are observed in (C) by the colocalization of F-actin punctae with degraded areas of gelatin, as seen in the merged image (actin/gelatin/nuclei). In (C), the cell nuclei were stained for using DAPI. In (B), no DAPI stain is provided because it obstructed the F-actin staining. Images were taken at 20x (macrophages, in (B)) and at 63x (BxPC-3 cells, in (C)) magnification. Scale bars = 20 μm. Images courtesy of Dr. A. Kuipers and Dr. P. Saini of Oregon Health and Science University, Portland, Oregon, USA
Not surprisingly, many of the studies on podosome biology have been conducted in cells of the immune system, which rely on invasion not only to navigate through the ECM, but also to degrade and remodel blood vessels for entry into circulation. Leukocytes [47], macrophages [47], megakaryocytes [48], eosinophils [49], and dendritic cells [50, 51], have all been documented to form podosomes, either constitutively or under specific stimulation. However, podosomes have also been observed in a number of other normal cell types, including osteoclasts [52, 53], aortic endothelial cells [31, 54], myoblasts [55], bronchial epithelial cells [56], neural crest cells [22], and in neuronal growth cones [57]. Similarly, many cancer cell types form invadopodia, including pancreatic [58], ovarian [59], prostate [16, 60], brain [61], skin [62], bladder [63–65], head and neck [66], breast [24], and melanoma [34].
Are invadopodia and podosomes distinct structures? There are several reports of differences. For example, invadopodia have been observed to last hours in culture, whereas podosomes typically have a lifespan of minutes [7, 67]. Moreover, it has been documented that podosomes do not protrude as far into the substrata as invadopodia, with protrusive lengths of 0.5–2 μm and greater than 2 μm, respectively [7]. Lastly, the cell membrane at invadopodia has been observed to undergo rapid morphological changes in the form of filament-like processes that extend into the underlying matrix; such a phenomenon was not observed at the cell membrane of podosomes [68]. We believe it is possible that differences in experimental conditions underlie many of the observed disparities between podosomes and invadopodia. A thorough examination on how culture conditions and matrix composition and rigidity impacts invadosome half-life and protrusion is lacking. However, one informative study suggests that invadopodia are able to sense and respond to a wide range of matrices, and that the initiation of invadopodia and the subsequent emergence of proteolytic activity are regulated by differences in substrate rigidity [69]. Furthermore, in one interesting study that used polycarbonate filters with pores that could be filled with ECM, it was shown that dendritic cell podosomes increased in length and width in response to ECM, suggesting that podosome formation is responsive to specific environments [50]. While minor differences in composition of invadopodia compared to podosomes (eg N-WASP rather than WASP) may be due to cell background differences, it seems most likely that invadopodia are the results of mimicry, or “hijacking” of the invasive behaviors of normal cell types. This concept was supported by a gene expression profiling study on cancer cells describing a host of differentially expressed genes involved in cell proliferation, apoptosis, survival, chemotaxis, and motility [70].
What triggers the formation of invadosomes? They can form in response to a variety of molecules, signaling pathways, and microenvironmental cues. For example, growth factors such as vascular endothelial growth factor (VEGF) [71, 72], platelet-derived growth factor (PDGF) [73], TGF-β [31, 74], and epidermal growth factors (EGF) [75–77], have all been shown to induce invadosomes. Other mechanisms that influence podosome and invadopodia formation include acidic pH [78–81], increased substrate rigidity and density [82–87], hypoxia [73, 76, 88–90], and reactive oxygen species (ROS) [91], each of which is considered a hallmark of aggressive tumors [6, 92–96].
In this review, we highlight the functions of both podosomes and invadopodia in diverse cellular processes. We also discuss invadopodia as critical players in multiple steps of metastasis, highlighting the recently found, novel role for invadopodia in 3D tumor growth. Throughout this review, we pay special attention to how the function of podosomes and invadopodia may be targeted in numerous disease states, including cancer initiation and the progression to metastasis, and developmental disorders, including Frank-ter Haar syndrome (FTHS) and Borrone dermato-cardio-skeletal syndrome (BDCS). Lastly, we end with a brief discussion on recent findings that implicate exosomes and microvesicles in invadopodia function.
Podosomes and invadopodia are present in diverse cell types, but have common functional themes
Podosomes in cells of the hematopoietic lineage
Osteoclasts, derived from monocytes, are large, multinucleated cells [97] that function to resorb bone matrix [98]. Using vesicles, endocytosis and exocytosis mechanisms, osteoclasts deliver proteases and hydrochloric acid to digest and degrade bone matrix and subsequently, rid themselves of the degradation products [98]. Osteoclasts were the first normal cell type described to make podosomes. In two independent studies, the addition of vitamin A to osteoclasts was shown to increase the adhesion capability both to glass substratum [99] as well as to bone laminae matrix [100], specifically by modulating the frequency of podosomes. This was the first evidence to suggest that podosomes may play significant roles in allowing the osteoclast to form a tight attachment to the bone matrix to be degraded and resorbed.
It was subsequently found that pH and calcium levels regulated podosome formation and bone resorptive activity in osteoclasts. In one study, increased extracellular pH surrounding osteoclasts resulted in decreased intracellular pH and calcium, and enhanced the formation of podosomes and thus adhesion to the matrix substrate [101]. On the other hand, exposure of osteoclasts to high extracellular calcium concentrations inhibited the formation of podosomes [102]. One pivotal study used bright-field and fluorescence microscopy to provide clear evidence for the function of podosomes in osteoclasts [53]. Rabbit osteoclasts were cultured on thin bone slices, and the resorption lacuna was visualized with bright-field microscopy. Subsequently, the osteoclasts were fixed and stained with rhodamine-conjugated phalloidin to identify areas of F-actin. By superimposing the bright-field images of the resorption lacuna with the fluorescent images of F-actin, it was clear that areas of F-actin correlated perfectly with the degraded bone, thus revealing podosomes as integral functional components in this pathway [53].
Since this early research, many studies have analyzed the contribution of podosomes to ECM degradation, as well as migration in three-dimensional environments, in a large variety of hematopoietic cell types, including those of both the myeloid and lymphoid lineage [47–51]. All of these cells are derived from hematopoietic stem cells, and terminally differentiate in the bone marrow. Once differentiated, these cells must make their way to the bloodstream and gain access to the vasculature to carry out their respective functions. It makes sense that many of these cell types would rely on podosomes for such migratory behavior, as they are primed to facilitate such activity.
The loss of podosomes in cells of the hematopoietic lineage has also been implicated in human disease. Wiskott-Aldrich syndrome (WAS) arises from mutations within the Wiskott-Aldrich syndrome protein (WASp) gene [103, 104], a hematopoietic-specific protein localized to the actin-core of podosomes [105]. WAS patients present with severe immunodeficiency, eczema and thrombocytopenia [106]. In one early defining study, it was observed that WASp deficiency lead to defects in podosome formation in patient-derived macrophages from WAS patients [105], strongly suggesting that impaired podosome formation is a contributing factor to the WAS immunodeficiency phenotype. Later, in studies with WASp knockout mice, it was shown that the re-introduction of WASp was sufficient to reinstate podosome formation in bone-marrow derived cells and to correct the immune deficiencies that both occurred in the knockout mice [107]. These key studies were instrumental in linking podosomes to the function of diverse cell types of the hematopoietic lineage, the lack of which can lead to various disease states [105, 108].
Podosomes in embryonic and postnatal development
During mammalian embryogenesis and postnatal development, cell migration is required to achieve the appropriate positioning of cells of differing lineages throughout the body [109]. Recent research has implicated podosomes in both processes, the disruption of which leads to numerous human disease states involving skeletal, ocular and cardiac tissues.
The first description and analysis of podosomes during embryogenesis comes from studies using zebrafish embryos. Morpholino targeting of the invadosome scaffold protein Tks5 resulted in numerous developmental defects, including decreased head and eye size, cardiac malformation, as well as irregularities in the lateral line and pigmentation [110]. Interestingly, the majority of the abnormalities in the Tks5 morphants were attributable to defects in tissues derived from neural crest cells (NCCs) [110]. These cells are derived from the ectoderm as a result of epidermal to mesenchymal transition, and give rise to pigment-producing melanocytes, skeletal and connective tissues found in the head and neck, medulla cells of the adrenal gland, and neurons and glial cells apart of the sensory, sympathetic and parasympathetic nervous systems [111, 112]. A combination of whole-mount in situ staining and confocal time-lapse microscopy experiments revealed that Tks5 morphants possessed significantly fewer migrating NCCs that were unable to migrate to the correct location within the developing embryo [110]. Additionally, the Tks5 morphant NCCs had a reduced number of protrusions compared to normal control cells. A murine NCC line was used to reveal podosome formation as marked by the colocalization of F-actin with cortactin, Tks5 and Arp2/3, in response to TGF-β or PMA, two molecules known to stimulate cell migration [110]. Inhibition of Src family kinases and depletion of Tks5 with RNAi both inhibited the formation of TGF-β and PMA-induced podosomes, indicating that the Src-Tks5 signaling pathway is likely needed for proper podosome function in NCCs. Taken together, this suggests that podosomes play important roles in embryonic development, specifically in NCCs and their ability to migrate correctly in three-dimensional space, to pattern the embryo and to generate tissues in the appropriate position. In keeping with this, the majority (90%) of homozygous Tks5 gene trap mice (Tks5trap/trap) died as neonates associated with complete cleft of the secondary palate, a neural crest-derived tissue [22].
Several studies have noted developmental defects caused by the loss of Tks4, the most highly related gene to Tks5. In one such study, homozygosity-mapping studies were used to map the genetic defect in 7 of 16 individuals with FTHS to a mutation of Tks4 [113]. FTHS is an autosomal recessive disease marked by skeletal, cardiac and ocular abnormalities [114]. Tks4 gene-trap mice, in which a gene-trap vector was placed between exons 3 and 4 in the Tks4 gene [113], showed striking phenotypic similarities to FTHS patients, including small body size, craniofacial defects, cardiac problems, loss of adipose tissue, and increased intraocular pressure accompanied by glaucoma and other ocular abnormalities, confirming that loss of Tks4 can cause FTHS. In a related study, linkage and sequence analysis identified a homozygous mutation in Tks4 as the underlying genetic cause for BDCS [115], a progressive disorder that affects the skin, joints, bone, and heart tissues, but unlike FTHS, no ocular abnormalities are observed [116, 117].
The nee mouse, first observed and identified by the Jackson Laboratory as a spontaneous mutant mouse, was initially characterized in an effort to find novel mouse models of human disease [118]. These mice exhibit numerous defects, including small body and skeleton size, infertility, abnormalities of the skull, hearing impairment, ocular abnormalities, and reduced bone mineral density. Linkage and sequence analysis on the nee mutant mice identified a single base pair deletion in the last exon of Tks4 as the cause [118]. In an effort to better understand the implications of the deletion, GFP fusion constructs for wild-type Tks4 and the nee mutant Tks4 were transfected into HEK239T cells, and the localization of each were assessed [118]. It was found that wild-type Tks4-GFP localized to the cytoplasm, while the nee mutant pervaded the entire cell, including the nucleus, suggesting that the localization of Tks4 is vital to its function and therefore successful mammalian development. Further studies on the nee mice showed that they develop early-onset glaucoma accompanied by high intraocular pressure and corneal opacity [119], consistent with the human syndromes described earlier. By three months of age, nee mice also demonstrated severe loss of retinal ganglion cells, a type of neuron located in the retina of the eye, along with the complete degeneration of optic nerve axons and a significant reduction in in the cross-sectional area of the optic nerve [119].
Despite the similarities between Tks4 and Tks5, and the fact that Tks4 is also an important component of invadosome formation and function [30, 34, 120], care must be taken in ascribing all these developmental phenotypes to loss of podosomes. For example, recent research using a Tks4 knockout mouse revealed a differentiation defect in mesenchymal stromal cells (MSCs), caused by the reduced expression of RunX2 and Osterix, two osteogenic transcription factors that are normally up-regulated during differentiation [121]. In an adipogenic assay, Tks4 knockout MSCs were unable to form lipid droplets in the cytoplasm [121]. While these studies could be interpreted to mean that podosomes somehow influence the expression of key genes involved in the differentiation of MSCs to osteoblasts and adipocytes, it is also possible that Tks4 has non-invadosome functions. This will require further investigation.
In ocular developmental processes, NCCs from the developing forebrain and midbrain migrate and differentiate into the corneal endothelium and stroma, trabecular meshwork, the iris stroma, and the ciliary body and stroma [111]. Severe congenital eye diseases affecting the anterior segment of the eye result when NCCs are unable to migrate properly within the ocular structure, including Axenfeld-Rieger syndrome, primary congenital glaucoma, and congenital ocular coloboma [111]. Moreover, recent studies have shown that NCCs can also differentiate into pericytes and vascular smooth muscle cells, two cell types that play significant roles in maintaining proper vasculature [111]. Within the eye, both pericytes and vascular smooth muscle cells surround endothelial cells within the hyaloid artery and the surrounding blood vessels of the optic nerve [111, 122–124] Disruption of proper blood vessel formation within ocular tissues results in congenital eye diseases, including colobomas and microphthalmia, while depletion of pericytes within retinal vasculature is a prominent cause of diabetic retinopathy [111]. As will be discussed later, endothelial cell podosomes function during angiogenic sprouting [31, 54, 71, 72, 74]. Notably, the Tks5 morphants had small eyes, with three-fold less volume when compared to control embryos [110]. Likewise, the nee mutant mice [118, 119] and the Tks4 gene trap mice [113] demonstrated multiple ocular abnormalities, suggesting that podosome function is vital to eye development, the disruption of which is responsible for ocular disease. It will be interesting to determine whether the NCC-derived pericytes and vascular smooth muscle cells form podosomes that perhaps function in blood vessel maintenance and remodeling within the eye, especially considering the fact that endothelial cells [31, 54], vascular smooth muscle cells [125–127], and NCCs [110] have been shown to form podosomes. It is also possible that congenital eye defects are partly caused by an underlying lack of NCC podosome function, which may inhibit the proper cellular migration and subsequent differentiation of these stem cells into their proper derivatives. Further research is needed to determine whether the Tks proteins and podosomes can be targeted therapeutically in congenital eye diseases, including glaucoma, coloboma, microphthalmia, and Axenfeld-Rieger syndrome.
Podosomes in angiogenesis and vasculature remodeling
Endothelial cells, derived from mesoderm, are a type of epithelial cell that form the interior surface of all blood and lymphatic vessels [128]. These cells function in barrier maintenance, blood clotting, angiogenesis, vasoconstriction and vasodilation [128]. Recent research has implicated podosomes in endothelial cell biology, where they function in degradation of basement membrane, and facilitate the formation of new blood vessels in sprouting angiogenesis mechanisms.
The first evidence that podosomes play roles in endothelial cell biology came from experiments on aortic endothelial cells under exposure to cytotoxic necrotizing factor 1 (CNF1), a Rho GTPase activator [54]. Rho GTPase has previously been implicated in mechanisms of cytoskeleton rearrangements, and this study aimed to understand what role it played in endothelial cells. Punctate podosome-like structures were observed on the ventral side of the endothelial cells, marked by the characteristic F-actin surrounded by vinculin [54]. This study also unveiled the presence of Arp2/3, WIP and N-Wasp in the podosome architecture of endothelial cells.
Podosomes in endothelial cells, like those in cells of hematopoietic origin, are capable of degradation. Multiple studies have described the presence of podosomes in vascular smooth muscle cells in vitro [125, 129–133]. We showed that A7r5 vascular smooth muscle cells formed functional podosomes when exposed to PDGF, in a process involving microRNA (miR) regulation of p53 and Src [127]. In vivo immunoelectron microscopy analysis of aortas from miR-143 KO mice showed the formation of podosomes, as marked by the presence of cortactin and Tks5 [127], providing the first evidence that podosomes may play roles in various vascular pathologies, including atherosclerosis, aneurysm, and restenosis. Aortic endothelial cells exposed to TGF-β form podosomes containing MT1-MMP and MMP9, with MT1-MMP being necessary for local degradation and invasion through a collagen matrix [31]. These findings were expanded in an ex vivo endothelium observation model [74]. These data suggest that podosomes are key components in vasculature remodeling and healing mechanisms. In fact, podosomes have recently been implicated in sprouting angiogenesis, whereby endothelial cells, responding to specific environmental cues, break down the vascular basement membrane and sprout to form new blood vessels [71]. Treatment of endothelial cells with vascular endothelial growth factor A (VEGF-A) induced the formation of podosome rosettes, specifically through the up regulation of integrin α6β1 [71]. Moreover, using a mouse aortic ring assay, it was shown that the formation of podosome-like rosettes, as measured by the colocalization of F-actin, cortactin and MT1-MMP, directly preceded the creation of new blood vessel branch points [71].
How do podosomes accomplish this task? It is possible that after facilitating the breakdown of the blood vessel basement membrane and ECM through the coordinated actions of proteases, podosomes may somehow serve as a recognition site for the synthesis of new blood vessels. Or, it may be that podosomes are only needed to create an open space for the creation of new blood vessels, and depletion of ECM stimulates sprouting. In a very recent in vivo approach using a mouse retinal neovascularization model, the endothelial tip cells, which conduct the formation of growing capillaries in angiogenesis, were shown to form podosomes in response to VEGF-A/Notch signaling [72]. Collagen-IV was degraded under areas of podosome presence, indicating that tip cell podosomes break down the underlying basement membrane in the endothelium, thus allowing for the expansion and growth of new vessels [72]. Taken together, such research implies that podosomes could be potential therapeutic targets to block inappropriate angiogenesis, including tumor angiogenesis.
Podosomes in the maturation of the post-synaptic membrane
Synaptogenesis, or the creation of new synapses, is a highly complex process that has been studied extensively in the neuromuscular junction (NMJ) [134]. The NMJ is comprised of three cell types: the motor neuron, the myofiber, and the Schwann cell [134]. When a motor neuron receives extracellular cues, it depolarizes and releases acetylcholine (Ach), a neurotransmitter. ACh migrates across the synaptic cleft to bind to acetylcholine receptors (AChRs) on the membrane of the myofiber, causing membrane depolarization and subsequent muscle contraction [134]. During the formation and maturation of the synapse, AChRs are made and assembled into small plaques in the membrane of the myotube, a multinucleated muscle fiber comprised of fused myoblasts [134]. In a surprising finding, it was recently discovered that cultured myotubes form podosomes at sites of AChR clusters [135]. Such podosomes are thought to remodel the clusters into pretzel-shaped patterns, an integral step in synapse maturation. Intriguingly, the synapse podosomes contained many canonical podosome proteins, including F-actin, Arp2/3, cortactin, Tks5, vinculin, and Src [135]. Moreover, synapse podosomes acted as sites of attachment to the underlying substratum, and were capable of remodeling ECM [135]. What role do podosomes play in the remodeling of the post-synaptic membrane? It is possible that podosome degradation of matrix components stimulates the appropriate distribution of the AChRs laterally across the membrane to form the appropriate pretzel shape of a mature synapse [135].
There is additional evidence to suggest that podosomes are involved in the NMJ, and likely in neuron development. Interestingly, neuronal growth cones have been observed to form podosome-like protrusions that are capable of degradation and that contain F-actin colocalized with Tks5, cortactin, Arp2/3, N-WASP, Mena, and α-actinin [57]. Such growth cone invadosomes are necessary for the proper extension of the motor neuron axons into the peripheral myotome tissue during development in vivo [57]. Moreover, the dystroglycan receptor, a transmembrane adhesion receptor found in skeletal muscle cells that links the actin cytoskeleton of the cell to extracellular laminins, was shown to interact with Tks5 via its SH3 domain to regulate podosome formation in myoblasts [55]. Dystroglycan also interacts with Grb2 [136], a protein localized to the invadosome [137]. In a dystroglycan-null chimeric mouse model, the dystroglycan complex was shown to play a major role in the formation of the synaptic basement membrane, as well as in the organization and stabilization of AChR clusters at NMJs [138]. Taken together, these data suggest that podosomes may play a significant role in pathways that form and stabilize the post-synaptic membrane, possibly through interactions between Tks5 and the dystroglycan complex.
Podosomes in antigen sampling and recognition
The function of the adaptive immune system is twofold: to eliminate foreign pathogens from the host, and to create a molecular memory of the pathogen in order to provide an enhanced response to it, should re-infection ever occur [139]. In the adaptive immune system, dendritic cells function to engulf foreign pathogens, (including bacteria, toxins and parasites) and subsequently present pathogen antigens to T cells. Dendritic cells form podosomes [51, 140–143], which play a role in antigen processing [50, 144]. Dendritic cells express pattern recognition receptors (PRRs), which efficiently scan and recognize pathogen-associated molecular patterns (PAMPs) on foreign invaders. The molecular interaction between a PRR and an Ag triggers endocytosis and/or phagocytosis to engulf the pathogen. In transmission electron microscopy (TEM) studies, dendritic cells grown on a gelatin-gold matrix produced sites of degradation coupled with the collection of gold particles in membrane-bound organelles within the podosome [50]. In cells lacking podosomes, there was less uptake of gold particles. Furthermore, MMP14-null dendritic cells were incapable of degrading gelatin-gold matrix and showed no internalized gold particles [50]. In similar studies, dendritic cells grown on filters with 1 μm pores were immunostained for various PRRs known to play roles in Ag uptake [144]. Interestingly, all PRRs in question localized to the podosomes, as well as CD71, a receptor involved in endocytosis [144]. Live cell imaging combined with fluorescence microscopy showed that the presence of podosomes corresponded with the uptake of ovalbumin, a well-characterized Ag, through the filter pores; such ovalbumin uptake was abolished when endocytosis was inhibited [144]. Taken together, these studies suggest that podosomes may be involved in the endocytosis required for antigen uptake.
In addition to dendritic cells, cells of the vascular endothelium also act as antigen presenting cells (APCs) [145]. In one recent study, an endothelial cell APC model was used to better understand how T cells create close connections with other cells in order to facilitate Ag sampling [146]. Live cell imaging analysis demonstrated that endothelium exposed to Ag was sufficient to activate CD4 T cells, a type of memory T cell of the adaptive immune system [146]. Next, using fluorescent membrane markers in the endothelium, it was shown that antigen-activated T cells produce cylindrical invaginations in the cell surface as they migrate, termed “podo-prints” [146]. Intriguingly, the “invadosome-like protrusions” (ILPs) formed by the T-cells that produce the podo-prints arranged themselves into rosettes [146], which are often observed in podosome formation [7]. Confocal microscopy showed that the ILPs formed by the T-cells were rich in F-actin and talin [146], two proteins implicated in, but not limited to the invadosome. Regardless, ILPs likely serve a role in the initial Ag recognition and subsequent T-cell receptor activation. Furthermore, when activated CD8 cytotoxic T lymphocytes were exposed to murine heart microvascular cells exposed to Ag, lysis in the endothelium was observed after 4 hours, suggestive of podosome-like proteolytic activity [146].
ILPs might be considered “lymphocyte equivalents” to podosomes and invadopodia, although ILPs were observed on cellular substrates (endothelium), whereas podosomes and invadopodia form on ECM. It is tempting to speculate that ILPs are podosomes that form on activated T cells in response to Ag stimulation, allowing for efficient Ag scanning by re-arranging the cytoskeletal components of both cells coming into close contact with one another. The thick proteoglycan coat found on all cell types represents an important energy barrier in the process of Ag recognition [147]. It is plausible that the lytic capabilities of ILPs may in essence re-arrange the architecture of both cell surfaces as they approach each other.
Podosomes in cell-cell fusion mechanisms
Cell fusion is an energy-requiring process by which two or more cells fuse, leading to the formation of multinucleated cells [148]. Fusion must occur during fertilization [149], osteoclastogenesis [150] and myotube formation [151]. It is hypothesized that in the case of osteoclastogenesis, the fusion of two or more osteoclasts results in much higher efficiency during bone resorption processes, although this is still not fully understood [152]. In a microarray analysis, non-treated and RANKL-treated macrophages (osteoclasts differentiate from macrophages under the influence of RANKL) were analyzed with respect to gene expression changes, with an effort to identify genes containing either a PH or PX domain [153]. This analysis identified Tks5, which was found to be present in the plasma membrane of osteoclasts undergoing fusion events [153]. Knockdown of Tks5 resulted in a significant decrease in podosome formation, and prevented osteoclast fusion [153]. In a co-culture assay with RANKL-treated macrophages and GFP-nuclear B16F9 melanoma cells, multinucleated osteoclasts with nuclear GFP were often observed [153]. Such heterocellular fusion was eradicated upon Tks5 depletion, demonstrating that Tks5-mediated invadosomes likely play vital roles in the fusion machinery responsible for cell-cell fusion. Although the mechanism for how this is accomplished remains unknown, it opens an avenue for further investigation into invadosomes in cell-cell fusion in cancer as a potential therapeutic target [154–158]. As myoblasts must fuse to form the myotube, and podosomes have recently been implicated in the appropriate positioning of AChRs on the myotube membrane, it will be interesting to determine whether podosomes also play a role in myoblast fusion.
Invadopodia function throughout metastasis
Metastasis is the process by which tumor cells disseminate from the primary tumor site to distant organs and tissues [159, 160] (Figure 2). It is comprised of five main steps: local invasion, intravasation into a blood vessel, survival in the blood vessel, extravasation out of the blood vessel, and colonization of a new site (Figure 2). In the metastatic cascade, tumor cells must first degrade the underlying basement membrane and invade the ECM in order to reach the circulatory system (Figure 2). During intravasation, tumor cells use MMPs and uPA to proteolytically break down the basal membrane of a blood vessel and enter into it [161–163]. Here, we focus on the involvement of invadopodia in numerous steps throughout metastasis.
Figure 2. Invadopodia function throughout metastasis.

A simplified illustration of the metastatic cascade is shown, with numbered steps. Cancer begins when a normal cell, like an epithelial cell, undergoes a transformative event, and becomes a non-invasive tumor cell. Upon acquiring additional mutations, the primary tumor grows in size (1), and some cells become invasive. Invasive tumor cells use the proteolytic activity of invadopodia to degrade the underlying basement membrane (2) and invade through the ECM (represented in the illustration by the presence of fragmented ECM proteins) (3). It is well established that some tumor cells can undergo EMT and are mesenchymal-like during migration and invasion, as represented in the illustration. By using protease activity to break down the basement membrane of the blood vessel, invadopodia function in intravasation of the tumor cell into the blood stream (4). The tumor cell survives and travels in circulation (5), until, through unknown mechanisms, it arrests and uses invadopodia to extravasate from the blood vessel (6). Tumor cells use invadopodia to invade at distant tissue locations. Upon colonization, tumor cells can undergo MET and become more epithelial-like as they form a metastatic growth, which relies upon invadopodia (7), although the mechanisms remain unknown. Angiogenesis occurs at the metastatic site to allow tumor cells access to circulation (8). Specific steps in metastasis, including (1) primary tumor growth, (5) survival in circulation, and (8) angiogenesis at metastatic sites, require further research to understand whether invadopodia are involved or not, and are marked by a question mark. The other steps, including (2) basement membrane degradation, (3) ECM remodeling and invasion, (4) intravasation into vasculature, (6) extravasation out of vasculature, and (7) growth of tumor cells at metastatic sites, have all been shown to involve invadopodia, and are marked with blue lightning bolts.
N-WASP functions to integrate output from diverse signaling pathways to control actin polymerization in the cell, by binding to and activating the actin related protein 2 and 3 (Arp2/3) complex [164]. N-WASP is an obligate component of invadopodia [165]; thus, modulating its activity is one method by which invadopodia function can be studied, although it should be noted that N-WASP also has other functions, for example in lamellipodium spreading [166]. In one elegant study, the role of N-WASP in the metastatic MTLn3 rat mammary adenocarcinoma cell line was evaluated [167]. Knockdown of N-WASP with shRNA or expression of a dominant negative form of N-WASP resulted in fewer tumor cells circulating in the blood, and fewer sites of metastases in the lung [167]. This provided the first evidence that invadopodia are likely important in the intravasation process. Further studies using intravital imaging and multiparametric analysis demonstrated the formation of Tks5-dependent invadopodia in slow moving cells migrating away from the primary tumor and towards blood vessels in MDA-MB-231 cells implanted orthotopically [168].
Although the exact process remains unknown, it is hypothesized that one way in which extravasation is initiated occurs when endothelial cells cluster around an arrested tumor cell in a small capillary and induce vasculature remodeling, thus allowing the tumor cell to escape from the blood vessel into the underlying stroma [169], although other mechanisms have also been described [170]. Two studies have shown that invadopodia function in extravasation. In the first, an in vivo chicken embryo model was used to demonstrate the pivotal importance of invadopodia in the extravasation of tumor cells from blood vessels into the extravascular stroma, as defined by the requirement for Tks adaptor proteins [171]. This study showed that invadopodia form early in extravasation, and then protrude into the layers of the endothelial vessel at endothelial junctions. In keeping with this, the genetic or pharmacological inhibition of invadopodia components (cortactin, Tks4 or Tks5) significantly impacted the number of micrometastases formed in the lung of an experimental mouse metastatic lung model [171]. In another related study, cortactin was knocked down in a bladder cancer cell model [63]. Invadopodia formation was significantly decreased in the cortactin knockdown cell lines, and upon tail vein injection into nude mice for an in vivo lung metastasis assay, significantly fewer metastases were detected in the lungs, providing more support for the importance of invadopodia in the extravasation process [63]; although it should be noted that cortactin is not exclusively localized to invadopodia.
Do invadopodia have functions beyond intravasation and extravasation (Figure 2)? We have shown in 3 different model systems (mouse fibrosarcoma, human breast cancer, and mouse and human melanoma) that knockdown of Tks5 reduces the in vivo growth of tumors, in subcutaneous, orthotopic and metastatic sites [24, 34, 172]. Intriguingly, the Tks5-KD tumors formed by both subcutaneous and tail-vein injection displayed lower blood vessel density and blood vessel diameter when compared to controls, raising the possibility that tumor cell invadopodia may also play roles in tumor angiogenesis [24]. In our breast cancer and melanoma studies, we also observed that cells with reduced Tks5 expression grew less well in matrices of type I collagen in vitro [24, 34]. It is of course possible that the reduced tumor growth observed in vitro and in vivo could be due to loss of invadopodia-independent functions of Tks5. But, a survey of the literature reveals that loss of other invadopodia regulators also results in similar phenotypes. The earliest in vitro studies in support of invadopodia playing roles in regulating 3D growth also used a matrix of type-I collagen as a 3D environment [173]. Here, squamous carcinoma cells, pancreatic carcinoma cells and fibrosarcoma cells all exhibited a 50–80% reduction in 3D proliferative capacity when exposed to the MMP inhibitors, TIMP-2 and BB-94 [173]. The next study involved cortactin, which is a Src-substrate that plays integral roles in cell migration and invasion by controlling branched actin assembly through multiple diverse mechanisms, as well as protease trafficking [174]. Cortactin is an important component of invadopodia [174], although it is also found in lamellipodia [175, 176] and filopodia [176]. In studies using a semi-orthotopic rat trachea model, three different head and neck squamous cell carcinoma cell lines with cortactin knockdown developed significantly smaller tumors in vivo when compared to control tumors [177]. Finally, cyclin dependent kinase 5 (Cdk5) is a serine/threonine protein kinase that has diverse roles in neuronal growth cone support, cell motility, cell cycle progression, insulin secretion, immune system function, angiogenesis, lymph valve formation, and cancer progression [178]. Cdk5 is required for invadopodia formation and function, and for invasion in several cancer cell lines, by phosphorylating and inactivating caldesmon, an inhibitor of invadosome function [179]. One recent study implicated Cdk5 in the 3D growth of prostate cancer cells [180], and another in pancreatic cancer cells in vitro and in vivo [181]. Taken together, even though each of these proteins have other functions, these studies suggest the strong possibility that invadopodia function in the regulation of tumor growth in 3D. We are actively pursuing the mechanism of this growth regulation.
Exosomes and microvesicles: an independent mode of invasion, or necessary for invadopodia-mediated invasion?
Exosomes and microvesicles are membrane-bound vesicles derived from multivesicular bodies (MVBs) of endosomes and the plasma membrane, respectively [182]. As carriers of proteins, lipids and RNA, these vesicles are involved in cell-cell communication [182–184]. In studies aimed at identifying necessary components for invadosome biogenesis and function, it was observed that the exocyst (comprised of 8 subunits, and involved in tethering of post-Golgi and recycling endosomes at the plasma membrane) [185] is necessary for invadopodia formation, and for the secretion of MMPs into the extracellular environment [186, 187]. Around the same time, it was also shown that late lyosomal/endosomal v-SNARE TI-VAMP/VAMP7, a mediator of vesicle fusion with the plasma membrane, was necessary for the transport of MT1-MMP to the sites of invadopodia [188]. More recent studies show that the v-SNARE, VAMP3, is important in the delivery of MT1-MMP to shedding microvesicles [189]. And lastly, MT1-MMP has previously been observed within both exosomes and microvesicles [190, 191]. These studies provided the initial evidence suggesting that membrane-bound vesicles may have significant roles in invadosome function. However, two recent defining studies, both of which describe the importance of membrane-bound vesicles in the delivery of cargo necessary for invasion, raise important questions regarding whether there are one or two separate mechanisms at play during cellular invasion.
In the first of these studies, which used the invasive HNSCC line SCC61 cultured on fluorescent gelatin, it was apparent that invadopodia appeared localized at, or directly adjacent to, multivesicular endosomes (MVEs), as distinguished by the exosome marker, CD63 [192]. Exosomes isolated from conditioned media contained both MT1-MMP and MMP2, while knockdown of Rab27a, a critical docking factor for MVEs, decreased exosome secretion, matrix degradation, and the number of invadopodia per cell, suggesting that exosomes are critical for invadopodia biogenesis and activity. Inhibition of N-WASP with the drug Wikostatin or knockdown of Tks5 with shRNA produced a significant decrease in the number of exosomes released per cell, providing evidence that invadopodia may also be critical sites of release for exosomes [192]. The second study, which made use of the invasive LOX melanoma cell line, showed that tumor cells switch from an amoeboid-phenotype, invadopodia-independent mechanism to a mesenchymal-phenotype, invadopodia-dependent means of invasion, as matrix stiffness increases [193], consistent with other studies [82, 194]. It was further shown that melanoma cells on more flexible gelatin substrates, in addition to adopting an amoeboid shape with blebbing membrane protrusions, released microvesicles [193]. These amoeboid-like cells tunneled through the gelatin, leaving trails in their wake. Conversely, melanoma cells seeded onto firmer gelatin matrices formed invadopodia, released significantly fewer microvesicles and did not form membrane blebs [193]. Degradation in these cells was typical for invadopodia-dot like punctae in the gelatin. This study suggests that tumor cells may employ two distinct mechanisms of invasion in response to the degree of matrix rigidity, which changes in the microenvironment as metastasis progresses. It will be interesting to determine whether the tumor-released microvesicles contain proteases, and whether the cargo within the microvesicles is necessary for degradation. Can tumor cells use exosomes and microvesicles in separate ways, and in different modes of invasion? It will also be of interest to determine whether exosomes and/or microvesicles are important for podosome function.
Conclusions and Future Perspectives
How might invadopodia regulate tumor growth? It is possible that they function, through proteolysis, to create more space in the ECM for tumors to grow. Alternatively, the pericellular proteolytic activity at invadopodia could activate various growth factors to drive tumor growth. Both ideas require further investigation. Regardless, if invadopodia do function in controlling tumor growth, this opens up the intriguing possibility of using invadopodia inhibitors to treat sites of metastatic growth.
Do invadopodia function at all stages of tumor progression (Figure 2)? Available evidence suggests that they are involved in local degradation and ECM invasion, as well as in intravasation, extravasation, and in growth of metastatic tumor cells. However, it will be important to determine whether invadopodia are critical for primary tumor growth, as well as for the survival of tumor cells in the bloodstream.
Although invadopodia and podosomes share many common functions, no studies to date have investigated whether loss of podosomes affects 3D growth in normal cells. Is it possible that 3D growth is a distinct function of only invadopodia, and observable only in cancer cells? Or, does cell division accompany podosome function? Cell types derived from myeloid progenitors, including osteoclasts and macrophages, are terminally differentiated and thus cannot be used to assess this question. However, endothelial cells and vascular smooth muscle cells, which do divide, represent an ideal cell model for this analysis. It will be interesting to determine whether invadopodia and podosomes do have differences in function, or whether invadopodia are true mimics of podosomes in all respects.
Acknowledgments
EKP is supported by the National Cancer Institute of the National Institutes of Health under award number T32CA106195. Work in the Courtneidge lab in recent years has been supported by R01 CA129686, R01 CA154002 and R21 CA177382 from the NCI, and by funds from the Knight Cancer Institute. We thank Dr. Arthur Kuipers and Dr. Priyanka Saini of the Courtneidge lab for the microscopy images used in Figure 1.
Abbreviations
- ECM
extracellular matrix
- F-actin
filamentous-actin
- N-WASP
neural Wiskott-Aldrich syndrome protein
- WASP
Wiskott-Aldrich syndrome protein
- WAS
Wiskott-Aldrich syndrome
- MT1-MMP or MMP14
membrane type I matrix metalloprotease
- SH3
Src homology 3 domain
- Tks5
tyrosine kinase substrate with 5 SH3 domains
- Tks4
tyrosine kinase substrate with 4 SH3 domains
- PX
Phox homology
- MMP
matrix metalloprotease
- uPA
urokinase-type plasminogen activator
- FGF
fibroblast growth factor
- TGF-β
transforming-growth factor β
- FAs
focal adhesions
- Arp2/3
actin-related protein 2 and 3
- VEGF
vascular endothelial growth factor
- PDGF
platelet-derived growth factor
- EGF
epidermal growth factor
- ROS
reactive oxygen species
- 3D
three-dimensional
- FTHS
Frank-ter Haar syndrome
- BDCS
Borrone dermato-cardio-skeletal syndrome
- NCCs
neural crest cells
- PMA
phorbol 12-myristate 13-acetate
- RNAi
RNA interference
- MSCs
mesenchymal stromal cells
- CNF1
cytotoxic necrotizing factor 1
- NMJ
neuromuscular junction
- ACh
acetylcholine
- PRRs
pattern recognition receptors
- PAMPs
pathogen-associated molecular patterns
- APCs
antigen presenting cells
- ILPs
invadosome-like protrusions
- GFP
green fluorescent protein
- KD
knockdown
- TIMP-2
tissue inhibitor of metalloproteases 2
- Cdk5
cyclin dependent kinase 5
- HNSCC
head and neck squamous cell carcinoma
- MVEs
multivesicular endosomes
- FITC
fluorescein isothiocyanate
Footnotes
Conflict of Interest: None
AUTHOR CONTRIBUTIONS
Both authors discussed the concepts for the review and composed the manuscript. EKP made the figures with guidance from SAC.
References
- 1.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 2.Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol. 2010;26:315–33. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 4.Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200:423–8. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- 5.Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15:771–85. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–26. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Sci. 2013;126:2979–89. doi: 10.1242/jcs.079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linder S, Wiesner C. Feel the force: Podosomes in mechanosensing. Exp Cell Res. 2016;343:67–72. doi: 10.1016/j.yexcr.2015.11.026. [DOI] [PubMed] [Google Scholar]
- 10.Linder S, Wiesner C. Tools of the trade: podosomes as multipurpose organelles of monocytic cells. Cell Mol Life Sci. 2015;72:121–35. doi: 10.1007/s00018-014-1731-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veillat V, Spuul P, Daubon T, Egana I, Kramer I, Genot E. Podosomes: Multipurpose organelles? Int J Biochem Cell Biol. 2015;65:52–60. doi: 10.1016/j.biocel.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 12.Foxall E, Pipili A, Jones GE, Wells CM. Significance of kinase activity in the dynamic invadosome. Eur J Cell Biol. 2016;95:483–492. doi: 10.1016/j.ejcb.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Di Martino J, Henriet E, Ezzoukhry Z, Goetz JG, Moreau V, Saltel F. The microenvironment controls invadosome plasticity. J Cell Sci. 2016;129:1759–68. doi: 10.1242/jcs.182329. [DOI] [PubMed] [Google Scholar]
- 14.Lock P, Abram CL, Gibson T, Courtneidge SA. A new method for isolating tyrosine kinase substrates used to identify fish, an SH3 and PX domain-containing protein, and Src substrate. EMBO J. 1998;17:4346–57. doi: 10.1093/emboj/17.15.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seals DF, Azucena EF, Jr, Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, Courtneidge SA. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–65. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Burger KL, Learman BS, Boucherle AK, Sirintrapun SJ, Isom S, Diaz B, Courtneidge SA, Seals DF. Src-dependent Tks5 phosphorylation regulates invadopodia-associated invasion in prostate cancer cells. Prostate. 2014;74:134–48. doi: 10.1002/pros.22735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–51. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- 18.Pawson T, Gish GD. SH2 and SH3 domains: from structure to function. Cell. 1992;71:359–62. doi: 10.1016/0092-8674(92)90504-6. [DOI] [PubMed] [Google Scholar]
- 19.Di Martino J, Paysan L, Gest C, Lagree V, Juin A, Saltel F, Moreau V. Cdc42 and Tks5: a minimal and universal molecular signature for functional invadosomes. Cell Adh Migr. 2014;8:280–92. doi: 10.4161/cam.28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oikawa T, Itoh T, Takenawa T. Sequential signals toward podosome formation in NIH-src cells. J Cell Biol. 2008;182:157–69. doi: 10.1083/jcb.200801042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci. 2009;122:2727–40. doi: 10.1242/jcs.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cejudo-Martin P, Yuen A, Vlahovich N, Lock P, Courtneidge SA, Diaz B. Genetic disruption of the sh3pxd2a gene reveals an essential role in mouse development and the existence of a novel isoform of tks5. PLoS One. 2014;9:e107674. doi: 10.1371/journal.pone.0107674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li CM, Chen G, Dayton TL, Kim-Kiselak C, Hoersch S, Whittaker CA, Bronson RT, Beer DG, Winslow MM, Jacks T. Differential Tks5 isoform expression contributes to metastatic invasion of lung adenocarcinoma. Genes Dev. 2013;27:1557–67. doi: 10.1101/gad.222745.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blouw B, Patel M, Iizuka S, Abdullah C, You WK, Huang X, Li J-L, Diaz B, Stallcup WB, Courtneidge SA. The invadopodia scaffold protein Tks5 is required for the growth of human breast cancer cells in vitro and in vivo. PloS one. 2015;10:e0121003. doi: 10.1371/journal.pone.0121003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stylli SS, IST, Kaye AH, Lock P. Prognostic significance of Tks5 expression in gliomas. J Clin Neurosci. 2012;19:436–42. doi: 10.1016/j.jocn.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Stylli SS, Luwor RB, Kaye AH, IST, Hovens CM, Lock P. Expression of the adaptor protein Tks5 in human cancer: prognostic potential. Oncol Rep. 2014;32:989–1002. doi: 10.3892/or.2014.3310. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol. 2012;196:375–85. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–17. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Itoh Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015;44–46:207–23. doi: 10.1016/j.matbio.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Buschman MD, Bromann PA, Cejudo-Martin P, Wen F, Pass I, Courtneidge SA. The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol Biol Cell. 2009;20:1302–11. doi: 10.1091/mbc.E08-09-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varon C, Tatin F, Moreau V, Van Obberghen-Schilling E, Fernandez-Sauze S, Reuzeau E, Kramer I, Genot E. Transforming growth factor beta induces rosettes of podosomes in primary aortic endothelial cells. Mol Cell Biol. 2006;26:3582–94. doi: 10.1128/MCB.26.9.3582-3594.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe A, Hoshino D, Koshikawa N, Seiki M, Suzuki T, Ichikawa K. Critical role of transient activity of MT1-MMP for ECM degradation in invadopodia. PLoS Comput Biol. 2013;9:e1003086. doi: 10.1371/journal.pcbi.1003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams KC, McNeilly RE, Coppolino MG. SNAP23, Syntaxin4, and vesicle-associated membrane protein 7 (VAMP7) mediate trafficking of membrane type 1-matrix metalloproteinase (MT1-MMP) during invadopodium formation and tumor cell invasion. Mol Biol Cell. 2014;25:2061–70. doi: 10.1091/mbc.E13-10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iizuka S, Abdullah C, Buschman MD, Diaz B, Courtneidge SA. The role of Tks adaptor proteins in invadopodia formation, growth and metastasis of melanoma. Oncotarget. 2016 doi: 10.18632/oncotarget.12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lagoutte E, Villeneuve C, Lafanechere L, Wells CM, Jones GE, Chavrier P, Rosse C. LIMK Regulates Tumor-Cell Invasion and Matrix Degradation Through Tyrosine Phosphorylation of MT1-MMP. Sci Rep. 2016;6:24925. doi: 10.1038/srep24925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rosse C, Chavrier P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu Rev Cell Dev Biol. 2016;32:555–576. doi: 10.1146/annurev-cellbio-111315-125227. [DOI] [PubMed] [Google Scholar]
- 37.McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13:534–40. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 38.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Wehrle-Haller B, Imhof B. The inner lives of focal adhesions. Trends Cell Biol. 2002;12:382–9. doi: 10.1016/s0962-8924(02)02321-8. [DOI] [PubMed] [Google Scholar]
- 41.Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–20. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 42.Cramer LP. Molecular mechanism of actin-dependent retrograde flow in lamellipodia of motile cells. Front Biosci. 1997;2:d260–70. doi: 10.2741/a189. [DOI] [PubMed] [Google Scholar]
- 43.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–54. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 44.Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol. 2007;179:1043–57. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–26. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Machesky LM. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 2008;582:2102–11. doi: 10.1016/j.febslet.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 47.Cougoule C, Van Goethem E, Le Cabec V, Lafouresse F, Dupre L, Mehraj V, Mege JL, Lastrucci C, Maridonneau-Parini I. Blood leukocytes and macrophages of various phenotypes have distinct abilities to form podosomes and to migrate in 3D environments. Eur J Cell Biol. 2012;91:938–49. doi: 10.1016/j.ejcb.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 48.Schachtner H, Calaminus SD, Sinclair A, Monypenny J, Blundell MP, Leon C, Holyoake TL, Thrasher AJ, Michie AM, Vukovic M, Gachet C, Jones GE, Thomas SG, Watson SP, Machesky LM. Megakaryocytes assemble podosomes that degrade matrix and protrude through basement membrane. Blood. 2013;121:2542–52. doi: 10.1182/blood-2012-07-443457. [DOI] [PubMed] [Google Scholar]
- 49.Johansson MW, Lye MH, Barthel SR, Duffy AK, Annis DS, Mosher DF. Eosinophils adhere to vascular cell adhesion molecule-1 via podosomes. Am J Respir Cell Mol Biol. 2004;31:413–22. doi: 10.1165/rcmb.2004-0099OC. [DOI] [PubMed] [Google Scholar]
- 50.Gawden-Bone C, Zhou Z, King E, Prescott A, Watts C, Lucocq J. Dendritic cell podosomes are protrusive and invade the extracellular matrix using metalloproteinase MMP-14. J Cell Sci. 2010;123:1427–37. doi: 10.1242/jcs.056515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banon-Rodriguez I, Monypenny J, Ragazzini C, Franco A, Calle Y, Jones GE, Anton IM. The cortactin-binding domain of WIP is essential for podosome formation and extracellular matrix degradation by murine dendritic cells. Eur J Cell Biol. 2011;90:213–23. doi: 10.1016/j.ejcb.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Miyauchi A, Hruska KA, Greenfield EM, Duncan R, Alvarez J, Barattolo R, Colucci S, Zambonin-Zallone A, Teitelbaum SL, Teti A. Osteoclast cytosolic calcium, regulated by voltage-gated calcium channels and extracellular calcium, controls podosome assembly and bone resorption. J Cell Biol. 1990;111:2543–52. doi: 10.1083/jcb.111.6.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanehisa J, Yamanaka T, Doi S, Turksen K, Heersche JN, Aubin JE, Takeuchi H. A band of F-actin containing podosomes is involved in bone resorption by osteoclasts. Bone. 1990;11:287–93. doi: 10.1016/8756-3282(90)90082-a. [DOI] [PubMed] [Google Scholar]
- 54.Moreau V, Tatin F, Varon C, Genot E. Actin can reorganize into podosomes in aortic endothelial cells, a process controlled by Cdc42 and RhoA. Mol Cell Biol. 2003;23:6809–22. doi: 10.1128/MCB.23.19.6809-6822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson O, Kleino I, Crimaldi L, Gimona M, Saksela K, Winder SJ. Dystroglycan, Tks5 and Src mediated assembly of podosomes in myoblasts. PLoS One. 2008;3:e3638. doi: 10.1371/journal.pone.0003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao H, Eves R, Yeh C, Kan W, Xu F, Mak AS, Liu M. Phorbol ester-induced podosomes in normal human bronchial epithelial cells. J Cell Physiol. 2009;218:366–75. doi: 10.1002/jcp.21609. [DOI] [PubMed] [Google Scholar]
- 57.Santiago-Medina M, Gregus KA, Nichol RH, O’Toole SM, Gomez TM. Regulation of ECM degradation and axon guidance by growth cone invadosomes. Development. 2015;142:486–96. doi: 10.1242/dev.108266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Botta GP, Reginato MJ, Reichert M, Rustgi AK, Lelkes PI. Constitutive K-RasG12D activation of ERK2 specifically regulates 3D invasion of human pancreatic cancer cells via MMP-1. Mol Cancer Res. 2012;10:183–96. doi: 10.1158/1541-7786.MCR-11-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Semprucci E, Tocci P, Cianfrocca R, Sestito R, Caprara V, Veglione M, Castro VD, Spadaro F, Ferrandina G, Bagnato A, Rosano L. Endothelin A receptor drives invadopodia function and cell motility through the beta-arrestin/PDZ-RhoGEF pathway in ovarian carcinoma. Oncogene. 2016;35:3432–42. doi: 10.1038/onc.2015.403. [DOI] [PubMed] [Google Scholar]
- 60.Desai B, Ma T, Chellaiah MA. Invadopodia and matrix degradation, a new property of prostate cancer cells during migration and invasion. J Biol Chem. 2008;283:13856–66. doi: 10.1074/jbc.M709401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mallawaaratchy DM, Buckland ME, McDonald KL, Li CC, Ly L, Sykes EK, Christopherson RI, Kaufman KL. Membrane proteome analysis of glioblastoma cell invasion. J Neuropathol Exp Neurol. 2015;74:425–41. doi: 10.1097/NEN.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 62.Martin-Villar E, Borda-d’Agua B, Carrasco-Ramirez P, Renart J, Parsons M, Quintanilla M, Jones GE. Podoplanin mediates ECM degradation by squamous carcinoma cells through control of invadopodia stability. Oncogene. 2015;34:4531–44. doi: 10.1038/onc.2014.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tokui N, Yoneyama MS, Hatakeyama S, Yamamoto H, Koie T, Saitoh H, Yamaya K, Funyu T, Nakamura T, Ohyama C, Tsuboi S. Extravasation during bladder cancer metastasis requires cortactinmediated invadopodia formation. Mol Med Rep. 2014;9:1142–6. doi: 10.3892/mmr.2014.1965. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto H, Sutoh M, Hatakeyama S, Hashimoto Y, Yoneyama T, Koie T, Saitoh H, Yamaya K, Funyu T, Nakamura T, Ohyama C, Tsuboi S. Requirement for FBP17 in invadopodia formation by invasive bladder tumor cells. J Urol. 2011;185:1930–8. doi: 10.1016/j.juro.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 65.Sutoh M, Hashimoto Y, Yoneyama T, Yamamoto H, Hatakeyama S, Koie T, Okamoto A, Yamaya K, Saitoh H, Funyu T, Nakamura T, Sato T, Ohyama C, Tsuboi S. Invadopodia formation by bladder tumor cells. Oncol Res. 2010;19:85–92. doi: 10.3727/096504010x12875107808008. [DOI] [PubMed] [Google Scholar]
- 66.Ammer AG, Kelley LC, Hayes KE, Evans JV, Lopez-Skinner LA, Martin KH, Frederick B, Rothschild BL, Raben D, Elvin P, Green TP, Weed SA. Saracatinib Impairs Head and Neck Squamous Cell Carcinoma Invasion by Disrupting Invadopodia Function. J Cancer Sci Ther. 2009;1:52–61. doi: 10.4172/1948-5956.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Destaing O, Saltel F, Geminard JC, Jurdic P, Bard F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol Biol Cell. 2003;14:407–16. doi: 10.1091/mbc.E02-07-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Artym VV, Matsumoto K, Mueller SC, Yamada KM. Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur J Cell Biol. 2011;90:172–80. doi: 10.1016/j.ejcb.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parekh A, Ruppender NS, Branch KM, Sewell-Loftin MK, Lin J, Boyer PD, Candiello JE, Merryman WD, Guelcher SA, Weaver AM. Sensing and modulation of invadopodia across a wide range of rigidities. Biophys J. 2011;100:573–82. doi: 10.1016/j.bpj.2010.12.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Condeelis J, Singer RH, Segall JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 71.Seano G, Chiaverina G, Gagliardi PA, di Blasio L, Puliafito A, Bouvard C, Sessa R, Tarone G, Sorokin L, Helley D, Jain RK, Serini G, Bussolino F, Primo L. Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat Cell Biol. 2014;16:931–41. 1–8. doi: 10.1038/ncb3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spuul P, Daubon T, Pitter B, Alonso F, Fremaux I, Kramer I, Montanez E, Genot E. VEGF-A/Notch-Induced Podosomes Proteolyse Basement Membrane Collagen-IV during Retinal Sprouting Angiogenesis. Cell Rep. 2016;17:484–500. doi: 10.1016/j.celrep.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 73.Hanna SC, Krishnan B, Bailey ST, Moschos SJ, Kuan PF, Shimamura T, Osborne LD, Siegel MB, Duncan LM, O’Brien ET, 3rd, Superfine R, Miller CR, Simon MC, Wong KK, Kim WY. HIF1alpha and HIF2alpha independently activate SRC to promote melanoma metastases. J Clin Invest. 2013;123:2078–93. doi: 10.1172/JCI66715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rottiers P, Saltel F, Daubon T, Chaigne-Delalande B, Tridon V, Billottet C, Reuzeau E, Genot E. TGFbeta-induced endothelial podosomes mediate basement membrane collagen degradation in arterial vessels. J Cell Sci. 2009;122:4311–8. doi: 10.1242/jcs.057448. [DOI] [PubMed] [Google Scholar]
- 75.Hwang YS, Park KK, Chung WY. Invadopodia formation in oral squamous cell carcinoma: the role of epidermal growth factor receptor signalling. Arch Oral Biol. 2012;57:335–43. doi: 10.1016/j.archoralbio.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 76.Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201:279–92. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou ZN, Sharma VP, Beaty BT, Roh-Johnson M, Peterson EA, Van Rooijen N, Kenny PA, Wiley HS, Condeelis JS, Segall JE. Autocrine HBEGF expression promotes breast cancer intravasation, metastasis and macrophage-independent invasion in vivo. Oncogene. 2014;33:3784–93. doi: 10.1038/onc.2013.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Busco G, Cardone RA, Greco MR, Bellizzi A, Colella M, Antelmi E, Mancini MT, Dell’Aquila ME, Casavola V, Paradiso A, Reshkin SJ. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010;24:3903–15. doi: 10.1096/fj.09-149518. [DOI] [PubMed] [Google Scholar]
- 79.Magalhaes MA, Larson DR, Mader CC, Bravo-Cordero JJ, Gil-Henn H, Oser M, Chen X, Koleske AJ, Condeelis J. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J Cell Biol. 2011;195:903–20. doi: 10.1083/jcb.201103045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brisson L, Driffort V, Benoist L, Poet M, Counillon L, Antelmi E, Rubino R, Besson P, Labbal F, Chevalier S, Reshkin SJ, Gore J, Roger S. NaV1.5 Na(+) channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J Cell Sci. 2013;126:4835–42. doi: 10.1242/jcs.123901. [DOI] [PubMed] [Google Scholar]
- 81.Greco MR, Antelmi E, Busco G, Guerra L, Rubino R, Casavola V, Reshkin SJ, Cardone RA. Protease activity at invadopodial focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol Rep. 2014;31:940–6. doi: 10.3892/or.2013.2923. [DOI] [PubMed] [Google Scholar]
- 82.Alexander NR, Branch KM, Parekh A, Clark ES, Iwueke IC, Guelcher SA, Weaver AM. Extracellular matrix rigidity promotes invadopodia activity. Curr Biol. 2008;18:1295–9. doi: 10.1016/j.cub.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Juin A, Planus E, Guillemot F, Horakova P, Albiges-Rizo C, Genot E, Rosenbaum J, Moreau V, Saltel F. Extracellular matrix rigidity controls podosome induction in microvascular endothelial cells. Biol Cell. 2013;105:46–57. doi: 10.1111/boc.201200037. [DOI] [PubMed] [Google Scholar]
- 84.Artym VV, Swatkoski S, Matsumoto K, Campbell CB, Petrie RJ, Dimitriadis EK, Li X, Mueller SC, Bugge TH, Gucek M, Yamada KM. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J Cell Biol. 2015;208:331–50. doi: 10.1083/jcb.201405099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jerrell RJ, Parekh A. Matrix rigidity differentially regulates invadopodia activity through ROCK1 and ROCK2. Biomaterials. 2016;84:119–29. doi: 10.1016/j.biomaterials.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Collin O, Tracqui P, Stephanou A, Usson Y, Clement-Lacroix J, Planus E. Spatiotemporal dynamics of actin-rich adhesion microdomains: influence of substrate flexibility. J Cell Sci. 2006;119:1914–25. doi: 10.1242/jcs.02838. [DOI] [PubMed] [Google Scholar]
- 87.Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, Charriere GM, Maridonneau-Parini I. Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat Commun. 2014;5:5343. doi: 10.1038/ncomms6343. [DOI] [PubMed] [Google Scholar]
- 88.Lucien F, Brochu-Gaudreau K, Arsenault D, Harper K, Dubois CM. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK) PLoS One. 2011;6:e28851. doi: 10.1371/journal.pone.0028851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arsenault D, Brochu-Gaudreau K, Charbonneau M, Dubois CM. HDAC6 deacetylase activity is required for hypoxia-induced invadopodia formation and cell invasion. PLoS One. 2013;8:e55529. doi: 10.1371/journal.pone.0055529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Md Hashim NF, Nicholas NS, Dart AE, Kiriakidis S, Paleolog E, Wells CM. Hypoxia-induced invadopodia formation: a role for beta-PIX. Open Biol. 2013;3:120159. doi: 10.1098/rsob.120159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal. 2009;2:ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krouskop TA, Wheeler TM, Kallel F, Garra BS, Hall T. Elastic moduli of breast and prostate tissues under compression. Ultrason Imaging. 1998;20:260–74. doi: 10.1177/016173469802000403. [DOI] [PubMed] [Google Scholar]
- 93.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 94.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 95.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–96. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Koga T, Martin TJ, Suda T. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A. 1990;87:7260–4. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vaananen HK, Zhao H, Mulari M, Halleen JM. The cell biology of osteoclast function. J Cell Sci. 2000;113(Pt 3):377–81. doi: 10.1242/jcs.113.3.377. [DOI] [PubMed] [Google Scholar]
- 99.Oreffo RO, Teti A, Triffitt JT, Francis MJ, Carano A, Zallone AZ. Effect of vitamin A on bone resorption: evidence for direct stimulation of isolated chicken osteoclasts by retinol and retinoic acid. J Bone Miner Res. 1988;3:203–10. doi: 10.1002/jbmr.5650030213. [DOI] [PubMed] [Google Scholar]
- 100.Zambonin-Zallone A, Teti A, Carano A, Marchisio PC. The distribution of podosomes in osteoclasts cultured on bone laminae: effect of retinol. J Bone Miner Res. 1988;3:517–23. doi: 10.1002/jbmr.5650030507. [DOI] [PubMed] [Google Scholar]
- 101.Teti A, Blair HC, Schlesinger P, Grano M, Zambonin-Zallone A, Kahn AJ, Teitelbaum SL, Hruska KA. Extracellular protons acidify osteoclasts, reduce cytosolic calcium, and promote expression of cell-matrix attachment structures. J Clin Invest. 1989;84:773–80. doi: 10.1172/JCI114235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Teti A, Barattolo R, Grano M, Colucci S, Argentino L, Teitelbaum SL, Hruska KA, Santacroce G, Zambonin Zallone A. Podosome expression in osteoclasts: influence of high extracellular calcium concentration. Boll Soc Ital Biol Sper. 1989;65:1039–43. [PubMed] [Google Scholar]
- 103.Villa A, Notarangelo L, Macchi P, Mantuano E, Cavagni G, Brugnoni D, Strina D, Patrosso MC, Ramenghi U, Sacco MG, et al. X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet. 1995;9:414–7. doi: 10.1038/ng0495-414. [DOI] [PubMed] [Google Scholar]
- 104.Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285:26–43. doi: 10.1111/nyas.12049. [DOI] [PubMed] [Google Scholar]
- 105.Linder S, Nelson D, Weiss M, Aepfelbacher M. Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci U S A. 1999;96:9648–53. doi: 10.1073/pnas.96.17.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117:725–38. doi: 10.1016/j.jaci.2006.02.005. quiz 739. [DOI] [PubMed] [Google Scholar]
- 107.Charrier S, Stockholm D, Seye K, Opolon P, Taveau M, Gross DA, Bucher-Laurent S, Delenda C, Vainchenker W, Danos O, Galy A. A lentiviral vector encoding the human Wiskott-Aldrich syndrome protein corrects immune and cytoskeletal defects in WASP knockout mice. Gene Ther. 2005;12:597–606. doi: 10.1038/sj.gt.3302440. [DOI] [PubMed] [Google Scholar]
- 108.Linder S, Wintergerst U, Bender-Gotze C, Schwarz K, Pannicke U, Aepfelbacher M. Macrophages of patients with X-linked thrombocytopenia display an attenuated Wiskott-Aldrich syndrome phenotype. Immunol Cell Biol. 2003;81:130–6. doi: 10.1046/j.0818-9641.2002.01147.x. [DOI] [PubMed] [Google Scholar]
- 109.Kurosaka S, Kashina A. Cell biology of embryonic migration. Birth Defects Res C Embryo Today. 2008;84:102–22. doi: 10.1002/bdrc.20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Murphy DA, Diaz B, Bromann PA, Tsai JH, Kawakami Y, Maurer J, Stewart RA, Izpisua-Belmonte JC, Courtneidge SA. A Src-Tks5 pathway is required for neural crest cell migration during embryonic development. PLoS One. 2011;6:e22499. doi: 10.1371/journal.pone.0022499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Williams AL, Bohnsack BL. Neural crest derivatives in ocular development: discerning the eye of the storm. Birth Defects Res C Embryo Today. 2015;105:87–95. doi: 10.1002/bdrc.21095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sauka-Spengler T, Bronner-Fraser M. Development and evolution of the migratory neural crest: a gene regulatory perspective. Curr Opin Genet Dev. 2006;16:360–6. doi: 10.1016/j.gde.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 113.Iqbal Z, Cejudo-Martin P, de Brouwer A, van der Zwaag B, Ruiz-Lozano P, Scimia MC, Lindsey JD, Weinreb R, Albrecht B, Megarbane A, Alanay Y, Ben-Neriah Z, Amenduni M, Artuso R, Veltman JA, van Beusekom E, Oudakker A, Millan JL, Hennekam R, Hamel B, Courtneidge SA, van Bokhoven H. Disruption of the podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of Frank-Ter Haar Syndrome. Am J Hum Genet. 2010;86:254–61. doi: 10.1016/j.ajhg.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maas SM, Kayserili H, Lam J, Apak MY, Hennekam RC. Further delineation of Frank-ter Haar syndrome. Am J Med Genet A. 2004;131:127–33. doi: 10.1002/ajmg.a.30244. [DOI] [PubMed] [Google Scholar]
- 115.Wilson GR, Sunley J, Smith KR, Pope K, Bromhead CJ, Fitzpatrick E, Di Rocco M, van Steensel M, Coman DJ, Leventer RJ, Delatycki MB, Amor DJ, Bahlo M, Lockhart PJ. Mutations in SH3PXD2B cause Borrone dermato-cardio-skeletal syndrome. Eur J Hum Genet. 2014;22:741–7. doi: 10.1038/ejhg.2013.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Borrone C, Di Rocco M, Crovato F, Camera G, Gambini C. New multisystemic disorder involving heart valves, skin, bones, and joints in two brothers. Am J Med Genet. 1993;46:228–34. doi: 10.1002/ajmg.1320460225. [DOI] [PubMed] [Google Scholar]
- 117.van Steensel MA, Ceulen RP, Delhaas T, de Die-Smulders C. Two Dutch brothers with Borrone dermato-cardio-skeletal syndrome. Am J Med Genet A. 2007;143A:1223–6. doi: 10.1002/ajmg.a.31719. [DOI] [PubMed] [Google Scholar]
- 118.Mao M, Thedens DR, Chang B, Harris BS, Zheng QY, Johnson KR, Donahue LR, Anderson MG. The podosomal-adaptor protein SH3PXD2B is essential for normal postnatal development. Mamm Genome. 2009;20:462–75. doi: 10.1007/s00335-009-9210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mao M, Hedberg-Buenz A, Koehn D, John SW, Anderson MG. Anterior segment dysgenesis and early-onset glaucoma in nee mice with mutation of Sh3pxd2b. Invest Ophthalmol Vis Sci. 2011;52:2679–88. doi: 10.1167/iovs.10-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gianni D, Taulet N, DerMardirossian C, Bokoch GM. c-Src-mediated phosphorylation of NoxA1 and Tks4 induces the reactive oxygen species (ROS)-dependent formation of functional invadopodia in human colon cancer cells. Mol Biol Cell. 2010;21:4287–98. doi: 10.1091/mbc.E10-08-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dulk M, Kudlik G, Fekete A, Ernszt D, Kvell K, Pongracz JE, Mero BL, Szeder B, Radnai L, Geiszt M, Csecsy DE, Kovacs T, Uher F, Lanyi A, Vas V, Buday L. The scaffold protein Tks4 is required for the differentiation of mesenchymal stromal cells (MSCs) into adipogenic and osteogenic lineages. Sci Rep. 2016;6:34280. doi: 10.1038/srep34280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Etchevers HC, Vincent C, Le Douarin NM, Couly GF. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 2001;128:1059–68. doi: 10.1242/dev.128.7.1059. [DOI] [PubMed] [Google Scholar]
- 123.Gage PJ, Rhoades W, Prucka SK, Hjalt T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005;46:4200–8. doi: 10.1167/iovs.05-0691. [DOI] [PubMed] [Google Scholar]
- 124.Trost A, Schroedl F, Lange S, Rivera FJ, Tempfer H, Korntner S, Stolt CC, Wegner M, Bogner B, Kaser-Eichberger A, Krefft K, Runge C, Aigner L, Reitsamer HA. Neural crest origin of retinal and choroidal pericytes. Invest Ophthalmol Vis Sci. 2013;54:7910–21. doi: 10.1167/iovs.13-12946. [DOI] [PubMed] [Google Scholar]
- 125.Zhou S, Webb BA, Eves R, Mak AS. Effects of tyrosine phosphorylation of cortactin on podosome formation in A7r5 vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;290:C463–71. doi: 10.1152/ajpcell.00350.2005. [DOI] [PubMed] [Google Scholar]
- 126.Furmaniak-Kazmierczak E, Crawley SW, Carter RL, Maurice DH, Cote GP. Formation of extracellular matrix-digesting invadopodia by primary aortic smooth muscle cells. Circ Res. 2007;100:1328–36. doi: 10.1161/CIRCRESAHA.106.147744. [DOI] [PubMed] [Google Scholar]
- 127.Quintavalle M, Elia L, Condorelli G, Courtneidge SA. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J Cell Biol. 2010;189:13–22. doi: 10.1083/jcb.200912096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61. [PubMed] [Google Scholar]
- 129.Webb BA, Eves R, Crawley SW, Zhou S, Cote GP, Mak AS. PAK1 induces podosome formation in A7r5 vascular smooth muscle cells in a PAK-interacting exchange factor-dependent manner. Am J Physiol Cell Physiol. 2005;289:C898–907. doi: 10.1152/ajpcell.00095.2005. [DOI] [PubMed] [Google Scholar]
- 130.Kaverina I, Stradal TE, Gimona M. Podosome formation in cultured A7r5 vascular smooth muscle cells requires Arp2/3-dependent de-novo actin polymerization at discrete microdomains. J Cell Sci. 2003;116:4915–24. doi: 10.1242/jcs.00818. [DOI] [PubMed] [Google Scholar]
- 131.Eves R, Webb BA, Zhou S, Mak AS. Caldesmon is an integral component of podosomes in smooth muscle cells. J Cell Sci. 2006;119:1691–702. doi: 10.1242/jcs.02881. [DOI] [PubMed] [Google Scholar]
- 132.Gu Z, Kordowska J, Williams GL, Wang CL, Hai CM. Erk1/2 MAPK and caldesmon differentially regulate podosome dynamics in A7r5 vascular smooth muscle cells. Exp Cell Res. 2007;313:849–66. doi: 10.1016/j.yexcr.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dorfleutner A, Cho Y, Vincent D, Cunnick J, Lin H, Weed SA, Stehlik C, Flynn DC. Phosphorylation of AFAP-110 affects podosome lifespan in A7r5 cells. J Cell Sci. 2008;121:2394–405. doi: 10.1242/jcs.026187. [DOI] [PubMed] [Google Scholar]
- 134.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 135.Proszynski TJ, Gingras J, Valdez G, Krzewski K, Sanes JR. Podosomes are present in a postsynaptic apparatus and participate in its maturation. Proc Natl Acad Sci U S A. 2009;106:18373–8. doi: 10.1073/pnas.0910391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–4. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- 137.Oser M, Dovas A, Cox D, Condeelis J. Nck1 and Grb2 localization patterns can distinguish invadopodia from podosomes. Eur J Cell Biol. 2011;90:181–8. doi: 10.1016/j.ejcb.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jacobson C, Cote PD, Rossi SG, Rotundo RL, Carbonetto S. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J Cell Biol. 2001;152:435–50. doi: 10.1083/jcb.152.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dempsey PW, Vaidya SA, Cheng G. The art of war: Innate and adaptive immune responses. Cell Mol Life Sci. 2003;60:2604–21. doi: 10.1007/s00018-003-3180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Burns S, Hardy SJ, Buddle J, Yong KL, Jones GE, Thrasher AJ. Maturation of DC is associated with changes in motile characteristics and adherence. Cell Motil Cytoskeleton. 2004;57:118–32. doi: 10.1002/cm.10163. [DOI] [PubMed] [Google Scholar]
- 141.Dehring DA, Clarke F, Ricart BG, Huang Y, Gomez TS, Williamson EK, Hammer DA, Billadeau DD, Argon Y, Burkhardt JK. Hematopoietic lineage cell-specific protein 1 functions in concert with the Wiskott-Aldrich syndrome protein to promote podosome array organization and chemotaxis in dendritic cells. J Immunol. 2011;186:4805–18. doi: 10.4049/jimmunol.1003102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van Helden SF, Krooshoop DJ, Broers KC, Raymakers RA, Figdor CG, van Leeuwen FN. A critical role for prostaglandin E2 in podosome dissolution and induction of high-speed migration during dendritic cell maturation. J Immunol. 2006;177:1567–74. doi: 10.4049/jimmunol.177.3.1567. [DOI] [PubMed] [Google Scholar]
- 143.Olivier A, Jeanson-Leh L, Bouma G, Compagno D, Blondeau J, Seye K, Charrier S, Burns S, Thrasher AJ, Danos O, Vainchenker W, Galy A. A partial down-regulation of WASP is sufficient to inhibit podosome formation in dendritic cells. Mol Ther. 2006;13:729–37. doi: 10.1016/j.ymthe.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 144.Baranov MV, Ter Beest M, Reinieren-Beeren I, Cambi A, Figdor CG, van den Bogaart G. Podosomes of dendritic cells facilitate antigen sampling. J Cell Sci. 2014;127:1052–64. doi: 10.1242/jcs.141226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Savage CO, Brooks CJ, Harcourt GC, Picard JK, King W, Sansom DM, Willcox N. Human vascular endothelial cells process and present autoantigen to human T cell lines. Int Immunol. 1995;7:471–9. doi: 10.1093/intimm/7.3.471. [DOI] [PubMed] [Google Scholar]
- 146.Sage PT, Varghese LM, Martinelli R, Sciuto TE, Kamei M, Dvorak AM, Springer TA, Sharpe AH, Carman CV. Antigen recognition is facilitated by invadosome-like protrusions formed by memory/effector T cells. J Immunol. 2012;188:3686–99. doi: 10.4049/jimmunol.1102594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bell GI, Dembo M, Bongrand P. Cell adhesion. Competition between nonspecific repulsion and specific bonding. Biophys J. 1984;45:1051–64. doi: 10.1016/S0006-3495(84)84252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen EH, Olson EN. Unveiling the mechanisms of cell-cell fusion. Science. 2005;308:369–73. doi: 10.1126/science.1104799. [DOI] [PubMed] [Google Scholar]
- 149.Primakoff P, Myles DG. Cell-cell membrane fusion during mammalian fertilization. FEBS Lett. 2007;581:2174–80. doi: 10.1016/j.febslet.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 150.Ishii M, Saeki Y. Osteoclast cell fusion: mechanisms and molecules. Mod Rheumatol. 2008;18:220–7. doi: 10.1007/s10165-008-0051-2. [DOI] [PubMed] [Google Scholar]
- 151.Abmayr SM, Pavlath GK. Myoblast fusion: lessons from flies and mice. Development. 2012;139:641–56. doi: 10.1242/dev.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bar-Shavit Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem. 2007;102:1130–9. doi: 10.1002/jcb.21553. [DOI] [PubMed] [Google Scholar]
- 153.Oikawa T, Oyama M, Kozuka-Hata H, Uehara S, Udagawa N, Saya H, Matsuo K. Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J Cell Biol. 2012;197:553–68. doi: 10.1083/jcb.201111116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bastida-Ruiz D, Van Hoesen K, Cohen M. The Dark Side of Cell Fusion. Int J Mol Sci. 2016:17. doi: 10.3390/ijms17050638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dittmar T, Nagler C, Niggemann B, Zanker KS. The dark side of stem cells: triggering cancer progression by cell fusion. Curr Mol Med. 2013;13:735–50. doi: 10.2174/1566524011313050005. [DOI] [PubMed] [Google Scholar]
- 156.Lu X, Kang Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009;69:8536–9. doi: 10.1158/0008-5472.CAN-09-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goldenberg DM. Horizontal transmission of malignancy by cell-cell fusion. Expert Opin Biol Ther. 2012;12(Suppl 1):S133–9. doi: 10.1517/14712598.2012.671807. [DOI] [PubMed] [Google Scholar]
- 158.Powell AE, Anderson EC, Davies PS, Silk AD, Pelz C, Impey S, Wong MH. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011;71:1497–505. doi: 10.1158/0008-5472.CAN-10-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–18. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nature Clinical Practice Oncology. 2008;5:206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chiang SP, Cabrera RM, Segall JE. Tumor cell intravasation. Am J Physiol Cell Physiol. 2016;311:C1–C14. doi: 10.1152/ajpcell.00238.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–62. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 163.Juncker-Jensen A, Deryugina EI, Rimann I, Zajac E, Kupriyanova TA, Engelholm LH, Quigley JP. Tumor MMP-1 activates endothelial PAR1 to facilitate vascular intravasation and metastatic dissemination. Cancer Res. 2013;73:4196–211. doi: 10.1158/0008-5472.CAN-12-4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Padrick SB, Rosen MK. Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem. 2010;79:707–35. doi: 10.1146/annurev.biochem.77.060407.135452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lorenz M, Yamaguchi H, Wang Y, Singer RH, Condeelis J. Imaging sites of N-wasp activity in lamellipodia and invadopodia of carcinoma cells. Curr Biol. 2004;14:697–703. doi: 10.1016/j.cub.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 166.Zhang X, Moore SW, Iskratsch T, Sheetz MP. N-WASP-directed actin polymerization activates Cas phosphorylation and lamellipodium spreading. J Cell Sci. 2014;127:1394–405. doi: 10.1242/jcs.134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125:724–34. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gligorijevic B, Bergman A, Condeelis J. Multiparametric classification links tumor microenvironments with tumor cell phenotype. PLoS Biol. 2014;12:e1001995. doi: 10.1371/journal.pbio.1001995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci. 2010;123:2332–41. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Azevedo AS, Follain G, Patthabhiraman S, Harlepp S, Goetz JG. Metastasis of circulating tumor cells: favorable soil or suitable biomechanics, or both? Cell Adh Migr. 2015;9:345–56. doi: 10.1080/19336918.2015.1059563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, Postenka CO, Turley EA, Courtneidge SA, Chambers AF, Lewis JD. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014;8:1558–70. doi: 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 172.Blouw B, Seals DF, Pass I, Diaz B, Courtneidge SA. A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. Eur J Cell Biol. 2008;87:555–67. doi: 10.1016/j.ejcb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 174.Kirkbride KC, Sung BH, Sinha S, Weaver AM. Cortactin: a multifunctional regulator of cellular invasiveness. Cell adhesion & migration. 2011;5:187–198. doi: 10.4161/cam.5.2.14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol. 2000;151:29–40. doi: 10.1083/jcb.151.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.He Y, Ren Y, Wu B, Decourt B, Lee AC, Taylor A, Suter DM. Src and cortactin promote lamellipodia protrusion and filopodia formation and stability in growth cones. Mol Biol Cell. 2015;26:3229–44. doi: 10.1091/mbc.E15-03-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Clark ES, Brown B, Whigham AS, Kochaishvili A, Yarbrough WG, Weaver AM. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene. 2009;28:431–444. doi: 10.1038/onc.2008.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shupp A, Casimiro MC, Pestell RG. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget. 2017 doi: 10.18632/oncotarget.14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Quintavalle M, Elia L, Price JH, Heynen-Genel S, Courtneidge SA. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci Signal. 2011;4:ra49. doi: 10.1126/scisignal.2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hsu FN, Chen MC, Chiang MC, Lin E, Lee YT, Huang PH, Lee GS, Lin H. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J Biol Chem. 2011;286:33141–9. doi: 10.1074/jbc.M111.252080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW, Goggins M, Maitra A, Nelkin BD. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70:4460–9. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125–34. doi: 10.1093/hmg/dds317. [DOI] [PubMed] [Google Scholar]
- 184.Lawson C, Vicencio JM, Yellon DM, Davidson SM. Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol. 2016;228:R57–71. doi: 10.1530/JOE-15-0201. [DOI] [PubMed] [Google Scholar]
- 185.Hsu SC, TerBush D, Abraham M, Guo W. The exocyst complex in polarized exocytosis. Int Rev Cytol. 2004;233:243–65. doi: 10.1016/S0074-7696(04)33006-8. [DOI] [PubMed] [Google Scholar]
- 186.Sakurai-Yageta M, Recchi C, Le Dez G, Sibarita JB, Daviet L, Camonis J, D’Souza-Schorey C, Chavrier P. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J Cell Biol. 2008;181:985–98. doi: 10.1083/jcb.200709076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Liu J, Yue P, Artym VV, Mueller SC, Guo W. The role of the exocyst in matrix metalloproteinase secretion and actin dynamics during tumor cell invadopodia formation. Mol Biol Cell. 2009;20:3763–71. doi: 10.1091/mbc.E08-09-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Steffen A, Le Dez G, Poincloux R, Recchi C, Nassoy P, Rottner K, Galli T, Chavrier P. MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr Biol. 2008;18:926–31. doi: 10.1016/j.cub.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 189.Clancy JW, Sedgwick A, Rosse C, Muralidharan-Chari V, Raposo G, Method M, Chavrier P, D’Souza-Schorey C. Regulated delivery of molecular cargo to invasive tumour-derived microvesicles. Nat Commun. 2015;6:6919. doi: 10.1038/ncomms7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem. 2008;105:1211–8. doi: 10.1002/jcb.21923. [DOI] [PubMed] [Google Scholar]
- 191.Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D’Souza-Schorey C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol. 2009;19:1875–85. doi: 10.1016/j.cub.2009.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hoshino D, Kirkbride KC, Costello K, Clark ES, Sinha S, Grega-Larson N, Tyska MJ, Weaver AM. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013;5:1159–68. doi: 10.1016/j.celrep.2013.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sedgwick AE, Clancy JW, Olivia Balmert M, D’Souza-Schorey C. Extracellular microvesicles and invadopodia mediate non-overlapping modes of tumor cell invasion. Sci Rep. 2015;5:14748. doi: 10.1038/srep14748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Aung A, Seo YN, Lu S, Wang Y, Jamora C, del Alamo JC, Varghese S. 3D traction stresses activate protease-dependent invasion of cancer cells. Biophys J. 2014;107:2528–37. doi: 10.1016/j.bpj.2014.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]